 Peter Meinke
Peter Meinke Stefan Hintze
Stefan Hintze Sarah Limmer
Sarah Limmer Benedikt Schoser
Benedikt Schoser
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 25 July 2018
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 9 - 2018 | https://doi.org/10.3389/fneur.2018.00601
This article is part of the Research TopicBeyond Borders: Myotonic Dystrophies – A European PerceptionView all 11 articles
Myotonic dystrophies (DM) are slowly progressing multisystemic disorders caused by repeat expansions in the DMPK or CNBP genes. The multisystemic involvement in DM patients often reflects the appearance of accelerated aging. This is partly due to visible features such as cataracts, muscle weakness, and frontal baldness, but there are also less obvious features like cardiac arrhythmia, diabetes or hypogammaglobulinemia. These aging features suggest the hypothesis that DM could be a segmental progeroid disease. To identify the molecular cause of this characteristic appearance of accelerated aging we compare clinical features of DM to “typical” segmental progeroid disorders caused by mutations in DNA repair or nuclear envelope proteins. Furthermore, we characterize if this premature aging effect is also reflected on the cellular level in DM and investigate overlaps with “classical” progeroid disorders. To investigate the molecular similarities at the cellular level we use primary DM and control cell lines. This analysis reveals many similarities to progeroid syndromes linked to the nuclear envelope. Our comparison on both clinical and molecular levels argues for qualification of DM as a segmental progeroid disorder.
Myotonic dystrophies (DM) are slowly progressing multisystemic disorders characterized by myotonia, muscle weakness, cataracts, and cardiac arrhythmia that can evolve into cardiomyopathy, insulin insensitivity and diabetes, testicular failure, and hypogammaglobulinemia (1, 2). The spectrum of DMs includes two types: type 1 (DM1) and type 2 (DM2) which are caused by mutations in two different genes. The age of onset of DMs ranges from congenital forms at birth to late onset at ~70 years. Clinical symptoms cover muscular weakness, cataracts, balding, skin changes, and diabetes mellitus, often mirroring the appearance of accelerated aging. While the pathomechanism of DM has been shown to be a general splicing defect, it remains unclear what genes and splice variants yield particular pathologies in the affected tissues. Here, we provide a clinical description of aging symptoms in DM and compare this to “typical” progeroid disorders which mimic physiological aging and are caused by mutations in nuclear envelope (NE) proteins or DNA-repair proteins. Furthermore, we directly investigate some molecular hallmarks of aging in primary cell lines of DM patients.
Myotonic dystrophy (DM) can be caused by mutations in two genes: DMPK and CNBP. In both cases the disease is caused by an expansion of repeat elements within non-coding regions of the genes. Those repeats are transcribed and therefore contained within the pre-spliced mRNA. It is thought that RNA containing the expanded repeat forms hairpin structures and accumulates in foci in the nucleus. Several RNA-binding proteins are then recruited to these foci where they interact strongly with the mutant RNA. Among these proteins are MBNL proteins which are involved in alternative splicing (3). The accumulation of these proteins in the mRNA foci is thought to result in their depletion from the rest of the nucleus, resulting in turn in general mis-splicing and toxicity. This mis-splicing tends to revert the splicing pattern to comprise many embryonic splice variants. Phenotypically myotonic dystrophy is an autosomal dominant disease with predominant myotonia and muscle wasting. Furthermore eyes, heart, bone, skin, the endocrine system, gastrointestinal organs, and the central as well as the peripheral nervous system can be affected. The repeat length can vary between different tissues significantly (4). This complicates a prediction of the clinical development of patients as the repeat length is usually measured in DNA gained from blood.
DM1 causing mutations are an expansion of a CTG repeat in the 3' UTR of the DMPK gene (5). The general tendency is that the longer the expanded repeat the more severe the resulting phenotype is. Anticipation is commonly observed; the number of repeats typically increases in offspring over their parents so that the heritable disease tends to be of increasing generational severity. Up to 35 CTG triplets are considered normal, a repeat length between 35 and 49 is considered to be a premutation. Between 50 and ~150 repeats have been observed in a mild expression of the phenotype and ~100 to 1000 CTG repeats were identified in patients with classical DM. Repeats consisting of more than 1,000 CTG-triplets result in congenital DM, the most severe expression of the disease. The sexual inheritance also affects the severity of the disease: maternal inheritance results in more severe clinical features than paternal inheritance (6, 7).
Milder DM phenotypes can encompass cataracts, mild myotonia, or diabetes mellitus only, and the age of onset ranges between 20 and 70 years (8). Additional symptoms described in classical DM1 include distal muscle weakness, fatigue, cardiac conduction defects, neuropathy, endocrinopathies (on top of diabetes mellitus), and alopecia. Age of onset for the classical phenotype ranges between 10 and 30 years. In congenital DM1 affected children suffer severe and generalized weakness, hypotonia and respiratory problems after birth. One study further found that DM1 patients may have an increased risk of skin cancer (9).
DM2 mutations are located within intron 1 of the CNBP gene: more than 75 CCTG repeats have been described as disease causing (10). Unlike DM1 there is no described correlation between repeat length and disease severity in DM2. DM2 is considered a clinically more benign disorder than DM1 (11) and can be distinguished by a proximal muscular dystrophy and sparing of facial muscles (11), and the lack of a congenital form or the severe central nervous system involvement observed in DM1 (10, 12).
The fact that DM1 and DM2 are not clinically identical indicates that there are additional factors contributing to the disease pathomechanism besides the sequestration of splicing factors. In DM2 the repeat expansions tend to be longer than in DM1 so that one would expect DM2 to be more severe, but the opposite is the case: DM2 is clinically more benign. Thus it is possible that apart from the RNA toxicity the respective gene loci are contributing in different ways to the phenotype.
Due to its multisystemic involvement DM was suggested decades ago to be a segmental progeroid syndrome (13). Later it was also proposed as a model for premature muscle aging (14) and it is possible that in some mild cases DM might mimic sarcopenia (15, 16). Skin abnormalities frequently observed in DM1 and DM2 are also regarded as indicators for premature aging (17).
A group of progeroid disorders is caused by mutations in proteins of the nuclear envelope (NE) and also proteins involved in their processing. The NE is a double membrane system enclosing the genome in eukaryotic cells (18). Nuclear envelope transmembrane proteins (NETs) reside within the NE and it is underlaid by a meshwork of intermediate filament proteins, the nuclear lamina (19, 20). NE proteins lost or mutated in progeroid syndromes include lamin A and BAF. Apart from progeroid syndromes mutations in the lamin A encoding gene LMNA cause several tissue specific diseases (including muscular dystrophy, neuropathy and lipodystrophy). The progeroid syndromes caused by LMNA mutations encompass Hutchinson-Gilford progeria syndrome (HGPS), mandibuloacral dysplasia (MAD), Malouf syndrome, and several atypical progeroid syndromes that cannot be assigned clearly. Lamin A is an intermediate filament protein which undergoes post-translational processing for farnesylation. It has functions in mechanical stability, higher-order genome organization, chromatin regulation, transcription, DNA replication, and DNA repair (21). The major protein involved in its post-translational processing is the zinc metalloprotease STE24, encoded by the ZMPSTE24 gene. Mutations in the ZMPSTE24 gene cause mandibuloacral dysplasia (MAD) and restrictive dermopathy (RD). BAF (barrier to autointegration factor), a DNA binding protein, is encoded by the BANF1 gene. Its functions include chromatin remodeling, gene expression, and DNA damage repair (22). It has been shown to interact with the LEM domain containing NETs emerin, MAN1 and Lap2β as well as with lamin A (23–25).
Hutchinson-Gilford progeria syndrome (HGPS) is most commonly caused by the de novo heterozygous LMNA mutation c.1824C>T; p.G608G which activates a cryptic splice site and causes the deletion of 50 amino acids. This deletion includes the cleavage site necessary for maturation of lamin A by post-translational processing (26, 27). Affected individuals appear healthy at birth, but develop a progeroid phenotype within 1–2 years. This comprises a short stature, low body weight, early loss of hair, loss of subcutaneous fat, localized scleroderma-like skin conditions, osteolysis, and facial features resembling aging (small face and jaw, prominent eyes, pinched nose, thin lips, and protruding ears). In most cases cardiovascular problems are the reason for death in the second decade of life (28, 29).
Mandibuloacral dysplasia (MAD) can be caused by recessive mutations in LMNA [MADA, (30)] or ZMPSTE24 [MADB, (31)]. While the LMNA mutations tend to be homozygous or compound heterozygous missense mutations, ZMPSTE24 mutations resulting in MADB tend to be a combination of missense and nonsense mutations (32). Patients are characterized by postnatal growth retardation, craniofacial anomalies with mandibular hypoplasia, skeletal malformations, osteolysis of distal phalanges, and clavicles, skin changes such as atrophy, and speckled hyperpigmentation, insulin resistance, and diabetes, and lipodystrophy which appears to be partial in MADA and generalized in MADB (32).
Restrictive dermopathy (RD) is caused by homozygous or compound heterozygous nonsense ZMPSTE24 mutations resulting in a loss of the protein (33). The term RD describes a rare, lethal, genodermatosis. Affected children die before birth or within the first week of life. Clinical features include tightly adherent thin skin, prominent vessels, characteristic facial features (“O” shaped mouth), generalized joint contractures, dysplasia of clavicles and respiratory insufficiency (32, 34).
Malouf syndrome is caused by heterozygous LMNA mutations within the N-terminal parts of lamin A. In 2003 mutations were identified in patients originally described as suffering from Werner syndrome, but with no mutation in the RECQL2 gene and therefore named atypical Werner syndrome (35)–though if the clinical phenotypes were actually matching Werner syndrome was not absolutely clear (36, 37). Later work (38) noted the phenotypic similarity to patients described by Malouf et al. (39). Described clinical findings include hypergonadotropic hypogonadism, cardiomyopathy, blepharoptosis, mild mental retardation, prominent nasal bones, scleroderma-like skin, and lipodystrophy (35, 38, 39).
In addition to these progeroid disorders there are several cases of so called atypical progeroid syndromes caused by LMNA mutations. These cases are often linked to a specific mutation and show overlaps between well described LMNA or ZMPSTE24 linked diseases. Therefore it's not possible to assign them clearly to a syndrome (40–44).
Nestor-Guillermo progeria syndrome (NGPS) is caused by recessive mutations in the BANF1 gene (45). Patients start to develop a failure to thrive at age of 2, the skin becomes dry and atrophic and they develop a generalized lipoatrophy, osteoporosis, and osteolysis.
Amongst these several NE-linked progeroid syndromes, the age of onset and life expectancy vary, but similarities include skin abnormalities (scleroderma-like, atrophy or speckled hyperpigmentation), osteolysis/osteoporosis, loss of hair, cardiac involvement, insulin resistance, typical facies, and in some cases muscular weakness. No noteworthy increased risk of cancer amongst these disorders has been reported (Table 1).
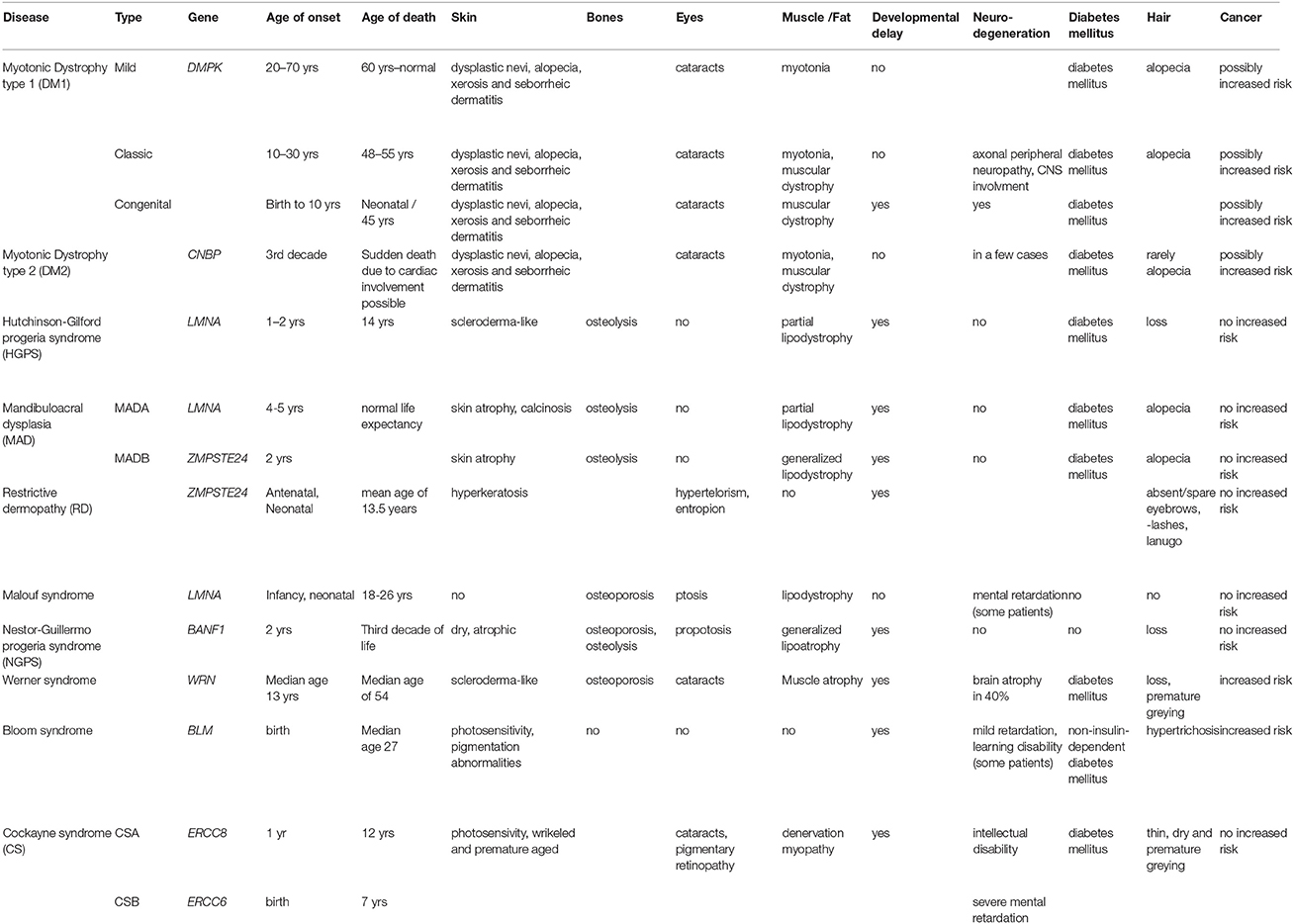
Table 1. Overview comparing myotonic dystrophies with selected nuclear envelope- and DNA repair- linked progeroid syndromes.
Another group of progeroid diseases are caused by mutations in DNA-repair proteins. Those encompass mutations in RecQ protein-like helicases (RECQL) and nuclear excision repair (NER) proteins. RecQ helicases play major roles in genome maintenance and stability (46). Mutations in genes encoding members of this protein family are causative for the premature aging disorders Werner syndrome and Bloom syndrome. NER proteins repair single stranded DNA damage—particular UV-induced DNA damage. Progeroid syndromes caused by mutations in NER protein encoding genes include Cockayne syndrome, Xeroderma pigmentosum, and Trichothiodystrophy.
Werner syndrome is caused by recessive mutations in the RECQL2 protein encoding WRN gene that result in a loss of protein by creating new stop codons or cause frameshifts resulting in a premature stop codon (47). RECQL2 is involved in DNA double-strand break repair where it regulates the pathway choice between classical and alternative non-homologous end joining (48) and relocalizes from the nucleolus to other nuclear regions upon DNA damage (49). It is suggested to be involved in telomere replication (50). Werner syndrome patients have scleroderma-like skin changes, cataracts, osteoporosis, arteriosclerosis, diabetes mellitus, cancer, characteristic “birdlike” facies, and can have alopecia (51).
Mutations in the BLM gene, encoding RECQL3, cause Bloom syndrome. The inheritance is recessive, and mutations result in a loss of protein or loss of function (52, 53). RECQL3 is involved in DNA replication and repair, where it acts in several steps during homologous recombination during DNA double-strand break repair (54). Patients present with pre- and postnatal growth deficiency, UV-sensitivity, hypo-, and hyperpigmented skin, and predisposition to malignancy (55).
Cockayne syndrome (CS) is distinguished into type A [CSA, caused by recessive ERCC8 mutations (56)] and type B [CSB, caused by recessive ERCC6 mutations (57)]. CSA patients show a progeroid appearance with slow growth and development, skin photosensitivity, thin and dry hair, pigmentary retinopathy, sensorineural hearing loss and dental caries (58). CSB patients are characterized by failure to thrive, severe mental retardation, congenital cataracts, loss of adipose tissue, joint contractures, distinctive face with small, deep-set eyes, and prominent nasal bridge, kyphosis, sensorineural hearing loss, and cachectic dwarfism (59).
Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder with patients showing acute photosensitivity and a predisposition to skin cancer on sun-exposed areas of the body (60). XP is caused by mutations in the XPA, ERCC3, XPC, ERCC2, DDB2, ERCC4, ERCC5, and POLH genes. Another NER protein associated disease is Trichothiodystrophy (TTD). Patients display a wide variety of clinical features which includes cutaneous, neurologic and growth abnormalities as well as intellectual/developmental disabilities, ocular abnormalities and decreased fertility (61). Causative mutations have been described in the ERCC3, GTF2H5, MPLKIP, GTF2E2, ERCC2, and RNF113A genes.
In general these DNA repair linked progeroid disorders exhibit a frequent involvement of the skin (scleroderma-like, hyperpigmentation, increased photosensitivity), osteoporosis, and cataracts occur, and there is also frequently neuronal involvement. In addition this group of disorders tends to have an increased risk of cancer (Table 1).
Aging related defects can be observed at the cellular level in cells from patients with premature-aging disorders. Cellular hallmarks of aging include senescence, telomere attrition, genomic instability, mitochondrial dysfunction, and loss of proteostasis (62). There are observations of premature senescence in DM cells — cells obtained from distal muscle of congenital DM1 patients show a reduced proliferative capacity and an increased rate of telomere shortening. The reduced proliferative capacity observed in these cells was thought to be caused by a p16 dependent premature senescence (63, 64). DM2 myoblasts have also been shown to reach premature senescence, but in a p16 independent manner (65). Congenital DM1, but not DM2 myoblasts show differentiation defects (66, 67). There are also changes in epigenetic marks in both DM1 and DM2 patient cells, suggesting possible changes to genome organization: DM2 myoblasts exhibit heterochromatin accumulation (68) and the DM1 locus is methylated to varying degrees across the expanded repeats (69) and especially in congenital samples (70).
Another aging-associated feature is mitochondrial dysfunction. It is proposed that mitochondrial free radicals cause oxidative damage which is a driving force in cellular aging (71). Mutations in mitochondrial DNA (mtDNA) can lead to premature aging (72, 73). Increased mtDNA deletions have been reported in DM (74). Furthermore mis-regulation of the mitochondrial protein CoQ10 has been described in DM in generell (75) and EFTu, HSP60, GRP75 as well as Dienoyl-CoA-Isomerase specifically in DM2 (76). Another aging-linked feature is the loss of proteostasis. Proteostasis has been shown to collapse during aging (77) and there are indications that this occurs in DM1 and DM2: CTG repeat expressing mice activate the ubiquitin-proteasome pathway (78) and altered protein degradation has been shown in DM2 myotubes (76).
DNA repair failure is certainly involved in the expansion of the repeats in both DM1 and DM2; this is potentially caused by slippage of DNA polymerase (79, 80). NER has also been shown to promote repeat expansion (81) and a polymorphism the MSH3 mismatch repair gene has been associated with somatic repeat instability (82). However, there are also reports indicating NE abnormalities in DM. In DM1 derived fibroblasts an altered localization of lamin A, lamin B1, and the NET emerin have been described (83). This altered localization includes distribution to invaginations of the NE, also known as nucleoplasmic reticuli (84). Knockdown of the zinc metalloprotease STE24, which is mutated in the progeroid syndromes MADB and RD and is a major player in the processing of prelamin A to mature lamin A, results in an enrichment of nucleoplasmic reticuli (85).
Primary human myoblast were obtained from the Muscle Tissue Culture Collection (MTCC) at the Friedrich-Baur-Institute (Department of Neurology, Ludwig-Maximilians-University, Munich, Germany). All control and patient materials were obtained with written informed consent of the donor. Ethical approval for this study was obtained from the ethical review committee at the Ludwig-Maximilians-University, Munich, Germany (reference 45-14). Repeat length was diagnosed on DNA extracted from blood. Age and sex of patients and controls are listed in Table 2.
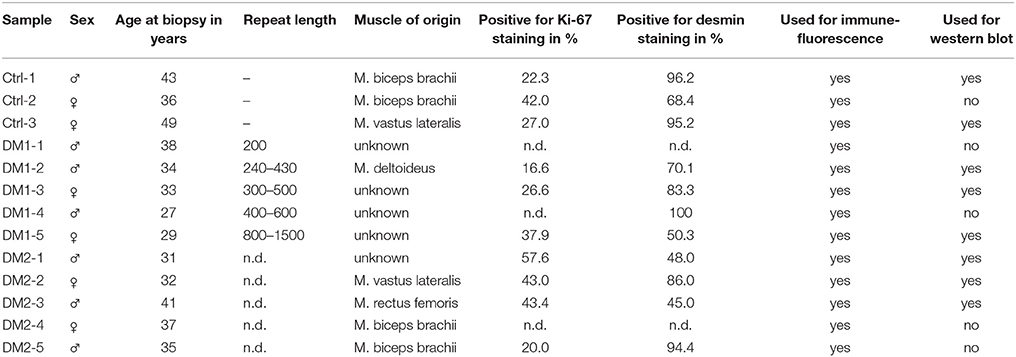
Table 2. Primary myoblast cell lines used and characterization.
Myoblasts were grown in tissue culture using skeletal muscle cell growth medium (PeloBiotec, Munich, Germany). Cells were kept from reaching confluency to avoid differentiation. Passage numbers were matched for controls and patient cells for the respective experiments, throughout all experiments passage numbers 8 to 10 have been used. For differentiation DMEM containing 5% HS, was used. Myotubes were differentiated for 7 days. Cells were grown at 37°C in a 5% CO2 incubator.
Myoblasts were fixed with methanol (−20°C). Following primary antibodies were used for staining: Ki-67 (Thermo Scientific, RM-9106-S0), emerin 5D10 and lamin A/C 4A7 (both provided by Glenn E. Morris). All secondary antibodies were Alexafluor conjugated and generated in donkey with minimal species cross-reactivity. DNA was visualized with DAPI (4,6-diamidino-2 phenylindole, dihydrochloride).
All images were obtained using an Olympus FluoView FV1000/BX 61microscope equipped with a 1.42 NA 60x objective and 3x zoom magnification. Image analysis was performed using ImageJ software. For quantification of nuclear invaginations at least 100 nuclei were counted for each measurement with the Olympus FluoView FV1000/BX 61 confocal microscope using the Z-drive to investigate the whole nucleus. For each sample at least two or three (depending on the fitness of each individual cell line) biological replicates were analyzed.
Whole protein extracts were generated from myoblast cell cultures using an ultrasonic sonicator with a MS73 tip (Bandelin Sonopuls). The proteins were separated by SDS gel electrophoresis using 4–15% TGX gels (BioRad #456–8087). Western blotting was performed using the Trans-Blot® TurboTM system (BioRad). Proteins were transferred to low fluorescent PVDF membranes (part of Trans-Blot® TurboTM RTA Transfer Kit #170-4274). Membranes were blocked with 5% BSA or 5% skim milk in 1xTBS/0, 1% Tween® 20. Following primary antibodies were used: lamin A/C 4A7 (provided by Glenn E. Morris), lamin B1 (D4Q4Z, Cell Signaling) p16INK4A (ab108349, Abcam), p21 (Cell Signaling #2947). For quantification mouse antiGAPDH (Milipore #MAB374) or rabbit antiGAPDH (Cell Signaling GAPDH (D16H11) XP #5174) were used. As secondary antibodies we used donkey anti-mouse IRDye 680RD, donkey anti-mouse IRDye 800CW, donkey anti-rabbit IRDye 680RD and donkey anti-rabbit IRDye 800 CW. All western blot images were obtained using a Licor FC. Quantification was done using the Licor ImageStudio Software. Western blots were repeated at least two times to confirm the results.
As cellular senescence is a hallmark of aging we decided to initially analyze cell cycle proteins linked to senescence in our primary human myoblast cell lines. First, we used Western blot analysis to quantify the expression of p21 and p16 (Figure 1). P21 is an inhibitor of the cell cycle (86), which fails to be up-regulated in DM2 myoblasts during differentiation (87). Western blot analysis of our myoblast cell lines shows only small changes in p21 expression (Figure 1A), however this may be within the observed expression level variations in primary myoblasts. P16 is a tumor suppressor protein that has previously been shown to be mis-regulated in congenital DM1 samples (63). There were no changes in p16 expression except in the DM1 cell line with the longest repeat where the expression was elevated. The next step was to quantify lamin B1, another senescence associated biomarker (88) which has not been investigated in DM before. While we do not see lamin B1 changes in DM2 myoblasts, it was down-regulated in all DM1 myoblasts tested (Figure 2B).
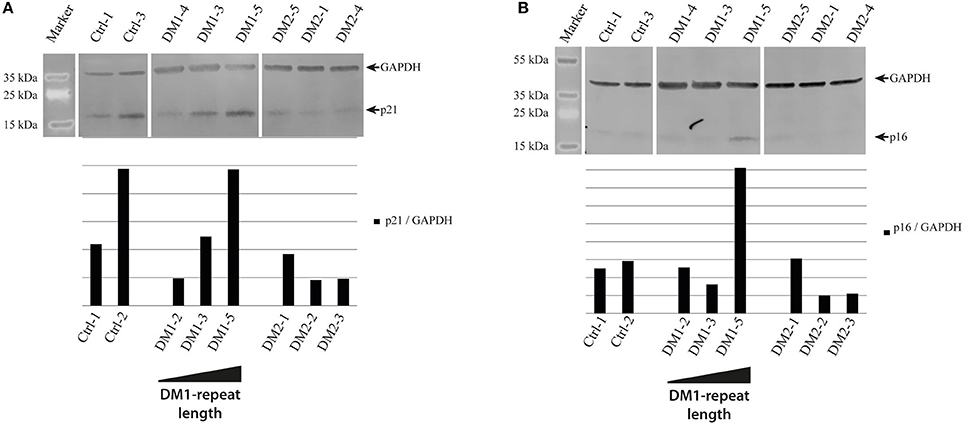
Figure 1. Cell cycle regulatory proteins in myotonic dystrophy. Western Blot and quantification of primary control, DM1 and DM2 myoblasts for cell cycle regulatory proteins p21 (A) and p16 (B). DM1 samples are ordered according their diagnosed repeat length from left (small repeat) to right (long repeat).
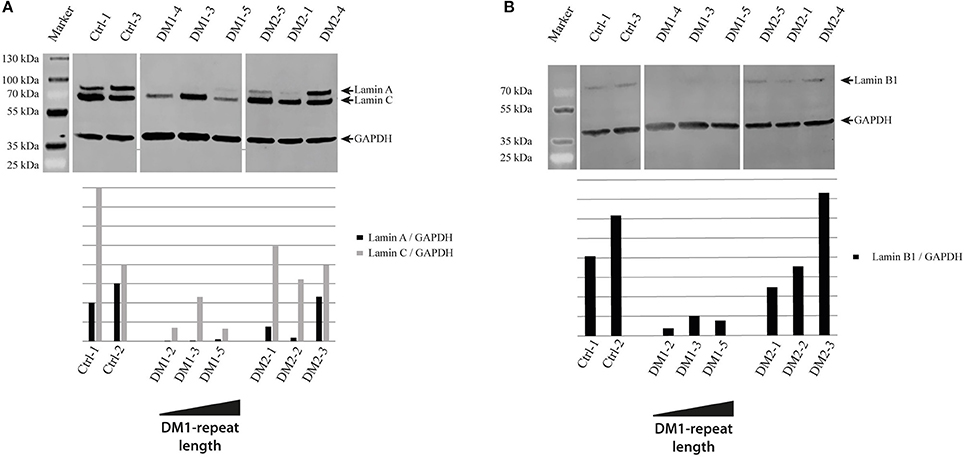
Figure 2. Lamina proteins in myotonic dystrophy. Western Blot and quantification of primary control, DM1 and DM2 myoblasts for lamin A and lamin C (A) and lamin B1 (B). DM1 samples are ordered according their diagnosed repeat length from left (small repeat) to right (long repeat).
The results of the lamin B1 quantification led us to further investigate effects on NE proteins. For this we quantified the expression of the other lamin subtypes A and C, finding that lamin A is strongly down-regulated in DM1 (Figure 2A). Immunofluorescence staining for lamin A/C shows an increased number of nuclei with invaginations in DM1 and DM2 myoblasts (Figure 3B). This has been confirmed and quantified by staining with the NET emerin (Figures 3A,C). For DM1, primary patient cell lines with longer CTG repeats show a greater percentage of nuclei with nuclear invaginations (Figure 3C). Co-staining with the proliferation marker Ki-67 revealed that all cells with nuclear invaginations are negative for Ki-67 and hence senescent. Staining of differentiated myotubes with lamin A/C and emerin shows the presence of nuclear invaginations in DM1 myotubes (Figures 4A,B).
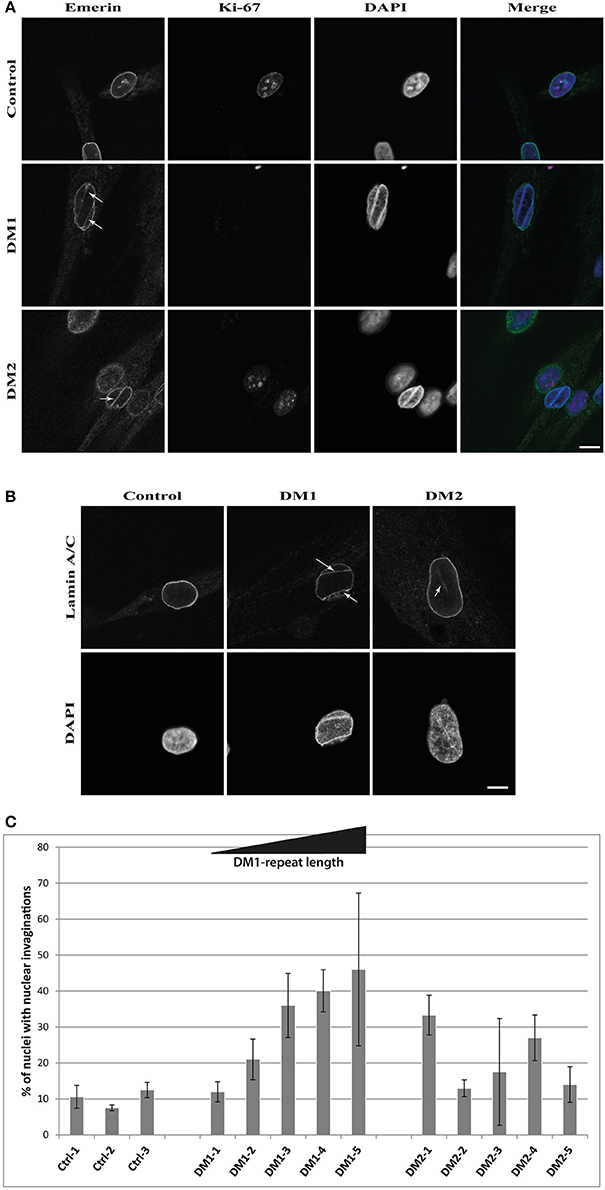
Figure 3. Nuclear envelope invaginations in myotonic dystrophy. Immunofluorescence staining of primary control, DM1 and DM2 myoblasts for (A) emerin and Ki-67 showing nuclear envelope invagination in DM1 and DM2 myoblasts, (B) confirmation of nuclear envelope invaginations by lamin A/C staining and (C) quantification of these structures in DM and control cell lines—standard deviation is shown. White arrows indicate invaginations of the nuclear envelope. Scale bar 10 μm.
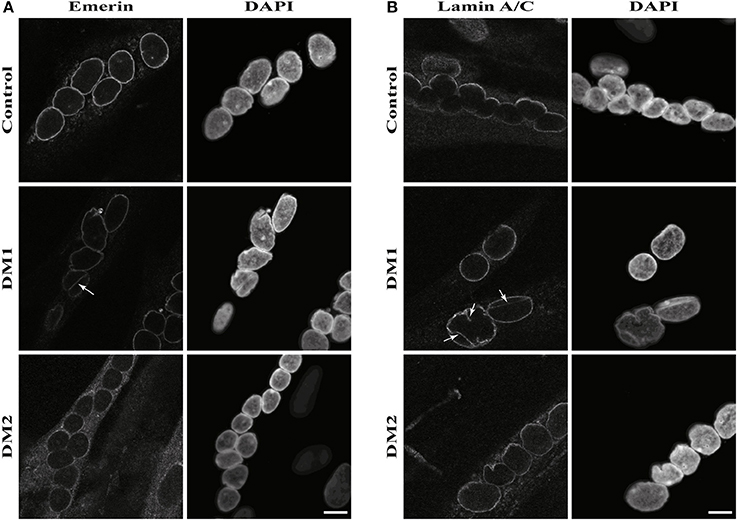
Figure 4. Nuclear envelope invaginations in myotonic dystrophy and control myotubes. Immunofluorescence staining of primary control, DM1 and DM2 myotubes for (A) emerin and (B) lamin A/C showing nuclear envelope invagination in DM1 myotubes. White arrows indicate invaginations of the nuclear envelope. Scale bar 10 μm.
The differences in cell cycle control described in DM1 and DM2 myoblasts gave rise to the idea that there are different pathomechanisms in each DM type. While for DM2 a down-regulation of the cell cycle inhibitor p21 has been shown (87), there is a reported mis-regulation of p16 in congenital DM1 (63). We can confirm both effects in our DM patient myoblasts, although the effects observed by us are not very strong and the mis-regulation of p16 in DM1 seems to be restricted to longer repeats (Figure 1).
Looking for a better senescence marker we quantified lamin B1, a protein of the NE that has been shown to be down-regulated in HGPS (89), cellular senescence (90) and normal aging (91). The down-regulation of lamin B1 occurs specifically in DM1 myoblasts and is independent of the repeat length (Figure 2B). This indicates that it could be a more relevant marker for DM1 in general than p16. Furthermore, the strong down-regulation of lamin A (Figure 2A) confirms that there are strong effects on the expression of nuclear lamina proteins in DM1. The composition of the nuclear lamina is important to achieve its multiple functions including mechanical stability, chromatin organization, transcriptional regulation, and response to oxidative stress (92, 93)–and the down-regulation of the lamins A and B1 is potentially having an effect on all these functions thus contributing to the phenotype.
Further evidence of NE involvement in DM comes from the enrichment of nuclear envelope invaginations in DM myoblasts (Figures 3, 4). DM myoblasts show more nuclei with these structures, with the strongest effects being observed in DM1. Moreover, all nuclei with invaginations have exited the cell cycle. In contrast to the lamin B1 levels, there appears to be a correlation of repeat length in DM1 and the percentage of nuclei positive for NE invaginations. The longer the diagnosed DM1 repeat the more myoblast nuclei in primary cell lines gained from those patients have invaginations of the nuclear envelope. This could be another overlap to NE-linked progeroid syndromes: knockdown of ZMPSTE24 also results in an enrichment of NE invaginations (85) and the loss of ZMPSTE24 in human results in RD, the most severe NE-linked progeroid syndrome (32). A possible explanation of how this contributes to the disease pathology is that mis-regulation of lamina proteins results in NE aberrations which force the cell to exit the cell cycle and enter senescence. Consequently, this might deplete the pool of myoblasts during muscle regeneration and contribute to the muscular dystrophy in DM via failed regeneration.
Taken together our results suggest that there is on both the clinical and molecular level clear evidence that DM reflects facets of segmental progeroid disorders (Figure 5).
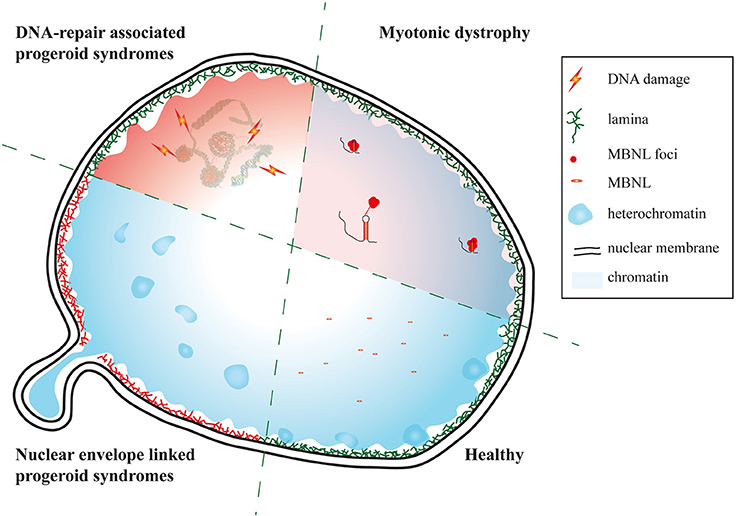
Figure 5. Myotonic dystrophy as a facet of progeroid syndromes? Schematic of affected nuclear regions and pathways in DM and nuclear envelope as well as DNA repair linked progeroid syndromes.
DMs qualify by clinical phenotypes as well as molecular features as segmental progeroid syndromes. In DM1 the composition of the NE is altered and there is an enrichment of nuclear invaginations likely contributing to the phenotype. As several NE-linked syndromes have muscle involvement, this further suggests the possibility of an overlap between NE-linked progseroid syndromes and DM1.
PM and BS contributed to the conception and design of the experiments. PM wrote the manuscript, SH and SL performed the experiments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work is supported by the generosity of Dr. Metin Colpan and by the FöFoLe Reg.-Nr. 959 (LMU). The Muscular Dystrophy Association (USA) supports monoclonal antibody development in the laboratory of Glenn E. Morris, who we want to thank for providing lamin A/C and emerin antibodies. We thank Daina Sadurska and Dr. Eric Schirmer for critical reading of the manuscript.
1. Ricker K, Koch MC, Lehmann-Horn F, Pongratz D, Speich N, Reiners K, et al. Proximal myotonic myopathy. Clinical features of a multisystem disorder similar to myotonic dystrophy. Arch Neurol. (1995) 52:25–31. doi: 10.1001/archneur.1995.00540250029009
2. Day JW, Roelofs R, Leroy B, Pech I, Benzow K, Ranum LP. Clinical and genetic characteristics of a five-generation family with a novel form of myotonic dystrophy (DM2). Neuromuscul Disord. (1999) 9:19–27. doi: 10.1016/S0960-8966(98)00094-7
3. Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. (2004) 13:3079–88. doi: 10.1093/hmg/ddh327
4. Monckton DG, Wong LJ, Ashizawa T, Caskey CT. Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analyses. Hum Mol Genet. (1995) 4:1–8. doi: 10.1093/hmg/4.1.1
5. Fu YH, Pizzuti A, Fenwick, R. G. Jr., King J, Rajnarayan S, Dunne PW, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science (1992) 255:1256–8. doi: 10.1126/science.1546326
6. Rakocevic-Stojanovic V, Savic D, Pavlovic S, Lavrnic D, Stevic Z, Basta I, et al. Intergenerational changes of CTG repeat depending on the sex of the transmitting parent in myotonic dystrophy type 1. Eur J Neurol. (2005) 12:236–7. doi: 10.1111/j.1468-1331.2004.01075.x
7. Martorell L, Cobo AM, Baiget M, Naudo M, Poza JJ, Parra J. Prenatal diagnosis in myotonic dystrophy type 1. Thirteen years of experience: implications for reproductive counselling in DM1 families. Prenatal Diagnosis (2007) 27:68–72. doi: 10.1002/pd.1627
8. Arsenault ME, Prevost C, Lescault A, Laberge C, Puymirat J, Mathieu J. Clinical characteristics of myotonic dystrophy type 1 patients with small CTG expansions. Neurology (2006) 66:1248–50. doi: 10.1212/01.wnl.0000208513.48550.08
9. Wang Y, Pfeiffer RM, Alsaggaf R, Meeraus W, Gage JC, Anderson LA, et al. Risk of skin cancer among patients with myotonic dystrophy type 1 based on primary care physician data from the U.K. Clin Pract Res Datalink Int J Cancer (2018) 142:1174–81. doi: 10.1002/ijc.31143
10. Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science (2001) 293:864–7. doi: 10.1126/science.1062125
11. Ricker K. Myotonic dystrophy and proximal myotonic myophathy. J Neurol. (1999) 246:334–8. doi: 10.1007/s004150050359
12. Montagnese F, Mondello S, Wenninger S, Kress W, Schoser B. Assessing the influence of age and gender on the phenotype of myotonic dystrophy type 2. J Neurol. (2017) 264:2472–80. doi: 10.1007/s00415-017-8653-2
13. Martin GM. Genetic syndromes in man with potential relevance to the pathobiology of aging. Birth Defects Orig Artic Ser. (1978) 14:5–39.
14. Mateos-Aierdi AJ, Goicoechea M, Aiastui A, Fernandez-Torron R, Garcia-Puga M, Matheu A, et al. Muscle wasting in myotonic dystrophies: a model of premature aging. Front Aging Neurosci. (2015) 7:125. doi: 10.3389/fnagi.2015.00125
15. Evans WJ, Campbell WW. Sarcopenia and age-related changes in body composition and functional capacity. J Nutr. (1993) 123:465–8. doi: 10.1093/jn/123.suppl_2.465
16. Fielding RA, Vellas B, Evans WJ, Bhasin S, Morley JE, Newman AB, et al. Sarcopenia: an undiagnosed condition in older adults. Current consensus definition: prevalence, etiology, and consequences International working group on sarcopenia J Am Med Direct Assoc. (2011) 12:249–56. doi: 10.1016/j.jamda.2011.01.003
17. Campione E, Botta A, Di Prete M, Rastelli E, Gibellini M, Petrucci A, et al. Cutaneous features of myotonic dystrophy types 1 and 2: implication of premature aging and vitamin D homeostasis. Neuromuscul Disord. (2017) 27:163–9. doi: 10.1016/j.nmd.2016.11.004
18. Callan HG, Randall JT, Tomlin SG. An electron microscope study of the nuclear membrane. Nature (1949) 163:280. doi: 10.1038/163280a0
19. Gerace L, Comeau C, Benson M. Organization and modulation of nuclear lamina structure. J Cell Sci Supplement (1984) 1:137–60. doi: 10.1242/jcs.1984.Supplement_1.10
20. de Las Heras JI, Meinke P, Batrakou DG, Srsen V, Zuleger N, Kerr AR, et al. Tissue specificity in the nuclear envelope supports its functional complexity. Nucleus (2013) 4:460–77. doi: 10.4161/nucl.26872
21. Dittmer TA, Misteli T. The lamin protein family. Genome Biol. (2011) 12:222. doi: 10.1186/gb-2011-12-5-222
22. Loi M, Cenni V, Duchi S, Squarzoni S, Lopez-Otin C, Foisner R, et al. Barrier-to-autointegration factor (BAF) involvement in prelamin A-related chromatin organization changes. Oncotarget (2016) 7:15662–77. doi: 10.18632/oncotarget.6697
23. Lee KK, Haraguchi T, Lee RS, Koujin T, Hiraoka Y, Wilson KL. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J Cell Sci. (2001) 114:4567–73.
24. Mattout-Drubezki A, Gruenbaum Y. Dynamic interactions of nuclear lamina proteins with chromatin and transcriptional machinery. Cell Mol Life Sci. (2003) 60:2053–63. doi: 10.1007/s00018-003-3038-3
25. Segura-Totten M, Wilson KL. BAF: roles in chromatin, nuclear structure and retrovirus integration. Trends Cell Biol. (2004) 14:261–6. doi: 10.1016/j.tcb.2004.03.004
26. De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, et al. Lamin a truncation in hutchinson-gilford progeria. Science (2003) 300:2055. doi: 10.1126/science.1084125
27. Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature (2003) 423:293–8. doi: 10.1038/nature01629
28. Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet Part A (2006) 140:2603–24. doi: 10.1002/ajmg.a.31346
29. Coutinho HD, Falcao-Silva VS, Goncalves GF, da Nobrega RB. Molecular ageing in progeroid syndromes: Hutchinson-Gilford progeria syndrome as a model. Immun Ageing (2009) 6:4. doi: 10.1186/1742-4933-6-4
30. Novelli G, Muchir A, Sangiuolo F, Helbling-Leclerc A, D'Apice MR, Massart C, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. (2002) 71:426–31. doi: 10.1086/341908
31. Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. (2003) 12:1995–2001. doi: 10.1093/hmg/ddg213
32. Navarro CL, Esteves-Vieira V, Courrier S, Boyer A, Duong Nguyen T, Huong le TT, et al. New ZMPSTE24 (FACE1) mutations in patients affected with restrictive dermopathy or related progeroid syndromes and mutation update. Eur J Hum Genet. (2014) 22:1002–11. doi: 10.1038/ejhg.2013.258
33. Navarro CL, De Sandre-Giovannoli A, Bernard R, Boccaccio I, Boyer A, Genevieve D, et al. Lamin A, and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. (2004) 13:2493–503. doi: 10.1093/hmg/ddh265
34. Smigiel R, Jakubiak A, Esteves-Vieira V, Szela K, Halon A, Jurek T, et al. Novel frameshifting mutations of the ZMPSTE24 gene in two siblings affected with restrictive dermopathy and review of the mutations described in the literature. Am J Med Genet Part A (2010) 152A:447–52. doi: 10.1002/ajmg.a.33221
35. Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, et al. LMNA mutations in atypical Werner's syndrome. Lancet (2003) 362:440–5. doi: 10.1016/S0140-6736(03)14069-X
36. Bonne G, Levy N. LMNA mutations in atypical Werner's syndrome. Lancet (2003) 362:1585–6. doi: 10.1016/S0140-6736(03)14761-7
37. Vigouroux C, Caux F, Capeau J, Christin-Maitre S, Cohen A. LMNA mutations in atypical Werner's syndrome. Lancet (2003) 362:1585–6. doi: 10.1016/S0140-6736(03)14760-5
38. McPherson E, Turner L, Zador I, Reynolds K, Macgregor D, Giampietro PF. Ovarian failure and dilated cardiomyopathy due to a novel lamin mutation. Am J Med Genet Part A (2009) 149A:567–72. doi: 10.1002/ajmg.a.32627
39. Malouf J, Alam S, Kanj H, Mufarrij A, Der Kaloustian VM. Hypergonadotropic hypogonadism with congestive cardiomyopathy: an autosomal-recessive disorder? Am J Med Genet. (1985) 20:483–9. doi: 10.1002/ajmg.1320200309
40. Kirschner J, Brune T, Wehnert M, Denecke J, Wasner C, Feuer A, et al. p.S143F mutation in lamin A/C: a new phenotype combining myopathy and progeria. Ann Neurol. (2005) 57:148–51. doi: 10.1002/ana.20359
41. Garg A, Subramanyam L, Agarwal AK, Simha V, Levine B, D'Apice MR, et al. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J Clin Endocrinol Metab. (2009) 94:4971–83. doi: 10.1210/jc.2009-0472
42. Madej-Pilarczyk A, Rosinska-Borkowska D, Rekawek J, Marchel M, Szalus E, Jablonska S, et al. Progeroid syndrome with scleroderma-like skin changes associated with homozygous R435C LMNA mutation. Am J Med Genet Part A (2009) 149A:2387–92. doi: 10.1002/ajmg.a.33018
43. Starke S, Meinke P, Camozzi D, Mattioli E, Pfaeffle R, Siekmeyer M, et al. Progeroid laminopathy with restrictive dermopathy-like features caused by an isodisomic LMNA mutation p.R435C. Aging (2013) 5:445–59. doi: 10.18632/aging.100566
44. Guo H, Luo N, Hao F, Bai Y. p.Pro4Arg mutation in LMNA gene: a new atypical progeria phenotype without metabolism abnormalities. Gene (2014) 546:35–9. doi: 10.1016/j.gene.2014.05.042
45. Puente XS, Quesada V, Osorio FG, Cabanillas R, Cadinanos J, Fraile JM, et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet. (2011) 88:650–6. doi: 10.1016/j.ajhg.2011.04.010
46. Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. (2014) 83:519–52. doi: 10.1146/annurev-biochem-060713-035428
47. Oshima J, Yu CE, Piussan C, Klein G, Jabkowski J, Balci S, et al. Homozygous and compound heterozygous mutations at the Werner syndrome locus. Hum Mol Genet. (1996) 5:1909–13. doi: 10.1093/hmg/5.12.1909
48. Shamanna RA, Lu H, de Freitas JK, Tian J, Croteau DL, Bohr VA. WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat Commun. (2016) 7:13785. doi: 10.1038/ncomms13785
49. Lan L, Nakajima S, Komatsu K, Nussenzweig A, Shimamoto A, Oshima J, et al. Accumulation of Werner protein at DNA double-strand breaks in human cells. J Cell Sci. (2005) 118:4153–62. doi: 10.1242/jcs.02544
50. Opresko PL, Otterlei M, Graakjaer J, Bruheim P, Dawut L, Kolvraa S, et al. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol Cell (2004) 14:763–74. doi: 10.1016/j.molcel.2004.05.023
51. Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner's syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (1966) 45:177–221. doi: 10.1097/00005792-196605000-00001
52. Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, et al. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell (1995) 83:655–66. doi: 10.1016/0092-8674(95)90105-1
53. Mirzaei H, Schmidt KH. Non-Bloom syndrome-associated partial and total loss-of-function variants of BLM helicase. Proc Natl Acad Sci USA. (2012) 109:19357–62. doi: 10.1073/pnas.1210304109
54. Larsen NB, Hickson ID. RecQ helicases: conserved guardians of genomic integrity. Adv Exp Med Biol. (2013) 767:161–84. doi: 10.1007/978-1-4614-5037-5_8
55. German J, Bloom D, Passarge E. Bloom's syndrome XI. Progress report for 1983. Clinical genetics (1984) 25:166–74. doi: 10.1111/j.1399-0004.1984.tb00480.x
56. Henning KA, Li L, Iyer N, McDaniel LD, Reagan MS, Legerski R, et al. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell (1995) 82:555–64. doi: 10.1016/0092-8674(95)90028-4
57. Mallery DL, Tanganelli B, Colella S, Steingrimsdottir H, van Gool AJ, Troelstra C, et al. Molecular analysis of mutations in the CSB (ERCC6) gene in patients with Cockayne syndrome. Am J Hum Genet. (1998) 62:77–85. doi: 10.1086/301686
58. Nance MA, Berry SA. Cockayne syndrome: review of 140 cases. Am J Med Genet. (1992) 42:68–84. doi: 10.1002/ajmg.1320420115
59. Falik-Zaccai TC, Laskar M, Kfir N, Nasser W, Slor H, Khayat M. Cockayne syndrome type II in a Druze isolate in Northern Israel in association with an insertion mutation in ERCC6. Am J Med Genet Part A (2008) 146A:1423–9. doi: 10.1002/ajmg.a.32309
60. Flejter WL, McDaniel LD, Johns D, Friedberg EC, Schultz RA. Correction of xeroderma pigmentosum complementation group D mutant cell phenotypes by chromosome and gene transfer: involvement of the human ERCC2 DNA repair gene. Proc Natl Acad Sci USA. (1992) 89:261–5. doi: 10.1073/pnas.89.1.261
61. Faghri S, Tamura D, Kraemer KH, Digiovanna JJ. Trichothiodystrophy: a systematic review of 112 published cases characterises a wide spectrum of clinical manifestations. J Med Genet. (2008) 45:609–21. doi: 10.1136/jmg.2008.058743
62. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
63. Bigot A, Klein AF, Gasnier E, Jacquemin V, Ravassard P, Butler-Browne G, et al. Large CTG repeats trigger p16-dependent premature senescence in myotonic dystrophy type 1 muscle precursor cells. Am J Pathol. (2009) 174:1435–42. doi: 10.2353/ajpath.2009.080560
64. Thornell LE, Lindstom M, Renault V, Klein A, Mouly V, Ansved T, et al. Satellite cell dysfunction contributes to the progressive muscle atrophy in myotonic dystrophy type 1. Neuropathol Appl Neurobiol. (2009) 35:603–13. doi: 10.1111/j.1365-2990.2009.01014.x
65. Renna LV, Cardani R, Botta A, Rossi G, Fossati B, Costa E, et al. Premature senescence in primary muscle cultures of myotonic dystrophy type 2 is not associated with p16 induction. Eur J Histochem. (2014) 58:2444. doi: 10.4081/ejh.2014.2444
66. Furling D, Coiffier L, Mouly V, Barbet JP, St Guily JL, Taneja K, et al. Defective satellite cells in congenital myotonic dystrophy. Hum Mol Genet. (2001) 10:2079–87. doi: 10.1093/hmg/10.19.2079
67. Pelletier R, Hamel F, Beaulieu D, Patry L, Haineault C, Tarnopolsky M, et al. Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiol Dis. (2009) 36:181–90. doi: 10.1016/j.nbd.2009.07.009
68. Malatesta M, Giagnacovo M, Renna LV, Cardani R, Meola G, Pellicciari C. Cultured myoblasts from patients affected by myotonic dystrophy type 2 exhibit senescence-related features: ultrastructural evidence. Eur J Histochem. (2011) 55:e26. doi: 10.4081/ejh.2011.e26
69. Ghorbani M, Taylor SJ, Pook MA, Payne A. Comparative (computational) analysis of the DNA methylation status of trinucleotide repeat expansion diseases. J Nucleic Acids (2013) 2013:689798. doi: 10.1155/2013/689798
70. Lopez Castel A, Nakamori M, Tome S, Chitayat D, Gourdon G, Thornton CA, et al. Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum Mol Genet. (2011) 20:1–15. doi: 10.1093/hmg/ddq427
71. Sanz A, Stefanatos RK. The mitochondrial free radical theory of aging: a critical view. Curr Aging Sci. (2008) 1:10–21. doi: 10.2174/1874609810801010010
72. Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature (2004) 429:417–23. doi: 10.1038/nature02517
73. Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science (2005) 309:481–4. doi: 10.1126/science.1112125
74. Sahashi K, Tanaka M, Tashiro M, Ohno K, Ibi T, Takahashi A, et al. Increased mitochondrial DNA deletions in the skeletal muscle of myotonic dystrophy. Gerontology (1992) 38:18–29. doi: 10.1159/000213303
75. Siciliano G, Mancuso M, Tedeschi D, Manca ML, Renna MR, Lombardi V, et al. Coenzyme Q10, exercise lactate and CTG trinucleotide expansion in myotonic dystrophy. Brain Res Bull. (2001) 56:405–10. doi: 10.1016/S0361-9230(01)00653-0
76. Rusconi F, Mancinelli E, Colombo G, Cardani R, Da Riva L, Bongarzone I, et al. Proteome profile in Myotonic Dystrophy type 2 myotubes reveals dysfunction in protein processing and mitochondrial pathways. Neurobiol Dis. (2010) 38:273–80. doi: 10.1016/j.nbd.2010.01.017
77. Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. (2009) 78:959–91. doi: 10.1146/annurev.biochem.052308.114844
78. Vignaud A, Ferry A, Huguet A, Baraibar M, Trollet C, Hyzewicz J, et al. Progressive skeletal muscle weakness in transgenic mice expressing CTG expansions is associated with the activation of the ubiquitin-proteasome pathway. Neuromuscul Disord. (2010) 20:319–25. doi: 10.1016/j.nmd.2010.03.006
79. Kovtun IV, Spiro C, McMurray CT. Triplet repeats and DNA repair: germ cell and somatic cell instability in transgenic mice. Methods Mol Biol. (2004) 277:309–19. doi: 10.1385/1-59259-804-8:309
80. Gomes-Pereira M, Hilley JD, Morales F, Adam B, James HE, Monckton DG. Disease-associated CAG.CTG triplet repeats expand rapidly in non-dividing mouse cells, but cell cycle arrest is insufficient to drive expansion. Nucleic Acids Res. (2014) 42:7047–56. doi: 10.1093/nar/gku285
81. Concannon C, Lahue RS. Nucleotide excision repair and the 26S proteasome function together to promote trinucleotide repeat expansions. DNA Repair. (2014) 13:42–9. doi: 10.1016/j.dnarep.2013.11.004
82. Morales F, Vasquez M, Santamaria C, Cuenca P, Corrales E, Monckton DG. A polymorphism in the MSH3 mismatch repair gene is associated with the levels of somatic instability of the expanded CTG repeat in the blood DNA of myotonic dystrophy type 1 patients. DNA Repair. (2016) 40:57–66. doi: 10.1016/j.dnarep.2016.01.001
83. Rodriguez R, Hernandez-Hernandez O, Magana JJ, Gonzalez-Ramirez R, Garcia-Lopez ES, Cisneros B. Altered nuclear structure in myotonic dystrophy type 1-derived fibroblasts. Mol Biol Rep. (2015) 42:479–88. doi: 10.1007/s11033-014-3791-4
84. Malhas A, Goulbourne C, Vaux DJ. The nucleoplasmic reticulum: form and function. Trends Cell Biol. (2011) 21:362–73. doi: 10.1016/j.tcb.2011.03.008
85. Goulbourne CN, Malhas AN, Vaux DJ. The induction of a nucleoplasmic reticulum by prelamin A accumulation requires CTP:phosphocholine cytidylyltransferase-alpha. J Cell Sci. (2011) 124:4253–66. doi: 10.1242/jcs.091009
86. El-Deiry WS. p21(WAF1) mediates cell-cycle inhibition, relevant to cancer suppression and therapy. Cancer Res. (2016) 76:5189–91. doi: 10.1158/0008-5472.CAN-16-2055
87. Timchenko NA, Iakova P, Cai ZJ, Smith JR, Timchenko LT. Molecular basis for impaired muscle differentiation in myotonic dystrophy. Mol Cell Biol. (2001) 21:6927–38. doi: 10.1128/MCB.21.20.6927-6938.2001
88. Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell (2012) 23:2066–75. doi: 10.1091/mbc.e11-10-0884
89. Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. (2005) 11:440–5. doi: 10.1038/nm1204
90. Shimi T, V., Butin-Israeli SA, Adam RB, Hamanaka AE, Goldman CA, et al. Goldman The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. (2011) 25:2579–93. doi: 10.1101/gad.179515.111
91. Golubtsova NN, Filippov FN, Gunin AG. Lamin B1 and lamin B2 in human skin in the process of aging. Adv Gerontol. (2016) 29:222–8. doi: 10.1134/S2079057016040068
92. Zuela N, Bar DZ, Gruenbaum Y. Lamins in development, tissue maintenance and stress. EMBO Rep. (2012) 13:1070–8. doi: 10.1038/embor.2012.167
Keywords: myotonic dystrophy, segmental progeroid disorder, nuclear envelope, premature aging, DNA repair
Citation: Meinke P, Hintze S, Limmer S and Schoser B (2018) Myotonic Dystrophy—A Progeroid Disease? Front. Neurol. 9:601. doi: 10.3389/fneur.2018.00601
Received: 19 March 2018; Accepted: 06 July 2018;
Published: 25 July 2018.
Edited by:
Nicolas Sergeant, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Rosanna Cardani, Policlinico San Donato (IRCCS), ItalyCopyright © 2018 Meinke, Hintze, Limmer and Schoser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Meinke, UGV0ZXIuTWVpbmtlQG1lZC51bmktbXVlbmNoZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.