 Rita L. Vaz
Rita L. Vaz Tiago F. Outeiro
Tiago F. Outeiro Joaquim J. Ferreira
Joaquim J. Ferreira- 1TechnoPhage, SA, Lisboa, Portugal
- 2Faculdade de Medicina, Universidade de Lisboa, Lisboa, Portugal
- 3Department of Experimental Neurodegeneration, Center for Nanoscale Microscopy and Molecular Physiology of the Brain, University Medical Center Göttingen, Göttingen, Germany
- 4Department of Experimental Neurodegeneration, Center for Biostructural Imaging of Neurodegeneration, University Medical Center Göttingen, Göttingen, Germany
- 5CEDOC, Chronic Diseases Research Centre, Faculdade de Ciências Médicas, NOVA Medical School, Universidade NOVA de Lisboa, Lisboa, Portugal
- 6The Medical School, Institute of Neuroscience, Newcastle University, Newcastle upon Tyne, United Kingdom
- 7Faculdade de Medicina, Instituto de Medicina Molecular, Universidade de Lisboa, Lisboa, Portugal
- 8Laboratory of Clinical Pharmacology and Therapeutics, Faculdade de Medicina, Universidade de Lisboa, Lisboa, Portugal
- 9CNS-Campus Neurológico Sénior, Torres Vedras, Portugal
Movement disorders can be primarily divided into hypokinetic and hyperkinetic. Most of the hypokinetic syndromes are associated with the neurodegenerative disorder Parkinson’s disease (PD). By contrast, hyperkinetic syndromes encompass a broader array of diseases, including dystonia, essential tremor, or Huntington’s disease. The discovery of effective therapies for these disorders has been challenging and has also involved the development and characterization of accurate animal models for the screening of new drugs. Zebrafish constitutes an alternative vertebrate model for the study of movement disorders. The neuronal circuitries involved in movement in zebrafish are well characterized, and most of the associated molecular mechanisms are highly conserved. Particularly, zebrafish models of PD have contributed to a better understanding of the role of several genes implicated in the disease. Furthermore, zebrafish is a vertebrate model particularly suited for large-scale drug screenings. The relatively small size of zebrafish, optical transparency, and lifecycle, are key characteristics that facilitate the study of multiple compounds at the same time. Several transgenic, knockdown, and mutant zebrafish lines have been generated and characterized. Therefore, it is central to critically analyze these zebrafish lines and understand their suitability as models of movement disorders. Here, we revise the pathogenic mechanisms, phenotypes, and responsiveness to pharmacotherapies of zebrafish lines of the most common movement disorders. A systematic review of the literature was conducted by including all studies reporting the characterization of zebrafish models of the movement disorders selected from five bibliographic databases. A total of 63 studies were analyzed, and the most relevant data within the scope of this review were gathered. The majority (62%) of the studies were focused in the characterization of zebrafish models of PD. Overall, the zebrafish models included display conserved biochemical and neurobehavioral features of the phenomenology in humans. Nevertheless, in light of what is known for all animal models available, the use of zebrafish as a model for drug discovery requires further optimization. Future technological developments alongside with a deeper understanding of the molecular bases of these disorders should enable the development of novel zebrafish lines that can prove useful for drug discovery for movement disorders.
Introduction
Movement disorders are a heterogeneous group of neurological conditions characterized by the inability to produce or control movement. The typical clinical features include either paucity of voluntary movements, referred to as hypokinesia, bradykinesia and akinesia, or excess of movement, commonly denoted as hyperkinesia, dyskinesia, and abnormal involuntary movements (1). These two major groups have been dynamic, including different categories over time. Most movement disorders lack effective pharmacological therapies, because their complex etiology and pathological mechanisms remain largely unknown. This complicates the development of adequate animal models and, ultimately, of therapeutic compounds. In this context, zebrafish (Danio rerio) (Figure 1) has become an attractive tool for drug discovery. Zebrafish presents a compromise between the scalability of invertebrate models and overall homology to vertebrates. In the last 20 years, several zebrafish models of brain disorders have been generated (2). Many discoveries were reported, but an overall analysis of zebrafish as an alternative model of movement disorders is lacking. Therefore, the scope of this review was to systematically analyze the latest developments in the generation and characterization of zebrafish models of the most common movement disorders. This highlights the translational value of zebrafish to model these diseases and, ultimately, for drug discovery. The pathogenic mechanisms, disease hallmarks, phenotypic effects of mutations or neurotoxins, and responsiveness to pharmacological interventions are covered for zebrafish models of two hypokinetic and five hyperkinetic disorders (Table 1).

Figure 1. Representative images of zebrafish at embryonic (A), larval (B), and adult (C) stage.
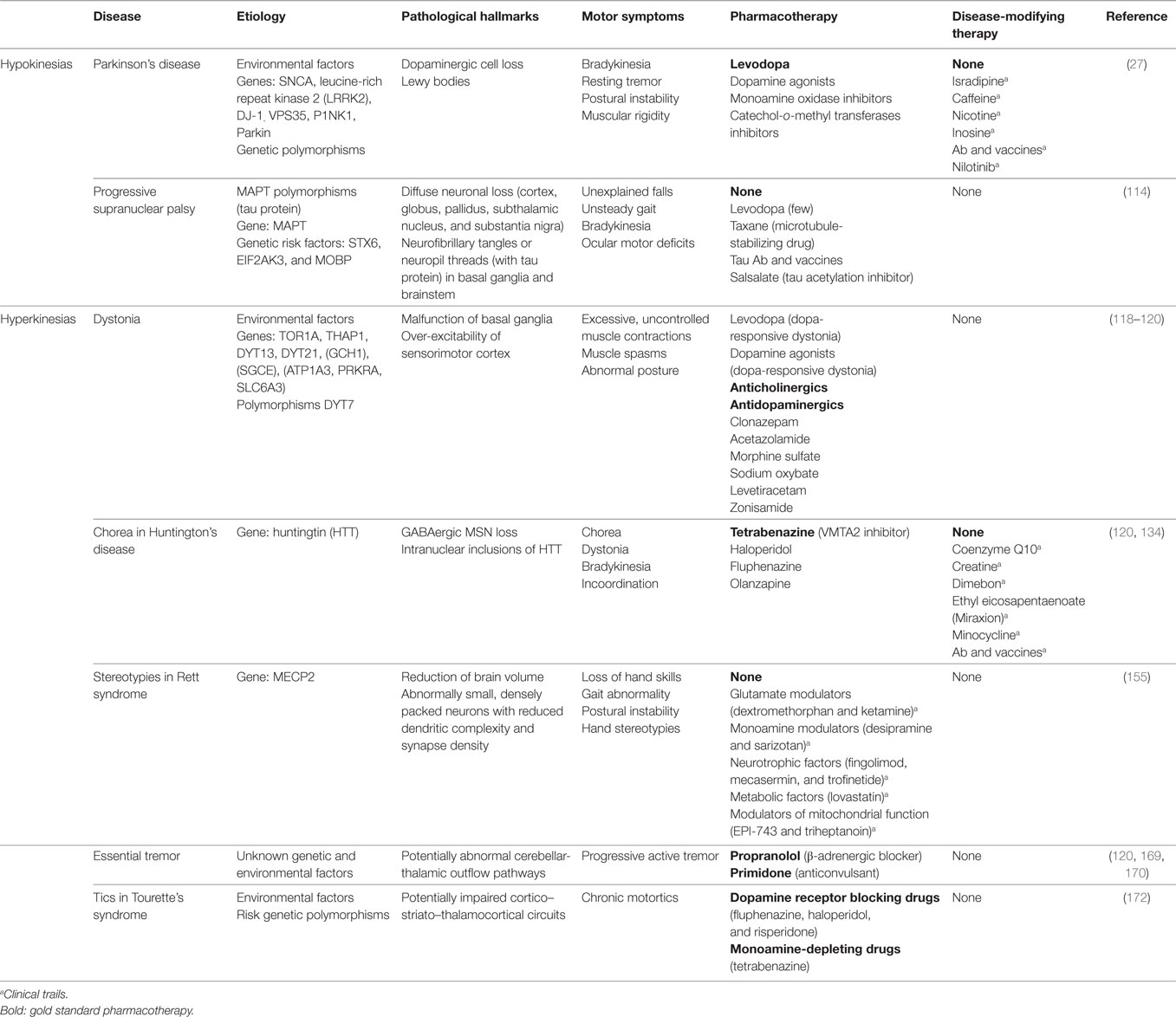
Table 1. Etiology, phenomenology, and pharmacotherapies of the movement disorders covered in this review.
Zebrafish as a Model for Translational Research
The utilization of zebrafish for drug discovery increased in the beginning of the twenty-first century (Figure 2) (3, 4). Due to its small size and fast reproduction, zebrafish is suitable for large-scale in vivo assays. Drug administration is facilitated through the aqueous environment, and the efficacy, bioavailability and toxicity can be readily determined. Importantly, zebrafish is a vertebrate, in contrast to other commonly used organisms, such as Drosophila melanogaster or Caenorhabditis elegans, in which the anatomical similarity with humans is much lower (5). The optical transparency is another advantage of this teleost, as it enables the direct observation of cellular and physiological processes in vivo and in real time. These and other practical features rendered zebrafish the mainstream model for investigation in developmental biology. In addition, it is now also widely used as a disease model and, more recently, it became an important tool for the screening of drugs (Figure 3) (6).
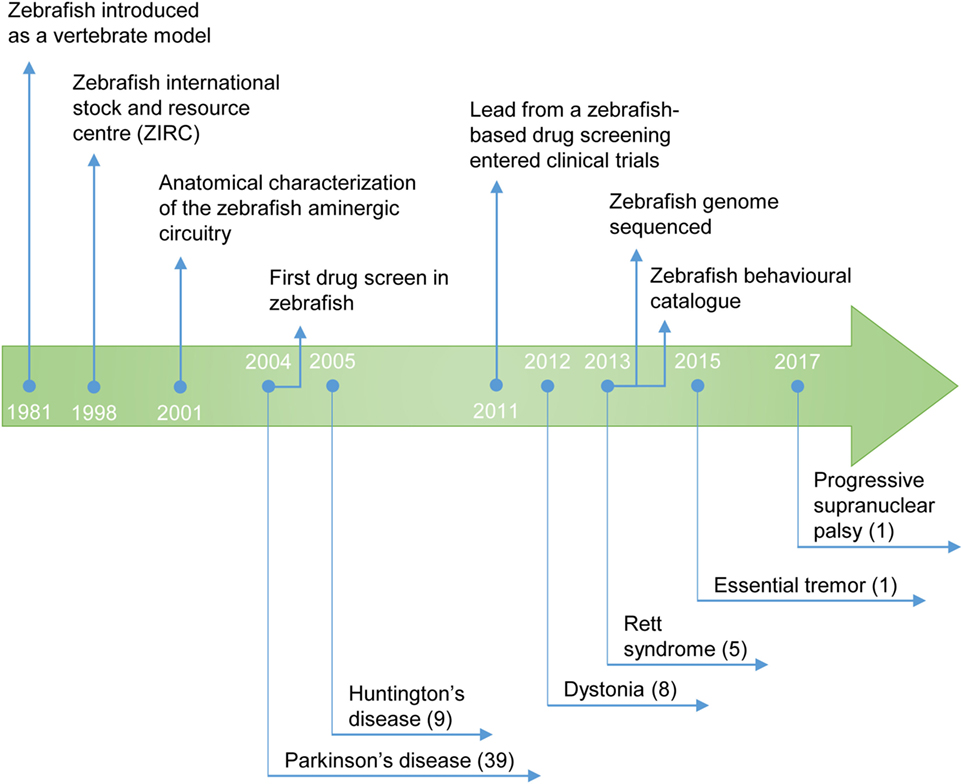
Figure 2. Timeline of the use of zebrafish as a model for the study of movement disorders and drug discovery. The publication year of the first study describing a zebrafish model of the movement disorder is highlighted. (#) Number of studies published to date.
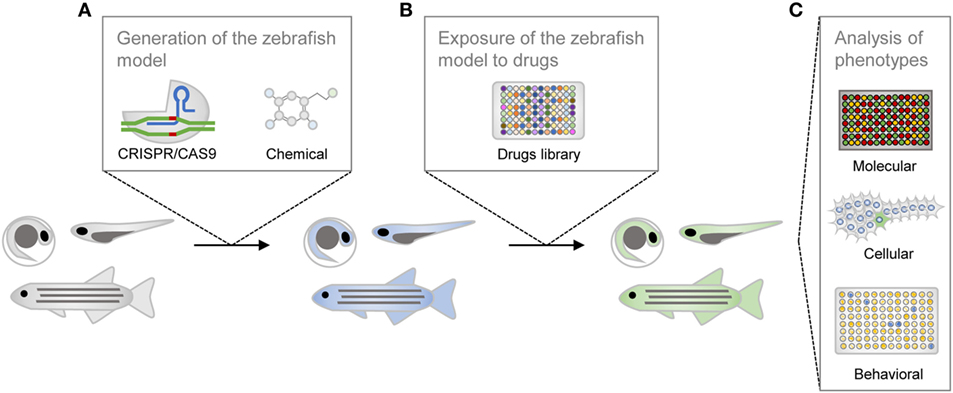
Figure 3. Schematic representation of the drug screening process in zebrafish. The zebrafish line is generated with genetic or chemical tools (A), is incubated with compounds from the library of small molecules (B), and is then phenotypically characterized (C).
Despite the evident differences between fish and mammals, zebrafish hold genomic and physiological homology to humans (7). Moreover, the genome of zebrafish is sequenced and available for annotation in databases. The genome of zebrafish includes orthologs of 71% of human genes, and a high degree of conservation in the functional properties of many of the encoded proteins (8). Physiological and anatomical homology is also evidenced in most of the organs, including the nervous system (7). The basic anatomical structure, the cellular populations, and the chemistry of the zebrafish and human nervous system are evolutionarily conserved. The nervous system of zebrafish is anatomically divided into the fore-, mid-, and hindbrain, including the diencephalon, telencephalon, cerebellum, and spinal cord (2, 6). The blood–brain barrier (BBB) is structurally and functionally similar to that of higher vertebrates and developed by 3 days post fertilization (dpf) (9, 10).
Dissection of the Monoaminergic System in Zebrafish
Specifically, the monoaminergic system is involved in the adjustment of movement and is predominantly conserved in vertebrates (Figure 4) (11). The tyrosine hydroxylase (TH) is an important marker of catecholaminergic neurons. Two genes, th1 and th2, encode the TH enzyme in zebrafish, and both proteins are highly similar to the mammalian TH (12). The neuronal populations that express TH1 can be found in the olfactory bulb, telencephalon, diencephalon, locus coeruleus, and caudal lobe (13). The neurons that express TH2 are found in the ventral preoptic region, hypothalamus and colocalize with TH1-positive neurons in the diencephalic dopaminergic cluster. The dopamine transporter (DAT) is also detected in this neuronal population (14). The major difference of the zebrafish catecholaminergic system is the absence of dopaminergic neuronal populations in the midbrain. The diencephalic dopaminergic cluster located in the posterior tuberculum of zebrafish has been suggested to be the functional homolog of substantia nigra in mammals (15). Like in mammals, the noradrenergic population is predominantly located in the locus coeruleus of zebrafish. In turn, the catecholamines, dopamine and noradrenaline, are detectable in zebrafish larvae with 5 dpf (16). Zebrafish encode one monoamine oxidase (MAO) with homology to the human MAO-A and MAO-B, and two putative catechol-o-methyl transferases (17, 18). The catecholaminergic receptors and transporters are also conserved among vertebrates. Eight subtypes of the dopamine receptor (17), five alpha-2-adrenergic receptors, and one noradrenaline transporter (19) have been identified in zebrafish so far.
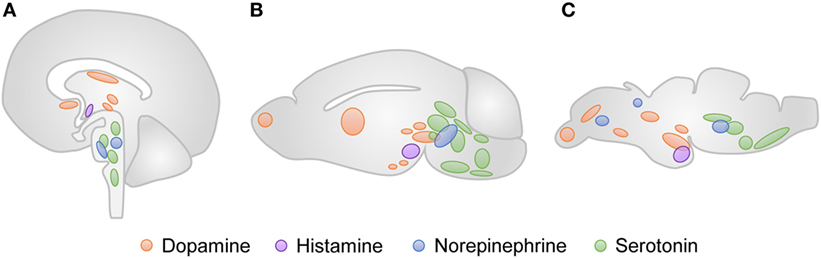
Figure 4. Neuronal clusters that modulate movement in vertebrates. The approximate anatomical location of the dopaminergic (orange), histaminergic (purple), noradrenergic (blue), and serotonergic (green) regions are represented for human (A), rodent (B), and zebrafish (C) brains.
The serotonergic and histaminergic systems of zebrafish also present homologies with the corresponding circuits in mammals. The zebrafish serotonergic neuronal groups can be found in the raphe nuclei, pretectum, posterior paraventricular organ, vagal lobe, and reticular formation (20). Particularly, the serotonergic pretectal and paraventricular neuronal populations are not found in higher vertebrates. Three orthologs of the mammalian serotonin receptors have been identified in the genome of zebrafish, along with two genes, slc6a4a and slc6a4b, that encode serotonin transporters (21, 22). The zebrafish histaminergic system consists of a posterior hypothalamic neuronal cluster, from which all histaminergic projections derive (11). Histamine can be detected at 100 hpf (23), and three histamine receptors have been identified in zebrafish (24).
Overall, the expression of monoaminergic proteins and the spatial distribution of monoaminergic neuronal populations are well characterized. Although zebrafish suffered genome duplication, it seems that the distribution of the duplicated proteins is complementary to that observed in mammals. This helps to explain why, despite the consistent differences, the drugs that target transporters, receptors or enzymes involved in the modulation of neurotransmitters have rather conserved effects (25, 26).
Methods
The aim of this review was to analyze the studies reported to date on the use of zebrafish models for movement disorders, and assess their potential for modeling the human pathologies. To achieve this, we conducted a systematic search of the literature using BiomedCentral, EBSCO host, PubMed/Medline, ScienceDirect, and Web of knowledge, in August and September 2017. The following search strategy was used for each of the five bibliographic databases: Title, abstract, keywords, or topic: (“Parkinson’s disease” OR “Parkinsons disease” OR “parkinsonism”) AND (“zebrafish”); (“progressive supranuclear palsy” OR “supranuclear palsy”) AND (“zebrafish”); (“dystonia” OR “dystonic”) AND (“zebrafish”); (“Tremor” OR “Tremors”) AND (“zebrafish”); (“Tourette’s syndrome” OR “Tourettes syndrome”) AND (“zebrafish”); (“Huntington’s disease” OR “Huntingtons disease”) AND (“zebrafish”); (“Rett syndrome” OR “Rett” OR “RTT”) AND (“zebrafish”). All dates were included in the search criteria. Only published, peer-reviewed studies written in English were considered. The studies with the description of the phenomenology of zebrafish models of movement disorders were included in the review, through scrutiny of the title and abstract of the papers identified during the systematic search. Studies with no description of the pathological mechanisms AND/OR phenotypes of the diseases were excluded from analysis.
For Parkinson’s disease (PD), a total of 39 studies (Figure 2) met the inclusion criteria, from 110 returned by BiomedCentral, 111 by EBSCO host, 221 by PubMed/Medline, 4,015 by ScienceDirect, and 181 by Web of knowledge. For progressive supranuclear palsy (PSP), 1 study fulfilled the inclusion criteria, from 8 returned by BiomedCentral, 2 by EBSCO host, 1 by PubMed/Medline, 207 by ScienceDirect, and 2 by Web of knowledge. For dystonia, 8 studies were in agreement with the inclusion criteria, from 15 returned by BiomedCentral, 7 by EBSCO host, 11 by PubMed/Medline, 363 by ScienceDirect, and 15 by Web of knowledge. For tremor, 1 study met the inclusion criteria, from 23 returned by BiomedCentral, 6 by EBSCO host, 8 by PubMed/Medline, 618 by ScienceDirect, and 9 by Web of knowledge. For Tourette’s syndrome, no study fulfilled the inclusion criteria, from 7 returned by BiomedCentral, 1 by PubMed/Medline, 245 by ScienceDirect, and 2 by Web of knowledge. For Huntington’s disease (HD), 9 studies were in line with the inclusion criteria, from 54 returned by BiomedCentral, 22 by EBSCO host, 31 by PubMed/Medline, 964 by ScienceDirect, and 52 by Web of knowledge. Finally, for Rett syndrome (RTT), 5 studies met the inclusion criteria, from 20 returned by BiomedCentral, 5 by PubMed/Medline, 638 by ScienceDirect, and 13 by Web of knowledge.
Zebrafish as a Model of Hypokinetic Movement Disorders
Parkinson’s disease and parkinsonism represent the most frequent hypokinetic syndromes. These include akinesia (inability to initiate voluntary movements), bradykinesia (slowness of voluntary movements), gait and balance disturbances (falls), freezing phenomenon (absence or marked reduction of forward stepping during walking), and rigidity (resistance to externally imposed joint movements).
Parkinson’s Disease
Parkinson’s disease is the most prevalent movement disorder, affecting 100–200 per 100,000 people (27). The etiology of PD is a combination of genetic and environmental factors that, at some point during disease progression, lead to dopaminergic cell loss in the substantia nigra pars compacta and to the accumulation of protein inclusions known as Lewy bodies. These inclusions are primarily composed of the protein α-synuclein (Table 1). Mutations in the alpha-synuclein (snca), leucine-rich repeat kinase 2 (lrrk2), vps35, PTEN induced putative kinase 1 (pink1), parkinsonism associated deglycase (dj-1), and parkin RBR E3 ubiquitin protein ligase (parkin) are associated with familial cases. However, the vast majority of the cases (~90–95%) are affected by sporadic PD. The treatment of motor symptoms aims at replacing dopamine and includes levodopa, among other dopaminergic modulators (Table 1). There are still no disease-modifying agents for PD, but several drugs are under clinical trials.
Chemical Zebrafish Models of PD
1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP)-Induced Models
Parkinson’s disease is the most studied movement disorder in zebrafish (Table 2). The effects of exposure to MPTP, known to cause loss of dopaminergic neurons and parkinsonism in humans (28), have been studied in zebrafish at all developmental stages (embryonic, larval, and adulthood). MPTP causes specific loss of dopaminergic neurons, a decrease of the dopamine, norepinephrine, and serotonin levels, and motility impairments in zebrafish larvae (29–32). By labeling monoaminergic neurons with GFP, Wen et al. (33) showed that the toxic effects of MPTP are more severe in the dopaminergic neurons located in the posterior tuberculum of the ventral diencephalon, corresponding to the mammalian midbrain dopaminergic neurons (33). Other neuronal clusters, including the serotonergic, also seem to be affected, but in less extent (32). By contrast, MPP+, the metabolite of MPTP, does not affect the serotonergic cluster, indicating a more specific action (32). MPP+ inhibits the multi-subunit enzyme complex I of the mitochondrial electron chain and impairs mitochondrial respiration (34). The subacute exposure to a low dose of MPP+ causes dramatic retrograde mitochondrial transport, before the appearance of neuronal and locomotor changes in zebrafish larvae (35). This suggests that MPP+ damages the mitochondria that are transported back to the cell body for degradation. This mechanism of bioenergetic homeostasis seems to be impaired at higher concentrations of MPP+. The MPTP-induced parkinsonian-like phenotype can be reverted by l-deprenyl (selegiline), an MAO-B inhibitor, and nomifensine, a DAT inhibitor (29, 30, 32). This indicates that the transport of MPTP and its conversion into the active metabolite, MPP+, is mediated by the same mechanisms in zebrafish and in mammals (34). Although selegiline is used as an anti-parkinsonian agent, its restorative effects in MPTP-lesioned zebrafish do not totally mimic the therapeutic effects in PD patients.
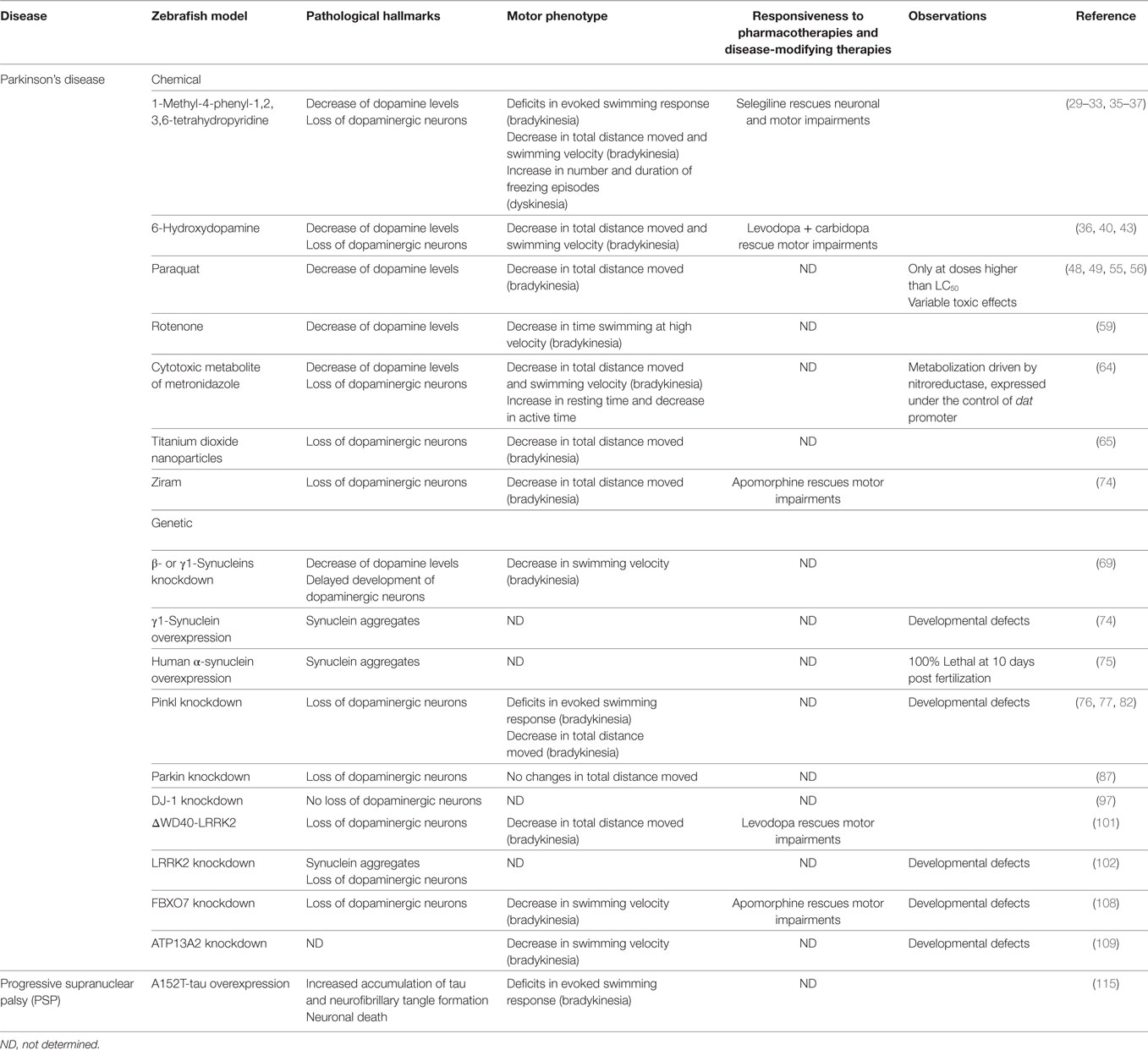
Table 2. General characteristics of the zebrafish models of hypokinetic movement disorders reviewed.
In adult zebrafish, MPTP induces a reduction of the levels of dopamine and norepinephrine, which results in marked decrease of the motor performance (31, 36). MPTP-lesioned adult zebrafish exhibit a significant decrease of the swimming velocity (bradykinesia), an erratic swimming pattern and increase of the freezing episodes (dyskinesia) (37). Strikingly, there is no reduction in the number of dopaminergic neurons, neither the activation of pathways that lead to cell death (31, 36). In mice, the activity of the TH decreases after exposure to MPTP (38). This could in part explain the loss of dopamine and norepinephrine in zebrafish, despite the lack of cellular death. In turn, MPTP could cause a transient loss of function of the dopaminergic neurons, instead of its death. A proteomic analysis in MPTP-lesioned zebrafish revealed altered transcriptional regulation of several genes, including lrrk2, dj-1, park2, and pink1. In addition, the expression of 73 proteins, some of which associated with neurological pathways, was also changed (37). For instance, the neurofilament light polypeptide-like (NEFL) protein, involved in the glutamatergic and GABAergic signaling in presynaptic nerve terminals, was found downregulated in the brain of MPTP-lesioned zebrafish. As demonstrated in zebrafish larvae, selegiline induces recovery of the PD-like symptoms in MPTP-lesioned adult zebrafish (37). On the other hand, adult zebrafish submitted to an MPP+ injection do not exhibit abnormal phenotype, in contrast to zebrafish larvae (31). MPP+ cannot cross the mammalian BBB (39), suggesting that in adult zebrafish the mature BBB prevents the entry of MMP+ into the central nervous system (CNS). The differences observed in the several developmental stages could result from a lower sensitivity of adult zebrafish to the neurotoxin. The access of MPTP to the brain is very different in each of the developmental stages, which in part could result from the distinct routes of administration adopted. Larvae were exposed to the neurotoxin through the water, because of its extreme permeability, whereas, adults received a single intramuscular or intraperitoneal injection.
6-Hydroxydopamine (6-OHDA)-Induced Models
6-Hydroxydopamine is a hydroxylated compound of dopamine that has been extensively used to induce dopaminergic lesions in rodents (34). Intramuscular injection of 6-OHDA causes a decrease of the dopamine and norepinephrine levels in adult zebrafish (36). Motor impairments are also observed, despite the lack of loss of dopaminergic neurons. By contrast, when administered into the ventral diencephalon of adult zebrafish, 6-OHDA induces significant ablation (>85%) of dopaminergic neurons in the posterior tuberculum and two other dopaminergic clusters, causing bradykinesia (40). The neuronal population in the olfactory bulb is one of the clusters affected. This mimics the phenotype in rats that exhibit olfactory impairments when lesioned with 6-OHDA into the substantia nigra (41). The inability to cross the BBB, explains the different phenotypes observed when 6-OHDA is administered intramuscularly or intracranially in zebrafish. A recovery of the dopaminergic neurons was observed 30 days post-lesion, which can be attributed to the neuro-regenerative capacity of the adult zebrafish brain (42). Therefore, the highly regenerative nature of zebrafish compromises the study of the progressive degenerative process in PD. On the other hand, the intracerebral administration of 6-OHDA revealed to be a laborious and meticulous protocol.
In the zebrafish larval stage, exposure to 6-OHDA in the water induces a decrease in the expression of TH, reduction in the locomotor activity and anxiogenic behavior (43). The locomotor impairments can be rescued by vitamin E, minocycline, and levodopa + carbidopa, the most effective drug used in patients with PD. Vitamin E is also able to normalize the expression of TH, while minocycline attenuates the increase of the expression of TNF-α and cd11b mRNA in 6-OHDA-lesioned zebrafish larvae. Vitamin E has antioxidant properties (44) and minocycline has shown anti-neuroinflammatory activity in rodents (45). This suggests that the oxidative stress and inflammatory process induced by 6-OHDA in zebrafish larvae share functional features with mammals.
Paraquat-Induced Models
Chronic exposure to pesticides, used in agriculture, has been recognized as a risk factor for development of parkinsonian syndromes. Paraquat induces oxidative stress and cytotoxicity in neurons (46). This herbicide is structurally similar to MPP+ and is associated with increased risk of developing PD (47). In adult zebrafish, the systemic administration of paraquat causes locomotor changes, but no anxiety-like behavior (48). In a different study, anxiolytic and aggressive behavior were observed, but no motor impairments in paraquat-lesioned adult zebrafish (49). While the former mimics the locomotor impairments, the later resembles the anxiogenic behavior in paraquat-lesioned rodents (50–52). The authors suggested that the results may be influenced by the genotype or gender of fish. Adult zebrafish lesioned with paraquat also reveal impairments in spatial memory, a decrease in the ratio of DOPAC/dopamine levels, a decrease in the expression of DAT, lowered mitochondrial viability, and an increase in the expression of antioxidant enzymes (48, 49). TH expression and reactive oxygen species (ROS) levels were unaltered. Despite the lack of cellular hallmarks of PD, the neurobehavioral syndrome in zebrafish is very similar to the one observed in paraquat-lesioned rodents (50, 51, 53). The neurotoxic effects of paraquat in this model are also variable (50, 51, 54).
Alternatively, when added to the water, paraquat seems to induce no parkinsonian-like phenotypes in larvae and adult zebrafish. These exhibit a normal number of dopaminergic neurons, normal behavior, and no developmental defects (31). Instead, when the LC50 of paraquat is used, zebrafish larvae exhibit a reduction of the dopamine and serotonin levels, activation of antioxidant and oxidative stress related genes, and distinct macrophage activation and migration (55, 56). In addition, significant increase of apoptotic cells in the head, trunk, and tail, and motor deficits are observed. However, the authors did not rule out general toxicity, leaving doubts about the specificity of the phenotypes observed. In addition, the toxic effects of paraquat seem to have high variability. Zebrafish larvae with 48 hpf exposed to 600 mg/L of paraquat do not show morphological defects, while 0.04 and 100 mg/L of paraquat were determined as LC50 in zebrafish larvae with 18 and 72 hpf, respectively. The successful induction of dopaminergic neuronal death by pesticides in rodents has also been striking. While paraquat is associated with high variability (57), rotenone induces weakly reproducible phenotypes and high mortality rates (58).
Rotenone-Induced Models
Exposure to rotenone has been linked to a higher risk of PD (47). In zebrafish, the phenotypes induced by rotenone are incongruent. One study found no cellular or behavioral parkinsonian-like phenotypes in larvae and adult zebrafish exposed to rotenone (31). By contrast, using the same concentration of the pesticide, time of exposure, and route of administration, a different study reported that adult zebrafish have decreased levels of dopamine and TH, deficits in motor function, anxiety and depression-like behavior, and olfactory dysfunction (59). Differences in TH expression and motor performance could be justified by the different protocols used to determine each of the parameters. In one study, the authors determined mRNA expression of TH by in situ hybridization and locomotor performance through the mean velocity of swimming. In the other, the authors analyzed TH expression by western blot and motor capacity through monitorization of freezing, swimming at low speed, and swimming at high speed. The difference between rotenone and vehicle treated zebrafish was detectable only at high speed swimming. The phenotypes observed are characteristic of PD patients (60, 61), and some mirror the phenotypes found in rodents (62, 63).
Other Neurotoxic Agents
Other less conventional methods have been used to induce dopaminergic neurotoxicity in zebrafish. This is the case of the transgenic line that expresses the reporter cyan fluorescent protein and the nitroreductase enzyme under the control of the dat promoter [Tg(dat:CFP-NTR)]. When transgenic larvae are exposed to metronidazole, the nitroreductase metabolizes it into a cytotoxic product that activates the apoptotic pathway and induces dopaminergic cell loss. The process can be monitored in real time, by detection of the reporter protein. Tg(dat:CFP-NTR) zebrafish larvae exposed to metronidazole show a reduction of the number of neurons in several dopaminergic clusters. This coincides with a decrease of dopamine levels and the appearance of locomotor impairments (64). The zebrafish line seems to maintain a persistent decrease of dopaminergic neurons that lasts longer than the other toxin-induced models.
Alternatively, a recent study showed that titanium dioxide nanoparticles (TiO2 NPs) cause parkinsonian-like phenotypes in zebrafish larvae (65). Exposure to TiO2 NPs induces premature hatching and abnormal development, but no lethality. TiO2 NPs accumulate in the brain of zebrafish larvae, resulting in the generation of ROS, loss of dopaminergic neurons, cell death in the hypothalamus and locomotor impairments. An increase in the expression of pink1, parkin, and uchl1 genes was also observed. Surprisingly, the authors also described an increase of the expression of the α-synuclein gene. Since zebrafish lack the ortholog of the human α-synuclein, the authors must have wanted to refer to the other two synucleins expressed in zebrafish (66). In rats, TiO2 NPs accumulate in the brain, stimulate oxidative stress and inflammatory responses, and cause impairments in the CNS (67). Despite the similarities of the phenotypes observed in zebrafish with the molecular and cellular mechanisms in PD, there is still no association of TiO2 NPs with increased risk of PD.
Genetic Zebrafish Models of PD
Synucleins
Among the several zebrafish genes with homology to human PD genes, an ortholog of the human α-synuclein appears not to be present in the zebrafish genome (68). Instead, zebrafish express three synuclein isoforms, β-, γ1-, and γ2-synucleins, that seem to compensate the absence of α-synuclein. Functionally, the zebrafish γ1-synuclein appears to be the closest to the human α-synuclein. Knockdown of the β- or γ1-synucleins induces motor impairments in zebrafish, which are even more severe when the expression of both synucleins is abrogated (69). Zebrafish lacking both synucleins have an abnormal development of the dopaminergic system, including delayed differentiation of dopaminergic neurons and reduced levels of dopamine. The phenotype can be reverted by the expression of human α-synuclein. Strikingly, the knockdown of the zebrafish β- and γ1-synucleins results in phenotypes that recapitulate the aspects observed in rodents lacking all synucleins (70–73). In zebrafish, overexpression of γ1-synuclein leads to the formation of neuronal aggregates and neurotoxicity, similarly to the human α-synuclein (74). On the other hand, downregulation of γ1-synuclein protects zebrafish from the toxicity of ziram. Exposure to ziram dramatically increases the risk to develop PD. This pesticide causes loss of dopaminergic neurons and impaired swimming behavior in zebrafish (74). Treatment with apomorphin recues the motor impairments. Moreover, CLR01, an inhibitor of amyloidogenic proteins self-assembly, protects zebrafish against ziram-induced neurotoxicity. These data suggest that ziram might induce toxicity on dopaminergic neurons through the formation of γ1-synuclein toxic oligomers. Still, none of the above zebrafish lines exhibit as a severe phenotype as zebrafish overexpressing human α-synuclein. During embryonic development, this line presents neuronal apoptosis, which results in severe deformities and death within 48–72 h (75). The neurotoxic effect of α-synuclein is mediated by the inhibition of the ubiquitin proteasome system and accumulation of α-synuclein. Treatment with CLR01 reduces the aggregation of α-synuclein and neuronal apoptosis, increasing viability. The devastating effects of the overexpression of human α-synuclein may hinder the successful generation of transgenic zebrafish lines. Perhaps, by restricting the expression of the protein to the dopaminergic neurons or to a transient manner could decrease the lethality, while maintaining the pathological mechanisms.
PTEN Induced Putative Kinase 1 (PINK1)
The gene PINK1 is implicated in genetic and sporadic cases of PD. Morpholino knockdown of the zebrafish PINK1 ortholog has added evidence to the importance of this gene in the control of oxidative stress and mitochondrial function. Downregulation or total abrogation of the expression of PINK1 results in mitochondrial dysfunction that leads to augmented levels of ROS and activation of the apoptotic signaling pathway in zebrafish (76). The PINK1 null mutant zebrafish line also presents mitochondrial impairments (77). The zebrafish Pink1 influences the expression of other proteins that are critical contributors to the pathogenic process. For instance, the activity of the mitochondrial protein GSK3β is increased and its inhibition, with LiCl and SB216763, partially rescues the phenotypes in PINK1 morphant zebrafish (76). TigarB, the zebrafish ortholog of the human glycolysis and apoptosis regulator Tigar, is also markedly increased in Pink1 null mutants (77). Tigar has been identified as a negative regulator of mitophagy, considered to be crucial in the pathogenesis of early-onset PD (78). The expression of other 177 genes, from the hypoxia-inducible factor (HIF) signaling, TGFβ-signaling, and several key toxicological responses (mitochondrial dysfunction, RAR activation, and biogenesis of mitochondria), is also altered (79). Particularly, the HIF pathway is the most affected pathway in PINK1 knockdown zebrafish. This is known to participate in the regulation of oxidative stress and neuronal differentiation in vitro (80, 81).
Whereas, the molecular mechanisms seem to be consistent, the phenotypes induced by the alteration of Pink1 expression in zebrafish vary. PINK1 morphant zebrafish exhibit general developmental delay, severe mispatterning of the axonal scaffold, and moderate decrease of the number of neurons, mainly in the dopaminergic system (76). The phenotype could be rescued by wild-type human pink1 mRNA. Further supporting these results, PINK1 knockdown zebrafish show changes in neuronal patterning and axonal projections (82). This line also presents mild loss of dopaminergic neurons in the diencephalon, which leads to spontaneous or evoked locomotor impairments. Motor performance could be rescued by the dopamine agonist SFK-38393. Moreover, the expression of exogenous PINK1 rescued all phenotypes. By contrast, a subsequent study reported no morphological or behavioral deficits in PINK1 morphant zebrafish (83). Instead, increased vulnerability to MPTP-induced toxicity was observed. The authors described a reduction in the expression of both, th1 and th2 mRNA forms, but normal levels of dat mRNA. Although there was a mild decrease in the number of TH-positive neurons in the dopaminergic diencephalic cluster, the normal levels of dat suggest that the downregulation of PINK1 may cause a decline in important mRNAs and proteins, instead of neuronal death. Still, the increased susceptibility to MPTP strengthens the importance of PINK1 in oxidative stress. In accordance, the exposure to H2O2 dramatically increases the expression of pink1 mRNA in zebrafish, which can be reverted by the antioxidant l-glutathione reduced (84). In PINK1 mutant zebrafish larvae, the loss of dopaminergic neurons is more evident and accompanied by marked microglial activation (77). In fact, PINK1 deficiency causes different phenotypes in mammals, as well. In humans, mutations in PINK1 can result in early-onset PD and are associated with mitochondrial dysfunction (85). In turn, PINK1 knockout mice have a mild phenotype, with no neuronal death, no changes in the levels of striatal dopamine, nor in the number and morphology of mitochondria (86). Interestingly, it seems that PINK1 knockdown zebrafish recapitulate better than mice the human phenotypes.
Parkin RBR E3 Ubiquitin Protein Ligase (Parkin)
Mutations in PINK1 and Parkin are implicated in mitochondrial dysfunction and seem to share several pathogenic mechanisms in PD. Consistently, Parkin knockdown zebrafish exhibit a phenotype that resembles PINK1 knockdown zebrafish. Abrogation of the expression of Parkin leads to impaired mitochondrial function, specific loss of dopaminergic neurons in the posterior tuberculum, and increased sensitivity to the toxic effects of MPP+ (87). Neither the serotonergic nor the motor neurons are affected, and the extent of dopaminergic loss is not enough to cause behavioral defects. Parkin knockdown zebrafish present two important phenotypes, mitochondrial dysfunction and dopaminergic cells loss, described in PD patients with mutations in Parkin (88, 89). Once again, it seems that the zebrafish model mimics better than mice models the pathological mechanisms in humans. Parkin knockout mice do not show any robust morphological changes, neither increased susceptibility to MPP+ (90, 91). Notwithstanding, morpholino-mediated knockdown of the zebrafish Parkin generates very dissimilar phenotypes. In a different study, no loss of dopaminergic neurons, neither morphological nor behavioral alterations, was observed upon Parkin knockdown in zebrafish (92). This result may be the consequence of a partial ablation (around 50%) of Parkin expression. Still, this line maintained the increased vulnerability to stress-induced cell death. Importantly, the authors also described that the overexpression of Parkin in a transgenic zebrafish line protects from proteotoxic stress-induced cell death. Similarly to PINK1, Parkin is suggested to have a protective role in PD (93). In zebrafish, it seems that the presenilin-associated rhomboid-like (PARL) protein is also part of the PINK1 and Parkin pathway. PARL is a component of the mitochondrial membrane involved in mitochondrial morphology and apoptosis. Morpholino knockdown of both zebrafish paralogs, parla and parlb, results in high mortality, whereas loss of PARLb leads to the mildest phenotype (94). Loss of one of the PARL’s results in mild neurodegeneration and disarranged dopaminergic neurons. Although changes in survival were not reported, generalized cell death was observed. Interestingly, the phenotype can be rescued by human parl mRNA and by zebrafish and human pink1 mRNA. The PARL gene has been linked to familial cases of PD. PARL is suggested to be important in the normal trafficking and processing of PINK1 and Parkin in mitochondria (95).
DJ-1
Mutations in DJ-1 are associated with early-onset PD. The inactivation of DJ-1 in zebrafish leads to an increase in the expression of p53 and Bax, but no cellular or morphological changes (96, 97). Moreover, the concomitant knockdown of DJ-1 and mdm2, a negative regulator of p53, results in dopaminergic neuronal death (96). This suggests that p53 may mediate cell loss in the absence of DJ-1. DJ-1 knockdown zebrafish exhibit loss of dopaminergic neurons after exposure to H2O2 and to the proteasome inhibitor MG132. The phenotype can be prevented with pharmacological inhibition of p53, by pifithrin-alpha (96, 97). This demonstrates that DJ-1 knockdown zebrafish are susceptible to programmed cell death and that DJ-1 may mediate the stress response machinery. In accordance, DJ-1-deficient mice only exhibit dopaminergic cell death after toxin exposure (98). The p53-glycerylaldehyde-3-phosphate dehydrogenase (GAPDH)–Bax pathway has been suggested to be involved in PD (99, 100).
Leucine-Rich Repeat Kinase 2 (LRRK2)
The phenotypes of LRRK2 morphant zebrafish have been characterized. The first study describing the consequences of the inhibition of the expression of LRRK2, showed embryonic lethality and severe developmental defects, such as brain developmental retardation, in zebrafish (101). A more recent study, described neuronal loss, affecting the dopaminergic system, upregulation of the expression of β-synuclein, Park13, and SOD1, and β-synuclein aggregation in the CNS (102). The authors also described a wide range of organ abnormalities but did not report such overt toxicity as the former study. On the other hand, the deletion of the WD40 domain of LRRK2 (ΔWD40-LRRK2) causes little impact on embryonic development, in zebrafish (101). Instead, this mutant zebrafish line presents a reduction and disorganization of the axonal tracts, predominantly in the midbrain. In addition, significant loss of the diencephalic dopaminergic neurons and locomotor defects were observed. The phenotype can be rescued by zebrafish and human lrrk2 mRNA overexpression. The administration of levodopa rescues the motor impairments, but not neurodegeneration, in line with the therapeutic effects in humans. Surprisingly, a subsequent study was not able to replicate the phenotypes described in the ΔWD40-LRRK2 zebrafish line (103). Nevertheless, it has been reported that mutations in the LRRK2-WD40 domain increase neuronal apoptosis under cellular stress (104). In mice, LRRK2 knockout and G2019S LRRK2 transgenesis do not induce neuropathological abnormalities, but LRRK2 seems to interfere with normal neurite outgrowth (105, 106).
Other PD Genes
Genes implicated in atypical PD, have also been characterized in zebrafish. Park15 is caused by loss of function of the protein encoded by the gene fbxo7 (107). Morpholino knockdown of the zebrafish Fbxo7 ortholog results in abnormal patterning and loss of dopaminergic neurons, which lead to severe motor impairments (108). Treatment with apomorphine, a dopamine agonist, can revert the locomotor defects. Surprisingly, the human fbxo7 mRNA failed to rescue the morphological phenotypes. This observation was justified with an atypical timing and localization of expression, as compared with the expression of the Fbxo7 endogenous zebrafish gene. Fbxo7 morphant zebrafish also exhibit developmental defects, as heart deformations, suggesting that Fbxo7 must have an important role in zebrafish development. The zebrafish ortholog of another gene linked to atypical PD, atp13a2, has also shown a crucial role during embryonic development. Complete abrogation of the expression of ATP13A2 leads to embryonic lethality, whereas partial knockdown results in abnormal splicing of atp13a2 mRNA and obvious behavioral impairments (109). The results are in line with experiments in mice, which have also revealed the importance of ATP13A2 during the early stages of embryonic development and neurogenesis (110).
Effect of Dopaminergic Modulators in Zebrafish
When assessing the validity of an animal model, beyond the pathological hallmarks of the disease, it is important to explore the pharmacologically evoked changes. Studies that report such experiments in zebrafish models of PD are scarce, but there exist some reports exploring the effects of drugs known to modulate movement on healthy zebrafish. For instance, haloperidol and chlorpromazine, two dopamine receptor antagonists, have been tested on zebrafish larvae. The suppression of the dopaminergic signaling by both compounds induces akinetic-like behavior (111). Another study supporting these data showed that the selective dopamine agonists, SFK-38393 and quinpirole, increase motor activity (112). By contrast, the dopamine antagonists, SCH-23390 and haloperidol, decrease motor activity in zebrafish larvae. Interestingly, the non-selective dopamine agonist, apomorphine, and dopamine antagonist, butaclamol, induce biphasic dose-response patterns. This may be attributed to the action of the drugs on multiple dopaminergic receptors. On the other hand, the dopamine antagonists, SCH-23390 and haloperidol, induce different dose-response profiles dependent on the lighting conditions. The authors suggested that the blockade of dopamine receptors in the retinal ganglion cells may have perturbed the adaptation to light/dark conditions. Alternatively, the effects of haloperidol were studied in catalepsy (muscular rigidity). Similarly to rats, it was observed that haloperidol causes the increase of catalepsy in zebrafish (113). This can be reverted by bromocriptine and pramipexole, two dopamine agonists commonly used to improve rigidity. Importantly, this study introduced a new core motor symptom of PD on zebrafish, in alternative to bradykinesia. All the above-mentioned studies demonstrated that the drugs that target the dopaminergic system in mammals elicit similar outcomes in zebrafish, suggesting that the underlying mechanisms that regulate movement are shared by both models. Nevertheless, the demonstration that the phenotypes observed in zebrafish actually result from modulation of the dopaminergic pathway is needed.
Overall, chemical and genetic zebrafish models of PD reproduce several of the biochemical, neurochemical, morphological, and neurobehavioral features of the disease in humans. Importantly, the pharmacological response to drugs used in the clinic is also conserved. The limitations inherent to each model do not seem to surpass the limitations also described in rodents and, in some cases, zebrafish resemble better than rodents, the human features. Finally, the zebrafish genes orthologs to the human genes associated with PD seem to be particularly conserved in terms of sequence and function, as well as, the role of the respective protein in the cellular pathways.
Other Parkinsonian Syndromes
Progressive supranuclear palsy is a PD-plus syndrome associated with tau neuropathology, which affects about 5–7 per 100,000 people (114). The neuropathological hallmarks include the presence of neurofibrillary tangles (insoluble 4-repeat tau protein) or neuropil threads in the basal ganglia and brainstem. Neuronal loss is diffuse, affecting different neuronal structures (Table 1). Initially, the clinical presentation is heterogeneous but tends to develop to unsteady gait, bradykinesia, unexplained falls, and ocular motor deficits (vertical supranuclear gaze palsy is used to confirm the diagnose). Most of the cases of PSP are sporadic and associated with polymorphisms in the gene that encodes the tau protein, MAPT. Mutations in the MAPT gene have been identified in several familial cases, as well, but are rarer.
Recently, zebrafish was used to assess the functional and biochemical consequences of a tau variant, p.A152T, identified to increase the risk of PSP in a cohort study (115). The A152T-tau transgenic zebrafish exhibit increased accumulation and phosphorylation of tau, with formation of neurofibrillary tangles (Table 2). The phenotype possibly results from impairments in the proteasome system. The overexpression of A152T-tau also causes neurodegeneration, associated with behavioral deficits in zebrafish. The phenomenology is compatible with the phenotypes observed in A152T transgenic rodent models (116, 117). The use of zebrafish to model PSP is at its beginning and needs further developments. Nevertheless, the data described so far indicate that zebrafish models of PSP may exhibit several neuropathological hallmarks of the disease. Effective pharmacotherapeutic options for PSP are null at the moment, and zebrafish may help to boost the discovery of new drugs.
Zebrafish as a Model of Hyperkinetic Movement Disorders
Hyperkinetic movement disorders have a more diverse phenomenology and include tremors, dystonia (sustained, repetitive, and patterned muscle contractions), tics (sudden, rapid, repetitive, and non-rhythmic movements), chorea (brief, irregular, abrupt, and non-repetitive movements), and stereotypies (repetitive or ritualistic movements), among others.
Dystonia
Dystonia is a common and clinically heterogeneous disorder, which can be manifested as an isolated clinical condition (primary dystonias) or associated with other neurological disorders (secondary dystonias) (118). The cause is diverse and includes genetic and environmental factors (Table 1). This phenomenon is believed to result from malfunction of the basal ganglia and, consequently, abnormal plasticity of the sensorimotor cortex (119). The pharmacotherapy for dystonia is solely symptomatic, mostly empirical, and adapted to each case (Table 1) (120). In the last 5 years, zebrafish have been used to understand the mechanisms of dystonia.
Genetic Zebrafish Models of Dystonia
The most common cause of early-onset primary dystonia is a mutation in the TOR1A gene (121). In zebrafish, the ortholog gene, tor1, is not essential for early development of the motor system. The morpholino-mediated knockdown zebrafish line presents normal viability, morphology, development, and behavior (Table 3) (122). Zebrafish TOR1 may have an important role in later events, but the transient effect of morpholino-mediated knockdown did not allow to confirm this fact. Accordingly, in TOR1A knockout mice, the first phenotypic abnormalities are only observed in a later developmental stage (123). Other genes implicated in dystonia are involved in neuronal development and brain maturation in zebrafish. For instance, dystonia in early childhood can be caused by an autosomal recessive mutation in the pantothenate kinase 2 (PANK2) gene (124). Morpholino knockdown of the zebrafish ortholog perturbs the neuronal development and brain morphology and induces hydrocephalus (125). The phenotype can be rescued by pantethine and coenzyme A. To further test the implication of mutations identified in the col6a3 gene of subjects with primary dystonia, a study found that morpholino knockdown of the zebrafish ortholog gene causes deficits in axonal outgrowth (126). The authors suggested that Col6a3 may participate in the structural organization of neurons. Therefore, its disruption can hamper the establishment of correct neuronal circuitries and synaptic remodeling processes, during brain development and maturation. Finally, knockdown of the ortholog of the human Atp1a3 leads to brain ventricle dilation and depolarization of Rohon–Beard neurons in zebrafish (127). Although response to tactile stimuli and motility are altered, the dopaminergic neurons seemed to be unaffected. Mutations in the Atp1a3 are implicated in rapid-onset dystonia parkinsonism (RDP) (128). Ventricle dilation is not observed in patients with RDP, but numerous symptoms reported in dystonic patients suggest the involvement of the somatosensory system. In turn, the depolarization of Rohon–Beard neurons in zebrafish is indicative of altered neuronal excitability, also described in rats (129).
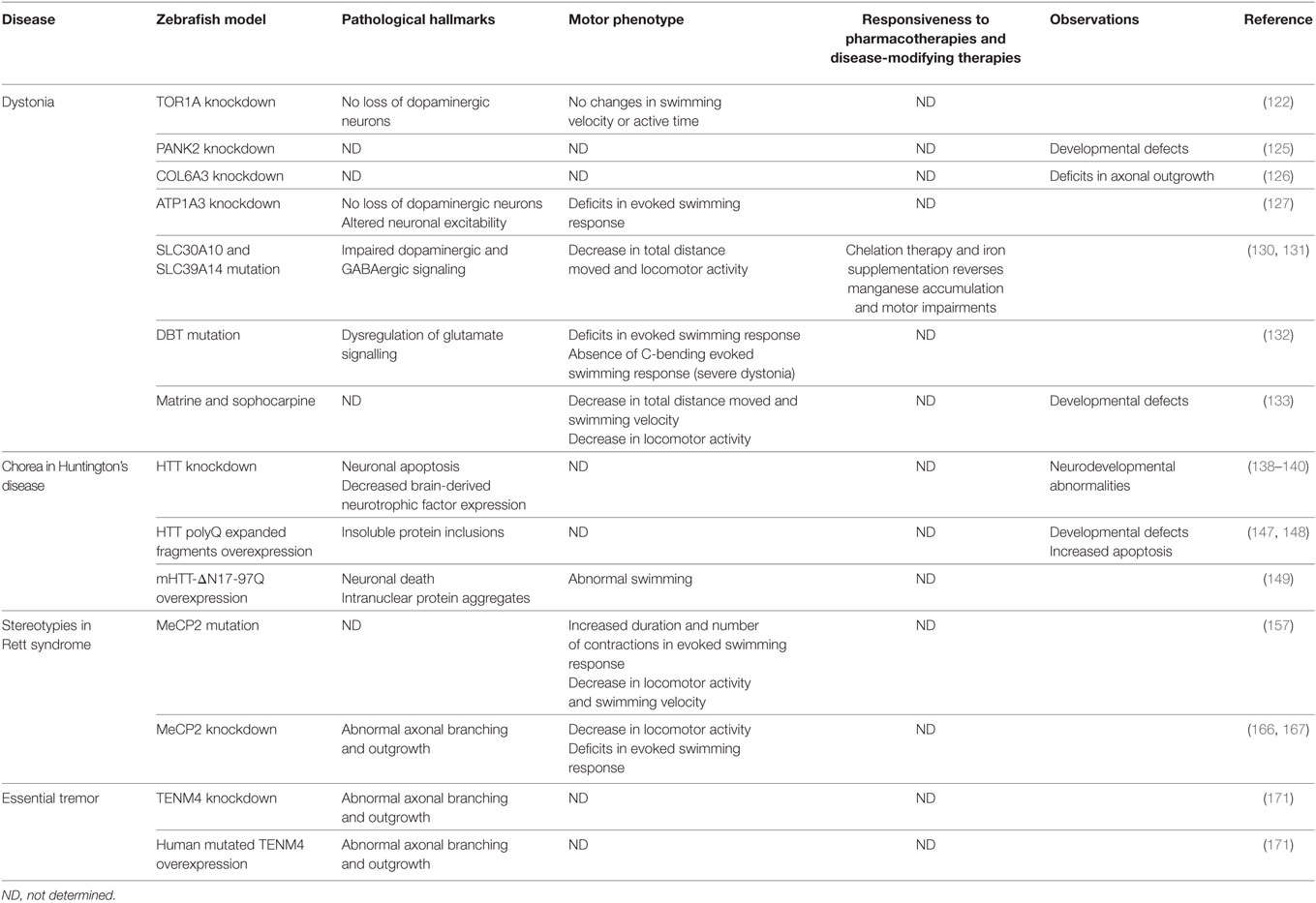
Table 3. General characteristics of the zebrafish models of hyperkinetic movement disorders reviewed.
In metabolic disorders, the risk to develop dystonia increases when the manganese homeostasis is compromised. In zebrafish, the mutation of the orthologs of the human manganese transporters, slc30a10 and slc39a14, results in manganese accumulation in the brain (130, 131). This leads to impaired dopaminergic and GABAergic signaling. Changes in the swimming pattern are also visible upon exposure to manganese. The phenotype can be reverted by chelation therapy and iron supplementation, currently used in the clinical practice. Maple syrup urine disease is another metabolic disorder, caused by mutations in the dihydrolipoamide branched-chain transacylase E2 (DBT) gene, which can result in severe dystonia. In zebrafish, disruption of the ortholog gene results in elevated levels of branched-chain amino acids (BCAA) (132). This phenomenon is also evidenced in mammalian models and patients. The increase of BCAA leads to the dysregulation of the neurotransmitter glutamate in the brain and spinal cord of zebrafish, which probably contributes to the progressive aberrant motility behavior evidenced. This phenotype was suggested to represent severe dystonia in zebrafish larvae.
Chemical Zebrafish Models of Dystonia
Zebrafish are sensitive to neurotoxic drugs that may cause dystonia. Matrine and sophocarpine, two drugs responsible for poisoning juvenile and infant patients, induce growth retardation in zebrafish (133). Exposure to these drugs also led to changes in spontaneous movements and locomotor performance. The authors suggested that this phenotype results from the neurotoxic effects of the drugs but did not show evidence of neurotoxicity.
In summary, it appears that the heterogeneous nature of dystonia can be reproduced in zebrafish. Zebrafish offers an excellent opportunity to understand the pathogenic mechanisms behind the vast number of genetic and environmental factors linked to dystonia. Nevertheless, this heterogeneity seems to result in a large number of studies, with minor characterization and no consolidation of the observed phenotypes. This may undermine a correct judgment about the validity of zebrafish as a vertebrate model of dystonia. Most certainly, further studies are essential.
Chorea in Huntington’s Disease
Chorea is the most common symptom of HD, which affects around 4–10 per 100,000 people in the western world (134). HD is an autosomal-dominant neurodegenerative disorder, which results from abnormal expansion of the CAG repeat in the huntingtin (HTT) gene (polyglutamine disease) (Table 1). The pathogenesis of HD results from the toxic effects of the mutant HTT RNA and protein, HTT aggregation (intranuclear inclusions of abnormal HTT are pathological hallmark) and impairments in protein homeostasis and clearance. These events lead to the death of GABAergic medium spiny neurons in the striatum. Chorea can be improved by tetrabenazine, and the therapeutic effects of other drugs are only empirical (120). There is currently no disease-modifying treatment for HD.
Genetic Zebrafish Models of HD
The zebrafish ortholog of the human HTT only encodes four glutamines, compared with up to 35 in humans (135). The zebrafish HTT protein is essential for iron, lipid, and cholesterol homeostasis (136, 137), energy metabolism (136), and brain development (138–140). Its knockdown leads to diverse phenotypes, including hemoglobulin deficiency (136), neuronal apoptosis in the midbrain and hindbrain (138, 139), neurophysiologic abnormalities (138–140), decreased expression of brain-derived neurotrophic factor (BDNF) (138), deficient formation of neural tubes and cell adhesion (140), increased activity of metalloproteinases (ADAM10 and Ncadherin) (140), and severe reduction in cartilage biogenesis (Table 3) (137). Consistently, patients with HD exhibit deficits in iron homeostasis (141, 142), energy metabolism (143), BDNF expression (144), and metalloproteinases activity (145). As observed in HTT knockout mice, the complete abrogation of the expression of zebrafish HTT results in embryonic lethality (138). In fact, the mouse models of HD either lack overt phenotypes or exhibit premature death (146). This makes the HTT morphant zebrafish a valuable alternative model to study the cellular function of HTT and its role in the pathological mechanisms of HD. Nevertheless, morpholino-mediated knockdown may result in variable phenotypes and must be considered cautiously.
Zebrafish lines expressing normal and expanded polyglutamine (polyQ) fragments of HTT have been reported (147, 148). In these lines, the misfolding, oligomerization, aggregation, and toxicity of the polyQ fragments are length dependent, in a manner similar to that observed in other animal models and in patients. Zebrafish embryos overexpressing fragments with more than 35Q repeats (mutant form) display insoluble protein inclusions and increased apoptosis (148). This leads to abnormal morphology and development. Strikingly, apoptosis can be detected in cells with no visible inclusions, suggesting that the oligomeric forms of the HTT may be the toxic components. The ubiquitous expression of the polyQ proteins may contribute to the severe phenotype observed. In alternative, it would be interesting to observe the effects of polyQ expression restricted to the CNS. The toxic effects and aggregation of the mutant fragments can be suppressed either by the chaperones Hsp40 and Hsp70 or by the ubiquitin ligase C-terminal Hsp70-interacting protein, resembling other HD models.
Remarkably, the expression of a mutated polyQ fragment lacking the 17 amino acids of the HTT N-terminal tail (mHTT-ΔN17-97Q) elicits toxicity only in neuronal cells of a transgenic zebrafish line (149). Particularly in neurons, mHTT-ΔN17-97Q fragments rapidly form massive intranuclear aggregates. This demonstrates that the neuronal cells have lower capacity to maintain the proteostasis of the expanded polyQ fragments. Moreover, the mHTT-ΔN17-97Q fragments tend to aggregate more and induce a more severe phenotype than the polyQ fragments with an intact N17 terminal. In mice, the expression of the htt-97Q gene lacking the N17 causes dramatic accumulation of nuclear mutant HTT aggregates and a robust striatal neurodegeneration that leads to adult-onset movement disorder (150). These results suggest that the N17 portion of the HTT protein substantially prevents the translocation of mutant HTT into the nucleus and plays an important role in the molecular mechanisms of the pathogenesis of HD. mHTT-ΔN17-97Q transgenic zebrafish are the first to recapitulate one of the pathological hallmarks of HD.
Chemical Zebrafish Models of HD
The administration of quinolinic acid (QA) into the striatum of adult rodents has been used to induce brain injury that replicates HD (151). Interestingly, while inducing injury, this excitotoxin also stimulates the subventricular neurogenesis zone and neuroblast migration (152, 153). This observation encouraged Skaggs and colleagues (154) to lesion the telencephalon of adult zebrafish with QA and study its neuronal effects. The QA induces cell death and microglial infiltration in the zebrafish CNS (154). However, it also stimulates cell proliferation and neurogenesis that results in total repair of the damage. The authors suggested that this zebrafish model is a powerful tool to study neuronal regeneration in an adult vertebrate and to test potential disease-modifying therapies. Still, the neurogenesis process in the CNS of adult zebrafish is very different from mammals, which might render the translation of the observations puzzling.
In general, the zebrafish HTT shares several important functions with the mammalian ortholog. Zebrafish lines that overexpress mutant HTT are proving to be useful to model HD. Nevertheless, there is only a shallow description of the motor phenotypes, and the responsiveness to pharmacotherapies still needs to be tested on these models.
Stereotypies in Rett Syndrome
Rett syndrome is a non-neurodegenerative disorder, which affects 1 in 10,000 females by the age of 12 (155). Caused by either nonsense or missense mutations in the methyl-CpG-binding protein 2 (MECP2) gene, RTT is characterized by hand stereotypies (Table 1). MeCP2 is a nuclear protein that recognizes DNA methylation to, presumably, regulate gene expression and activation. The patients with RTT, exhibit abnormally small and densely packed neurons, with reduced dendritic complexity and synaptic density. At the cellular level, alterations in different signaling and homeostatic pathways are reported, along with mitochondrial dysfunction and oxidative stress. The options available for the treatment of RTT are currently limited, but several compounds are under clinical trials. These include modulators of neurotransmitters or regulators of cellular metabolism and homeostasis (Table 1).
Genetic Zebrafish Models of RTT
In mice, null mutations in MeCP2 drastically reduce lifespan (156). By contrast, zebrafish carrying a null mutation in MeCP2 show normal viability and fertility (157). Instead, MeCP2-null zebrafish exhibit clear motor impairments at early developmental stage (Table 3). These include spontaneous and sensory-evoked motor anomalies, and defective anxiety-like behavior. This zebrafish line has a nonsense mutation in the methyl-CpG binding domain (mecp2Q63X), crucial for protein function. The authors suggested that the modest phenotype observed may result from a compensatory mechanism triggered by other proteins belonging to the MeCP2 family or from gene duplication in zebrafish. On the other hand, the studies exploring thigmotaxis in mouse models of RTT have yielded confounding results, some of them contradictory to the ones observed in zebrafish. The authors advocated that the complexity of the neuronal circuitry in mice may have hampered the interpretation of the results in behavioral tests. Later proteomic analysis in the MeCP2-null zebrafish line revealed changes in the expression of proteins critical for energy metabolism, balance of redox status and muscle function (158). This is in line with the reported in RTT patients and experimental mouse models (159–164).
Additional studies in zebrafish confirmed the essential role of MECP2 in neuronal differentiation (165), axonal branching of primary motor neurons (166), and peripheral innervation of sensory neurons (167). The studies further proved that MeCP2 regulates the expression of several cell differentiating factors (Id1–Her2 axis), BDNF and axonal guidance cues (such as Sema5b and Robo2). The indirect disruption of the expression of these genes is involved in RTT-like phenotypes. Accordingly, downregulation of MeCP2 induces a decrease in motor activity and impairments in the sensory function of zebrafish, as observed in mice with partial loss of MeCP2 (168).
All these studies strengthened the notion that the zebrafish MeCP2 is crucial for the regulation of gene expression and activation. Therefore, Mecp2-deficient zebrafish have several phenotypes, reminiscent of the phenomenology observed in mouse models of RTT and in patients with RTT. The normal lifespan in MeCP2-null zebrafish may enable a more profound characterization of the pathophysiological dynamics of RTT and the screening of new drugs.
Other Hyperkinetic Syndromes
Tremor is a rhythmic oscillation of a body part. Besides resting tremor in PD, this phenomenology is mostly common in the neurologic disorder essential tremor (ET) (120). Progressive action tremor is the classic feature of ET. The most effective drugs for the treatment of ET are propranolol and primidone (Table 1) (169). However, these drugs induce highly variable therapeutic effects and are associated with several adverse effects. The anticonvulsant, topimarate, the GABA agonist, gabapentine, and several benzodiazepines have also shown to improve tremor. The pathological mechanisms and etiology of ET are highly heterogeneous (170). In addition, these are difficult to identify, because ET is commonly a comorbidity. Several genetic and environmental risk factors have been suggested, but none was consistently confirmed in larger cohort studies. This renders the discovery of effective pharmacologic treatments particularly difficult.
Zebrafish may be a practical choice to unravel some of the pathological mechanisms and risk factors implicated in ET. This model has been used to explore the physiological role of TENM4 and the pathological effects of mutations identified in the TENM4 of families with ET. Morpholino knockdown of the TENM4 zebrafish ortholog results in a modest reduction of myelination and aberrant extension, branching and architecture of small axons in the CNS of zebrafish (Table 3) (171). Zebrafish expressing mutated human TENM4 mRNA show a similar phenotype. These observations are concordant with studies in other animal models. Since 2015, no other study reported the generation of a zebrafish model of tremor, possibly because of the limited knowledge about the nature of ET.
Perhaps for the same reason, at the time of this review, no zebrafish model of Tourette’s syndrome has been reported. There is limited understanding of the etiology of this multifactorial syndrome (Table 1), because several genetic variants and mutations, and non-genetic determinants are implicated. Moreover, these are not exclusive of the disease. This heterogeneity results in a complex and variable neurological and clinical phenomenology. Tourette’s syndrome is typically manifested by various motor and phonic chronic tics (172). The cortico–striato–thalamocortical circuitry is potentially impaired, but the specific neuronal pathway(s) involved remain unknown. Neuroleptic drugs and the monoamine depleting drug, tetrabenazine, are the most effective for tic suppression (Table 1). There exist different rodent models of tics, but their validity is debatable. The inability to assess several human symptomatic features, such as premonitory sensation, is an important limitation of these animals. While the same skepticism can apply to zebrafish, zebrafish models of Tourette’s syndrome may help to elucidate the interplay between genetic and non-genetic risk factors. Ultimately, this may provide valuable clues about the neuropathology of this syndrome.
Discussion
Zebrafish have been extensively used in the study of the CNS. More recently, the use of zebrafish as a model of human brain diseases and for drug discovery has increased (2, 173). Here, we review several zebrafish models of movement disorders and discuss their translational value. Overall, these models exhibit conserved biochemical and neurobehavioral features. In retrospect, many advantages can be named, but pitfalls must also be highlighted.
First, all studies used zebrafish during embryonic, larval or young adult stage. Practical reasons can justify the use of zebrafish at these developmental stages. For instance, at embryonic and larval stage zebrafish are more permeable, enabling the delivery of drugs through the water. In higher vertebrates, some of the neurotoxic agents used to model movement disorders have to be delivered directly in the brain (174). This approach increases variability between animals and is much more invasive, resulting in the death of some animals. Furthermore, the time lapse between drug administration and appearance of the first phenotypes is much longer in rodents than in zebrafish. Nevertheless, the use of zebrafish larvae is not as accurate as the use of adult zebrafish, where the BBB is fully functional and better mimics the mammalian physiology. The use of zebrafish at early developmental stages also allows to explore the function of specific genes during system development and maturation. However, it must be considered carefully when extrapolating the phenotypes observed to chronic and late-onset disorders. Moreover, the mutant and transgenic lines described here were characterized very early during development, and whether these lines display any pathology in adulthood was not reported. Inevitably, the brain of zebrafish is more complex at adulthood and may mimic more accurately the physiologic features of the mammalian brain.
In turn, morpholino-mediated knockdown is extensively used, because it is a practical tool to reveal the phenotypes induced by downregulation of a gene in zebrafish, but it also has many pitfalls. The extension of abrogation of the expression of the gene can vary drastically, depending on the knockdown strategy and efficiency. Zebrafish possess duplicates of several genes, which can result in a differentiated regulation of gene expression and different phenotypes. In addition, morpholino-mediated knockdown has an acute and transient effect, which does not mimic the chronic effects of downregulation of a gene. On the other hand, non-specific and off-target effects are common, most of the studies lacked the right controls to rule out these effects and, therefore, could not exclude that other systems could be affected. Finally, the knockdown of a gene may not be suited to model some diseases. This is the case of HD, as loss of function of the HTT protein alone does not seem to lead to the disorder (175). The genetic manipulation of zebrafish is particularly easy and should be more exhaustively explored. The mutated or transgenic zebrafish lines develop rapidly, which is an advantage when compared with the time-consuming development of rodent models. When developing transgenic lines, neuronal promoters should be used, to prevent overt toxicity of the transgene. This has been observed for zebrafish lines that overexpress human α-synuclein or HTT polyQ fragments with non-specific promoters (75, 148). In rodents, neuronal promoters are commonly used to restrict the expression of the proteins to the CNS (176).
Another limitation of most of the studies is the restriction of the characterization of the zebrafish model to a single cellular biomarker or behavioral parameter. For instance, most of the studies in zebrafish models of PD determined the expression of TH to assess the integrity of the dopaminergic system, but DAT would be a more specific biomarker for this neuronal population. The zebrafish proteomics is conserved, which allows the use of commercially available biomarkers of other species. Several of these markers have already been tested on zebrafish targets, and many present similar reactivity. In turn, almost all studies limited the evaluation of behavioral changes to total locomotor activity and/or speed, which does not represent the multi symptomatic nature of movement disorders. The assessment of these parameters in zebrafish is a sound strategy, because they are related to the parameters used in rodents to depict bradykinesia. Nevertheless, zebrafish possess a diverse repertoire of behaviors with homology to humans, which have been cataloged and can be easily explored by experimenter-independent behavioral tracking systems (177). Several behavioral tests have been optimized to evaluate motor performance, motor coordination, balance, escape responses, exploratory behavior, reward/punishment-related behavior, learning, memory, social interaction, and aggressive or anxious behaviors (178–183). It is now crucial to overview the array of behavioral tests available for zebrafish, as it has been systematically done for rodents (184, 185).
Furthermore, similar to other models, a deeper characterization of zebrafish will certainly improve the validity of this model system. Regardless the differences between the zebrafish brain and mammalian brain, homologous functions have been attributed to different neuronal regions of each vertebrate. This is the case of the zebrafish diencephalic dopaminergic region (15). However, other neuronal components and physiologic events that modulate movement are still highly unknown. It would be relevant to understand the cerebral components of the zebrafish brain that correspond to the constituents of the mammalian basal ganglia–thalamocortical circuits and to investigate how they are interconnect. Is there a direct and indirect pathway-like system in zebrafish? How do zebrafish control movement features like velocity or direction? Perhaps, to dissect these processes in simpler brain circuitries, as the zebrafish ones, will help to understand more complex mechanisms in mammals. For instance, the small size of the zebrafish brain is useful for three-dimensional mapping of brain structures. With the up-to-date microscopic techniques, whole-brain neuronal connectivity can be easily performed in zebrafish and reveal anatomical relationships, that in larger brains may not be as facilitated. Another particularity that should be further explored is the regenerative capacity of the zebrafish CNS. Several neuronal proliferating sites were identified in the zebrafish brain as compared with two found in the mammalian brain (186). Notwithstanding, many of the molecular and cellular factors that drive regeneration in the brain of adult zebrafish are poorly understood and yet unknown. This may difficult the translation of disease-modifying drugs identified using zebrafish.
Finally, the distinctive and reproducible behaviors of zebrafish exposed to certain neuroactive drugs are a powerful evidence of the conserved functional properties of the neuronal circuitries in vertebrates (187–189). In addition, it highlights the translational value of zebrafish. To improve the validity of this model, it is now important to explore the mechanisms triggered by these drugs on the zebrafish targets. This will increase our understanding of the zebrafish neuronal modulation, and most importantly, enlighten the pharmacodynamic properties of the compounds in zebrafish. In fact, the factors that influence the pharmacodynamic and pharmacokinetic properties of drugs in zebrafish are poorly understood. For instance, previously, it has been assumed that zebrafish totally absorb and distribute through the system the small molecules present in the water (190). However, a recent review has suggested that the absorption of chemicals, as well as distribution through the BBB in zebrafish is comparable to mammals (191). Therefore, the chemical properties of the compounds should be considered when extrapolating concentrations between these two models. The metabolism and excretion of drugs in zebrafish are also difficult to predict. Despite zebrafish have important metabolic enzymes also found in mammals (192), these are not fully characterized. In addition, several differences in the metabolism of chemicals have been reported (193). The compounds used in the clinic are extensively characterized and are particularly adequate to investigate the differences of these compounds on fish and mammals. This would also create a basis to more precisely extrapolate doses between both models.
Probably, a key pitfall in the discovery of new drugs in zebrafish is the absence of general guidelines to calculate mammalian equivalent doses from zebrafish doses. While between mammalian models there exist established formulas (194, 195), the dose extrapolation from zebrafish to mammals is still empirical nowadays. Moreover, much of the existing literature has omitted this rationale (196–199). The only way to understand this rational is by increasing the number of studies where the properties of drugs in zebrafish are translated to mammals. Until now, it seems that only a minority has reached this far. It is not clear dough, whether there is little interest on these drugs, or they actually did not show the same properties in rodents and were not reported. The translation of the discoveries will allow the elaboration of a meta-analysis where the effective doses in zebrafish and mammals can be compared. Ultimately, it would boost the generation of formulas that rule dose extrapolation from zebrafish to different mammalian models and increase the validity of zebrafish models.
Conclusion
This review underscores the strengths and limitations of the zebrafish models of movement disorders developed to date. Importantly, it raises awareness that zebrafish can mimic the phenomenology of different movement disorders but needs further characterization. To date, there are a substantial number of studies reporting the use of zebrafish as a model for PD. However, for other movement disorders, this number is still limited. Considering the pathological hallmarks, motor phenotypes, and responsiveness to pharmacotherapies, from the seven movement disorders reviewed here, zebrafish models have only been fully characterized in the context of PD. Notwithstanding, the use of zebrafish to model human disorders dates back to the beginning of the twenty-first century, which compared with the 500 years of use of rodent models, is at embryonic stage. This difference of half a century may explain the skepticism that still exists about the use of zebrafish as an animal model of human diseases. The number of studies reporting the use of zebrafish as an animal model is growing and, therefore, the analysis of the pros and cons of the use of this vertebrate model for drug discovery is important.
This study also provides a comprehensive assessment of the methodologies adopted and emphasizes that most of the limitations are inherent to it. Many techniques are available to surpass these limitations and generate consistent and well-characterized models of movement disorders. With the next-generation sequencing, to couple genomic approaches with in vivo studies will not only improve our ability to understand the pathogenic mechanisms of complex diseases, as movement disorders but also precipitate the discovery of novel drugs for these disorders. Zebrafish should be considered a practical and inexpensive tool for this approach, provided that, as with any other non-mammalian model, the potential molecules selected in it are further validated by studies in mammals. It is not expected of zebrafish models of movement disorders to fully recapitulate such complex human phenomenology. Even mammalian models have their flaws and do not precisely mimic the symptomatology evidenced in patients. Furthermore, several compounds selected using the traditional models have also failed to demonstrate therapeutic effects in humans. Finally, it is vital to create a comprehensive correlation between zebrafish and mammalian models and, ultimately, be able to translate the findings to humans.
Overall, while it is already evident that zebrafish models of movement disorders share many cellular and physiologic mechanisms with mammalian models and patients, this model is still showing its usefulness for drug discovery. It was not until 2011 that a positive hit from a zebrafish-based drug screening entered phase I clinical trials (200). The usefulness of zebrafish to model human diseases will only be unquestionable when a drug selected in this model proves efficacy in human patients. Meanwhile, the use of zebrafish to study movement disorders will certainly result in a better understanding of their mechanisms and, hopefully, in the discovery of better therapies.
Author Contributions
RV and JF conceived and outlined the study. RV wrote the first draft. TO and JF did critical revision.
Conflict of Interest Statement
RV was affiliated with TechnoPhage SA, under the scope of a PhD student scholarship financed by FCT. All other authors declare no conflicts of interest.
Acknowledgments
The authors would like to thank Mariana Trigo Pereira, who kindly provided the pictures of zebrafish. RV was supported by a grant (SFRH/BD/78077/2011) from Fundação para e a Ciência e Tecnologia. TO is supported by the DFG Center for Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB).
References
2. Kalueff AV, Stewart AM, Gerlai R. Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol Sci (2014) 35:63–75. doi:10.1016/j.tips.2013.12.002
3. Goldsmith P. Zebrafish as a pharmacological tool: the how, why and when. Curr Opin Pharmacol (2004) 4:504–12. doi:10.1016/j.coph.2004.04.005
4. Rennekamp AJ, Peterson RT. 15 Years of zebrafish chemical screening. Curr Opin Chem Biol (2015) 24:58–70. doi:10.1016/j.cbpa.2014.10.025
5. Giacomotto J, Ségalat L. High-throughput screening and small animal models, where are we? Br J Pharmacol (2010) 160:204–16. doi:10.1111/j.1476-5381.2010.00725.x
6. Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet (2007) 8:353–67. doi:10.1038/nrg2091
7. Williams CH, Hong CC. Multi-step usage of in vivo models during rational drug design and discovery. Int J Mol Sci (2011) 12:2262–74. doi:10.3390/ijms12042262
8. Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature (2013) 496:498–503. doi:10.1038/nature12111
9. Jeong J, Kwon H, Ahn J, Kang D, Kwon S, Park JA, et al. Functional and developmental analysis of the blood-brain barrier in zebrafish. Brain Res Bull (2008) 75:619–28. doi:10.1016/j.brainresbull.2007.10.043
10. Xie J, Farage E, Sugimoto M, Anand-Apte B. A novel transgenic zebrafish model for blood-brain and blood-retinal barrier development. BMC Dev Biol (2010) 10:76. doi:10.1186/1471-213X-10-76
11. Kaslin J, Panula P. Comparative anatomy of the histaminergic and other aminergic systems in zebrafish (Danio rerio). J Comp Neurol (2001) 440:342–77. doi:10.1002/cne.1390
12. Candy J, Collet C. Two tyrosine hydroxylase genes in teleosts. Biochim Biophys Acta (2005) 1727:35–44. doi:10.1016/j.bbaexp.2004.11.005
13. Chen Y-C, Priyadarshini M, Panula P. Complementary developmental expression of the two tyrosine hydroxylase transcripts in zebrafish. Histochem Cell Biol (2009) 132:375–81. doi:10.1007/s00418-009-0619-8
14. Holzschuh J, Ryu S, Aberger F, Driever W. Dopamine transporter expression distinguishes dopaminergic neurons from other catecholaminergic neurons in the developing zebrafish embryo. Mech Dev (2001) 101:237–43. doi:10.1016/S0925-4773(01)00287-8
15. Rink E, Wullimann MF. The teleostean (zebrafish) dopaminergic system ascending to the subpallium (striatum) is located in the basal diencephalon (posterior tuberculum). Brain Res (2001) 889:316–30. doi:10.1016/S0006-8993(00)03174-7
16. Sallinen V, Sundvik M, Reenilä I, Peitsaro N, Khrustalyov D, Anichtchik O, et al. Hyperserotonergic phenotype after monoamine oxidase inhibition in larval zebrafish. J Neurochem (2009) 109:403–15. doi:10.1111/j.1471-4159.2009.05986.x
17. Panula P, Chen YC, Priyadarshini M, Kudo H, Semenova S, Sundvik M, et al. The comparative neuroanatomy and neurochemistry of zebrafish CNS systems of relevance to human neuropsychiatric diseases. Neurobiol Dis (2010) 40:46–57. doi:10.1016/j.nbd.2010.05.010
18. Anichtchik O, Sallinen V, Peitsaro N, Panula P. Distinct structure and activity of monoamine oxidase in the brain of zebrafish (Danio rerio). J Comp Neurol (2006) 498:593–610. doi:10.1002/cne.21057
19. Ruuskanen JO, Laurila J, Xhaard H, Rantanen V-V, Vuoriluoto K, Wurster S, et al. Conserved structural, pharmacological and functional properties among the three human and five zebrafish alpha 2-adrenoceptors. Br J Pharmacol (2005) 144:165–77. doi:10.1038/sj.bjp.0706057
20. Rink E, Guo S. The too few mutant selectively affects subgroups of monoaminergic neurons in the zebrafish forebrain. Neuroscience (2004) 127:147–54. doi:10.1016/j.neuroscience.2004.05.004
21. Norton WHJ, Folchert A, Bally-Cuif L. Comparative analysis of serotonin receptor (HTR1A/HTR1B families) and transporter (slc6a4a/b) gene expression in the zebrafish brain. J Comp Neurol (2008) 511:521–42. doi:10.1002/cne.21831
22. Wang Y, Takai R, Yoshioka H, Shirabe K. Characterization and expression of serotonin transporter genes in zebrafish. Tohoku J Exp Med (2006) 208:267–74. doi:10.1620/tjem.208.267
23. Eriksson KS, Peitsaro N, Karlstedt K, Kaslin J, Panula P. Development of the histaminergic neurons and expression of histidine decarboxylase mRNA in the zebrafish brain in the absence of all peripheral histaminergic systems. Eur J Neurosci (1998) 10:3799–812. doi:10.1046/j.1460-9568.1998.00394.x
24. Peitsaro N, Sundvik M, Anichtchik OV, Kaslin J, Panula P. Identification of zebrafish histamine H1, H2 and H3 receptors and effects of histaminergic ligands on behavior. Biochem Pharmacol (2007) 73:1205–14. doi:10.1016/j.bcp.2007.01.014
25. Maximino C, Herculano AM. A review of monoaminergic neuropsychopharmacology in zebrafish. Zebrafish (2010) 7:359–78. doi:10.1089/zeb.2010.0669
26. Rico EP, Rosemberg DB, Seibt KJ, Capiotti KM, Da Silva RS, Bonan CD. Zebrafish neurotransmitter systems as potential pharmacological and toxicological targets. Neurotoxicol Teratol (2011) 33:608–17. doi:10.1016/j.ntt.2011.07.007
27. Kalia LV, Lang AE. Parkinson’s disease. Lancet (2015) 386:896–912. doi:10.1016/S0140-6736(14)61393-3
28. Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science (1983) 219:979–80. doi:10.1126/science.6823561
29. Lam CS, Korzh V, Strahle U. Zebrafish embryos are susceptible to the dopaminergic neurotoxin MPTP. Eur J Neurosci (2005) 21:1758–62. doi:10.1111/j.1460-9568.2005.03988.x
30. McKinley ET, Baranowski TC, Blavo DO, Cato C, Doan TN, Rubinstein AL. Neuroprotection of MPTP-induced toxicity in zebrafish dopaminergic neurons. Brain Res Mol Brain Res (2005) 141:128–37. doi:10.1016/j.molbrainres.2005.08.014
31. Bretaud S, Lee S, Guo S. Sensitivity of zebrafish to environmental toxins implicated in Parkinson’s disease. Neurotoxicol Teratol (2004) 26:857–64. doi:10.1016/j.ntt.2004.06.014
32. Sallinen V, Torkko V, Sundvik M, Reenilä I, Khrustalyov D, Kaslin J, et al. MPTP and MPP+ target specific aminergic cell populations in larval zebrafish. J Neurochem (2009) 108:719–31. doi:10.1111/j.1471-4159.2008.05793.x
33. Wen L, Wei W, Gu W, Huang P, Ren X, Zhang Z, et al. Visualization of monoaminergic neurons and neurotoxicity of MPTP in live transgenic zebrafish. Dev Biol (2008) 314:84–92. doi:10.1016/j.ydbio.2007.11.012
34. Bové J, Perier C. Neurotoxin-based models of Parkinson’s disease. Neuroscience (2012) 211:51–76. doi:10.1016/j.neuroscience.2011.10.057
35. Dukes AA, Bai Q, Van Laar VS, Zhou Y, Ilin V, David CN, et al. Live imaging of mitochondrial dynamics in CNS dopaminergic neurons in vivo demonstrates early reversal of mitochondrial transport following MPP+ exposure. Neurobiol Dis (2016) 95:238–49. doi:10.1016/j.nbd.2016.07.020
36. Anichtchik OV, Kaslin J, Peitsaro N, Scheinin M, Panula P. Neurochemical and behavioural changes in zebrafish Danio rerio after systemic administration of 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurochem (2004) 88:443–53. doi:10.1111/j.1471-4159.2004.02190.x
37. Babu NS, Murthy CLN, Kakara S, Sharma R, Swamy CVB, Idris MM. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine induced Parkinson’s disease in zebrafish. Proteomics (2016) 16:1407–20. doi:10.1002/pmic.201500291
38. Serra PA, Sciola L, Delogu MR, Spano A, Monaco G, Miele E, et al. The neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine induces apoptosis in mouse nigrostriatal glia. Relevance to nigral neuronal death and striatal neurochemical changes. J Biol Chem (2002) 277:34451–61. doi:10.1074/jbc.M202099200
39. Perry TL, Yong VW, Jones K, Wall RA, Clavier RM, Foulks JG, et al. Effects of N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and its metabolite, N-methyl-4-phenylpyridinium ion, on dopaminergic nigrostriatal neurons in the mouse. Neurosci Lett (1985) 58:321–6. doi:10.1016/0304-3940(85)90074-6
40. Vijayanathan Y, Lim FT, Lim SM, Long CM, Tan MP, Majeed ABA, et al. 6-OHDA-lesioned adult zebrafish as a useful Parkinson’s disease model for dopaminergic neuroregeneration. Neurotox Res (2017) 32(3):496–508. doi:10.1007/s12640-017-9778-x
41. Höglinger GU, Alvarez-Fischer D, Arias-Carrión O, Djufri M, Windolph A, Keber U, et al. A new dopaminergic nigro-olfactory projection. Acta Neuropathol (2015) 130:333–48. doi:10.1007/s00401-015-1451-y
42. Zupanc GKH, Hinsch K, Gage FH. Proliferation, migration, neuronal differentiation, and long-term survival of new cells in the adult zebrafish brain. J Comp Neurol (2005) 488:290–319. doi:10.1002/cne.20571
43. Feng C, Wen Z, Huang S, Hung H, Chen C, Yang S, et al. Effects of 6-hydroxydopamine exposure on motor activity and biochemical expression in zebrafish (Danio rerio) larvae. Zebrafish (2014) 11:227–39. doi:10.1089/zeb.2013.0950
44. Yamauchi R. Vitamin E: mechanisms of its antioxidant activity. Food Sci Technol Int Tokyo (1997) 3:301–9. doi:10.3136/fsti9596t9798.3.301
45. Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci U S A (2001) 98:14669–74. doi:10.1073/pnas.251341998
46. Day BJ, Patel M, Calavetta L, Chang LY, Stamler JS. A mechanism of paraquat toxicity involving nitric oxide synthase. Proc Natl Acad Sci U S A (1999) 96:12760–5. doi:10.1073/pnas.96.22.12760
47. Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, et al. Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect (2011) 119:866–72. doi:10.1289/ehp.1002839
48. Bortolotto JW, Cognato GP, Christoff RR, Roesler LN, Leite CE, Kist LW, et al. Long-term exposure to paraquat alters behavioral parameters and dopamine levels in adult zebrafish (Danio rerio). Zebrafish (2014) 11:142–53. doi:10.1089/zeb.2013.0923
49. Nunes ME, Müller TE, Braga MM, Fontana BD, Quadros VA, Marins A, et al. Chronic treatment with paraquat induces brain injury, changes in antioxidant defenses system, and modulates behavioral functions in zebrafish. Mol Neurobiol (2017) 54:3925–34. doi:10.1007/s12035-016-9919-x
50. Brooks AI, Chadwick CA, Gelbard HA, Cory-Slechta DA, Federoff HJ. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Res (1999) 823:1–10. doi:10.1016/S0006-8993(98)01192-5
51. Ren J, Zhao Y, Sun X. Toxic influence of chronic oral administration of paraquat on nigrostriatal dopaminergic neurons in C57BL/6 mice. Chin Med J (Engl) (2009) 122:2366–71.
52. Czerniczyniec A, Karadayian AG, Bustamante J, Cutrera RA, Lores-Arnaiz S. Paraquat induces behavioral changes and cortical and striatal mitochondrial dysfunction. Free Radic Biol Med (2011) 51:1428–36. doi:10.1016/j.freeradbiomed.2011.06.034
53. Songin M, Strosznajder JB, Fitał M, Kuter K, Kolasiewicz W, Nowak P, et al. Glycogen synthase kinase 3β and its phosphorylated form (Y216) in the paraquat-induced model of parkinsonism. Neurotox Res (2011) 19:162–71. doi:10.1007/s12640-010-9153-7
54. Breckenridge CB, Sturgess NC, Butt M, Wolf JC, Zadory D, Beck M, et al. Pharmacokinetic, neurochemical, stereological and neuropathological studies on the potential effects of paraquat in the substantia nigra pars compacta and striatum of male C57BL/6J mice. Neurotoxicology (2013) 37:1–14. doi:10.1016/j.neuro.2013.03.005
55. Nellore J, Nandita P. Paraquat exposure induces behavioral deficits in larval zebrafish during the window of dopamine neurogenesis. Toxicol Rep (2015) 2:950–6. doi:10.1016/j.toxrep.2015.06.007
56. Wang Q, Liu S, Hu D, Wang Z, Wang L, Wu T, et al. Identification of apoptosis and macrophage migration events in paraquat-induced oxidative stress using a zebrafish model. Life Sci (2016) 157:116–24. doi:10.1016/j.lfs.2016.06.009
57. Miller GW. Paraquat: the red herring of Parkinson’s disease research. Toxicol Sci (2007) 100:1–2. doi:10.1093/toxsci/kfm223
58. Fleming SM, Zhu C, Fernagut P-O, Mehta A, DiCarlo CD, Seaman RL, et al. Behavioral and immunohistochemical effects of chronic intravenous and subcutaneous infusions of varying doses of rotenone. Exp Neurol (2004) 187:418–29. doi:10.1016/j.expneurol.2004.01.023
59. Wang Y, Liu W, Yang J, Wang F, Sima Y, Zhong ZM, et al. Parkinson’s disease-like motor and non-motor symptoms in rotenone-treated zebrafish. Neurotoxicology (2017) 58:103–9. doi:10.1016/j.neuro.2016.11.006
60. Doty RL, Deems DA, Stellar S. Olfactory dysfunction in parkinsonism: a general deficit unrelated to neurologic signs, disease stage, or disease duration. Neurology (1988) 38:1237–44. doi:10.1212/WNL.38.8.1237
61. Ziemssen T, Reichmann H. Non-motor dysfunction in Parkinson’s disease. Parkinsonism Relat Disord (2007) 13:323–32. doi:10.1016/j.parkreldis.2006.12.014
62. Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci (2000) 3:1301–6. doi:10.1038/81834
63. Santiago RM, Barbieiro J, Lima MM, Dombrowski PA, Andreatini R, Vital MA. Depressive-like behaviors alterations induced by intranigral MPTP, 6-OHDA, LPS and rotenone models of Parkinson’s disease are predominantly associated with serotonin and dopamine. Prog Neuropsychopharmacol Biol Psychiatry (2010) 34:1104–14. doi:10.1016/j.pnpbp.2010.06.004
64. Godoy R, Noble S, Yoon K, Anisman H, Ekker M. Chemogenetic ablation of dopaminergic neurons leads to transient locomotor impairments in zebrafish larvae. J Neurochem (2015) 135:249–60. doi:10.1111/jnc.13214
65. Hu Q, Guo F, Zhao F, Fu Z. Effects of titanium dioxide nanoparticles exposure on parkinsonism in zebrafish larvae and PC12. Chemosphere (2017) 173:373–9. doi:10.1016/j.chemosphere.2017.01.063
66. Toni M, Cioni C. Fish synucleins: an update. Mar Drugs (2015) 13:6665–86. doi:10.3390/md13116665
67. Kumar S, Meena R, Paulraj R. Role of macrophage (M1 and M2) in titanium-dioxide nanoparticle-induced oxidative stress and inflammatory response in rat. Appl Biochem Biotechnol (2016) 180:1257–75. doi:10.1007/s12010-016-2165-x
68. Sun Z, Gitler AD. Discovery and characterization of three novel synuclein genes in zebrafish. Dev Dyn (2008) 237:2490–5. doi:10.1002/dvdy.21569
69. Milanese C, Sager JJ, Bais Q, Farrells TC, Cannons JR, Greenamyre JT, et al. Hypokinesia and reduced dopamine levels in zebrafish lacking beta- and gamma1-synucleins. J Biol Chem (2012) 287:2971–83. doi:10.1074/jbc.M111.308312
70. Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, et al. Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci U S A (2004) 101:14966–71. doi:10.1073/pnas.0406283101
71. Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, et al. Alpha beta gamma-synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A (2010) 107:19573–8. doi:10.1073/pnas.1005005107
72. Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science (2010) 329:1663–7. doi:10.1126/science.1195227
73. Anwar S, Peters O, Millership S, Ninkina N, Doig N, Connor-Robson N, et al. Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J Neurosci (2011) 31:7264–74. doi:10.1523/JNEUROSCI.6194-10.2011
74. Lulla A, Barnhill L, Bitan G, Ivanova MI, Nguyen B, O’Donnell K, et al. Neurotoxicity of the Parkinson’s disease-associated pesticide ziram is synuclein-dependent in zebrafish embryos. Environ Health Perspect (2016) 124:1766–75. doi:10.1289/EHP141
75. Prabhudesai S, Sinha S, Attar A, Kotagiri A, Fitzmaurice AG, Lakshmanan R, et al. A novel “molecular tweezer” inhibitor of α-synuclein neurotoxicity in vitro and in vivo. Neurotherapeutics (2012) 9:464–76. doi:10.1007/s13311-012-0120-2
76. Anichtchik O, Diekmann H, Fleming A, Roach A, Goldsmith P, Rubinsztein DC. Loss of PINK1 function affects development and results in neurodegeneration in zebrafish. J Neurosci (2008) 28:8199–207. doi:10.1523/JNEUROSCI.0979-08.2008
77. Flinn LJ, Keatinge M, Bretaud S, Mortiboys H, Matsui H, De Felice E, et al. TigarB causes mitochondrial dysfunction and neuronal loss in PINK1 deficiency. Ann Neurol (2013) 74:837–47. doi:10.1002/ana.23999
78. Vives-Bauza C, Przedborski S. Mitophagy: the latest problem for Parkinson’s disease. Trends Mol Med (2011) 17:158–65. doi:10.1016/j.molmed.2010.11.002
79. Priyadarshini M, Tuimala J, Chen YC, Panula P. A zebrafish model of PINK1 deficiency reveals key pathway dysfunction including HIF signaling. Neurobiol Dis (2013) 54:127–38. doi:10.1016/j.nbd.2013.02.002
80. Schroedl C, McClintock DS, Budinger GR, Chandel NS. Hypoxic but not anoxic stabilization of HIF-1α requires mitochondrial reactive oxygen species. Am J Physiol Lung Cell Mol Physiol (2002) 283:L922–31. doi:10.1152/ajplung.00014.2002
81. Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell (2005) 9:617–28. doi:10.1016/j.devcel.2005.09.010
82. Xi Y, Ryan J, Noble S, Yu M, Yilbas AE, Ekker M. Impaired dopaminergic neuron development and locomotor function in zebrafish with loss of pink1 function. Eur J Neurosci (2010) 31:623–33. doi:10.1111/j.1460-9568.2010.07091.x
83. Sallinen V, Kolehmainen J, Priyadarshini M, Toleikyte G, Chen YC, Panula P. Dopaminergic cell damage and vulnerability to MPTP in Pink1 knockdown zebrafish. Neurobiol Dis (2010) 40:93–101. doi:10.1016/j.nbd.2010.06.001
84. Priyadarshini M, Orosco LA, Panula PJ. Oxidative stress and regulation of pink1 in zebrafish (Danio rerio). PLoS One (2013) 8:e81851. doi:10.1371/journal.pone.0081851
85. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science (2004) 304:1158–60. doi:10.1126/science.1096284
86. Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, et al. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A (2007) 104:11441–6. doi:10.1073/pnas.0702717104
87. Flinn L, Mortiboys H, Volkmann K, Kster RW, Ingham PW, Bandmann O. Complex i deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain (2009) 132:1613–23. doi:10.1093/brain/awp108
88. Müftüoglu M, Elibol B, Dalmızrak Ö, Ercan A, Kulaksız G, Ögüs H, et al. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov Disord (2004) 19:544–8. doi:10.1002/mds.10695
89. Mortiboys H, Thomas KJ, Koopman WJ, Klaffke S, Abou-Sleiman P, Olpin S, et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol (2008) 64:555–65. doi:10.1002/ana.21492
90. Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci U S A (2005) 102:2174–9. doi:10.1073/pnas.0409598102
91. Thomas B, von Coelln R, Mandir AS, Trinkaus DB, Farah MH, Leong Lim K, et al. MPTP and DSP-4 susceptibility of substantia nigra and locus coeruleus catecholaminergic neurons in mice is independent of parkin activity. Neurobiol Dis (2007) 26:312–22. doi:10.1016/j.nbd.2006.12.021
92. Fett ME, Pilsl A, Paquet D, van Bebber F, Haass C, Tatzelt J, et al. Parkin is protective against proteotoxic stress in a transgenic zebrafish model. PLoS One (2010) 5:e11783. doi:10.1371/journal.pone.0011783
93. Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, et al. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron (2002) 36:1007–19. doi:10.1016/S0896-6273(02)01125-X
94. Noble S, Ismail A, Godoy R, Xi Y, Ekker M. Zebrafish Parla- and Parlb-deficiency affects dopaminergic neuron patterning and embryonic survival. J Neurochem (2012) 122:196–207. doi:10.1111/j.1471-4159.2012.07758.x
95. Shi G, Lee JR, Grimes DA, Racacho L, Ye D, Yang H, et al. Functional alteration of PARL contributes to mitochondrial dysregulation in Parkinson’s disease. Hum Mol Genet (2011) 20:1966–74. doi:10.1093/hmg/ddr077
96. Bretaud S, Allen C, Ingham PW, Bandmann O. p53-dependent neuronal cell death in a DJ-1-deficient zebrafish model of Parkinson’s disease. J Neurochem (2007) 100:1626–35. doi:10.1111/j.1471-4159.2006.04291.x
97. Baulac S, Lu H, Strahle J, Yang T, Goldberg MS, Shen J, et al. Increased DJ-1 expression under oxidative stress and in Alzheimer’s disease brains. Mol Neurodegener (2009) 4:1–12. doi:10.1186/1750-1326-4-12
98. Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci U S A (2005) 102:5215–20. doi:10.1073/pnas.0501282102
99. de la Monte SM, Sohn YK, Ganju N, Wands JR. P53- and CD95-associated apoptosis in neurodegenerative diseases. Lab Invest (1998) 78:401–11.
100. Hartmann A, Michel PP, Troadec JD, Mouatt-Prigent A, Faucheux BA, Ruberg M, et al. Is Bax a mitochondrial mediator in apoptotic death of dopaminergic neurons in Parkinson’s disease? J Neurochem (2001) 76:1785–93. doi:10.1046/j.1471-4159.2001.00160.x
101. Sheng D, Qu D, Kwok KH, Ng SS, Lim AY, Aw SS, et al. Deletion of the WD40 domain of LRRK2 in zebrafish causes parkinsonism-like loss of neurons and locomotive defect. PLoS Genet (2010) 6:e1000914. doi:10.1371/journal.pgen.1000914
102. Prabhudesai S, Bensabeur FZ, Abdullah R, Basak I, Baez S, Alves G, et al. LRRK2 knockdown in zebrafish causes developmental defects, neuronal loss, and synuclein aggregation. J Neurosci Res (2016) 94:717–35. doi:10.1002/jnr.23754
103. Ren G, Xin S, Li S, Zhong H, Lin S. Disruption of lrrk2 does not cause specific loss of dopaminergic neurons in zebrafish. PLoS One (2011) 6:e20630. doi:10.1371/journal.pone.0020630
104. Tan EK, Zhao Y, Skipper L, Tan MG, Di Fonzo A, Sun L, et al. The LRRK2 Gly2385Arg variant is associated with Parkinson’s disease: genetic and functional evidence. Hum Genet (2007) 120:857–63. doi:10.1007/s00439-006-0268-0
105. Wang L, Xie C, Greggio E, Parisiadou L, Shim H, Sun L, et al. The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J Neurosci (2008) 28:3384–91. doi:10.1523/JNEUROSCI.0185-08.2008
106. Andres-Mateos E, Mejias R, Sasaki M, Li X, Lin BM, Biskup S, et al. Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine). J Neurosci (2009) 29:15846–50. doi:10.1523/JNEUROSCI.4357-09.2009
107. Fonzo AD, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Guedes LC, et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology (2009) 72:240–5. doi:10.1212/01.wnl.0000338144.10967.2b
108. Zhao T, Zondervan-van der Linde H, Severijnen L-A, Oostra BA, Willemsen R, Bonifati V. Dopaminergic neuronal loss and dopamine-dependent locomotor defects in Fbxo7-deficient zebrafish. PLoS One (2012) 7:e48911. doi:10.1371/journal.pone.0048911
109. Lopes da Fonseca T, Correia A, Hasselaar W, van der Linde HC, Willemsen R, Outeiro TF. The zebrafish homologue of Parkinson’s disease ATP13A2 is essential for embryonic survival. Brain Res Bull (2013) 90:118–26. doi:10.1016/j.brainresbull.2012.09.017
110. Weingarten L, Dave H, Li H, Crawford D. Developmental expression of P5 ATPase mRNA in the mouse. Cell Mol Biol Lett (2012) 17:153–70. doi:10.2478/s11658-011-0039-3
111. Farrell TC, Cario CL, Milanese C, Vogt A, Jeong J-H, Burton EA. Evaluation of spontaneous propulsive movement as a screening tool to detect rescue of Parkinsonism phenotypes in zebrafish models. Neurobiol Dis (2011) 44:9–18. doi:10.1016/j.nbd.2011.05.016
112. Irons TD, Kelly PE, Hunter DL, MacPhail RC, Padilla S. Acute administration of dopaminergic drugs has differential effects on locomotion in larval zebrafish. Pharmacol Biochem Behav (2013) 103:792–813. doi:10.1016/j.pbb.2012.12.010
113. Makhija DT, Jagtap AG. Studies on sensitivity of zebrafish as a model organism for Parkinson’s disease: comparison with rat model. J Pharmacol Pharmacother (2014) 5:39–46. doi:10.4103/0976-500X.124422
114. Boxer AL, Yu J-T, Golbe LI, Litvan I, Lang AE, Höglinger GU. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol (2017) 16:552–63. doi:10.1016/S1474-4422(17)30157-6
115. Lopez A, Lee SE, Wojta K, Ramos EM, Klein E, Chen J, et al. A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain (2017) 140:1128–46. doi:10.1093/brain/awx005
116. Maeda S, Djukic B, Taneja P, Yu G-Q, Lo I, Davis A, et al. Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep (2016) 17:530–51. doi:10.15252/embr.201541438
117. Decker JM, Krüger L, Sydow A, Dennissen FJ, Siskova Z, Mandelkow E, et al. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep (2016) 17:552–69. doi:10.15252/embr.201541439
118. Phukan J, Albanese A, Gasser T, Warner T. Primary dystonia and dystonia-plus syndromes: clinical characteristics, diagnosis, and pathogenesis. Lancet Neurol (2011) 10:1074–85. doi:10.1016/S1474-4422(11)70232-0
119. Hallett M. Neurophysiology of dystonia: the role of inhibition. Neurobiol Dis (2011) 42:177–84. doi:10.1016/j.nbd.2010.08.025
120. Jankovic J. Treatment of hyperkinetic movement disorders. Lancet Neurol (2009) 8:844–56. doi:10.1016/S1474-4422(09)70183-8
121. Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet (1997) 17:40–8. doi:10.1038/ng0997-40
122. Sager JJ, Torres GE, Burton EA. The zebrafish homologue of the human DYT1 dystonia gene is widely expressed in CNS neurons but non-essential for early motor system development. PLoS One (2012) 7:e45175. doi:10.1371/journal.pone.0045175
123. Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron (2005) 48:923–32. doi:10.1016/j.neuron.2005.11.010
124. Kruer MC, Hiken M, Gregory A, Malandrini A, Clark D, Hogarth P, et al. Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain (2011) 134:947–58. doi:10.1093/brain/awr042
125. Zizioli D, Tiso N, Guglielmi A, Saraceno C, Busolin G, Giuliani R, et al. Knock-down of pantothenate kinase 2 severely affects the development of the nervous and vascular system in zebrafish, providing new insights into PKAN disease. Neurobiol Dis (2016) 85:35–48. doi:10.1016/j.nbd.2015.10.010
126. Zech M, Lam DD, Francescatto L, Schormair B, Salminen AV, Jochim A, et al. Recessive mutations in the alpha-3 (VI) collagen gene COL6A3 cause early-onset isolated dystonia. Am J Hum Genet (2015) 96:1–11. doi:10.1016/j.ajhg.2015.04.010
127. Doganli C, Beck HC, Ribera AB, Oxvig C, Lykke-Hartmann K. α3Na+/K+-ATPase deficiency causes brain ventricle dilation and abrupt embryonic motility in zebrafish. J Biol Chem (2013) 288:8862–74. doi:10.1074/jbc.M112.421529
128. de Carvalho Aguiar P, Sweadner KJ, Penniston JT, Zaremba J, Liu L, Caton M, et al. Mutations in the Na+/K+-ATPase α3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron (2004) 43:169–75. doi:10.1016/j.neuron.2004.06.028
129. Dobretsov M, Stimers JR. Neuronal function and alpha3 isoform of the Na/K-ATPase. Front Biosci (2005) 10:2373–96. doi:10.2741/1704
130. Tuschl K, Meyer E, Valdivia LE, Zhao N, Dadswell C, Abdul-Sada A, et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun (2016) 7:1–16. doi:10.1038/ncomms11601
131. Xia Z, Wei J, Li Y, Wang J, Li W, Wang K, et al. Zebrafish slc30a10 deficiency revealed a novel compensatory mechanism of Atp2c1 in maintaining manganese homeostasis. PLoS Genet (2017) 13:e1006892. doi:10.1371/journal.pgen.1006892
132. Friedrich T, Lambert AM, Masino MA, Downes GB. Mutation of zebrafish dihydrolipoamide branched-chain transacylase E2 results in motor dysfunction and models maple syrup urine disease. Dis Model Mech (2012) 5:248–58. doi:10.1242/dmm.008383
133. Lu ZG, Li MH, Wang JS, Wei DD, Liu QW, Kong LY. Developmental toxicity and neurotoxicity of two matrine-type alkaloids, matrine and sophocarpine, in zebrafish (Danio rerio) embryos/larvae. Reprod Toxicol (2014) 47:33–41. doi:10.1016/j.reprotox.2014.05.015
134. Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol (2011) 10:83–98. doi:10.1016/S1474-4422(10)70245-3
135. Karlovich CA, John RM, Ramirez L, Stainier DY, Myers RM. Characterization of the Huntington’s disease (HD) gene homologue in the zebrafish Danio rerio. Gene (1998) 217:117–25. doi:10.1016/S0378-1119(98)00342-4
136. Lumsden AL, Henshall TL, Dayan S, Lardelli MT, Richards RI. Huntingtin-deficient zebrafish exhibit defects in iron utilization and development. Hum Mol Genet (2007) 16:1905–20. doi:10.1093/hmg/ddm138
137. Futter M, Diekmann H, Schoenmakers E, Sadiq O, Chatterjee K, Rubinsztein DC. Wild-type but not mutant huntingtin modulates the transcriptional activity of liver X receptors. J Med Genet (2009) 46:438–46. doi:10.1136/jmg.2009.066399
138. Diekmann H, Anichtchik O, Fleming A, Futter M, Goldsmith P, Roach A, et al. Decreased BDNF levels are a major contributor to the embryonic phenotype of huntingtin knockdown zebrafish. J Neurosci (2009) 29:1343–9. doi:10.1523/JNEUROSCI.6039-08.2009
139. Henshall TL, Tucker B, Lumsden AL, Nornes S, Lardelli MT, Richards RI. Selective neuronal requirement for huntingtin in the developing zebrafish. Hum Mol Genet (2009) 18:4830–42. doi:10.1093/hmg/ddp455
140. Lo Sardo V, Zuccato C, Gaudenzi G, Vitali B, Ramos C, Tartari M, et al. An evolutionary recent neuroepithelial cell adhesion function of huntingtin implicates ADAM10-Ncadherin. Nat Neurosci (2012) 15:713–21. doi:10.1038/nn.3080
141. Morrison PJ, Nevin NC. Serum iron, total iron binding capacity and ferritin in early Huntington disease patients. Ir J Med Sci (1994) 163:236–7. doi:10.1007/BF02943258
142. Bartzokis G, Cummings J, Perlman S, Hance DB, Mintz J. Increased basal ganglia iron levels in Huntington disease. Arch Neurol (1999) 56:569–74. doi:10.1001/archneur.56.5.569
143. Browne SE, Beal MF. The energetics of Huntington’s disease. Neurochem Res (2004) 29:531–46. doi:10.1023/B:NERE.0000014824.04728.dd
144. Zuccato C, Marullo M, Conforti P, MacDonald ME, Tartari M, Cattaneo E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol (2008) 18:225–38. doi:10.1111/j.1750-3639.2007.00111.x
145. Miller JP, Holcomb J, Al-Ramahi I, de Haro M, Gafni J, Zhang N, et al. Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington’s disease. Neuron (2010) 67:199–212. doi:10.1016/j.neuron.2010.06.021
146. Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell (1995) 81:811–23. doi:10.1016/0092-8674(95)90542-1
147. Miller VM, Nelson RF, Gouvion CM, Williams A, Rodriguez-Lebron E, Harper SQ, et al. CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J Neurosci (2005) 25:9152–61. doi:10.1523/JNEUROSCI.3001-05.2005
148. Schiffer NW, Broadley SA, Hirschberger T, Tavan P, Kretzschmar HA, Giese A, et al. Identification of anti-prion compounds as efficient inhibitors of polyglutamine protein aggregation in a zebrafish model. J Biol Chem (2007) 282:9195–203. doi:10.1074/jbc.M607865200
149. Veldman MB, Rios-Galdamez Y, Lu X-H, Gu X, Qin W, Li S, et al. The N17 domain mitigates nuclear toxicity in a novel zebrafish Huntington’s disease model. Mol Neurodegener (2015) 10:67. doi:10.1186/s13024-015-0063-2
150. Gu X, Cantle JP, Greiner ER, Lee CYD, Barth AM, Gao F, et al. N17 modifies mutant huntingtin nuclear pathogenesis and severity of disease in HD BAC transgenic mice. Neuron (2015) 85:726–41. doi:10.1016/j.neuron.2015.01.008
151. Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature (1986) 321:168–71. doi:10.1038/321168a0
152. Tattersfield A, Croon R, Liu Y, Kells A, Faull RL, Connor B. Neurogenesis in the striatum of the quinolinic acid lesion model of Huntington’s disease. Neuroscience (2004) 127:319–32. doi:10.1016/j.neuroscience.2004.04.061
153. Collin T, Arvidsson A, Kokaia Z, Lindvall O. Quantitative analysis of the generation of different striatal neuronal subtypes in the adult brain following excitotoxic injury. Exp Neurol (2005) 195:71–80. doi:10.1016/j.expneurol.2005.03.017
154. Skaggs K, Goldman D, Parent JM. Excitotoxic brain injury in adult zebrafish stimulates neurogenesis and long-distance neuronal integration. Glia (2014) 62:2061–79. doi:10.1002/glia.22726
155. Leonard H, Cobb S, Downs J. Clinical and biological progress over 50 years in Rett syndrome. Nat Rev Neurol (2016) 13:37–51. doi:10.1038/nrneurol.2016.186
156. Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet (2001) 27:327–31. doi:10.1038/85906
157. Pietri T, Roman A-C, Guyon N, Romano SA, Washbourne P, Moens CB, et al. The first mecp2-null zebrafish model shows altered motor behaviors. Front Neural Circuits (2013) 7:118. doi:10.3389/fncir.2013.00118
158. Cortelazzo A, Pietri T, De Felice C, Leoncini S, Guerranti R, Signorini C, et al. Proteomic analysis of the Rett syndrome experimental model mecp2Q63X mutant zebrafish. J Proteomics (2017) 154:128–33. doi:10.1016/j.jprot.2016.12.010
159. Conti V, Gandaglia A, Galli F, Tirone M, Bellini E, Campana L, et al. MeCP2 affects skeletal muscle growth and morphology through non cell-autonomous mechanisms. PLoS One (2015) 10:e0130183. doi:10.1371/journal.pone.0130183
160. Segawa M. Early motor disturbances in Rett syndrome and its pathophysiological importance. Brain Dev (2005) 27:S54–8. doi:10.1016/j.braindev.2004.11.010
161. Kriaucionis S, Paterson A, Curtis J, Guy J, Macleod N, Bird A. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol Cell Biol (2006) 26:5033–42. doi:10.1128/MCB.01665-05
162. Viola A, Saywell V, Villard L, Cozzone PJ, Lutz NW. Metabolic fingerprints of altered brain growth, osmoregulation and neurotransmission in a Rett syndrome model. PLoS One (2007) 2:e157. doi:10.1371/journal.pone.0000157
163. Jin L-W, Horiuchi M, Wulff H, Liu X-B, Cortopassi GA, Erickson JD, et al. Dysregulation of glutamine transporter SNAT1 in Rett syndrome microglia: a mechanism for mitochondrial dysfunction and neurotoxicity. J Neurosci (2015) 35:2516–29. doi:10.1523/JNEUROSCI.2778-14.2015
164. Justice MJ, Buchovecky CM, Kyle SM, Djukic A. A role for metabolism in Rett syndrome pathogenesis. Rare Dis (2013) 1:e27265. doi:10.4161/rdis.27265
165. Gao H, Bu Y, Wu Q, Wang X, Chang N, Lei L, et al. Mecp2 regulates neural cell differentiation by suppressing the Id1-Her2/Hes5 axis in zebrafish. J Cell Sci (2015) 128:2340–50. doi:10.1242/jcs.167874
166. Nozawa K, Lin Y, Kubodera R, Shimizu Y, Tanaka H, Ohshima T. Zebrafish Mecp2 is required for proper axonal elongation of motor neurons and synapse formation. Dev Neurobiol (2017) 77:1101–13. doi:10.1002/dneu.22498
167. Leong WY, Lim ZH, Korzh V, Pietri T, Goh ELK. Methyl-CpG binding protein 2 (Mecp2) regulates sensory function through Sema5b and Robo2. Front Cell Neurosci (2015) 9:481. doi:10.3389/fncel.2015.00481
168. Samaco RC, Fryer JD, Ren J, Fyffe S, Chao H-T, Sun Y, et al. A partial loss of function allele of methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum Mol Genet (2008) 17:1718–27. doi:10.1093/hmg/ddn062
171. Hor H, Francescatto L, Bartesaghi L, Ortega-Cubero S, Kousi M, Lorenzo-Betancor O, et al. Missense mutations in TENM4, a regulator of axon guidance and central myelination, cause essential tremor. Hum Mol Genet (2015) 24:5677–86. doi:10.1093/hmg/ddv281
172. Robertson MM, Eapen V, Singer HS, Martino D, Scharf JM, Paschou P, et al. Gilles de la Tourette syndrome. Nat Rev Dis Primers (2017) 3:16097. doi:10.1038/nrdp.2016.97
173. Fontana BD, Mezzomo NJ, Kalueff AV, Rosemberg DB. The developing utility of zebrafish models of neurological and neuropsychiatric disorders: a critical review. Exp Neurol (2018) 299:157–71. doi:10.1016/j.expneurol.2017.10.004
174. Jagmag SA, Tripathi N, Shukla SD, Maiti S, Khurana S. Evaluation of models of Parkinson’s disease. Front Neurosci (2016) 9:503. doi:10.3389/fnins.2015.00503
175. Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science (1995) 269:407–10. doi:10.1126/science.7618107
176. Visanji NP, Brotchie JM, Kalia LV, Koprich JB, Tandon A, Watts JC, et al. α-Synuclein-based animal models of Parkinson’s disease: challenges and opportunities in a new era. Trends Neurosci (2016) 39:750–62. doi:10.1016/j.tins.2016.09.003
177. Kalueff AV, Gebhardt M, Stewart AM, Cachat JM, Brimmer M, Chawla JS, et al. Towards a comprehensive catalog of zebrafish behavior 1.0 and beyond. Zebrafish (2013) 10:70–86. doi:10.1089/zeb.2012.0861
178. Panula P, Sallinen V, Sundvik M, Kolehmainen J, Torkko V, Tiittula A, et al. Modulatory neurotransmitter systems and behavior: towards zebrafish models of neurodegenerative diseases. Zebrafish (2006) 3:235–47. doi:10.1089/zeb.2006.3.235
179. Stewart AM, Braubach O, Spitsbergen J, Gerlai R, Kalueff AV. Zebrafish models for translational neuroscience research: from tank to bedside. Trends Neurosci (2014) 37:264–78. doi:10.1016/j.tins.2014.02.011
180. Nicolson T, Rüsch A, Friedrich RW, Granato M, Ruppersberg JP, Nüsslein-Volhard C. Genetic analysis of vertebrate sensory hair cell mechanosensation: the zebrafish circler mutants. Neuron (1998) 20:271–83. doi:10.1016/S0896-6273(00)80455-9
181. Feng Y, Xu Q. Pivotal role of hmx2 and hmx3 in zebrafish inner ear and lateral line development. Dev Biol (2010) 339:507–18. doi:10.1016/j.ydbio.2009.12.028
182. Blazina AR, Vianna MR, Lara DR. The spinning task: a new protocol to easily assess motor coordination and resistance in zebrafish. Zebrafish (2013) 10:480–5. doi:10.1089/zeb.2012.0860
183. Chatterjee P, Padmanarayana M, Abdullah N, Holman CL, LaDu J, Tanguay RL, et al. Otoferlin deficiency in zebrafish results in defects in balance and hearing: rescue of the balance and hearing phenotype with full-length and truncated forms of mouse otoferlin. Mol Cell Biol (2015) 35:1043–54. doi:10.1128/MCB.01439-14
184. Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome (1997) 8:711–3. doi:10.1007/s003359900551
185. Brooks SP, Dunnett SB. Tests to assess motor phenotype in mice: a user’s guide. Nat Rev Neurosci (2009) 10:519–29. doi:10.1038/nrn2652
186. Zupanc GK. Adult neurogenesis and neuronal regeneration in the brain of teleost fish. J Physiol Paris (2008) 102:357–73. doi:10.1016/j.jphysparis.2008.10.007
187. Kokel D, Bryan J, Laggner C, White R, Cheung CY, Mateus R, et al. Rapid behavior-based identification of neuroactive small molecules in the zebrafish. Nat Chem Biol (2010) 6:231–7. doi:10.1038/nchembio.307
188. Rihel J, Prober DA, Arvanites A, Lam K, Zimmerman S, Jang S, et al. Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science (2010) 327:348–51. doi:10.1126/science.1183090
189. Bruni G, Lakhani P, Kokel D. Discovering novel neuroactive drugs through high-throughput behavior-based chemical screening in the zebrafish. Front Pharmacol (2014) 5:153. doi:10.3389/fphar.2014.00153
190. Mathias JR, Saxena MT, Mumm JS. Advances in zebrafish chemical screening technologies. Future Med Chem (2012) 4:1811–22. doi:10.4155/fmc.12.115
191. Nishimura Y, Murakami S, Ashikawa Y, Sasagawa S, Umemoto N, Shimada Y, et al. Zebrafish as a systems toxicology model for developmental neurotoxicity testing. Congenit Anom (Kyoto) (2015) 55:1–16. doi:10.1111/cga.12079
192. Goldstone JV, Mcarthur AG, Kubota A, Zanette J, Parente T, Jönsson ME, et al. Identification and developmental expression of the full complement of cytochrome P450 genes in zebrafish. BMC Genomics (2010) 11:643. doi:10.1186/1471-2164-11-643
193. Chng HT, Ho HK, Yap CW, Lam SH, Chan ECY. An investigation of the bioactivation potential and metabolism profile of zebrafish versus human. J Biomol Screen (2012) 17:974–86. doi:10.1177/1087057112447305
194. FDA Center for Drug Evaluation and Research. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Food and Drug Administration (FDA) (2005). Available from: https://www.fda.gov/downloads/drugs/guidances/ucm078932.pdf
195. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm (2016) 7:27–31. doi:10.4103/0976-0105.177703
196. Parng C, Ton C, Lin Y-X, Roy NM, McGrath P. A zebrafish assay for identifying neuroprotectants in vivo. Neurotoxicol Teratol (2006) 28:509–16. doi:10.1016/j.ntt.2006.04.003
197. Buckley CE, Marguerie A, Roach AG, Goldsmith P, Fleming A, Alderton WK, et al. Drug reprofiling using zebrafish identifies novel compounds with potential pro-myelination effects. Neuropharmacology (2010) 59:149–59. doi:10.1016/j.neuropharm.2010.04.014
198. Li G, Zhang Z, Quan Q, Jiang R-W, Szeto SS, Yuan S, et al. Discovery, synthesis, and functional characterization of a novel neuroprotective natural product from the fruit of alpinia oxyphylla for use in Parkinson’s disease through LC/MS-based multivariate data analysis-guided fractionation. J Proteome Res (2016) 15:2595–606. doi:10.1021/acs.jproteome.6b00152
199. Robertson AL, Ogryzko NV, Henry KM, Loynes CA, Foulkes MJ, Meloni MM, et al. Identification of benzopyrone as a common structural feature in compounds with anti-inflammatory activity in a zebrafish phenotypic screen. Dis Model Mech (2016) 9:621–32. doi:10.1242/dmm.024935
Keywords: drug discovery, hyperkinesia, hypokinesia, movement disorders, zebrafish models
Citation: Vaz RL, Outeiro TF and Ferreira JJ (2018) Zebrafish as an Animal Model for Drug Discovery in Parkinson’s Disease and Other Movement Disorders: A Systematic Review. Front. Neurol. 9:347. doi: 10.3389/fneur.2018.00347
Received: 01 February 2018; Accepted: 30 April 2018;
Published: 01 June 2018
Edited by:
Antonio Pisani, Università degli Studi di Roma Tor Vergata, ItalyReviewed by:
Graziella Madeo, National Institutes of Health (NIH), United StatesAlessandro Tozzi, University of Perugia, Italy
Fibrosis Gardoni, Università degli Studi di Milano, Italy
Copyright: © 2018 Vaz, Outeiro and Ferreira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiago F. Outeiro, dG91dGVpckBnd2RnLmRl;
Joaquim J. Ferreira, amZlcnJlaXJhQG1lZGljaW5hLnVsaXNib2EucHQ=