 Stephan Wenninger
Stephan Wenninger Federica Montagnese
Federica Montagnese Benedikt Schoser
Benedikt Schoser
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 02 May 2018
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 9 - 2018 | https://doi.org/10.3389/fneur.2018.00303
This article is part of the Research Topic Beyond Borders: Myotonic Dystrophies – A European Perception View all 11 articles
Myotonic dystrophy type 1 (DM1) and type 2 (DM2) represent the most frequent multisystemic muscular dystrophies in adulthood. They are progressive, autosomal dominant diseases caused by an abnormal expansion of an unstable nucleotide repeat located in the non-coding region of their respective genes DMPK for DM1 and CNBP in DM2. Clinically, these multisystemic disorders are characterized by a high variability of muscular and extramuscular symptoms, often causing a delay in diagnosis. For both subtypes, many symptoms overlap, but some differences allow their clinical distinction. This article highlights the clinical core features of myotonic dystrophies, thus facilitating their early recognition and diagnosis. Particular attention will be given to signs and symptoms of muscular involvement, to issues related to respiratory impairment, and to the multiorgan involvement. This article is part of a Special Issue entitled “Beyond Borders: Myotonic Dystrophies—A European Perception.”
The genetic background of myotonic dystrophies type 1 and 2 (DM1 and DM2) is due to repeat expansions of unstable nucleotides in untranslated DNA regions causing mis-splicing of mRNAs, which affects almost all cells and organs of the human body. This sum of alterations leads to an extremely heterogeneous phenotype with musltisystemic involvement. Many findings and symptoms of DM1 and DM2 overlap, but important differences usually allow their prompt clinical distinction (Table 2). Both types of myotonic dystrophies represent the most common inherited muscle disorders in adulthood with regional variations in prevalence and incidence. In general, DM1 occurs more frequently than DM2, with some exeptions in northern and mid European countries such as Finland, Germany, and Czech Republic, where DM1 and DM2 are almost equally represented. Table 1 provides a useful summary of the country-specific prevalences (1–4).
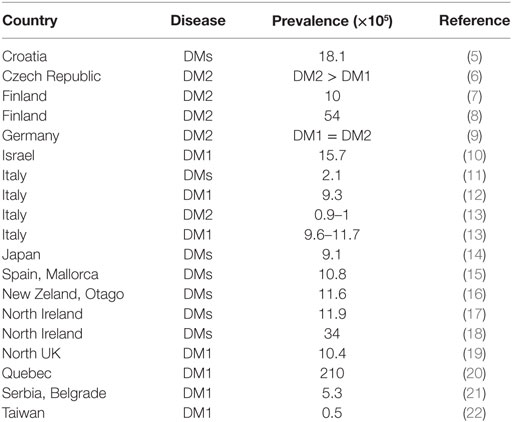
Table 1. Country-specific prevalences of DM1 and DM2.
For DM1, there is a rough correlation between the expansion of CTG-repeats and the onset of symptoms as well as the severity of the disease; nevertheless predictions about the clinical features and the progression of the disease based on CTG-repeat size should be made very carefully (23, 24). 5 to 37 CTG-repeats are physiologic in healthy individuals. An expansion between 38 and 49 repeats does typically not cause any symptoms and reflects the premutation phenotype (3). The following four phenotypes are based on CTG-repeat sizes and onset of symtoms. It is important that phenotypes and CTG-repeat sizes do not show a linear and strict relationship and thus may overlap (25, 26):
• a mild phenotype with an expansion of 50–150 CTG-repeats,
• a classic phenotype with a wide span from mild to severe symptoms and an expansion of 50–1,000 CTG-repeats,
• a childhood/juvenile phenotype with early-onset and typically >800 CTG-repeats, and
• the most severe “congenital form” with usually >1,000 CTG-repeats.
CTG-repeats will expand in every following generation, and fully penetrant alleles occur with >50 CTG-repeats. This results in the so called anticipation, a clinical term describing an earlier onset with a more severe phenotype in the next generations (26). Furthermore, the repeat instability leads notably to premature aging of almost all organs, so DM1 may be counted among the progeroid diseases (27). The most typical appearance of DM1 is the “adult-onset” or “classic” phenotype with a CTG-repeat size ranging from 50 to <1,000. It is characterized by a distinctive combination of muscular symptoms, such as facial weakness, ptosis, grip myotonia, and distal muscle weakness with muscular atrophy. The classic phenotype is typically accompanied by extramuscular symptoms like cognitive impairment, cataracts, and diabetes mellitus. Nevertheless, as this multisystem disorder often presents with a high variability, some patients may primarily show only non-specific extramuscular symptoms like fatigue, daytime sleepiness, gastrointestinal symptoms, or cardiac conduction defects in an early stage of the disease, which could delay the diagnosis. Mildly affected patients with CTG-repeat sizes 50–100 may have normal or only minimally shortened lifespan (28). Because of comorbidities, such as cardiac and pulmonary complications, life expectancy is, however, reduced in about 70% of the patients with the classic phenotype (25).
DM2 (also referred to as proximal myotonic myopathy) is caused by the expansion of the tetranucleotide CCTG-repeat in the first intron of CNBP (cellular nucleic acid-binding protein), formerly known as zinc finger protein 9 (ZNF9) gene (29). Similar to DM1, these expansions are extremely unstable, causing widespread cellular abnormalities of mRNA splicing. In DM2, the expansion ranges from 75 to 11,000 with a mean of 5,000 CCTG-repeats. In contrast to DM1, there is no correlation between clinical phenotype and CCTG-repeat length and no anticipation has been observed (29, 30).
The symptoms myotonia, muscular weakness, and muscular atrophy are the principal traits of DMs and gave the eponym for these two types of the disease. In DM1, patients present with characteristic distally predominant muscular atrophy and weakness mainly involving finger flexors, wrist flexors, and foot extensors (Figures 1A,B). The latter will cause foot drop and gait disturbance with repeated falls and injuries (31). In contrast to this, muscle weakness in DM2 is typically proximal and axial, affecting more consistently the neck flexors, hip flexors, and hip extensors (Figure 1C) (30, 32). This predominantly proximal muscular involvement has been documented also by MRI studies that showed an early degeneration of the erector spinae and gluteus maximus muscles (33, 34). Muscle weakness is one of the most frequently reported symptoms in DM1 (>45% of patients with adult phenotype) and DM2 (40–55% of patients) (26, 32). Figure 1D illustrates the predominantly affected muscle groups of patients with DM1 and DM2 (see Table 2 for differentiating DM1 and DM2).
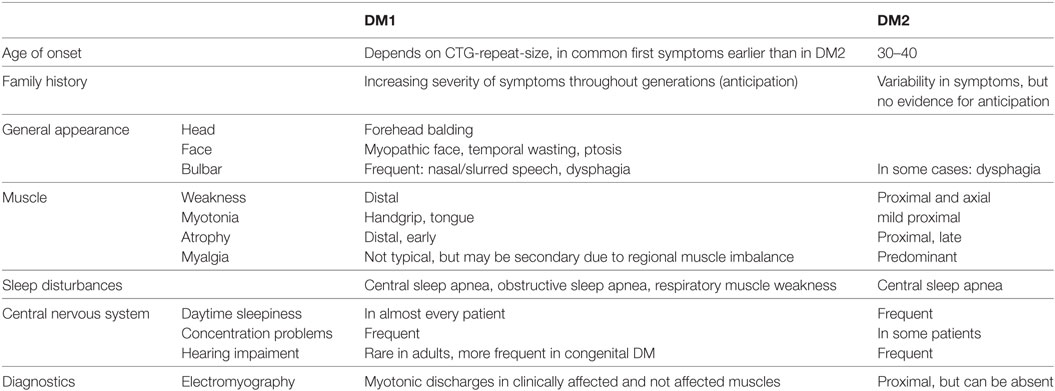
Table 2. Core Clinical Symptoms helpful for differentiating DM1 and DM2.
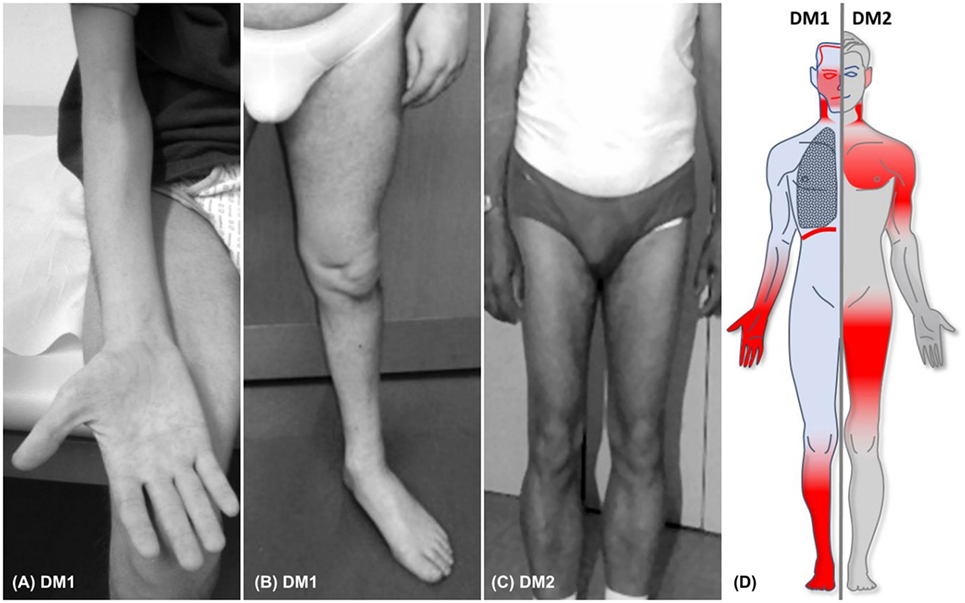
Figure 1. (A,B) Typical distal muscular atrophy in patients with DM1. (C) Atrophy of proximal muscles in a patient with DM2. (D) Figure illustrating the core phneotypes of DM1 (left) and DM2 (right). Regions of muscular involvement (weakness and atrophy) are highlighted in red.
The typical facial appearance of DM1 patients (“myopathic face”—“hatchet face”) is a prominent and early feature and is caused by weakness and atrophy of facial muscles and ptosis that might give the false impression of a tired, sad, or emotionless patient (35). Balding of the forehead and atrophying of the temporal muscle are often seen (Figure 2) and completes the overall picture of a patient with DM1. Severe weakness of orbiculari oculi muscles cause not only ptosis but also insufficient eyelid closure with risk of recurrent conjunctivitis. This facial muscle involvement is usually not seen in DM2 patients, thus it may help in differentiating DM1 from DM2 patients (Table 2).

Figure 2. The characteristic face of a patient with DM1: long face, temporomandibular wasting, balding forehead.
Especially in patients with DM1, the speech can be nasal and slurred, due to the weakness of oropharyngeal muscles, sometimes causing chewing and swallowing difficulties.
Myotonia is a more frequent symptom in DM1 mainly affecting the fingers (grip myotonia), the jaw, and the tongue (36). Clinically, a warm-up phenomenon is usually observed when myotonia improves with repeated contractions, which is mostly true for grip myotonia, but also for myotonia of the tongue and the jaw (37, 38). An increased excitability of muscle fibers is thought to be the cause for myotonia, leading to continuous discharges of repetitive action potentials after voluntary contraction or mechanical stimulation (39) in electromyography (EMG). These myotonic runs can be detected with EMG even in clinically unaffected muscles of DM1 patients, but can be rare or even be absent in DM2 (40). On a molecular basis, it has been suggested that myotonia is caused by mis-splicing of the chloride channel (CLCN1, ClC-1) due to misregulated MBNL1 and CUGBP1 (41). Recent studies have investigated the relationship between a central nervous system involvement and myotonia, suggesting that myotonia should no longer be considered as a solitary peripherally triggered muscular symptom (42, 43). In one fMRI study, higher cerebral blood oxygen level-dependent signals (BOLD) in specific primary and secondary motor areas were found during myotonia episodes. This was interpreted as a relationship between myotonia and high-order motor control areas (44). In another study, the severity of myotonia correlated with diffuse white matter alterations in specific primary and secondary motor areas (45). There are contradictory reports about the correlation between myotonia, grip strength, and CTG-repeat length (46, 47) for DM1 patients. Overall, grip strength correlates negatively with CTG-repeat length in most studies, but this is not necessarily true for myotonia. In one recent study, there was a statistically significant correlation between grip myotonia and CTG-repeat length, but this was not clinically meaningful and not predictive (47).
Myotonia seems to be usually mild to moderate or even absent in many DM2 patients, impacting only minimally their quality of life (36). However, its occurrence in different cohorts ranges between 24 and 75% (30, 32, 40). This variability is partly due to the discrepancy sometimes observed between a history of myotonia reported by patients and the clinical evidence of myotonic phenomenon, which is observed on neurological examination only in a minority of DM2 patients. Few patients may, however, display a severe myotonia and in some of these cases additional mutations in ion channel genes CLCN1 and SCN4A have been identified (48, 49). It is, therefore, advised to screen atypical cases with severe myotonia for mutation in these genes that act as phenotype modifier enhancing the myotonic phenomenon in DM2 (49). Particularly limb girdle myotonia is frequently neglected and underdiagnosed. With aging, the presence of myotonia gradually becomes less clinically relevant as it is overwhelmed by the gradually worsening of muscle weakness (32). This trend is also confirmed in studies assessing quality of life of DM2 patients, where significant predictors of worse QoL (quality of life) were older age, worse muscle strength, and higher level of fatigue (50).
Musculoskeletal pain or myalgia may be present in some DM1 patients, but is less frequent in comparison to DM2 (Table 2). However, with the progression of the disease, a muscular imbalance due to weakness may occur and secondary complications such as regional myofascial pain or joint pain syndromes may develop even in DM1. In DM2, about 60% of patients complain of diffuse myalgia. These are usually exercise-related and worsen in cold temperatures (33, 51). Some patients consider pain as the most disabling symptom of the disease also because of its poor response to common analgesics (7). The pathophysiological mechanism of myalgia in DM2 is yet to be elucidated, but it is probably related to specific molecular changes occurring in the muscles of DM2 patients (52).
For adult-onset DM1, the first muscular symptoms can become apparent in early adulthood, but some patients may exhibit subtle symptoms like grip myotonia, ptosis or slurred speech in childhood. Patients with classic DM1 are typically diagnosed at around 30 years, but mildly affected patients with CTG-length 50–100 may present solely some slight myotonia or cataracts and may have their diagnosis delayed until they are around 40 years old (28). The clinical onset of DM2 typically occurs later than DM1, around the third to fourth decade; it may, however, often go unrecognized for several years due to only mild or unspecific clinical symptoms like myalgia or muscle cramps.
Respiratory muscle weakness will occur in a high percentage of the patients with DM1 in an early stage of the disease and chronic respiratory failure may develop (53). Expiratory muscles seem to be affected sooner than inspiratory muscles, resulting in early recurrent pneumonia due to a weak cough and insufficient airway clearance. The exact prevalence of respiratory insufficiency in DM1 is unclear because symptoms of nocturnal hypoventilation overlap with typical neuropsychological symptoms like fatigue, daytime sleepiness, and concentration difficulties (54). As both respiratory muscle weakness and cardiac symptoms account most for the reduced survival of the patients, repeated testing for early diagnosis is essential. A pure respiratory muscle weakness rarely occurs in DM2 and only about 6–15% of patients require non-invasive ventilation (55).
Fatigue, daytime sleepiness, and concentration difficulties are frequently reported symptoms in DMs. In DM1, cognitive deficits were initially attributed to a low IQ or mental retardation, but recent studies show that this assumption was wrong for a large cohort of patients and mainly applies for cases of congenital myotonic dystrophy (CDM). In fact, for the classic phenotype of DM1, neuropsychological deficits are as variable as muscular symptoms, and even recent publications about the correlation of CTG-repeat size and neuropsychological deficits show contradictory study results (56–58). There seems to be a correlation between diffuse brain alterations in primarily white and secondary gray matter, linking the DM1 to the group of brain disconnection disorders (59). Caso et al. investigated 51 DM1 patients and found a correlation between changes in brain white matter and cognitive impairment (60). Cerebral white matter hyperintensities have been observed in both DM1 and DM2 patients, especially in those older than 40 years, but their clinical and functional significance still remains unclear (61–63). In a recent study about the educational profile of a large cohort of young DM1 patients, no significant differences compared to the healthy population were found (35), assuming that cognitive and concentration disturbances may occur later in the course of the disease in the context of a variable premature cognitive decline, as suggested by the study of Modoni et al. (56). Mild cognitive and behavioral symptoms are also present in DM2 patients. In particular, altered visuo-spatial and executive functions, reduced attention and flexibility of thinking, avoidant behavioral trait, and depression have been detected in these patients (63, 64). In many cases, neuropsychological disturbances jeopardize the ability to work and reduce the quality of life more than muscular symptoms.
Excessive daytime sleepiness, fatigue, and concentration difficulties may also be caused by central sleep disturbances or sleep apnea. Sleep-disordered breathing is one of the earliest manifestations and occurs in a high percentage of patients with DM1 (65), but overlapping symptoms of nocturnal hypoventilation and CNS symptoms may delay diagnosis and treatment. Chronic central sleep-disordered breathing has an impact on quality of life, morbidity, and mortality and should be assessed frequently in every patient with DM1 (54, 55, 66).
Until now, little is known about changes in CNS causing cognitive deficits and central sleep disrupted breathing. Almost every clinical study is conducted with the usually more affected DM1 patients, therefore data for DM2 patients are limited. On a molecular basis, MBNL1 and probably CELF may both be involved in CNS alterations, but little is known about molecular defects causing highly variable CNS symptoms in DM1 (42, 43). The above mentioned aspects lead to a discussion as to whether CNS dysfunction is caused by altered neurodevelopment, by neuro-dysfunction or by neurodegeneration within the definition of progeroid diseases (67). The hypothesis of a neurodegenerative disease is endorsed by findings of tau pathology and neurofibrillary degenerations, even if no correlations with CTG-repeat length were found (68). Overall, CNS dysfunction seems to be multifactorial.
The most frequent, early and typical extramuscular manifestation is the occurrence of early-onset cataract, observed in about 50–60% of patients (30, 32, 69). A medical history of cataract surgery in combination with muscular symptoms often leads to the diagnosis of DM, even in mildly affected patients without any sign of muscular impairment (70–72). The mechanisms underlying the pathophysiology of cataract in DMs are still largely unknown. At first, a potential effect of the CTG-mutation on the expression of neighboring genes such as SIX5 was considered in DM1 (73). But recent findings showed that SIX5 knock-out mice develop the nuclear type of cataract and not the posterior subcapsular/cortical type that are commonly observed in DMs. In addition, SIX5 is not adjacent to the DM2 repeat expansion so that this mechanism could not explain cataracts in DM2. More recent studies of global transcription performed on samples of lens epithelium in patients affected by DM1, DM2, and controls, identified a high similarity as regards the pattern of gene expression between DM1 and DM2 and hypothesized that common molecular mechanisms should be involved in cataract formation probably involving interferon signaling pathways (74, 75).
Endocrine dysfunctions such as diabetes, hypogonadism, and secondary hyperparathyroidism with decreased Vitamin-D levels are frequent in DMs and their occurrence increases with progression of the disease (76–78). A cross-sectional study on 68 DM1 patients showed at least one endocrine dysfunction in 44% at baseline and in 84% after 8 years (76). Diabetes mellitus, if not properly treated, may complicate and aggravate the clinical picture due to diabetic polyneuropathy with worsening of gait instability and distal weakness. Hyperparathyroidism may contribute to fatigue and muscle impairment (76). More rarely, abnormalities in growth hormone secretion and glucose intolerance may be observed (79).
Some degree of hearing loss has been described in DM2 since its first description (80). A recent systematic study on 56 Dutch and French DM2 patients then demonstrated that a mild to moderate hearing impairment was present in about 60% of examined patients. It is mostly a cochlear sensorineural hearing impairment which may be interpreted as an early presbycusis (81), well fitting in the interpretation of DMs as premature aging diseases. Similar features of cochlear impairment have also been described in some studies on DM1 patients (82).
In DMs, cardiac involvement is common. Cardiologic comorbidities include arrhythmias, atrial fibrillation, and conduction defects (e.g., AV-blocks) and often requires the implantation of pacemakers. Other infrequent manifestations are sudden death, heart failure, Brugada syndrome, ischemic heart disease, and mitral valve prolapse (83, 84). Dilated cardiomyopathies may also occur in some patients, but are not frequently found. Cardiac abnormalities in DM2 are similar to those observed in DM1 but occur less frequently. According to a recent observational case–control study on a large cohort of DM2/DM1 patients, it emerged that electrocardiographic abnormalities as PR > 200 ms and QRS > 100 ms were more frequent in DM1 (respectively, 31 and 48%) than DM2 patients (10 and 17%). Of those, 6 DM2 vs. 28 DM1 patients needed a pacemaker/implanted cardioverter (85). In the same study, echocardiography did not show any significant structural abnormalities but it was previously reported that a cardiomyopathy might occur in about 3% of DM2 patients. In DM1, the severity of cardiac involvement seems directly related with the size of CTG-expansion as recently studied by Chong-Nguyen et al. (9, 30, 85, 86). Atrial fibrillations and arrhythmias increase the risk of cerebral ischemia (87) and mortality and morbidity significantly depend on early cardiologic diagnosis and treatment (83).
Along with elevations of creatine kinase, elevations of AST and ALT are frequent in patients with DM (88). In some cases, liver biopsies are performed because of these elevated “hepatic” enzymes without retrieving any pathologic result. The elevation of gamma-GT is suggested to be caused by contractions of bile canaliculi and bile ductules, whereas elevated levels of AST and ALT have their origin in skeletal muscle and go along with elevations of creatine kinase (89).
Alternating constipation, pseudo-constipation, bloating, and diarrhea are frequently reported symptoms in DM1, accompanied by stomach cramps, reflux, and regurgitation. They are caused by involvement of smooth and striated muscles and endocrine dysfunctions (90, 91). Swallowing problems are typical for DM1 patients and due to reduced oral transport that is caused by myotonia and weakness of the tongue. Dysphagia is caused by reduced swallowing reflex and reduced esophageal motility (92) which causes the major clinical problem by risk of aspiration. In conjunction with weakness of early affected expiratory muscles, this results in recurrent pneumonia and increased risk of death. A reduced or absent gastrointestinal peristaltic movement was earlier shown in radiological studies as well as delayed intestinal transits (93). Megacolon with the risk of ileus, volvulus and rupture, is a significant and life-threatening complication. Delayed emptying of the gall bladder may increase the risk for gallstones.
A higher incidence for neoplasms was found in several studies (28, 94, 95), most of them showed a predisposition in patients with DM1 for cancers such as skin cancer (like benign calcifying cutaneous tumors, pilomatricomas), thyroid, testicular, and prostate cancer. Because of the limited number of high-quality surveys and studies about the prevalence of cancer in DM1, further research is needed. A survey from the UK DM registry showed that 12.4% of the DM1/DM2 patients reported at least one benign tumor and 6.2% reported at least one malignant tumor with a high incidence of skin tumors (96), but there was no epidemiologic correlation with a non-DM-population.
There is some debate as to whether peripheral neuropathy is a multisystemic manifestation of DMs or are caused by metabolic and endocrine dysfunctions. Its manifestation is not typical at early stages of the disease but may occur in about one-third of patients in later stages (97) of DM1 patients and contributes to balance impairment and increased risk of falls (31, 98). There were no significant correlations between age, duration of neuromuscular symptoms or CTG-repeat size (98, 99), suggesting that the affection of peripheral nerve system is secondary to metabolic and endocrine dysfunctions.
Patients with congenital DM1 have large CTG-expansions of more than 800, usually around 1,000. Characteristically, these large expansions are caused by maternal transmission, but CDM with paternal transmission is also known (23, 100–102). Clinically, CDM patients are severely affected and symptoms are often present before birth as polyhydramnios and reduced fetal movement. Hypotonia, generalized weakness, hyporeflexia, bilateral talipes, contractures, arthrogryposis, facial dysmorphia (carp mouth, ptosis, long neck and face, temporal muscle atrophy), and a weak cry are typical symptoms at birth or in the first days after delivery. Weak sucking and respiratory insufficiency often make ventilatory support unavoidable. Respiratory insufficiency is present in about 50% of newborns and is the main cause of dramatically reduced survival with a mortality rate of 30–40% (103). Infants who survive will typically reach their motor and cognitive milestones with some delay but might be able to walk independently. Similarly to DM1, a distal weakness is typical in CDM and a proximal involvement indicates a poor prognosis (104). Besides muscular symptoms, cognitive impairment, and neuropsychological disorders are the most common and variable manifestations in CDM. Symptoms range from intellectual impairment to selective cognitive impairment, apathy, and autism, as well as impaired attention, severe anxiety, and mood and depression syndromes (3, 102, 105–107). In the course of the disease, patients might require special schooling. In their third and fourth decades, patients may develop secondary complications, such as severe contractures, scoliosis, and worsening of cardiorespiratory symptoms (4).
The childhood and juvenile onset DM1 echoes the broad overlapping spectrum of symptoms of the congenital and the adult phenotypes. Commonly, there is an expansion of CTG-repeats of more than 800 repeats. First clinical symptoms may become apparent at age 1–10 for childhood onset and at age 10–20 for juvenile onset (3). Neurocognitive symptoms such as learning disability and learning difficulties are often prominent at age around 10 years and may become earlier apparent than muscular symptoms (107). In contrast to CDM, prenatal abnormalities or muscular symptoms right after delivery (neonatal hypotonia, sucking and swallowing difficulties and secondary dysmorphic features) are not typical, but a mild facial weakness or subtle facial dysmorphia may occur (3, 108). Early motor development is normal or only slightly delayed. Principal complaints in early childhood are speech and learning difficulties because of a mental handicap. At school, learning difficulties may become apparent and sometimes require special education. A study on 28 childhood-DM-patients showed that the full-scale IQ was significantly decreased (73.6) and 68% of the patients had repeated at least one school grade. 54% had additional psychiatric symptoms such as anxiety disorder, mood disorder, and attention-deficit-hyperactivity disorder (107). In adolescence, patients may show typical muscular and non-muscular symptoms of adult-onset DM1, e.g., like distal weakness, clinical myotonia, or gastrointestinal symptoms. Cardiologic symptoms, such as cardiac arrhythmias or cardiomyopathy, may occur, also leading to severe complications and sudden death (83). Life expectancy is not necessarily reduced, as long as core symptoms are recognized and treated sufficiently.
Myotonic dystrophies represent the most variable clinical phenotypes, so treatment stratification is key for any modern therapeutic approach. We still need much more understanding of the signs and symptoms of DM patients in correlation to their molecular origins.
SW: review of publications, writing, critical revision of manuscript for intellectual content, and final approval of the manuscript. FM: review of publications, writing, and critical revision of manuscript for intellectual content. BS: critical revision of manuscript for intellectual content.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Suominen T, Bachinski LL, Auvinen S, Hackman P, Baggerly KA, Angelini C, et al. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet (2011) 19(7):776–82. doi:10.1038/ejhg.2011.23
2. Udd B, Meola G, Krahe R, Wansink DG, Bassez G, Kress W, et al. Myotonic dystrophy type 2 (DM2) and related disorders report of the 180th ENMC workshop including guidelines on diagnostics and management 3-5 December 2010, Naarden, The Netherlands. Neuromuscul Disord (2011) 21(6):443–50. doi:10.1016/j.nmd.2011.03.013
3. Ho G, Cardamone M, Farrar M. Congenital and childhood myotonic dystrophy: current aspects of disease and future directions. World J Clin Pediatr (2015) 4(4):66–80. doi:10.5409/wjcp.v4.i4.66
4. Meola G, Cardani R. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta (2015) 1852(4):594–606. doi:10.1016/j.bbadis.2014.05.019
5. Medica I, Markovic D, Peterlin B. Genetic epidemiology of myotonic dystrophy in Istria, Croatia. Acta Neurol Scand (1997) 95(3):164–6. doi:10.1111/j.1600-0404.1997.tb00089.x
6. Vohanka S, Parmova O, Mazanec R, Strenkova J, Ridzon P, Ehler E, et al. Myotonic dystrophy in Czech Republic: data from the national registry. J Neurol Sci (2015) 357:e347–8. doi:10.1016/j.jns.2015.08.1232
7. Udd B, Meola G, Krahe R, Thornton C, Ranum LP, Bassez G, et al. 140th ENMC International Workshop: myotonic dystrophy DM2/PROMM and other myotonic dystrophies with guidelines on management. Neuromuscul Disord (2006) 16(6):403–13. doi:10.1016/j.nmd.2006.03.010
8. Suominen S, Koskenvuo K, Sillanmaki L, Vahtera J, Korkeila K, Kivimaki M, et al. Non-response in a nationwide follow-up postal survey in Finland: a register-based mortality analysis of respondents and non-respondents of the Health and Social Support (HeSSup) Study. BMJ Open (2012) 2(2):e000657. doi:10.1136/bmjopen-2011-000657
9. Schoser BG, Ricker K, Schneider-Gold C, Hengstenberg C, Durre J, Bultmann B, et al. Sudden cardiac death in myotonic dystrophy type 2. Neurology (2004) 63(12):2402–4. doi:10.1212/01.WNL.0000147335.10783.E4
10. Segel R, Silverstein S, Lerer I, Kahana E, Meir R, Sagi M, et al. Prevalence of myotonic dystrophy in Israeli Jewish communities: inter-community variation and founder premutations. Am J Med Genet A (2003) 119A(3):273–8. doi:10.1002/ajmg.a.20177
11. Pinessi L, Bergamini L, Cantello R, Di Tizio C. Myotonia congenita and myotonic dystrophy: descriptive epidemiological investigation in Turin, Italy (1955-1979). Ital J Neurol Sci (1982) 3(3):207–10. doi:10.1007/BF02043311
12. Siciliano G, Manca M, Gennarelli M, Angelini C, Rocchi A, Iudice A, et al. Epidemiology of myotonic dystrophy in Italy: re-apprisal after genetic diagnosis. Clin Genet (2001) 59(5):344–9. doi:10.1034/j.1399-0004.2001.590508.x
13. Vanacore N, Rastelli E, Antonini G, Bianchi ML, Botta A, Bucci E, et al. An age-standardized prevalence estimate and a sex and age distribution of myotonic dystrophy types 1 and 2 in the Rome province, Italy. Neuroepidemiology (2016) 46(3):191–7. doi:10.1159/000444018
14. Nakagawa M, Nakahara K, Yoshidome H, Suehara M, Higuchi I, Fujiyama J, et al. Epidemiology of progressive muscular dystrophy in Okinawa, Japan. Classification with molecular biological techniques. Neuroepidemiology (1991) 10(4):185–91. doi:10.1159/000110268
15. Burcet J, Canellas F, Cavaller G, Vich M. [Epidemiologic study of myotonic dystrophy on the island of Mallorca]. Neurologia (1992) 7(2):61–4.
16. Ford C, Kidd A, Hammond-Tooke G. Myotonic dystrophy in Otago, New Zealand. N Z Med J (2006) 119(1241):U2145.
17. Magee A, Nevin NC. The epidemiology of myotonic dystrophy in Northern Ireland. Community Genet (1999) 2(4):179–83. doi:10.1159/000016209
18. Hughes MI, Hicks EM, Nevin NC, Patterson VH. The prevalence of inherited neuromuscular disease in Northern Ireland. Neuromuscul Disord (1996) 6(1):69–73. doi:10.1016/0960-8966(94)00017-4
19. Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain (2009) 132(Pt 11):3175–86. doi:10.1093/brain/awp236
20. Bouchard G, Roy R, Declos M, Mathieu J, Kouladjian K. Origin and diffusion of the myotonic dystrophy gene in the Saguenay region (Quebec). Can J Neurol Sci (1989) 16(1):119–22. doi:10.1017/S0317167100028651
21. Mladenovic J, Pekmezovic T, Todorovic S, Rakocevic-Stojanovic V, Savic D, Romac S, et al. Epidemiology of myotonic dystrophy type 1 (Steinert disease) in Belgrade (Serbia). Clin Neurol Neurosurg (2006) 108(8):757–60. doi:10.1016/j.clineuro.2006.04.004
22. Hsiao KM, Chen SS, Li SY, Chiang SY, Lin HM, Pan H, et al. Epidemiological and genetic studies of myotonic dystrophy type 1 in Taiwan. Neuroepidemiology (2003) 22(5):283–9. doi:10.1159/000071191
23. Brunner HG, Bruggenwirth HT, Nillesen W, Jansen G, Hamel BC, Hoppe RL, et al. Influence of sex of the transmitting parent as well as of parental allele size on the CTG expansion in myotonic dystrophy (DM). Am J Hum Genet (1993) 53(5):1016–23.
24. Bouchard JP, Cossette L, Bassez G, Puymirat J. Natural history of skeletal muscle involvement in myotonic dystrophy type 1: a retrospective study in 204 cases. J Neurol (2015) 262(2):285–93. doi:10.1007/s00415-014-7570-x
25. Turner C, Hilton-Jones D. Myotonic dystrophy: diagnosis, management and new therapies. Curr Opin Neurol (2014) 27(5):599–606. doi:10.1097/WCO.0000000000000128
26. De Antonio M, Dogan C, Hamroun D, Mati M, Zerrouki S, Eymard B, et al. Unravelling the myotonic dystrophy type 1 clinical spectrum: a systematic registry-based study with implications for disease classification. Rev Neurol (2016) 172(10):572–80. doi:10.1016/j.neurol.2016.08.003
27. Mateos-Aierdi AJ, Goicoechea M, Aiastui A, Fernandez-Torron R, Garcia-Puga M, Matheu A, et al. Muscle wasting in myotonic dystrophies: a model of premature aging. Front Aging Neurosci (2015) 7:125. doi:10.3389/fnagi.2015.00125
28. Bird TD. Myotonic dystrophy type 1. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K et al. editors. GeneReviews® [Internet]. Seattle, WA: University of Washington 1993–2018.
29. Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science (2001) 293(5531):864–7. doi:10.1126/science.1062125
30. Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W, et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology (2003) 60(4):657–64. doi:10.1212/01.WNL.0000054481.84978.F9
31. Jiménez-Moreno AC, Raaphorst J, Babačić H, Wood L, van Engelen B, Lochmüller H, et al. Falls and resulting fractures in myotonic dystrophy: results from a multinational retrospective survey. Neuromuscul Disord (2018) 28(3):229–35. doi:10.1016/j.nmd.2017.12.010
32. Montagnese F, Mondello S, Wenninger S, Kress W, Schoser B. Assessing the influence of age and gender on the phenotype of myotonic dystrophy type 2. J Neurol (2017) 264(12):2472–80. doi:10.1007/s00415-017-8653-2
33. Ricker K, Koch MC, Lehmann-Horn F, Pongratz D, Otto M, Heine R, et al. Proximal myotonic myopathy: a new dominant disorder with myotonia, muscle weakness, and cataracts. Neurology (1994) 44(8):1448–52. doi:10.1212/WNL.44.8.1448
34. Kornblum C, Lutterbey G, Bogdanow M, Kesper K, Schild H, Schroder R, et al. Distinct neuromuscular phenotypes in myotonic dystrophy types 1 and 2: a whole body highfield MRI study. J Neurol (2006) 253(6):753–61. doi:10.1007/s00415-006-0111-5
35. Stahl K, Wenninger S, Schuller A, Montagnese F, Schoser B. [Educational and professional qualifications of adults with myotonic dystrophies – a misleading perception by the myopathic face?]. Fortschr Neurol Psychiatr (2016) 84(4):211–6. doi:10.1055/s-0042-104193
36. Heatwole C, Johnson N, Bode R, Dekdebrun J, Dilek N, Hilbert JE, et al. Patient-reported impact of symptoms in myotonic dystrophy type 2 (PRISM-2). Neurology (2015) 85(24):2136–46. doi:10.1212/WNL.0000000000002225
37. de Swart BJ, van Engelen BG, van de Kerkhof JP, Maassen BA. Myotonia and flaccid dysarthria in patients with adult onset myotonic dystrophy. J Neurol Neurosurg Psychiatry (2004) 75(10):1480–2. doi:10.1136/jnnp.2003.032151
38. Logigian EL, Blood CL, Dilek N, Martens WB, Moxley RT, Wiegner AW, et al. Quantitative analysis of the “warm-up” phenomenon in myotonic dystrophy type 1. Muscle Nerve (2005) 32(1):35–42. doi:10.1002/mus.20339
39. Hudson AJ, Ebers GC, Bulman DE. The skeletal muscle sodium and chloride channel diseases. Brain (1995) 118(Pt 2):547–63. doi:10.1093/brain/118.2.547
40. Young NP, Daube JR, Sorenson EJ, Milone M. Absent, unrecognized, and minimal myotonic discharges in myotonic dystrophy type 2. Muscle Nerve (2010) 41(6):758–62. doi:10.1002/mus.21615
41. Meola G. Clinical aspects, molecular pathomechanisms and management of myotonic dystrophies. Acta Myol (2013) 32(3):154–65.
42. Caillet-Boudin ML, Fernandez-Gomez FJ, Tran H, Dhaenens CM, Buee L, Sergeant N. Brain pathology in myotonic dystrophy: when tauopathy meets spliceopathy and RNAopathy. Front Mol Neurosci (2014) 6:57. doi:10.3389/fnmol.2013.00057
43. Goodwin M, Mohan A, Batra R, Lee KY, Charizanis K, Fernandez Gomez FJ, et al. MBNL sequestration by toxic RNAs and RNA misprocessing in the myotonic dystrophy brain. Cell Rep (2015) 12(7):1159–68. doi:10.1016/j.celrep.2015.07.029
44. Toth A, Lovadi E, Komoly S, Schwarcz A, Orsi G, Perlaki G, et al. Cortical involvement during myotonia in myotonic dystrophy: an fMRI study. Acta Neurol Scand (2015) 132(1):65–72. doi:10.1111/ane.12360
45. Zanigni S, Evangelisti S, Giannoccaro MP, Oppi F, Poda R, Giorgio A, et al. Relationship of white and gray matter abnormalities to clinical and genetic features in myotonic dystrophy type 1. Neuroimage Clin (2016) 11:678–85. doi:10.1016/j.nicl.2016.04.012
46. Andersen G, Orngreen MC, Preisler N, Colding-Jorgensen E, Clausen T, Duno M, et al. Muscle phenotype in patients with myotonic dystrophy type 1. Muscle Nerve (2013) 47(3):409–15. doi:10.1002/mus.23535
47. Hogrel JY, Ollivier G, Ledoux I, Hebert LJ, Eymard B, Puymirat J, et al. Relationships between grip strength, myotonia, and CTG expansion in myotonic dystrophy type 1. Ann Clin Transl Neurol (2017) 4(12):921–5. doi:10.1002/acn3.496
48. Cardani R, Giagnacovo M, Botta A, Rinaldi F, Morgante A, Udd B, et al. Co-segregation of DM2 with a recessive CLCN1 mutation in juvenile onset of myotonic dystrophy type 2. J Neurol (2012) 259(10):2090–9. doi:10.1007/s00415-012-6462-1
49. Bugiardini E, Rivolta I, Binda A, Soriano Caminero A, Cirillo F, Cinti A, et al. SCN4A mutation as modifying factor of myotonic dystrophy type 2 phenotype. Neuromuscul Disord (2015) 25(4):301–7. doi:10.1016/j.nmd.2015.01.006
50. Rakocevic Stojanovic V, Peric S, Paunic T, Pesovic J, Vujnic M, Peric M, et al. Quality of life in patients with myotonic dystrophy type 2. J Neurol Sci (2016) 365:158–61. doi:10.1016/j.jns.2016.04.018
51. Suokas KI, Haanpaa M, Kautiainen H, Udd B, Hietaharju AJ. Pain in patients with myotonic dystrophy type 2: a postal survey in Finland. Muscle Nerve (2012) 45(1):70–4. doi:10.1002/mus.22249
52. Moshourab R, Palada V, Grunwald S, Grieben U, Lewin GR, Spuler S. A molecular signature of myalgia in myotonic dystrophy 2. EBioMedicine (2016) 7:205–11. doi:10.1016/j.ebiom.2016.03.017
53. Reardon W, Newcombe R, Fenton I, Sibert J, Harper PS. The natural history of congenital myotonic dystrophy: mortality and long term clinical aspects. Arch Dis Child (1993) 68(2):177–81. doi:10.1136/adc.68.2.177
54. Boentert M, Wenninger S, Sansone VA. Respiratory involvement in neuromuscular disorders. Curr Opin Neurol (2017) 30(5):529–37. doi:10.1097/WCO.0000000000000470
55. Sansone VA, Gagnon C. 207th ENMC Workshop on chronic respiratory insufficiency in myotonic dystrophies: management and implications for research, 27-29 June 2014, Naarden, The Netherlands. Neuromuscul Disord (2015) 25(5):432–42. doi:10.1016/j.nmd.2015.01.011
56. Modoni A, Silvestri G, Pomponi MG, Mangiola F, Tonali PA, Marra C. Characterization of the pattern of cognitive impairment in myotonic dystrophy type 1. Arch Neurol (2004) 61(12):1943–7. doi:10.1001/archneur.61.12.1943
57. Winblad S, Lindberg C, Hansen S. Cognitive deficits and CTG repeat expansion size in classical myotonic dystrophy type 1 (DM1). Behav Brain Funct (2006) 2:16. doi:10.1186/1744-9081-2-16
58. Sugiyama A, Sone D, Sato N, Kimura Y, Ota M, Maikusa N, et al. Brain gray matter structural network in myotonic dystrophy type 1. PLoS One (2017) 12(11):e0187343. doi:10.1371/journal.pone.0187343
59. Minnerop M, Weber B, Schoene-Bake JC, Roeske S, Mirbach S, Anspach C, et al. The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain (2011) 134(Pt 12):3530–46. doi:10.1093/brain/awr299
60. Caso F, Agosta F, Peric S, Rakocevic-Stojanovic V, Copetti M, Kostic VS, et al. Cognitive impairment in myotonic dystrophy type 1 is associated with white matter damage. PLoS One (2014) 9(8):e104697. doi:10.1371/journal.pone.0104697
61. Kornblum C, Reul J, Kress W, Grothe C, Amanatidis N, Klockgether T, et al. Cranial magnetic resonance imaging in genetically proven myotonic dystrophy type 1 and 2. J Neurol (2004) 251(6):710–4. doi:10.1007/s00415-004-0408-1
62. Franc DT, Muetzel RL, Robinson PR, Rodriguez CP, Dalton JC, Naughton CE, et al. Cerebral and muscle MRI abnormalities in myotonic dystrophy. Neuromuscul Disord (2012) 22(6):483–91. doi:10.1016/j.nmd.2012.01.003
63. Schneider-Gold C, Bellenberg B, Prehn C, Krogias C, Schneider R, Klein J, et al. Cortical and subcortical grey and white matter atrophy in myotonic dystrophies type 1 and 2 is associated with cognitive impairment, depression and daytime sleepiness. PLoS One (2015) 10(6):e0130352. doi:10.1371/journal.pone.0130352
64. Meola G, Sansone V, Perani D, Scarone S, Cappa S, Dragoni C, et al. Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM-1) and in proximal myotonic myopathy (PROMM/DM-2). Neuromuscul Disord (2003) 13(10):813–21. doi:10.1016/S0960-8966(03)00137-8
65. Bianchi ML, Losurdo A, Di Blasi C, Santoro M, Masciullo M, Conte G, et al. Prevalence and clinical correlates of sleep disordered breathing in myotonic dystrophy types 1 and 2. Sleep Breath (2014) 18(3):579–89. doi:10.1007/s11325-013-0921-5
66. Ho G, Widger J, Cardamone M, Farrar MA. Quality of life and excessive daytime sleepiness in children and adolescents with myotonic dystrophy type 1. Sleep Med (2017) 32:92–6. doi:10.1016/j.sleep.2016.12.005
67. Gourdon G, Meola G. Myotonic dystrophies: state of the art of new therapeutic developments for the CNS. Front Cell Neurosci (2017) 11:101. doi:10.3389/fncel.2017.00101
68. Itoh K, Mitani M, Kawamoto K, Futamura N, Funakawa I, Jinnai K, et al. Neuropathology does not correlate with regional differences in the extent of expansion of CTG repeats in the brain with myotonic dystrophy type 1. Acta Histochem Cytochem (2010) 43(6):149–56. doi:10.1267/ahc.10019
69. Ekstrom AB, Tulinius M, Sjostrom A, Aring E. Visual function in congenital and childhood myotonic dystrophy type 1. Ophthalmology (2010) 117(5):976–82. doi:10.1016/j.ophtha.2010.01.055
70. Brunner HG, Nillesen W, van Oost BA, Jansen G, Wieringa B, Ropers HH, et al. Presymptomatic diagnosis of myotonic dystrophy. J Med Genet (1992) 29(11):780–4. doi:10.1136/jmg.29.11.780
71. Kidd A, Turnpenny P, Kelly K, Clark C, Church W, Hutchinson C, et al. Ascertainment of myotonic dystrophy through cataract by selective screening. J Med Genet (1995) 32(7):519–23. doi:10.1136/jmg.32.7.519
72. Rakocevic-Stojanovic V, Peric S, Pesovic J, Sencanic I, Bozic M, Svikovic S, et al. Genetic testing of individuals with pre-senile cataract identifies patients with myotonic dystrophy type 2. Eur J Neurol (2017) 24(11):e79–80. doi:10.1111/ene.13401
73. Sato S, Nakamura M, Cho DH, Tapscott SJ, Ozaki H, Kawakami K. Identification of transcriptional targets for Six5: implication for the pathogenesis of myotonic dystrophy type 1. Hum Mol Genet (2002) 11(9):1045–58. doi:10.1093/hmg/11.9.1045
74. Rhodes JD, Lott MC, Russell SL, Moulton V, Sanderson J, Wormstone IM, et al. Activation of the innate immune response and interferon signalling in myotonic dystrophy type 1 and type 2 cataracts. Hum Mol Genet (2012) 21(4):852–62. doi:10.1093/hmg/ddr515
75. Shao D, Zhu X, Sun W, Huo L, Chen W, Wang H, et al. Investigation of the molecular mechanisms underlying myotonic dystrophy types 1 and 2 cataracts using microRNAtarget gene networks. Mol Med Rep (2017) 16(4):3737–44. doi:10.3892/mmr.2017.7059
76. Passeri E, Bugiardini E, Sansone VA, Valaperta R, Costa E, Ambrosi B, et al. Vitamin D, parathyroid hormone and muscle impairment in myotonic dystrophies. J Neurol Sci (2013) 331(1–2):132–5. doi:10.1016/j.jns.2013.06.008
77. Terracciano C, Rastelli E, Morello M, Celi M, Bucci E, Antonini G, et al. Vitamin D deficiency in myotonic dystrophy type 1. J Neurol (2013) 260(9):2330–4. doi:10.1007/s00415-013-6984-1
78. Dahlqvist JR, Orngreen MC, Witting N, Vissing J. Endocrine function over time in patients with myotonic dystrophy type 1. Eur J Neurol (2015) 22(1):116–22. doi:10.1111/ene.12542
79. Matsumura T, Iwahashi H, Funahashi T, Takahashi MP, Saito T, Yasui K, et al. A cross-sectional study for glucose intolerance of myotonic dystrophy. J Neurol Sci (2009) 276(1–2):60–5. doi:10.1016/j.jns.2008.08.037
80. Thornton CA, Griggs RC, Moxley RT III. Myotonic dystrophy with no trinucleotide repeat expansion. Ann Neurol (1994) 35(3):269–72. doi:10.1002/ana.410350305
81. van Vliet J, Tieleman AA, van Engelen BGM, Bassez G, Servais L, Behin A, et al. Hearing impairment in patients with myotonic dystrophy type 2. Neurology (2018) 90(7):e615–22. doi:10.1212/WNL.0000000000004963
82. Pisani V, Tirabasso A, Mazzone S, Terracciano C, Botta A, Novelli G, et al. Early subclinical cochlear dysfunction in myotonic dystrophy type 1. Eur J Neurol (2011) 18(12):1412–6. doi:10.1111/j.1468-1331.2011.03470.x
83. Bassez G, Lazarus A, Desguerre I, Varin J, Laforet P, Becane HM, et al. Severe cardiac arrhythmias in young patients with myotonic dystrophy type 1. Neurology (2004) 63(10):1939–41. doi:10.1212/01.WNL.0000144343.91136.CF
84. Wahbi K, Algalarrondo V, Becane HM, Fressart V, Beldjord C, Azibi K, et al. Brugada syndrome and abnormal splicing of SCN5A in myotonic dystrophy type 1. Arch Cardiovasc Dis (2013) 106(12):635–43. doi:10.1016/j.acvd.2013.08.003
85. Sansone VA, Brigonzi E, Schoser B, Villani S, Gaeta M, De Ambroggi G, et al. The frequency and severity of cardiac involvement in myotonic dystrophy type 2 (DM2): long-term outcomes. Int J Cardiol (2013) 168(2):1147–53. doi:10.1016/j.ijcard.2012.11.076
86. Chong-Nguyen C, Wahbi K, Algalarrondo V, Becane HM, Radvanyi-Hoffman H, Arnaud P, et al. Association between mutation size and cardiac involvement in myotonic dystrophy type 1: an analysis of the DM1-Heart Registry. Circ Cardiovasc Genet (2017) 10(3):e001526. doi:10.1161/CIRCGENETICS.116.001526
87. Yoshida K, Aburakawa Y, Suzuki Y, Kuroda K, Kimura T. The frequency and risk factors for ischemic stroke in myotonic dystrophy type 1 patients. J Stroke Cerebrovasc Dis (2018) 27(4):914–8. doi:10.1016/j.jstrokecerebrovasdis.2017.10.030
88. Ronnemaa T, Alaranta H, Viikari J, Tilvis R, Falck B. Increased activity of serum gamma-glutamyltransferase in myotonic dystrophy. Acta Med Scand (1987) 222(3):267–73. doi:10.1111/j.0954-6820.1987.tb10669.x
89. Kalafateli M, Triantos C, Tsamandas A, Kounadis G, Labropoulou-Karatza C. Abnormal liver function tests in a patient with myotonic dystrophy type 1. Ann Hepatol (2012) 11(1):130–3.
90. Brunner HG, Hamel BC, Rieu P, Howeler CJ, Peters FT. Intestinal pseudo-obstruction in myotonic dystrophy. J Med Genet (1992) 29(11):791–3. doi:10.1136/jmg.29.11.791
91. Bellini M, Biagi S, Stasi C, Costa F, Mumolo MG, Ricchiuti A, et al. Gastrointestinal manifestations in myotonic muscular dystrophy. World J Gastroenterol (2006) 12(12):1821–8. doi:10.3748/wjg.v12.i12.1821
92. Ercolin B, Sassi FC, Mangilli LD, Mendonca LI, Limongi SC, de Andrade CR. Oral motor movements and swallowing in patients with myotonic dystrophy type 1. Dysphagia (2013) 28(3):446–54. doi:10.1007/s00455-013-9458-9
93. Goldberg HI, Sheft DJ. Esophageal and colon changes in myotonia dystrophica. Gastroenterology (1972) 63(1):134–9.
94. Gadalla SM, Lund M, Pfeiffer RM, Gortz S, Mueller CM, Moxley RT III, et al. Cancer risk among patients with myotonic muscular dystrophy. JAMA (2011) 306(22):2480–6. doi:10.1001/jama.2011.1796
95. Win AK, Perattur PG, Pulido JS, Pulido CM, Lindor NM. Increased cancer risks in myotonic dystrophy. Mayo Clin Proc (2012) 87(2):130–5. doi:10.1016/j.mayocp.2011.09.005
96. Alsaggaf R, Wang Y, Marini-Bettolo C, Wood L, Nikolenko N, Lochmuller H, et al. Benign and malignant tumors in the UK myotonic dystrophy patient registry. Muscle Nerve (2018) 57(2):316–20. doi:10.1002/mus.25736
97. Peric S, Stojanovic VR, Nikolic A, Kacar A, Basta I, Pavlovic S, et al. Peripheral neuropathy in patients with myotonic dystrophy type 1. Neurol Res (2013) 35(4):331–5. doi:10.1179/1743132812Y.0000000144
98. Hermans MC, Faber CG, Vanhoutte EK, Bakkers M, De Baets MH, de Die-Smulders CE, et al. Peripheral neuropathy in myotonic dystrophy type 1. J Peripher Nerv Syst (2011) 16(1):24–9. doi:10.1111/j.1529-8027.2011.00313.x
99. Bae JS, Kim OK, Kim SJ, Kim BJ. Abnormalities of nerve conduction studies in myotonic dystrophy type 1: primary involvement of nerves or incidental coexistence? J Clin Neurosci (2008) 15(10):1120–4. doi:10.1016/j.jocn.2007.11.002
100. Harper PS, Dyken PR. Early-onset dystrophia myotonica. Evidence supporting a maternal environmental factor. Lancet (1972) 2(7767):53–5. doi:10.1016/S0140-6736(72)91548-6
101. Zeesman S, Carson N, Whelan DT. Paternal transmission of the congenital form of myotonic dystrophy type 1: a new case and review of the literature. Am J Med Genet (2002) 107(3):222–6. doi:10.1002/ajmg.10141
102. Angeard N, Gargiulo M, Jacquette A, Radvanyi H, Eymard B, Heron D. Cognitive profile in childhood myotonic dystrophy type 1: is there a global impairment? Neuromuscul Disord (2007) 17(6):451–8. doi:10.1016/j.nmd.2007.02.012
103. Wallgren-Pettersson C, Bushby K, Mellies U, Simonds A. 117th ENMC workshop: ventilatory support in congenital neuromuscular disorders – congenital myopathies, congenital muscular dystrophies, congenital myotonic dystrophy and SMA (II) 4-6 April 2003, Naarden, The Netherlands. Neuromuscul Disord (2004) 14(1):56–69. doi:10.1016/j.nmd.2003.09.003
104. Mathieu J, Allard P, Potvin L, Prevost C, Begin P. A 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology (1999) 52(8):1658–62. doi:10.1212/WNL.52.8.1658
105. Ekstrom AB, Hakenas-Plate L, Samuelsson L, Tulinius M, Wentz E. Autism spectrum conditions in myotonic dystrophy type 1: a study on 57 individuals with congenital and childhood forms. Am J Med Genet B Neuropsychiatr Genet (2008) 147B(6):918–26. doi:10.1002/ajmg.b.30698
106. Douniol M, Jacquette A, Guile JM, Tanguy ML, Angeard N, Heron D, et al. Psychiatric and cognitive phenotype in children and adolescents with myotonic dystrophy. Eur Child Adolesc Psychiatry (2009) 18(12):705–15. doi:10.1007/s00787-009-0037-4
107. Douniol M, Jacquette A, Cohen D, Bodeau N, Rachidi L, Angeard N, et al. Psychiatric and cognitive phenotype of childhood myotonic dystrophy type 1. Dev Med Child Neurol (2012) 54(10):905–11. doi:10.1111/j.1469-8749.2012.04379.x
Keywords: myotonic dystrophies, DM1, DM2, phenotypes, myotonia, sleep disorders, repeat expansion diseases
Citation: Wenninger S, Montagnese F and Schoser B (2018) Core Clinical Phenotypes in Myotonic Dystrophies. Front. Neurol. 9:303. doi: 10.3389/fneur.2018.00303
Received: 09 March 2018; Accepted: 18 April 2018;
Published: 02 May 2018
Edited by:
Edoardo Malfatti, INSERM UMRS974 Institut de Myologie, FranceReviewed by:
Massimiliano Filosto, Asst degli Spedali Civili di Brescia, ItalyCopyright: © 2018 Wenninger, Montagnese and Schoser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephan Wenninger, c3RlcGhhbi53ZW5uaW5nZXJAbWVkLnVuaS1tdWVuY2hlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.