 Airton Leonardo de Oliveira Manoel
Airton Leonardo de Oliveira Manoel R. Loch Macdonald
R. Loch Macdonald- 1Adult Critical Care Unit, Hospital Paulistano – United Health Group, São Paulo, Brazil
- 2Keenan Research Center for Biomedical Science, Department of Surgery, Li Ka Shing Knowledge Institute, University of Toronto, Toronto, ON, Canada
- 3Division of Neurosurgery, St. Michael’s Hospital, Labatt Family Centre of Excellence in Brain Injury and Trauma Research, Keenan Research Centre for Biomedical Science, Department of Surgery, Li Ka Shing Knowledge Institute, University of Toronto, Toronto, ON, Canada
Aneurysmal subarachnoid hemorrhage (SAH) is a sub-type of hemorrhagic stroke associated with the highest rates of mortality and long-term neurological disabilities. Despite the improvement in the management of SAH patients and the reduction in case fatality in the last decades, disability and mortality remain high in this population. Brain injury can occur immediately and in the first days after SAH. This early brain injury can be due to physical effects on the brain such as increased intracranial pressure, herniations, intracerebral, intraventricular hemorrhage, and hydrocephalus. After the first 3 days, angiographic cerebral vasospasm (ACV) is a common neurological complication that in severe cases can lead to delayed cerebral ischemia and cerebral infarction. Consequently, the prevention and treatment of ACV continue to be a major goal. However, most treatments for ACV are vasodilators since ACV is due to arterial vasoconstriction. Other targets also have included those directed at the underlying biochemical mechanisms of brain injury such as inflammation and either independently or as a consequence, cerebral microthrombosis, cortical spreading ischemia, blood–brain barrier breakdown, and cerebral ischemia. Unfortunately, no pharmacologic treatment directed at these processes has yet shown efficacy in SAH. Enteral nimodipine and the endovascular treatment of the culprit aneurysm, remain the only treatment options supported by evidence from randomized clinical trials to improve patients’ outcome. Currently, there is no intervention directly developed and approved to target neuroinflammation after SAH. The goal of this review is to provide an overview on anti-inflammatory drugs tested after aneurysmal SAH.
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) is a complex cerebrovascular disease with profound systemic complications (1–3). Despite the advances achieved in the management of those patients, high disability and mortality rates continue to devastate this patients’ population (4).
Aneurysm rebleeding is the most threatening early neurological complication and usually occurs in the first 24 h after the initial hemorrhage (5, 6). However, delayed cerebral ischemia (DCI) remains the major cause of morbidity and mortality among patients who survive after repair of the ruptured aneurysm (7, 8).
For decades, angiographic cerebral vasospasm (ACV), a common neuroradiological finding after SAH, has been proposed as the primary cause of DCI and DCI-related cerebral infarction (9). In turn, severe angiographic vasospasm can cause DCI and DCI-related cerebral infarction, which made the prevention and treatment of ACV a major target in the management of SAH. Crowley et al. analyzed 381 patients who were part of a randomized clinical trial and all of whom underwent serial CT scanning and angiography (10). Angiographic vasospasm was graded as none/mild (209, 55%), moderate (118, 31%), or severe (54, 14%). Delayed cerebral infarctions occurred in 3% of those with no/mild, 10% with moderate, and 46% with severe vasospasm. The data also show that a minority of patients can develop DCI without ACV (10). Angiographic vasospasm occurs in approximately 70% of patients during the first 2 weeks after SAH, but the incidence of DCI is only around 30% (7). Some patients with severe ACV do not develop ischemia or infarction for the same reasons that a patient with a carotid artery occlusion may not, which includes (a) the recruitment of collateral blood flow through the anterior and posterior communicating arteries (i.e., the primary collateral pathway), or through the ophthalmic artery and leptomeningeal vessels (i.e., the secondary collateral pathway) (11, 12) and (b) the cerebral vasoreactivity status (i.e., intact versus impaired) (13). In addition, patients may develop cerebral infarction in vascular territories that are not affected by ACV (14, 15). Moreover, enteral nimodipine is the only pharmacological treatment shown to improve functional outcome, despite the fact that it does not lead to marked cerebral vasodilation (16, 17).
Also, a meta-analysis that included 14 randomized controlled trials in SAH, involving 4,235 patients, showed that the tested drugs significantly reduced angiographic vasospasm, but clinical outcomes remained unaffected (18).
Because of the failure of several pharmacological trials and the dissociation between vasospasm-related morbidity and functional outcomes, other possible mechanisms, such as early brain injury, cortical spreading ischemia, microthrombosis, cerebral autoregulation impairment, and capillary transit time heterogeneity, are thought to play a role in the pathophysiology of DCI and DCI-related cerebral infarction (1, 3, 19, 20). These additional mechanisms tend to be more difficult to assess clinically and the dissociation mentioned above could also be due to drug toxicity and off-target effects, effects of rescue therapy, inadequate trial sample size and other trial design problems, and testing of only one dose and dose regimen.
Another common biochemical mechanism after brain injury is neuroinflammation, which may also contribute to many of the causes of brain damage after SAH. Subarachnoid blood and subsequent hemoglobin degradation can in turn trigger the inflammatory cascade, which also develops in the brain probably secondary to ischemia, blood–brain barrier breakdown, and such. There is an increasing interest in the understanding of the role of neuroinflammation in the pathophysiology of DCI and other severe complications after SAH.
The main goal of this review article is to summarize the concept of neuroinflammation following SAH, and its potential contribution to ACV, DCI, and systemic complications. We also address the possible benefits of targeting the inflammatory cascade after aneurysmal SAH. Finally, we review studies of drugs directed at neuroinflammation after SAH (Table 1).
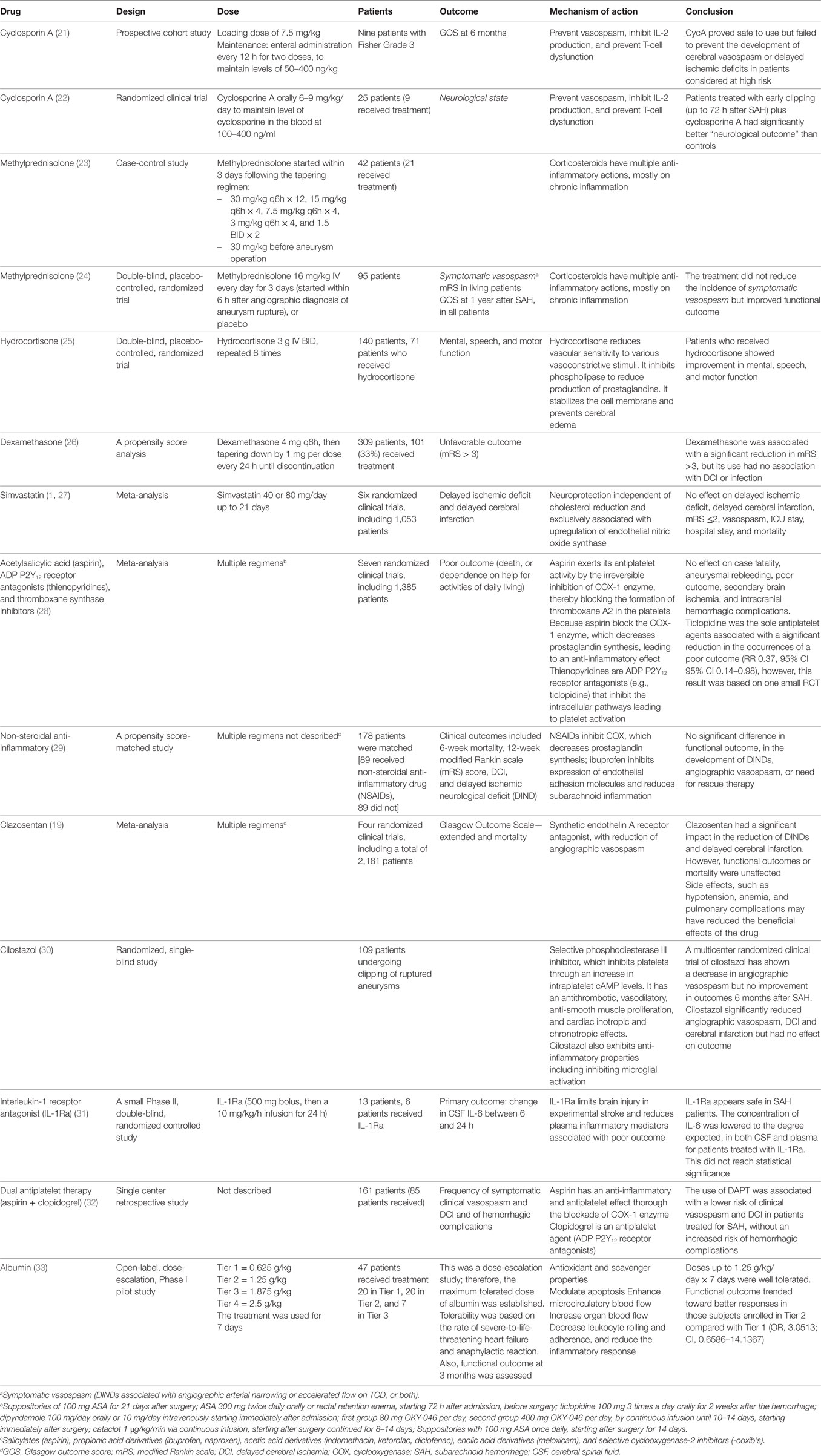
Table 1. Summary of medication with anti-inflammatory activity tested in SAH.
Search Strategy
A PubMed search was performed from inception to November 2017 for articles published in English by using the terms “Subarachnoid Hemorrhage” [Mesh] AND (“Neuroinflammation” [Title/Abstract] OR “inflammation” [Title/Abstract]). A total of 272 articles were found, including 155 articles on human subjects. The authors’ own databases additionally were used as a source for this review article. This review was limited to studies in humans, because the literature on experimental animal models is vast, and it is beyond the scope of this article.
DCI and Angiographic Vasospasm
The majority of patients (approximately 70%) develop some radiological degree of vasospasm between 3 and 14 days after a single SAH (9). However, only 30% of those will progress to develop DCI and even fewer to delayed cerebral infarction (15, 34). While ACV is an important contributor to DCI, there is evidence that other processes are involved (1, 3, 35–38).
Two major neurological consequences of aneurysmal SAH are currently described, early brain injury and ACV/DCI. Early brain injury refers to the acute effects of aneurysm rupture. Arterialized blood flows into the subarachnoid space, which suddenly increases the intracranial pressure. Consequently, the cerebral perfusion pressure is acutely reduced leading to a reduction in cerebral blood flow that produces global cerebral ischemia. Clinically, the phenomenon is characterized by transient loss of consciousness that can progress to intracranial circulatory arrest in severe cases (39, 40). There also are probably effects from the subarachnoid blood as well as brain injury from other physical and biochemical processes. Ischemia with its associated cell death causes influx of inflammatory cells, activation of microglia and neuroinflammation. Patients who survive the initial hemorrhage are at risk of DCI that is the main determinant of unfavorable outcome after SAH (41). DCI is a clinical syndrome characterized by “the occurrence of focal neurological impairment (such as hemiparesis, aphasia, apraxia, hemianopia, or neglect) and/or a decrease of at least two points on the Glasgow coma scale (either on the total score or on one of its individual components, such as eye, motor on either side, or verbal) (7). This should last for at least 1 h, is not apparent immediately after aneurysm occlusion, and cannot be attributed to other causes by means of clinical assessment, computed tomographic, or magnetic resonance imaging of the brain and appropriate laboratory studies”. The pathophysiology of DCI is multifactorial and remains to be completely elucidated, however, it is hypothesized that ACV is one if not the primary contributor, along with cortical spreading ischemia, impaired cerebral autoregulation and microcirculation constriction, microthrombosis, capillary transit time heterogeneity, and neuroinflammation (1, 3, 35–38, 42). The risk of DCI is increased by the amount of subarachnoid blood (i.e., the volume, density, and persistence of blood in the subarachnoid space) (43–45); by the initial level of consciousness (i.e., early brain injury) (46, 47); and by factors that alter the relationship between the brain oxygen/glucose supply and demand (1).
Inflammation as an Additional Contributor to DCI
There is recent evidence suggesting that neuroinflammation may play an important role in the damage of cerebral cells after SAH (42, 48). The products of erythrocyte degradation in the subarachnoid space lead to the accumulation of hemoglobin and its products (i.e., methemoglobin, heme, and hemin), which activate toll-like receptor 4, initiating the inflammatory cascade. Microglia, the resident inflammatory cells in the central nervous system, are activated, which triggers the upregulation of a large number of endothelial adhesion molecules, allowing the inflammatory cells to reach the subarachnoid space (49). Macrophages and neutrophils, once in the subarachnoid space, start the process of phagocytosis of the product of degraded red cells, with the attempt of removing the extravascular hemoglobin, the process of which may promote neuronal healing (50). The clearance of hemoglobin from subarachnoid space depends on and is accelerated by the ligation of hemoglobin to haptoglobin. The complex hemoglobin/haptoglobin is than phagocyted by immune cells (51).
Macrophages and neutrophils are indispensable for the clearance of subarachnoid blood, however, they may become imprisoned within the subarachnoid space after the eventual reestablishment of the blood–brain barrier, and also because of the changes occurring in the cerebral spinal fluid (CSF) flow. The main problem with the entrapment of inflammatory cell in the subarachnoid space is that they suffer degranulation, releasing several inflammatory and vasoactive factors, such as endothelins. Also, these inflammatory and vasoactive factors are not restricted to the subarachnoid space, but generalized throughout the central nervous system, inducing cerebritis, aseptic meningitis, and cerebral vasoconstriction (50).
Monitoring Inflammation After SAH
Several methods exist to monitor systemic and neurological inflammation in the patients with SAH. They include classic systemic signs of inflammation such as fever, leukocytosis, and systemic inflammatory response (SIRS), as well as the evaluation of pro- and anti-inflammatory cytokines in the peripheral blood, CSF, and cerebral extracellular fluid (52).
Increased levels of inflammatory cytokines are found in the CSF, cerebral extracellular fluid, and blood of patients with SAH (53). Two sets of authors reviewed published data reporting biomarkers for assessing outcome after SAH (54, 55). Neuron and astrocyte proteins, cell adhesion proteins, and extracellular matrix molecules, vascular, coagulation, and cardiac proteins have all been measured in various studies of CSF and blood after SAH. Inflammatory substances in blood that have been shown to be elevated after SAH included C-reactive protein (CRP), tumor necrosis factor α (TNF-α), and interleukin-1 receptor antagonist (IL-1Ra) and IL-6 (IL-6). Elevated levels of TNF-α, IL-1Ra, IL-6, and IL-8 were also described in the CSF (56–58). IL-1Ra concentrations were measured in blood and were significantly higher after SAH than in controls, in poor-grade SAH patients compared to good grades and in patients who had unfavorable as compared to favorable outcome (56). DCI further increased the concentration of IL-1Ra.
In one study, CSF TNF-α concentrations increased between 4 and 10 days after SAH in patients with unfavorable outcome whereas they did not if there was favorable outcome (56). The same rough correlation of increased CSF TNF-α and unfavorable outcome was reported by other investigators (59). Similarly, CSF levels of IL-6 and IL-8 were shown to be significantly elevated in SAH patients compared to controls, and this elevation was associated with the development of symptomatic vasospasm between day 5 and 7 (58).
Subarachnoid increases in IL-1, IL-6, and TNF-α may precede the hemodynamic abnormalities detected by transcranial Doppler and to be associated with the development of severe ACV diagnosed by transcranial Doppler ultrasound.
Elevated IL-6 levels also may be an early marker of DCI and unfavorable outcome. Muroi et al. showed in a cohort of 138 SAH patients that early elevation of IL-6 (days 3–7) was significantly associated with the occurrence of DCI and unfavorable outcome, after the adjustment for confounding factors such as infection and therapeutic hypothermia (60). In addition, elevated IL-6 levels detected in the cerebral extracellular fluid by cerebral microdialysis in patients of all grades predicted the occurrence of DCI in one study (61).
Complement system is another important component of inflammatory response after SAH. Complement activation and the formation of membrane attack complex contribute to the development of cerebral edema and the disruption in the blood–brain barrier. Interestingly, the process of complement activation and membrane attack complex formation is also implicated in the inflammation and degradation of the cerebral aneurysm wall, with consequent aneurysm rupture (62).
The complement activation in association with the formation of membrane attack complex may contribute to the development of angiographic vasospasm (63). In one study investigated the role of sequential measurement of serum complements (i.e., CH50, C3, C4) in the first 3 weeks of hemorrhage. Hunt and Hess grade was associated with C4 levels, and C4 levels were also markedly reduced in patients with severe angiographic vasospasm with neurological deficits (64).
Although, the measurement of cytokines and their receptors in the CSF, cerebral extracellular fluid, and blood seem to be feasible, their use remains experimental. Also, inflammation is the fundamental response of tissues to injury and is generally required for tissue healing. There are situations where inflammation is detrimental. Therefore, it is going to be very complex to determine which aspect of cerebral inflammation is beneficial or harmful (52). More adequate, well controlled and thus basically prospective multicenter studies would be needed before we could draw conclusions on the clinical utility of inflammatory biomarkers (53).
Inflammation, Brain Injury, and Systemic Complications
There is systemic immune response activation after SAH and it is commonly manifested by high levels of circulating cytokines, such as IL-1, IL-6, and TNF-α (65). These are key mediators of systemic inflammation. Clinically, this process is usually manifested by fever, leukocytosis, tachycardia, and tachypnea. These are components of SIRS, which has been defined by the presence of two or more of the following: temperature <36 or >38°C, heart rate >90 bpm, respiratory rate of >20 breaths/min, and white blood cell count of <4,000 or >12,000/mm3 (66, 67). In 2 reports that included 276 and 413 patients with SAH, respectively, SIRS was present on admission in over half and in 63–85% within 4 days (68–70). Its occurrence was associated with poorer neurological grade, larger amount of subarachnoid blood on computed tomography. In one of the two studies, it was an independent predictor of angiographic vasospasm, systemic complications, unfavorable outcome, and death.
Systemic inflammatory response is a relatively nonspecific clinical syndrome that reflects a complex interaction among inflammation, coagulation, sympathoadrenal activation, and endothelial cell activation and dysfunction (71). This complex process generates and perpetuates tissue hypoperfusion, ultimately culminating with microthrombosis and compromised microcirculation blood flow, which is manifested by the multi-organ dysfunction syndrome (71, 72). In addition, catecholamines released into the systemic circulation after SAH may induce cardiac and pulmonary dysfunction (i.e., myocardial stunning and neurogenic pulmonary edema) (73, 74). Sympathoadrenal activation, represented by high circulating catecholamine levels, has also been shown to be highly associated with coagulopathy, endotheliopathy, and functional outcome in patients with isolated traumatic brain injury (75, 76).
Other indirect surrogate markers of systemic inflammatory activity after SAH include hyperthermia, elevated white blood cell count, hyperglycemia, high erythrocyte sedimentation rate, high CRP, transthyretin, and negative nitrogen balance (60, 64, 66, 72–82). Those systemic inflammatory parameters are associated with angiographic vasospasm, DCI, and unfavorable outcome (66, 68).
Platelet Activation and Inflammation After SAH
Inflammation is intimately related to platelet adhesion, activation of coagulation cascade, and consequently the formation of microthrombi (82). Injury to a blood vessel leads to endothelial platelet adhesion, which triggers the coagulation cascade. Thrombin, von Willebrand factor, and collagen are capable of activating the platelets, which release a large number of procoagulant factors, responsible for the development of a hemostatic clot (82–84).
In SAH patients, microthrombosis was described in an autopsy study of seven patients who died within 3 days of SAH (85). The density of thrombi (microclot burden) was associated with clinical and radiological evidence of delayed ischemia. Also, microclot burden was significantly associated with histological evidence of ischemia.
Additional evidence for microthromboembolism includes a prospective study of SAH patients monitored with transcranial Doppler (86). Microembolic signals were detected in 16 of 23 patients (70%), and 44 of 138 arteries (32%) monitored. Microembolic signals were more common in patients with compared with patients without clinical vasospasm (83 versus 54%). These data at least show an association between inflammation, activation of coagulation cascade and formation of microthrombi after SAH and may be an additional component of DCI (83).
Inflammation and platelet activation have been associated with both early brain injury and DCI after SAH (82, 84). Frontera et al. (82), in a prospective study enrolled 106 consecutive SAH patients, comparing them with 26 control subjects. Compared to controls, SAH patients demonstrated higher levels o inflammatory biomarkers (i.e. higher levels of C-reactive protein), and higher levels of platelets activation by thromboelastography. Platelet activation and inflammation were related to the severity of early brain injury, and it was also associated with the development of DCI and unfavorable functional outcome (82).
Interestingly, nimodipine has been shown to increase endogenous fibrinolysis, which may partially explain the benefit of nimodipine use by the reduction in microthrombosis after SAH (87). However, the exact mechanism by which nimodipine exerts its beneficial effects remains to be determined, but its probably multifactorial, including fibrinolysis, inhibition of cortical spreading ischemia, and the blockage of calcium influx to neurons after ischemia (1, 20).
Antiplatelet Drugs After SAH
Cyclooxygenase (COX) enzymes are fundamental for the balance of vascular homeostasis (88). Thromboxane A2 (TxA2) is released by platelets as a result of COX-1 activity, and prostacyclin is synthetized mainly in vascular endothelium by COX-2 (Figure 1). TxA2 induces platelet aggregation, vasoconstriction, and smooth muscle proliferation, while prostacyclin antagonizes TxA2 effects in the macrovascular endothelium through smooth muscle relaxation and vasodilator effects. Prostacyclin also inhibits platelet aggregation through prostacyclin receptors.
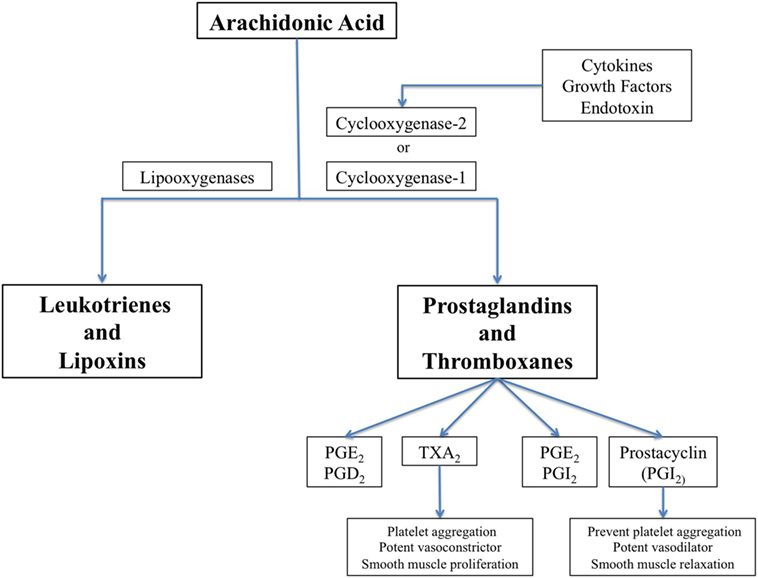
Figure 1. The biochemical pathway for synthesis of prostaglandins, thromboxanes, leucotrienes, and lipoxins, showing cyclooxygenase (COX)-1 which is constitutively expressed in many cells and COX-2 that is induced by inflammatory stimuli.
Acetylsalicylic acid [aspirin, a non-steroidal anti-inflammatory drug (NSAID)] inhibits cyclic prostanoid synthesis (TxA2, prostacyclin and other prostaglandins, Figure 2). The mechanism of its antithrombotic effect is by irreversible acetylation of COX-1, an enzyme constitutively expressed platelets and most other cells. This inhibits platelet aggregation. Its anti-inflammatory effects are due to inhibition of COX-2, which is induced by inflammation and generates inflammatory prostaglandins.
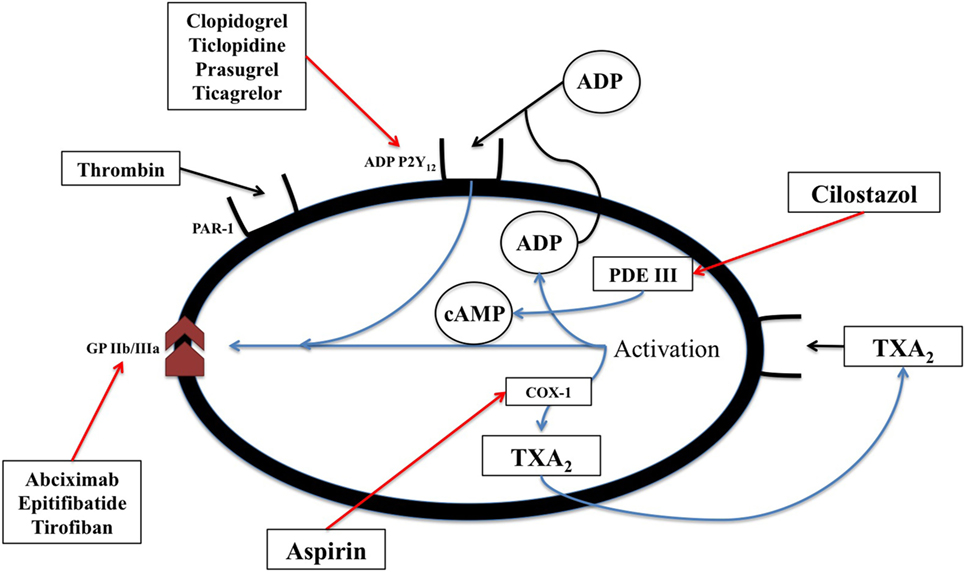
Figure 2. The biochemical pathways and receptors on platelets along with the site of action of non-steroidal anti-inflammatory drugs as well as some physiologic mediators of platelet degranulation and aggregation. This figure was modified from Angiolillo (89).
Dorhout Mees et al. conducted a meta-analysis of seven randomized clinical trials (1,385 patients) that evaluated the effect of antiplatelet drugs on outcome, case fatality, secondary ischemia, hemorrhagic intracranial complications, and aneurysm rebleeding in patients with SAH (28). The studies tested different drugs and regimens, including suppositories or oral aspirin, oral ticlopidine, oral or intravenous dipyridamole, continuous infusion of OKY-046, and continuous infusion of cataclot (90–94). There was no effect of the antiplatelet drugs on any of the outcome measures. Ticlopidine was associated with a significant reduction in poor outcome but this was based on one randomized trial of 135 patients (95).
Steroids for SAH
Corticosteroids are anti-inflammatory. They inhibit the formation of adhesion molecules, arachidonic acid metabolites, cytokines, and chemokines. The mechanism of action that accounts for their anti-inflammatory effects is direct binding of the glucocorticoid to the glucocorticoid receptor, forming a glucocorticoid/glucocorticoid receptor complex. This is transported in to the nucleus where it binds to glucocorticoid responsive elements in the promoter region of genes, or interacts with other transcription factors, in particular activating protein-1 or nuclear factor-κB (96).
Several different types doses and dose regimens of corticosteroids have been studied in patients with SAH. Overall, the studies show mixed results and sample sizes are too small to draw conclusions among other shortcomings of the studies (23–25). The largest study was a retrospective cohort that included 309 patients, 101 (33%) who received dexamethasone. Dexamethasone significantly reduced in the odds of unfavorable outcome although it had no effect on DCI or the rate of infections (26). Glucocorticoids are not recommended for routine use in patients with SAH.
Important to mention is that a large randomized clinical trial, including more than 10,000 patients with moderate-to-severe traumatic brain injury, compared the effect of 48 h infusion of methylprednisolone with placebo. The risk of death from all causes was increased in the intervention group (97, 98). The exact cause of worse outcome with the use of steroids after traumatic brain injury is not well established.
Immune-Suppressive Treatment in SAH
There are at least two studies that tested the use of immune-suppressive treatment in SAH. The first study was a randomized clinical trial, which included 25 patients (22). Nine received the intervention (cyclosporine A orally 6–9 mg/kg/day to maintain level of cyclosporine in the blood at 100–400 ng/ml). Patients treated with early clipping (up to 72 h after SAH) plus cyclosporine A had significantly better neurological outcome. The second study treated nine SAH patients with Fisher Grade 3 SAH with cyclosporine A (21). Despite the small number of patients, cyclosporine A was considered to be safe but to not prevent ACV or DCI.
Many SAH patients admitted to the critical care unit are exposed to invasive procedures and catheter placement, such as mechanical ventilation, external ventricular drain, invasive intracranial monitors, and vascular catheters. These invasive tools are fundamental for the management of poor-grade SAH patients (1), however, they may increase the risk of hospital-acquired infections (99). Therefore, the use of immunosuppressive treatment, including the use of glucocorticoids, might increase the risk of infection in this patient population, which increases the hospital morbidity and mortality.
NSAIDs for SAH
Non-steroidal anti-inflammatory drugs inhibit COX (88). Traditionally they inhibited COX-1 and COX-2 although there are some specific COX-2 inhibitors. Thus, they inhibit inflammation mediated by COX-2. While aspirin is the classic NSAID, the effects differ between the newer NSAIDs and from aspirin probably due to the relative potencies against COX-1 and COX-2 and their effects on other targets. For example, ibuprofen reduces the expression of adhesion molecules by the endothelium, which in turn decreases the inflammation in the subarachnoid space. In a propensity score-matched study, Nassiri et al. studied 178 patients with SAH who were matched by propensity scoring into 89 who received NSAIDS and 89 who did not (29). Use of NSAIDs was associated with lower in-hospital mortality and shorter intensive care and hospital stay. There was, however, no significant difference in functional outcome, in the development of delayed ischemic neurological deficits, angiographic vasospasm or need for rescue therapy. Muroi and colleagues found that NSAID use was associated with lower systemic IL-6 and CRP concentrations and with better outcome in 138 patients with SAH (100). There is at least one randomized clinical trial of NSAIDs in 81 patients with SAH (101). Meloxicam had no significant effect on ACV or clinical outcome compared to placebo. Currently, the treatment of SAH patients with NSAIDs cannot be recommended.
Other Potentially Anti-Inflammatory Drugs
Several other medications with anti-inflammatory activity have been tested in patients with SAH (Table 1). The statins are the best studied and have been completely ineffective at least in the doses and dose regimens studied (102). Albumin has multiple systemic and cerebral effects, including antioxidant and scavenger properties; the capacity to modulate cellular apoptosis; enhance of microcirculatory and organ blood flow; and an anti-inflammatory effect through the decrease in leukocyte rolling and adherence. Albumin has been studied in a dose-escalation, Phase I, pilot study (33). Doses up to 1.25 g/kg/day × 7 days were well tolerated, with a trend toward better 3-month functional outcome. A Phase III placebo control trial will be carried out in the near future.
Conclusion
There is an increasing interest in the understanding of the role of neuroinflammation in the pathophysiology of early brain injury and DCI after SAH. Inflammatory biomarkers are associated with the occurrence of ACV, DCI, and unfavorable outcome. However, the use of anti-inflammatory agents was studied in only a small numbers of subjects. Acetylsalicylic acid, other NSAIDs, thromboxane synthase inhibitors, steroids, nitric oxide donors, and immunosuppressant therapies have not shown beneficial clinical effects. On the other hand, none have been studied in enough detail or in adequate, well-controlled clinical trials to reach a definitive conclusion about safety and efficacy. Currently, there is no intervention directly developed and approved to target neuroinflammation after SAH, therefore anti-inflammatory treatments are not suggested in SAH, at least in the doses and dose regimens studied.
Author Contributions
AM wrote the first version of the manuscript. RM made crucial extensive revisions.
Conflict of Interest Statement
RM receives grant support from the Brain Aneurysm Foundation, Canadian Institutes for Health Research, Genome Canada, and the Ontario Genomics Institute; and he is Chief Scientific Officer of Edge Therapeutics, Inc. The other author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
RM receives grant support from the Brain Aneurysm Foundation, Canadian Institutes for Health Research, Genome Canada, and the Ontario Genomics.
References
1. de Oliveira Manoel AL, Goffi A, Marotta TR, Schweizer TA, Abrahamson S, Macdonald RL. The critical care management of poor-grade subarachnoid haemorrhage. Crit Care (2016) 20:21. doi:10.1186/s13054-016-1193-9
2. Macdonald RL, Schweizer TA. Spontaneous subarachnoid haemorrhage. Lancet (2017) 389(10069):655–66. doi:10.1016/S0140-6736(16)30668-7
3. Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol (2014) 10(1):44–58. doi:10.1038/nrneurol.2013.246
4. de Oliveira Manoel AL, Mansur A, Silva GS, Germans MR, Jaja BNR, Kouzmina E, et al. Functional outcome after poor-grade subarachnoid hemorrhage: a single-center study and systematic literature review. Neurocrit Care (2016) 25:338–50. doi:10.1007/s12028-016-0305-3
5. Germans MR, Coert BA, Vandertop WP, Verbaan D. Time intervals from subarachnoid hemorrhage to rebleed. J Neurol (2014) 261:1425–31. doi:10.1007/s00415-014-7365-0
6. van Donkelaar CE, Bakker NA, Veeger NJGM, Uyttenboogaart M, Metzemaekers JDM, Luijckx G-J, et al. Predictive factors for rebleeding after aneurysmal subarachnoid hemorrhage: rebleeding aneurysmal subarachnoid hemorrhage study. Stroke (2015) 46:2100–6. doi:10.1161/STROKEAHA.115.010037
7. Vergouwen MDI, Vermeulen M, van Gijn J, Rinkel GJE, Wijdicks EF, Muizelaar JP, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke (2010) 41:2391–5. doi:10.1161/STROKEAHA.110.589275
8. Jaja BNR, Saposnik G, Lingsma HF, Macdonald E, Thorpe KE, Mamdani M, et al. Development and validation of outcome prediction models for aneurysmal subarachnoid haemorrhage: the SAHIT multinational cohort study. BMJ (2018) 360:j5745–5717. doi:10.1136/bmj.j5745
9. Weir B, Grace M, Hansen J, Rothberg C. Time course of vasospasm in man. J Neurosurg (1978) 48:173–8. doi:10.3171/jns.1978.48.2.0173
10. Crowley RW, Medel R, Dumont AS, Ilodigwe D, Kassell NF, Mayer SA, et al. Angiographic vasospasm is strongly correlated with cerebral infarction after subarachnoid hemorrhage. Stroke (2011) 42:919–23. doi:10.1161/STROKEAHA.110.597005
11. Vernieri F, Pasqualetti P, Matteis M, Passarelli F, Troisi E, Rossini PM, et al. Effect of collateral blood flow and cerebral vasomotor reactivity on the outcome of carotid artery occlusion. Stroke (2001) 32:1552–8. doi:10.1161/01.STR.32.7.1552
12. Liebeskind DS. Collateral circulation. Stroke (2003) 34:2279–84. doi:10.1161/01.STR.0000086465.41263.06
13. Silvestrini M, Vernieri F, Pasqualetti P, Matteis M, Passarelli F, Troisi E, et al. Impaired cerebral vasoreactivity and risk of stroke in patients with asymptomatic carotid artery stenosis. JAMA (2000) 283:2122–7. doi:10.1001/jama.283.16.2122
14. Rabinstein AA, Weigand S, Atkinson JLD, Wijdicks EFM. Patterns of cerebral infarction in aneurysmal subarachnoid hemorrhage. Stroke (2005) 36:992–7. doi:10.1161/01.STR.0000163090.59350.5a
15. Vergouwen MDI, Ilodigwe D, Macdonald RL. Cerebral infarction after subarachnoid hemorrhage contributes to poor outcome by vasospasm-dependent and -independent effects. Stroke (2011) 42:924–9. doi:10.1161/STROKEAHA.110.597914
16. Petruk KC, West M, Mohr G, Weir BK, Benoit BG, Gentili F, et al. Nimodipine treatment in poor-grade aneurysm patients. Results of a multicenter double-blind placebo-controlled trial. J Neurosurg (1988) 68:505–17. doi:10.3171/jns.1988.68.4.0505
17. Dorhout Mees SM, Rinkel GJE, Feigin VL, Algra A, van den Bergh WM, Vermeulen M, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev (2007):CD000277. doi:10.1002/14651858.CD000277.pub3
18. Etminan N, Vergouwen MD, Ilodigwe D, Macdonald RL. Effect of pharmaceutical treatment on vasospasm, delayed cerebral ischemia, and clinical outcome in patients with aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. J Cereb Blood Flow Metab (2011) 31(6):1443–51. doi:10.1038/jcbfm.2011.7
19. Shen J, Pan J-W, Fan Z-X, Xiong X-X, Zhan R-Y. Dissociation of vasospasm-related morbidity and outcomes in patients with aneurysmal subarachnoid hemorrhage treated with clazosentan: a meta-analysis of randomized controlled trials. J Neurosurg (2013) 119:180–9. doi:10.3171/2013.3.JNS121436
20. Rowland MJ, Hadjipavlou G, Kelly M, Westbrook J, Pattinson KTS. Delayed cerebral ischaemia after subarachnoid haemorrhage: looking beyond vasospasm. Br J Anaesth (2012) 109:315–29. doi:10.1093/bja/aes264
21. Manno EM, Gress DR, Ogilvy CS, Stone CM, Zervas NT. The safety and efficacy of cyclosporine A in the prevention of vasospasm in patients with Fisher grade 3 subarachnoid hemorrhages: a pilot study. Neurosurgery (1997) 40:289–93. doi:10.1097/00006123-199702000-00010
22. Ryba M, Pastuszko M, Iwanska K, Bidzinski J, Dziewiecki C. Cyclosporine A prevents neurological deterioration of patients with SAH – a preliminary report. Acta Neurochir (1991) 112:25–7. doi:10.1007/BF01402450
23. Chyatte D, Fode NC, Nichols DA, Sundt TM. Preliminary report: effects of high dose methylprednisolone on delayed cerebral ischemia in patients at high risk for vasospasm after aneurysmal subarachnoid hemorrhage. Neurosurgery (1987) 21:157–60. doi:10.1097/00006123-198708000-00004
24. Gomis P, Graftieaux JP, Sercombe R, Hettler D, Scherpereel B, Rousseaux P. Randomized, double-blind, placebo-controlled, pilot trial of high-dose methylprednisolone in aneurysmal subarachnoid hemorrhage. J Neurosurg (2010) 112:681–8. doi:10.3171/2009.4.JNS081377
25. Hashi K, Takakura K, Sano K, Ohta T, Saito I, Okada K. [Intravenous hydrocortisone in large doses in the treatment of delayed ischemic neurological deficits following subarachnoid hemorrhage – results of a multi-center controlled double-blind clinical study]. No To Shinkei (1988) 40:373–82.
26. Mohney N, Williamson CA, Rothman E, Ball R, Sheehan KM, Pandey AS, et al. A propensity score analysis of the impact of dexamethasone use on delayed cerebral ischemia and poor functional outcomes after subarachnoid hemorrhage. World Neurosurg (2018) 109:e655–61. doi:10.1016/j.wneu.2017.10.051
27. Liu H, Xu X. Effect of simvastatin in patients with aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. Am J Emerg Med (2017) 35(12):1940–5. doi:10.1016/j.ajem.2017.09.001
28. Dorhout Mees SM, van den Bergh WM, Algra A, Rinkel GJ. Antiplatelet therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev (2007) (4):CD006184.
29. Nassiri F, Ibrahim GM, Badhiwala JH, Witiw CD, Mansouri A, Alotaibi NM, et al. Propensity score-matched study of the use of non-steroidal anti-inflammatory agents following aneurysmal subarachnoid hemorrhage. Neurocrit Care (2016) 25:351–8. doi:10.1007/s12028-016-0266-6
30. Senbokuya N, Kinouchi H, Kanemaru K, Ohashi Y, Fukamachi A, Yagi S, et al. Effects of cilostazol on cerebral vasospasm after aneurysmal subarachnoid hemorrhage: a multicenter prospective, randomized, open-label blinded end point trial. J Neurosurg (2013) 118:121–30. doi:10.3171/2012.9.JNS12492
31. Singh N, Hopkins SJ, Hulme S, Galea JP, Hoadley M, Vail A, et al. The effect of intravenous interleukin-1 receptor antagonist on inflammatory mediators in cerebrospinal fluid after subarachnoid haemorrhage: a phase II randomised controlled trial. J Neuroinflammation (2014) 11:1. doi:10.1186/1742-2094-11-1
32. Nagahama Y, Allan L, Nakagawa D, Zanaty M, Starke RM, Chalouhi N, et al. Dual antiplatelet therapy in aneurysmal subarachnoid hemorrhage: association with reduced risk of clinical vasospasm and delayed cerebral ischemia. J Neurosurg (2017) 1–9. doi:10.3171/2017.5.JNS17831
33. Suarez JI, Martin RH, Calvillo E, Dillon C, Bershad EM, Macdonald RL, et al. The albumin in subarachnoid hemorrhage (ALISAH) multicenter pilot clinical trial safety and neurologic outcomes. Stroke (2012) 43(3):683–90. doi:10.1161/STROKEAHA.111.633958
34. Shimoda M, Takeuchi M, Tominaga J, Oda S, Kumasaka A, Tsugane R. Asymptomatic versus symptomatic infarcts from vasospasm in patients with subarachnoid hemorrhage: serial magnetic resonance imaging. Neurosurgery (2001) 49:1341–8; discussion 1348–50. doi:10.1097/00006123-200112000-00010
35. Winkler MK, Dengler N, Hecht N, Hartings JA, Kang EJ, Major S, et al. Oxygen availability and spreading depolarizations provide complementary prognostic information in neuromonitoring of aneurysmal subarachnoid hemorrhage patients. J Cereb Blood Flow Metab (2017) 37(5):1841–56. doi:10.1177/0271678X16641424
36. Stein SC, Browne KD, Chen X-H, Smith DH, Graham DI. Thromboembolism and delayed cerebral ischemia after subarachnoid hemorrhage: an autopsy study. Neurosurgery (2006) 59:781–8. doi:10.1227/01.NEU.0000227519.27569.45
37. Budohoski KP, Czosnyka M, Kirkpatrick PJ, Smielewski P, Steiner LA, Pickard JD. Clinical relevance of cerebral autoregulation following subarachnoid haemorrhage. Nat Rev Neurol (2013) 9(3):152–63. doi:10.1038/nrneurol.2013.11
38. Ostergaard L, Aamand R, Karabegovic S, Tietze A, Blicher JU, Mikkelsen IK, et al. The role of the microcirculation in delayed cerebral ischemia and chronic degenerative changes after subarachnoid hemorrhage. J Cereb Blood Flow Metab (2013) 33(12):1825–37. doi:10.1038/jcbfm.2013.173
39. Hop JWJ, Rinkel GJG, Algra AA, van Gijn JJ. Initial loss of consciousness and risk of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Stroke (1999) 30:2268–71. doi:10.1161/01.STR.30.11.2268
40. Suwatcharangkoon S, Meyers E, Falo C, Schmidt JM, Agarwal S, Claassen J, et al. Loss of consciousness at onset of subarachnoid hemorrhage as an important marker of early brain injury. JAMA Neurol (2016) 73:28. doi:10.1001/jamaneurol.2015.3188
41. Jaja BNR, Cusimano MD, Etminan N, Hänggi D, Hasan D, Ilodigwe D, et al. Clinical prediction models for aneurysmal subarachnoid hemorrhage: a systematic review. Neurocrit Care (2012) 18:143–53. doi:10.1007/s12028-012-9792-z
42. Lucke-Wold BP, Logsdon AF, Manoranjan B, Turner RC, McConnell E, Vates GE, et al. Aneurysmal subarachnoid hemorrhage and neuroinflammation: a comprehensive review. Int J Mol Sci (2016) 17(4):497. doi:10.3390/ijms17040497
43. Frontera JA, Claassen J, Schmidt JM, Wartenberg KE, Temes R, Connolly ES, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified fisher scale. Neurosurgery (2006) 59:21–7. doi:10.1227/01.NEU.0000218821.34014.1B
44. Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery (1980) 6(1):1–9. doi:10.1097/00006123-198001000-00001
45. Reilly C, Amidei C, Tolentino J, Jahromi BS, Macdonald RL. Clot volume and clearance rate as independent predictors of vasospasm after aneurysmal subarachnoid hemorrhage. J Neurosurg (2004) 101:255–61. doi:10.3171/jns.2004.101.2.0255
46. de Oliveira Manoel AL, Jaja BN, Germans MR, Yan H, Qian W, Kouzmina E, et al. The VASOGRADE: a simple grading scale for prediction of delayed cerebral ischemia after subarachnoid hemorrhage. Stroke (2015) 46(7):1826–31. doi:10.1161/STROKEAHA.115.008728
47. Rosengart AJ, Schultheiss KE, Tolentino J, Macdonald RL. Prognostic factors for outcome in patients with aneurysmal subarachnoid hemorrhage. Stroke (2007) 38:2315–21. doi:10.1161/STROKEAHA.107.484360
48. Dumont AS, Dumont RJ, Chow MM, Lin C-L, Calisaneller T, Ley KF, et al. Cerebral vasospasm after subarachnoid hemorrhage: putative role of inflammation. Neurosurgery (2003) 53:123–35. doi:10.1227/01.NEU.0000068863.37133.9E
49. Gallia GL, Tamargo RJ. Leukocyte-endothelial cell interactions in chronic vasospasm after subarachnoid hemorrhage. Neurol Res (2006) 28(7):750–8. doi:10.1179/016164106X152025
50. Pradilla G, Chaichana KL, Hoang S, Huang J, Tamargo RJ. Inflammation and cerebral vasospasm after subarachnoid hemorrhage. Neurosurg Clin N Am (2010) 21(2):365–79. doi:10.1016/j.nec.2009.10.008
51. Galea J, Cruickshank G, Teeling JL, Boche D, Garland P, Perry VH, et al. The intrathecal CD163-haptoglobin-hemoglobin scavenging system in subarachnoid hemorrhage. J Neurochem (2012) 121:785–92. doi:10.1111/j.1471-4159.2012.07716.x
52. Thelin EP, Tajsic T, Zeiler FA, Menon DK, Hutchinson PJA, Carpenter KLH, et al. Monitoring the neuroinflammatory response following acute brain injury. Front Neurol (2017) 8:231–14. doi:10.3389/fneur.2017.00351
53. Zeiler FA, Thelin EP, Czosnyka M, Hutchinson PJ, Menon DK, Helmy A. Cerebrospinal fluid and microdialysis cytokines in aneurysmal subarachnoid hemorrhage: a scoping systematic review. Front Neurol (2017) 8:379. doi:10.3389/fneur.2017.00379
54. Hong CM, Tosun C, Kurland DB, Gerzanich V, Schreibman D, Simard JM. Biomarkers as outcome predictors in subarachnoid hemorrhage – a systematic review. Biomarkers (2014) 19:95–108. doi:10.3109/1354750X.2014.881418
55. Lad SP, Hegen H, Gupta G, Deisenhammer F, Steinberg GK. Proteomic biomarker discovery in cerebrospinal fluid for cerebral vasospasm following subarachnoid hemorrhage. J Stroke Cerebrovasc Dis (2012) 21(1):30–41. doi:10.1016/j.jstrokecerebrovasdis.2010.04.004
56. Mathiesen T, Edner G, Ulfarsson E, Andersson B. Cerebrospinal fluid interleukin-1 receptor antagonist and tumor necrosis factor-alpha following subarachnoid hemorrhage. J Neurosurg (1997) 87:215–20. doi:10.3171/jns.1997.87.2.0215
57. Fassbender K, Hodapp B, Rossol S, Bertsch T, Schmeck J, Schütt S, et al. Inflammatory cytokines in subarachnoid haemorrhage: association with abnormal blood flow velocities in basal cerebral arteries. J Neurol Neurosurg Psychiatry (2001) 70:534–7. doi:10.1136/jnnp.70.4.534
58. Osuka K, Suzuki Y, Tanazawa T, Hattori K, Yamamoto N, Takayasu M, et al. Interleukin-6 and development of vasospasm after subarachnoid haemorrhage. Acta Neurochir (1998) 140:943–51. doi:10.1007/s007010050197
59. Nakahara T, Tsuruta R, Kaneko T, Yamashita S, Fujita M, Kasaoka S, et al. High-mobility group box 1 protein in CSF of patients with subarachnoid hemorrhage. Neurocrit Care (2009) 11:362–8. doi:10.1007/s12028-009-9276-y
60. Muroi C, Hugelshofer M, Seule M, Tastan I, Fujioka M, Mishima K, et al. Correlation among systemic inflammatory parameter, occurrence of delayed neurological deficits, and outcome after aneurysmal subarachnoid hemorrhage. Neurosurgery (2013) 72:367–75. doi:10.1227/NEU.0b013e31828048ce
61. Sarrafzadeh A, Schlenk F, Gericke C, Vajkoczy P. Relevance of cerebral interleukin-6 after aneurysmal subarachnoid hemorrhage. Neurocrit Care (2010) 13:339–46. doi:10.1007/s12028-010-9432-4
62. Tulamo R, Frösen J, Junnikkala S, Paetau A, Pitkäniemi J, Kangasniemi M, et al. Complement activation associates with saccularcerebral artery aneurysm wall degeneration and rupture. Neurosurgery (2006) 59:1069–77. doi:10.1227/01.NEU.0000245598.84698.26
63. Park CC, Shin ML, Simard JM. The complement membrane attack complex and the bystander effect in cerebral vasospasm. J Neurosurg (1997) 87:294–300. doi:10.3171/jns.1997.87.2.0294
64. Kawano T, Yonekawa Y. Serum complements as indicator for predicting vasospasm and its severity after aneurysmal subarachnoid hemorrhage. Nihon Geka Hokan (1990) 59:189–97.
65. Jung CS, Lange B, Zimmermann M, Seifert V. CSF and serum biomarkers focusing on cerebral vasospasm and ischemia after subarachnoid hemorrhage. Stroke Res Treat (2013) 2013:560305. doi:10.1155/2013/560305
66. Yoshimoto Y, Tanaka Y, Hoya K. Acute systemic inflammatory response syndrome in subarachnoid hemorrhage. Stroke (2001) 32:1989–93. doi:10.1161/hs0901.095646
67. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest (1992) 101:1644–55. doi:10.1378/chest.101.6.1644
68. Dhar R, Diringer MN. The burden of the systemic inflammatory response predicts vasospasm and outcome after subarachnoid hemorrhage. Neurocrit Care (2008) 8:404–12. doi:10.1007/s12028-008-9054-2
69. Tam AKH, Ilodigwe D, Mocco J, Mayer S, Kassell N, Ruefenacht D, et al. Impact of systemic inflammatory response syndrome on vasospasm, cerebral infarction, and outcome after subarachnoid hemorrhage: exploratory analysis of CONSCIOUS-1 database. Neurocrit Care (2010) 13:182–9. doi:10.1007/s12028-010-9402-x
70. Nyström PO. The systemic inflammatory response syndrome: definitions and aetiology. J Antimicrob Chemother (1998) 41(Suppl A):1–7. doi:10.1093/jac/41.suppl_1.1
71. Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood (2003) 101:3765–77. doi:10.1182/blood-2002-06-1887
72. Malham GM, Souter MJ. Systemic inflammatory response syndrome and acute neurological disease. Br J Neurosurg (2001) 15:381–7. doi:10.1080/02688690120082378
73. Mashaly HA, Provencio JJ. Inflammation as a link between brain injury and heart damage: the model of subarachnoid hemorrhage. Cleve Clin J Med (2008) 75:26–30. doi:10.3949/ccjm.75.Suppl_2.S26
74. Naredi S, Lambert G, Eden E, Zäll S, Runnerstam M, Rydenhag B, et al. Increased sympathetic nervous activity in patients with nontraumatic subarachnoid hemorrhage. Stroke (2000) 31:901–6. doi:10.1161/01.STR.31.4.901
75. Di Battista AP, Rizoli SB, Lejnieks B, Min A, Shiu MY, Peng HT, et al. Sympathoadrenal activation is associated with acute traumatic coagulopathy and endotheliopathy in isolated brain injury. Shock (2016) 46:96–103. doi:10.1097/SHK.0000000000000642
76. Rizoli SB, Jaja BNR, Di Battista AP, Rhind SG, Neto AC, da Costa L, et al. Catecholamines as outcome markers in isolated traumatic brain injury: the COMA-TBI study. Crit Care (2017) 21:1–10. doi:10.1186/s13054-017-1620-6
77. Scaravilli V, Tinchero G, Citerio G; The Participants in the International Multi-disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Fever management in SAH. Neurocrit Care (2011) 15:287–94. doi:10.1007/s12028-011-9588-6
78. McMahon CJ, Hopkins S, Vail A, King AT, Smith D, Illingworth KJ, et al. Inflammation as a predictor for delayed cerebral ischemia after aneurysmal subarachnoid haemorrhage. J Neurointerv Surg (2013) 5:512–7. doi:10.1136/neurintsurg-2012-010386
79. McGirt MJ, Mavropoulos JC, McGirt LY, Alexander MJ, Friedman AH, Laskowitz DT, et al. Leukocytosis as an independent risk factor for cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg (2003) 98(6):1222–6. doi:10.3171/jns.2003.98.6.1222
80. The Participants in the International Multi-disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage, Schmutzhard E, Rabinstein AA. Spontaneous subarachnoid hemorrhage and glucose management. Neurocrit Care (2011) 15:281–6. doi:10.1007/s12028-011-9601-0
81. Badjatia N, Monahan A, Carpenter A, Zimmerman J, Schmidt JM, Claassen J, et al. Inflammation, negative nitrogen balance, and outcome after aneurysmal subarachnoid hemorrhage. Neurology (2015) 84:680–7. doi:10.1212/WNL.0000000000001259
82. Frontera JA, Provencio JJ, Sehba FA, McIntyre TM, Nowacki AS, Gordon E, et al. The role of platelet activation and inflammation in early brain injury following subarachnoid hemorrhage. Neurocrit Care (2016) 26:48–57. doi:10.1007/s12028-016-0292-4
83. Vergouwen MD, Vermeulen M, Coert BA, Stroes ES, Roos YB. Microthrombosis after aneurysmal subarachnoid hemorrhage: an additional explanation for delayed cerebral ischemia. J Cereb Blood Flow Metab (2008) 28(11):1761–70. doi:10.1038/jcbfm.2008.74
84. Boluijt J, Meijers JC, Rinkel GJ, Vergouwen MD. Hemostasis and fibrinolysis in delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: a systematic review. J Cereb Blood Flow Metab (2015) 35(5):724–33. doi:10.1038/jcbfm.2015.13
85. Stein SC, Graham DI, Chen X-H, Smith DH. Association between intravascular microthrombosis and cerebral ischemia in traumatic brain injury. Neurosurgery (2004) 54:687–91. doi:10.1227/01.NEU.0000108641.98845.88
86. Romano JG, Forteza AM, Concha M, Koch S, Heros RC, Morcos JJ, et al. Detection of microemboli by transcranial Doppler ultrasonography in aneurysmal subarachnoid hemorrhage. Neurosurgery (2002) 50:1026–30; discussion 1030–1. doi:10.1097/00006123-200205000-00016
87. Vergouwen M, Vermeulen M, de Haan RJ, Levi M, Roos YB. Dihydropyridine calcium antagonists increase fibrinolytic activity: a systematic review. J Cereb Blood Flow Metab (2006) 27:1293–308. doi:10.1038/sj.jcbfm.9600431
88. Young AMH, Karri SK, Ogilvy CS. Non-steroidal anti-inflammatory drugs used as a treatment modality in subarachnoid hemorrhage. Curr Drug Saf (2012) 7:197–201. doi:10.2174/157488612803251342
89. Angiolillo DJ. Antiplatelet therapy in diabetes: efficacy and limitations of current treatment strategies and future directions. Diabetes Care (2009) 32:531–40. doi:10.2337/dc08-2064
90. van den Bergh WM; MASH Study Group, Algra A, Dorhout Mees SM, van Kooten F, Dirven CMF, et al. Randomized controlled trial of acetylsalicylic acid in aneurysmal subarachnoid hemorrhage: the MASH Study. Stroke (2006) 37:2326–30. doi:10.1161/01.STR.0000236841.16055.0f
91. Mendelow AD, Stockdill G, Steers AJ, Hayes J, Gillingham FJ. Double-blind trial of aspirin in patient receiving tranexamic acid for subarachnoid hemorrhage. Acta Neurochir (1982) 62:195–202. doi:10.1007/BF01403624
92. Hop JW, Rinkel GJ, Algra A, Berkelbach van der Sprenkel JW, van Gijn J. Randomized pilot trial of postoperative aspirin in subarachnoid hemorrhage. Neurology (2000) 54:872–8. doi:10.1212/WNL.54.4.872
93. Shaw MD, Foy PM, Conway M, Pickard JD, Maloney P, Spillane JA, et al. Dipyridamole and postoperative ischemic deficits in aneurysmal subarachnoid hemorrhage. J Neurosurg (1985) 63:699–703. doi:10.3171/jns.1985.63.5.0699
94. Suzuki S, Sano K, Handa H, Asano T, Tamura A, Yonekawa Y, et al. Clinical study of OKY-046, a thromboxane synthetase inhibitor, in prevention of cerebral vasospasms and delayed cerebral ischaemic symptoms after subarachnoid haemorrhage due to aneurysmal rupture: a randomized double-blind study. Neurol Res (1989) 11(2):79–88.
95. Tokiyoshi K, Ohnishi T, Nii Y. Efficacy and toxicity of thromboxane synthetase inhibitor for cerebral vasospasm after subarachnoid hemorrhage. Surg Neurol (1991) 36:112–8. doi:10.1016/0090-3019(91)90228-2
96. van der Velden VH. Glucocorticoids: mechanisms of action and anti-inflammatory potential in asthma. Mediators Inflamm (1998) 7(4):229–37. doi:10.1080/09629359890910
97. Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G, et al. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomised placebo-controlled trial. Lancet (2004) 364:1321–8. doi:10.1016/S0140-6736(04)17188-2
98. Edwards P, Arango M, Balica L, Cottingham R, El-Sayed H, Farrell B, et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet (2005) 365:1957–9. doi:10.1016/S0140-6736(05)66552-X
99. Chatzi M, Karvouniaris M, Makris D, Tsimitrea E, Gatos C, Tasiou A, et al. Bundle of measures for external cerebral ventricular drainage-associated ventriculitis. Crit Care Med (2014) 42:66–73. doi:10.1097/CCM.0b013e31829a70a5
100. Muroi C, Hugelshofer M, Seule M, Keller E. The impact of nonsteroidal anti-inflammatory drugs on inflammatory response after aneurysmal subarachnoid hemorrhage. Neurocrit Care (2013) 20:240–6. doi:10.1007/s12028-013-9930-2
101. Ghodsi SM, Mohebbi N, Naderi S, Anbarloie M, Aoude A, Habibi Pasdar SS. Comparative efficacy of meloxicam and placebo in vasospasm of patients with subarachnoid hemorrhage. Iran J Pharm Res (2015) 14:125–30.
Keywords: neuroinflammation, subarachnoid hemorrhage, early brain injury, secondary brain injury, vasospasm, delayed cerebral ischemia
Citation: de Oliveira Manoel AL and Macdonald RL (2018) Neuroinflammation as a Target for Intervention in Subarachnoid Hemorrhage. Front. Neurol. 9:292. doi: 10.3389/fneur.2018.00292
Received: 15 February 2018; Accepted: 16 April 2018;
Published: 02 May 2018
Edited by:
Fernando Testai, University of Illinois at Chicago, United StatesReviewed by:
Adel Helmy, University of Cambridge, United KingdomElisa R. Zanier, Istituto Di Ricerche Farmacologiche Mario Negri, Italy
Copyright: © 2018 de Oliveira Manoel and Macdonald. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Airton Leonardo de Oliveira Manoel, YWlydG9ubGVvLm1hbm9lbEBnbWFpbC5jb20=