 Nikita M. Bajwa
Nikita M. Bajwa Chandrasekhar Kesavan
Chandrasekhar Kesavan Subburaman Mohan
Subburaman Mohan- 1Musculoskeletal Disease Center, VA Loma Linda Healthcare System, Loma Linda, CA, United States
- 2Department of Medicine, Loma Linda University, Loma Linda, CA, United States
- 3Department of Orthopedic Surgery, Loma Linda University, Loma Linda, CA, United States
Traumatic brain injury (TBI) leads to long-term cognitive, behavioral, affective deficits, and increase neurodegenerative diseases. It is only in recent years that there is growing awareness that TBI even in its milder form poses long-term health consequences to not only the brain but to other organ systems. Also, the concept that hormonal signals and neural circuits that originate in the hypothalamus play key roles in regulating skeletal system is gaining recognition based on recent mouse genetic studies. Accordingly, many TBI patients have also presented with hormonal dysfunction, increased skeletal fragility, and increased risk of skeletal diseases. Research from animal models suggests that TBI may exacerbate the activation and inactivation of molecular pathways leading to changes in both osteogenesis and bone destruction. TBI has also been found to induce the formation of heterotopic ossification and increased callus formation at sites of muscle or fracture injury through increased vascularization and activation of systemic factors. Recent studies also suggest that the disruption of endocrine factors and neuropeptides caused by TBI may induce adverse skeletal effects. This review will discuss the long-term consequences of TBI on the skeletal system and TBI-induced signaling pathways that contribute to the formation of ectopic bone, altered fracture healing, and reduced bone mass.
Introduction
Traumatic brain injury (TBI) is the disruption of brain activity due to an external force or violent blow to the head. TBI can lead to a series of physical, cognitive, social, emotional, and behavioral impairments (1) and is the leading cause of death and disability in both combat and civilian populations. More than 1.7 million people in the U.S. experience a TBI annually (1), and it is a major cause of death and disability worldwide, especially in children and young adults. Significant proportions of survivors require hospital care, extended rehabilitation, and may have long-term physical, cognitive, and psychological disorders. Many statistics do not account for individuals who have not reported an injury or received medical care, and disabilities may be significantly higher, often with long-term consequences. TBI may be classified based on severity as mild, moderate, or severe and location of injury and time of lost consciousness (2). Symptoms can range from mild concussions, with symptoms lasting from seconds, to more severe injuries with symptoms lasting years or even death. Individuals who have suffered mild TBI, or concussions, report adverse effects resulting from the TBI(s) months later (3) due to the rotational stress caused by head movement. In fact, TBI is the beginning of an ongoing, possibly lifelong, process that impacts multiple organ systems and bone that is proximal or distal to the site of injury. This review will discuss the consequences that TBI on the skeleton and its possible mechanisms.
Posttraumatic Mortality and Morbidity
The clinical severity of TBI has been associated with an increased risk for mortality. Per the CDC, there are over 50,000 deaths in the U.S. annually and approximately 22% die within the first 5 years after suffering a TBI. TBI patients (mild, moderate, or severe) with brain edema were eight times as likely to die compared to TBI patients without edema and surprisingly, were five times as likely to die for mild TBI-related edema (4). Those who survive moderate-to-severe TBI and receive rehabilitation have a shorter life expectancy by 9 years (5).
A diffusion tensor imaging study of the corpus callosum found that those with mild TBI had significant white matter abnormalities up to 3 months post-injury (6). These studies suggest that mild TBI, previously considered harmless, could in fact lead to more TBI-related morbidity.
Neurological Disorders and Neurodegenerative Diseases
Trauma-induced neurological and neurodegenerative sequalae are relatively common. An estimated 70% of adults in the United States have experienced a traumatic event in their lifetime, in which, approximately 20% go on to develop PTSD. These individuals are at an increased risk for psychological disorders (i.e., depression and anxiety), neurological disorders (i.e., epilepsy and sleep disorders), physical injuries, substance abuse, and fatigue, all of which can lead to poor decisions and actions and can manifest into physical symptoms.
Concussion, or mild TBI, accounts for approximately 90% of all brain traumas sustained (7). While many impairments resulting from a mild TBI tend to resolve within a few months, a subset of these individuals, in addition to moderate and severe TBI, exhibit gradual cognitive decline and motor deficits such as those that are characteristic in Alzheimer’s disease, Parkinson’s disease, and chronic traumatic encephalopathy (8).
TBI Effects on the Skeletal System
Traumatic brain injury and its associated physiological processes have been found to lead to significant skeletal abnormalities that often transpire over time (Table 1). While the consequences and mechanisms of trauma to the head and the pathophysiological and neurochemical events that occur during the course of initial hours and days are being extensively investigated, little is known regarding the long-term consequence of TBI on remote organs that are under the influence of neural and neuroendocrine humoral outflow from the brain via the pituitary. Recently, the discovery of bone regulation by neural signals represents an emerging area of study that is identifying novel regulatory axes between the nervous system and bone cells. Based on the key role for cells of the hypothalamic nuclei in the neuro(endo)crine regulation of bone remodeling, it is predictable that injury to the brain will have a severe impact on the regulatory molecules that control skeletal growth/maintenance. The rest of this review will focus on the long-term consequences of TBI on the skeletal system and the potential mechanisms for TBI effects on bone.
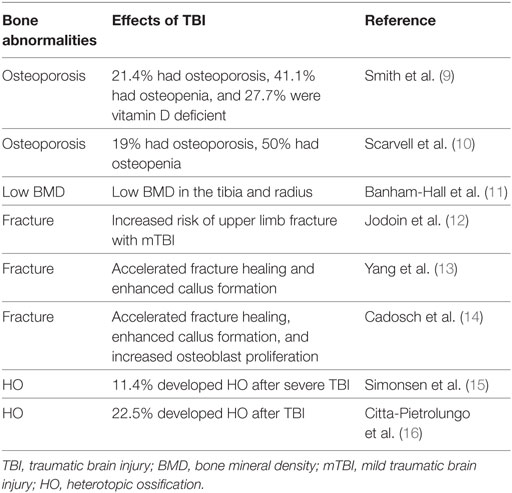
Table 1. Examples of clinical studies showing relationship between TBI and bone-related abnormalities.
Heterotopic Ossification (HO)
Heterotopic ossification is defined as the formation of mature, lamellar bone, in non-osseous tissue, typically between the muscle and joint capsule. Although, there are rare hereditary disorders associated with HO, it is well established that HO is usually acquired following trauma to soft tissues, bone, and neurological damage. The incidence of ectopic bone formation is frequently seen in patients with TBI (17–22). This ectopic deposition of bone elicits symptoms including pain with a significant loss of range of motion (17, 23, 24).
Three subtypes of HO have been identified: genetic, traumatic, and neurogenic. Genetic HO occurs in individuals with inherited conditions such as fibrodysplasia ossificans progressive and progressive osseous heteroplasia. Traumatic HO occurs in response to injuries such as acetabular fractures, fractures and joint dislocations, blast injuries, burns, combat, amputee injuries, and muscle trauma (25–28). Neurogenic HO occurs in response to TBIs and spinal cord injuries (17, 29). Those with TBI alone go on to develop HO throughout the body, distant from the primary injury, in locations such as the hip, knee, elbow, or shoulder (30). Many returning from military combat develop HO from the combination of trauma and neurogenic injuries (18, 31–33). TBI occurrence with local tissue injury has been associated with the pathogenesis of HO and suggests that both systemic and local mechanisms may contribute to HO. Recently, serum collected from TBI patients promoted conversion of muscle cells into osteoblasts, suggesting that TBI may accelerate the formation of ectopic bone at the sites of injury (34).
The identification of cellular and molecular events leading to HO continue to remain elusive. Recent studies suggest that the activation of the bone morphogenic protein (BMP) signaling may play a significant role in the pathogenesis of HO based on the established role for BMPs to promote de novo bone formation in non-skeletal sites (35–38). Specifically, HO induction with BMP-2 led to the expression of osteoblast-specific transcription factors in the endonerium, suggesting that endoneurial progenitors are osteogenic precursors that contribute to HO (39). Further, osteoblasts isolated from HO bone (rodent and clinical models) exhibited increased osteogenic differentiation compared to osteoblasts derived from normal bone (40).
Studies have also linked the release of pro-angiogenic factors to the onset of HO (Figure 1) (41–43). The vascularization of injured tissue represents an important step during the transition from hypertrophic cartilage to bone during the endochondral ossification of cartilage (44, 45). Neovascularization occurs within the injured tissue because of low oxygen tension that is caused by reduced activity of prolyl hydroxylase domain proteins (PHDs). The early response activity of PHD proteins are oxygen dependent and, under normoxia, hydrolyze hypoxia-inducible factor (HIF)-α subunits, resulting in their degradation and inactivity. Under hypoxia, PHD activity decreases due to reduced oxygen levels. Thus, the HIF-1α subunit avoids hydroxylation, accumulates in the nucleus, recruits HIF-1β, and together bind to penta-nucleotide hypoxia-responsive elements to promote transcription of key genes, such as VEGF, involved in the angiogenesis pathway (44, 46, 47). Hypoxic conditions also impact bone formation by stimulating oxygen-sensing factors that promote chondrocyte survival and differentiation (35, 48, 49). The increase in osteogenic precursor cells create a feedback loop to increase angiogenesis and exacerbate chondrocyte differentiation and hypertrophy. Conditional disruption of the Phd2 gene in chondrocytes promoted endochondral ossification and dramatic increase in trabecular bone mass (50). It was found that loss of PHD2 in chondrocytes promoted chondrocyte differentiation and osteoblast formation and that this effect was in part mediated via upregulation of HIF1α signaling. The extent to which hypoxia signaling contributes to ectopic bone formation caused by brain injury remains to be elucidated.
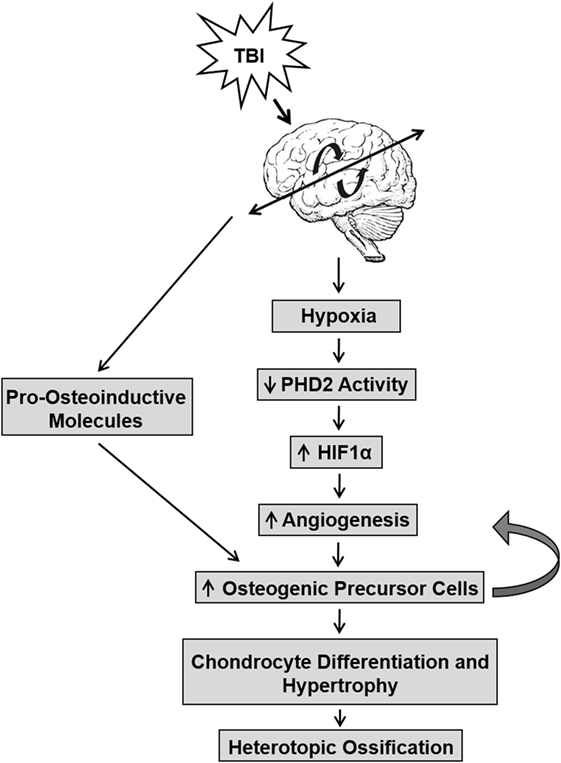
Figure 1. The interaction between traumatic brain injury (TBI) and hypoxic conditions that lead to the development of heterotopic ossification (HO). TBI induces a hypoxic environment in tissue that reduces PHD2 activity, which in turn, prevents the cleavage of hypoxia-inducible factor (HIF)1α and increases angiogenesis. This pathway increases osteogenic precursor cell activity, thereby promoting chondrocyte differentiation and hypertrophy in soft tissues and leading to bone formation in HO. TBI may also directly affect pro-osteoinductive molecules that promote increased osteogenic precursor cell activity.
Fracture Healing
Fracture healing can be defined as the physiological repair of bone tissue, structure, and function after injury. Patients who have sustained severe TBI commonly demonstrate alterations in the healing process of bone. There is mounting evidence linking the association between TBI and enhanced osteogenesis postfracture. TBI in combination with fracture results in higher bone volume, higher mineral density (51), and accelerated healing with enhanced callus formation (14, 52–55).
The physiology of fracture healing involves both local and systemic factors that can be divided into three overlapping phases: inflammatory phase, followed by reparation, and finally remodeling. During the inflammatory phase, bleeding from the fracture and adjacent soft tissues result in the formation of hematoma at the injured site. The hemopoietic cells at the hematoma secrete cytokines and growth factors and attract osteoprogenitor and mesenchymal cells that result in the proliferation of osteoblasts and fibroblasts. Cytokines such as interleukin (IL)-6, produced by stromal/osteoblastic cells, enhance angiogenesis and osteoclastogenesis and regulate bone resorption and callus formation (56). During the reparative phase, a primary soft callus develops, followed by a medullary (hard) callus several weeks later. During the remodeling phase, the fracture site is reshaped by the interactions between osteoclasts resorbing bone and osteoblasts forming bone that strengthens the bone.
The existence of whether humoral osteogenic factors released post-TBI and/or direct nervous action guides the induction of enhanced fracture healing is still being debated. Several studies have detected humoral osteogenic factors in the serum of TBI patients (57) and in cells (14, 58). There is also evidence in the literature to suggest that a combination of signaling cascades involving humoral, neuronal, and local bone markers may play a significant role in fracture repair after trauma (59, 60). Cell proliferation was significantly increased in rat osteoblast cells treated with serum from TBI patients, suggesting the role of a circulating growth factor (either systemic or local) that promoted osteogenic activity (61). In another study, growth hormone (GH) levels continued to increase during enhanced osteogenesis with a gradual increase in IGF-1 during fracture healing in patients with combined TBI and fracture (62). Other growth factors have also been implicated in the process of fracture healing. Serum levels of epidermal growth factor and nerve growth factor gradually increase up to 14 days post-TBI and fracture combined injury compared to fracture or TBI groups alone (63).
The central and peripheral nervous systems play important roles in the regulation of bone with many factors influencing this densely innervated tissue in the body. One of these factors is leptin, and it has been hypothesized to be an integral part of fracture healing (Figure 2). Leptin is an adipocyte-derived hormone expressed in several tissues that primarily controls energy and food intake (64) and is involved in the regulation of insulin homeostasis, reproduction, immune function, and brain development (65). More recently, in vivo studies have shown elevated levels of GH and IGF-I in serum of TBI and fractured rabbits (66) that could be caused by increase in leptin levels presumably due to disruption of blood–brain barrier. In addition, higher percentage of leptin-positive cells in the callus have also been detected in combined TBI and fractured rats (67, 68). Further, studies using knockout mice suggest that changes in callus formation were not detected in leptin-deficient ob/ob mice that underwent TBI and osteonomy (69); however, peripheral injections of leptin significantly increased bone mass and osteoblastic activity in ob/ob mice (65, 70). Neuropeptide Y (NPY), a neurotransmitter with regulatory functions in bone homeostasis, is downstream of leptin signaling. NPY levels increase and promote osteogenic differentiation in patients with TBI and fractures (71). NPY and leptin share an inverse relationship, and recent studies suggest that NPY may mediate part of the skeletal phenotype in ob/ob mice (72, 73). Another factor is Semaphorin 3A, a secreted cytokine regulated by neural injury and is known to guide axonal and dendritic growth and neural migration. Recent studies have suggested that Semaphorin 3A can suppress bone resorption by binding to neurophilin-1 that in turn inhibits receptor activator of nuclear factor κB ligand-induced osteoclast differentiation and other pathways and synchronously stimulates osteoblasts and enhanced osteogenesis via canonical Wnt-signaling (74). In addition, stromal cell-derived factor-1, a chemokine protein, has been found to contribute to endochondral bone repair in TBI and fracture healing via increased expression surrounding tissues of fractured bone (75). Thus, while a number of signaling molecules have been implicated in the accelerated fracture healing caused by TBI, the mechanism by which brain injury influences these molecules and the extent to which the identified molecules interact to regulate fracture healing remain to be established.
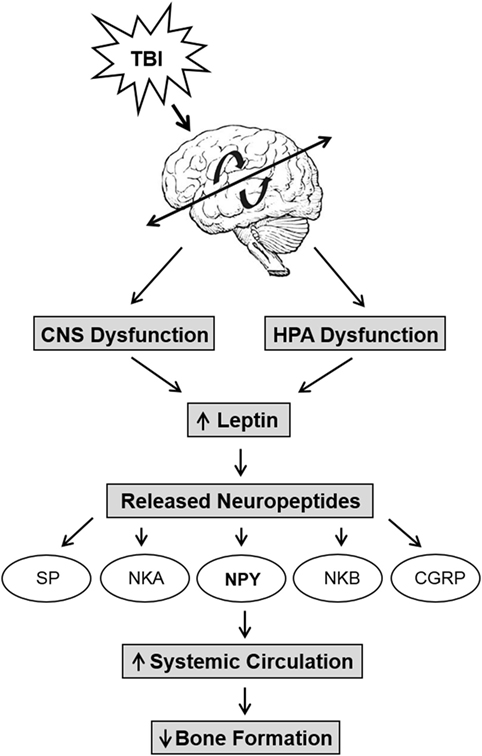
Figure 2. The relationship between trauma, neuropeptides, and decreased bone formation. Traumatic brain injury (TBI) induces central nervous system disruption and inflammation and causes an upregulation in leptin levels due to the compromised blood–brain barrier. TBI also causes hypothalamus–pituitary–adrenal axis (HPA) dysfunction that increases leptin and causes the release of neuropeptides such as substance P (SP), neurokinin A (NKA), neuropeptide Y (NPY), neurokinin B (NKB), and calcitonin gene-related peptide (CGRP). These neuropeptides propagate further inflammation that further increase systemic circulation of NPY and other neuropeptides that reduce bone formation via the leptin pathway.
Low Bone Mass
Patients who have experienced TBI may have an increased risk of osteopenia and osteoporosis (9, 11), which are asymptomatic systemic skeletal diseases result in the micro-architectural deterioration of bone tissue leading to bone fragility and ultimately, fractures. Changes in bone are characterized by reduced bone mineral density (BMD) due from immobility and other metabolism-related factors. In animal models, TBI significantly reduces BMD in cortical bone (2, 76) and decreases in tibial trabecular bone mass (77) irrespective of mobility. In stroke patients, the highest rate of bone loss occurs within the first year (78), particularly, greatest loss of BMD in the paretic hip and upper limbs. Adults with TBI have been shown to have higher BMD loss in the femur and vitamin D deficiency (9). Several groups of stroke patients have also been shown to have deficiency in vitamin D levels (25-hydroxyvitamin D or 1,25-dihydroxyvitamin D) (79, 80). Although low vitamin D levels in stroke patients may be attributed to lack of exposure to sunlight following stroke and the hypoparathyroidism induced by immobilization hypercalcemia, it may also be that many stroke patients have insufficient levels of vitamin D prior to stroke (81).
Traumatic brain injury-induced skeletal changes seems to be dependent in part on the variations in parathyroid hormone (PTH) and vitamin D axis. TBI reduces PTH activity, thereby reducing 1,25-dihydroxyvitamin D production and activity. The resulting hypoparathyroidism and reduced serum calcium levels leads to increased bone resorption and reduced microstructural integrity (82), and, thereby, increasing risk for fractures (9, 83, 84).
Mechanisms Detrimental Effects of TBI on Bone
Inflammation
Activation of the inflammatory response in the brain occurs within a few seconds after trauma and with the permeation of the blood–brain barrier and activation of several injury cascades (85, 86). Injury to the central nervous system (CNS) results in an increase of blood products, tissue debris, prostaglandins, reactive oxygen specials, and nitrogen species. These factors, in turn, trigger an innate response of resident immune cells (i.e., macrophages, mast cells, granulocytes, dendritic cells, and natural killer cells) through the activation of microglia and astrocytes, increased migration and recruitment of leukocytes, and the release of inflammatory mediators such as cytokines and chemokines (pro- and/or anti-inflammatory) and results in local and systemic immune responses (87, 88). Systemic inflammation shifts toward an adaptive immune response and to the chronic stages post-TBI (days, weeks, months, years), which may, in fact, exacerbate the onset of skeletal deterioration. For example, increased systemic IL-6 and IL-11 may directly stimulate osteoclastic activity or act via osteoblast lineage cells that increase osteoclast formation through receptor activator of nuclear factor kappa-B ligand (89). Similarly, increased levels of systemic IL-18 has been associated with poor clinical outcome in TBI patients (90).
Neuropeptides
Trauma also leads to neurogenic inflammation, where the activation of sensory unmyelinated neurons by noxious stimuli (i.e., TBI) causes the simultaneous release of neuropeptides such as substance P (SP), neurokinin A, neuropeptide Y (NPY), neurokinin B, and calcitonin gene-related peptide as shown in Figure 2. The release of peptides results in vasodilation, increased vascular and blood–brain barrier permeability (91). The compromised blood–brain barrier causes an influx of plasma proteins that enhances the inflammatory response, as well as creating a positive feedback with neurogenic inflammation to propagate further inflammation with the release of pro-inflammatory mediators, oxidative factors, etc., which causes further neural damage. In addition, the disruption of the hypothalamus may increase the circulation of peripheral NPY and reduce bone formation via leptin (72, 73).
Hypothalamus–Pituitary–Adrenal Axis (HPA)
Together with the central nervous and circulatory systems, the HPA axis, also the endocrine system, is critical for the integration and coordination for many bodily functions such as stress reactions, digestion, immune system, mood and emotions, sexuality, etc. The HPA axis acts as a complex integration and feedback network between three endocrine glands: hypothalamus, pituitary gland, and the adrenal gland. The hypothalamus synthesizes and secretes vasopressin and corticotropin-releasing hormone, which in turn stimulates the secretion of adrenocorticotropic hormone in the pituitary gland and further stimulates the production of glucocorticoid hormones. The glucocorticoid hormones in turn act back on the hypothalamus in a negative feedback loop, secreting hormones as necessary for function. In healthy individuals, HPA signaling maintains homeostasis. In response to brain injury, the function of the HPA axis is disrupted, causing a reduction in the production of many endocrine factors (hypopituitarism; Figure 1) (92, 93). It has been reported that 70% of patients with TBI have hypothalamic–pituitary dysfunction (94). For example, a 12-month prospective study found that the rate of hypopituitarism in post-TBI patients was reduced by 33 and 23% at 3 and 12 months, respectively (95). Another clinical study also reported anterior pituitary dysfunction in 56 and 36% of TBI patients at 3 and 12 months post injury, respectively (96). These studies indicate that HPA dysfunction is a key consequence of TBI.
Hypopituitarism is the most common manifestation of the HPA axis disruption via TBI that causes hormonal deficiencies (Figure 3) (97–101). Individuals suffering from TBI are at the risk of developing long-term negative consequences on the skeleton via deficiencies in GH, gonadotrophin, and thyroid-stimulating hormone (TSH), all of which significantly influence bone metabolism. For example, GH is known to have anabolic and catabolic mechanistic pathways that may be injury dependent in bone. GH interacts with insulin-like growth factor-1 and both contribute to the regulation of bone mass through the stimulation of osteoblast activity (102, 103). Deficiency in GH created a catabolic effect that increases skeletal fragility and low bone mass that leads to higher fracture risk (104) and delayed fracture healing (105). However, GH creates an anabolic effect by stimulating accelerated fracture healing (106, 107). TSH directly inhibits osteoclast skeletal remodeling and osteoblast bone formation (108) and lower levels of TSH significantly increases bone resorption and reduces bone osteogenesis (109). Pituitary hormone disturbances are frequently found after TBI and lead to changes in bone mineralization (110). These hypothalamic–pituitary changes may be exacerbated in repetitive trauma. We have found that mild repetitive TBI led to significant loss skeletal mass (2), and more recently, we have shown that these negative effects on bone microarchitecture and mechanical properties were mediated by osteoblast function via reduced endocrine IGF-1 actions (111). Recent studies also demonstrate an important role for central control of bone mass involving leptins and neuropeptides (112, 113). Thus, the hormonal disruption of the HPA leads to many detrimental skeletal effects.
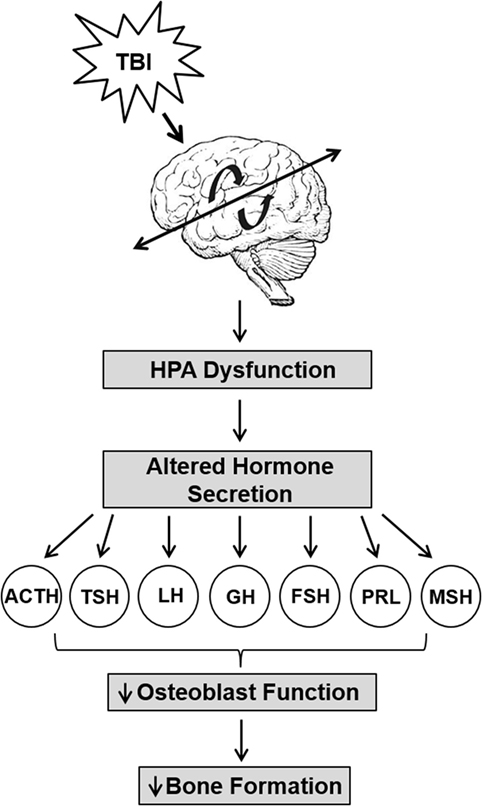
Figure 3. Trauma-induced hypothalamus–pituitary–adrenal axis (HPA) dysfunction lead to reduced bone formation. Traumatic brain injury causes significant HPA dysfunction that leads to increased levels of adrenocorticotropin (ACTH), prolactin (PRL), and growth hormone (GH), but decreased or unchanged levels in luteinizing hormone (LH), follicle-stimulating hormone (FSH), PRL, melanocyte-stimulating hormone (MSH), and thyrotropin (TSH) levels. The altered secretion of hormones impact osteoblast function and impair bone formation.
Clinical Recommendations
The physical presentation of bone metabolism abnormalities post-TBI may not be detected until considerable skeletal damage has already occurred. We recommend the assessment of serum from TBI patients for levels of biochemical markers of bone metabolism (i.e., bone-specific alkaline phosphatase, osteocalcin, N-terminal propeptide of type I procollagen, N-terminal telopeptide of type I collagen, and C-terminal telopeptide of type I collagen) and GH deficiency (GH, insulin-like growth factor-I, IGF binding protein-3, and acid labile subunit) (60) as the first line of preventative treatment. Patients with increased serum markers of bone turnover and GH deficiency would benefit from DXA imaging to monitor skeletal deficits across time. Changes in serum levels of bone markers can be used to further tailor pharmacological interventions toward promoting bone formation (PTH, sclerostin antibody) or inhibiting bone resorption (bisphosphonates). Thus, a multifaceted treatment approach is necessary for the detection and prevention of skeletal deficit in TBI patients.
Conclusion
Traumatic brain injury leads to significant functional impairments and structural alterations that can lead to permanent long-term changes in TBI survivors. These changes include sleep disturbances, impaired movement, cognitive dysfunction, and neurodegenerative diseases. Alterations in the CNS, as well as the disruption of the HPA axis alter skeletal remodeling processes and enhance resorption in sites distal to the site of injury, ultimately leading to low bone mass and skeletal fragility. Interestingly, TBI stimulates osteoblasts and enhanced osteogenesis in fractured healing bones. We conclude that TBI is a complex multifaceted process that leads to acute and long-term skeletal consequences that directly impacts bone integrity and function in TBI survivors.
Future Perspectives
Although several studies have revealed key mechanisms in the pathogenesis of TBI-induced bone alterations, particularly in the release of humoral and osteogenic factors, many mechanistic pathways remain to be explored to fully understand the processes. Further studies are needed to investigate the consequences of moderate to severe TBI and whether the mechanistic pathways leading to skeletal alterations are similar or different. These investigations would lead to pharmacological approaches targeting pathways simultaneously may aid in preventing adverse skeletal changes.
Author Contributions
NB, CK, and SM wrote the manuscript. NB prepared tables and figures. SM revised and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
SM is a recipient of a Senior Research Career Scientist Award from the Department of Veterans Affairs This publication was supported by funding from a Veterans Administration BLR&D merit review grant 1-101-BX-002717 to SM. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
References
1. Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil (2006) 21(5):375–8. doi:10.1097/00001199-200609000-00001
2. Yu H, Watt H, Mohan S. The negative impact of traumatic brain injury (TBI) on bone in a mouse model. Brain Inj (2014) 28(2):244–51. doi:10.3109/02699052.2013.859735
3. McMahon P, Hricik A, Yue JK, Puccio AM, Inoue T, Lingsma HF, et al. Symptomatology and functional outcome in mild traumatic brain injury: results from the prospective TRACK-TBI study. J Neurotrauma (2014) 31(1):26–33. doi:10.1089/neu.2013.2984
4. Tucker B, Aston J, Dines M, Caraman E, Yacyshyn M, McCarthy M, et al. Early brain edema is a predictor of in-hospital mortality in traumatic brain injury. J Emerg Med (2017) 53(1):18–29. doi:10.1016/j.jemermed.2017.02.010
5. Centers for Disese Control and Prevention. Traumatic Brain Injury & Concussion. (2017). Available from: https://www.cdc.gov/traumaticbraininjury/index.html
6. Rutgers DR, Fillard P, Paradot G, Tadie M, Lasjaunias P, Ducreux D. Diffusion tensor imaging characteristics of the corpus callosum in mild, moderate, and severe traumatic brain injury. AJNR Am J Neuroradiol (2008) 29(9):1730–5. doi:10.3174/ajnr.A1213
7. Fourtassi M, Hajjioui A, Ouahabi AE, Benmassaoud H, Hajjaj-Hassouni N, Khamlichi AE. Long term outcome following mild traumatic brain injury in Moroccan patients. Clin Neurol Neurosurg (2011) 113(9):716–20. doi:10.1016/j.clineuro.2011.07.010
8. Petraglia AL, Plog BA, Dayawansa S, Chen M, Dashnaw ML, Czerniecka K, et al. The spectrum of neurobehavioral sequelae after repetitive mild traumatic brain injury: a novel mouse model of chronic traumatic encephalopathy. J Neurotrauma (2014) 31(13):1211–24. doi:10.1089/neu.2013.3255
9. Smith E, Comiskey C, Carroll A. Prevalence of and risk factors for osteoporosis in adults with acquired brain injury. Ir J Med Sci (2016) 185(2):473–81. doi:10.1007/s11845-016-1399-5
10. Scarvell JM, Van Twest MS, Stanton SF, Burski G, Smith PN. Prevalence of undisclosed osteoporosis in patients with minimal trauma fractures: a prospective cohort study. Phys Sportsmed (2013) 41(2):38–43. doi:10.3810/psm.2013.05.2011
11. Banham-Hall N, Kothwal K, Pipkin J, Bentley J, Dickens GL. Prevalence of low bone mineral density in inpatients with traumatic brain injury receiving neurobehavioural rehabilitation: a postoperative, observational study. Physiotherapy (2013) 99(4):328–34. doi:10.1016/j.physio.2012.12.009
12. Jodoin M, Rouleau DM, Charlebois-Plante C, Benoit B, Leduc S, Laflamme GY, et al. Incidence rate of mild traumatic brain injury among patients who have suffered from an isolated limb fracture: upper limb fracture patients are more at risk. Injury (2016) 47(8):1835–40. doi:10.1016/j.injury.2016.05.036
13. Yang TY, Wang TC, Tsai YH, Huang KC. The effects of an injury to the brain on bone healing and callus formation in young adults with fractures of the femoral shaft. J Bone Joint Surg Br (2012) 94(2):227–30. doi:10.1302/0301-620X.94B2.28193
14. Cadosch D, Gautschi OP, Thyer M, Song S, Skirving AP, Filgueira L, et al. Humoral factors enhance fracture-healing and callus formation in patients with traumatic brain injury. J Bone Joint Surg Am (2009) 91(2):282–8. doi:10.2106/JBJS.G.01613
15. Simonsen LL, Sonne-Holm S, Krasheninnikoff M, Engberg AW. Symptomatic heterotopic ossification after very severe traumatic brain injury in 114 patients: incidence and risk factors. Injury (2007) 38(10):1146–50. doi:10.1016/j.injury.2007.03.019
16. Citta-Pietrolungo TJ, Alexander MA, Steg NL. Early detection of heterotopic ossification in young patients with traumatic brain injury. Arch Phys Med Rehabil (1992) 73(3):258–62.
17. Sullivan MP, Torres SJ, Mehta S, Ahn J. Heterotopic ossification after central nervous system trauma: a current review. Bone Joint Res (2013) 2(3):51–7. doi:10.1302/2046-3758.23.2000152
18. Isaacson BM, Potter BK, Bloebaum RD, Epperson RT, Kawaguchi BS, Swanson TM, et al. Link between clinical predictors of heterotopic ossification and histological analysis in combat-injured service members. J Bone Joint Surg Am (2016) 98(8):647–57. doi:10.2106/JBJS.15.00895
19. Ippolito E, Formisano R, Caterini R, Farsetti P, Penta F. Resection of elbow ossification and continuous passive motion in postcomatose patients. J Hand Surg Am (1999) 24(3):546–53. doi:10.1053/jhsu.1999.0360
20. Ippolito E, Formisano R, Caterini R, Farsetti P, Penta F. Operative treatment of heterotopic hip ossification in patients with coma after brain injury. Clin Orthop Relat Res (1999) (365):130–8. doi:10.1097/00003086-199908000-00018
21. Ippolito E, Formisano R, Farsetti P, Caterini R, Penta F. Excision for the treatment of periarticular ossification of the knee in patients who have a traumatic brain injury. J Bone Joint Surg Am (1999) 81(6):783–9. doi:10.2106/00004623-199906000-00005
22. Garland DE. Periarticular ossification. J Bone Joint Surg Am (2000) 82-A(8):1206–7. doi:10.2106/00004623-200008000-00046
23. Garland DE. Clinical observations on fractures and heterotopic ossification in the spinal cord and traumatic brain injured populations. Clin Orthop Relat Res (1988) (233):86–101.
24. Sawyer JR, Myers MA, Rosier RN, Puzas JE. Heterotopic ossification: clinical and cellular aspects. Calcif Tissue Int (1991) 49(3):208–15. doi:10.1007/BF02556120
25. Melcer T, Belnap B, Walker GJ, Konoske P, Galarneau M. Heterotopic ossification in combat amputees from Afghanistan and Iraq wars: five case histories and results from a small series of patients. J Rehabil Res Dev (2011) 48(1):1–12. doi:10.1682/JRRD.2010.03.0033
26. Baschera D, Rad H, Collopy D, Zellweger R. Incidence and clinical relevance of heterotopic ossification after internal fixation of acetabular fractures: retrospective cohort and case control study. J Orthop Surg Res (2015) 10:60. doi:10.1186/s13018-015-0202-z
27. Alfieri KA, Forsberg JA, Potter BK. Blast injuries and heterotopic ossification. Bone Joint Res (2012) 1(8):192–7. doi:10.1302/2046-3758.18.2000102
28. Evans EB. Heterotopic bone formation in thermal burns. Clin Orthop Relat Res (1991) (263):94–101.
29. Coelho CV, Beraldo PS. Risk factors of heterotopic ossification in traumatic spinal cord injury. Arq Neuropsiquiatr (2009) 67(2b):382–7. doi:10.1590/S0004-282X2009000300002
30. Ranganathan K, Loder S, Agarwal S, Wong VW, Forsberg J, Davis TA, et al. Heterotopic ossification: basic-science principles and clinical correlates. J Bone Joint Surg Am (2015) 97(13):1101–11. doi:10.2106/JBJS.N.01056
31. Potter BK, Burns TC, Lacap AP, Granville RR, Gajewski DA. Heterotopic ossification following traumatic and combat-related amputations. Prevalence, risk factors, and preliminary results of excision. J Bone Joint Surg Am (2007) 89(3):476–86. doi:10.2106/JBJS.F.00412
32. Forsberg JA, Pepek JM, Wagner S, Wilson K, Flint J, Andersen RC, et al. Heterotopic ossification in high-energy wartime extremity injuries: prevalence and risk factors. J Bone Joint Surg Am (2009) 91(5):1084–91. doi:10.2106/JBJS.H.00792
33. Brown KV, Dharm-Datta S, Potter BK, Etherington J, Mistlin A, Hsu JR, et al. Comparison of development of heterotopic ossification in injured US and UK Armed Services personnel with combat-related amputations: preliminary findings and hypotheses regarding causality. J Trauma (2010) 69(Suppl 1):S116–22. doi:10.1097/TA.0b013e3181e44cc7
34. Cadosch D, Toffoli AM, Gautschi OP, Frey SP, Zellweger R, Skirving AP, et al. Serum after traumatic brain injury increases proliferation and supports expression of osteoblast markers in muscle cells. J Bone Joint Surg Am (2010) 92(3):645–53. doi:10.2106/JBJS.I.00097
35. Wang H, Lindborg C, Lounev V, Kim JH, McCarrick-Walmsley R, Xu M, et al. Cellular hypoxia promotes heterotopic ossification by amplifying BMP signaling. J Bone Miner Res (2016) 31(9):1652–65. doi:10.1002/jbmr.2848
36. Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet (2006) 38(5):525–7. doi:10.1038/ng1783
37. Shi L, Sun W, Gao F, Cheng L, Li Z. Heterotopic ossification related to the use of recombinant human BMP-2 in osteonecrosis of femoral head. Medicine (2017) 96(27):e7413. doi:10.1097/MD.0000000000007413
38. Liu K, Tripp S, Layfield LJ. Heterotopic ossification: review of histologic findings and tissue distribution in a 10-year experience. Pathol Res Pract (2007) 203(9):633–40. doi:10.1016/j.prp.2007.05.014
39. Lazard ZW, Olmsted-Davis EA, Salisbury EA, Gugala Z, Sonnet C, Davis EL, et al. Osteoblasts have a neural origin in heterotopic ossification. Clin Orthop Relat Res (2015) 473(9):2790–806. doi:10.1007/s11999-015-4323-9
40. Agarwal S, Drake J, Qureshi AT, Loder S, Li S, Shigemori K, et al. Characterization of cells isolated from genetic and trauma-induced heterotopic ossification. PLoS One (2016) 11(8):e0156253. doi:10.1371/journal.pone.0156253
41. Wang Y, Wan C, Deng L, Liu X, Cao X, Gilbert SR, et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest (2007) 117(6):1616–26. doi:10.1172/JCI31581
42. Riddle RC, Khatri R, Schipani E, Clemens TL. Role of hypoxia-inducible factor-1alpha in angiogenic-osteogenic coupling. J Mol Med (2009) 87(6):583–90. doi:10.1007/s00109-009-0477-9
43. Zelzer E, Mamluk R, Ferrara N, Johnson RS, Schipani E, Olsen BR. VEGFA is necessary for chondrocyte survival during bone development. Development (2004) 131(9):2161–71. doi:10.1242/dev.01053
44. Carlevaro MF, Cermelli S, Cancedda R, Descalzi Cancedda F. Vascular endothelial growth factor (VEGF) in cartilage neovascularization and chondrocyte differentiation: auto-paracrine role during endochondral bone formation. J Cell Sci (2000) 113(1):59–69.
45. Ortega N, Behonick DJ, Werb Z. Matrix remodeling during endochondral ossification. Trends Cell Biol (2004) 14(2):86–93. doi:10.1016/j.tcb.2003.12.003
46. Maes C, Araldi E, Haigh K, Khatri R, Van Looveren R, Giaccia AJ, et al. VEGF-independent cell-autonomous functions of HIF-1alpha regulating oxygen consumption in fetal cartilage are critical for chondrocyte survival. J Bone Miner Res (2012) 27(3):596–609. doi:10.1002/jbmr.1487
47. Pfander D, Cramer T, Schipani E, Johnson RS. HIF-1alpha controls extracellular matrix synthesis by epiphyseal chondrocytes. J Cell Sci (2003) 116(Pt 9):1819–26. doi:10.1242/jcs.00385
48. Wang Y, Roche O, Yan MS, Finak G, Evans AJ, Metcalf JL, et al. Regulation of endocytosis via the oxygen-sensing pathway. Nat Med (2009) 15(3):319–24. doi:10.1038/nm.1922
49. Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev (2001) 15(21):2865–76. doi:10.1101/gad.934301
50. Cheng S, Xing W, Pourteymoor S, Schulte J, Mohan S. Conditional deletion of prolyl hydroxylase domain-containing protein 2 (Phd2) gene reveals its essential role in chondrocyte function and endochondral bone formation. Endocrinology (2016) 157(1):127–40. doi:10.1210/en.2015-1473
51. Locher RJ, Lunnemann T, Garbe A, Schaser K, Schmidt-Bleek K, Duda G, et al. Traumatic brain injury and bone healing: radiographic and biomechanical analyses of bone formation and stability in a combined murine trauma model. J Musculoskelet Neuronal Interact (2015) 15(4):309–15.
52. Giannoudis PV, Mushtaq S, Harwood P, Kambhampati S, Dimoutsos M, Stavrou Z, et al. Accelerated bone healing and excessive callus formation in patients with femoral fracture and head injury. Injury (2006) 37(Suppl 3):S18–24. doi:10.1016/j.injury.2006.08.020
53. Newman RJ, Stone MH, Mukherjee SK. Accelerated fracture union in association with severe head injury. Injury (1987) 18(4):241–6. doi:10.1016/0020-1383(87)90006-4
54. Perkins R, Skirving AP. Callus formation and the rate of healing of femoral fractures in patients with head injuries. J Bone Joint Surg Br (1987) 69(4):521–4.
55. Garland DE, Rothi B, Waters RL. Femoral fractures in head-injuries adults. Clin Orthop Relat Res (1982) (166):219–25.
56. Lee SK, Lorenzo J. Cytokines regulating osteoclast formation and function. Curr Opin Rheumatol (2006) 18(4):411–8. doi:10.1097/01.bor.0000231911.42666.78
57. Boes M, Kain M, Kakar S, Nicholls F, Cullinane D, Gerstenfeld L, et al. Osteogenic effects of traumatic brain injury on experimental fracture-healing. J Bone Joint Surg Am (2006) 88(4):738–43. doi:10.2106/JBJS.D.02648
58. Eid K, Labler L, Ertel W, Trentz O, Keel M. Systemic effects of severe trauma on the function and apoptosis of human skeletal cells. J Bone Joint Surg Br (2006) 88(10):1394–400. doi:10.1302/0301-620X.88B10.17139
59. van Kuijk AA, Geurts AC, van Kuppevelt HJ. Neurogenic heterotopic ossification in spinal cord injury. Spinal Cord (2002) 40(7):313–26. doi:10.1038/sj.sc.3101309
60. Trentz OA, Handschin AE, Bestmann L, Hoerstrup SP, Trentz OL, Platz A. Influence of brain injury on early posttraumatic bone metabolism. Crit Care Med (2005) 33(2):399–406. doi:10.1097/01.CCM.0000152221.87477.21
61. Bidner SM, Rubins IM, Desjardins JV, Zukor DJ, Goltzman D. Evidence for a humoral mechanism for enhanced osteogenesis after head injury. J Bone Joint Surg Am (1990) 72(8):1144–9. doi:10.2106/00004623-199072080-00004
62. Wildburger R, Zarkovic N, Leb G, Borovic S, Zarkovic K, Tatzber F. Post-traumatic changes in insulin-like growth factor type 1 and growth hormone in patients with bone fractures and traumatic brain injury. Wien Klin Wochenschr (2001) 113(3–4):119–26.
63. Zhuang YF, Li J. Serum EGF and NGF levels of patients with brain injury and limb fracture. Asian Pac J Trop Med (2013) 6(5):383–6. doi:10.1016/S1995-7645(13)60043-7
64. Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science (1995) 269(5223):546–9. doi:10.1126/science.7624778
65. Steppan CM, Crawford DT, Chidsey-Frink KL, Ke H, Swick AG. Leptin is a potent stimulator of bone growth in ob/ob mice. Regul Pept (2000) 92(1–3):73–8. doi:10.1016/S0167-0115(00)00152-X
66. Yan H, Zhang HW, Fu P, Liu BL, Jin WZ, Duan SB, et al. Leptin’s effect on accelerated fracture healing after traumatic brain injury. Neurol Res (2013) 35(5):537–44. doi:10.1179/1743132813Y.0000000201
67. Wei Y, Wang L, Clark JC, Dass CR, Choong PF. Elevated leptin expression in a rat model of fracture and traumatic brain injury. J Pharm Pharmacol (2008) 60(12):1667–72. doi:10.1211/jpp/60.12.0013
68. Wang L, Yuan JS, Zhang HX, Ding H, Tang XG, Wei YZ. Effect of leptin on bone metabolism in rat model of traumatic brain injury and femoral fracture. Chin J Traumatol (2011) 14(1):7–13.
69. Graef F, Seemann R, Garbe A, Schmidt-Bleek K, Schaser KD, Keller J, et al. Impaired fracture healing with high non-union rates remains irreversible after traumatic brain injury in leptin-deficient mice. J Musculoskelet Neuronal Interact (2017) 17(2):78–85.
70. Turner RT, Kalra SP, Wong CP, Philbrick KA, Lindenmaier LB, Boghossian S, et al. Peripheral leptin regulates bone formation. J Bone Miner Res (2013) 28(1):22–34. doi:10.1002/jbmr.1734
71. Gu XC, Zhang XB, Hu B, Zi Y, Li M. Neuropeptide Y accelerates post-fracture bone healing by promoting osteogenesis of mesenchymal stem cells. Neuropeptides (2016) 60:61–6. doi:10.1016/j.npep.2016.09.005
72. Baldock PA, Lee NJ, Driessler F, Lin S, Allison S, Stehrer B, et al. Neuropeptide Y knockout mice reveal a central role of NPY in the coordination of bone mass to body weight. PLoS One (2009) 4(12):e8415. doi:10.1371/journal.pone.0008415
73. Baldock PA, Sainsbury A, Allison S, Lin EJ, Couzens M, Boey D, et al. Hypothalamic control of bone formation: distinct actions of leptin and y2 receptor pathways. J Bone Miner Res (2005) 20(10):1851–7. doi:10.1359/JBMR.050523
74. Hayashi M, Nakashima T, Taniguchi M, Kodama T, Kumanogoh A, Takayanagi H. Osteoprotection by semaphorin 3A. Nature (2012) 485(7396):69–74. doi:10.1038/nature11000
75. Liu X, Zhou C, Li Y, Ji Y, Xu G, Wang X, et al. SDF-1 promotes endochondral bone repair during fracture healing at the traumatic brain injury condition. PLoS One (2013) 8(1):e54077. doi:10.1371/journal.pone.0054077
76. Brady RD, Shultz SR, Sun M, Romano T, van der Poel C, Wright DK, et al. Experimental Traumatic Brain Injury Induces Bone Loss in Rats. J Neurotrauma (2016) 33(23):2154–60. doi:10.1089/neu.2014.3836
77. Yu H, Wergedal JE, Rundle CH, Mohan S. Reduced bone mass accrual in mouse model of repetitive mild traumatic brain injury. J Rehabil Res Dev (2014) 51(9):1427–37. doi:10.1682/JRRD.2014.04.0095
78. Jorgensen L, Jacobsen BK, Wilsgaard T, Magnus JH. Walking after stroke: does it matter? Changes in bone mineral density within the first 12 months after stroke. A longitudinal study. Osteoporos Int (2000) 11(5):381–7. doi:10.1007/s001980070103
79. Poole KE, Loveridge N, Barker PJ, Halsall DJ, Rose C, Reeve J, et al. Reduced vitamin D in acute stroke. Stroke (2006) 37(1):243–5. doi:10.1161/01.STR.0000195184.24297.c1
80. Sato Y, Asoh T, Kondo I, Satoh K. Vitamin D deficiency and risk of hip fractures among disabled elderly stroke patients. Stroke (2001) 32(7):1673–7. doi:10.1161/01.STR.32.7.1673
81. Hochwald O, Harman-Boehm I, Castel H. Hypovitaminosis D among inpatients in a sunny country. Isr Med Assoc J (2004) 6(2):82–7.
82. Rubin MR, Bilezikian JP. Hypoparathyroidism: clinical features, skeletal microstructure and parathyroid hormone replacement. Arq Bras Endocrinol Metabol (2010) 54(2):220–6. doi:10.1590/S0004-27302010000200019
83. Ruohola JP, Laaksi I, Ylikomi T, Haataja R, Mattila VM, Sahi T, et al. Association between serum 25(OH)D concentrations and bone stress fractures in Finnish young men. J Bone Miner Res (2006) 21(9):1483–8. doi:10.1359/jbmr.060607
84. Davey T, Lanham-New SA, Shaw AM, Hale B, Cobley R, Berry JL, et al. Low serum 25-hydroxyvitamin D is associated with increased risk of stress fracture during Royal Marine recruit training. Osteoporos Int (2016) 27(1):171–9. doi:10.1007/s00198-015-3228-5
85. Greve MW, Zink BJ. Pathophysiology of traumatic brain injury. Mt Sinai J Med (2009) 76(2):97–104. doi:10.1002/msj.20104
86. Laskowski RA, Creed JA, Raghupathi R. Frontiers in neuroengineering pathophysiology of mild TBI: implications for altered signaling pathways. In: Kobeissy FH, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects, Chapter 4. Boca Raton, FL: CRC Press/Taylor & Francis (c) 2015 by Taylor & Francis Group, LLC (2015).
87. Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics (2010) 7(1):22–30. doi:10.1016/j.nurt.2009.10.016
88. Lu J, Goh SJ, Tng PY, Deng YY, Ling EA, Moochhala S. Systemic inflammatory response following acute traumatic brain injury. Front Biosci (Landmark Ed) (2009) 14:3795–813. doi:10.2741/3489
89. Sims NA. Cell-specific paracrine actions of IL-6 family cytokines from bone, marrow and muscle that control bone formation and resorption. Int J Biochem Cell Biol (2016) 79:14–23. doi:10.1016/j.biocel.2016.08.003
90. Ciaramella A, Della Vedova C, Salani F, Viganotti M, D’Ippolito M, Caltagirone C, et al. Increased levels of serum IL-18 are associated with the long-term outcome of severe traumatic brain injury. Neuroimmunomodulation (2014) 21(1):8–12. doi:10.1159/000354764
91. Sorby-Adams AJ, Marcoionni AM, Dempsey ER, Woenig JA, Turner RJ. The role of neurogenic inflammation in blood-brain barrier disruption and development of cerebral oedema following acute central nervous system (CNS) injury. Int J Mol Sci (2017) 18(8):1788. doi:10.3390/ijms18081788
92. Mesquita J, Varela A, Medina JL. Trauma and the endocrine system. Endocrinol Nutr (2010) 57(10):492–9. doi:10.1016/j.endonu.2010.06.012
93. Formisano R, Grelli S, Matteucci C, Santilli VV, Vinicola VV, Scivoletto G, et al. Immunological and endocrinological disturbances in patients after prolonged coma following head injury. Eur J Neurol (1998) 5(2):151–8. doi:10.1046/j.1468-1331.1998.520151.x
94. Hari Kumar KVS, Swamy MN, Khan MA. Prevalence of hypothalamo pituitary dysfunction in patients of traumatic brain injury. Indian J Endocrinol Metab (2016) 20(6):772–8. doi:10.4103/2230-8210.192917
95. Scranton RA, Baskin DS. Impaired pituitary axes following traumatic brain injury. J Clin Med (2015) 4(7):1463–79. doi:10.3390/jcm4071463
96. Schneider HJ, Schneider M, Saller B, Petersenn S, Uhr M, Husemann B, et al. Prevalence of anterior pituitary insufficiency 3 and 12 months after traumatic brain injury. Eur J Endocrinol (2006) 154(2):259–65. doi:10.1530/eje.1.02071
97. Kopczak A, Kilimann I, von Rosen F, Krewer C, Schneider HJ, Stalla GK, et al. Screening for hypopituitarism in 509 patients with traumatic brain injury or subarachnoid hemorrhage. J Neurotrauma (2014) 31(1):99–107. doi:10.1089/neu.2013.3002
98. Cuesta M, Hannon MJ, Crowley RK, Behan LA, Tormey W, Rawluk D, et al. Symptoms of gonadal dysfunction are more predictive of hypopituitarism than nonspecific symptoms in screening for pituitary dysfunction following moderate or severe traumatic brain injury. Clin Endocrinol (2016) 84(1):92–8. doi:10.1111/cen.12874
99. Leal-Cerro A, Flores JM, Rincon M, Murillo F, Pujol M, Garcia-Pesquera F, et al. Prevalence of hypopituitarism and growth hormone deficiency in adults long-term after severe traumatic brain injury. Clin Endocrinol (2005) 62(5):525–32. doi:10.1111/j.1365-2265.2005.02250.x
100. Zheng P, He B, Tong W. Dynamic pituitary hormones change after traumatic brain injury. Neurol India (2014) 62(3):280–4. doi:10.4103/0028-3886.136922
101. Srinivasan L, Roberts B, Bushnik T, Englander J, Spain DA, Steinberg GK, et al. The impact of hypopituitarism on function and performance in subjects with recent history of traumatic brain injury and aneurysmal subarachnoid haemorrhage. Brain Inj (2009) 23(7):639–48. doi:10.1080/02699050902970778
102. Olney RC. Regulation of bone mass by growth hormone. Med Pediatr Oncol (2003) 41(3):228–34. doi:10.1002/mpo.10342
103. Bouillon R, Prodonova A. Growth and hormone deficiency and peak bone mass. J Pediatr Endocrinol Metab (2000) 13(Suppl 6):1327–36. doi:10.1515/jpem-2000-s604
104. Mormando M, Chiloiro S, Bianchi A, Giampietro A, Angelini F, Tartaglione L, et al. Growth hormone receptor isoforms and fracture risk in adult-onset growth hormone-deficient patients. Clin Endocrinol (2016) 85(5):717–24. doi:10.1111/cen.13161
105. Weiss S, Henle P, Bidlingmaier M, Moghaddam A, Kasten P, Zimmermann G. Systemic response of the GH/IGF-I axis in timely versus delayed fracture healing. Growth Horm IGF Res (2008) 18(3):205–12. doi:10.1016/j.ghir.2007.09.002
106. Andreassen TT, Oxlund H. Local anabolic effects of growth hormone on intact bone and healing fractures in rats. Calcif Tissue Int (2003) 73(3):258–64. doi:10.1007/s00223-002-2074-6
107. Bail HJ, Raschke MJ, Kolbeck S, Krummrey G, Windhagen HJ, Weiler A, et al. Recombinant species-specific growth hormone increases hard callus formation in distraction osteogenesis. Bone (2002) 30(1):117–24. doi:10.1016/S8756-3282(01)00628-7
108. Abe E, Marians RC, Yu W, Wu XB, Ando T, Li Y, et al. TSH is a negative regulator of skeletal remodeling. Cell (2003) 115(2):151–62. doi:10.1016/S0092-8674(03)00771-2
109. Dumic-Cule I, Draca N, Luetic AT, Jezek D, Rogic D, Grgurevic L, et al. TSH prevents bone resorption and with calcitriol synergistically stimulates bone formation in rats with low levels of calciotropic hormones. Horm Metab Res (2014) 46(5):305–12. doi:10.1055/s-0033-1363989
110. Locatelli V, Bianchi VE. Effect of GH/IGF-1 on bone metabolism and osteoporsosis. Int J Endocrinol (2014) 2014:235060. doi:10.1155/2014/235060
111. Kesavan CBN, Watt H, Mohan S. Experimental repetitive mild traumatic brain injury induces deficits in trabecular bone microarchitecture and strength in mice. Bone Res (2017) 5:17042. doi:10.1038/boneres.2017.42
112. Hatton J, Kryscio R, Ryan M, Ott L, Young B. Systemic metabolic effects of combined insulin-like growth factor-I and growth hormone therapy in patients who have sustained acute traumatic brain injury. J Neurosurg (2006) 105(6):843–52. doi:10.3171/jns.2006.105.6.843
Keywords: osteoporosis, growth hormone, bone formation, bone resorption, heterotopic ossification, fracture repair, neuropeptides
Citation: Bajwa NM, Kesavan C and Mohan S (2018) Long-term Consequences of Traumatic Brain Injury in Bone Metabolism. Front. Neurol. 9:115. doi: 10.3389/fneur.2018.00115
Received: 08 December 2017; Accepted: 15 February 2018;
Published: 05 March 2018
Edited by:
Elham Rostami, Academic Hospital, SwedenReviewed by:
Eric Peter Thelin, University of Cambridge, United KingdomRita Formisano, Fondazione Santa Lucia (IRCCS), Italy
Marco Fidel Avila-Rodriguez, Universidad del Tolima, Colombia
Copyright: © 2018 Bajwa, Kesavan and Mohan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Subburaman Mohan, c3ViYnVyYW1hbi5tb2hhbiYjeDAwMDQwO3ZhLmdvdg==