 Paola Imbriani
Paola Imbriani Tommaso Schirinzi
Tommaso Schirinzi Maria Meringolo
Maria Meringolo Nicola B. Mercuri
Nicola B. Mercuri Antonio Pisani
Antonio Pisani- 1Department of Systems Medicine, University of Rome “Tor Vergata”, Rome, Italy
- 2Laboratory of Neurophysiology and Plasticity, Fondazione Santa Lucia (IRCCS), Rome, Italy
Significant advances have been made in the understanding of the numerous mechanisms involved in Parkinson’s disease (PD) pathogenesis. The identification of PD pathogenic mutations and the use of different animal models have contributed to better elucidate the processes underlying the disease. Here, we report a brief survey of some relevant cellular mechanisms, including autophagic–lysosomal dysfunction, endoplasmic reticulum stress, and mitochondrial impairment, with the main aim to focus on their potential convergent roles in determining early alterations at the synaptic level, mainly consisting in a decrease in dopamine release at nigrostriatal terminals and loss of synaptic plasticity at corticostriatal synapses. In a number of experimental models, this synaptopathy has been shown to be an initial, central event in PD pathogenesis, preceding neuronal damage, thereby representing a valuable tool for testing potential disease-modifying treatments.
Introduction
Parkinson’s disease (PD) is a common neurodegenerative disorder, currently affecting 1% of the population above 60 years (1) and characterized by progressive motor deficits, including akinesia (or bradykinesia), rigidity, resting tremor, and postural instability (2). The neuropathological hallmarks of PD are the progressive loss of substantia nigra pars compacta (SNpc) dopaminergic neurons (DAns) and the presence of intraneuronal α-synuclein cytoplasmic inclusions, termed Lewy bodies (3). In the past two decades, a number of pathogenic mutations associated with PD have been identified (4), improving our understanding of pathogenic disease mechanisms. Many PD-related genes, such as SNCA, PINK1, GBA1, have a crucial role in different cellular mechanisms that have proven to be involved in PD, including autophagy/lysosome pathway, endoplasmic reticulum (ER) stress, and mitochondrial impairment (5). In this regard, the purpose of this review is to provide a brief overview of the most relevant pathogenic mechanisms, but with a specific focus on clues supporting early synaptic dysfunction as a functional and structural event that could represent a final convergent phenomenon for multiple distinct processes. Comprehensive review of PD-related pathogenic mechanisms is beyond the scope of this survey, and we refer the readers to other recent excellent reviews (5, 6).
Synaptopathy in PD
Despite considerable progress in our understanding of the aberrant mechanisms involved in PD pathogenesis, some key questions remain unanswered. Among these, a central issue is to establish the precise sequence of events at the cellular level and where the pathogenic process begins. Multiple lines of evidence suggest that the primary site of α-synucleinopathy is represented by the synaptic terminal, with the occurrence of an early synaptic impairment that precedes axon degeneration and with subsequent retrograde progression through a dying-back mechanism (7) (Figure 1). This “synaptopathy” described at the cellular level could correspond to an early, presymptomatic time window in patients, when only a 30–50% decrease in striatal dopamine levels can be detected, providing evidence for a powerful ability of the motor system to compensate (8). The concept of synaptopathy is closely related to α-synuclein, the major constituent of Lewy body, which, in physiological conditions, is primarily localized to the presynaptic terminals, where it affects the fusion and clustering of synaptic vesicles, thus influencing neurotransmitter release (9–11). Evidence on how α-synuclein plays a crucial role in synaptic function and plasticity comes from several studies on animal models. In a transgenic mouse model of α-synucleinopathy (αSyn 1-120 mice), α-synuclein aggregates were detected at striatal dopaminergic terminals, with an impairment of dopamine release from nigrostriatal synaptic terminals, even in the absence of nigral DAn loss (12). In another model, a bacterial artificial chromosome (BAC) transgenic mouse with overexpression of human wild-type α-synuclein (SNCA-OVX), a clear time-dependent progression was observed: in 3-month-old mice, in spite of the absence of overt neuropathology, early deficits in dopamine release in the dorsal striatum and increased clustering of vesicles in dopamine terminals were found (13). Conversely, at 18 months, mice showed motor deficits, loss of dopamine neurons, and a reduced firing rate in the remaining SNpc dopamine neurons, further indicating synaptic dysfunction as an early event. Of relevance, this feature does not apply only to α-synuclein models. Many other PD animal models, including those based on the administration of the “classical” neurotoxins 6-OHDA and MPTP, and the ones with genetic mutations not involving α-synuclein, have contributed to identify synaptic dysfunctions occurring at early stages of the disease. In a 6-OHDA model of early PD with partial denervation and mild motor alterations, the decreased level of dopamine observed was responsible for a selective impairment of corticostriatal synaptic plasticity recorded from spiny projection neurons (SPNs), with a specific deficit of long-term potentiation (LTP) and with sparing of long-term depression (LTD) (14). Similar corticostriatal synaptic plasticity impairments were also found by Chou et al. (15), who performed electrophysiological recordings from SPNs and from SNpc dopaminergic cells of 8–9-month-old LRRK2 (G2019S mutation) transgenic mice. LRRK2 is a multidomain protein with kinase activity, whose mutations are involved in autosomal dominant forms of PD (16). The function of LRRK2 has not been fully elucidated, although strong evidence implicates a role in intracellular trafficking, vesicular recycling, and modulation of synaptic transmission (17). In line with this, Chou et al. identified an early decrease in spontaneous firing frequency of SNpc dopaminergic cells, without gross degeneration of nigrostriatal terminals, and impaired evoked dopamine release, with subsequent deficit in LTD induction in striatal neurons (15). Moreover, a recent study on LRRK2 BAC transgenic rats revealed alterations to dopamine circuit function, in the form of L-DOPA-responsive motor dysfunction, a reduction in SNpc dopamine neurons burst firing, and an impaired striatal dopamine release, occurring in the absence of neurodegeneration or abnormal protein accumulation (18). Besides the examples reported so far, the concept of synaptopathy can be extended to various PD models and appears to be linked to different cellular mechanisms, as discussed in the following sections.
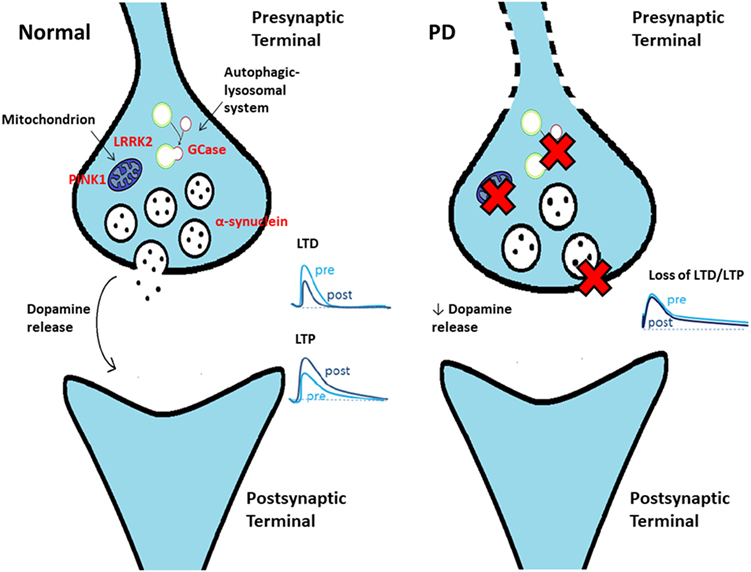
Figure 1. Schematic cartoon showing a synaptic terminal in control conditions (left) and in Parkinson’s disease (PD) (right). Several mechanisms contribute to early dysfunction at the terminal level, causing an impaired dopamine release. As a consequence, synaptic plasticity at corticostriatal synapses [long-term depression (LTD) and long-term potentiation (LTP)] is impaired in PD.
Dysfunctional Autophagy–Lysosome System and the Role of GBA1 Mutations
Autophagy, acting through lysosomal degradation, represents the main proteolytic system in neurons (19). An abnormal autophagic activity leads to the accumulation of aberrant proteins and toxic components (20), contributing to the neurodegeneration observed in several diseases, including PD (21). In PD, dysfunctional autophagy is responsible for the accumulation of α-synuclein: specifically, two types of autophagy (macroautophagy and chaperone-mediated autophagy), both involved in α-synuclein degradation, appear to be impaired in PD (22). In addition, growing evidence indicates that excessive α-synuclein itself blocks these degradation pathways, promoting α-synuclein aggregation (23). Experimental findings show that Lewy body-like aggregates are able to resist to macroautophagy degradation by impairing clearance of autophagosomes (24) and that two α-synuclein familial mutations, SNCA-A30P and SNCA-A53T, can alter the chaperone-mediated autophagy pathway (25). In the recent past, a growing body of evidence suggests a prominent role for decreased glucocerebrosidase (GCase) activity in autophagic failure and subsequent α-synuclein accumulation in PD (26). GCase is a lysosomal enzyme encoded by the GBA1 gene, ubiquitously expressed in the brain with some regional variations (27), whose homozygous or heterozygous compound mutations cause Gaucher disease (GD), with the accumulation of glycolipid substrate (28). Of note, heterozygous GBA1 mutations represent the most relevant risk factor for PD, since they can be found in approximately 5–10% of idiopathic PD patients (29). The majority of GBA1 mutations are associated with reduced GCase levels (30), with milder mutations responsible for slightly diminished enzyme levels, conferring a much lower risk of PD than mutations causing severe enzymatic dysfunction (31). The molecular mechanisms underlying the increased PD risk in GBA1 mutation carriers have not been fully clarified. A dual interplay has been proposed for GCase and α-synuclein. On the one hand, GCase loss-of-function would lead to aberrant lysosomal protein degradation and neurotoxicity, whereas on the other hand α-synuclein may inhibit the activity of normal GCase (32). Accordingly, in postmortem samples of PD patients without GBA1 mutations, a reduced GCase activity has been reported (33). The scenario is even more complex, considering that significant loss of GCase activity can cause neurodegeneration even in the absence of α-synuclein (34). To better elucidate the precise mechanisms linking GBA1 with parkinsonism, a number of disease models have been developed, including both animal models and cell-based models. GBA1 knockout mice and transgenic mouse lines carrying GBA1 point mutations well recapitulate GD phenotype, with accumulation of α-synuclein and ubiquitinated proteins, the presence of typical “Gaucher cells” and inflammation (28), but they also prove to be appropriate for the study of the effects of GCase reduction in PD pathogenesis. In the knockout mice, for instance, the lysosomal defect demonstrated in neurons and astrocytes lacking GBA1 was correlated with dysfunctional and fragmented mitochondria, pointing out a possible relationship between decreased GCase activity and impaired mitophagy, due to the inhibition of the degradation of mitochondria by the autophagy–lysosome pathway (35). However, it is important to note that GBA1 mutations do not act only through defective lysosome pathway: on the contrary, GCase loss-of-function may be associated with multiple other pathogenic cellular mechanisms, including ER stress, calcium metabolism dysregulation, and neuroinflammation (36), that, altogether, might contribute to the aggregation of misfolded α-synuclein. In this context, any disease-modifying therapy designed to increase GCase levels would act at different pathogenic levels, with the final target to slow down the progressive aggregation of α-synuclein (37, 38). In view of the close linkage between GCase function and α-synuclein deposition, it could be questioned whether this lysosomal enzyme deficiency can also affect synaptic function. A relatively recent experimental study gives interesting insights about this issue: in a murine model of PD, where a subchronic conduritol-β-epoxide exposure induced GCase inhibition, Ginns and colleagues identified an early synaptic impairment, in the form of a reduction of striatal evoked dopamine release and altered synaptic plasticity markers, including post-synaptic density size and miRNA expression levels, together with glial activation within nigrostriatal pathway and abnormal α-synuclein accumulation (39). The effects of GBA1 insufficiency on dopaminergic neurotransmission and synaptic function documented in this animal model fit with clinical observations. Indeed, patients carrying GBA1 mutations show early striatal presynaptic dopaminergic dysfunction even before the onset of motor symptoms (40, 41). Further studies of individuals carrying a mutant GBA1 allele, together with the development and characterization of different GBA1 models, will help to clarify the mechanisms underlying the Parkinson’s–GD connection and to provide novel insights into the influence of diminished GCase activity on synaptic transmission, which could lead to develop novel therapeutic interventions.
ER Stress
The ER represents a quality control system to check the correct protein folding, while misfolded or unfolded proteins are directed toward cytosol for the degradation by ER-associated degradation system (42). The accumulation of misfolded proteins inside the ER lumen, defined ER stress, is a toxic process to which the cell reacts by activating the unfolded protein response (UPR) (43), with the aim to restore ER homeostasis. Conversely, if the adaptive response is insufficient, the cell undergoes apoptosis. The involvement of ER stress has been demonstrated in several neurodegenerative conditions, including PD (44), and many reports prove the role of some PD-related genes in this cellular process. For example, differentiated PC12 cells with expression of A53T mutant α-synuclein show decreased proteasome activity and increased ER stress (45). Moreover, a recent study performed on iPSC-derived DAns carrying GBA-N370S mutation demonstrates the activation of UPR with upregulation of ER-resident chaperones (46). Given the existence of an interplay between mitochondrial and ER stress (47), also mitochondrial proteins such as Parkin and PTEN-induced putative kinase 1 (PINK1) have a crucial role in this mechanism. Activation of ER stress, mediated by mitofusin bridges occurring between defective mitochondria and the ER, has been reported in Drosophila PINK1 and Parkin mutants (48). Despite the growing interest in this field, the contribution of ER stress to neuronal death still needs further investigation, in view of a potential application in PD therapeutics (49). In this regard, interesting inputs come from a very recent study performed on three different rodent models of PD, in which the authors described an activation of RNA-like ER kinase (PERK) signaling, as well as in postmortem brain tissue derived from parkinsonian patients (50). PERK is a crucial ER stress sensor, whose chronic signaling blocks the translation of essential synaptic proteins, impacting neuronal survival and synaptic function. In these experimental settings, PERK inhibition exerted a neuroprotective effect, as evidenced by an increase in dopamine levels and in the expression of synaptic proteins. This once again highlights the relevance of synaptopathy in multiple aberrant mechanisms in PD.
Mitochondrial Impairment
Most neurodegenerative diseases share a mitochondrial impairment as a major pathophysiological hallmark. Mitochondria are the main source of chemical energy for the cell and, as a consequence, their dysfunction leads to decreased levels of ATP and production of reactive oxygen species, which negatively impact neuronal physiology and, ultimately, cell survival (49). An impaired mitochondrial complex I activity has been demonstrated in the SNpc of PD patients (51). It is well established that many environmental toxins act as potent mitochondrial complex I inhibitors, and accordingly, different toxin-based experimental models of PD have been developed in order to reproduce mitochondrial dysfunction. MPTP, acting through its metabolite MPP+, causes loss of dopaminergic SNpc neurons, inducing parkinsonian features in mice and non-human primates (52). The same toxin has been used to develop MPTP-treated models with only partial dopaminergic deafferentation, in which an early reduction in spine density in both the caudate nucleus and putamen could be detected, as an early pathological hallmark of the disease (53). Rotenone, a largely used pesticide, is another mitochondrial complex I toxin, and its administration reproduces many histochemical and behavioral features of human PD in rodents and non-human primates, including selective nigrostriatal dopaminergic lesions and α-synuclein-positive cytoplasmic aggregates in nigral neurons (54). Such experimental evidence has been further confirmed by a number of epidemiological studies (55). Mitochondrial dysfunction and oxidative stress are tightly connected to PINK1, a serine/threonine kinase located in the intermembrane mitochondrial space and involved in the mitophagic pathway (56). PINK1 loss-of-function mutations are linked to inherited early-onset forms of PD (57). In a mouse model carrying heterozygous PINK1 mutations, we identified early rearrangement within corticostriatal circuitry, expressed by selective impairment of LTP with a physiologically expressed LTD, in the absence of motor phenotype and dopaminergic neuronal loss (58). These observations were in line with the results of neuroimaging and physiological studies performed on PINK1 heterozygous mutation carriers manifesting initial alterations in the nigrostriatal circuit (59, 60). Accordingly, the heterozygous condition related to familial parkinsonism represents an ideal preclinical model, which allows us to study early alterations occurring before the onset of motor signs, in a time window suitable to test potential novel disease modifying therapy (61). A number of attempts have been made to recreate the gene–environment interaction that might underlie disease pathogenesis. Recently, we exposed PINK1 heterozygous knockout mice to rotenone, which was chronically administered at very low doses. Of interest, combination of gene mutation with minimal rotenone exposure was able to cause severe alterations of corticostriatal synaptic plasticity, to an extent similar to that observed in the PINK1 homozygous knockout model (62, 63). The experimental use of toxins inducing mitochondrial impairment has contributed over the past decades to improve our knowledge on the pathogenic mechanisms of neurodegenerative diseases, in an attempt to develop neuroprotective agents and etiologic treatments.
Conclusion and Future Directions
Over the past decades, major advances have been made in the understanding of the mechanisms involved in PD pathogenesis and the mechanisms that, furthermore, share common elements and contribute synergistically to neuronal dysfunction. Growing evidence attributes an undisputed central role to α-synuclein aggregation, also in view of its interaction with multiple processes, including intracellular trafficking, mitochondrial dysfunction, ER stress, and lysosomal dysfunction. However, there remain many unclear issues. The captivating prion-like hypothesis, according to which aggregated α-synuclein is trans-synaptically spread through the brain connectome, is still debated, as it does not fully recapitulate PD pathogenesis (64). Indeed, the pattern of spreading of Lewy-body pathology does not precisely match Braak’s theory, according to a number of studies examining postmortem PD samples (65). Yet, at cellular level, the pattern of distribution of α-synuclein aggregates appears to spare brainstem GABAergic neurons (66). Such evidence highlights some limitations to the “prion-like” theory and supports the need for a more comprehensive hypothesis that could take into consideration the selective neuronal susceptibility (64). In this complex scenario, many experimental models point toward the synapse as the primary site of PD pathology and considering synapse failure as a putative common denominator. Indeed, synaptopathy is an early event in PD pathogenesis in most phenotypic and genetic models reported so far (Table 1). Impairment of synaptic activity and plasticity at corticostriatal synapses represents a peculiar endophenotype, in distinct models of human movement disorders (67–69). In addition, these alterations have been shown to parallel time-dependent progression of cellular demise, thereby mimicking a very important disease stage, where potential disease-modifying treatments could be tested. Understanding the molecular events leading to synaptic dysfunction, achieved by the use of suitable PD animal models, will encourage the development of potential synapse-target therapies, in the hope of actively intervene on one of the mechanisms leading to PD pathogenesis.
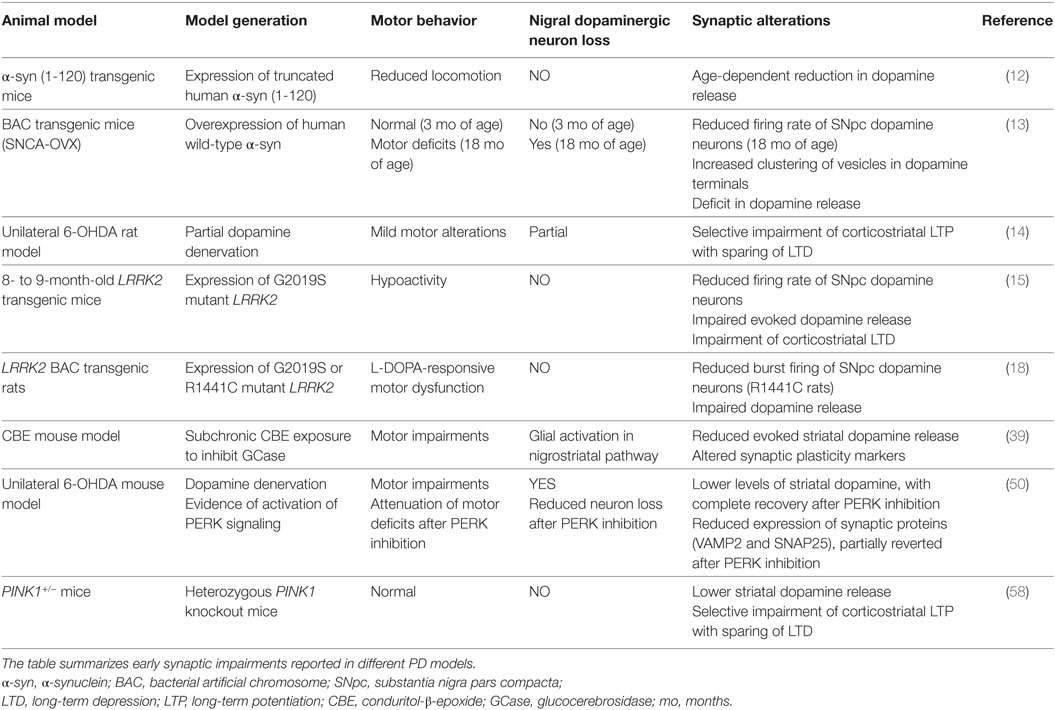
Table 1. Synaptopathy in different animal models of Parkinson’s disease.
Author Contributions
PI and AP designed the study and wrote the paper. TS and MM prepared illustrations and revised the text. NM revised the text.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was partially supported by Ministero dell’Istruzione, dell’Università e della Ricerca (PRIN 2015).
References
1. Tysnes OB, Storstein A. Epidemiology of Parkinson’s disease. J Neural Transm (Vienna) (2017) 124:901–5. doi:10.1007/s00702-017-1686-y
2. Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry (2008) 79:368–76. doi:10.1136/jnnp.2007.131045
3. Dauer W, Przedborski S. Parkinson’s disease: mechanisms and model. Neuron (2003) 39:889–909. doi:10.1016/S0896-6273(03)00568-3
4. Puschmann A. Monogenic Parkinson’s disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord (2013) 19:407–15. doi:10.1016/j.parkreldis.2013.01.020
5. Cherubini M, Wade-Martins R. Convergent pathways in Parkinson’s disease. Cell Tissue Res (2017). doi:10.1007/s00441-017-2700-2
6. Surmeier DJ, Halliday GM, Simuni T. Calcium, mitochondrial dysfunction and slowing the progression of Parkinson’s disease. Exp Neurol (2017) 298:202–9. doi:10.1016/j.expneurol.2017.08.001
7. Calo L, Wegrzynowicz M, Santivañez-Perez J, Grazia Spillantini M. Synaptic failure and α-synuclein. Mov Disord (2016) 31:169–77. doi:10.1002/mds.26479
8. Hilker R, Schweitzer K, Coburger S, Ghaemi M, Weisenbach S, Jacobs AH, et al. Nonlinear progression of Parkinson disease as determined by serial positron emission tomographic imaging of striatal fluorodopa F 18 activity. Arch Neurol (2005) 62(3):378–82. doi:10.1001/archneur.62.3.378
9. Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci (1988) 8:2804–15.
10. Picconi B, Piccoli G, Calabresi P. Synaptic dysfunction in Parkinson’s disease. Adv Exp Med Biol (2012) 970:553–72. doi:10.1007/978-3-7091-0932-8_24
11. Burré J, Sharma M, Südhof TC. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A (2014) 111:E4274–83. doi:10.1073/pnas.1416598111
12. Garcia-Reitbock P, Anichtchik O, Bellucci A, Iovino M, Ballini C, Fineberg E, et al. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain (2010) 133:2032–44. doi:10.1093/brain/awq132
13. Janezic S, Threlfell S, Dodson PD, Dowie MJ, Taylor TN, Potgieter D, et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci U S A (2013) 110:E4016–25. doi:10.1073/pnas.1309143110
14. Paillé V, Picconi B, Bagetta V, Ghiglieri V, Sgobio C, Di Filippo M, et al. Distinct levels of dopamine denervation differentially alter striatal synaptic plasticity and NMDA receptor subunit composition. J Neurosci (2010) 30:14182–93. doi:10.1523/JNEUROSCI.2149-10.2010
15. Chou JS, Chen CY, Chen YL, Weng YH, Yeh TH, Lu CS, et al. (G2019S) LRRK2 causes early-phase dysfunction of SNpc dopaminergic neurons and impairment of corticostriatal long-term depression in the PD transgenic mouse. Neurobiol Dis (2014) 68:190–9. doi:10.1016/j.nbd.2014.04.021
16. Li JQ, Tan L, Yu JT. The role of the LRRK2 gene in parkinsonism. Mol Neurodegener (2014) 9:47. doi:10.1186/1750-1326-9-47
17. Berwick DC, Harvey K. LRRK2: an éminence grise of Wnt-mediated neurogenesis? Front Cell Neurosci (2013) 7:82. doi:10.3389/fncel.2013.00082
18. Sloan M, Alegre-Abarrategui J, Potgieter D, Kaufmann AK, Exley R, Deltheil T, et al. LRRK2 BAC transgenic rats develop progressive, L-DOPA-responsive motor impairment, and deficits in dopamine circuit function. Hum Mol Genet (2016) 25:951–63. doi:10.1093/hmg/ddv628
19. Nikoletopoulou V, Papandreou ME, Tavernarakis N. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ (2015) 22:398–407. doi:10.1038/cdd.2014.204
20. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature (2006) 441:885–9. doi:10.1038/nature04724
21. Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature (2006) 441:880–4. doi:10.1038/nature04723
22. Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alphasynuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem (2008) 283:23542–56. doi:10.1074/jbc.M801992200
23. Moors T, Paciotti S, Chiasserini D, Calabresi P, Parnetti L, Beccari T, et al. Lysosomal dysfunction and α-synuclein aggregation in Parkinson’s disease: diagnostic links. Mov Disord (2016) 31:791–801. doi:10.1002/mds.26562
24. Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR, Lee VM. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem (2013) 288:15194–210. doi:10.1074/jbc.M113.457408
25. Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science (2004) 305:1292–5. doi:10.1126/science.1101738
26. Bae EJ, Yang NY, Lee C, Lee HJ, Kim S, Sardi SP, et al. Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and alpha-synuclein aggregation. Exp Mol Med (2015) 47:e188. doi:10.1038/emm.2015.84
27. Dopeso-Reyes IG, Sucunza D, Rico A, Pignataro D, Marín-Ramos D, Roda E. Glucocerebrosidase expression patterns in the non-human primate brain. Brain Struct Funct (2017) 223:343–55. doi:10.1007/s00429-017-1504-1
28. O’Regan G, deSouza RM, Balestrino R, Schapira AH. Glucocerebrosidase mutations in Parkinson disease. J Parkinsons Dis (2017) 7:411–22. doi:10.3233/JPD-171092
29. Schapira AH. Glucocerebrosidase and Parkinson’s disease: recent advances. Mol Cell Neurosci (2015) 66:37–42. doi:10.1016/j.mcn.2015.03.013
30. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson disease. N Engl J Med (2009) 361:1651–61. doi:10.1056/NEJMoa0901281
31. Swan M, Saunders-Pullman R. The association between beta-glucocerebrosidase mutations and parkinsonism. Curr Neurol Neurosci Rep (2013) 13:368. doi:10.1007/s11910-013-0368-x
32. Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell (2011) 146:37–52. doi:10.1016/j.cell.2011.06.001
33. Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain (2015) 138:2648–58. doi:10.1093/brain/awv179
34. Kinghorn KJ, Grönke S, Castillo-Quan JI, Woodling NS, Li L, Sirka E, et al. A drosophila model of neuronopathic Gaucher disease demonstrates lysosomal-autophagic defects and altered motor signalling and is functionally rescued by rapamycin. J Neurosci (2016) 36:11654–70. doi:10.1523/JNEUROSCI.4527-15.2016
35. Osellame LD, Rahim AA, Hargreaves IP, Gegg ME, Richard-Londt A, Brandner S, et al. Mitochondria and quality control defects in a mouse model of Gaucher disease – links to Parkinson’s disease. Cell Metab (2013) 17:941–53. doi:10.1016/j.cmet.2013.04.014
36. Gegg ME, Schapira AH. Mitochondrial dysfunction associated with glucocerebrosidase deficiency. Neurobiol Dis (2016) 90:43–50. doi:10.1016/j.nbd.2015.09.006
37. Sardi SP, Clarke J, Viel C, Chan M, Tamsett TJ, Treleaven CM, et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci U S A (2013) 110:3537–42. doi:10.1073/pnas.1220464110
38. Migdalska-Richards A, Daly L, Bezard E, Schapira AH. Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann Neurol (2016) 80:766–75. doi:10.1002/ana.24790
39. Ginns EI, Mak SK, Ko N, Karlgren J, Akbarian S, Chou VP, et al. Neuroinflammation and α-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol Genet Metab (2014) 111:152–62. doi:10.1016/j.ymgme.2013.12.003
40. Goker-Alpan O, Masdeu JC, Kohn PD, Ianni A, Lopez G, Groden C, et al. The neurobiology of glucocerebrosidase-associated parkinsonism: a positron emission tomography study of dopamine synthesis and regional cerebral blood flow. Brain (2012) 135:2440–8. doi:10.1093/brain/aws174
41. McNeill A, Wu RM, Tzen KY, Aguiar PC, Arbelo JM, Barone P, et al. Dopaminergic neuronal imaging in genetic Parkinson’s disease: insights into pathogenesis. PLoS One (2013) 8:e69190. doi:10.1371/journal.pone.0069190
42. McCaffrey K, Braakman I. Protein quality control at the endoplasmic reticulum. Essays Biochem (2016) 60:227–35. doi:10.1042/EBC20160003
43. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol (2012) 13:89–102. doi:10.1038/nrm3270
44. Remondelli P, Renna M. The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front Mol Neurosci (2017) 10:187. doi:10.3389/fnmol.2017.00187
45. Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, et al. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet (2005) 14:3801–11. doi:10.1093/hmg/ddi396
46. Fernandes HJ, Hartfield EM, Christian HC, Emmanoulidou E, Zheng Y, Booth H, et al. ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Reports (2016) 6:342–56. doi:10.1016/j.stemcr.2016.01.013
47. Bouman L, Schlierf A, Lutz AK, Shan J, Deinlein A, Kast J, et al. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ (2011) 18:769–82. doi:10.1038/cdd.2010.142
48. Celardo I, Costa AC, Lehmann S, Jones C, Wood N, Mencacci NE, et al. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis (2016) 7:e2271. doi:10.1038/cddis.2016.173
49. Goswami P, Joshi N, Singh S. Neurodegenerative signaling factors and mechanisms in Parkinson’s pathology. Toxicol In Vitro (2017) 43:104–12. doi:10.1016/j.tiv.2017.06.008
50. Mercado G, Castillo V, Soto P, López N, Axten JM, Sardi P, et al. Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol Dis (2018) 112:136–48. doi:10.1016/j.nbd.2018.01.004
51. Mann VM, Cooper JM, Daniel SE, Srai K, Jenner P, Marsden CD, et al. Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann Neurol (1994) 36:876–81. doi:10.1002/ana.410360612
52. Bovè J, Prou D, Perier C, Przedborski S. Toxin-induced models of Parkinson’s disease. NeuroRx (2005) 2:484–94. doi:10.1602/neurorx.2.3.484
53. Villalba RM, Lee H, Smith Y. Dopaminergic denervation and spine loss in the striatum of MPTP-treated monkeys. Exp Neurol (2009) 215:220–7. doi:10.1016/j.expneurol.2008.09.025
54. Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol (2003) 179:9–16. doi:10.1006/exnr.2002.8072
55. Johnson ME, Bobrovskaya L. An update on the rotenone models of Parkinson’s disease: their ability to reproduce the features of clinical disease and model gene-environment interactions. Neurotoxicology (2015) 46:101–16. doi:10.1016/j.neuro.2014.12.002
56. Park J, Lee G, Chung J. The PINK1-parkin pathway is involved in the regulation of mitochondrial remodeling process. Biochem Biophys Res Commun (2009) 378:518–23. doi:10.1016/j.bbrc.2008.11.086
57. Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol (2013) 9:445–54. doi:10.1038/nrneurol.2013.132
58. Madeo G, Schirinzi T, Martella G, Latagliata EC, Puglisi F, Shen J, et al. PINK1 heterozygous mutations induce subtle alterations in dopamine-dependent synaptic plasticity. Mov Disord (2014) 29:41–53. doi:10.1002/mds.25724
59. Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol (2007) 6:652–62. doi:10.1016/S1474-4422(07)70174-6
60. Eggers C, Schmidt A, Hagenah J, Brüggemann N, Klein JC, Tadic V, et al. Progression of subtle motor signs in PINK1 mutation carriers with mild dopaminergic deficit. Neurology (2010) 74:1798–805. doi:10.1212/WNL.0b013e3181e0f79c
61. Schirinzi T, Madeo G, Martella G, Maltese M, Picconi B, Calabresi P, et al. Early synaptic dysfunction in Parkinson’s disease: insights from animal models. Mov Disord (2016) 31:802–13. doi:10.1002/mds.26620
62. Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, et al. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A (2007) 104:11441–6. doi:10.1073/pnas.0702717104
63. Martella G, Madeo G, Maltese M, Vanni V, Puglisi F, Ferraro E, et al. Exposure to low-dose rotenone precipitates synaptic plasticity alterations in PINK1 heterozygous knockout mice. Neurobiol Dis (2016) 91:21–36. doi:10.1016/j.nbd.2015.12.020
64. Surmeier DJ, Obeso JA, Halliday GM. Parkinson’s disease is not simply a prion disorder. J Neurosci (2017) 37:9799–807. doi:10.1523/JNEUROSCI.1787-16.2017
65. Halliday G, McCann H, Shepherd C. Evaluation of the Braak hypothesis: how far can it explain the pathogenesis of Parkinson’s disease? Expert Rev Neurother (2012) 12:673–86. doi:10.1586/ern.12.47
66. Kingsbury AE, Bandopadhyay R, Silveira-Moriyama L, Ayling H, Kallis C, Sterlacci W, et al. Brain stem pathology in Parkinson’s disease: an evaluation of the Braak staging model. Mov Disord (2010) 25:2508–15. doi:10.1002/mds.23305
67. Quartarone A, Pisani A. Abnormal plasticity in dystonia: disruption of synaptic homeostasis. Neurobiol Dis (2011) 42:162–70. doi:10.1016/j.nbd.2010.12.011
68. Calabresi P, Pisani A, Rothwell J, Ghiglieri V, Obeso JA, Picconi B. Hyperkinetic disorders and loss of synaptic downscaling. Nat Neurosci (2016) 19:868–75. doi:10.1038/nn.4306
Keywords: Parkinson’s disease, cellular mechanisms, synaptopathy, dopamine transmission, animal models
Citation: Imbriani P, Schirinzi T, Meringolo M, Mercuri NB and Pisani A (2018) Centrality of Early Synaptopathy in Parkinson’s Disease. Front. Neurol. 9:103. doi: 10.3389/fneur.2018.00103
Received: 14 December 2017; Accepted: 13 February 2018;
Published: 01 March 2018
Edited by:
Fabio Blandini, Fondazione Istituto Neurologico Nazionale Casimiro Mondino (IRCCS), ItalyReviewed by:
Jose L. Lanciego, Universidad de Navarra, SpainMarie Therese Fuzzati-Armentero, France Parkinson, France
Copyright: © 2018 Imbriani, Schirinzi, Meringolo, Mercuri and Pisani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Pisani, cGlzYW5pJiN4MDAwNDA7dW5pcm9tYTIuaXQ=