 Michel Goedert
Michel Goedert Yoshiki Yamaguchi
Yoshiki Yamaguchi Sushil K. Mishra
Sushil K. Mishra Makoto Higuchi3
Makoto Higuchi3 Naruhiko Sahara
Naruhiko Sahara
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 15 February 2018
Sec. Neurodegeneration
Volume 9 - 2018 | https://doi.org/10.3389/fneur.2018.00070
This article is part of the Research TopicBrain Protein Aging and Dementia ControlView all 23 articles
A pathological pathway leading from soluble, monomeric to insoluble, filamentous Tau, is believed to underlie human Tauopathies. Cases of frontotemporal dementia are caused by dominantly inherited mutations in MAPT, the Tau gene. They show that dysfunction of Tau protein is sufficient to cause neurodegeneration and dementia. Extrapolation to the more common sporadic Tauopathies leads one to conclude that the pathological pathway is central to the development of all cases of disease, even if there are multiple reasons for Tau assembly. These findings are conceptually similar to those reported for beta-amyloid, alpha-synuclein and prion protein. Here, we provide an overview of Tau filaments and their positron emission tomography ligands.
Neurofibrillary lesions strongly correlate with cognitive deficits, making them an important therapeutic target for Alzheimer’s disease (AD) (1, 2). Dominantly inherited mutations in MAPT, the Tau gene, cause a form of frontotemporal dementia that can be associated with parkinsonism (FTDP-17T), showing that dysfunction of Tau protein is sufficient to cause neurodegeneration and dementia (3). In FTDP-17T, abundant filamentous Tau inclusions are present in either nerve cells or in both nerve cells and glial cells. Aβ deposits, a defining feature of AD, are not characteristic of FTDP-17T. However, there are many similarities between cases of FTDP-17T and other pure Tauopathies, such as sporadic progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD), and Pick’s disease, especially with regard to the isoform composition of Tau filaments.
Tau is expressed predominantly in the central and peripheral nervous systems, where it is most abundant in nerve cell axons. It belongs to the family of Tau/MAP2/MAP4 microtubule-associated proteins. Tau is natively unfolded with a low content of secondary structure (4, 5). However, long-range contacts between N- and C-termini, as well as between both termini and the repeats (i.e., paperclip conformation), have been described (5, 6). Using single-molecule Förster resonance energy transfer, it has been shown that upon tubulin binding the repeats expand and long-range contacts between both termini and the repeats are reduced (7).
Tau can be divided into an N-terminal domain, a proline-rich region, the repeat region, and a C-terminal domain (2). The N-terminal domain projects away from microtubules (8). Residues 2–18 have been shown to be involved in a signaling cascade that inhibits axonal transport (9). The N-terminal region also binds to the C-terminus of the p150 subunit of the dynactin complex (10). The proline-rich region has seven PXXP motifs, which provide recognition sites for SH3 domain-containing proteins of the Src family of non-receptor tyrosine kinases, such as Fyn (11). Its interaction with Tau may regulate the targeting of Fyn and thereby mediate beta-amyloid-induced toxicity (12). It has been reported that the proline-rich region of Tau also mediates binding to other proteins, including bridging integrator 1 and peptidyl-prolyl cis/trans isomerases (13). Interactions between Tau and microtubules are mediated through the repeats and some adjoining sequences (2). Less is known about the role of the C-terminal region. Tau belongs to the family of intrinsically disordered proteins, which have many interaction partners and are commonly implicated in neurodegenerative diseases (14). Theoretical calculations have estimated that more than 70 different binding partners of tau may exist (14).
Six Tau isoforms ranging from 352 to 441 amino acids in length are expressed in adult human brain (Figure 1A) (15). They are produced by alternative mRNA splicing of transcripts from MAPT and differ by the presence or absence of inserts of 29 or 58 amino acids (encoded by exons 2 and 3 of MAPT, with exon 3 being only transcribed in conjunction with exon 2) in the N-terminal half, and the inclusion, or not, of the 31 amino acid microtubule-binding repeat, encoded by exon 10, in the C-terminal half. Inclusion of exon 10 results in the production of three Tau isoforms with four repeats each (4R) and its exclusion in a further three isoforms with three repeats each (3R). The repeats comprise residues 244–368 of Tau, in the numbering of the 441 amino acid isoform. The N-terminal inserts are not believed to play an active role in Tau aggregation, but the insert encoded by exon 10 is important. In adult human brain, similar levels of 3R and 4R Tau are expressed (16), and the finding that a correct isoform ratio is essential for preventing neurodegeneration (17, 18) came as a surprise. Inclusion of exons 2 and 3, giving rise to 2N isoforms, is relatively underrepresented in comparison with inclusion of exon 2 and exclusion of exons 2 and 3, such that 2N, 1N, and 0N Tau isoforms make up 9, 54, and 37% of the total.
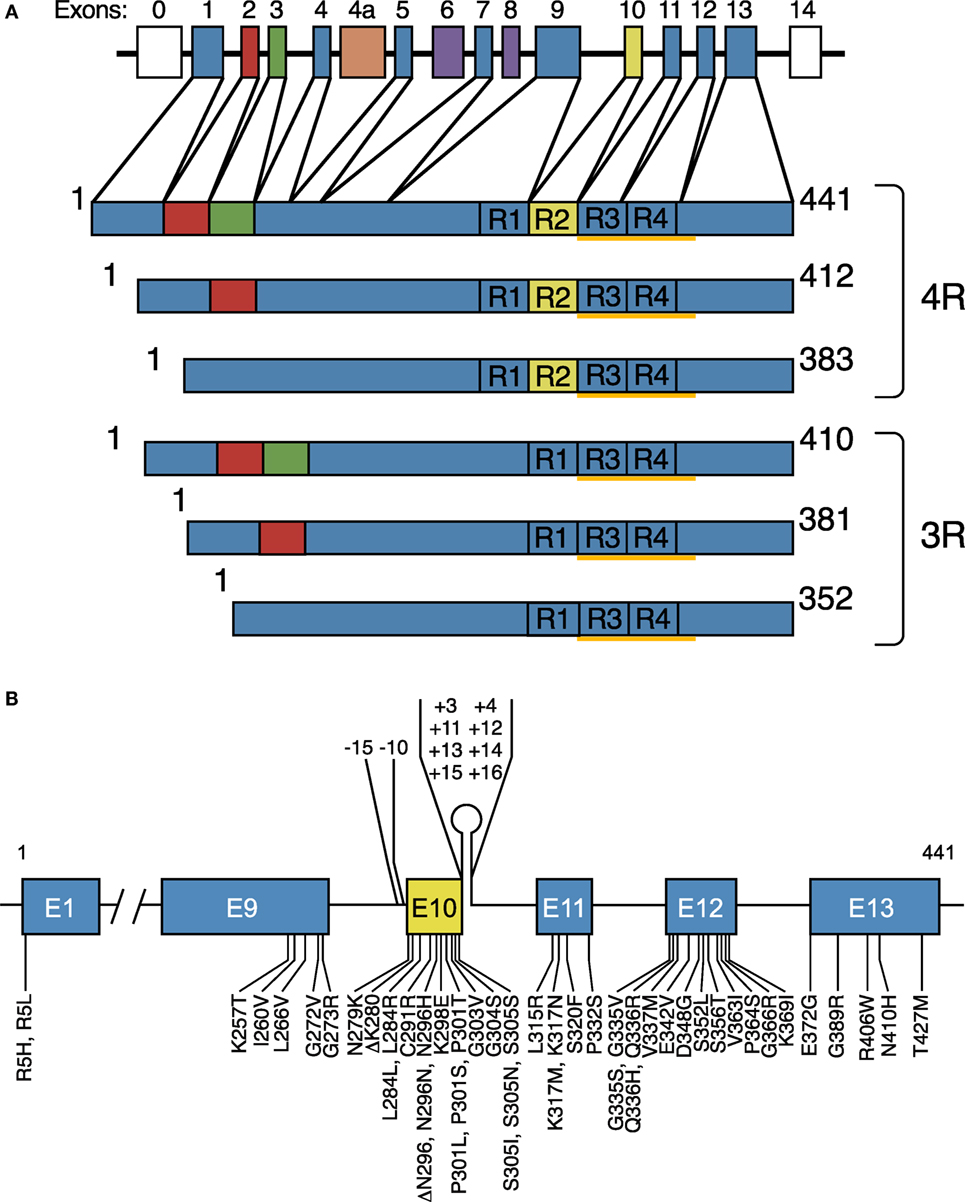
Figure 1. Human brain Tau isoforms and disease-causing MAPT mutations. (A) MAPT and the six Tau isoforms expressed in adult human brain. MAPT consists of 16 exons (E). Alternative mRNA splicing of E2 (red), E3 (green), and E10 (yellow) gives rise to six Tau isoforms (352–441 amino acids). The constitutively spliced exons (E1, E4, E5, E7, E9, E11, E12, and E13) are shown in blue. E0, which is part of the promoter, and E14 are non-coding (white). E6 and E8 (violet) are not transcribed in human brain. E4a (orange) is only expressed in the peripheral nervous system. The repeats (R1–R4) are shown, with three isoforms having four repeats each (4R) and three isoforms having three repeats each (3R). The core sequences of the Tau filaments from Alzheimer’s disease brain (V306-F378) determined by cryo-EM are underlined. (B), Mutations in MAPT in cases of frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17T). Forty-nine coding region mutations and 10 intronic mutations flanking E10 are shown.
Why six Tau isoforms are found in adult human brain is not known. Isoform expression is not conserved between species (19–22). Thus, in adult mouse brains, 4R Tau isoforms are almost exclusively present, whereas adult chicken brains express 3R, 4R, and 5R Tau isoforms. However, what is conserved is the expression of one hyperphosphorylated 3R Tau isoform lacking N-terminal repeats during vertebrate development. In mice the switch from 3R to 4R Tau occurs between postnatal days 9 and 18, with Tau phosphorylation also decreasing during that time (23). However, isoform switching and phosphorylation are regulated differently. Adult Tau isoforms with 4R are better at promoting microtubule assembly and binding to microtubules than the fetal 3R Tau isoform (16). This is consistent with the need for a more dynamic cytoskeleton during the development of nerve cells.
The repeats and some adjoining sequences constitute the microtubule-binding domains of Tau. Single-molecule tracking revealed a kiss-and-hop mechanism, with a dwell time of Tau on individual microtubules of only about 40 ms (24, 25). Isoform differences did not influence this interaction. Despite these rapid dynamics, Tau promoted microtubule assembly. It remains to be seen if microtubules were also stabilized. In brain, Tau is subject to a large number of posttranslational modifications, including phosphorylation, acetylation, methylation, glycation, isomerization, O-GlcNAcylation, nitration, sumoylation, ubiquitination, and truncation (26–28). Big Tau, which carries an additional large exon in the N-terminal half, is only expressed in the peripheral nervous system (29, 30). Several structural models have been put forward for the binding of Tau to microtubules (31–33), but there is no consensus. Overall, it appears that the microtubule-bound conformation of Tau may delay aggregation. Cryogenic electron microscopy (cryo-EM) is bound to provide atomic structures of Tau bound to microtubules that were assembled from tubulin in different ways (34).
Full-length Tau assembles into filaments through its repeats, with the N-terminal half and the C-terminus forming the fuzzy coat (35–38). Tau filaments from human brain and those assembled from expressed protein have a cross-β structure characteristic of amyloid fibrils, with their cores consisting of approximately 90 amino acids (39). The region of Tau that binds to microtubules also forms the core of Tau filaments, suggesting that physiological function and pathological assembly are mutually exclusive.
Phosphorylation of Tau negatively regulates its ability to interact with microtubules, and filamentous Tau is abnormally hyperphosphorylated (40). However, it remains to be proved that phosphorylation is the trigger for aggregation in human diseases. Alternatively, a conformational change in Tau arising from assembly may cause its hyperphosphorylation. Recombinant Tau assembles in bulk into filaments when incubated with heparin, in the absence of phosphorylation (41, 42). However, it has also been shown that recombinant S262A 4R Tau assembled into filaments following incubation with brain extracts from adult rats (43). Other posttranslational modifications may also be involved. Initial studies on Tau acetylation reported that it promoted phosphorylation and aggregation (44, 45). However, subsequent work has suggested that an inverse correlation exists between Tau acetylation and phosphorylation, with acetylation inhibiting Tau assembly (46, 47). Unlike phosphorylation, acetylation occurs on lysine residues, as do glycation, ubiquitination, and methylation.
Many publications equate Tau phosphorylation with aggregation. This is probably not correct. Although aggregated Tau is heavily phosphorylated in human brain, not all phosphorylated Tau is aggregated or on its way to aggregation. For instance, highly phosphorylated Tau forms during hibernation, in the absence of aggregation (48). There is substantial overlap between the phosphorylation of Tau during development and its hyperphosphorylation in disease. However, some Tau phosphorylation, such as that at T212, S214, and T217 detected by antibody AT100, is pathological (49). Antibody AT8 has been used to detect both physiologically and pathologically phosphorylated Tau. It was recently shown that it recognizes triply phosphorylated Tau (S202, T205, and S208) better than doubly phosphorylated protein (S202 andT205), raising the possibility of differential phosphorylation of pathologically and physiologically phosphorylated Tau at the AT8 epitope (50).
In AD, chronic traumatic encephalopathy, postencephalitic parkinsonism, and many other Tauopathies, all six isoforms are present in the disease filaments (Table 1) (2). They are either paired helical (PHFs) or straight (SFs) and contain both 3R and 4R Tau isoforms in a one-to-one ratio, similar to the isoform composition and relative abundance of the six isoforms in soluble Tau from normal human brain. By cryo-EM, the cores of Tau filaments from AD are made of two identical protofilaments consisting of residues V306-F378 of Tau, which adopt a combined cross-β/β-helix structure, possibly defining the seed for Tau aggregation (51). The N-terminal part of the cross-β structure is formed by the hexapeptide 306VQIVYK311 (PHF6), which is essential for the oligomerization of Tau and its assembly into filaments (52, 53). It packs through a heterotypic, non-staggered interface with the opposing residues 373–378. The same packing interface is absent in the widely used K18 and K19 proteins, which span three or four repeat domains of recombinantly expressed Tau, and end at E372 (54). Therefore, filaments made of K18 and K19 proteins cannot represent the complete core structure of PHFs and SFs from the brains of individuals with AD. The second hexapeptide motif 275VQIINK280 (PHF6*) that is required for filament assembly (55) does not form part of the core of Tau filaments from AD brain. However, inhibitors of the PHF6* motif have been shown to reduce the heparin-induced assembly of 4R Tau (56). Both hexapeptide motifs were required for the seeded aggregation of mutant human Tau in transfected non-neuronal cells (57). It remains to be seen if PHF6 and PHF6* are required for the assembly of Tau in human brain.

Table 1. Neurodegenerative diseases with abundant tau inclusions.
Each protofilament contains eight β-strands, five of which give rise to two pairs of anti-parallel β-sheets, with the other three forming a β-helix. PHFs and SFs differ in their inter-protofilament packing, showing that they are ultrastructural polymorphs. The protofilaments of PHFs are arranged base-to-base, whereas those of SFs are arranged back-to-base. These findings do not explain why all six Tau isoforms are found in PHFs and SFs. However, a less ordered β-sheet is present upstream of V306; it can accommodate an additional 16 amino acids, which probably correspond to a mixture of residues 259–274 (R1) from 3R Tau and 290–305 (R2) from 4R Tau.
In other diseases, such as PSP, CBD, AGD, globular glial Tauopathy, and aging-related Tau astrogliopathy, isoforms with 4R Tau are found in the filaments (Table 1) (3), but the presence of 3R Tau-positive neuronal inclusions has also been reported in PSP and CBD (58, 59). The Pick bodies of Pick’s disease are only made of 3R Tau (Table 1) (60). The morphologies of Tau filaments in different diseases vary, even when they are made of the same isoforms. Silver staining can also detect these differences (61). Inclusions made of all six Tau isoforms stain with Gallyas–Braak and Campbell–Switzer. Those made of 4R Tau are only positive with Gallyas–Braak, whereas those made of 3R Tau stain only with Campbell–Switzer. It remains to be seen if the cores of filaments made of 3R or 4R Tau differ structurally from those of AD, which are made of 3R + 4R Tau isoforms.
The specificity of antibodies Alz50 and MC-1 for assembled Tau relies on a conformation that all isoforms can undergo and which requires two discontinuous intramolecular epitopes separated by almost 300 amino acids (62, 63). They are 7EFE9 in the N-terminus and 313VDLSKVTSKC322 in R3. MC-1 staining is one of the earliest markers of misfolded Tau. NMR experiments using heparin-induced filaments of 4R Tau also provided evidence for an interaction between the N-terminus and residues 313–322 of the structured core (64). Moreover, the cryo-EM structures of Tau filaments from AD brain showed a density consistent with 7EFE9 contacting K317 and K321 in the protofilament core (51). These electrostatic interactions may be essential for Tau filament formation, implying that acetylation of K317 and/or K321 might protect against aggregation. The only known disease-causing mutations in MAPT that are located outside the repeats and the C-terminus (R5H and R5L) (3) are close to 7EFE9.
Fifty-nine different mutations in MAPT have been identified in FTDP-17T (Figure 1B) (3). The filaments consist of 3R, 4R, or 3R + 4R Tau (Figure 2) (65). MAPT mutations account for approximately 5% of cases of frontotemporal dementia and are concentrated in exons 9–12 (encoding R1–R4) and the introns flanking exon 10. They can be divided into those with a primary effect at the protein level and those affecting the alternative splicing of Tau pre-mRNA. There is no obvious correlation between known mutations and posttranslational modifications of Tau.
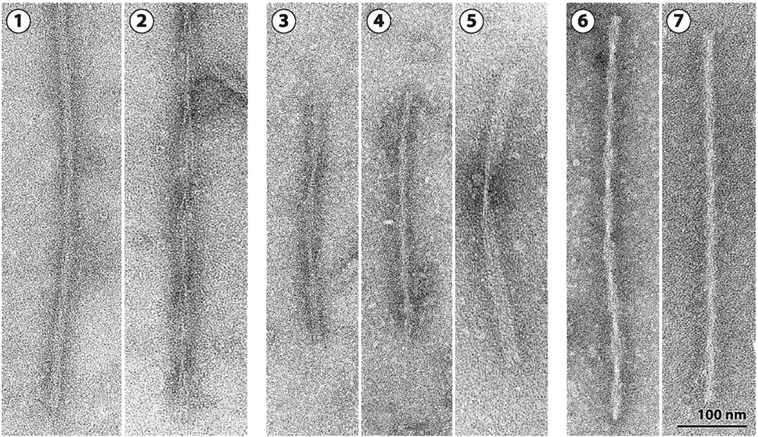
Figure 2. Tau filaments from FTDP-17T. [1,2] Neuronal Tau filaments from a case with abundant Pick body-like inclusions and a G389R mutation in MAPT (66). [1] Straight filaments form the majority species and [2] strongly stranded twisted filaments are in the minority. [3–5] Tau filaments from cases with neuronal and glial inclusions and a P301L mutation in MAPT or an intronic mutation [from Ref. (67, 68)]. [3] Narrow twisted ribbons and [4] occasional rope-like filaments. [5] Familial multiple system Tauopathy with presenile dementia and other cases caused by MAPT mutations in the intron after E10 are characterized by wide twisted ribbons and neuronal and glial Tau inclusions. The filaments in [3–5] are made of 4R Tau. [6,7] Tau filaments from a case with a V337M mutation in MAPT [from Ref. (69)]. [6] Paired helical and [7] straight filaments are present as in Alzheimer’s disease. Tau inclusions are largely neuronal, and filaments in [6] and [7] are made of 3R and 4R Tau.
It has been suggested that patients with AD-type neurofibrillary degeneration restricted to hippocampus and medial temporal lobe, who lack Aβ deposits, suffer from primary age-related Tauopathy (PART), a condition that differs from AD (70). Tangle-only dementia, a rare form of dementia, may represent a severe form of PART (71). However, the view that PART is different from AD has been challenged, because it is clinically and neuropathologically similar to what appear to be the early stages of the Tau pathology of AD (72).
In AD, following the death of tangle-bearing cells, Tau filaments can remain in the extracellular space as ghost tangles, which consist largely of Tau repeats that have lost their fuzzy coat through proteolysis. In Pick’s disease, PSP, CBD, and most cases caused by MAPT mutations, Tau filaments do not accumulate to a significant extent in the extracellular space following the death of aggregate-bearing cells. The reasons why Tau filaments from AD brain are less soluble remain to be established (2).
Monomeric Tau assembles into filaments through oligomerization (1, 2). In tissue sections, filamentous Tau aggregates are labeled by amyloid-binding dyes, such as Congo red, thioflavins, and some luminescent conjugated oligothiophenes (2, 73). These dyes appear to bind to both intra- and extracellular Tau deposits. They are useful for cross-sectional studies but require the availability of brain tissue.
To perform longitudinal studies and to assess the effects of treatments on the level of aggregates, one needs to be able to visualize Tau inclusions repeatedly in the living human nervous system. The field of PET imaging of brain inclusions characteristic of human neurodegenerative diseases started with the development of [11C]Pittsburgh compound B ([11C]PIB), a derivative of thioflavin T, which detects β-amyloid deposits in the living brain (74). Subsequently, several PET tracers for aggregated Tau, such as [11C]PBB3, [18F]PM-PBB3, [18F]AV-1451, [18F]THK5351, [18F]MK-6240, [18F]R06958948, [18F]GTP-1, and [18F]PI-2620, were developed and are currently being tested in humans (75–81). Most tracers show a high affinity for Tau inclusions and recognize β-amyloid deposits less well (79, 81). However, some off-target effects have also been described. Thus, non-specific retention of [11C]PBB3 was seen in the dural venous sinuses (75). In vitro studies have shown that [18F]AV-1451 can bind to monoamine oxidase (MAO)-A, as well as to pigmented and mineralized vascular structures (82). Retention of [18F]AV-1451 in the choroid plexus of control individuals also reflected off-target binding (83). Age-related, off-target effects of [18F]AV-1451 binding in the basal ganglia closely correlated with iron accumulation (84). Selegiline, a MAO-B inhibitor, reduced [18F]THK5351 signal in basal ganglia and neocortex (85). Moreover, an in vitro study confirmed that MAO-B was an off-target binding substrate for [18F]THK5351 (86). Perhaps most worryingly, elevated binding of [18F]AV-1451 and [18F]THK5351 has been described in the semantic variant of primary progressive aphasia, a form of frontotemporal dementia that is consistently associated with assembled TDP-43, but not with Tau inclusions (87–89). Where studied, second generation Tau PET tracers ([18F]PM-PBB3, [18F]MK-6240, [18F]R06958948, [18F]GTP-1, and [18F]Pl-2620) have shown less off-target binding than the first generation of tracers. Future autopsy studies are needed to identify the binding targets of these ligands. On the other hand, the distribution of [18F]AV-1451 binding, a first generation tracer, recapitulated Braak staging in AD brain (90). Moreover, a combination of PET imaging with [18F]AV-1451 and graph theory supported the view that tau pathology can undergo transneuronal spread (91), consistent with experimental studies (92, 93).
To develop more specific and selective ligands, it is important to determine where in the structured cores of Tau filaments PET ligands bind. Recent advances in cryo-EM, which have resulted in the determination of the high-resolution structures of Tau filaments from AD brain (51), have made this possible in principle. We used this information, together with molecular docking (94), to study the binding of PBB3 to the protofilament core of Tau filaments from AD (Figure 3A). As shown in Figure 3, PBB3 bound in a perpendicular manner to a high-affinity site (S1) in the C-shaped part of the protofilament, which includes residues 349–351 (RVQ) of Tau (Figure 3B). Two lower affinity binding sites were also detected, at residues 364–369 (PGGGNK) (S2) and 351–353 (QSK) (S3) (Figure 3B).
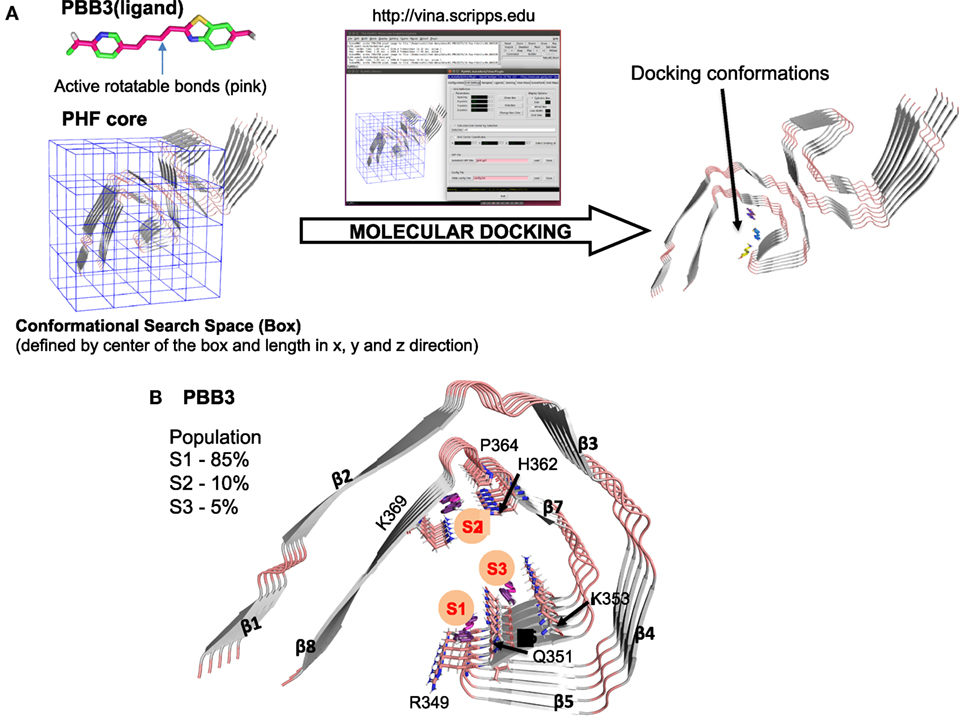
Figure 3. Molecular docking of PBB3 to paired helical Tau filaments (PHFs) from Alzheimer’s disease brain. (A) Schematic representation of the docking process using Pymol and AutoDock Vina. The PHF core structure was from Ref. (51) (PDB ID: 503L). Out of 100 docking conformations, the top 20 were selected for further analysis. (B) Molecular docking of PBB3 into the PHF protofilament core structure. The top 20 conformations distributed into three clusters (S1, S2, and S3). S1 had the highest affinity for the Tau filament, followed by S2 and S3.
PBB3 visualizes the Tau pathologies of AD and non-AD Tauopathies (75, 95, 96). Unlike PBB3, previous in vitro and in vivo studies have shown that AV-1451 binds only with low-affinity to filaments from non-AD Tauopathies (95, 97, 98). It has been reported that AV-1451 and its lead compound failed to visualize Tau inclusions in a mouse line transgenic for human P301L Tau (76). However, using [11C]PBB3, it was possible to image Tau inclusions in mouse models of Tauopathy (lines PS19 and Tg4510) (75, 99). These findings further support the view that AV-1451 recognizes Tau inclusions made of 3R or 4R Tau with lower affinity than those made of 3R + 4R Tau. It will be interesting to obtain cryo-EM structures of the cores of Tau filaments from AD and other Tauopathies with bound PET ligands. One cannot exclude that high-affinity binding sites exist in the “fuzzy coat” of human brain Tau filaments. However, both AV-1451 and PBB3 have been shown to detect extracellular Tau inclusions in AD brain (95). We believe that the aggregated Tau in extracellular tangles corresponds closely to the structured filament cores.
The determination of high-resolution structures of Tau filaments by cryo-EM has opened the way for elucidating the structures of other amyloid filaments from human brain. Future work will tell what the differences between morphotypes of amyloid filaments are, which will in turn inform the mechanisms underlying the prion-like propagation of protein aggregates. Perhaps most importantly, cryo-EM will make it possible to relate mechanisms of amyloid formation of recombinant proteins to those in human brain.
MG and NS wrote the manuscript; YY and SM analyzed the molecular docking simulation; and MH critiqued the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
MG is an Honorary Professor in the Department of Clinical Neurosciences of the University of Cambridge, UK. This work was supported by the UK Medical Research Council (MC_U105184291 to MG) and the European Union (Joint Programme-Neurodegeneration Research REfrAME to MG and Horizon 2020 IMPRiND to MG). This work was also supported by grants from the Japanese Ministry of Education, Culture, Sports, Science and Technology (26117001 and 15K06793 to NS) and the Strategic Research Program for Brain Sciences of the Japan Agency for Medical Research and Development (to NS). MH holds a patent on compounds including PBB3 (JP 5422782/EP 12 844 742.3).
1. Sahara N, Avila J. “Tau oligomers,” what we know and what we don’t know. Front Neurol (2014) 5:1. doi:10.3389/fneur.2014.00001
2. Goedert M, Eisenberg DS, Crowther RA. Propagation of Tau aggregates and neurodegeneration. Annu Rev Neurosci (2017) 40:189–210. doi:10.1146/annurev-neuro-072116-031153
3. Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol (2015) 41:24–46. doi:10.1111/nan.12213
4. Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol (1977) 116:227–47. doi:10.1016/0022-2836(77)90214-5
5. Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry (2006) 45:2283–93. doi:10.1021/bi0521543
6. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci (2016) 17:5–21. doi:10.1038/nrn.2015.1
7. Melo AM, Coraor J, Alpha-Cobb G, Elbaum-Garfinkle S, Nath A, Rhoades E. A functional role for intrinsic disorder in the tau-tubulin complex. Proc Natl Acad Sci U S A (2016) 113:14336–41. doi:10.1073/pnas.1610137113
8. Chen J, Kanai Y, Cowan NJ, Hirokawa N. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature (1992) 360:674–7. doi:10.1038/360674a0
9. Kanaan NM, Morfini GA, Lapointe NE, Pigino GF, Patterson KR, Song Y, et al. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci (2011) 31:9858–68. doi:10.1523/JNEUROSCI.0560-11.2011
10. Magnani E, Fan J, Gasparini L, Golding M, Williams M, Schiavo G, et al. Interaction of tau protein with the dynactin complex. EMBO J (2007) 26:4546–54. doi:10.1038/sj.emboj.7601878
11. Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci (1998) 111(Pt 21):3167–77.
12. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, Van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell (2010) 142:387–97. doi:10.1016/j.cell.2010.06.036
13. Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron (2011) 70:410–26. doi:10.1016/j.neuron.2011.04.009
14. Uversky VN. Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front Aging Neurosci (2015) 7:18. doi:10.3389/fnagi.2015.00018
15. Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron (1989) 3:519–26. doi:10.1016/0896-6273(89)90210-9
16. Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J (1990) 9:4225–30.
17. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature (1998) 393:702–5. doi:10.1038/31508
18. Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A (1998) 95:7737–41. doi:10.1073/pnas.95.13.7737
19. Kosik KS, Orecchio LD, Bakalis S, Neve RL. Developmentally regulated expression of specific tau sequences. Neuron (1989) 2:1389–97. doi:10.1016/0896-6273(89)90077-9
20. Janke C, Beck M, Stahl T, Holzer M, Brauer K, Bigl V, et al. Phylogenetic diversity of the expression of the microtubule-associated protein tau: implications for neurodegenerative disorders. Brain Res Mol Brain Res (1999) 68:119–28. doi:10.1016/S0169-328X(99)00079-0
21. Yoshida H, Goedert M. Molecular cloning and functional characterization of chicken brain tau: isoforms with up to five tandem repeats. Biochemistry (2002) 41:15203–11. doi:10.1021/bi026464m
22. Takuma H, Arawaka S, Mori H. Isoform changes of tau protein during development in various species. Brain Res Dev Brain Res (2003) 142:121–7. doi:10.1016/S0165-3806(03)00056-7
23. Tuerde D, Kimura T, Miyasaka T, Furusawa K, Shimozawa A, Hasegawa M, et al. Isoform-independent and -dependent phosphorylation of microtubule-associated protein tau in mouse brain during postnatal development. J Biol Chem (2018) 293:1781–93. doi:10.1074/jbc.M117.798918
24. Janning D, Igaev M, Sündermann F, Bruhmann J, Beutel O, Heinisch JJ, et al. Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons. Mol Biol Cell (2014) 25:3541–51. doi:10.1091/mbc.E14-06-1099
25. Niewidok B, Igaev M, Sündermann F, Janning D, Bakota L, Brandt R. Presence of a carboxy-terminal pseudorepeat and disease-like pseudohyperphosphorylation critically influence tau’s interaction with microtubules in axon-like processes. Mol Biol Cell (2016) 27:3537–49. doi:10.1091/mbc.E16-06-0402
26. Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol (2013) 12:609–22. doi:10.1016/S1474-4422(13)70090-5
27. Morris M, Knudsen GM, Maeda S, Trinidad JC, Ioanoviciu A, Burlingame AL, et al. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci (2015) 18:1183–9. doi:10.1038/nn.4067
28. Mair W, Muntel J, Tepper K, Tang S, Biernat J, Seeley WW, et al. FLEXITau: quantifying post-translational modifications of Tau protein in vitro and in human disease. Anal Chem (2016) 88:3704–14. doi:10.1021/acs.analchem.5b04509
29. Couchie D, Mavilia C, Georgieff IS, Liem RK, Shelanski ML, Nunez J. Primary structure of high molecular weight tau present in the peripheral nervous system. Proc Natl Acad Sci U S A (1992) 89:4378–81. doi:10.1073/pnas.89.10.4378
30. Goedert M, Spillantini MG, Crowther RA. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc Natl Acad Sci U S A (1992) 89:1983–7. doi:10.1073/pnas.89.5.1983
31. Al-Bassam J, Ozer RS, Safer D, Halpain S, Milligan RA. MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J Cell Biol (2002) 157:1187–96. doi:10.1083/jcb.200201048
32. Kar S, Fan J, Smith MJ, Goedert M, Amos LA. Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. EMBO J (2003) 22:70–7. doi:10.1093/emboj/cdg001
33. Kadavath H, Höfele RV, Biernat J, Kumar S, Tepper K, Urlaub H, et al. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci U S A (2015) 112:7501–6. doi:10.1073/pnas.1504081112
34. Nogales E, Kellogg EH. Challenges and opportunities in the high-resolution cryo-EM visualization of microtubules and their binding partners. Curr Opin Struct Biol (2017) 46:65–70. doi:10.1016/j.sbi.2017.06.003
35. Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A (1988) 85:4051–5. doi:10.1073/pnas.85.11.4051
36. Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A (1988) 85:4884–8.
37. Wischik CM, Novak M, Thogersen HC, Edwards PC, Runswick MJ, Jakes R, et al. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A (1988) 85:4506–10.
38. Lee VM, Balin BJ, Otvos L, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science (1991) 251:675–8. doi:10.1126/science.1899488
39. Berriman J, Serpell LC, Oberg KA, Fink AL, Goedert M, Crowther RA. Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-beta structure. Proc Natl Acad Sci U S A (2003) 100:9034–8. doi:10.1073/pnas.1530287100
40. Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol (2016) 12:15–27. doi:10.1038/nrneurol.2015.225
41. Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature (1996) 383:550–3. doi:10.1038/383550a0
42. Perez M, Valpuesta JM, Medina M, Montejo De Garcini E, Avila J. Polymerization of tau into filaments in the presence of heparin: the minimal sequence required for tau-tau interaction. J Neurochem (1996) 67:1183–90. doi:10.1046/j.1471-4159.1996.67031183.x
43. Despres C, Byrne C, Qi H, Cantrelle FX, Huvent I, Chambraud B, et al. Identification of the Tau phosphorylation pattern that drives its aggregation. Proc Natl Acad Sci U S A (2017) 114:9080–5. doi:10.1073/pnas.1708448114
44. Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron (2010) 67:953–66. doi:10.1016/j.neuron.2010.08.044
45. Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun (2011) 2:252. doi:10.1038/ncomms1255
46. Cook C, Carlomagno Y, Gendron TF, Dunmore J, Scheffel K, Stetler C, et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum Mol Genet (2014) 23:104–16. doi:10.1093/hmg/ddt402
47. Carlomagno Y, Chung DC, Yue M, Castanedes-Casey M, Madden BJ, Dunmore J, et al. An acetylation-phosphorylation switch that regulates tau aggregation propensity and function. J Biol Chem (2017) 292:15277–86. doi:10.1074/jbc.M117.794602
48. Arendt T, Stieler J, Strijkstra AM, Hut RA, Rüdiger J, Van Der Zee EA, et al. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci (2003) 23:6972–81.
49. Yoshida H, Goedert M. Sequential phosphorylation of tau protein by cAMP-dependent protein kinase and SAPK4/p38delta or JNK2 in the presence of heparin generates the AT100 epitope. J Neurochem (2006) 99:154–64. doi:10.1111/j.1471-4159.2006.04052.x
50. Malia TJ, Teplyakov A, Ernst R, Wu SJ, Lacy ER, Liu X, et al. Epitope mapping and structural basis for the recognition of phosphorylated tau by the anti-tau antibody AT8. Proteins (2016) 84:427–34. doi:10.1002/prot.24988
51. Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature (2017) 547:185–90. doi:10.1038/nature23002
52. Von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming beta structure. Proc Natl Acad Sci U S A (2000) 97:5129–34. doi:10.1073/pnas.97.10.5129
53. Sahara N, Maeda S, Murayama M, Suzuki T, Dohmae N, Yen SH, et al. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci (2007) 25:3020–9. doi:10.1111/j.1460-9568.2007.05555.x
54. Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry (1994) 33:9511–22. doi:10.1021/bi00198a017
55. Von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow EM, et al. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J Biol Chem (2001) 276:48165–74. doi:10.1074/jbc.M105196200
56. Seidler PM, Boyer DR, Rodriguez JA, Sawaya MR, Cascio D, Murray K, et al. Structure-based inhibitors of tau aggregation. Nat Chem (2018) 10(2):170–6. doi:10.1038/nchem.2889
57. Falcon B, Cavallini A, Angers R, Glover S, Murray TK, Barnham L, et al. Conformation determines the seeding potencies of native and recombinant Tau aggregates. J Biol Chem (2015) 290:1049–65. doi:10.1074/jbc.M114.589309
58. Uchihara T, Nakamura A, Shibuya K, Yagishita S. Specific detection of pathological three-repeat tau after pretreatment with potassium permanganate and oxalic acid in PSP/CBD brains. Brain Pathol (2011) 21:180–8. doi:10.1111/j.1750-3639.2010.00433.x
59. Uchihara T, Hara M, Nakamura A, Hirokawa K. Tangle evolution linked to differential 3- and 4-repeat tau isoform deposition: a double immunofluorolabeling study using two monoclonal antibodies. Histochem Cell Biol (2012) 137:261–7. doi:10.1007/s00418-011-0891-2
60. De Silva R, Lashley T, Strand C, Shiarli AM, Shi J, Tian J, et al. An immunohistochemical study of cases of sporadic and inherited frontotemporal lobar degeneration using 3R- and 4R-specific tau monoclonal antibodies. Acta Neuropathol (2006) 111:329–40. doi:10.1007/s00401-006-0048-x
61. Uchihara T, Tsuchiya K, Nakamura A, Akiyama H. Argyrophilic grains are not always argyrophilic-distinction from neurofibrillary tangles of diffuse neurofibrillary tangles with calcification revealed by comparison between Gallyas and Campbell-Switzer methods. Acta Neuropathol (2005) 110:158–64. doi:10.1007/s00401-005-1031-7
62. Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem (1996) 271:32789–95. doi:10.1074/jbc.271.51.32789
63. Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res (1997) 48:128–32. doi:10.1002/(SICI)1097-4547(19970415)48:2<128::AID-JNR5>3.0.CO;2-E
64. Bibow S, Mukrasch MD, Chinnathambi S, Biernat J, Griesinger C, Mandelkow E, et al. The dynamic structure of filamentous tau. Angew Chem Int Ed Engl (2011) 50:11520–4. doi:10.1002/anie.201105493
65. Crowther RA, Goedert M. Abnormal tau-containing filaments in neurodegenerative diseases. J Struct Biol (2000) 130:271–9. doi:10.1006/jsbi.2000.4270
66. Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M, et al. Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol (1999) 58:1207–26. doi:10.1097/00005072-199912000-00002
67. Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci U S A (1997) 94:4113–8. doi:10.1073/pnas.94.8.4113
68. Spillantini MG, Crowther RA, Kamphorst W, Heutink P, Van Swieten JC. Tau pathology in two Dutch families with mutations in the microtubule-binding region of tau. Am J Pathol (1998) 153:1359–63. doi:10.1016/S0002-9440(10)65721-5
69. Spillantini MG, Crowther RA, Goedert M. Comparison of the neurofibrillary pathology in Alzheimer’s disease and familial presenile dementia with tangles. Acta Neuropathol (1996) 92:42–8. doi:10.1007/s004010050487
70. Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol (2014) 128:755–66. doi:10.1007/s00401-014-1349-0
71. Ulrich J, Spillantini MG, Goedert M, Dukas L, Stähelin HB. Abundant neurofibrillary tangles without senile plaques in a subset of patients with senile dementia. Neurodegeneration (1992) 1:257–64.
72. Duyckaerts C, Braak H, Brion JP, Buée L, Del Tredici K, Goedert M, et al. PART is part of Alzheimer disease. Acta Neuropathol (2015) 129:749–56. doi:10.1007/s00401-015-1390-7
73. Shirani H, Appelqvist H, Back M, Klingstedt T, Cairns NJ, Nilsson KPR. Synthesis of thiophene-based optical ligands that selectively detect tau pathology in Alzheimer’s disease. Chemistry (2017) 23:17127–35. doi:10.1002/chem.201703846
74. Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol (2004) 55:306–19. doi:10.1002/ana.20009
75. Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron (2013) 79:1094–108. doi:10.1016/j.neuron.2013.07.037
76. Xia CF, Arteaga J, Chen G, Gangadharmath U, Gomez LF, Kasi D, et al. [18F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement (2013) 9:666–76. doi:10.1016/j.jalz.2012.11.008
77. Hostetler ED, Walji AM, Zeng Z, Miller P, Bennacef I, Salinas C, et al. Preclinical characterization of 18F-MK-6240, a promising PET tracer for in vivo quantification of human neurofibrillary tangles. J Nucl Med (2016) 57:1599–606. doi:10.2967/jnumed.115.171678
78. Walji AM, Hostetler ED, Selnick H, Zeng Z, Miller P, Bennacef I, et al. Discovery of 6-(Fluoro-18F)-3-(1H-pyrrolo[2,3-c]pyridin-1-yl)isoquinolin-5-amine ([18F]-MK-6240): a positron emission tomography (PET) imaging agent for quantification of neurofibrillary tangles (NFTs). J Med Chem (2016) 59:4778–89. doi:10.1021/acs.jmedchem.6b00166
79. Hall B, Mak E, Cervenka S, Aigbirhio FI, Rowe JB, O’Brien JT. In vivo tau PET imaging in dementia: pathophysiology, radiotracer quantification, and a systematic review of clinical findings. Ageing Res Rev (2017) 36:50–63. doi:10.1016/j.arr.2017.03.002
80. Mueller A, Kroth H, Schieferstein H, Berndt M, Oden F, Capotosti F, et al. Preclinical characterization of PI-2620, a novel tau PET tracer for detection of tau in AD and other tauopathies. Alzheimer Dement (2017) 13:141–2. doi:10.1016/j.jalz.2017.06.2567
81. Saint-Aubert L, Lemoine L, Chiotis K, Leuzy A, Rodriguez-Vieitez E, Nordberg A. Tau PET imaging: present and future directions. Mol Neurodegener (2017) 12:19. doi:10.1186/s13024-017-0162-3
82. Lowe VJ, Curran G, Fang P, Liesinger AM, Josephs KA, Parisi JE, et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun (2016) 4:58. doi:10.1186/s40478-016-0315-6
83. Marquie M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol (2015) 78:787–800. doi:10.1002/ana.24517
84. Choi JY, Cho H, Ahn SJ, Lee JH, Ryu YH, Lee MS, et al. Off-target 18F-AV-1451 binding in the basal ganglia correlates with age-related iron accumulation. J Nucl Med (2018) 59:117–20. doi:10.2967/jnumed.117.195248
85. Ng KP, Pascoal TA, Mathotaarachchi S, Therriault J, Kang MS, Shin M, et al. Monoamine oxidase B inhibitor, selegiline, reduces 18F-THK5351 uptake in the human brain. Alzheimers Res Ther (2017) 9:25. doi:10.1186/s13195-017-0253-y
86. Harada R, Ishiki A, Kai H, Sato N, Furukawa K, Furumoto S, et al. Correlations of 18F-THK5351 PET with post-mortem burden of tau and astrogliosis in Alzheimer’s disease. J Nucl Med (2017). doi:10.2967/jnumed.117.197426
87. Bevan-Jones WR, Cope TE, Jones PS, Passamonti L, Hong YT, Fryer TD, et al. [(18)F]AV-1451 binding in vivo mirrors the expected distribution of TDP-43 pathology in the semantic variant of primary progressive aphasia. J Neurol Neurosurg Psychiatry (2017). doi:10.1136/jnnp-2017-316402
88. Lee H, Seo S, Lee SY, Jeong HJ, Woo SH, Lee KM, et al. [18F]-THK5351 PET Imaging in patients with semantic variant primary progressive aphasia. Alzheimer Dis Assoc Disord (2017). doi:10.1097/WAD.0000000000000216
89. Makaretz SJ, Quimby M, Collins J, Makris N, McGinnis S, Schultz A, et al. Flortaucipir tau PET imaging in semantic variant primary progressive aphasia. J Neurol Neurosurg Psychiatry (2017). doi:10.1136/jnnp-2017-316409
90. Schwarz AJ, Yu P, Miller BB, Shcherbinin S, Dickson J, Navitsky M, et al. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain (2016) 139:1539–50. doi:10.1093/brain/aww023
91. Cope TE, Rittman T, Borchert RJ, Jones PS, Vatansever D, Allinson K, et al. Tau burden and the functional connectome in Alzheimer’s disease and progressive supranuclear palsy. Brain (2018) 141:550–67. doi:10.1093/brain/awx347
92. Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol (2009) 11:909–13. doi:10.1038/ncb1901
93. He Z, Guo JL, Mcbride JD, Narasimhan S, Kim H, Changolkar L, et al. Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med (2018) 24:29–38. doi:10.1038/nm.4443
94. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem (2010) 31:455–61. doi:10.1002/jcc.21334
95. Ono M, Sahara N, Kumata K, Ji B, Ni R, Koga S, et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain (2017) 140:764–80. doi:10.1093/brain/aww339
96. Shimada H, Kitamura S, Shinotoh H, Endo H, Niwa F, Hirano S, et al. Association between Abeta and tau accumulations and their influence on clinical features in aging and Alzheimer’s disease spectrum brains: a [11C]PBB3-PET study. Alzheimers Dement (Amst) (2017) 6:11–20. doi:10.1016/j.dadm.2016.12.009
97. Marquie M, Normandin MD, Meltzer AC, Siao Tick Chong M, Andrea NV, Anton-Fernandez A, et al. Pathological correlations of [F-18]-AV-1451 imaging in non-Alzheimer tauopathies. Ann Neurol (2017) 81:117–28. doi:10.1002/ana.24844
98. Smith R, Schöll M, Honer M, Nilsson CF, Englund E, Hansson O. Tau neuropathology correlates with FDG-PET, but not AV-1451-PET, in progressive supranuclear palsy. Acta Neuropathol (2017) 133:149–51. doi:10.1007/s00401-016-1650-1
Keywords: tau protein, Tauopathy, tau isoform, filamentous tau aggregate, cryo-electron microscopy, positron emission tomography ligand
Citation: Goedert M, Yamaguchi Y, Mishra SK, Higuchi M and Sahara N (2018) Tau Filaments and the Development of Positron Emission Tomography Tracers. Front. Neurol. 9:70. doi: 10.3389/fneur.2018.00070
Received: 08 December 2017; Accepted: 30 January 2018;
Published: 15 February 2018
Edited by:
Emmanuel Planel, Laval University, CanadaReviewed by:
Michal Novak, Slovak Academy of Sciences (SAS), SlovakiaCopyright: © 2018 Goedert, Yamaguchi, Mishra, Higuchi and Sahara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michel Goedert, bWdAbXJjLWxtYi5jYW0uYWMudWs=;
Naruhiko Sahara, c2FoYXJhLm5hcnVoaWtvQHFzdC5nby5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.