 Peter N. Alexandrov1
Peter N. Alexandrov1 Yuhai Zhao
Yuhai Zhao Walter J. Lukiw
Walter J. Lukiw- 1Russian Academy of Medical Sciences, Moscow, Russia
- 2LSU Neuroscience Center, Louisiana State University Health Sciences Center New Orleans, New Orleans, LA, United States
- 3Department of Anatomy and Cell Biology, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 4Department of Neurology, Shengjing Hospital, China Medical University, Shenyang, China
- 5Department of Ophthalmology, Louisiana State University Health Sciences Center New Orleans, New Orleans, LA, United States
- 6Department of Neurology, Louisiana State University Health Sciences Center New Orleans, New Orleans, LA, United States
Signaling between neurons in the human central nervous system (CNS) is accomplished through a highly interconnected network of presynaptic and postsynaptic elements essential in the conveyance of electrical and neurochemical information. One recently characterized core postsynaptic element essential to the efficient operation of this complex network is a relatively abundant ~184.7 kDa proline-rich synapse-associated cytoskeletal protein known as Shank3 (SH3-ankyrin repeat domain; encoded at human chr 22q13.33). In this “Perspectives” article, we review and comment on current advances in Shank3 research and include some original data that show common Shank3 deficits in a number of seemingly unrelated human neurological disorders that include sporadic Alzheimer’s disease (AD), autism spectrum disorder (ASD), bipolar disorder (BD), Phelan–McDermid syndrome (PMS; 22q13.3 deletion syndrome), and schizophrenia (SZ). Shank3 was also found to be downregulated in the CNS of the transgenic AD (TgAD) 5x familial Alzheimer’s disease murine model engineered to overexpress the 42 amino acid amyloid-beta (Aβ42) peptide. Interestingly, the application of known pro-inflammatory stressors, such as the Aβ42 peptide and the metal-neurotoxin aluminum sulfate, to human neuronal–glial cells in primary culture resulted in a significant decrease in the expression of Shank3. These data indicate that deficits in Shank3-expression may be one common denominator linking a wide-range of human neurological disorders that exhibit a progressive or developmental synaptic disorganization that is temporally associated with cognitive decline.
Shank3—An Essential Postsynaptic Scaffolding Protein
A small gene family of proline-enriched synapse-associated “SH3 and multiple ankyrin repeat domain” proteins known as Shank1, Shank2, and Shank3 encode abundant postsynaptic scaffolding proteins highly enriched at glutamatergic synapses in the human and murine central nervous system (CNS) (1–5). All three Shank genes have alternative promoter options and complex intron/exon arrangements resulting in the generation of a complex array of mRNA transcripts and protein isoforms. For example, Shank3 (also known as ProSAP2), the best studied of the three Shank proteins, is encoded at mouse chromosome 15E3 (analogous to human chr 22q13.3), spans ~60 kb of genomic DNA, has 22 exons and multiple intragenic promoters and several alternative splicing exons, and is highly expressed in CNS neurons (1, 2, 4, 6). As part of this small Shank gene family, the neuronal-enriched, multi-domain integral scaffolding protein Shank3 functions: (i) to organize and interconnect multiple postsynaptic-membrane proteins, ionic-channel and neurotransmitter receptors to the β-actin-enriched microfilament cytoskeletal-system; (ii) to regulate synaptic development, function, and plasticity by orchestrating the assembly of postsynaptic-signaling complexes, maintaining dendritic spine and synaptic architecture and supporting G-protein-coupled signaling-pathways (1–3, 5, 7). To add to this complexity, Shank3 gene expression appears to be regulated by epigenetic mechanisms that include DNA methylation, histone acetylation, and posttranscriptional regulators involving microRNA activities, thus contributing to the temporal and tissue-specific expression of different Shank3 isoforms (8, 9). Different Shank3 protein isoforms are alternately expressed according to brain region, cell type, and developmental stage; for example, (i) all five major Shank3 isoforms (Shank 3A-3E) are highly abundant in mouse hippocampal neurons throughout development and aging (1–3, 7); (ii) full length human Shank3 (1,731 amino acids; 184,667 Da) contains six highly interactive domains in tandem conducive to engagement in multiple protein–protein interactions at the postsynaptic density (PSD) (1–4); (iii) posttranscriptional regulation of Shank3 expression may be mediated by microRNAs, such as miRNA-34a, that itself has been implicated in multiple neuropsychiatric disorders involving synaptic disruption (8–11); and (iv) Shank3 possesses a remarkably complex interactome, conducive to Shank3’s role as a master organizer of a highly interconnected synaptic and cytoskeletal network (12–17). This involves the 4–7 nm diameter β-actin microfilaments, the major cytoskeletal protein found in the PSD, and the 10 nm diameter intermediate filaments within the soma, neurites, and synapses of neuronal cells (1–4, 6, 11, 14, 18, 19).
Alzheimer’s Disease (AD), Autism Spectrum Disorder (ASD), Bipolar Disorder (BD), Phelan–McDermid Syndrome (PMS), and Schizophrenia (SZ)—Shank3-Mediated Synaptic Degeneration and Cognitive Disability
As forementioned alternate Shank3 protein isoforms are differentially expressed according to developmental stage, cell type, and course of aging, suggesting the existence of isoform-specific roles for Shank3 at varying subcellular localizations, along different CNS neurites and synaptic endings at different stages of development and disease. Shank3 mutations that include gene breakpoints, deletions, point mutations, missense mutations, microdeletions, and nonsense mutations are associated with moderate to severe intellectual disabilities (2–7). AD with an incidence of one in seven people at age of 65–74 years and one in three people at age of 85 years and older, and the most common cause of intellectual decline associated with aging, is characterized by neuronal cell atrophy, swelling of neuronal nuclei, synaptic atrophy and loss, inflammatory neurodegeneration, progressive memory impairment, and a devastating cognitive decline (20–24). In AD brain tissues, in parallel, occurs the appearance of the 42 amino acid amyloid-beta (Aβ42) peptide-containing extracellular lesions, and although controversial, the accumulation of metallic neurotoxins such as aluminum (23–27). Shank3 was found to be reduced to ~0.25-fold of controls in AD neocortex (Figure 1). Shank3 gene mutations were first implicated (i) in ASD, a developmental disorder involving the disruption of social skills, speech and nonverbal communication, repetitive behaviors, and cognitive disabilities characterized pathologically by disordered gray and white matter, consistent changes in the densities of dendritic spines, region-specific abnormalities in neuronal morphology and cytoarchitectural organization, impairment in synaptic plasticity and progressive synaptic disorganization (28–30); (ii) in PMS, a developmental disorder that is caused by a 22q13.3 deletion and is characterized by ASD-like behavior, hypotonia, delayed or absent speech, intellectual disability, and cognitive impairment (28, 29, 31). Interestingly, patients with Shank3 mutations appear to have more-severe cognitive deficits than those with Shank1 or Shank2 mutations, and suggest that Shank3 mutation screening in clinical practice, and perhaps the restoration of the Shank3 gene activity may selectively rescue pathogenic synaptic defects of some ASD-associated behavioral phenotypes (7, 28, 29). Similar attributes in BD and SZ involving malformations in neuronal morphology, abnormalities in cytoarchitectural organization, impairment in synaptic plasticity and synaptic disorganization, impairment or insufficiency in a large portfolio of synaptic adhesion/organizing molecules, and cognitive disability are widely reported, and three extremely comprehensive studies have recently appeared (32–34). Also very recently, genetic- and bioinformatics-based analysis of brain region-specific Shank3 interactomes has been generated, which may be useful for understanding the heterogeneity of neuronal pathophysiology related to Shank3 genetic mutations and alternate Shank3 isoforms (12–15, 33–35). Here, we report the common downregulation in the expression of Shank3 in sporadic AD, ASD, BD, PMS, and SZ in 7-month-old 5x familial Alzheimer’s disease (5xFAD) transgenic animals exhibiting extensive amyloid deposition, compared with age-matched controls and in stressed human neuronal–glial (HNG) cell cocultures (see below; Figures 1 and 2). Ongoing work is currently in progress to further compare other neurological diseases involving progressive synaptic disorganization and intellectual disability, including attention deficit hyperactivity disorder, epilepsy, prion disease and others for Shank3 deficits, and how different “loss-of-function” mutations or altered Shank3 expression can lead to such phenotypic diversity.
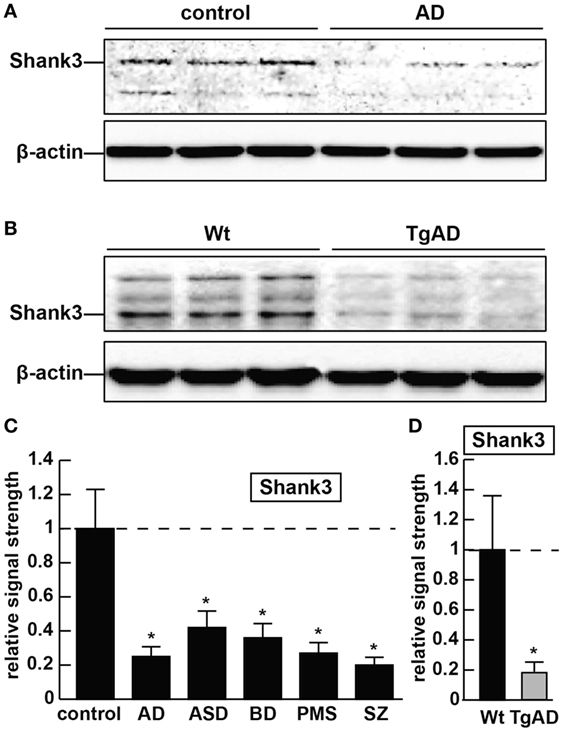
Figure 1. The abundance of the ~184.7 kDa Shank3 protein is decreased in synaptosome preparations from multiple human neurological disorders; (A) Shank3 protein levels are reduced in age- and gender-matched (all female) human brain temporal lobe neocortex from sporadic Alzheimer’s disease (AD) patients (control and AD; upper panel) when compared with control β-actin signals in the same sample [mean age ± 1 SD; control mean age 75 ± 8.3 years, N = 6; AD mean age 77 ± 6.5 years, N = 3]; all postmortem intervals (PMI; interval of death-to brain-freezing at −81°C) were 3 h or less; β-actin antibody 3598-100; Sigma-Aldrich, St. Louis, MO, USA and human Shank3 monoclonal antibody (C-4; sc-10479: H-75; sc-377088; Santa Cruz Biotechnologies, Santa Cruz, CA, USA; the main band at ~184.7 kDa is Shank3 protein); (B) similarly the levels of Shank3 protein are reduced in murine brain cortex from 7-month-old 5x familial Alzheimer’s disease (5xFAD) transgenic animals versus wild-type C57BL/6 age-matched controls; wild-type (Wt) and TgAD (5xFAD) murine models compared with β-actin signals in the same sample, as demonstrated by representative Western blot analysis using methods previously described in detail by our laboratory and as suggested by the manufacturer (11, 36, 37); modulation of actin dynamics at the synapse is likely to drive the cytoarchitectural changes that are associated with synaptic plasticity; in panels (A,B) multiple bands for Shank3 protein on Western gels may be indicative of alternate translation products from differentially spliced Shank3 mRNAs and/or amino acid side-chain modification [(1–5, 7); see text]; (C) bar graphs representative of Shank3 protein levels in age- and gender-matched (all female) human brain temporal lobe neocortex in control, AD, autism spectrum disorder (ASD), bipolar disorder (BD), Phelan–McDermid syndrome (PMS) (22q13.3 deletion syndrome), and schizophrenia (SZ) using Western blot analysis of synaptosome-enriched fractions [prepared using differential gradient centrifugation (12, 38–40)]; for control and AD, mean age ± 1 SD is given above; for all six tissue types PMIs were 3 h or less; ASD, BD, PMS, or SZ cases each had their own individual age-matched controls set to 1.0 in panel (C); there were no significant differences in age between ASD, BD, PMS, or SZ cases and their individual controls; mean ages for ASD, BD, PMS, and SZ cases were 7.2 ± 2.7, 41.1 ± 7.1, 38.4 ± 6.8, and 44.3 ± 6.5 years, respectively; in these neurological disorders, Shank3 protein abundance was observed to be reduced from 0.21-fold (SZ) to 0.42-fold (ASD) of controls; (D) Shank3 protein abundance is similarly reduced to approximately 0.2-fold of control in the cortex of 7-month-old 5xFAD TgAD models; N = 3–6 samples of each neurological deficit or control; a dashed horizontal line at 1.0 has been included for ease of comparison; bars are the mean ± 1 SD of that mean; *p < 0.01 (ANOVA).
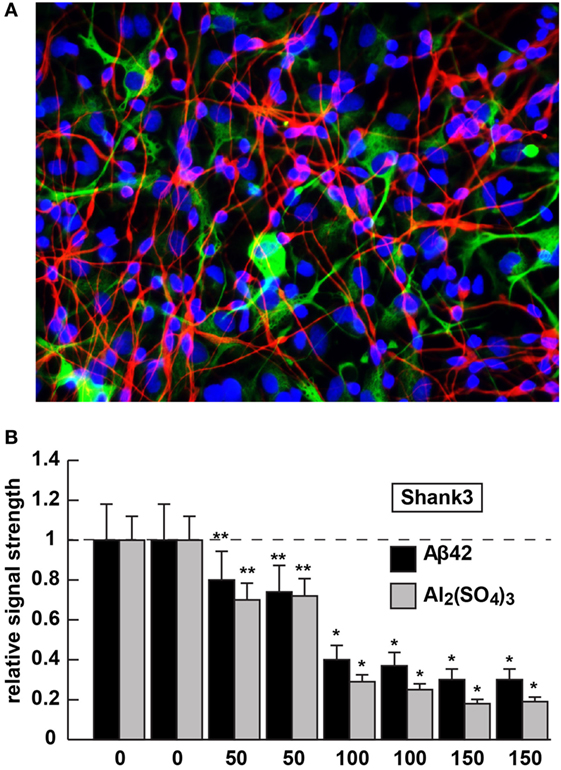
Figure 2. Shank3 downregulation in 42 amino acid amyloid-beta (Aβ42) peptide- or aluminum sulfate [Al2(SO4)3]-stressed human neuronal–glial (HNG) cells in primary coculture; (A) 2-week-old HNG cocultures (approximately 60% neurons and 40% astroglia) stained with the neuron-specific marker β tubulin 3 (red signal; λmax ~680 nm) or the astroglial-specific marker GFAP (green signal; λmax ~550 nm); HNG nuclei have been also stained with DAPI (blue; λmax ~430 nm); culture of HNG cells has been previously described in detail by our laboratory (19, 41, 42); human brain neurons do not culture well without the presence of astroglial cells; magnification 40×; HNG cocultures were incubated with Aβ42 peptide (0, 50, 100, or 150 nM for 36 h) or ultrapure aluminum sulfate [Al2(SO4)3; 0, 50, 100 or 150 nM for 36 h]; these concentrations of stressors and times were selected from previous reports of Aβ42 peptide- and aluminum sulfate-induced inflammatory neurodegeneration and other relevant reports on neurotoxicity toward HNG cells, human brain microvessel endothelial cells that line the cerebral vasculature and other brain cell types (25–27, 36, 42–47); methodologies involving the application of Aβ42 peptide- or aluminum sulfate as physiologically realistic stressors to HNG cells in primary coculture have been explained in detail in previously published reports from our laboratory (11, 36, 42, 45–47); HNG whole-cell protein extracts were prepared, and Shank3 abundance was quantified using Western analysis and ImageQuant as described in Figures 1A,B (10, 36, 37, 43); (B) results are quantified in bar-graph format; at 100 nM Aβ42 peptide- or aluminum sulfate-treatment Shank3 levels were reduced between 0.2- and 0.3-fold of untreated control values; N = 3–5 samples of each treatment or condition; in Figure 2B, a dashed horizontal line at 1.0 has been included for ease of comparison; bars represent the mean ± 1 SD; *p < 0.01; **p < 0.05 (ANOVA).
Aβ42 Peptide or Aluminum Sulfate [Al2(SO4)3] Downregulate Shank3 Expression
The 42 amino acid amyloid-beta peptide and aluminum (sulfate) were chosen as highly relevant pathogenic neurological stressors for the experimental treatment of HNG cells and assessment of Shank3 expression for the following reasons (see also Figure 2). First, the highly amyloidogenic Aβ42 peptides are one of the two key molecular lesions associated with AD (the other being and hyperphosphorylated tau), and multiple transgenic murine models for AD (TgAD) and involving massive amyloid overexpression have been generated (23, 24). The 5xFAD murine TgAD model containing five familial AD mutations; including three amyloid mutations and two presenilin 1 (PSEN1) mutations [APP KM670/671NL (Swedish), APP I716V (Florida), APP V717I (London), PSEN1 M146L (A>C), and PSEN1 L286V] exhibit substantial Aβ42 peptide generation, amyloid plaque deposition, astrogliosis, and cognitive impairment by 7 months of age and also exhibit deficits in Shank3 at this time point [(25); Figure 1].
Aluminum is a ubiquitous metallic neurotoxin and extremely potent genotoxin (25–27). Human intake of an average of 10 mg Al/day (range 10–1,000 mg Al/day) occurs chiefly via the ingestion of drinking water, food, medicine, and inhalation of airborne dust (43, 44, 48). Fortunately, the low solubility of aluminum at biological pH and highly evolved endothelial- and epithelial cell-based gastrointestinal and blood–brain barriers prevent this potent and ubiquitous metallotoxin from easy access to human biological compartments. However, aluminum that does gain entry into the CNS in animal models has been shown to induce NF-κB and specific microRNA-mediated inflammatory and neuro-immune pathogenic gene expression programs that closely emulate many aspects of CNS pathology and progressive memory dysfunction, including synaptic disorganization, as seen in advanced late stage AD brain (43, 44, 48). Our laboratory and others, for example, have further shown (i) the aluminum-mediated downregulation of several key brain essential genes in multiple neurodegenerative disorders by a pro-inflammatory NF-κB-regulated microRNA-146a (27, 36, 43, 45, 46, 48); (ii) a robust upregulation of microRNA-34a expression by reactive oxygen species and NF-κB increases in the presence of aluminum sulfate in HNG cocultures that decreases the expression of Shank3 [see Figure 2; (36, 46)]. We speculate that this may contribute to altered neurotransmission in multiple neuropsychiatric disorders [(25–27); unpublished observations]. The Shank3 downregulation in Aβ42 peptide- or aluminum sulfate-stressed HNG cells in primary culture (at concentrations of 0, 50, 100, or 150 nM for 36 h) is shown in Figure 2. Altered neurotransmission and synaptic dysfunction in the presence of amyloid peptides or neurotoxic metal salts are widely documented in very recent reviews in this subject area (25–27, 31, 41, 43, 44, 48–50).
Concluding Remarks
Multifactorial diseases that include sporadic AD, ASD, BD, PMS, and SZ exhibit considerable heterogeneity in their presentation, and on the surface appear to exhibit more neuropathological variability than commonality. However, current research has advanced our understanding of the dynamics of the postsynaptic multi-domain proline-rich synapse-associated Shank3 protein and, perhaps surprisingly, has revealed a common Shank3-mediated “generic” disruption of synaptic organization in each of these neurological disorders. Interestingly, in neurological diseases involving synaptic disruption and cognitive decline, not all synaptic proteins appear to be equally affected. For example, the PSD-, β-actin-, and Shank3-associated scaffolding protein Homer 1 is decreased in AD but not in SZ brains [unpublished observations; (35)], and the relatively abundant PSD-95-associated DLGAP scaffold protein subtype DLGAP4 is significantly decreased in SZ but not in BD (15). Recently published studies including our own provides at least five novel and significant findings: (i) that the essential scaffolding protein Shank3 appears to be commonly and significantly reduced in synaptosomal preparations of brain tissues obtained from AD, ASD, BD, PMS, and SZ patients (2, 6–8, 37); (ii) that in the 5xFAD amyloid-overexpressing TgAD model [bearing the APP KM670/671NL (Swedish), APP I716V (Florida), APP V717I (London), PSEN1 M146L (A>C), and PSEN1 L286V mutations], the accumulation of Aβ42 peptides in the brain appears to be accompanied by reduced bioavailability of Shank3 (Figures 1 and 2); (iii) that research implicating Shank3 as an essential postsynaptic cytoskeletal organizing protein infers that the presence of Aβ42 peptides driving Shank3 downregulation would also be disruptive toward normal synaptic structure and signaling capabilities (12–17); (iv) that deficits in Shank3 abundance can be induced in HNG cells in primary coculture from the peripheral application of AD-relevant stressor substances, at physiologically realistic concentrations, that include Aβ42 peptides or environmentally abundant neurotoxins such as aluminum sulfate (42–44, 48); and (v) that Aβ42 peptide, aluminum sulfate, and perhaps other related intrinsic or environmental neurotoxins via depletion of essential synaptic proteins such as Shank3 can be detrimental to the homeostatic maintenance of synaptic structure, function, and plasticity, and may be conducive to a phenotype involving progressive cognitive insufficiency (42–44, 47, 48, 51).
Importantly, these experimental interpretations should remain be somewhat speculative as they are derived from preliminary studies, and more research is required to further clarify the significance of Shank3 deficits in other neurological disorders and experimental models. Whether Shank3 downregulation is a direct cause or consequence of these neurological disorders, or an unrelated epiphenomenon remains open to question. However, several recent studies of Shank3’s extensive and remarkably diverse interactome further underscore the idea that this highly interconnected synaptic protein is a “master cytoskeletal hub” within the PSD network. This further suggests that Shank3 disruption or deficiency could contribute to multiple neurological disorders associated with: (i) altered neurogenesis and disrupted expression of synaptic proteins that affect synaptic organization and plasticity; (ii) altered activation of cell adhesion molecules such as integrin; (iii) developmental and morphogenetic abnormalities; (iv) changes in biometal abundances and transporters such as those for zinc; (v) alterations in G protein-coupled metabotropic glutamate receptor 5 (mGluR5)-Homer scaffolds and mGlu5 receptors; and (vi) peripheral aberrations such as abnormally heightened sensitivity to pain in diseases associated with Shank3 deficits or in Shank3 animal models (12–17, 47, 51–61). From these most recent findings, we may further speculate (i) that deficits in Shank3 expression and synaptic structural disorganization represent a common underlying mechanism for neurological disorders, which exhibit abnormal synaptic dynamics and a neurodegenerative phenotype; (ii) predict that detectable alterations in Shank3 gene structure and/or alternate Shank3 mRNA isoforms screened in utero or in newborns may be diagnostic for the onset of neurological disorders such as AD, ASD, BD, PMS, and SZ in later life; and (iii) propose that pharmacological approaches including anti-microRNA strategies directed toward the manipulation of Shank3 and/or related synaptic protein expression may be useful in the clinical management of multiple neuropsychiatric disorders, which exhibit an underlying commonality in intellectual disability and/or progressive cognitive decline.
Ethics Statement
All procedures involving murine and postmortem human tissues were followed and handled in strict accordance with the ethics review board policies at donor institutions and the Institutional Biosafety Committee/Institutional Review Board (IBC/IRB) ethical guidelines at the LSU Health Sciences Center, LA 70112 (IBC# 12323; IRB# 6774).
Author Contributions
PA, LC, VJ, and YZ performed the experiments; YZ and WL analyzed the data; WL organized the data and wrote the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The work in this paper was presented in part at the Vavilov Institute of General Genetics Autumn 2016 Seminar Series (Институт общей генетики имени Вавилова Осень 2016 Семинар серии) in Moscow, Russia, October 2016, at the Society for Neuroscience (SFN) Annual Meeting, San Diego CA, USA, November 2016, and will be presented in Abstract-Special Symposium platform format at the Society for Neuroscience (SFN) Annual Meeting, Washington, DC, USA, November 2017. Sincere thanks are extended to Drs. L. Carver, J. G. Cui, F. Culicchia, C. Eicken, K. Navel, W. Poon, and the late Drs. J. M. Hill and T. P. Kruck for helpful discussions in this research area, for short postmortem interval (PMI) human brain tissues or extracts, for initial bioinformatics and data interpretation, and to D. Guillot and A. I. Pogue for expert technical assistance and medical artwork. Thanks are also extended to the University of California at Irvine Brain Bank, the University of Maryland Brain Bank, the Harvard Brain Tissue Resource Center, the LSU School of Medicine, and the many neuropathologists, physicians, and researchers of the US and Canada who have provided high quality, short PMI human CNS, or extracted brain tissue fractions for scientific analysis and study. Research on the microRNAs, pro-inflammatory, and pathogenic signaling in the Lukiw laboratory involving the microbiome, the innate-immune response, amyloidogenesis, synaptogenesis, and neuroinflammation in AD, prion, and in other neurological diseases was supported through an unrestricted grant to the LSU Eye Center from Research to Prevent Blindness (RPB); the Louisiana Biotechnology Research Network (LBRN) and NIH grants NEI EY006311, NIA AG18031, and NIA AG038834 (WL). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Aging, the National Center for Research Resources, or the National Institutes of Health.
Abbreviations
5xFAD, 5x familial Alzheimer’s disease (a TgAD model carrying 2 PSEN1 and 3βAPP mutations; see text); AD, Alzheimer’s disease; ASD, autism spectrum disorder; BD, bipolar disorder; mGlu5 miRNA, microRNA; PMS, Phelan–McDermid syndrome (22q13.3 deletion syndrome); SZ, schizophrenia.
References
1. Available from: http://www.genecards.org/cgi-bin/carddisp.pl?gene=SHANK3 (accessed December 6, 2017).
2. Monteiro P, Feng G. SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat Rev Neurosci (2017) 18:147–57. doi: 10.1038/nrn.2016.183
3. Sarowar T, Grabrucker AM. Actin-dependent alterations of dendritic spine morphology in shankopathies. Neural Plast (2016) 2016:8051861.
4. Leblond CS, Nava C, Polge A, Gauthier J, Huguet G, Lumbroso S, et al. Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet (2014) 10:e1004580. doi:10.1371/journal.pgen.1004580
5. Sala C, Vicidomini C, Bigi I, Mossa A, Verpelli C. Shank synaptic scaffold proteins: keys to understanding the pathogenesis of autism and other synaptic disorders. J Neurochem (2015) 135:849–58. doi:10.1111/jnc.13232
6. Fu AK, Ip NY. Regulation of postsynaptic signaling in structural synaptic plasticity. Curr Opin Neurobiol (2017) 45:148–55. doi:10.1016/j.conb.2017.05.016
7. Mei Y, Monteiro P, Zhou Y, Kim JA, Gao X, Fu Z, et al. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature (2016) 530:481–4. doi:10.1038/nature16971
8. Hill JM, Lukiw WJ. MicroRNA (miRNA)-mediated pathogenetic signaling in Alzheimer’s disease (AD). Neurochem Res (2016) 41:96–100. doi:10.1007/s11064-015-1734-7
9. Choi SY, Pang K, Kim JY, Ryu JR, Kang H, Liu Z, et al. Post-transcriptional regulation of SHANK3 expression by miRNAs related to multiple neuropsychiatric disorders. Mol Brain (2015) 8:74. doi:10.1186/s13041-015-0165
10. Zhao Y, Jaber V, Lukiw WJ. Over-expressed pathogenic miRNAs in Alzheimer’s disease (AD) and prion disease (PrD) drive deficits in TREM2-mediated Aβ42 peptide clearance. Front Aging Neurosci (2016) 8:140. doi:10.3389/fnagi.2016.00140
11. Bhattacharjee S, Zhao Y, Dua P, Rogaev EI, Lukiw WJ. microRNA-34a-mediated down-regulation of the microglial-enriched triggering receptor and phagocytosis-sensor TREM2 in age-related macular degeneration. PLoS One (2016) 11:e0150211. doi:10.1371/journal.pone.0150211
12. Sundararajan T, Manzardo AM, Butler MG. Functional analysis of schizophrenia genes using GeneAnalytics program and integrated databases. Gene (2018) 641:25–34. doi:10.1016/j.gene.2017.10.035
13. Li J, Wilkinson B, Clementel VA, Hou J, O’Dell TJ, Coba MP. Long-term potentiation modulates synaptic phosphorylation networks and reshapes the structure of the postsynaptic interactome. Sci Signal (2016) 9(440):rs8. doi:10.1126/scisignal.aaf6716
14. Lee Y, Kang H, Lee B, Zhang Y, Kim Y, Kim S, et al. Integrative analysis of brain region-specific Shank3 interactomes for understanding the heterogeneity of neuronal pathophysiology related to Shank3 mutations. Front Mol Neurosci (2017) 10:110. doi:10.3389/fnmol.2017.00110
15. Rasmussen AH, Rasmussen HB, Silahtaroglu A. The DLGAP family: neuronal expression, function and role in brain disorders. Mol Brain (2017) 10:43. doi:10.1186/s13041-017-0324-9
16. Richards C, Powis L, Moss J, Stinton C, Nelson L, Oliver C. Prospective study of autism phenomenology and the behavioral phenotype of Phelan-McDermid syndrome: comparison to fragile X syndrome, Down syndrome and idiopathic autism spectrum disorder. J Neurodev Disord (2017) 9:37. doi:10.1186/s11689-017-9217-6
17. Sungur AÖ, Schwarting RKW, Wöhr M. Behavioral phenotypes and neurobiological mechanisms in the Shank mouse model for autism spectrum disorder: a translational perspective. Behav Brain Res (2017). doi:10.1016/j.bbr.2017.09.038
18. Cajigas IJ, Tushev G, Will TJ, tom Dieck S, Fuerst N, Schuman EM. The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron (2012) 74:453–66. doi:10.1016/j.neuron.2012.02.036
19. Buxbaum AR, Wu B, Singer RH. Single β-actin mRNA detection in neurons reveals a mechanism for regulating its translatability. Science (2014) 343:419–22. doi:10.1126/science.1242939
20. (2017). Available from: https://www.alz.org/documents_custom/2016-facts-and-figures.pdf; http://www.alz.org/facts/?gclid (accessed December 6, 2017).
21. Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM. Synaptic impairment in Alzheimer’s disease: a dysregulated symphony. Trends Neurosci (2017) 40:347–57. doi:10.1016/j.tins.2017.04.002
22. Kozlov S, Afonin A, Evsyukov I, Bondarenko A. Alzheimer’s disease: as it was in the beginning. Rev Neurosci (2017). doi:10.1515/revneuro-2017-0006
23. Brody DL, Jiang H, Wildburger N, Esparza TJ. Non-canonical soluble amyloid-beta aggregates and plaque buffering: controversies and future directions for target discovery in Alzheimer’s disease. Alzheimers Res Ther (2017) 9:62. doi:10.1186/s13195-017-0293-3
24. (2017). Available from: http://www.alzforum.org/research-models/alzheimers-disease (accessed December 6, 2017).
25. Pogue AI, Lukiw WJ. Natural and synthetic neurotoxins in our environment: from Alzheimer’s disease (AD) to autism spectrum disorder (ASD). J Alzheimers Dis Parkinsonism (2016) 6:249.
26. Morris G, Puri BK, Frye RE. The putative role of environmental aluminum in the development of chronic neuropathology in adults and children. How strong is the evidence and what could be the mechanisms involved? Metab Brain Dis (2017). doi:10.1007/s11011-017-0077-2
27. Walton JR. Chronic aluminum intake causes Alzheimer’s disease: applying Sir Austin Bradford Hill’s causality criteria. J Alzheimers Dis (2014) 40:765–838. doi:10.3233/JAD-132204
28. Schroeder JC, Reim D, Boeckers TM, Schmeisser MJ. Genetic animal models for autism spectrum disorder. Curr Top Behav Neurosci (2017) 30:311–24. doi:10.1007/7854_2015_407
29. Ismail FY, Fatemi A, Johnston MV. Cerebral plasticity: windows of opportunity in the developing brain. Eur J Paediatr Neurol (2017) 21:23–48. doi:10.1016/j.ejpn.2016.07.007
30. Fukata Y, Yokoi N, Miyazaki Y, Fukata M. The LGI1-ADAM22 protein complex in synaptic transmission and synaptic disorders. Neurosci Res (2017) 116:39–45. doi:10.1016/j.neures.2016.09.011
31. Varghese M, Keshav N, Jacot-Descombes S, Warda T, Wicinski B, Dickstein DL, et al. Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol (2017). doi:10.1007/s00401-017-1736-4
32. Ashok AH, Marques TR, Jauhar S, Nour MM, Goodwin GM, Young AH, et al. The dopamine hypothesis of bipolar affective disorder: the state of the art and implications for treatment. Mol Psychiatry (2017) 22:666–79. doi:10.1038/mp.2017.16
33. Al-Diwani A, Pollak TA, Langford AE, Lennox BR. Synaptic and neuronal autoantibody-associated psychiatric syndromes: controversies and hypotheses. Front Psychiatry (2017) 8:13. doi:10.3389/fpsyt.2017.00013
34. Wang X, Xu Q, Bey AL, Lee Y, Jiang YH. Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Mol Autism (2014) 5:30. doi:10.1186/2040-2392-5-30
35. Lee Y, Kim SG, Lee B, Zhang Y, Kim Y, Kim S, et al. Striatal transcriptome and interactome analysis of Shank3-overexpressing mice reveals the connectivity between Shank3 and mTORC1 Signaling. Front Mol Neurosci (2017) 10:201. doi:10.3389/fnmol.2017.00201
36. Lukiw WJ, Percy ME, Kruck TP. Nanomolar aluminum induces pro-inflammatory and pro-apoptotic gene expression in human brain cells in primary culture. J Inorg Biochem (2005) 99:1895–8. doi:10.1016/j.jinorgbio.2005.04.021
37. Zhao Y, Jaber V, Lukiw WJ. Secretory products of the human GI tract microbiome and their potential impact on Alzheimer’s disease (AD): detection of lipopolysaccharide (LPS) in AD hippocampus. Front Cell Infect Microbiol (2017) 7:318. doi:10.3389/fcimb.2017.00318
38. (2017). Available from: https://www.thermofisher.com/us/en/home/life-science/protein-biology/ (accessed December 6, 2017).
39. Kamat PK, Kalani A, Tyagi N. Method and validation of synaptosomal preparation for isolation of synaptic membrane proteins from rat brain. MethodsX (2014) 1:102–7. doi:10.1016/j.mex.2014.08.002
40. Evans GJ. Subcellular fractionation of the brain: preparation of synaptosomes and synaptic vesicles. Cold Spring Harb Protoc (2015) 2015:462–6. doi:10.1101/pdb.prot083469
41. Müller UC, Deller T, Korte M. Not just amyloid: physiological functions of the amyloid precursor protein family. Nat Rev Neurosci (2017) 18:281–98. doi:10.1038/nrn.2017.29
42. Pogue AI, Zhao Y, Jaber V, Percy ME, Cong L, Lukiw WJ. Selective targeting and accumulation of aluminum in tissues of C57BL/6J mice fed aluminum sulfate activates a pro-inflammatory NF-kB-microRNA-146a signaling program. J Neurol Neurotoxicol (2017) (in press).
43. Pogue AI, Lukiw WJ. Aluminum, the genetic apparatus of the human CNS and Alzheimer’s disease (AD). Morphologie (2016) 100:56–64. doi:10.1016/j.morpho.2016.01.001
44. Pogue AI, Dua P, Hill JM, Lukiw WJ. Progressive inflammatory pathology in the retina of aluminum-fed 5xFAD transgenic mice. J Inorg Biochem (2015) 152:206–9. doi:10.1016/j.jinorgbio.2015.07.009
45. Zhao Y, Hill JM, Bhattacharjee S, Percy ME, Pogue AI, Lukiw WJ. Aluminum-induced amyloidogenesis and impairment in the clearance of amyloidpeptides from the central nervous system in Alzheimer’s disease. Front Neurol (2014) 5:167. doi:10.3389/fneur.2014.00167
46. Alexandrov PN, Kruck TP, Lukiw WJ. Nanomolar aluminum induces expression of the inflammatory systemic biomarker C-reactive protein (CRP) in human brain microvessel endothelial cells (hBMECs). J Inorg Biochem (2015) 152:210–3. doi:10.1016/j.jinorgbio.2015.07.013
47. Pogue AI, Li YY, Cui JG, Zhao Y, Kruck TPA, Percy ME, et al. Characterization of an NF-κB-regulated, miRNA-146a-mediated down-regulation of complement factor H (CFH) in metal-sulfate-stressed human brain cells. J Inorg Biochem (2009) 103:1591–5. doi:10.1016/j.jinorgbio.2009.05.012
48. Wang P, Wang ZY. Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res Rev (2017) 35:265–90. doi:10.1016/j.arr.2016.10.003
49. Nixon RA. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. FASEB J (2017) 31:2729–43. doi:10.1096/fj.201700359
50. Musardo S, Marcello E. Synaptic dysfunction in Alzheimer’s disease: from the role of amyloid β-peptide to the α-secretase ADAM10. Eur J Pharmacol (2017). doi:10.1016/j.ejphar.2017.06.018
51. Li K, Li L, Cui B, Gai Z, Li Q, Wang S, et al. Early postnatal exposure to airborne fine particulate matter induces autism-like phenotypes in male rats. Toxicol Sci(2017). doi:10.1093/toxsci/kfx240
52. Wei SC, Yang-Yen HF, Tsao PN, Weng MT, Tung CC, Yu LCH, et al. SHANK3 regulates intestinal barrier function through modulating ZO-1 expression through the PKCε-dependent pathway. Inflamm Bowel Dis (2017). doi:10.1097/MIB.0000000000001250
53. Zhao H, Tu Z, Xu H, Yan S, Yan H, Zheng Y, et al. Altered neurogenesis and disrupted expression of synaptic proteins in prefrontal cortex of SHANK3-deficient non-human primate. Cell Res (2017) 27:1293–7. doi:10.1038/cr.2017.95
54. Lilja J, Zacharchenko T, Georgiadou M, Jacquemet G, De Franceschi N, Peuhu E, et al. SHANK proteins limit integrin activation by directly interacting with Rap1 and R-Ras. Nat Cell Biol (2017) 19:292–305. doi:10.1038/ncb3487
55. Han Q, Kim YH, Wang X, Liu D, Zhang ZJ, Bey AL, et al. SHANK3 deficiency impairs heat hyperalgesia and TRPV1 signaling in primary sensory neurons. Neuron (2016) 92:1279–93. doi:10.1016/j.neuron.2016.11.007
56. Zhang C, Wu Z, Hong W, Peng D, Fang Y. Evaluating the association between the SHANK3 gene and bipolar disorder. Psychiatry Res (2016) 244:284–8. doi:10.1016/j.psychres.2016.07.058
57. Dhamne SC, Silverman JL, Super CE, Lammers SHT, Hameed MQ, Modi ME, et al. Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol Autism (2017) 8:26. doi:10.1186/s13229-017-0142-z
58. Kathuria A, Nowosiad P, Jagasia R, Aigner S, Taylor RD, Andreae LC, et al. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol Psychiatry (2017). doi:10.1038/mp.2017.185
59. Pfaender S, Sauer AK, Hagmeyer S, Mangus K, Linta L, Liebau S, et al. Zinc deficiency and low enterocyte zinc transporter expression in human patients with autism related mutations in SHANK3. Sci Rep (2017) 7:45190. doi:10.1038/srep45190
60. Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK, et al. Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat Commun (2016) 7:11459. doi:10.1038/ncomms11459
Keywords: 5x familial Alzheimer’s disease transgenic mice, 42 amino acid amyloid-beta peptides, aluminum sulfate, Alzheimer’s disease, autism spectrum disorder, bipolar disorder-schizophrenia, neurotransmission, Shank3 protein
Citation: Alexandrov PN, Zhao Y, Jaber V, Cong L and Lukiw WJ (2017) Deficits in the Proline-Rich Synapse-Associated Shank3 Protein in Multiple Neuropsychiatric Disorders. Front. Neurol. 8:670. doi: 10.3389/fneur.2017.00670
Received: 28 August 2017; Accepted: 27 November 2017;
Published: 11 December 2017
Edited by:
Giuseppe Di Fede, Istituto Neurologico Carlo Besta (IRCCS), ItalyReviewed by:
Alessandro Bertoli, Università degli Studi di Padova, ItalyCarlo Sala, Istituto di Neuroscienze (CNR), Italy
Copyright: © 2017 Alexandrov, Zhao, Jaber, Cong and Lukiw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Walter J. Lukiw, d2x1a2l3QGxzdWhzYy5lZHU=