 Gyaninder Pal Singh1
Gyaninder Pal Singh1 Tumul Chowdhury
Tumul Chowdhury Bernhard Schaller
Bernhard Schaller
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurol. , 05 December 2016
Sec. Autonomic Neuroscience
Volume 7 - 2016 | https://doi.org/10.3389/fneur.2016.00221
This article is part of the Research Topic The trigemino-cardiac reflex: beyond the diving reflex View all 9 articles
Sudden infant death syndrome (SIDS) is an unexplained death in infants, which usually occurs during sleep. The cause of SIDS remains unknown and multifactorial. In this regard, the diving reflex (DR), a peripheral subtype of trigeminocardiac reflex (TCR), is also hypothesized as one of the possible mechanisms for this condition. The TCR is a well-established neurogenic reflex that manifests as bradycardia, hypotension, apnea, and gastric hypermotility. The TCR shares many similarities with the DR, which is a significant physiological adaptation to withstand hypoxia during apnea in many animal species including humans in clinical manifestation and mechanism of action. The DR is characterized by breath holding (apnea), bradycardia, and vasoconstriction, leading to increase in blood pressure. Several studies have described congenital anomalies of autonomic nervous system in the pathogenesis of SIDS such as hypoplasia, delayed neuronal maturation, or decreased neuronal density of arcuate nucleus, hypoplasia, and neuronal immaturity of the hypoglossal nucleus. The abnormalities of autonomic nervous system in SIDS may explain the role of TCR in this syndrome involving sympathetic and parasympathetic nervous system. We reviewed the available literature to identify the role of TCR in the etiopathogenesis of SIDS and the pathways and cellular mechanism involved in it. This synthesis will help to update our knowledge and improve our understanding about this mysterious, yet common condition and will open the door for further research in this field.
Sudden infant death syndrome (SIDS) is defined as the sudden unexplained death of a seemingly healthy child less than 1 year of age, usually during sleep. For the diagnosis of SIDS, the death should remain unexplained even after the autopsy, investigation of mortality scene, and review of clinical history (1). SIDS remains a leading cause of death in infants between ages of 1 month and 1 year. The incidence of SIDS varies between regions and among racial and ethnic subgroups (2, 3). It is a multifactorial disorder, the cause of which is still not fully elucidated. The exact cause of death in SIDS remains unclear; however, the exaggeration of parasympathetic activity and cardiorespiratory response to hypoxia has been suggested as a possible underlying mechanism (4–9). In addition, postnatal age, gestational age at birth, and level of arousability are also linked with SIDS (10, 11).
Infants who succumb to SIDS typically experience a severe bradycardia, which is the most shared and predictive event in infants monitored for life-threatening incidents (12, 13). It may be preceded or is accompanied by centrally mediated apnea. Such abnormal and exaggerated response to sensory trigeminal nerve stimulation has also been implicated in the etiopathogenesis of SIDS (14, 15).
Stimulation of trigeminal nerve leads to consecutive reflex bradycardia, hypotension, apnea, and gastric hypermotility, commonly known as the trigeminocardiac reflex (TCR). This reflex is most often transient, but sometimes may be pronounced and sustainable, particularly, in infants. The diving reflex (DR) (a subtype of TCR) is triggered as a result of stimulation of one of the sensory branches of the trigeminal nerve and leads to inhibition of cardiorespiratory center, thereby causes bradycardia and apnea (16–19). An exaggerated response to hypoxia (i.e., augmented TCR response) causing lethal bradycardia and apnea can be accused of sudden death in the victims of SIDS. In this article, we reviewed the available literature on SIDS to identify the evidence and explore the role of TCR in the pathogenesis of SIDS.
Trigeminocardiac reflex has been classified into various subtypes including central, peripheral, and ganglionic TCR (17, 20–24). The central TCR is triggered by the stimulation of the intracranial part of trigeminal nerve proximal to Gasserian ganglion, and the peripheral TCR is triggered by the stimulation of the ophthalmic, maxillary, or mandibular branches of trigeminal nerve (25–30). TCR triggered due to the direct stimulation of Gasserian ganglion is classified as a separate entity (19).
The branches of the trigeminal nerve, Gasserian ganglion, the sensory nucleus of the trigeminal nerve forms the afferent pathway of the reflex (26, 31–33) (Figure 1). The short internuncial nerve fibers of the reticular formation connect the afferent pathway to the efferent pathway, which is predominantly formed by the parasympathetic neurons of the dorsal motor nucleus of the vagus nerve and nucleus ambiguus (19). Animal studies have shown the involvement of several other brainstem nuclei in the TCR pathway, which includes trigeminal nucleus caudalis, paratrigeminal nucleus, parabrachial nucleus, rostral ventrolateral medulla oblongata, and dorsal medullary reticular field (34–36). Also, various subtypes of TCR show a difference in their reflex arc. While peripherally originated TCR is relayed via the spinal nucleus of the trigeminal nerve to the Kölliker–Fuse nucleus, the centrally originated TCR is conveyed via the nucleus of the solitary tract to the lateral parabrachial nucleus (35).
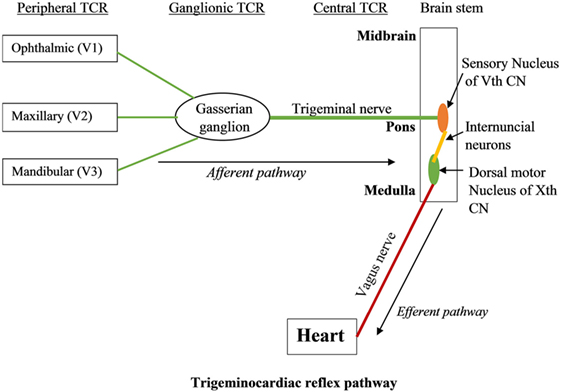
Figure 1. The trigeminocardiac reflex pathway.
Activation of the sympathetic nervous system has been implicated for the other less common manifestations of TCR, such as tachycardia and hypertension, which are seen in some subtypes of TCR. Studies have revealed that stimulation of the anterior ethmoidal nerve in the nasal mucosa (peripheral TCR) may simultaneously activate the sympathetic and vagal responses. This may result in parasympathetically mediated bradycardia along with sympathetically mediated peripheral vasoconstriction and hypertension (37, 38). In contrast to this, the centrally stimulated TCR manifests as bradycardia and hypotension due to activation of cardioinhibitory vagal response, whereas the ganglionic TCR is clinically present as either increase or decrease in heart rate (bradycardia/tachycardia) and blood pressure (hypotension/hypertension) (39). These varied presentations of TCR are due to co-activation of parasympathetic and sympathetic nervous system (39).
The DR is a powerful autonomic reflex that manifests as the reflex bradycardia, apnea, peripheral vasoconstriction, and hypertension triggered by submersion of a face in cold water through branches of the trigeminal nerve (40). Both TCR and DR are phylogenetically oxygen-conserving reflexes, and researchers reveal a similar reflex arch in both (41). Thus, DR appears to be another subtype of TCR (39). The difference between DR and peripheral TCR shows different effect on blood pressure. Although peripheral TCR causes normotension or hypotension, DR leads to hypertension. This is due to intense peripheral vasoconstriction caused by more strong sympathetic stimulation during DR than during peripheral TCR (42). This reflex also persists in humans and is probably inherited from diving birds and amphibians (18, 32, 43–45). It is particularly prominent in infants and manifests as severe bradycardia upon a single submersion of the face in the water (46, 47). Activation of this reflex due to the stimulation of sensory trigeminal fibers over the face and nasal mucosa causes apnea, a sudden drop in heart rate (due to parasympathetic activation), and a gradual increase in blood pressure as a result of peripheral vasoconstriction (due to increase in sympathetic tone) (44, 45). Also, there is a contraction of spleen releasing erythrocytes in the circulation (48, 49). Thus, the blood flow is redirected to vital organs (the brain and the heart) from the periphery and visceral organs. The heart rate is reduced, thereby lowering O2 requirement of myocardium and the blood flow to the brain is increased without an increase in the cerebral metabolic oxygen demand. Thus, DR is a protective oxygen-conserving reflex (18, 32, 44, 45). However, an exaggerated response that results in profound bradycardia can sometimes prove harmful or even fatal (14, 15, 50) and is often said to be associated with sudden death in infants.
Sudden infant death syndrome is the leading cause of death in the postneonatal period (12, 13, 51, 52). The exaggeration of parasympathetic activity and cardiorespiratory response to hypoxia has been suggested as the possible mechanism for these events (4–9). Similarly, trigeminal air-stream stimulation (TAS) model also showed that the TAS may induce apnea and bradycardia in premature infants (53). In infants monitored for apparent life-threatening events, severe bradycardia was the most prevalent and predictive event seen in infants who succumbed to SIDS (12, 13, 54), and hypoxia is a frequent event that precedes death in infants of SIDS (55).
Interestingly, the laryngeal chemo-reflex (LCR), a protective mechanism, causes closure of the glottis, coughing, and apnea during aspiration of the fluid into larynx/trachea and, therefore, has also postulated as one of the causes of SIDS (56, 57). On the other hand, DR, a subtype of TCR, has also been implicated to have a role in SIDS (14, 15, 58). The TCR is regulated by many brainstem nuclei and endogenously modulated by many neurotransmitters, the important one being serotonergic (5-HT), cholinergic (ACh), and nicotinergic (42, 59). Abnormalities in the modulation of these neurotransmitters along with defect in brainstem nuclei maturation may lead to exaggerated TCR response. Therefore, we summarize pieces of evidence in four hypotheses.
Abnormalities of serotonergic neurons have been observed in victims of SIDS (60–67). These victims had a higher number of 5-HT neurons in the medulla and cerebrospinal fluid (68, 69). Also, these medullary 5-HT neurons have been proposed to act as central respiratory chemoreceptors that are involved in the facilitation of respiration in response to hypoxic episode and generation of respiratory rhythm (70–73). These observations suggest that medullary 5-HT dysfunction may result in loss of respiratory and autonomic response to hypoxia and hypercarbia leading to sudden death in SIDS victims during sleep. Interestingly, the 5-HT1A-binding density was more reduced in males compared to females’ SIDS victims, which also explains why males are more vulnerable to SIDS (68, 74). Notably, these conditions (hypoxia, hypercarbia, and male gender) are also common risk factors for inciting the TCR. In animal experiment models, investigators have shown that the serotonin modulation is linked with TCR mechanism that further explains the possible role of TCR in SIDS (42, 75).
The decrease in cholinergic receptors density, as well as binding dysfunction of cholinergic receptors, has also been implicated as a risk factor for SIDS. Investigators have found a reduction in some choline acetyltransferase (ChAT) neurons as well as their binding capacities in hypoglossal nucleus and dorsal motor nucleus of vagus in SIDS cases (76–84). Also, hypoplasia of the arcuate nucleus has also been observed in these infants (76, 85, 86). These findings suggest a specific defect in cholinergic neurons in the brainstem of SIDS infants, which could cause abnormal control of cardiovascular and respiratory functions in these babies and contribute to the etiology of SIDS (76). They observed that cholinergic neurons endogenously inhibit the excitatory glutamatergic transmission to parasympathetic cardiac vagal neurons in response to trigeminal nerve stimulation via mAChRs. Neostigmine (an acetylcholinesterase inhibitor) significantly inhibited, whereas atropine (muscarinic receptor antagonist) enhanced this transmission, thus demonstrating the role of muscarinic (m4 type mACh) receptors. A decreased cholinergic activity could result in reduced inhibition of excitatory neurotransmission to cardiac vagal neurons in response to trigeminal nerve stimulation and thus an exaggerated TCR response in SIDS infants (59). Cholinergic receptors also play a significant role in sleep-dependent changes. The cholinergic neurotransmission in the brainstem is an important integral component of rapid eyeball movement sleep generation (87, 88). Change in cholinergic receptor activity is associated with potentiation of TCR and trigeminally evoked respiratory suppression (89) as well as with altered sleep–awake cycles in infants both of which are also seen in victims of SIDS. Studies have identified incomplete and less frequent arousal from sleep in response to hypoxia in SIDS victims. Kato et al. studied the characteristics of arousal from sleep in 16 infants who were being monitored for some days or weeks before they died of SIDS. The polygraphic sleep recordings of these infants were compared with those of matched control infants. The result of this study showed significantly fewer cortical arousal (complete arousal) in an infant who eventually died of SIDS later than in the control infants. Victims of SIDS had more frequent and longer duration of subcortical arousal (incomplete arousal) than controls. This study suggested an incomplete arousal process from sleep in infants who succumb to SIDS (90). Sensory stimulation of the trigeminal nerve during REM sleep has been shown to cause REM sleep-associated respiratory failures in SIDS infants (89, 91).
Prenatal exposure of the fetus to nicotine alters the density and binding capacity of serotonin and cholinergic receptors (61, 63, 68, 80, 81, 92, 93) and is one of the major risk factors contributing to SIDS (94, 95). Gorini et al. used a rat model to study the effects of prenatal nicotine exposure in the offsprings of the mothers who were exposed to clinically significant nicotine levels during gestation (75). The results of this study showed an exaggerated TCR response in animals exposed to nicotine during the prenatal period. They observed that prenatal exposure to nicotine significantly facilitates excitatory glutamatergic neurotransmission to cardiac vagal neurons in the nucleus ambiguus upon stimulation of trigeminal sensory afferents compared to their unexposed counterparts. The prenatal nicotine exposure also enhanced the endogenous serotonergic facilitation of TCR. All these effects thus lead to heightened TCR response (75). Also, a reduction in the number and function of AChRs has also been found in infants exposed to prenatal nicotine. Fetal exposure to nicotine suppresses mRNA expression and thus decreases brainstem mAChR binding. This again contributes to exaggeration of TCR by reduced inhibition of cardiac vagal neurons. Exposure to nicotine during the prenatal period facilitates modulation of inhibitory and excitatory pathways to the vagal nucleus in response to hypoxia or hypercapnia (96, 97). Prenatal exposure to nicotine decreases inhibitory GABAergic signals to the vagal nucleus during hypoxia (98) as well as hypercapnia (99). This decreased inhibitory GABAergic inputs to vagal nucleus cause increase in vagal activity to heart, thereby causing severe and sometimes lethal bradycardia in these animals (100–102). These findings suggest the likely cellular mechanism that causes an exaggerated response and pronounced bradycardia in victims of SIDS. Fetal exposure to nicotine also causes dysfunction of brainstem monoaminergic pathway. It leads to downregulation of 5-HT receptors and enhancing the risk of death due to SIDS (85). Prenatal nicotine exposure modulates 5-HT receptors in areas of brainstem regulating cardiorespiratory function that results in exaggerated TCR response and lethal outcome (75, 103).
Frequent developmental abnormalities in the brain stem, particularly in the arcuate nucleus, have been identified in SIDS (85, 86, 104–106). The arcuate nucleus is an important cardiorespiratory center in the medulla and hypoplasia of this nucleus has been detected in over 50% of infants dying of SIDS (105). Alterations in another brainstem nucleus have also been demonstrated (85, 107–112). Some of these nuclei (e.g., nucleus ambiguus, parabrachial nucleus) also participate in the reflex arc of TCR (40, 42). Besides, Lavezzi et al. observed an association between tobacco use and decreased in the functional activity of trigeminal nucleus that can trigger sudden death in babies (108). On the other hand, SIDS may occur due to a lack of sufficient development and plasticity of glutamatergic synapses (insufficient glutamate signaling) in the mesencephalic nucleus of the trigeminal nerve and reticular formation of the brainstem (113). All these findings thus suggest the role of developmental defects (i.e., neuronal deficiency and immaturity) in the brainstem nuclei regulating cardiorespiratory and other autonomic function in infants who die of SIDS. Therefore, the TCR may be a missing link in the etiopathogenesis of this subgroup of patients as well.
Serotonergic or/and cholinergic dysfunction in the brainstem autonomic nuclei causes an exaggerated TCR response and thus culminates in sudden intense bradycardia, apnea, and death and, therefore, can be linked with the etiopathogenesis of SIDS. However, whether the exaggerated TCR response is the cause in all cases of SIDS is a subject for future research.
TC made substantial contributions to conception and design, and/or acquisition of data, and/or analysis and interpretation of data, and helped in writing the manuscript. GS participated in drafting and writing the article. BB participated in writing the article. BS participated in writing and gave final approval of the version. All the authors have given final approval for submission of this version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Willinger M, James LS, Catz C. Defining the sudden infant death syndrome (SIDS): deliberations of an expert panel convened by the National Institute of Child Health and Human Development. Pediatr Pathol (1991) 11:677–84. doi:10.3109/15513819109065465
2. Moon RY, Horne RS, Hauck FR. Sudden infant death syndrome. Lancet (2007) 370:1578–87. doi:10.1016/S0140-6736(07)61662-6
3. Hoyert DL. Mortality associated with birth defects: influence of successive disease classification revisions. Birth Defects Res A Clin Mol Teratol (2003) 67:651–5. doi:10.1002/bdra.10117
4. Misurya VK, Singh SP, Kulshrestha VK. Prevention of oculocardiac reflex (O.C.R) during extraocular muscle surgery. Indian J Ophthalmol (1990) 38:85–7.
5. Gilani SM, Jamil M, Akbar F, Jehangir R. Anticholinergic premedication for prevention of oculocardiac reflex during squint surgery. J Ayub Med Coll Abbottabad (2005) 17:57–9.
6. Hunsley JE, Bush GH, Jones CJ. A study of glycopyrrolate and atropine in suppression of oculocardiac reflex during strabismus surgery in children. Br J Anaesth (1982) 54:459–64. doi:10.1093/bja/54.4.459
7. Karhunen U, Cozanitis DA, Brander P. The oculocardiac reflex in adults. A dose response study of glycopyrrolate and atropine. Anaesthesia (1984) 39:524. doi:10.1111/j.1365-2044.1984.tb07354.x
8. Schaller B. Ketamine and decrease of oculocardiac reflex. Acta Anesth Scand (2008) 52:446. doi:10.1111/j.1399-6576.2007.01513.x
9. Gandevia SC, McCloskey DI, Potter EK. Reflex bradycardia occurring in response to diving, nasopharyngeal stimulation and ocular pressure, and its modification by respiration and swallowing. J Physiol (1978) 276:383–94. doi:10.1113/jphysiol.1978.sp012241
10. Horne RS, Parslow PM, Ferens D, Bandopadhayay P, Osborne A, Watts AM, et al. Arousal responses and risk factors for sudden infant death syndrome. Sleep Med (2002) 2:61–5. doi:10.1016/S1389-9457(02)00168-5
11. Horne RS, Parslow PM, Harding R. Respiratory control and arousal in sleeping infants. Pediatr Respir Rev (2004) 3:190–8. doi:10.1016/j.prrv.2004.04.011
12. Meny RG, Carroll JL, Carbone MT, Kelly DH. Cardiorespiratory recordings from infants dying suddenly and unexpectedly at home. Pediatrics (1994) 93:44–9.
13. Poets CF, Meny RG, Chobanian MR, Bonofiglo RE. Gasping and other cardiorespiratory patterns during sudden infant deaths. Pediatr Res (1999) 45:350–4. doi:10.1203/00006450-199903000-00010
14. Lobban CD. The human dive reflex as a primary cause of SIDS. A review of the literature. Med J Aust (1991) 155:561–3.
15. Lobban CD. The oxygen-conserving dive reflex re-examined as the principal contributory factor in sudden infant death. Med Hypotheses (1995) 44:273–7. doi:10.1016/0306-9877(95)90179-5
16. Lemaitre F, Chowdhury T, Schaller B. The trigeminocardiac reflex – a comparison with the diving reflex in humans. Arch Med Sci (2015) 11(2):419–26. doi:10.5114/aoms.2015.50974
17. Chowdhury T, Mendelowitz D, Golanov E, Spiriev T, Arasho B, Sandu N, et al. Trigeminocardiac reflex: the current clinical and physiological knowledge. J Neurosurg Anesthesiol (2015) 27(2):136–47. doi:10.1097/ANA.0000000000000065
18. Schaller B, Cornelius JF, Sandu N, Ottaviani G, Perez-Pinzon MA. Oxygen-conserving reflexes of the brain: the current molecular knowledge. J Cell Mol Med (2009) 13(4):644–7. doi:10.1111/j.1582-4934.2009.00659.x
20. Meuwly C, Golanov E, Chowdhury T, Erne P, Schaller B. Trigeminal cardiac reflex: new thinking model about the definition based on a literature review. Medicine (2015) 94:1–9. doi:10.1097/MD.0000000000000484
21. Sandu N, Chowdhury T, Schaller BJ; Trigemino-Cardiac Reflex Examination Group (TCREG). How to apply case reports in clinical practice using surrogate models via example of the trigeminocardiac reflex. J Med Case Rep (2016) 6(10):8. doi:10.1186/s13256-016-0849-z
22. Arasho B, Sandu N, Spiriev T, Prabhakar H, Schaller B. Management of the trigeminocardiac reflex: facts and own experience. Neurol India (2009) 57(4):375–80. doi:10.4103/0028-3886.55577
23. Sandu N, Sadr-Eshkevari P, Schaller BJ; Trigemino-Cardiac Reflex Examination Group (TCREG). Usefulness of case reports to improve medical knowledge regarding trigemino-cardiac reflex in skull base surgery. J Med Case Rep (2011) 15(5):149. doi:10.1186/1752-1947-5-149
24. Sandu N, Schaller B. The trigemino-cardiac reflex: a view to the future. Arch Med Sci (2010) 6(1):138–9. doi:10.5114/aoms.2010.13523
25. Schaller B, Filis A, Sandu N, Buchfelder M. Peripheral trigemino-cardiac reflex. Acta Neurochir (Wien) (2009) 151(12):1727. doi:10.1007/s00701-009-0390-6
26. Schaller B, Filis A, Sandu N, Rasper J, Noethen C, Buchfelder M. Trigemino-cardiac reflex may be refractory to conventional management in adults. Acta Neurochir (Wien) (2008) 150(9):929–30. doi:10.1007/s00701-008-0010-x
27. Schaller BJ, Filis A, Buchfelder M. Trigemino-cardiac reflex in humans initiated by peripheral stimulation during neurosurgical skull-base operations. Its first description. Acta Neurochir (Wien) (2008) 150(7):715–7. doi:10.1007/s00701-008-1602-1
28. Schaller BJ, Filis A, Buchfelder M. Detection and prevention of the trigeminocardiac reflex during skull base surgery. Acta Neurochir (Wien) (2007) 149(3):331. doi:10.1007/s00701-006-1088-7
30. Spiriev T, Sandu N, Kondoff S, Tzekov C, Schaller B. Tic and autonomic symptoms. J Neurosurg (2012) 116(6):1397–8. doi:10.3171/2011.9.JNS111532
31. Chowdhury T, Sandu N, Sadr-Eshkevari P, Meuwly C, Schaller B. Trigeminocardiac reflex: current trends. Expert Rev Cardiovasc Ther (2014) 12(1):9–11. doi:10.1586/14779072.2014.862498
32. Schaller BJ, Sandu N, Cornelius JF, Filis A, Perez-Pinzon MA. Oxygen-conserving implications of the trigemino-cardiac reflex in the brain: the molecular basis of neuroprotection? Mol Med (2009) 15(5–6):125–6. doi:10.2119/molmed.2009.00013
33. Spiriev T, Tzekov C, Laleva L, Kostadinova C, Kondoff S, Sandu N, et al. Central trigeminocardiac reflex in pediatric neurosurgery: a case report and review of the literature. J Med Case Rep (2012) 6:372. doi:10.1186/1752-1947-6-372
34. Schaller BJ, Buchfelder M. Trigemino-cardiac reflex in skull base surgery: from a better understanding to a better outcome? Acta Neurochir (Wien) (2006) 148(10):1029–31. doi:10.1007/s00701-006-0889-z
35. Schaller B, Cornelius JF, Prabhakar H, Koerbel A, Gnanalingham K, Sandu N, et al. The trigeminocardiac reflex: an update of the current knowledge. J Neurosurg Anesthesiol (2009) 21:187–95. doi:10.1097/ANA.0b013e3181a2bf22
36. Ohshita N, Nakajo N, Takemura M. Characteristics of the trigeminal depressor response in cats. J Neurosci Res (2004) 76:891–901. doi:10.1002/jnr.20131
37. McCulloch PF, Faber KM, Panneton WM. Electrical stimulation of the anterior ethmoidal nerve produces the diving response. Brain Res (1999) 830:24–31. doi:10.1016/S0006-8993(99)01374-8
38. Nalivaiko E, De Pasquale CG, Blessing WW. Electrocardiographic changes associated with the nasopharyngeal reflex in conscious rabbits: vago-sympathetic co-activation. Auton Neurosci (2003) 105:101–4. doi:10.1016/S1566-0702(03)00048-1
39. Chowdhury T, Sandu N, Meuwly C, Cappellani RB, Schaller B. Trigeminocardiac reflex: differential behavior and risk factors around the course of the trigeminal nerve. Future Neurol (2014) 9:41–7. doi:10.2217/fnl.13.62
40. Schaller B. Trigeminocardiac reflex: a clinical phenomenon or a new physiological entity. Neurology (2004) 251:658–65. doi:10.1007/s00415-004-0458-4
41. Cornelius JF, Sadr-Eshkevari P, Arasho BD, Sandu N, Spiriev T, Lemaitre F, et al. The trigemino-cardiac reflex in adults: own experience. Expert Rev Cardiovasc Ther (2010) 8:895–8. doi:10.1586/erc.10.74
42. Gorini C, Jameson HS, Mendelowitz D. Serotonergic modulation of the trigeminocardiac reflex neurotransmission to cardiac vagal neurons in the nucleus ambiguus. J Neurophysiol (2009) 102:1443–50. doi:10.1152/jn.00287.2009
43. Alboni P, Alboni M, Gianfranchi L. Diving bradycardia: a mechanism of defence against hypoxic damage. J Cardiovasc Med (Hagerstown) (2011) 12:422–7. doi:10.2459/JCM.0b013e328344bcdc
44. Sandu N, Cornelius J, Filis A, Nöthen C, Rasper J, Kulinsky VI, et al. Cerebral hemodynamic changes during the trigeminocardiac reflex: description of a new animal model protocol. ScientificWorldJournal (2010) 10:1416–23. doi:10.1100/tsw.2010.136
45. Sandu N, Spiriev T, Lemaitre F, Filis A, Schaller B. New molecular knowledge towards the trigemino-cardiac reflex as a cerebral oxygen-conserving reflex. ScientificWorldJournal (2010) 4(10):811–7. doi:10.1100/tsw.2010.71
46. Goksor E, Rosengren L, Wennergren G. Bradycardic response during submersion in infant swimming. Acta Paediatr (2002) 91:307–12. doi:10.1111/j.1651-2227.2002.tb01720.x
47. Foster GE, Sheel AW. The human diving response, its function, and its control. Scand J Med Sci Sports (2005) 15:3–12. doi:10.1111/j.1600-0838.2005.00440.x
48. Gooden BA. Mechanism of the human diving response. Integr Physiol Behav Sci (1994) 29(1):6–16. doi:10.1007/BF02691277
49. Liner MH. Tissue gas stores of the body and head-out immersion in humans. J Appl Physiol (1993) 75(3):1285–93.
50. Matturri L, Ottaviani G, Lavezzi AM. Sudden infant death triggered by dive reflex. J Clin Pathol (2005) 58:77–80. doi:10.1136/jcp.2004.020867
51. Fewell JE, Smith FG, Ng VK. Prenatal exposure to nicotine impairs protective responses of rat pups to hypoxia in an age-dependent manner. Respir Physiol (2001) 127:61–73. doi:10.1016/S0034-5687(01)00232-8
52. Nachmanoff DB, Panigrahy A, Filiano JJ, Mandell F, Sleeper LA, Valdes-Dapena M, et al. Brainstem 3H-nicotine receptor binding in the sudden infant death syndrome. J Neuropathol Exp Neurol (1998) 57:1018–25. doi:10.1097/00005072-199811000-00004
53. Ramet J, Praud JP, D’Allest AM, Dehan M, Gaultier C. Trigeminal airstream stimulation. Maturation-related cardiac and respiratory responses during REM sleep in human infants. Chest (1990) 98(1):92–6.
54. Cote A, Hum C, Brouillette RT, Themens M. Frequency and timing of recurrent events in infants using home cardiorespiratory monitors. J Pediatr (1998) 132:783–9. doi:10.1016/S0022-3476(98)70304-X
55. Jones KL, Krous HF, Nadeau J, Blackbourne B, Zielke HR, Gozal D. Vascular endothelial growth factor in the cerebrospinal fluid of infants who died of sudden infant death syndrome: evidence for antecedent hypoxia. Pediatrics (2003) 111:358–63. doi:10.1542/peds.111.2.358
56. Thach BT. Maturation and transformation of reflexes that protect the laryngeal airway from liquid aspiration from fetal to adult life. Am J Med (2003) 111(Suppl 8A):69S–77S. doi:10.1016/S0002-9343(01)00860-9
57. Leiter JC, Böhm I. Mechanisms of pathogenesis in the sudden infant death syndrome. Respir Physiol Neurobiol (2007) 159(2):127–38. doi:10.1016/j.resp.2007.05.014
58. Morpurgo CV, Lavezzi AM, Ottaviani G, Rossi L. Bulbo-spinal pathology and sudden respiratory infant death syndrome. Eur J Anaesthesiol (2004) 21:589–93. doi:10.1097/00003643-200408000-00001
59. Gorini C, Philbin K, Bateman R, Mendelowitz D. Endogenous inhibition of the trigeminally evoked neurotransmission to cardiac vagal neurons by muscarinic acetylcholine receptors. J Neurophysiol (2010) 104:1841–8. doi:10.1152/jn.00442.2010
60. Kinney HC, Randall LL, Sleeper LA, Willinger M, Belliveau RA, Zec N, et al. Serotonergic brainstem abnormalities in Northern Plains Indians with the sudden infant death syndrome. J Neuropathol Exp Neurol (2003) 62:1178–91. doi:10.1093/jnen/62.11.1178
61. Panigrahy A, Filiano J, Sleeper LA, Mandell F, Valdes-Dapena M, Krous HF, et al. Decreased serotonergic receptor binding in rhombic lip-derived regions of the medulla oblongata in the sudden infant death syndrome. J Neuropathol Exp Neurol (2000) 59:377–84. doi:10.1093/jnen/59.5.377
62. Kinney HC, Myers MM, Belliveau RA, Randall LL, Trachtenberg FL, Fingers ST, et al. Subtle autonomic and respiratory dysfunction in sudden infant death syndrome associated with serotonergic brainstem abnormalities: a case report. J Neuropathol Exp Neurol (2005) 64:689–94. doi:10.1097/01.jnen.0000174334.27708.43
63. Hunt CE. Gene-environment interactions: implications for sudden unexpected deaths in infancy. Arch Dis Child (2005) 90:48–53. doi:10.1136/adc.2004.051458
64. Narita N, Narita M, Takashima S, Nakayama M, Nagai T, Okado N. Serotonin transporter gene variation is a risk factor for sudden infant death syndrome in the Japanese population. Pediatrics (2001) 107:690–2. doi:10.1542/peds.107.4.690
65. Weese-Mayer DE, Berry-Kravis EM, Maher BS, Silvestri JM, Curran ME, Marazita ML. Sudden infant death syndrome: association with a promoter polymorphism of the serotonin transporter gene. Am J Med Genet (2003) 117:268–74. doi:10.1002/ajmg.a.20005
66. Weese-Mayer DE, Zhou L, Berry-Kravis EM, Maher BS, Silvestri JM, Marazita ML. Association of the serotonin transporter gene with sudden infant death syndrome: a haplotype analysis. Am J Med Genet (2003) 122:238–45. doi:10.1002/ajmg.a.20427
67. Weese-Mayer DE, Berry-Kravis EM, Zhou L, Maher BS, Curran ME, Silvestri JM, et al. Sudden infant death syndrome: case-control frequency differences at genes pertinent to early autonomic nervous system embryologic development. Pediatr Res (2004) 56:391–6. doi:10.1203/01.PDR.0000136285.91048.4A
68. Paterson DS, Trachtenberg FL, Thompson EG, Belliveau RA, Beggs AH, Darnall R, et al. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA (2006) 296:2124–32. doi:10.1001/jama.296.17.2124
69. Caroff J, Girin E, Alix D, Cann-Moisan C, Sizun J, Barthelemy L. Neurotransmission and sudden infant death. Study of cerebrospinal fluid. CR Acad Sci III (1992) 314:451–4.
70. Richerson GB. Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat Rev Neurosci (2004) 5:449–61. doi:10.1038/nrn1409
71. Messier ML, Li A, Nattie EE. Inhibition of medullary raphe serotonergic neurons has age-dependent effects on the CO2 response in newborn piglets. J Appl Physiol (2004) 96:1909–19. doi:10.1152/japplphysiol.00805.2003
72. Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, et al. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci (2004) 7:48–55. doi:10.1038/nn1166
73. Pena F, Ramirez JM. Endogenous activation of serotonin-2A receptors is required for respiratory rhythm generation in vitro. J Neurosci (2002) 22:11055–64.
74. American Academy of Pediatrics Task Force on Sudden Infant Death Syndrome. The changing concept of sudden infant death syndrome: diagnostic coding shifts, controversies regarding the sleeping environment, and new variables to consider in reducing risk. Pediatrics (2005) 116:1245–55. doi:10.1542/peds.2005-1499
75. Gorini C, Jameson H, Woerman AL, Perry DC, Mendelowitz D. Prenatal nicotine exposure enhances the trigeminocardiac reflex via serotonin receptor facilitation in brainstem pathways. J Appl Physiol (2013) 115:415–21. doi:10.1152/japplphysiol.00552.2013
76. Mallard C, Tolcos M, Leditschke J, Campbell P, Rees S. Reduction in choline acetyltransferase immunoreactivity but not muscarinic-m2 receptor immunoreactivity in the brainstem of SIDS infants. J Neuropathol Exp Neurol (1999) 58:255–64. doi:10.1097/00005072-199903000-00005
77. Kalaria RN, Fiedler C, Hunsaker JC III, Sparks DL. Synaptic neurochemistry of human striatum during development: changes in sudden infant death syndrome. J Neurochem (1993) 60:2098–105. doi:10.1111/j.1471-4159.1993.tb03494.x
78. Sparks DL, Hunsaker JC III. Sudden infant death syndrome: altered aminergic-cholinergic synaptic markers in hypothalamus. J Child Neurol (1991) 6:335–9. doi:10.1177/088307389100600409
79. Oda Y. Choline acetyltransferase: the structure, distribution and pathologic changes in the central nervous system. Pathol Int (1999) 49:921–37. doi:10.1046/j.1440-1827.1999.00977.x
80. Kinney HC. Brainstem mechanisms underlying the sudden infant death syndrome: evidence from human pathologic studies. Dev Psychobiol (2009) 51:223–33. doi:10.1002/dev.20367
81. Kinney HC, Filiano JJ, Sleeper LA, Mandell F, Valdes-Dapena M, White WF. Decreased muscarinic receptor binding in the arcuate nucleus in sudden infant death syndrome. Science (1995) 269:1446–50. doi:10.1126/science.7660131
82. Slotkin TA, Epps TA, Stenger ML, Sawyer KJ, Seidler FJ. Cholinergic receptors in heart and brainstem of rats exposed to nicotine during development: implications for hypoxia tolerance and perinatal mortality. Brain Res Dev Brain Res (1999) 113:1–12. doi:10.1016/S0165-3806(98)00173-4
83. Zhu J, Taniguchi T, Konishi Y, Mayumi M, Muramatsu I. Nicotine administration decreases the number of binding sites and mRNA of M1 and M2 muscarinic receptors in specific brain regions of rat neonates. Life Sci (1998) 62:1089–98.
84. Kubo S, Orihara Y, Gotohda T, Tokunaga I, Tsuda R, Ikematsu K, et al. Immunohistochemical studies on neuronal changes in brain stem nucleus of forensic autopsied cases. II. Sudden infant death syndrome. Nihon Hoigaku Zasshi (1998) 52:350–4.
85. Matturri L, Ottaviani G, Alfonsi G, Crippa M, Rossi L, Lavezzi AM. Study of the brainstem, particularly the arcuate nucleus, in sudden infant death syndrome (SIDS) and sudden intra-uterine unexplained death (SIUD). Am J Forensic Med Pathol (2004) 25:44–8. doi:10.1097/01.paf.0000113813.83779.21
86. Filiano JJ, Kinney HC. Arcuate nucleus hypoplasia in the sudden infant death syndrome. J Neuropathol Exp Neurol (1992) 51:394–403. doi:10.1097/00005072-199207000-00002
87. Vazquez J, Baghdoyan HA. GABAA receptors inhibit acetylcholine release in cat pontine reticular formation: implications for REM sleep regulation. J Neurophysiol (2004) 92:2198–206. doi:10.1152/jn.00099.2004
88. Verret L, Leger L, Fort P, Luppi PH. Cholinergic and noncholinergic brainstem neurons expressing Fos after paradoxical (REM) sleep deprivation and recovery. Eur J Neurosci (2005) 21:2488–504. doi:10.1111/j.1460-9568.2005.04060.x
89. Dutschmann M, Herbert H. Pontine cholinergic mechanisms enhance trigeminally evoked respiratory suppression in the anesthetized rat. J Appl Physiol (1999) 87:1059–65.
90. Kato I, Franco P, Groswasser J, Scaillet S, Kelmanson I, Togari H, et al. Incomplete arousal processes in infants who were victims of sudden death. Am J Respir Crit Care Med (2003) 168:1298–303. doi:10.1164/rccm.200301-134OC
91. Gould JB, Lee AF, Morelock S. The relationship between sleep and sudden infant death. Ann N Y Acad Sci (1988) 533:62–77. doi:10.1111/j.1749-6632.1988.tb37234.x
92. Hunt CE, Hauck FR. Sudden infant death syndrome. CMAJ (2006) 174:1861–9. doi:10.1503/cmaj.051671
93. Frank MG, Srere H, Ledezma C, O’Hara B, Heller HC. Prenatal nicotine alters vigilance states and AChR gene expression in the neonatal rat: implications for SIDS. Am J Physiol Regul Integr Comp Physiol (2001) 280:R1134–40.
94. Duncan JR, Garland M, Myers MM, Fifer WP, Yang M, Kinney HC, et al. Prenatal nicotine-exposure alters fetal autonomic activity and medullary neurotransmitter receptors: implications for sudden infant death syndrome. J Appl Physiol (2009) 107:1579–90. doi:10.1152/japplphysiol.91629.2008
95. Slotkin TA, Seidler FJ. Mimicking maternal smoking and pharmacotherapy of preterm labor: fetal nicotine exposure enhances the effect of late gestational dexamethasone treatment on noradrenergic circuits. Brain Res Bull (2011) 86:435–40. doi:10.1016/j.brainresbull.2011.08.009
96. Huang ZG, Griffioen KJ, Wang X, Dergacheva O, Kamendi H, Gorini C, et al. Nicotinic receptor activation occludes purinergic control of central cardiorespiratory network responses to hypoxia/hypercapnia. J Neurophysiol (2007) 98:2429–38. doi:10.1152/jn.00448.2007
97. Kamendi H, Stephens C, Dergacheva O, Wang X, Huang ZG, Bouairi E, et al. Prenatal nicotine exposure alters the nicotinic receptor subtypes that modulate excitation of parasympathetic cardiac neurons in the nucleus ambiguus from primarily alpha3beta2 and/or alpha6betaX to alpha3beta4. Neuropharmacology (2006) 51:60–6. doi:10.1016/j.neuropharm.2006.03.001
98. Neff RA, Simmens SJ, Evans C, Mendelowitz D. Prenatal nicotine exposure alters central cardiorespiratory responses to hypoxia in rats: implications for sudden infant death syndrome. J Neurosci (2004) 24:9261–8. doi:10.1523/JNEUROSCI.1918-04.2004
99. Huang ZG, Griffioen KJ, Wang X, Dergacheva O, Kamendi H, Gorini C, et al. Differential control of central cardiorespiratory interactions by hypercapnia and the effect of prenatal nicotine. J Neurosci (2006) 26:21–9. doi:10.1523/JNEUROSCI.4221-05.2006
100. Huang ZG, Wang X, Evans C, Gold A, Bouairi E, Mendelowitz D. Prenatal nicotine exposure alters the types of nicotinic receptors that facilitate excitatory inputs to cardiac vagal neurons. J Neurophysiol (2004) 92:2548–54. doi:10.1152/jn.00500.2004
101. Huang ZG, Wang X, Dergacheva O, Mendelowitz D. Prenatal nicotine exposure recruits an excitatory pathway to brainstem parasympathetic cardioinhibitory neurons during hypoxia/hypercapnia in the rat: implications for sudden infant death syndrome. Pediatr Res (2005) 58:562–7. doi:10.1203/01.PDR.0000179380.41355.FC
102. Evans C, Wang J, Neff R, Mendelowitz D. Hypoxia recruits a respiratory-related excitatory pathway to brainstem premotor cardiac vagal neurons in animals exposed to prenatal nicotine. Neuroscience (2005) 133:1073–9. doi:10.1016/j.neuroscience.2005.03.053
103. Slotkin TA, Seidler FJ. Mimicking maternal smoking and pharmacotherapy of preterm labor: interactions of fetal nicotine and dexamethasone on serotonin and dopamine synaptic function in adolescence and adulthood. Brain Res Bull (2010) 82:124–34. doi:10.1016/j.brainresbull.2010.02.015
104. Matturri L, Biondo B, Mercurio P, Rossi L. Severe hypoplasia of medullary arcuate nucleus: quantitative analysis in sudden infant death syndrome. Acta Neuropathol (2000) 99:371–5. doi:10.1007/s004010051138
105. Matturri L, Biondo B, Suarez-Mier MP, Rossi L. Brainstem lesions in the sudden infant death syndrome: variability in the hypoplasia of the arcuate nucleus. Acta Neuropathol (2002) 104:12–20. doi:10.1007/s00401-001-0511-7
106. Lavezzi AM, Ottaviani G, Mauri M, Matturri L. Hypoplasia of the arcuate nucleus and maternal smoking during pregnancy in sudden unexplained perinatal and infant death. Neuropathology (2004) 24:284–9. doi:10.1111/j.1440-1789.2004.00558.x
107. Ottaviani G, Matturri L, Mingrone R, Lavezzi AM. Hypoplasia and neuronal immaturity of the hypoglossal nucleus in sudden infant death. J Clin Pathol (2006) 59:497–500. doi:10.1136/jcp.2005.032037
108. Lavezzi AM, Mehboob R, Matturri L. Developmental alterations of the spinal trigeminal nucleus disclosed by substance P immunohistochemistry in fetal and infant sudden unexplained deaths. Neuropathology (2011) 31:405–13. doi:10.1111/j.1440-1789.2010.01190.x
109. Lavezzi AM, Ottaviani G, Mingrone R, Matturri L. Analysis of the human locus coeruleus in perinatal and infant sudden unexplained deaths. Possible role of the cigarette smoking in the development of this nucleus. Brain Res Dev Brain Res (2005) 154:71–80. doi:10.1016/j.devbrainres.2004.10.007
110. Matturri L, Ottaviani G, Lavezzi AM. Techniques and criteria in pathologic and forensic-medical diagnostics of sudden unexpected infant and perinatal death. Am J Clin Pathol (2005) 124:259–68. doi:10.1309/J6AR-EY41-HKBE-YVHX
111. Matturri L, Ottaviani G, Benedetti G, Agosta E, Lavezzi AM. Unexpected perinatal death and sudden infant death syndrome (SIDS): anatomopathological and legal aspects. Am J Forensic Med Pathol (2005) 26:155–60.
112. Lavezzi AM, Ottaviani G, Rossi L, Matturri L. Hypoplasia of the parabrachial/Kölliker-fuse complex in perinatal death. Biol Neonate (2004) 86:92–7. doi:10.1159/000078310
Keywords: sudden infant death syndrome, trigeminocardiac reflex, diving reflex, oxygen-conserving reflex, bradycardia, asystole, smoking, prenatal nicotine exposure
Citation: Singh GP, Chowdhury T, Bindu B and Schaller B (2016) Sudden Infant Death Syndrome – Role of Trigeminocardiac Reflex: A Review. Front. Neurol. 7:221. doi: 10.3389/fneur.2016.00221
Received: 13 August 2016; Accepted: 22 November 2016;
Published: 05 December 2016
Edited by:
Erwin Lemche, King’s College London, UKReviewed by:
Graham J. Galloway, Translational Research Institute, AustraliaCopyright: © 2016 Singh, Chowdhury, Bindu and Schaller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tumul Chowdhury, dHVtdWx0aHVuZGVyQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.