 Kevin Pendo
Kevin Pendo Christopher M. DeGiorgio
Christopher M. DeGiorgio- 1Princeton University, Princeton, NJ, USA
- 2Department of Neurology, University of California Los Angeles, Los Angeles, CA, USA
There is increasing evidence supporting dietary and alternative therapies for epilepsy, including the ketogenic diet, modified Atkins diet, and omega-3 fatty acids. Vitamin D3 is actively under investigation as a potential intervention for epilepsy. Vitamin D3 is fat-soluble steroid, which shows promise in animal models of epilepsy. Basic research has shed light on the possible mechanisms by which Vitamin D3 may reduce seizures, and animal data support the efficacy of Vitamin D3 in rat and mouse models of epilepsy. Very little clinical data exist to support the treatment of human epilepsy with Vitamin D3, but positive findings from preliminary clinical trials warrant larger Phase I and II clinical trials in order to more rigorously determine the potential therapeutic value of Vitamin D3 as a treatment for human epilepsy.
Introduction
Epilepsy affects approximately two million Americans and 65 million people worldwide (1). Among those with epilepsy, 22–30% have drug-resistant epilepsy (DRE) (1, 2). DRE causes cognitive and mood impairment, injuries, and increased risk of death including sudden death in epilepsy (SUDEP) (1–3). Antiepileptic drugs (AEDs) are the primary medical treatment for epilepsy. However, even for those whose seizures are well controlled by AEDs, allergies, neurological and systemic toxicity, depression, memory loss, and osteoporosis are common problems (4, 5). Because of the limitations and potential toxicity of existing AEDs, there is significant clinical interest in finding alternative therapies for epilepsy.
In the search for alternative epilepsy treatments, Vitamin D3 is an intriguing candidate (6). As early as 1974, Christiansen postulated that supplementation of Vitamin D might improve calcium and magnesium levels and may decrease hyperexcitability in patients with epilepsy. In the four decades since, progress has been made in understanding the biochemical and cellular mechanisms of Vitamin D3’s anticonvulsant properties. Animal data have supported the anticonvulsant effects of Vitamin D3 in mice and rats (7–11). Existing evidence for the use of Vitamin D3 in treating human epilepsy is very limited (6, 12). There is a critical need for larger clinical trials to establish the safety and efficacy of vitamin D3 in epilepsy. In this review, we will critically analyze the animal and human evidence to date supporting the use of Vitamin D3 as a treatment for epilepsy.
Vitamin D3 Overview: Biochemistry and Role in Human Health
The most biologically active form of Vitamin D in humans is Vitamin D3 (cholecalciferol), which is a fat-soluble steroid hormone (13). Dietary sources of Vitamin D3 include dairy, meat, fish, and mushrooms (14). The primary source of Vitamin D3 is exposure of the skin to ultraviolet sunlight (14). The metabolic pathway of Vitamin D3 is summarized in Figure 1. 7-dehydrocholesterol is converted to Vitamin D3 in the skin after exposure to sunlight. Vitamin D3 is converted to 25-hydroxy-cholecalciferol (25-OH Vitamin D3) in the liver. 25-OH Vitamin D3 is the major circulating form of Vitamin D, but it itself is biologically inactive and must be converted to the active form 1,25-dihydroxy-Vitamin D3 (1,25 Vitamin D3) in the kidneys (13–15). Vitamin D3 is important for calcium metabolism, bone health, cardiac function, and blood pressure maintenance, among other health benefits (14, 16, 17). Vitamin D3 deficiency is a marker of poor health and overall mortality (16). However, 40–50% of Americans have insufficient Vitamin D3 levels, and insufficiency is even more prevalent in underserved populations, including Hispanics (69%) and African Americans (82%) (18).
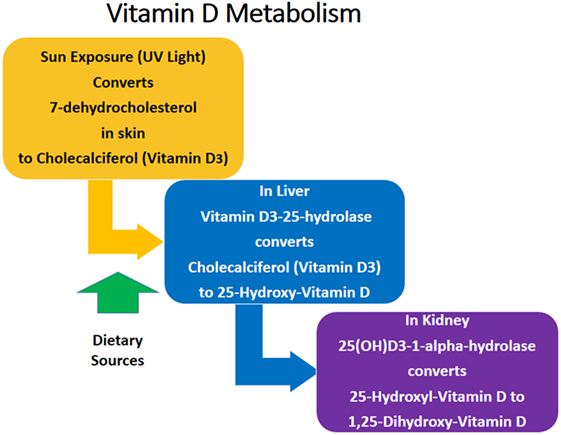
Figure 1. Vitamin D metabolism.
Vitamin D3 in the Brain and Nervous System
Among its variety of health benefits, Vitamin D3 plays an important role in the human brain and nervous system, as indicated by increasing evidence gathered over the past several decades. Researchers have explored the role of Vitamin D3 in Alzheimer’s disease and dementias (19, 20), Parkinson’s disease (19, 21), multiple sclerosis (22–24), schizophrenia (25), affective disorders (13, 26), cognitive decline (13, 27), and epilepsy (6, 12). Vitamin D3 is also involved in neuroprotection (15, 28, 29), brain cell proliferation and differentiation (30, 31), and brain development (30, 32, 33). A neurological role of Vitamin D3 is further supported by the presence of Vitamin D3-specific receptors and enzymes in neurons and glial cells throughout the brain, in the spinal cord, and in the peripheral nervous system (34–37). The broad role of Vitamin D3 in the nervous system has engendered research into Vitamin D3’s anticonvulsant action in the brain, and the proposed mechanisms of action can generally be categorized as either genomic or non-genomic.
Genomic Mechanisms of Action
Genomic mechanisms behind Vitamin D3’s anticonvulsant effect are based on Vitamin D3’s ability to regulate the expression of genes, a process that is mediated by a nuclear Vitamin D3 receptor (VDR) (38). VDR is a ligand-specific transcription factor, which is activated by Vitamin D3 and subsequently alters gene expression (28, 29). Through this mechanism, Vitamin D3 lowers the expression of certain proconvulsant cytokines, such as IL-1β and TNF-α. These cytokines can increase seizure susceptibility in several ways. IL-1β is involved in a pathway that results in phosphorylation of the NR2B subunit of the NMDA receptor, which is a glutamate receptor that is important in the generation of seizures (39). The phosphorylation of this NMDA receptor subunit increases Ca2+ influx into neurons (40) and stabilizes the receptor in the membrane (41), leading to the hyperexcitability of neurons that can cause seizures (39, 42). IL-1β can also cause neuronal hyperexcitability by increasing the release probability of glutamate (43), an excitatory neurotransmitter, and inhibiting its reuptake (39, 44). In addition, IL-1β can reduce inhibitory GABA-ergic Cl− flux (45), furthering the proconvulsant effect of this cytokine (39). The TNF-α cytokine acts as a proconvulsant because it initiates both the recruitment of AMPA receptors to the neuronal membrane and the endocytosis of GABAA receptors away from the membrane (46, 47). The TNF-α-induced overexpression of AMPA receptors and under-expression of GABAA receptors on the neuronal membranes results in more excitatory synaptic transmission and less inhibitory signaling, which increases the likelihood of epileptic activity.
Through its nuclear VDR, Vitamin D3 can also increase the expression of anticonvulsant growth factors GDNF and NT3 (15, 29, 48–50). NT3 leads to an anticonvulsant effect by downregulating TrkA and TrkC receptors, which are receptors that regulate synaptic strength (50). The mechanism behind GDNF’s anticonvulsant action remains largely unknown, but it is speculated that, similar to that of NT3, it involves some modulation of synaptic transmission (51). Vitamin D3-activated VDR also promotes expression of the calcium-binding proteins parvalbumin and calbindins, which inhibit epileptic episodes (15, 29, 52). By binding to Ca2+ in the presynaptic terminal, these calcium-binding proteins prevent excessive Ca2+-induced neurotransmitter release and thus protect against epileptic activity (52, 53).
Non-Genomic Mechanisms of Action
Faster, non-genomic mechanisms of Vitamin D3’s anticonvulsant effect have been proposed as well. Vitamin D3’s ability to increase calcium uptake from the intestine can alter plasma and brain Ca2+ concentrations, which may decrease neuronal excitability and prevent seizures. However, evidence suggests that Vitamin D3’s anticonvulsant effect is not wholly attributable to its role in altering calcium levels (6, 8, 9). Rather, it is more likely that Vitamin D3’s rapid, anticonvulsant effect results from its ability to fine-tune Ca2+ and Cl− currents across neuronal membranes (54, 55). Vitamin D3 initiates non-genomic signal transduction pathways that ultimately alter the conductance of L-type calcium channels and chloride channels, therefore affecting neuronal excitability and seizure susceptibility at the threshold level (55–57). The details of these non-genomic signal transduction pathways are debated, and although it used to be thought that they were mediated by a distinct membrane Vitamin D3 receptor (VDRmem) (58), more recent evidence suggests that these rapid, non-genomic anticonvulsant pathways are actually mediated by the same protein – VDR – that mediates Vitamin D3’s genomic actions (54, 57, 59–61), with different domains of VDR being involved in the genomic and non-genomic pathways that lead to Vitamin D3’s anticonvulsant effects.
Vitamin D3 in Animal Models of Seizures
Rat Models
In 1984, Siegel et al. published a seminal paper describing the effect of Vitamin D3 on seizure thresholds in rat hippocampi (7). Using artificial electrical stimulation to model seizures, they found that stereotactic injection of 50 or 100 μg of 1,25 Vitamin D3 into the hippocampus of rats significantly elevated the seizure threshold in all rats treated. This elevation in threshold was noticeable 5–10 min after the injection of 1,25 Vitamin D3, and the effect lasted at least 120–180 min. Intravenous injection of 1,25 Vitamin D3 also significantly elevated seizure threshold, but the effect was transient, lasting only 30 min, perhaps due to limited uptake of 1,25 Vitamin D3 in the brain. Most rats were Vitamin D3-sufficient, but they found that in one Vitamin D3-deficient rat, a lower dose of 1,25 Vitamin D3 was required to raise the seizure threshold to a similar extent.
Mouse Models
Over two decades after Siegel et al.’s rat study, Kalueff et al. explored the anticonvulsant effects of Vitamin D3 in a mouse model of seizures (8). Subcutaneous injection of 33 μg of 1,25 Vitamin D3 incurred an anticonvulsant effect in a chemically induced model of seizures. Compared to controls, mice injected with 1,25 Vitamin D3 40 min prior to the injection of pentylenetetrazol (PTZ), a seizure-inducing chemical, exhibited longer mean latency to seizure onset (77 vs. 55 s), shorter mean duration of tonic–clonic seizures (10 vs. 32 s), and lower mortality (18 vs. 55%). However, the anticonvulsant effects of 1,25 Vitamin D3 were nearly gone if Vitamin D3 injection occurred 3, 6, 12, or 24 h before PTZ injection. The acute efficacy of 1,25 Vitamin D3 suggests that the anticonvulsant effect in this model was due to non-genomic actions of the steroid. In addition, differences in Ca2+ levels between control and experimental mice were non-significant, suggesting that 1,25 Vitamin D3 exerted an anticonvulsant effect independent of its role in calcium metabolism (8).
In a separate study, Kalueff et al. found that the partial deletion of the VDR gene in mice led to increased seizure severity in the model of PTZ-induced seizures (9). Compared to wild-type mice, VDR-knockout mice demonstrated significantly shorter latencies to seizure onset (50.4 vs. 66.9 s), higher Racine scores of seizure severity (5.9 vs. 4.9), and increased mortality (90 vs. 40%). Of note, none of the mice in either the control or experimental condition showed spontaneous seizure activity, suggesting that the VDR gene acts at the threshold level of seizures. Both wild-type and VDR-knockout mice had normal calcium levels, suggesting that the partial deletion of the VDR gene increases seizure intensity via a non-calcium mechanism and providing further evidence of an anticonvulsant effect of Vitamin D3 that is independent from its role in calcium metabolism (9).
In two studies, Borowicz et al. have shown that certain doses of Vitamin D3 enhance the efficacy of several AEDs in a mouse electroshock model of epilepsy without altering the concentrations of the drugs, suggesting a synergistic pharmacological interaction (10, 11). The authors also reported some anticonvulsant action of Vitamin D3 in its own right (10), and they found that treatment with Vitamin D3 led to no deleterious changes in motor coordination, long-term memory, or anxiety (10, 11).
Overall, existing evidence from rat and mouse studies supports an acute anticonvulsant effect of Vitamin D3 in electric shock and chemically induced models of seizure. Further research is needed to explore the longer-term effects of Vitamin D3 therapy in diverse animal models of epilepsy.
Vitamin D3 in Human Epilepsy
People with epilepsy are often Vitamin D3-deficient, along with having decreased bone density and higher rates of osteoporosis (62). Furthermore, certain AEDs, such as carbamazepine and phenytoin, are known to decrease Vitamin D3 levels in people who are taking them due to increased metabolic clearance of Vitamin D3 and conversion to inactive forms (63, 64). People with epilepsy face a sixfold risk for bone fracture compared to the normal population, likely an interplay between frequent falls, reduced bone density, and low levels of Vitamin D3 (62). Maternal Vitamin D3 deficiency during pregnancy has also been associated with hypocalcemia-induced seizures in neonates, which have been successfully treated with calcium and Vitamin D3 supplementation in several case studies (65–68).
In humans, little clinical data exist about the effect of Vitamin D3 supplementation on seizures. In 1974, Christiansen et al. conducted a pilot study in which they treated 23 epilepsy patients with Vitamin D3 (6). Subjects were divided into two groups (A and B) for the duration of the 12-week study, which was divided into a 4-week observation phase (T1) followed by two 4-week treatment periods (T2 and T3). Group A (n = 9) received 4,000 IU/day of Vitamin D3 during T2, followed by 16,000 IU/day during T3. Group B (n = 14) received placebo during T2, followed by 8,000 IU/day of Vitamin D3 during T3. During T2, Group A (treated with 4,000 IU/day of Vitamin D3) experienced a mean reduction in seizure frequency of 32% from baseline, while Group B (placebo) experienced an 8% reduction in mean seizure frequency from baseline. During T3, Group A (treated with 16,000 IU/day of Vitamin D3) experienced a 29% reduction in mean seizure frequency from baseline, while Group B (being treated with 8,000 IU/day of Vitamin D3) experienced a similar. In both groups, high dose vitamin D3 (8000 to 16000 IU/day) was associated with reductions in seizure frequency 33% reduction in mean seizure frequency from baseline. The authors concluded that high dose Vitamin D3 significantly reduced the number of seizures in patients with poorly controlled epilepsy, and, contrary to the authors’ hypothesis, it did so independently of calcium or magnesium levels (6).
Nearly 40 years after Christiansen et al.’s findings, Holló et al. conducted the most recent clinical study of Vitamin D3 therapy in human epilepsy (12). Their subjects consisted of 13 patients with DRE. At baseline, low 25-OH-Vitamin D3 levels <30 ng/ml were present in 12/13 patients and deficient levels (<12 ng/ml) were present in 8/13 patients; 1/13 patients had a normal Vitamin D3 level at baseline. Treatment consisted of Vitamin D3 supplementation aimed at normalizing the serum Vitamin D3 levels of all the subjects. To the 12 patients with low or deficient Vitamin D3 levels at baseline, an oral dose of 40,000–200,000 IU bolus of Vitamin D3 was administered, and treatment was continued with a daily maintenance dose of 2,000–2,600 IU/day of Vitamin D3. The one subject with normal baseline Vitamin D3 level only received the daily maintenance doses. Vitamin D3 levels were rechecked 3 months after treatment onset to determine successful normalization of Vitamin D3 levels and rule out potential Vitamin D3 toxicity. Vitamin D3 supplementation was determined to be safe, as no subjects showed toxic levels of Vitamin D3 at the 3-month follow-up (12). Median Vitamin D3 level rose from 11.8 ng/ml at baseline (range: <4–34.2 ng/ml) to 38.0 ng/ml at 3-month follow-up (range: 23.3–45.0 ng/ml). This elevation in Vitamin D3 levels was significant (p = 0.001, sign test), and the posttreatment Vitamin D3 levels of all subjects were within or close to the normal range (12). The efficacy of the Vitamin D3 treatment in reducing seizures was determined by comparing the number of seizures experienced during the 90 days prior to treatment onset to the number of seizures experienced in the 90 days after treatment onset. Among all subjects, 10/13 experienced fewer seizures after initialization of Vitamin D3 treatment, 2/13 had more seizures, and 1/13 had the same number of seizures. The median reduction in seizure number following treatment onset was 40% and was significant (p = 0.04). In addition, 5/13 patients experienced a ≥50% reduction in number of seizures. The existing clinical evidence suggests a therapeutic effect of Vitamin D3 in human epilepsy, but there is a need for larger Phase I trials and Phase II randomized, placebo-controlled trials to investigate optimal dosing and short-term and long-term efficacy.
Does Vitamin D3 have a Potential Role in Reducing Sudep Risk?
Vitamin D3 status is strongly associated with risk of sudden cardiac death in heart disease and patients with severe kidney disease on hemodialysis. In a large prospective study of 2,300 patients in the Cardiovascular Health Study, the risk of sudden cardiac death was 2-times higher in those with Vitamin D3 levels <20 ng/ml (4 events/1,000) than in those with Vitamin D3 levels >20 ng/ml (2 events/1,000) (69). Similarly, in a study of 1,108 patients with chronic kidney disease, very low levels of Vitamin D3 (25-hydroxy-Vitamin D3 levels <25 nmol/l) were 3-times more likely to sustain sudden cardiac death than those with high levels >75 nmol/l (hazard ratio = 2.99) (70).
Common to severe heart and kidney disease is impaired heart rate variability (HRV), particularly vagus-mediated high-frequency HRV (69–72). Patients with DRE, who are at high risk for SUDEP, have impaired vagus-mediated HRV, similar in magnitude to patients with heart failure (69, 70, 73, 74). Recently, subjects with DRE, at high risk of SUDEP, as measured by the SUDEP-7 inventory, were found to have severe impairment in RMSSD, a measure of vagus-mediated HRV (73, 74). In a recent study linking SUDEP risk in patients with DRE, those with the highest SUDEP-7 inventory risk scores in the highest quartile had RMSSD values of 17.6 ms, vs. 32.0 ms for those with the lowest SUDEP-7 inventory scores (p = 0.03, trend test) (74). This finding is relevant since Vitamin D3 supplementation improves vagus-mediated HRV (71, 72, 75). Recently, Vitamin D3 supplementation ranging from 5,000 to 10,000 IUs in normal controls resulted in significant improvements in high-frequency HRV, as measured by the low-frequency/high-frequency HRV ratio (75). A similar result was recently found in patients with IGA nephropathy, where high-frequency HRV, as measured by the LF/HF HRV ratio, also increased after Vitamin D3 supplementation (71).
Conclusion and Future Directions
The weight of evidence from basic research and animal models over the past several decades supports an anticonvulsant effect of Vitamin D3. Vitamin D3’s anticonvulsant action may be via genomic and non-genomic mechanisms. Epidemiological data as well as a variety of case studies also point to a connection between Vitamin D3 and epilepsy and support the use of Vitamin D3 as a potential therapy for human epilepsy, both in its own right and in conjunction with existing AEDs. However, the clinical data that exist are limited by small sample size and/or lack of randomization and double-blind placebo control. Despite these limitations, existing clinical data have, in the opinion of this review, been positive enough to warrant larger Phase I and Phase II clinical trials in order to more rigorously determine the potential therapeutic value of Vitamin D3 as a treatment for human epilepsy. Recently, our group has received an IND for a Phase I study of Vitamin D3 in DRE to study the safety, preliminary efficacy, and potential cardiac benefits of Vitamin D3 5,000 IU/day in DRE.
Author Contributions
The authors have contributed to the preparation, research, and writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by a generous grant from Beverly and James Peters and family, as well as from Linda and Robert Brill and family.
References
1. Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med (2000) 342:314–9. doi: 10.1056/NEJM200002033420503
2. Kwan P, Schachter SC, Brodie MJ. Drug-resistant epilepsy. N Engl J Med (2011) 365:919–26. doi:10.1056/NEJMra1004418
3. Chen Z, Liew D, Kwan P. Excess mortality and hospitalized morbidity in newly treated epilepsy patients. Neurology (2016) 87:718–25. doi:10.1212/WNL.0000000000002984
4. Meier C, Kraenzlin ME. Antiepileptics and bone health. Ther Adv Musculoskelet Dis (2011) 3:235–43. doi:10.1177/1759720X11410769
5. Reynolds EH. Chronic antiepileptic toxicity: a review. Epilepsia (1975) 16:319–52. doi:10.1111/j.1528-1157.1975.tb06062.x
6. Christiansen C, Rodbro P, Sjo O. “Anticonvulsant action” of vitamin D in epileptic patients? A controlled pilot study. Br Med J (1974) 2:258–9. doi:10.1136/bmj.2.5913.258
7. Siegel A, Malkowitz L, Moskovits MJ, Christakos S. Administration of 1, 25-dihydroxyvitamin D 3 results in the elevation of hippocampal seizure threshold levels in rats. Brain Res (1984) 298:125–9. doi:10.1016/0006-8993(84)91153-3
8. Kalueff AV, Minasyan A, Tuohimaa P. Anticonvulsant effects of 1, 25-dihydroxyvitamin D in chemically induced seizures in mice. Brain Res Bull (2005) 67:156–60. doi:10.1016/j.brainresbull.2005.06.022
9. Kalueff AV, Minasyan A, Keisala T, Kuuslahti M, Miettinen S, Tuohimaa P. Increased severity of chemically induced seizures in mice with partially deleted vitamin D receptor gene. Neurosci Lett (2006) 394:69–73. doi:10.1016/j.neulet.2005.10.007
10. Borowicz KK, Morawska M, Furmanek-Karwowska K, Luszczki JJ, Czuczwar SJ. Cholecalciferol enhances the anticonvulsant effect of conventional antiepileptic drugs in the mouse model of maximal electroshock. Eur J Pharmacol (2007) 573:111–5. doi:10.1016/j.ejphar.2007.07.002
11. Borowicz KK, Morawska D, Morawska M. Effect of cholecalciferol on the anticonvulsant action of some second generation antiepileptic drugs in the mouse model of maximal electroshock. Pharmacol Rep (2015) 67:875–80. doi:10.1016/j.pharep.2015.01.012
12. Holló A, Clemens Z, Kamondi A, Lakatos P, Szűcs A. Correction of vitamin D deficiency improves seizure control in epilepsy: a pilot study. Epilepsy Behav (2012) 24:131–3. doi:10.1016/j.yebeh.2012.03.011
13. Stewart A, Wong K, Cachat J, Elegante M, Gilder T, Mohnot S, et al. Neurosteroid vitamin D system as a nontraditional drug target in neuropsychopharmacology. Behav Pharmacol (2010) 21:420–6. doi:10.1097/FBP.0b013e32833c850f
14. NIH office of Dietary Supplements. Vitamin D. Fact Sheet for Professionals. NIH Office of Dietary Supplements – Vitamin D. (2016). Available from: https://ods.od.nih.gov/factsheets/VitaminD-HealthProfessional/
15. Garcion E, Wion-Barbot N, Montero-Menei CN, Berger F, Wion D. New clues about vitamin D functions in the nervous system. Trends Endocrinol Metab (2002) 13:100–5. doi:10.1016/S1043-2760(01)00547-1
16. Lugg ST, Howells PA, Thicket DR. Optimal vitamin D supplemental levels for cardiovascular disease protection. Dis Markers (2015) 8:1–10. doi:10.1155/2015/864370
17. Kalueff AV, Tuohimaa P. Neurosteroid hormone vitamin D and its utility in clinical nutrition. Curr Opin Clin Nutr Metab Care (2007) 10:12–9. doi:10.1097/MCO.0b013e328010ca18
18. Zadhir A, Tareen N, Pan D, Norris K, Martins D. The prevalence of hypovitaminosis D among US adults: data from the NHANES III. Ethn Dis (2005) 15:S5–97.
19. Evatt ML, Delong MR, Khazai N, Rosen A, Triche S, Tangpricha V. Prevalence of vitamin D insufficiency in patients with Parkinson’s disease and Alzheimer disease. Arch Neurol (2008) 65:1348–52. doi:10.1001/archneur.65.10.1348
20. Buell JS, Dawson-Hughes B, Scott TM, Weiner DE, Dallal GE, Qui WQ, et al. 25-Hydroxyvitamin D, dementia, and cerebrovascular pathology in elders receiving home services. Neurology (2010) 74:18–26. doi:10.1212/WNL.0b013e3181beecb7
21. Knekt P, Kilkkinen A, Rissanen H, Marniemi J, Sääksjärvi K, Heliövaara M. Serum vitamin D and the risk of Parkinson disease. Arch Neurol (2010) 67:808–11. doi:10.1001/archneurol.2010.120
22. Mowry EM. Vitamin D: evidence for its role as a prognostic factor in multiple sclerosis. J Neurol Sci (2011) 311:19–22. doi:10.1016/j.jns.2011.06.035
23. Munger KL, Zhang SM, O’Reilly E, Hernán MA, Olek MJ, Willett WC, et al. Vitamin D intake and incidence of multiple sclerosis. Neurology (2004) 62:60–5. doi:10.1212/01.WNL.0000101723.79681.38
24. Burton JM, Kimball S, Vieth R, Bar-Or A, Dosch HM, Cheung R, et al. A phase I/II dose-escalation trial of vitamin D3 and calcium in multiple sclerosis. Neurology (2010) 74:1852–9. doi:10.1212/WNL.0b013e3181e1cec2
25. Mackay-Sim A, Feron F, Eyles D, Burne T, McGrath J. Schizophrenia, vitamin D, and brain development. Int Rev Neurobiol (2004) 59:351–80. doi:10.1016/S0074-7742(04)59014-1
26. Kalueff AV, Lou YR, Laaksi I, Tuohimaa P. Increased anxiety in mice lacking Vitamin D receptor gene. Neuroreport (2004) 15:1241–4. doi:10.1097/01.wnr.0000129370.04248.92
27. Miller JW, Harvey DJ, Beckett LA, Green R, Farias ST, Reed BR, et al. Vitamin D status and rates of cognitive decline in a multiethnic cohort of older adults. JAMA Neurol (2015) 72:1295–303. doi:10.1001/jamaneurol.2015.2115
28. Brewer LD, Thibault V, Chen KC, Langub MC, Landfield PW, Porter NM. Vitamin D hormone confers neuroprotection in parallel with down regulation of L-type calcium channel expression in hippocampal neurons. J Neurosci (2001) 21:98–108.
29. Kalueff AV, Eremin KO, Tuohimaa P. Mechanisms of neuroprotective action of vitamin D3. Biochemistry (2004) 69:738–41.
30. Eyles D, Brown J, Mackay-Sim A, McGrath J, Feron F. Vitamin D3 and brain development. Neuroscience (2003) 118:641–53. doi:10.1016/S0306-4522(03)00040-X
31. Ko P, Burkert R, McGrath J, Eyles D. Maternal vitamin D3 deprivation and the regulation of apoptosis and cell cycle during rat brain development. Brain Res Dev Brain Res (2004) 153:61–8. doi:10.1016/j.devbrainres.2004.07.013
32. Banerjee P, Chaterjee M. Anti-proliferative role of vitamin D and its analogs – a brief overview. Mol Cell Biochem (2003) 253:247–54. doi:10.1023/A:1026058311857
33. Féron F, Burne TH, Brown J, Smith E, McGrath JJ, Mackay-Sim A, et al. Developmental vitamin D3 deficiency alters the adult rat brain. Brain Res Bull (2005) 65:141–8. doi:10.1016/j.brainresbull.2004.12.007
34. Langub MC, Herman JP, Malluche HH, Koszewski NJ. Evidence of functional vitamin D receptors in rat hippocampus. Neuroscience (2001) 104:49–56. doi:10.1016/S0306-4522(01)00049-5
35. Baas D, Prüfer K, Ittel ME, Kuchler-Bopp S, Labourdette G, Sarliève LL, et al. Rat oligodendrocytes express the vitamin D(3) receptor and respond to 1,25-dihydroxyvitamin D(3). Glia (2000) 31:59–68. doi:10.1002/(SICI)1098-1136(200007)31:1<59::AID-GLIA60>3.0.CO;2-Y
36. Prüfer K, Veenstra TD, Jirikowski GF, Kumar R. Distribution of 1,25-dihydroxyvitamin D3 receptor immunoreactivity in the rat brain and spinal cord. J Chem Neuroanat (1999) 16:135–45. doi:10.1016/S0891-0618(99)00002-2
37. Walbert T, Jirikowski GF, Prufer K. Distribution of 1,25-dihydroxyvitamin D3 receptor immunoreactivity in the limbic system of the rat. Horm Metab Res (2001) 33:525–31. doi:10.1055/s-2001-17210
38. Ramagopalan SV, Heger A, Berlanga AJ, Maugeri NJ, Lincoln MR, Burrell A, et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res (2010) 20:1352–60. doi:10.1101/gr.107920.110
39. Vezzani A, Balosso S, Ravizza T. The role of cytokines in the pathophysiology of epilepsy. Brain Behav Immun (2008) 22:797–803. doi:10.1016/j.bbi.2008.03.009
40. Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci (2003) 23:8692–700.
41. Viviani B, Gardoni F, Marinovich M. Cytokines and neuronal ion channels in health and disease. Int Rev Neurobiol (2007) 82:247–63. doi:10.1016/S0074-7742(07)82013-7
42. Vezzani A, Baram TZ. New roles of interleukin-1 beta in the mechanisms of epilepsy. Epilepsy Curr (2007) 7:45–50. doi:10.1111/j.1535-7511.2007.00165.x
43. Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci (2001) 4:702–10. doi:10.1038/89490
44. Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation (2000) 7:153–9. doi:10.1159/000026433
45. Wang S, Cheng Q, Malik S, Yang J. Interleukin-1beta inhibits gamma-aminobutyric acid type A (GABA(A)) receptor current in cultured hippocampal neurons. J Pharmacol Exp Ther (2000) 292:497–504.
46. Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, et al. Control of synaptic strength by glial TNFalpha. Science (2002) 295:22282–5. doi:10.1126/science.1067859
47. Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci (2005) 25:3219–28. doi:10.1523/JNEUROSCI.4486-04.2005
48. Kalueff AV, Lehtimaki KA, Ylinen A, Honkaniemi J, Peltola J. Intranasal administration of human IL-6 increases the severity of chemically induced seizures in rats. Neurosci Lett (2004) 365:106–10. doi:10.1016/j.neulet.2004.04.061
49. Martin D, Miller G, Rosendahl M, Russell DA. Potent inhibitory effects of glial-derived neurotrophic factor against kainic acid mediated seizures in the rat. Brain Res (1995) 683:172–8. doi:10.1016/0006-8993(95)00369-2
50. Xu B, Michalski B, Racine RJ, Fahnestock M. Continuous induction of neurotrophin-3 triggers sprouting, decreases the levels of TrkA and TrkC, and inhibits epileptogenesis and activity-dependent axonal growth in adult rats. Neuroscience (2002) 115:1295–308. doi:10.1016/S0306-4522(02)00384-6
51. Kanter-Schlifke I, Georgievska B, Kirik D, Kokaia M. Seizure suppression by GDNF gene therapy in animal models of epilepsy. Mol Ther (2007) 15:1106–13.
52. Leranth C, Ribak CE. Calcium-binding proteins are concentrated in the CA2 field of the monkey hippocampus: a possible key to this region’s resistance to epileptic damage. Exp Brain Res (1991) 85:129–36. doi:10.1007/BF00229993
53. Caillard O, Moreno H, Schwaller B, Llano I, Celio MR, Marty A. Role of the calcium-binding protein parvalbumin in short-term synaptic plasticity. Proc Natl Acad Sci U S A (2000) 97:13372–7. doi:10.1073/pnas.230362997
54. Nguyen TM, Lieberherr M, Fritsch J, Guillozo H, Alvarez ML, Fitouri Z, et al. The rapid effects of 1,25-dihydroxyvitamin D3 require the vitamin D receptor and influence 24-hydroxylase activity: studies in human skin fibroblasts bearing vitamin D receptor mutations. J Biol Chem (2004) 279:7591–7. doi:10.1074/jbc.M309517200
55. Norman AW, Okamura WH, Bishop JE, Henry HL. Update on biological actions of 1alpha,25(OH)2-vitamin D3 (rapid effects) and 24R,25(OH)2-vitamin D3. Mol Cell Endocrinol (2002) 197:1–13. doi:10.1016/S0303-7207(02)00273-3
56. Caffrey JM, Farach-Carson MC. Vitamin D3 metabolites modulate dihydropyridine-sensitive calcium currents in clonal rat osteosarcoma cells. J Biol Chem (1989) 264:20265–74.
57. Zanello LP, Norman AW. Rapid modulation of osteoblast ion channel response by 1alpha,25(OH) 2-vitamin D3 requires the presence of a functional vitamin D nuclear receptor. Proc Natl Acad Sci U S A (2004) 101:1589–94. doi:10.1073/pnas.0305802101
58. Jia Z, Nemere I. Immunochemical studies on the putative plasmalemmal receptor for 1, 25-dihydroxyvitamin D3 II. Chick kidney and brain. Steroids (1999) 64:541–50. doi:10.1016/S0039-128X(99)00030-6
59. Norman AW. Vitamin D receptor: new assignments for an already busy receptor. Endocrinology (2011) 147:5542–8. doi:10.1210/en.2006-0946
60. Huhtakangas JA, Olivera CJ, Bishop JE, Zanello LP, Norman AW. The vitamin D receptor is present in caveolae-enriched plasma membranes and binds 1 alpha,25(OH)2-vitamin D3 in vivo and in vitro. Mol Endocrinol (2004) 18:2660–71. doi:10.1210/me.2004-0116
61. DeLuca HF. Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr (2004) 80:1689–96.
62. Sheth RD, Gidal BE, Hermann BP. Pathological fractures in epilepsy. Epilepsy Behav (2006) 9:601–5. doi:10.1016/j.yebeh.2006.08.003
63. Hahn TJ. Drug-induced disorders of vitamin D and mineral metabolism. Clin Endocrinol Metab (1980) 9:107–27. doi:10.1016/S0300-595X(80)80023-5
64. Ali FE, Al-Bustan MA, Al-Busairi WA, Al-Mulla FA. Loss of seizure control due to anticonvulsant-induced hypocalcemia. Ann Pharmacother (2004) 38:1002–5. doi:10.1345/aph.1D467
65. Oki J, Takedatsu M, Itoh J, Yano K, Cho K, Okuno A. Hypocalcemic focal seizures in a one-month-old infant of a mother with a low circulating level of vitamin D. Brain Dev (1991) 13:132–4. doi:10.1016/S0387-7604(12)80122-7
66. Agarwal KS, Beri RS, Puliyel JM. Repeated unifocal seizure in post neonatal infants with hypocalcemia. Indian Pediatr (2000) 37:203–5.
67. Dijkstra SH, Arpaci G, Huijsman WA, Boot AM, van den Akker EL. [Seizures in foreign newborns due to maternal vitamin-D deficiency]. Ned Tijdschr Geneeskd (2005) 149:257–60.
68. Camadoo L, Tibbott R, Isaza F. Maternal vitamin D deficiency associated with neonatal hypocalcemic convulsion. Nutr J (2007) 6:1. doi:10.1186/1475-2891-6-23
69. Deo R, Katz R, Shlipak MG, Sotoodehnia N, Psaty BM, Sarnak MJ, et al. Vitamin D, parathyroid hormone, and sudden cardiac death: results from the Cardiovascular Health Study. Hypertension (2011) 58:1021–8. doi:10.1161/HYPERTENSIONAHA.111.179135
70. Drechsler C, Pilz S, Obermayer-Pietsch B, Verduijn M, Tomaschitz A, Krane V, et al. Vitamin D deficiency is associated with sudden cardiac death, combined cardiovascular events, and mortality in hemodialysis patients. Eur Heart J (2010) 31:2253–61. doi:10.1093/eurheartj/ehq246
71. Mann MC, Hemmelgarn BR, Exner DV, Hanley DA, Turin TC, Wheeler DC, et al. Vitamin D supplementation is associated with stabilization of cardiac autonomic tone in IgA nephropathy. Hypertension (2015) 66:e4–6. doi:10.1161/HYPERTENSIONAHA.115.05688
72. Mann MC, Exner DV, Hemmelgarn BR, Hanley DA, Turin TC, MacRae JM, et al. The VITAH trial VITamin D supplementation and cardiac autonomic tone in hemodialysis: a blinded, randomized controlled trial. BMC Nephrol (2014) 6(15):129. doi:10.1186/1471-2369-15-129
73. DeGiorgio CM, Miller P, Meymandi S, Chin A, Epps J, Gordon S, et al. RMSSD, a measure of vagus-mediated heart rate variability, is associated with risk factors for SUDEP: the SUDEP-7 inventory. Epilepsy Behav (2010) 19:78–81. doi:10.1016/j.yebeh.2010.06.011
74. Novak JL, Miller PR, Markovic D, Meymandi SK, DeGiorgio CM. Risk assessment for sudden death in epilepsy: the SUDEP-7 inventory. Front Neurol (2015) 6:252. doi:10.3389/fneur.2015.00252
Keywords: cholecalciferol, vitamin D3, epilepsy, SUDEP, animal models, clinical trials
Citation: Pendo K and DeGiorgio CM (2016) Vitamin D3 for the Treatment of Epilepsy: Basic Mechanisms, Animal Models, and Clinical Trials. Front. Neurol. 7:218. doi: 10.3389/fneur.2016.00218
Received: 15 September 2016; Accepted: 21 November 2016;
Published: 08 December 2016
Edited by:
Jeremy Daniel Slater, University of Texas Medical School at Houston, USAReviewed by:
Patricia Braga, School of Medicine, Montevideo, UruguayDetlev Boison, Legacy Health, USA
Copyright: © 2016 Pendo and DeGiorgio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher M. DeGiorgio, Y2RlZ2lvcmdpb0BkaHMubGFjb3VudHkuZ292