 Aaron Ritter
Aaron Ritter Jeffrey Cummings
Jeffrey Cummings- Cleveland Clinic Lou Ruvo Center for Brain Health, Las Vegas, NV, USA
With the demographic shift of the global population toward longer life expectancy, the number of people living with Alzheimer’s disease (AD) has rapidly expanded and is projected to triple by the year 2050. Current treatments provide symptomatic relief but do not affect the underlying pathology of the disease. Therapies that prevent or slow the progression of the disease are urgently needed to avoid this growing public health emergency. Insights gained from decades of research have begun to unlock the pathophysiology of this complex disease and have provided targets for disease-modifying therapies. In the last decade, few therapeutic agents designed to modify the underlying disease process have progressed to clinical trials and none have been brought to market. With the focus on disease modification, biomarkers promise to play an increasingly important role in clinical trials. Six biomarkers have now been included in diagnostic criteria for AD and are regularly incorporated into clinical trials. Three biomarkers are neuroimaging measures – hippocampal atrophy measured by magnetic resonance imaging (MRI), amyloid uptake as measured by Pittsburg compound B positron emission tomography (PiB-PET), and decreased fluorodeoxyglucose (18F) uptake as measured by PET (FDG-PET) – and three are sampled from fluid sources – cerebrospinal fluid levels of amyloid β42 (Aβ42), total tau, and phosphorylated tau. Fluid biomarkers are important because they can provide information regarding the underlying biochemical processes that are occurring in the brain. The purpose of this paper is to review the literature regarding the existing and emerging fluid biomarkers and to examine how fluid biomarkers have been incorporated into clinical trials.
Introduction
Alzheimer’s disease (AD), the most common cause of dementia, is a progressive neurodegenerative disorder that becomes more prevalent with increasing age. Currently, there are more than 44 million people worldwide living with dementia (1). As the demographics of the global population shift toward longer life, it is projected that this number will be more than triple by the year 2050. With the estimated cost of dementia already exceeding 1% of the world’s gross domestic product (1), this rapid increase constitutes a looming public health emergency. Available therapies for AD were approved based on their ability to improve the symptoms of the disease but do not alter underlying pathophysiologic processes (2). In order to ease the public health burden posed by AD, drugs with disease-modifying properties are urgently needed.
Insights gained from decades of AD research have begun to elucidate the pathophysiology underlying this complex disease. It is now widely accepted that the chain of biochemical events thought to be responsible for AD are triggered many years prior to symptom onset (3). While an enhanced understanding of the two characteristic pathological changes seen in AD – plaques composed of amyloid β (Aβ) and neurofibrillary tangles (NFTs) composed of hyperphoshorylated tau – have yielded targets that may be amenable to pharmacological intervention, no therapeutics with potentially disease-modifying properties have advanced past Phase III trials. A number of theories have been proposed to explain this failure: (1) selection of patients based on clinical diagnosis can be inaccurate, leading to the inclusion of large number of patients without AD in clinical trials (4): (2) the timing of interventions designed to clear amyloid – at stages when subjects have already begun to manifest the symptoms of mild to moderate dementia – is too late in the disease course to affect cognitive change (5, 6): (3) the progression of the disease is too gradual to demonstrate drug–placebo differences in “typical length” drug trials (7): (4) candidate agents have been permitted to advance to Phase III trials without strong evidence of target engagement or disease modification from preclinical models or early clinical trials (8).
New strategies are needed to address the high failure rate in AD drug development. New trial designs, centralized rating and review, more predictive models in preclinical testing, improved clinical outcome measures, and more stringent testing of drugs in Phase II are all strategies that may improve success rates. While proof of efficacy of AD treatments will ultimately depend on demonstration of benefit on clinical measures, biological markers (biomarkers) of underlying disease processes will take on enhanced significance, especially as trials move toward enrolling subjects earlier in the disease process.
Aided by the development of biomarkers, AD is now considered one clinical disease with a continuum through several clinical stages (5). Reflecting this change in disease conception, several biomarkers have now been accepted widely enough that they have been incorporated into the two most recent research criteria (9–12). Three of these biomarkers are imaging biomarkers: hippocampal atrophy as detected by structural magnetic resonance imaging (MRI); decreased uptake of (18F) in characteristic regions on positron emission tomography (FDG-PET); and increased amyloid tracer retention on PET (PiB-PET). Three biomarkers are cerebrospinal fluid (CSF) protein levels: low CSF levels of amyloid β42 (Aβ42) and elevated CSF levels of total (t-tau) and phosphorylated tau (p-tau). Imaging biomarkers are important because they can provide crucial information about topographical changes in the brain. There are a number of excellent reviews describing their use in both clinical practice and drug trials (13). They will not be described here. The focus of this contribution is fluid biomarkers. The purpose of this paper is to review the literature regarding the existing and emerging fluid biomarkers and to examine how fluid biomarkers have been incorporated into clinical trials.
Fluid Biomarkers Regularly Incorporated into Clinical Trials
CSF Aβ42
A picture of the complex chain of events leading to AD has emerged over the last three decades. The leading theory to explain the pathophysiological changes in AD is the amyloid cascade hypothesis (14). Based largely on models derived from familial cases of AD – in which, one of three autosomal dominantly inherited mutations results in pathological aggregation and accumulation of Aβ – the amyloid cascade hypothesis posits that the pathological accumulation of amyloid triggers a complex sequence of biochemical events ultimately leading to widespread synaptic dysfunction, neuronal dysfunction, and cell death. An overview of the initial steps involved in Aβ production is provided in Figure 1.
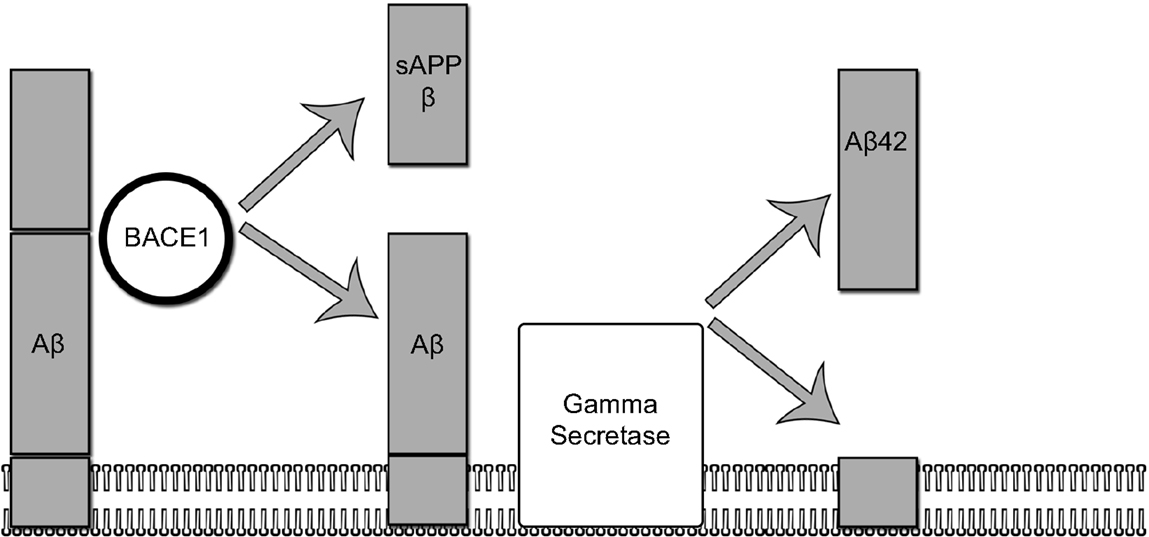
Figure 1. The amyloidogenic pathway. In the amyloidogenic pathway, The amyloid precursor protein (APP) is processed in two sequential steps: (1) in the first step, APP is cleaved by BACE1 yielding a membrane-bound fragment and releasing sAPP into the interstitial space. (2) In the second step, gamma secretase cleaves the remaining membrane-bound fragment releasing an abeta 42 fragment.
The amount of Aβ in the brain is determined by a balance between Aβ production and degradation/clearance mechanisms (15). Several enzymes, such as neprilysin, insulin-degrading enzyme, plasminogen inhibitor, break down Aβ in the interstitial space (16). Fragments that are not degraded in the brain are actively transported across the blood–brain barrier (BBB) or diffuse into the CSF space (17). The two transport proteins responsible for Aβ efflux from the brain are the low density lipoprotein receptor related protein-1 (LRP-1) and Apo J (15). Once in blood, Aβ is rapidly taken up by plasma proteins and transported to the liver for further degradation. A dynamic equilibrium exists between the amount of Aβ in the CSF and the amount of Aβ in the plasma space, and a small amount of non-neuronal Aβ is found in the CSF. A transport protein known as the receptor for advanced end products (RAGE) is responsible for the influx of Aβ from the serum into the CNS. The amount of amyloid in the brain is a highly regulated process and it is estimated that the entire load of soluble Aβ is turned over twice per day (17).
In AD, there is a significant decrease in Aβ clearance (18) resulting in dramatic increases (100–1,000 fold) in the amount of Aβ in the brain (17). Aβ fragments consisting of 42 amino acids (Aβ42) are particularly prone to aggregation (19). As amyloid concentrations rise, Aβ42 fragments rapidly aggregate into oligomers of various sizes and conformations (20). Aβ oligomers are neurotoxic and have been shown to inhibit memory, disrupt long-term potentiation, and impair synaptic function in animal models (21, 22). Emerging data is beginning to clarify the role that Aβ oligomers play in triggering AD pathophysiology (23). In addition to oligomerizing, Aβ fragments also fibrillize into cross-β-sheets, forming the insoluble plaques that constitute the main neuropathological finding in AD. The primary role of amyloid plaques seems to be to serve as large reservoirs of soluble amyloid (the amount of insoluble fibrillar Aβ is 100-fold greater than the amount of soluble Aβ in the brain) (24). Plaques may serve to buffer any changes in the amount of circulating amyloid. Plaques, however, are not entirely benign species as array tomography has revealed that they are surrounded by a ring of dystrophic and disfigured neurons (25), implying that they exert local neurotoxic effects (26). Plaque burden, however, correlates poorly with disease severity (27, 28) and it is now widely thought that Aβ’s primary role in the pathogenesis of AD is by triggering another pathological process (29).
Several commercially available, CSF enzyme-linked immunosorbent assays (ELISAs) have been developed that detect CSF Aβ. CSF assays for Aβ detect soluble monomeric species. In AD, levels of CSF Aβ 40 remain stable while Aβ42 levels have consistently been shown drop to <50% of normal (30). The reduction in CSF Aβ42 levels is generally thought to reflect both the sequestration of Aβ42 in insoluble plaques (27) and aggregation into oligomeric species (31). Post-mortem studies have also reported correlations between low CSF Aβ42 and increased amyloid plaque load (32, 33). With the development of amyloid PET imaging (which allows for the direct visualization of fibrillar amyloid), the relationship between low CSF Aβ42 levels and amyloid plaque has been established in vivo (34) and has been confirmed in many different studies (35, 36). Although low CSF Aβ42 levels and increased fibrillar uptake on PET scan generally correspond with one another and are often used interchangeably to diagnose AD, it is important to note that they are not detecting the same form of amyloid (CSF assays detect monomeric, soluble amyloid while PET imaging detects fibrillar plaque). The discrepancy between the two measures has been illustrated in several studies (37, 38). A recent study using cross-sectional data found that 20% of cognitively normal subjects had low CSF Aβ42 levels but negative PET scans. This discrepancy was seen in only 6% of subjects with dementia (38). PET scan positivity was also found to correlate closely with increased CSF tau levels. The authors interpreted these findings to suggest that CSF Aβ42 “positivity” comes earlier in the disease progression than amyloid uptake on PET scan. If this finding is verified in longitudinal studies, it would suggest that low levels of CSF Aβ42 may be a marker of early disease processes while amyloid scanning would have utility as a marker of disease progression.
CSF Tau
Neurofibrillary tangles composed of hyperphosphorylated tau are the second major neuropathologic finding in AD. Tau is a ubiquitous intracellular protein that promotes cellular stability through interactions with microtubule proteins (39). Consequently, tau plays a key role in maintaining neuronal integrity, cellular signaling, and axonal transport. The dynamic relationship that exists between tau and microtubule proteins is driven by the phosphorylation state of tau, which is under the control of a variety of kinases and phosphatases (40, 41). In AD, for reasons that remain to be elucidated, the phosphorylation state of tau increases (42). Various theories have been proposed to explain this phenomenon. A leading theory is that it is a direct response to the toxic effects of Aβ accumulation (43); however, other potential causes include neuroinflammation (44), oxidative stress (45), genetic factors (46), or even infection (47). Tau hyperphosphorylation is a key step in the pathogenesis of AD because hyperphorsphorylated tau no longer binds to microtubule proteins (48). This leads to higher cytosolic concentrations of unbound tau. Unbound, hyperphosphorylated tau is susceptible to aggregation, protein trapping, and misfolding (49, 50). Aggregated fibrils consisting of hyperphosphorylaed tau comprise the helical filaments in NFTs. The accumulation of NFTs within neuronal axons is toxic to cells. Both the loss of normal physiological function (i.e., loss of cellular integrity) and the gain of toxicity induced by NFT accretion are thought to contribute to neuronal dysfunction in AD (50).
In AD, NFT accumulation proceeds through the brain in a stereotypical pattern, appearing first in the locus coeruleus and the entorhinal cortex, proceeding next to the hippocampus, and then spreading to the temporal cortex and neocortical association areas (51). Neuropathological studies have reported correlations between NFT formation and neuronal loss, both of which increase in parallel with AD disease progression (52). Understanding the intercellular spread of NFT as it progresses through the brain has been the focus of recent investigation (53, 54). In mouse models, injection of filamentous tau induces NFT formation at the injection site that over time progresses to neighboring and synaptically connected brain regions (55). This finding suggests that tau exhibits prion-like behavior as it spreads from highly focal brain regions to involvement of limbic, paralimbic, and neocortical regions (56).
In AD, CSF levels of t-tau increase to 3× normal (57). Increases in CSF t-tau have been associated with both NFT burden and Braak staging (33). Elevations in CSF t-tau, however, are not specific to AD as transient elevations are found following stroke (58) and traumatic brain injury (TBI) (59). This finding suggests that elevated CSF t-tau levels are reflective of non-specific neuronal injury and cell death. The highest levels of CSF t-tau are found in Creutzfeldt–Jakob disease (CJD), a disease characterized by accelerated neurodegeneration (60). It is also important to note that tau secretion is an active physiological process, occurring independently of neuronal injury (56). In AD, an additional source of CSF tau is the residence of this molecule in extracellular space during its passage from neuron to neuron. More research is needed to fully understand the composition of CSF t-tau levels in AD.
In addition to detecting total tau (t-tau), several ELISAs have been developed that reflect the phosphorylation state of tau. In AD, CSF levels of p-tau increase to approximately twice normal levels. Commonly used assays measure tau phosphorylation at residue either 181 or 231, both of which increase to similar levels in AD (61). Autopsy studies reveal that CSF p-tau correlates with NFT burden in AD (62). Because levels of p-tau are thought to reflect both NFT load and phosphorylation state, elevations in p-tau are generally thought to be a more specific finding in AD than elevations in CSF t-tau (61, 63). Dissociations between high t-tau and normal p-tau levels have been reported in several dementing diseases including CJD (64), frontotemporal dementia, and vascular dementia (61).
Utility of CSF Aβ42, t-Tau, and p-Tau
Used individually, CSF markers (CSF Aβ42 or tau) demonstrate good sensitivity in distinguishing subjects with AD from non-controls (41); however, several studies have reported poor specificity in distinguishing subjects with AD from non-AD dementias (65–67). Diagnostic precision has also been shown to decrease with increasing age (68). Diagnostic accuracy increases considerably when these measures are combined into a so-called “AD signature” consisting of low Aβ42 and elevated total and p-tau. This signature demonstrates 80–95% sensitivity and specificity in identifying subjects with AD in the dementia phase of disease (5) and has been shown to be highly predictive of AD pathology at autopsy (28). The ability of CSF biomarkers to identify subjects harboring AD pathology is considerably better than the accuracy of a diagnosis made on clinical grounds alone. In a study looking at 919 autopsy-confirmed cases of AD that comprise the National Alzheimer’s Coordinating Center (NACC) database, clinical diagnosis was 71–88% sensitive but only 44–71% specific in predicting AD pathology at autopsy (69). The challenge of accurately identifying subjects with AD pathology based on clinical diagnosis alone has also been demonstrated in clinical trials that have incorporated amyloid PET scans (4, 70, 71). Data from several clinical trials suggest that a substantial percentage of subjects enrolled in clinical trials do not actually have evidence of AD pathology on PET scan. For example, in the Phase III trial of bapineuzumab >35% of APOE ε4 non-carriers had negative amyloid scans (70). As it is unlikely that compounds with putative anti-AD properties will produce clinical benefits in subjects without AD pathology, inaccurate inclusion rates increase the likelihood of trial failure. Incorporating CSF biomarkers into inclusion criteria is a strategy that can be used to enrich patient samples, increase a trial’s statistical power, and ensure that candidate compounds are being accurately tested against the AD substrates they are designed to ameliorate.
The temporal relationship among Aβ42, t-tau, and p-tau levels has been the subject of much exploration and several models have been proposed to explain the complex dynamics that exist between CSF biomarkers and disease progression (43, 72). There is now convincing evidence that CSF Aβ42 and tau levels convert from normal to “pathologic” years before the onset of clinical symptoms, providing a powerful tool to assess which individuals are at risk for developing AD dementia (73). Decreases in CSF Aβ42 are typically appreciated before changes in CSF tau, and in accordance with the amyloid cascade hypothesis, suggest that amyloid accumulation drives tau pathology. Examining a cohort of subjects with autosomal dominant AD, Bateman et al. demonstrated that changes in Aβ42 can be fully appreciated 25 years before expected symptom onset and changes in tau 15 years before expected symptoms onset (3). In cohorts without AD mutations, several studies have reported that decreases in CSF Aβ42 (with or without changes in CSF tau) can be detected in cognitively normal subjects and predict the development of cognitive decline (74) and dementia (75, 76). CSF biomarkers have also showed good sensitivity (83–95%) and specificity (71–90%) in predicting which subjects with mild cognitive impairment (MCI) will progress to develop AD dementia (77–80). The accurate identification of patients in this early stage of the disease is important because MCI is a non-specific syndrome and only around 50% of subjects with MCI are thought to have AD (81). Using CSF biomarkers to accurately identify subjects harboring AD pathology as early as possible in the disease course will allow for testing of candidate compounds earlier in the disease course and at time points that may prove more amenable to pharmacological intervention.
While the CSF biomarkers discussed above provide a powerful window into the pathological processes occurring in AD, several limitations deserve mention. An innate limitation of all fluid biomarkers is that they lack anatomical precision (82). Unlike imaging biomarkers, CSF biomarkers do not provide insight into the topographic distribution of pathological changes in the brain. Another limitation of current CSF biomarkers is that aside from small increases in t-tau (83), they remain fairly stable during the dementia phase of disease (84). Therefore, current CSF biomarkers have limited utility in disease staging or prognosis (73). Furthermore, because only weak associations between CSF biomarkers and clinical measures have been reported (85), it is unknown if drug-induced changes in these measures will result in clinically meaningful effects (16). Unknown variables include when interventions need to be timed and to what degree a biomarker change may be correlated with a clinical outcome (86). An additional limitation of CSF biomarkers is the high degree of variability and lack of assay standardization that exists among laboratories. A 2013 study analyzing data from Alzheimer’s Association quality control program reported a 20–30% discrepancy among laboratories in measuring CSF biomarkers (68). This is too high for globally accepted reference ranges to be assigned (87). Quality control and standardization projects have been initiated with the intent of improving precision and reproducibility across laboratories (5).
Emerging CSF Biomarkers
Given the limitations of the currently used CSF biomarkers, substantial research has been devoted to finding and validating additional CSF biomarkers. Guided by an enhanced understanding of the neurobiological changes in AD, several promising candidate markers have been identified. Table 1 summarizes the development of CSF candidates.
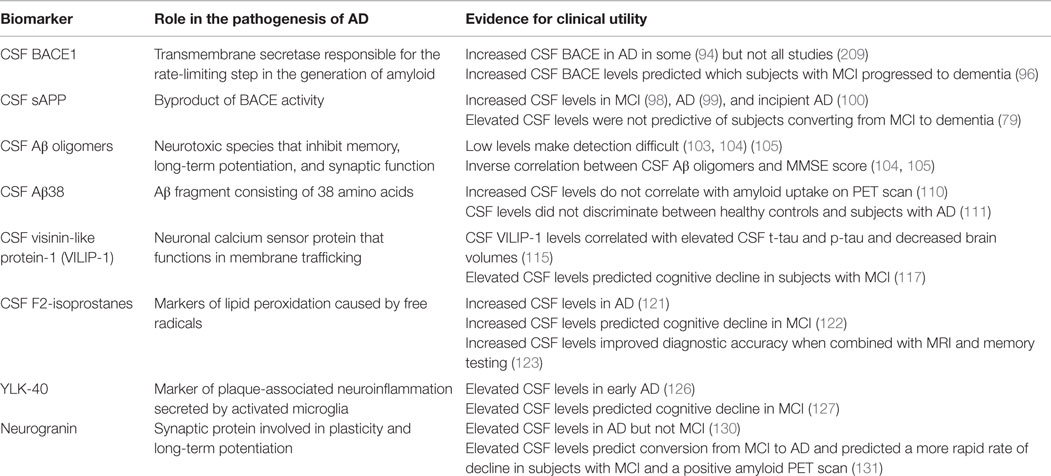
Table 1. Candidate CSF biomarkers.
Amyloid-Related CSF Biomarker Candidates
BACE1
BACE1 is an aspartic protease that catalyzes the rate-limiting step in the generation of Aβ42 (Figure 1). BACE1 also plays a role in the processing of other membrane proteins, such as neuregulin (88), and is thought to influence myelination (89) and synaptic plasticity (90). Because of its diverse and important role in normal brain functioning, BACE1 activity is synchronized by a variety of complicated regulatory mechanisms at both the transcriptional and translational levels (91). Increased levels of BACE1 and indicators of BACE1 activity have been found in the brains of patients with AD (92, 93). Elevations in CSF BACE1 have also been detected in the CSF of patients with AD (94, 95) and subjects with MCI who later went on to develop AD (96). Several explanations have been proposed to account for the increases in CSF BACE1 in AD. Increased CSF BACE1 levels have been found to correlate with increases in CSF t-tau (96) and one possibility is that BACE1 release into the CSF is a product of a non-specific release of proteins from injured or dying neurons. New research, however, suggests a more complicated picture, in which, normal regulatory controls on BACE1 activity are lost. Faghihi et al., for example, has reported that a non-coding antisense RNA that stabilizes BACE1 mRNA and results in increased BACE1 activity is increased in the brains of subjects with AD. Furthermore, in vitro exposure of cells to Aβ42 induces this antisense RNA, laying the groundwork for a deleterious feed-forward cycle of AD disease progression, in which, increased levels of Aβ induce the expression of increased BACE1 activity and further Aβ production (97). CSF BACE1 will be important in establishing target engagement in compounds with putative BACE1 inhibiting properties.
sAPP-β
The first step in APP processing is the proteolytic cleavage by BACE1. This cleavage yields two products, one of which is the membrane bound fragment (which then undergoes further processing by gamma secretase to eventually form Aβ) and the other, a larger amino acid fragment, sAPP-β, which is secreted into the interstitial space. Levels of CSF sAPP-β may serve as an indirect marker of BACE activity and Aβ production. Studies looking at the clinical correlation between CSF sAPP-β have generally been positive and elevated levels of sAPP-β have been reported in MCI (98), AD (99), and patients with incipient AD (100). However, not all studies have demonstrated meaningful clinical correlations (79). Changes in CSF levels of sAPP-β may eventually be used in clinical trials to provide evidence of target engagement and to monitor for drug effects.
Aβ Oligomers
In vitro exposure of Aβ oligomers to hippocampal neurons quickly impairs synaptic function and is more toxic than exposure to monomeric or fibrillar forms of amyloid (101). This finding, in conjunction with reports from several animal models that demonstrate neuroanatomical and behavioral abnormalities before the appearance of plaques (25), has led the field to consider the role of Aβ oligomers in AD pathogenesis. The steady state of Aβ oligomers in the CSF is very low – <0.02% of total CSF Aβ levels (102) – and attempts to detect them standard assays have failed (101) while other attempts have produced variable results (103–105). Recently, Hong et al. were able to demonstrate that Aβ oligomers in the interstitial fluid were quickly sequestered onto cellular membranes, displaying a particular affinity for GM1 gangliosides (102). In this study, Aβ oligomers demonstrated a higher binding affinity for cell membranes than monomeric Aβ species, potentially explaining the low contribution of oligomers to the overall composition of CSF Aβ levels. The authors were also able to detect low levels of GM1-bound Aβ in human CSF. These levels correlated with CSF Aβ42. Further investigation is needed to determine if CSF GM1-bound Aβ will prove useful as a biomarker in AD. It is also important to note that soluble Aβ oligomers may have utility as a progression biomarker, as two studies – one using flow cytometry (105) and the other using ELISA (104) – have reported an inverse correlation between levels of CSF Aβ oligomers and score on MMSE. The challenges of reliably quantifying Aβ oligomers in CSF will need to be overcome before the potential of this biomarker can be fully realized.
Aβ Isoforms
While most Aβ species exist as peptide fragments consisting of either 40 or 42 amino acids, isoforms of varying length have also been detected in the CSF of patients with AD (106–108). One small study reported that a particular CSF amyloid “signature” consisting of Aβ16, Aβ33, Aβ39, and Aβ42 could distinguish subjects with AD from controls with an accuracy of 86% (106). The performance of Aβ38 has been investigated in a number of studies and as an exploratory measure in a phase II trial of avagacestat (109). The utility of CSF Aβ38 appears to be limited given that levels do not correlate with amyloid uptake on PET (110) and did not discriminate controls from subjects with AD in another study (111).
Non-Amyloid CSF Biomarker Candidates
Cerebrospinal fluid markers that reflect processes that occur after amyloid deposition, including neurodegeneration, synapse loss, neuroinflammation, oxidative stress, etc. may also provide diagnostic and prognostic utility. A select group of candidates will be discussed here. For a comprehensive review, the reader is directed to the review by Fagan and Perrin (112).
Visinin-Like Protein-1
Visinin-like protein-1 (VILIP-1) is a neuronal calcium sensor protein that can be detected in most regions of the brain (sparing the caudate and putamen) (113). It belongs to a family of proteins thought to play a role in membrane trafficking (Braunewell Cell Tissue Res) and is thought to play a role in calcium-mediated neuronal death (114). CSF levels of VILIP-1 have shown to correlate with CSF t-tau, p-tau, and brain volumes (115, 116). High levels of CSF VILIP-1 have also been reported to predict the cognitive decline in a cohort of patients with mild AD followed over a period of 2.6 years (117). Several studies have shown that higher levels of CSF VILIP-1 are seen in AD than other dementing diseases, such as dementia with Lewy bodies (114), frontotemporal dementia, and progressive supranuclear palsy (117).
F2-Isoprostanes
There is a growing body of evidence suggesting that oxidative damage plays a key role in the pathogenesis of AD (118). F2-isoprostanes are markers of lipid peroxidation caused by free radicals (119). Increased levels of F2-isoprostanes are found in AD brains (120) and in the CSF of patients with AD (121). Elevated levels of CSF F2-isoprostanes have also been shown to correlate with eventual cognitive decline in MCI (122) and improve diagnostic accuracy of AD when combined with memory testing and MRI (123).
YKL-40
Neuropathological, biochemical, and genetic studies indicate that alterations in neuroinflammatory pathways play a role in the pathogenesis of AD (124). YKL-40 is a marker of plaque-associated neuroinflammation that is secreted by activated microglia (125). Several studies suggest that YKL-40 may be an early marker of AD as levels have been shown to be increased in the preclinical phase (116, 126) and to predict cognitive decline in early stage dementia (127).
Neurogranin
Neurogranin is a synaptic protein that is enriched in forebrain areas (128). It is thought to be involved in synaptic plasticity and long-term potentiation (129). Elevated levels of neurogranin have been reported in the CSF of subjects with AD (but not MCI) (130). Elevated levels of CSF neurogranin have been shown to predict conversion from MCI to AD and to predict a more rapid rate of decline in subjects with MCI and a positive amyloid PET scan (131).
Serum Biomarkers
The process of obtaining CSF fluid by lumbar puncture (LP) is invasive and associated with a small but significant risk of post-LP headache (132). Given the negative public perception of the LP procedure, it is unlikely that all patients in a clinical trial would agree to have CSF sampling. Serum samples are easily obtained and readily accepted by patients. The development of a reliable serum biomarker could potentially be integrated into a multi-stage screening and diagnostic process, to provide valuable information about which patients should proceed to more expensive/invasive testing, and to monitor disease progression (133). Currently, there has been little success in finding reliable serum biomarkers in AD or MCI (41). Table 2 summarizes the findings regarding candidate serum biomarkers in AD.
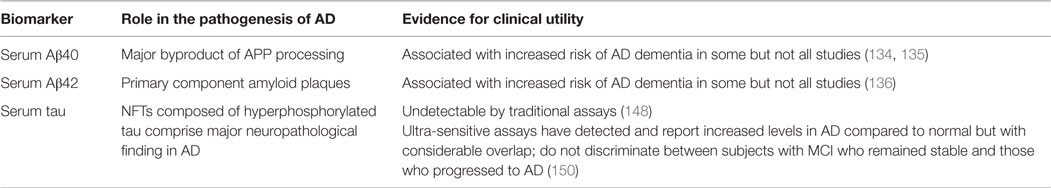
Table 2. Candidate non-CSF biomarkers.
Serum Aβ
Despite being the focus of intense investigation, the utility of serum Aβ as AD biomarkers has not been fully defined. Serum Aβ (40,42) levels in AD show considerable overlap with non-AD controls, which limits its use as a diagnostic marker (92). The use of serum Aβ as a marker of risk is also unclear as some studies have reported an increased risk with increased Aβ40 (134, 135) or Aβ42 (136) while others have reported that increased risk is associated with low levels of Aβ42 (137). In addition, several studies have failed to find an association between serum Aβ levels and AD risk (138, 139). One meta-analysis reported that a low Aβ42:Aβ40 ratio was associated with an increased risk of AD (140); however, the generalizability of this analysis is limited by the heterogeneity of included studies. Little is known about the prognostic value of serum levels of Aβ. One study has reported that higher baseline levels of serum Aβ42 were associated with faster rates of cognitive decline over a 1-year period in subjects with AD (141). The small sample size and the lack of follow-up analysis of plasma levels means that additional research is needed to determine if serum levels can be used for patient stratification. Changes in serum Aβ levels have also been detected in several clinical trials and have been used as evidence to support claims of target engagement (71, 142). Further investigation is needed to clarify the association between serum Aβ levels and AD pathophysiology.
One potential explanation for the discrepancy between the performance of CSF Aβ and serum Aβ is that serum levels do not accurately reflect CSF Aβ levels (143). The majority of CSF Aβ is of neuronal origin and is thought to directly reflect Aβ production in the brain. Serum Aβ, on the other hand, is derived from a variety of non-neuronal sources including the liver, bone, muscle, kidney, pancreas, and platelets (66). The physiologic milieu in the CSF is also drastically different from the serum compartment. In the serum, there are 300× more Aβ binding proteins than in the CSF (15) and the majority of Aβ in the serum is protein bound (144).
Serum Tau
Transient elevations in serum tau are detected in response to neuronal injury from ischemic stroke (145), hypoxic brain injury during cardiac arrest (146), and TBI (147). There is considerable evidence that the biochemical regulation of tau is dependent on which biological compartment it resides. For example, following neuronal injury, CSF tau may stay elevated for weeks while in the serum, tau is cleared rapidly, returning to normal levels within hours (58). As a result, serum tau levels are not thought to accurately reflect CSF tau levels. In a small study using a sandwich ELISA, serum tau levels were essentially undetectable in patients with AD despite having elevated CSF t-tau levels (148). More recently, ultra-sensitive assays have been developed that have captured changes in serum tau levels following TBI (146) and cardiac arrest (149). This assay has been tested in one cohort with AD (150). In this study, higher serum tau levels were seen in patients with AD as compared to subjects with MCI and controls; however, a considerable degree of overlap was noted across the three groups, limiting its diagnostic utility (150). Additionally, serum tau levels did not discriminate between subjects with MCI who remained stable and those with MCI who went on to develop AD.
Other Serum Markers
Other novel serum targets for development include F2-isoprostanes (151) and plasma complement factor H (152); however, the results of studies looking at these candidates have been disappointing and do not support their application as diagnostic or prognostic factors at this time.
Proteomic Approaches
An alternative approach to developing serum biomarkers in AD is to identify a characteristic profile of protein markers, which, taken together, would constitute a pathological “fingerprint” (133). Significant interest in proteomic strategies was generated following a study, which identified a characteristic pattern of 18 abnormal plasma signaling and inflammatory proteins in a sample of patients with AD (153). Applied to a pre-existing data set, this profile correctly identified subjects with AD from healthy controls with 90% accuracy. In addition, this profile predicted conversion from MCI to dementia in 20 of 22 patients (followed up to 6 years). With advances in bioinformatics, the numbers of trials employing proteomic approaches have increased. Using pre-existing data sets, a number of proteomic profiles have been identified, which have shown high diagnostic accuracy (154–157). Challenges to the proteomic approach include successful replication of findings across studies (154) and whether profiles can reach appropriate standardization levels to be replicated across laboratories (133). Guidelines designed to approach these challenges have recently been published (158). No consensus has been reached on a specific proteomic profile that provides reliable information in AD.
Urine and Saliva
Urine and saliva are appealing targets for biomarker development due to their ease of collection. Molecules sampled from these sources, however, are subjected to filtration and metabolic processing and may not reflect biochemical changes occurring in the brain. For this reason, AD research has largely ignored these biological compartments (159). One small study detected reduced acetylcholinesterase activity in the saliva of patients with AD compared to normal controls (160) while another found no difference (161). Increased levels of salivary Aβ42 have been demonstrated in patients with mild AD compared to normal controls and patients with Parkinson’s disease (162). In another study using mass spectroscopy, an increased salivary p-tau to t-tau ratio was found in AD patients compared to normal controls (163). More research is needed on these readily accessible fluids to determine if they contain meaningful information on brain states.
Use of Fluid Biomarkers in Clinical Trials
The scope of use of fluid biomarkers in clinical trials is described below. Here, we describe the results of several clinical trials in which fluid biomarkers were included among outcome measures. Table 3 summarizes the results of these studies as well as others that are not described.
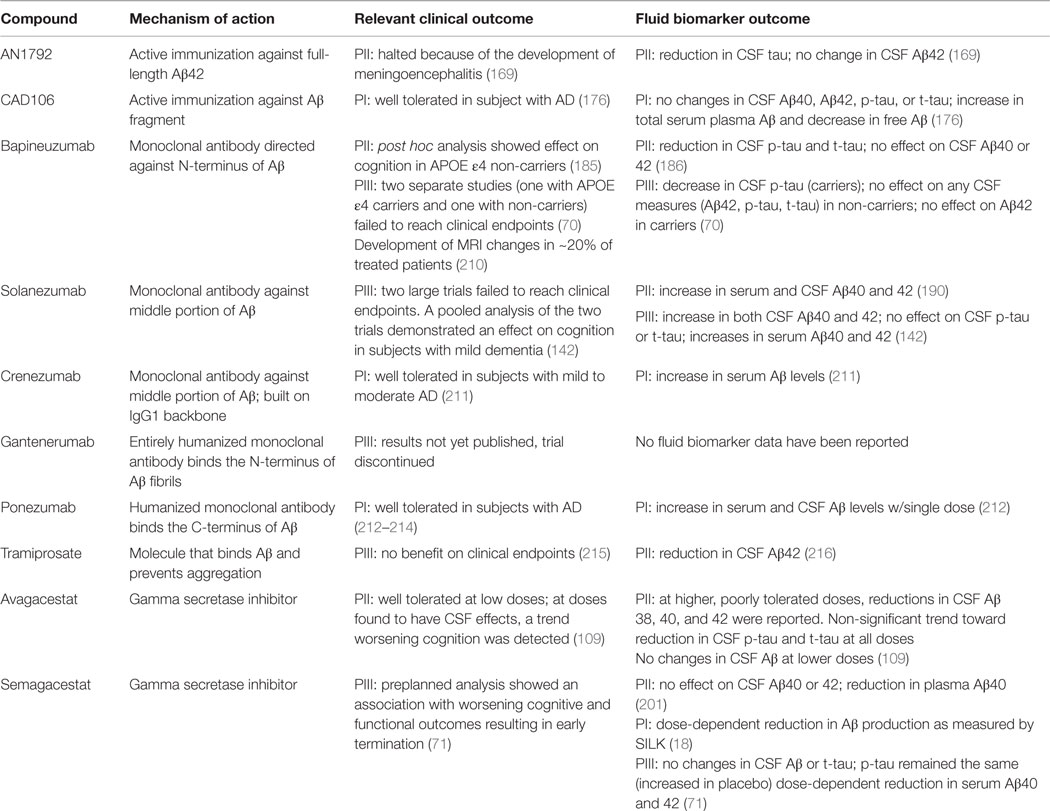
Table 3. Fluid biomarkers in clinical trials.
Active Amyloid Immunization Strategies
The impetus for the development of amyloid immunotherapy strategies came from a landmark study involving the PDAPP transgenic mouse, which overexpresses mutant human APP. In this study, it was shown that amyloid plaque deposition could be prevented by immunizing mice against Aβ42 (164). Subsequent studies reported that active immunization attenuated memory changes and reduced behavioral impairment (165, 166). Testing in several different models revealed that the greatest benefit was seen when immunization was achieved before the expected age of amyloid deposition (164, 167), signifying that immunization strategies work best in a clearance paradigm (167).
Composed of a full-length synthetic Aβ42 molecule, AN1792 was the first anti-amyloid vaccine evaluated in clinical trials. Despite appearing safe and demonstrating efficacy on an exploratory measure of functional decline in Phase I (168), further development of AN1792 was halted after 6% of subjects developed meningoencephalitis during Phase II testing (169). While the exact cause of this response remains unknown, the type of T-cell response (Th2-biased in the Phase I study and Th1-biased in the Phase II study) differed between the two studies (170). Treatment was terminated early (only 20% developed the predetermined antibody response), but double-blind assessments were continued during the entire 12-month period. Antibody response was associated with two positive clinical effects: improvement on composite scores of memory function and, in an extended follow-up study, significantly less functional decline (171). CSF monitoring in a subset of 11 subjects deemed “antibody responders” showed significant reductions in CSF t-tau (−204 ± pg/mL) at 1 year. Changes in CSF Aβ42 levels were not appreciated (169).
Several post-mortem neuropathological studies have been completed on subjects receiving the AN1792 vaccine (172–175). Because of the small number of participants and lack of information about baseline (or pretreatment) plaque burden, it is difficult to make definitive conclusions about these studies (8). Nonetheless, several interesting findings have been reported including reductions in plaque load (174) and decreased microglial activation (173). Evidence of pathological change was not, however, associated with improvement in survival time or time to severe dementia (174). Only one study (examining five brains) reported evidence of a reduction in tau pathology (175).
It is difficult to make accurate assessments regarding the CSF and neuropathological data from the AN1792 trials given the small sample sizes and the heterogeneity of the reported findings. According to the amyloid hypothesis, an active immune response would likely only be beneficial if achieved prior to the event that triggers the cascade (29). From a fluid biomarker perspective, it is unknown if the dramatic changes in CSF t-tau had any association with the positive signal seen on several clinical metrics. This is one of many unanswered questions that remain after this trial. Clearly, additional study is required to fully inform decisions about whether active immunization strategies can be efficacious in the treatment or prevention of AD. Several vaccines designed to illicit a safer B-cell response, including ACC-001, CAD106, V950, and Affitope AD02, are in various stages of clinical testing (86). The results of both Phase I and IIa testing have been published for CAD106 (176, 177). Although the vaccine appears much safer than AN1792, neither study demonstrated a significant biomarker or clinical effect.
Passive Amyloid Immunization Strategies
Passive immunization strategies involve the infusion of humanized monoclonal antibodies designed to bind amyloid species. Preclinical studies have shown that passively administered antibodies can enter the CNS and bind to various forms of amyloid (178). Compounds in this class differ depending on what domain within the Aβ fragment they bind (179).
Bapineuzumab
Bapineuzumab is a humanized monoclonal antibody directed against the N-terminus of Aβ. Recognition of the N-terminus ensures that bapineuzumab can attach to both soluble and insoluble amyloid species. Several theories have been proposed to explain bapineuzumab’s mechanism of action including direct inhibition of plaque formation (180) and antibody-mediated triggering of microglial cells to clear plaques (181). In preclinical models, bapineuzumab-treated PDAPP mice show reduced cortical amyloid plaque burdens (178). As with other amyloid therapies, treatment with bapineuzumab appears most effective for preventing rather than clearing pre-existing plaques (6). One potential explanation for the inability of bapineuzumab to clear existing plaques is proposed by Demattos et al. who hypothesize that in advanced disease, bapineuzumab is unable to bind plaques because it is saturated by soluble amyloid species that surround mature plaques (182). Infusion of bapineuzumab has also been associated with an increased incidence of microhemorrhage, which is thought to be due to its binding to vascular amyloid (183).
A Phase II study was undertaken to assess the safety of bapineuzumab in subjects with mild to moderate AD dementia (184). Higher rates of edema known as amyloid-related imaging abnormalities (ARIA) were seen at higher infusion doses and in subjects possessing the APOE ε4 genotype. Although clinical benefits were not initially detected, a post hoc analysis using multiple comparisons suggested possible benefits on both cognition and function (185). The biomarker data from Phase II testing also detected a possible disease-modifying signal as CSF data (n = 27) showed significant reductions in p-tau (−9.9 pg/mL) and a trend toward reduction in t-tau (−72.3 pg/mL) (186). In a smaller trial using an identical protocol, change in amyloid uptake as measured by PET scan was assessed as a primary outcome. In this trial, treatment with bapineuzumab (N = 20) was associated with reduced cortical binding compared with baseline (4).
Based on the positive signals seen in the Phase II trials, bapineuzumab advanced to Phase III testing (9). To reduce the risk of ARIA-E, dose selection was based on APOE ε4 status. Included in the secondary analysis was amyloid PET, volumetric MRI, and CSF biomarkers. Results of this study were disappointing as primary endpoints were not met. Although there were some signs of a positive biomarker effect, the signal was much weaker in Phase III testing than had been seen in the Phase II trial. APOE ε4 carriers (N = 127) experienced significant but small reductions in CSF p-tau (−5.8 pg/mL) compared to the placebo comparison group. In non-carriers, significant reductions in CSF p-tau were reported but only at the highest dose (−8.17 pg/mL). No significant changes were noted in CSF Aβ42 levels or t-tau levels. In both APOE ε4 carriers and non-carriers, amyloid uptake (as measured by PET scan) remained unchanged during the course of the trial.
The interpretation of outcome data from the bapineuzumab trials is complicated by the finding that a significant percentage of participants (6% of APOE ε4 carriers and 36% of APOE ε4 non-carriers) did not have evidence of amyloid pathology on PET scan. Nonetheless, the reduction of CSF p-tau is notable and suggests that passive immunization strategies targeting amyloid may be able to effect key pathological processes. Additional studies are needed to replicate this finding. The preclinical data suggest that bapineuzumab may be more effective when timed earlier in the disease course or at higher doses (182). The candidacy of bapineuzumab, however, is limited by ARIA-E.
Solanezumab
Solanezumab is a humanized monoclonal antibody directed against the middle amino acid section of Aβ. Because this epitope is not accessible on amyloid plaques, solanezumab only binds soluble Aβ species and does not bind Aβ plaques (187) or oligomers (188). In mouse models, infused solanezumab rapidly binds and completely sequesters plasma Aβ (187). By capturing the entire pool of soluble Aβ, solanezumab prevents this pool of amyloid from re-entering the brain, potentially shifting the amyloid gradient toward plaque dissolution and efflux out of the brain (29). According to this hypothesis, solanezumab acts as a “peripheral sink” as it draws amyloid out of the brain. In mouse models, peripheral administration of solanezumab results in rapid, 1,000-fold increases in plasma Aβ and significant reductions in plaque deposition (187). Not all preclinical data on solanezumab has been positive as one study found that treatment neither prevented nor reduced amyloid deposition (189). Unlike bapinezumab, solanezumab has not been associated with ARIA-E in either preclinical or human testing.
In a Phase II testing, treatment with solanezumab was associated with dose-related increases in both plasma and CSF levels of Aβ40 and 42 (190). Notably, both antibody-bound and antibody-free levels of CSF Aβ42 increased. Increases in unbound CSF Aβ42 could be interpreted as evidence of Aβ42 leaving plaques and diffusing down the gradient to replace sequestered plasma Aβ species consistent with the peripheral sink hypothesis. Amyloid PET scanning would have been informative in determining if the source of the increased unbound Aβ42 was in fact from plaque.
Solanezumab advanced to two large Phase III trials known as EXPEDITION 1 and 2 (142). Although both trials failed to meet primary endpoints, identical study designs allowed for pooling of data across the two studies. In the pooled analysis, the subgroup identified as having mild AD showed statistically significant slower rates of cognitive decline and positive trends on functional measures (185). Consistent with the Phase II trial, serum levels of both Aβ40 and 42 increased following infusion and remained significantly elevated during the entire trial. In a smaller subset of patients with CSF data (N = 44), significant increases were seen in both total CSF Aβ40 and 42, but unlike the Phase II trial, there were no significant changes in unbound Aβ42. Treatment was also not associated with changes in CSF tau, volumetric MRI, or amyloid PET.
Any interpretation of outcome data from the Phase III study of solanezumab must be tempered by the finding that a significant percentage (>20%) of enrollees who underwent amyloid PET scanning during the trial had negative scans (29). The dramatic increases in both serum and CSF levels of Aβ species in those treated with solanezumab could be interpreted as evidence of amyloid mobilization in the CNS. Whether antibody-mediated sequestration of soluble amyloid is enough to drive deposited amyloid out of plaque is still unknown and was not demonstrated in this trial with PET scanning (187). Clearly, the preclinical evidence regarding solanezumab has suggested a more profound effect on amyloid plaque prevention than clearance, and, as with other anti-amyloid therapies, treatment may prove more effective earlier in the disease course. Two ongoing trials of solanezumab – one enrolling patients with mild AD and the other enrolling cognitively subjects – will hope to shed light on these lingering issues.
Gamma Secretase Inhibitors
Gamma secretase is a multi-unit enzyme complex that facilitates the second enzymatic step in the processing of APP to Aβ. It consists of four subunits: nicastrin, presenilin-1 (PSEN1), anterior pharynx-defective-1, and presenilin-2 (PSEN2). Mutations in the genes that code for PSEN1 or PSEN2 cause early-onset AD by increasing the fractional production of Aβ42 (27). In animal models, compounds that decrease gamma secretase activity have been shown to reduce Aβ42 synthesis and improve behavioral and cognitive symptoms (191, 192). Development of safe gamma secretase inhibitors is complicated by the enzyme’s crucial role in the regulation of Notch protein signaling pathways. Notch signaling is involved in cell fate pathways in rapidly dividing cells and disruption of normal Notch protein function can result in adverse gastrointestinal, hematologic, and dermatologic effects (193). Safe gamma secretase inhibitors must show a selective preference for Aβ inhibition over disruption of Notch signaling pathways.
Semagacestat
Semagacestat is a gamma secretase inhibitor that demonstrates selective inhibition of APP processing over Notch inhibition in several in vitro studies (194, 195). Not all studies have reported this preference, and in the most recent study (published after the Phase III trials were completed) semagacestat showed greater affinitiy for inhibiting Notch signaling pathways than BACE (196). In animal models, semagacestat reduces soluble Aβ in brain, CSF, and serum. Because studies using microdialysis show significant reductions in interstitial amyloid, there was also hope that gamma secretase inhibition would drive the amyloid gradient and promote the dissolution of amyloid out of plaques and into the interstitial space (197). Data from several mouse models suggested that although gamma secretase reduced soluble Aβ levels and prevented the formation of new plaques, there was little evidence that treatment promoted the clearance of pre-existing plaques (198, 199).
Early human testing of semagacestat was enriched by the use of stable isotope labeling kinetics (SILK) (18). By continuously labeling and monitoring soluble Aβ in the CSF, SILK provides an estimation of the production and clearance of Aβ over a specified period of time (200). Using SILK, it was shown that single doses of semagacestat caused dramatic reductions in Aβ production in healthy human subjects. This finding provided convincing evidence of target engagement and semagacestat advanced to additional testing. In a 14 week Phase II study powered to detect safety, treatment was associated with significant reductions in serum Aβ40, but somewhat surprisingly, not with significant changes in either CSF Aβ40 or Aβ42 (post hoc analyses suggested a trend toward CSF Aβ40 reduction) (201).
Two large multicenter trials enrolling more than 2,000 patients have been conducted (71). Known as the IDENTITY 1 and IDENTITY 2, both trials were terminated early after a preplanned interim analysis revealed that treatment was associated with an increased incidence of adverse side effects. Patients receiving active treatment experienced skin cancers, GI symptoms, and dermatological side effects at twice the rate of those receiving placebo. In the modified intention-to-treat population, treatment was associated with worsening cognition and functional status. Biomarker from IDENTIY included both serum and CSF biomarkers as well as neuroimaging. Significant dose-dependent reductions in both serum Aβ40 and 42 were seen with treatment. Notably, the reduction in serum Aβ40 was more than twice that seen for Aβ42. CSF monitoring of Aβ (40,42) and tau was done in a smaller subset of patients (N = 47). Although no significant changes were seen in either Aβ or t-tau, there was a significant reduction in p-tau levels, which was greater in the lower dose group (8% vs. 4%). Changes in amyloid uptake were not appreciated in 59 patients with multiple amyloid PET scans. Worsening cognition and an increased rate of side effects were also seen in Phase II testing of avagacestat, another gamma secretase inhibitor (109).
Unless gamma secretase inhibitors without Notch signaling inhibition can be developed (and definitively proven in vitro), it is unwise to devote further resources to gamma secretase inhibition as a viable treatment for AD. Inhibition of Aβ production, however, remains a promising option for AD therapies. Biomarker data from the semagacestat trial, which showed significant (albeit, modest) reductions in CSF p-tau levels, may indicate that reducing Aβ production may alter the neuropathological process of AD. An alternative pathway to reduce Aβ production is with BACE1 inhibition. Several lines of research support the role of BACE1 activity in the pathogenesis of AD including two studies that have reported allelic variations, that reduce BACE1 activity, are protective against AD (202, 203). A significant barrier to BACE1 inhibitor development is that its large active site requires the development of bulky compounds that do not pass through the BBB into the brain (204). Nonetheless, several BACE1 inhibitors have been developed and are entering clinical testing. Preliminary data suggest that BACE1 inhibitors significantly reduce CSF Aβ42 levels (205).
Conclusion
Aided by the development of several validated biomarkers, the concept of AD has drastically changed over the past 30 years. Reflected in new research criteria, AD is now seen as a disease that progress through several stages (ranging from a prodromal/asymptomatic stage to mildly symptomatic to frank dementia) (5). We now know that the biological processes that lead to the disease are triggered years to decades before the onset of symptoms (9). Fluid biomarkers, which provide a window into the complex biochemical process in the brain, will take on an enhanced role in overcoming the challenges of developing therapeutic agents with disease-modifying properties. Three CSF fluid biomarkers (consisting of low Aβ42 and elevated t-tau and p-tau) are now widely accepted and commonly used in both clinical practice and research. When combined, these three biomarkers constitute an “AD signature” that better predicts the presence of AD pathology on autopsy than a diagnosis made on clinical grounds (73). Because changes in these biomarkers can be detected years before the dementia phase of disease, they have also been shown to demonstrate good accuracy in identifying individuals at risk for disease progression (77). As a result, they should be used to enhance clinical trial enrichment strategies, especially as trials move toward enrolling patients earlier in the disease course. Less is known about their utility in tracking disease progression or monitoring therapeutic responses. There are some data to suggest that CSF tau tracks more closely with disease progression (52) and may be better suited in this role than Aβ. It is still unknown if drug-induced changes in these markers will result in clinically meaningful benefits.
Due to several shortcomings in the current fluid biomarkers, it is imperative that new biomarkers be developed. Several promising new candidates have emerged with good preliminary data to support their further development. These include CSF BACE1 (96), VILIP-1, and YLK-40 (116). The matching of a biomarker with a particular drug designed to modulate that aspect of AD pathophysiology (CSF BACE1 with a BACE1 inhibitor) has the potential to provide information about target engagement, inform dosing decisions, and to monitor for drug effects. Perhaps, the most promising of all emerging approaches is the development of proteomics. With further development of biotechnology that promises to increase the capacity to analyze larger datasets, it seems likely that an “AD fingerprint” composed of several fluid biomarkers will emerge that will enhance our ability to identify, stage, and maybe even chose appropriate treatments for AD.
Several candidate agents with potential disease-modifying properties have advanced to Phase III testing, each has failed to meet clinical endpoints. A few trials have included biomarker data as secondary outcomes. Owing to the heterogeneity of the findings and lack of correlation with clinical metrics, these results are difficult to interpret. The slow progression of the disease, complicated pathophysiology, and difficulty in accurately modeling the pathology of sporadic AD in animal models present formidable challenges to clinical trial design and implementation. Biomarkers, however, have the ability to answer questions more quickly and effectively about target engagement, patient selection, and disease monitoring. In preclinical studies, biomarkers can be used to verify that a candidate agent is having its proposed effect on the biological systems it is designed to target. Because animal models are limited in their ability to replicate all of the behavioral and pathological features of AD (206), testing in multiple animals may improve the predictive value of clinical testing. Preclinical testing should also include biomarker data that are translatable to humans (including both CSF and serum). CSF testing in larger animals like guinea pigs and canines can provide valuable information about a candidate drug’s effects in the CSF and may improve upon information derived from mouse models (207). As a candidate compound advances to early clinical testing in humans, an early priority should be to confirm that biomarker changes demonstrated in preclinical testing are seen in humans (8). This can be tested with smaller, proof of concept trials that are powered to pre-specified endpoints. It is at this stage that go, no-go decisions can be made about advancing to longer, more expensive trials. If an agent is to be labeled with a claim of disease modification, support may come from biomarker data in Phase III trials. Figure 2 illustrates a potential model for a standard parallel group design. Groups receiving active treatment and placebo would be compared based on clinical measures and a biomarker known to exert an effect on the underlying pathophysiology. A drug–placebo difference would be supported by differences on clinical measures (cognition, function, or global outcomes); while an effect on disease pathology would be demonstrated by showing a significant difference on biomarker measures of disease progression (for example, CSF t-tau). A statistically significant correlation between these two measures could potentially be used to support a claim for disease modification (208).
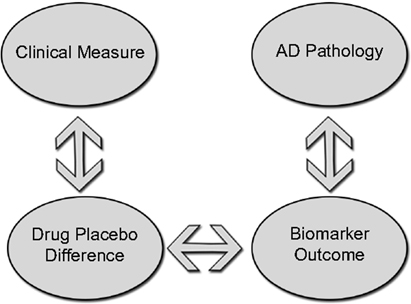
Figure 2. Standard parallel group design to demonstrate disease modification groups receiving active treatment and placebo would be compared on clinical measures while an effect on disease pathology would be demonstrated by showing differences on a biomarker measure of disease progression. A correlation between drug–placebo difference and a biomarker outcome could potentially support a claim of disease modification.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. 2013 Alzheimer’s disease facts and figures. Alzheimers Dement (2013) 9(2):208–45. doi:10.1016/j.jalz.2013.02.003
3. Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med (2012) 367(9):795–804. doi:10.1056/NEJMoa1202753
4. Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol (2010) 9(4):363–72. doi:10.1016/S1474-4422(10)70043-0
5. Hampel H, Lista S, Teipel SJ, Garaci F, Nistico R, Blennow K, et al. Perspective on future role of biological markers in clinical therapy trials of Alzheimer’s disease: a long-range point of view beyond 2020. Biochem Pharmacol (2014) 88(4):426–49. doi:10.1016/j.bcp.2013.11.009
6. Karran E, Hardy J. Antiamyloid therapy for Alzheimer’s disease – are we on the right road? N Engl J Med (2014) 370(4):377–8. doi:10.1056/NEJMe1313943
7. Siemers ER. How can we recognize “disease modification” effects? J Nutr Health Aging (2009) 13(4):341–3. doi:10.1007/s12603-009-0036-7
8. Karran E, Hardy J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann Neurol (2014) 76(2):185–205. doi:10.1002/ana.24188
9. Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol (2014) 13(6):614–29. doi:10.1016/S1474-4422(14)70090-0
10. Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement (2011) 7(3):280–92. doi:10.1016/j.jalz.2011.03.003
11. Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement (2011) 7(3):270–9. doi:10.1016/j.jalz.2011.03.008
12. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement (2011) 7(3):263–9. doi:10.1016/j.jalz.2011.03.005
13. Petrella JR. Neuroimaging and the search for a cure for Alzheimer disease. Radiology (2013) 269(3):671–91. doi:10.1148/radiol.13122503
14. Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science (1992) 256(5054):184–5. doi:10.1126/science.1566067
15. Bates KA, Verdile G, Li QX, Ames D, Hudson P, Masters CL, et al. Clearance mechanisms of Alzheimer’s amyloid-beta peptide: implications for therapeutic design and diagnostic tests. Mol Psychiatry (2009) 14(5):469–86. doi:10.1038/mp.2008.96
16. Cummings JL. Biomarkers in Alzheimer’s disease drug development. Alzheimers Dement (2011) 7(3):e13–44. doi:10.1016/j.jalz.2010.06.004
17. Bateman RJ, Klunk WE. Measuring target effect of proposed disease-modifying therapies in Alzheimer’s disease. Neurotherapeutics (2008) 5(3):381–90. doi:10.1016/j.nurt.2008.05.009
18. Bateman RJ, Siemers ER, Mawuenyega KG, Wen G, Browning KR, Sigurdson WC, et al. A gamma-secretase inhibitor decreases amyloid-beta production in the central nervous system. Ann Neurol (2009) 66(1):48–54. doi:10.1002/ana.21623
19. Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, et al. Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J Biol Chem (1992) 267(1):546–54.
20. Lee J, Culyba EK, Powers ET, Kelly JW. Amyloid-beta forms fibrils by nucleated conformational conversion of oligomers. Nat Chem Biol (2011) 7(9), 602–9. doi:10.1038/nchembio.624
21. Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature (2006) 440(7082):352–7. doi:10.1038/nature04533
22. Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci (2005) 8(8):1051–8. doi:10.1038/nn1503
23. Lesne SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, et al. Brain amyloid-beta oligomers in ageing and Alzheimer’s disease. Brain (2013) 136(Pt 5):1383–98. doi:10.1093/brain/awt062
24. Mathis CA, Lopresti BJ, Klunk WE. Impact of amyloid imaging on drug development in Alzheimer’s disease. Nucl Med Biol (2007) 34(7):809–22. doi:10.1016/j.nucmedbio.2007.06.015
25. Mucke L, Selkoe DJ. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med (2012) 2(7):a006338. doi:10.1101/cshperspect.a006338
26. Bezprozvanny I. Amyloid goes global. Sci Signal (2009) 2(63):pe16. doi:10.1126/scisignal.263pe16
27. Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol (2007) 8(2):101–12. doi:10.1038/nrm2101
28. Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C. Age, neuropathology, and dementia. N Engl J Med (2009) 360(22):2302–9. doi:10.1056/NEJMoa0806142
29. Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov (2011) 10(9):698–712. doi:10.1038/nrd3505
30. Zetterberg H, Blennow K, Hanse E. Amyloid beta and APP as biomarkers for Alzheimer’s disease. Exp Gerontol (2010) 45(1):23–9. doi:10.1016/j.exger.2009.08.002
31. Englund H, Degerman Gunnarsson M, Brundin RM, Hedlund M, Kilander L, Lannfelt L, et al. Oligomerization partially explains the lowering of Abeta42 in Alzheimer’s disease cerebrospinal fluid. Neurodegener Dis (2009) 6(4):139–47. doi:10.1159/000225376
32. Strozyk D, Blennow K, White LR, Launer LJ. CSF Abeta 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology (2003) 60(4):652–6. doi:10.1212/01.WNL.0000046581.81650.D0
33. Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, et al. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol (2009) 66(3):382–9. doi:10.1001/archneurol.2008.596
34. Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol (2006) 59(3):512–9. doi:10.1002/ana.20730
35. Weigand SD, Vemuri P, Wiste HJ, Senjem ML, Pankratz VS, Aisen PS, et al. Transforming cerebrospinal fluid Abeta42 measures into calculated Pittsburgh compound B units of brain Abeta amyloid. Alzheimers Dement (2011) 7(2):133–41. doi:10.1016/j.jalz.2010.08.230
36. Tolboom N, van der Flier WM, Yaqub M, Boellaard R, Verwey NA, Blankenstein MA, et al. Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. J Nucl Med (2009) 50(9):1464–70. doi:10.2967/jnumed.109.064360
37. Fagan AM. What does it mean to be ‘amyloid-positive’? Brain (2015) 138(Pt 3):514–6. doi:10.1093/brain/awu387
38. Mattsson N, Insel PS, Donohue M, Landau S, Jagust WJ, Shaw LM, et al. Independent information from cerebrospinal fluid amyloid-beta and florbetapir imaging in Alzheimer’s disease. Brain (2015) 138(Pt 3):772–83. doi:10.1093/brain/awu367
39. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A (1975) 72(5):1858–62. doi:10.1073/pnas.72.5.1858
40. Imahori K, Uchida T. Physiology and pathology of tau protein kinases in relation to Alzheimer’s disease. J Biochem (1997) 121(2):179–88.
41. Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol (2010) 6(3):131–44. doi:10.1038/nrneurol.2010.4
42. Tepper K, Biernat J, Kumar S, Wegmann S, Timm T, Hubschmann S, et al. Oligomer formation of tau protein hyperphosphorylated in cells. J Biol Chem (2014) 289(49):34389–407. doi:10.1074/jbc.M114.611368
43. Jack CR Jr, Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron (2013) 80(6):1347–58. doi:10.1016/j.neuron.2013.12.003
44. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol (2015) 14(4):388–405. doi:10.1016/S1474-4422(15)70016-5
45. Bonda DJ, Wang X, Lee HG, Smith MA, Perry G, Zhu X. Neuronal failure in Alzheimer’s disease: a view through the oxidative stress looking-glass. Neurosci Bull (2014) 30(2):243–52. doi:10.1007/s12264-013-1424-x
46. Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissection of Alzheimer’s disease and related dementias: amyloid and its relationship to tau. Nat Neurosci (1998) 1(5):355–8. doi:10.1038/1565
47. Honjo K, van Reekum R, Verhoeff NP. Alzheimer’s disease and infection: do infectious agents contribute to progression of Alzheimer’s disease? Alzheimers Dement (2009) 5(4):348–60. doi:10.1016/j.jalz.2008.12.001
48. Iqbal K, Gong CX, Liu F. Hyperphosphorylation-induced tau oligomers. Front Neurol (2013) 4:112. doi:10.3389/fneur.2013.00112
49. Brandt R, Hundelt M, Shahani N. Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim Biophys Acta (2005) 1739(2–3):331–54. doi:10.1016/j.bbadis.2004.06.018
50. Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci (2007) 8(9):663–72. doi:10.1038/nrn2194
51. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (1991) 82(4):239–59. doi:10.1007/BF00308809
52. Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol (1997) 41(1):17–24. doi:10.1002/ana.410410106
53. Dujardin S, Begard S, Caillierez R, Lachaud C, Delattre L, Carrier S, et al. Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS One (2014) 9(6):e100760. doi:10.1371/journal.pone.0100760
54. Stohr J, Condello C, Watts JC, Bloch L, Oehler A, Nick M, et al. Distinct synthetic Abeta prion strains producing different amyloid deposits in bigenic mice. Proc Natl Acad Sci U S A (2014) 111(28):10329–34. doi:10.1073/pnas.1408968111
55. Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol (2009) 11(7):909–13. doi:10.1038/ncb1901
56. Clavaguera F, Hench J, Goedert M, Tolnay M. Invited review: prion-like transmission and spreading of tau pathology. Neuropathol Appl Neurobiol (2015) 41(1):47–58. doi:10.1111/nan.12197
57. Blennow K. Cerebrospinal fluid protein biomarkers for Alzheimer’s disease. NeuroRx (2004) 1(2):213–25. doi:10.1602/neurorx.1.2.213
58. Hesse C, Rosengren L, Andreasen N, Davidsson P, Vanderstichele H, Vanmechelen E, et al. Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neurosci Lett (2001) 297(3):187–90. doi:10.1016/S0304-3940(00)01697-9
59. Franz G, Beer R, Kampfl A, Engelhardt K, Schmutzhard E, Ulmer H, et al. Amyloid beta 1-42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology (2003) 60(9):1457–61. doi:10.1212/01.WNL.0000063313.57292.00
60. Otto M, Wiltfang J, Cepek L, Neumann M, Mollenhauer B, Steinacker P, et al. Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology (2002) 58(2):192–7. doi:10.1212/WNL.58.2.192
61. Hampel H, Buerger K, Zinkowski R, Teipel SJ, Goernitz A, Andreasen N, et al. Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: a comparative cerebrospinal fluid study. Arch Gen Psychiatry (2004) 61(1):95–102. doi:10.1001/archpsyc.61.1.95
62. Buerger K, Ewers M, Pirttila T, Zinkowski R, Alafuzoff I, Teipel SJ, et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain (2006) 129(Pt 11):3035–41. doi:10.1093/brain/awl269
63. van Harten AC, Kester MI, Visser PJ, Blankenstein MA, Pijnenburg YA, van der Flier WM, et al. Tau and p-tau as CSF biomarkers in dementia: a meta-analysis. Clin Chem Lab Med (2011) 49(3):353–66. doi:10.1515/CCLM.2011.086
64. Buerger K, Otto M, Teipel SJ, Zinkowski R, Blennow K, DeBernardis J, et al. Dissociation between CSF total tau and tau protein phosphorylated at threonine 231 in Creutzfeldt-Jakob disease. Neurobiol Aging (2006) 27(1):10–5. doi:10.1016/j.neurobiolaging.2004.12.003
65. Grossman M, Farmer J, Leight S, Work M, Moore P, Van Deerlin V, et al. Cerebrospinal fluid profile in frontotemporal dementia and Alzheimer’s disease. Ann Neurol (2005) 57(5):721–9. doi:10.1002/ana.20477
66. Roher AE, Esh CL, Kokjohn TA, Castano EM, Van Vickle GD, Kalback WM, et al. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimers Dement (2009) 5(1):18–29. doi:10.1016/j.jalz.2008.10.004
67. Engelborghs S, De Vreese K, Van de Casteele T, Vanderstichele H, Van Everbroeck B, Cras P, et al. Diagnostic performance of a CSF-biomarker panel in autopsy-confirmed dementia. Neurobiol Aging (2008) 29(8):1143–59. doi:10.1016/j.neurobiolaging.2007.02.016
68. Mattsson N, Andreasson U, Persson S, Carrillo MC, Collins S, Chalbot S, et al. CSF biomarker variability in the Alzheimer’s Association quality control program. Alzheimers Dement (2013) 9(3):251–61. doi:10.1016/j.jalz.2013.01.010
69. Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J Neuropathol Exp Neurol (2012) 71(4):266–73. doi:10.1097/NEN.0b013e31824b211b
70. Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med (2014) 370(4):322–33. doi:10.1056/NEJMoa1304839
71. Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med (2013) 369(4):341–50. doi:10.1056/NEJMoa1210951
72. Young AL, Oxtoby NP, Daga P, Cash DM, Fox NC, Ourselin S, et al. A data-driven model of biomarker changes in sporadic Alzheimer’s disease. Brain (2014) 137(Pt 9):2564–77. doi:10.1093/brain/awu176
73. Blennow K, Dubois B, Fagan AM, Lewczuk P, de Leon MJ, Hampel H. Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease. Alzheimers Dement (2015) 11(1):58–69. doi:10.1016/j.jalz.2014.02.004
74. Stomrud E, Hansson O, Blennow K, Minthon L, Londos E. Cerebrospinal fluid biomarkers predict decline in subjective cognitive function over 3 years in healthy elderly. Dement Geriatr Cogn Disord (2007) 24(2):118–24. doi:10.1159/000105017
75. Skoog I, Davidsson P, Aevarsson O, Vanderstichele H, Vanmechelen E, Blennow K. Cerebrospinal fluid beta-amyloid 42 is reduced before the onset of sporadic dementia: a population-based study in 85-year-olds. Dement Geriatr Cogn Disord (2003) 15(3):169–76. doi:10.1159/000068478
76. van Harten AC, Visser PJ, Pijnenburg YA, Teunissen CE, Blankenstein MA, Scheltens P, et al. Cerebrospinal fluid Abeta42 is the best predictor of clinical progression in patients with subjective complaints. Alzheimers Dement (2013) 9(5):481–7. doi:10.1016/j.jalz.2012.08.004
77. Buchhave P, Minthon L, Zetterberg H, Wallin AK, Blennow K, Hansson O. Cerebrospinal fluid levels of beta-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry (2012) 69(1):98–106. doi:10.1001/archgenpsychiatry.2011.155
78. Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA (2009) 302(4):385–93. doi:10.1001/jama.2009.1064
79. Hertze J, Minthon L, Zetterberg H, Vanmechelen E, Blennow K, Hansson O. Evaluation of CSF biomarkers as predictors of Alzheimer’s disease: a clinical follow-up study of 4.7 years. J Alzheimers Dis (2010) 21(4):1119–28. doi:10.3233/JAD2010100207
80. Hampel H, Teipel SJ, Fuchsberger T, Andreasen N, Wiltfang J, Otto M, et al. Value of CSF beta-amyloid1-42 and tau as predictors of Alzheimer’s disease in patients with mild cognitive impairment. Mol Psychiatry (2004) 9(7):705–10. doi:10.1038/sj.mp.4001473
81. DeCarli C. Mild cognitive impairment: prevalence, prognosis, aetiology, and treatment. Lancet Neurol (2003) 2(1):15–21. doi:10.1016/S1474-4422(03)00262-X
82. Rosen C, Hansson O, Blennow K, Zetterberg H. Fluid biomarkers in Alzheimer’s disease – current concepts. Mol Neurodegener (2013) 8:20. doi:10.1186/1750-1326-8-20
83. Andreasen N, Minthon L, Clarberg A, Davidsson P, Gottfries J, Vanmechelen E, et al. Sensitivity, specificity, and stability of CSF-tau in AD in a community-based patient sample. Neurology (1999) 53(7):1488–94. doi:10.1212/WNL.53.7.1488
84. Buchhave P, Blennow K, Zetterberg H, Stomrud E, Londos E, Andreasen N, et al. Longitudinal study of CSF biomarkers in patients with Alzheimer’s disease. PLoS One (2009) 4(7):e6294. doi:10.1371/journal.pone.0006294
85. Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, et al. MRI and CSF biomarkers in normal, MCI, and AD subjects: diagnostic discrimination and cognitive correlations. Neurology (2009) 73(4):287–93. doi:10.1212/WNL.0b013e3181af79e5
86. Kang JH, Ryoo NY, Shin DW, Trojanowski JQ, Shaw LM. Role of cerebrospinal fluid biomarkers in clinical trials for Alzheimer’s disease modifying therapies. Korean J Physiol Pharmacol (2014) 18(6):447–56. doi:10.4196/kjpp.2014.18.6.447
87. Bjerke M, Portelius E, Minthon L, Wallin A, Anckarsater H, Anckarsater R, et al. Confounding factors influencing amyloid beta concentration in cerebrospinal fluid. Int J Alzheimers Dis (2010) 2010:1–11. doi:10.4061/2010/986310
88. Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, et al. Control of peripheral nerve myelination by the beta-secretase BACE1. Science (2006) 314(5799):664–6. doi:10.1126/science.1132341
89. Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, et al. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci (2006) 9(12):1520–5. doi:10.1038/nn1797
90. Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci (2005) 25(50):11693–709. doi:10.1523/JNEUROSCI.2766-05.2005
91. Rossner S, Sastre M, Bourne K, Lichtenthaler SF. Transcriptional and translational regulation of BACE1 expression – implications for Alzheimer’s disease. Prog Neurobiol (2006) 79(2):95–111. doi:10.1016/j.pneurobio.2006.06.001
92. Fukumoto H, Tennis M, Locascio JJ, Hyman BT, Growdon JH, Irizarry MC. Age but not diagnosis is the main predictor of plasma amyloid beta-protein levels. Arch Neurol (2003) 60(7):958–64. doi:10.1001/archneur.60.7.958
93. Holsinger RM, McLean CA, Beyreuther K, Masters, CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer’s disease. Ann Neurol (2002) 51(6):783–6. doi:10.1002/ana.10208
94. Verheijen JH, Huisman LG, van Lent N, Neumann U, Paganetti P, Hack CE, et al. Detection of a soluble form of BACE-1 in human cerebrospinal fluid by a sensitive activity assay. Clin Chem (2006) 52(6):1168–74. doi:10.1373/clinchem.2006.066720
95. Holsinger RM, Lee JS, Boyd A, Masters CL, Collins SJ. CSF BACE1 activity is increased in CJD and Alzheimer disease versus [corrected] other dementias. Neurology (2006) 67(4):710–2. doi:10.1212/01.wnl.0000229925.52203.4c
96. Zetterberg H, Andreasson U, Hansson O, Wu G, Sankaranarayanan S, Andersson ME, et al. Elevated cerebrospinal fluid BACE1 activity in incipient Alzheimer disease. Arch Neurol (2008) 65(8):1102–7. doi:10.1001/archneur.65.8.1102
97. Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, et al. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med (2008) 14(7):723–30. doi:10.1038/nm1784
98. Olsson A, Hoglund K, Sjogren M, Andreasen N, Minthon L, Lannfelt L, et al. Measurement of alpha- and beta-secretase cleaved amyloid precursor protein in cerebrospinal fluid from Alzheimer patients. Exp Neurol (2003) 183(1):74–80. doi:10.1016/S0014-4886(03)00027-X
99. Lewczuk P, Kamrowski-Kruck H, Peters O, Heuser I, Jessen F, Popp J, et al. Soluble amyloid precursor proteins in the cerebrospinal fluid as novel potential biomarkers of Alzheimer’s disease: a multicenter study. Mol Psychiatry (2010) 15(2):138–45. doi:10.1038/mp.2008.84
100. Perneczky R, Tsolakidou A, Arnold A, Diehl-Schmid J, Grimmer T, Forstl H, et al. CSF soluble amyloid precursor proteins in the diagnosis of incipient Alzheimer disease. Neurology (2011) 77(1):35–8. doi:10.1212/WNL.0b013e318221ad47
101. Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM, et al. Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol (2013) 73(1):104–19. doi:10.1002/ana.23748
102. Hong S, Ostaszewski BL, Yang T, O’Malley TT, Jin M, Yanagisawa K, et al. Soluble Abeta oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron (2014) 82(2):308–19. doi:10.1016/j.neuron.2014.02.027
103. Georganopoulou DG, Chang L, Nam JM, Thaxton CS, Mufson EJ, Klein WL, et al. Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer’s disease. Proc Natl Acad Sci U S A (2005) 102(7):2273–6. doi:10.1073/pnas.0409336102
104. Fukumoto H, Tokuda T, Kasai T, Ishigami N, Hidaka H, Kondo M, et al. High-molecular-weight beta-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J (2010) 24(8):2716–26. doi:10.1096/fj.09-150359
105. Santos AN, Ewers M, Minthon L, Simm A, Silber RE, Blennow K, et al. Amyloid-beta oligomers in cerebrospinal fluid are associated with cognitive decline in patients with Alzheimer’s disease. J Alzheimers Dis (2012) 29(1):171–6. doi:10.3233/JAD-2012-111361
106. Portelius E, Zetterberg H, Andreasson U, Brinkmalm G, Andreasen N, Wallin A, et al. An Alzheimer’s disease-specific beta-amyloid fragment signature in cerebrospinal fluid. Neurosci Lett (2006) 409(3):215–9. doi:10.1016/j.neulet.2006.09.044
107. Lewczuk P, Esselmann H, Meyer M, Wollscheid V, Neumann M, Otto M, et al. The amyloid-beta (Abeta) peptide pattern in cerebrospinal fluid in Alzheimer’s disease: evidence of a novel carboxyterminally elongated Abeta peptide. Rapid Commun Mass Spectrom (2003) 17(12):1291–6. doi:10.1002/rcm.1048
108. Schoonenboom NS, Mulder C, Van Kamp GJ, Mehta SP, Scheltens P, Blankenstein MA, et al. Amyloid beta 38, 40, and 42 species in cerebrospinal fluid: more of the same? Ann Neurol (2005) 58(1):139–42. doi:10.1002/ana.20508
109. Coric V, van Dyck CH, Salloway S, Andreasen N, Brody M, Richter RW, et al. Safety and tolerability of the gamma-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch Neurol (2012) 69(11):1430–40. doi:10.1001/archneurol.2012.2194
110. Fagan AM, Mintun MA, Shah AR, Aldea P, Roe CM, Mach RH, et al. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer’s disease. EMBO Mol Med (2009) 1(8–9):371–80. doi:10.1002/emmm.200900048
111. Mehta PD, Pirttila T. Increased cerebrospinal fluid A beta38/A beta42 ratio in Alzheimer disease. Neurodegener Dis (2005) 2(5):242–5. doi:10.1159/000090363
112. Fagan AM, Perrin RJ. Upcoming candidate cerebrospinal fluid biomarkers of Alzheimer’s disease. Biomark Med (2012) 6(4):455–76. doi:10.2217/bmm.12.42
113. Paterlini M, Revilla V, Grant AL, Wisden W. Expression of the neuronal calcium sensor protein family in the rat brain. Neuroscience (2000) 99(2):205–16. doi:10.1016/S0306-4522(00)00201-3
114. Stejskal D, Sporova L, Svestak M, Karpisek M. Determination of serum visinin like protein-1 and its potential for the diagnosis of brain injury due to the stroke: a pilot study. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub (2011) 155(3):263–8. doi:10.5507/bp.2011.049
115. Tarawneh R, D’Angelo G, Macy E, Xiong C, Carter D, Cairns NJ, et al. Visinin-like protein-1: diagnostic and prognostic biomarker in Alzheimer disease. Ann Neurol (2011) 70(2):274–85. doi:10.1002/ana.22448
116. Sutphen CL, Jasielec MS, Shah AR, Macy EM, Xiong C, Vlassenko AG, et al. Longitudinal cerebrospinal fluid biomarker changes in preclinical Alzheimer disease during middle age. JAMA Neurol (2015). doi:10.1001/jamaneurol.2015.1285
117. Tarawneh R, Lee JM, Ladenson JH, Morris JC, Holtzman DM. CSF VILIP-1 predicts rates of cognitive decline in early Alzheimer disease. Neurology (2012) 78(10):709–19. doi:10.1212/WNL.0b013e318248e568
118. Sultana R, Baglioni M, Cecchetti R, Cai J, Klein JB, Bastiani P, et al. Lymphocyte mitochondria: toward identification of peripheral biomarkers in the progression of Alzheimer disease. Free Radic Biol Med (2013) 65:595–606. doi:10.1016/j.freeradbiomed.2013.08.001
119. Miller E, Morel A, Saso L, Saluk J. Isoprostanes and neuroprostanes as biomarkers of oxidative stress in neurodegenerative diseases. Oxid Med Cell Longev (2014) 2014:572491. doi:10.1155/2014/572491
120. Pratico D, Lee V MY, Trojanowski JQ, Rokach J, Fitzgerald GA. Increased F2-isoprostanes in Alzheimer’s disease: evidence for enhanced lipid peroxidation in vivo. FASEB J (1998) 12(15):1777–83.
121. Montine TJ, Kaye JA, Montine KS, McFarland L, Morrow JD, Quinn JF. Cerebrospinal fluid abeta42, tau, and f2-isoprostane concentrations in patients with Alzheimer disease, other dementias, and in age-matched controls. Arch Pathol Lab Med (2001) 125(4):510–2. doi:10.1043/0003-9985(2001)125<0510:cfataf>2.0.co;2
122. Brys M, Pirraglia E, Rich K, Rolstad S, Mosconi L, Switalski R, et al. Prediction and longitudinal study of CSF biomarkers in mild cognitive impairment. Neurobiol Aging (2009) 30(5):682–90. doi:10.1016/j.neurobiolaging.2007.08.010
123. de Leon MJ, Mosconi L, Li J, De Santi S, Yao Y, Tsui WH, et al. Longitudinal CSF isoprostane and MRI atrophy in the progression to AD. J Neurol (2007) 254(12):1666–75. doi:10.1007/s00415-007-0610-z
124. Eikelenboom P, van Exel E, Hoozemans JJ, Veerhuis R, Rozemuller AJ, van Gool WA. Neuroinflammation – an early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegener Dis (2010) 7(1–3):38–41. doi:10.1159/000283480
125. Bonneh-Barkay D, Bissel SJ, Wang G, Fish KN, Nicholl GC, Darko SW, et al. YKL-40, a marker of simian immunodeficiency virus encephalitis, modulates the biological activity of basic fibroblast growth factor. Am J Pathol (2008) 173(1):130–43. doi:10.2353/ajpath.2008.080045
126. Perrin RJ, Craig-Schapiro R, Malone JP, Shah AR, Gilmore P, Davis AE, et al. Identification and validation of novel cerebrospinal fluid biomarkers for staging early Alzheimer’s disease. PLoS One (2011) 6(1):e16032. doi:10.1371/journal.pone.0016032
127. Craig-Schapiro R, Perrin RJ, Roe CM, Xiong C, Carter D, Cairns NJ, et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol Psychiatry (2010) 68(10):903–12. doi:10.1016/j.biopsych.2010.08.025
128. Gerendasy DD, Herron SR, Watson JB, Sutcliffe JG. Mutational and biophysical studies suggest RC3/neurogranin regulates calmodulin availability. J Biol Chem (1994) 269(35):22420–6.
129. Huang KP, Huang FL, Jager T, Li J, Reymann KG, Balschun D. Neurogranin/RC3 enhances long-term potentiation and learning by promoting calcium-mediated signaling. J Neurosci (2004) 24(47):10660–9. doi:10.1523/JNEUROSCI.2213-04.2004
130. Thorsell A, Bjerke M, Gobom J, Brunhage E, Vanmechelen E, Andreasen N, et al. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer’s disease. Brain Res (2010) 1362:13–22. doi:10.1016/j.brainres.2010.09.073
131. Kvartsberg H, Duits FH, Ingelsson M, Andreasen N, Ohrfelt A, Andersson K, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimers Dement (2014). doi:10.1016/j.jalz.2014.10.009
132. de Almeida SM, Shumaker SD, LeBlanc SK, Delaney P, Marquie-Beck J, Ueland S, et al. Incidence of post-dural puncture headache in research volunteers. Headache (2011) 51(10):1503–10. doi:10.1111/j.1526-4610.2011.01959.x
133. Henriksen K, O’Bryant SE, Hampel H, Trojanowski JQ, Montine TJ, Jeromin A, et al. The future of blood-based biomarkers for Alzheimer’s disease. Alzheimers Dement (2014) 10(1):115–31. doi:10.1016/j.jalz.2013.01.013
134. van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM. Plasma Abeta(1-40) and Abeta(1-42) and the risk of dementia: a prospective case-cohort study. Lancet Neurol (2006) 5(8):655–60. doi:10.1016/S1474-4422(06)70501-4
135. Mayeux R, Honig LS, Tang MX, Manly J, Stern Y, Schupf N, et al. Plasma A[beta]40 and A[beta]42 and Alzheimer’s disease: relation to age, mortality, and risk. Neurology (2003) 61(9):1185–90. doi:10.1212/01.WNL.0000091890.32140.8F
136. Schupf N, Tang MX, Fukuyama H, Manly J, Andrews H, Mehta P, et al. Peripheral Abeta subspecies as risk biomarkers of Alzheimer’s disease. Proc Natl Acad Sci U S A (2008) 105(37):14052–7. doi:10.1073/pnas.0805902105
137. Sundelof J, Giedraitis V, Irizarry MC, Sundstrom J, Ingelsson E, Ronnemaa E, et al. Plasma beta amyloid and the risk of Alzheimer disease and dementia in elderly men: a prospective, population-based cohort study. Arch Neurol (2008) 65(2):256–63. doi:10.1001/archneurol.2007.57
138. Lopez OL, Kuller LH, Mehta PD, Becker JT, Gach HM, Sweet RA, et al. Plasma amyloid levels and the risk of AD in normal subjects in the Cardiovascular Health Study. Neurology (2008) 70(19):1664–71. doi:10.1212/01.wnl.0000306696.82017.66
139. Hansson O, Zetterberg H, Vanmechelen E, Vanderstichele H, Andreasson U, Londos E, et al. Evaluation of plasma Abeta(40) and Abeta(42) as predictors of conversion to Alzheimer’s disease in patients with mild cognitive impairment. Neurobiol Aging (2010) 31(3):357–67. doi:10.1016/j.neurobiolaging.2008.03.027
140. Koyama A, Okereke OI, Yang T, Blacker D, Selkoe DJ, Grodstein F. Plasma amyloid-beta as a predictor of dementia and cognitive decline: a systematic review and meta-analysis. Arch Neurol (2012) 69(7):824–31. doi:10.1001/archneurol.2011.1841
141. Laske C, Sopova K, Gkotsis C, Eschweiler GW, Straten G, Gawaz M, et al. Amyloid-beta peptides in plasma and cognitive decline after 1 year follow-up in Alzheimer’s disease patients. J Alzheimers Dis (2010) 21(4):1263–9. doi:10.3233/JAD2010100510
142. Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med (2014) 370(4):311–21. doi:10.1056/NEJMoa1312889
143. Toledo JB, Shaw LM, Trojanowski JQ. Plasma amyloid beta measurements – a desired but elusive Alzheimer’s disease biomarker. Alzheimers Res Ther (2013) 5(2):8. doi:10.1186/alzrt162
144. Kuo YM, Emmerling MR, Lampert HC, Hempelman SR, Kokjohn TA, Woods AS, et al. High levels of circulating Abeta42 are sequestered by plasma proteins in Alzheimer’s disease. Biochem Biophys Res Commun (1999) 257(3):787–91. doi:10.1006/bbrc.1999.0552
145. Wunderlich MT, Lins H, Skalej M, Wallesch CW, Goertler M. Neuron-specific enolase and tau protein as neurobiochemical markers of neuronal damage are related to early clinical course and long-term outcome in acute ischemic stroke. Clin Neurol Neurosurg (2006) 108(6):558–63. doi:10.1016/j.clineuro.2005.12.006
146. Randall J, Mortberg E, Provuncher GK, Fournier DR, Duffy DC, Rubertsson S, et al. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: results of a pilot study. Resuscitation (2013) 84(3):351–6. doi:10.1016/j.resuscitation.2012.07.027
147. Liliang PC, Liang CL, Weng HC, Lu K, Wang KW, Chen HJ, et al. Tau proteins in serum predict outcome after severe traumatic brain injury. J Surg Res (2010) 160(2):302–7. doi:10.1016/j.jss.2008.12.022
148. Noguchi-Shinohara M, Hamaguchi T, Nozaki I, Sakai K, Yamada M. Serum tau protein as a marker for the diagnosis of Creutzfeldt-Jakob disease. J Neurol (2011) 258(8):1464–8. doi:10.1007/s00415-011-5960-x
149. Shahim P, Linemann T, Inekci D, Karsdal MA, Blennow K, Tegner Y, et al. Serum tau fragments predict return to play in concussed professional ice hockey players. J Neurotrauma (2015). doi:10.1089/neu.2014.3741
150. Zetterberg H, Wilson D, Andreasson U, Minthon L, Blennow K, Randall J, et al. Plasma tau levels in Alzheimer’s disease. Alzheimers Res Ther (2013) 5(2):9. doi:10.1186/alzrt163
151. Montine TJ, Quinn J, Kaye J, Morrow JD. F(2)-isoprostanes as biomarkers of late-onset Alzheimer’s disease. J Mol Neurosci (2007) 33(1):114–9. doi:10.1007/s12031-007-0044-1
152. Williams MA, Haughton D, Stevenson M, Craig D, Passmore AP, Silvestri G. Plasma complement factor H in Alzheimer’s Disease. J Alzheimers Dis (2015). doi:10.3233/JAD-142742
153. Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med (2007) 13(11):1359–62. doi:10.1038/nm1653
154. Hu WT, Holtzman DM, Fagan AM, Shaw LM, Perrin R, Arnold SE, et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology (2012) 79(9):897–905. doi:10.1212/WNL.0b013e318266fa70
155. Doecke JD, Laws SM, Faux NG, Wilson W, Burnham SC, Lam CP, et al. Blood-based protein biomarkers for diagnosis of Alzheimer disease. Arch Neurol (2012) 69(10):1318–25. doi:10.1001/archneurol.2012.1282
156. O’Bryant SE, Xiao G, Barber R, Huebinger R, Wilhelmsen K, Edwards M, et al. A blood-based screening tool for Alzheimer’s disease that spans serum and plasma: findings from TARC and ADNI. PLoS One (2011) 6(12):e28092. doi:10.1371/journal.pone.0028092
157. Llano DA, Devanarayan V, Simon AJ. Evaluation of plasma proteomic data for Alzheimer disease state classification and for the prediction of progression from mild cognitive impairment to Alzheimer disease. Alzheimer Dis Assoc Disord (2013) 27(3):233–43. doi:10.1097/WAD.0b013e31826d597a
158. O’Bryant SE, Gupta V, Henriksen K, Edwards M, Jeromin A, Lista S, et al. Guidelines for the standardization of preanalytic variables for blood-based biomarker studies in Alzheimer’s disease research. Alzheimers Dement (2014). doi:10.1016/j.jalz.2014.08.099
159. Zurbig P, Jahn H. Use of proteomic methods in the analysis of human body fluids in Alzheimer research. Electrophoresis (2012) 33(24):3617–30. doi:10.1002/elps.201200360
160. Sayer R, Law E, Connelly PJ, Breen KC. Association of a salivary acetylcholinesterase with Alzheimer’s disease and response to cholinesterase inhibitors. Clin Biochem (2004) 37(2):98–104. doi:10.1016/j.clinbiochem.2003.10.007
161. Boston PF, Gopalkaje K, Manning L, Middleton L, Loxley M. Developing a simple laboratory test for Alzheimer’s disease: measuring acetylcholinesterase in saliva – a pilot study. Int J Geriatr Psychiatry (2008) 23(4):439–40. doi:10.1002/gps.1882
162. Bermejo-Pareja F, Antequera D, Vargas T, Molina JA, Carro E. Saliva levels of Abeta1-42 as potential biomarker of Alzheimer’s disease: a pilot study. BMC Neurol (2010) 10:108. doi:10.1186/1471-2377-10-108
163. Shi M, Sui YT, Peskind ER, Li G, Hwang H, Devic I, et al. Salivary tau species are potential biomarkers of Alzheimer’s disease. J Alzheimers Dis (2011) 27(2):299–305. doi:10.3233/JAD-2011-110731
164. Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature (1999) 400(6740):173–7. doi:10.1038/22124
165. Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature (2000) 408(6815):982–5. doi:10.1038/35050116
166. Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature (2000) 408(6815):979–82. doi:10.1038/35050110
167. Das P, Murphy MP, Younkin LH, Younkin SG, Golde TE. Reduced effectiveness of Abeta1-42 immunization in APP transgenic mice with significant amyloid deposition. Neurobiol Aging (2001) 22(5):721–7. doi:10.1016/S0197-4580(01)00245-7
168. Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, et al. Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology (2005) 64(1):94–101. doi:10.1212/01.WNL.0000148604.77591.67
169. Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology (2005) 64(9):1553–62. doi:10.1212/01.WNL.0000159740.16984.3C
170. Pride M, Seubert P, Grundman M, Hagen M, Eldridge J, Black RS. Progress in the active immunotherapeutic approach to Alzheimer’s disease: clinical investigations into AN1792-associated meningoencephalitis. Neurodegener Dis (2008) 5(3–4):194–6. doi:10.1159/000113700
171. Vellas B, Black R, Thal LJ, Fox NC, Daniels M, McLennan G, et al. Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res (2009) 6(2):144–51. doi:10.2174/156720509787602852
172. Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, et al. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology (2005) 64(1):129–31. doi:10.1212/01.WNL.0000148590.39911.DF
173. Zotova E, Bharambe V, Cheaveau M, Morgan W, Holmes C, Harris S, et al. Inflammatory components in human Alzheimer’s disease and after active amyloid-beta42 immunization. Brain (2013) 136(Pt 9):2677–96. doi:10.1093/brain/awt210
174. Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet (2008) 372(9634):216–23. doi:10.1016/S0140-6736(08)61075-2
175. Serrano-Pozo A, William CM, Ferrer I, Uro-Coste E, Delisle MB, Maurage CA, et al. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain (2010) 133(Pt 5):1312–27. doi:10.1093/brain/awq056
176. Winblad B, Andreasen N, Minthon L, Floesser A, Imbert G, Dumortier T, et al. Safety, tolerability, and antibody response of active Abeta immunotherapy with CAD106 in patients with Alzheimer’s disease: randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol (2012) 11(7):597–604. doi:10.1016/S1474-4422(12)70140-0
177. Farlow MR, Andreasen N, Riviere ME, Vostiar I, Vitaliti A, Sovago J, et al. Long-term treatment with active Abeta immunotherapy with CAD106 in mild Alzheimer’s disease. Alzheimers Res Ther (2015) 7(1):23. doi:10.1186/s13195-015-0108-3
178. Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med (2000) 6(8):916–9. doi:10.1038/78682
179. Blennow K, Hampel H, Zetterberg H. Biomarkers in amyloid-beta immunotherapy trials in Alzheimer’s disease. Neuropsychopharmacology (2014) 39(1):189–201. doi:10.1038/npp.2013.154
180. Legleiter J, Czilli DL, Gitter B, DeMattos RB, Holtzman DM, Kowalewski T. Effect of different anti-Abeta antibodies on Abeta fibrillogenesis as assessed by atomic force microscopy. J Mol Biol (2004) 335(4):997–1006. doi:10.1016/j.jmb.2003.11.019
181. Lemere CA. Developing novel immunogens for a safe and effective Alzheimer’s disease vaccine. Prog Brain Res (2009) 175:83–93. doi:10.1016/S0079-6123(09)17506-4
182. Demattos RB, Lu J, Tang Y, Racke MM, Delong CA, Tzaferis JA, et al. A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer’s disease mice. Neuron (2012) 76(5):908–20. doi:10.1016/j.neuron.2012.10.029
183. Schroeter S, Khan K, Barbour R, Doan M, Chen M, Guido T, et al. Immunotherapy reduces vascular amyloid-beta in PDAPP mice. J Neurosci (2008) 28(27):6787–93. doi:10.1523/JNEUROSCI.2377-07.2008
184. Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology (2009) 73(24):2061–70. doi:10.1212/WNL.0b013e3181c67808
185. Vellas B, Carrillo MC, Sampaio C, Brashear HR, Siemers E, Hampel H, et al. Designing drug trials for Alzheimer’s disease: what we have learned from the release of the phase III antibody trials: a report from the EU/US/CTAD task force. Alzheimers Dement (2013) 9(4):438–44. doi:10.1016/j.jalz.2013.03.007
186. Blennow K, Zetterberg H, Rinne JO, Salloway S, Wei J, Black R, et al. Effect of immunotherapy with bapineuzumab on cerebrospinal fluid biomarker levels in patients with mild to moderate Alzheimer disease. Arch Neurol (2012) 69(8):1002–10. doi:10.1001/archneurol.2012.90
187. DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A (2001) 98(15):8850–5. doi:10.1073/pnas.151261398
188. Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci (2005) 25(3):629–36. doi:10.1523/JNEUROSCI.4337-04.2005
189. Seubert P, Barbour R, Khan K, Motter R, Tang P, Kholodenko D, et al. Antibody capture of soluble Abeta does not reduce cortical Abeta amyloidosis in the PDAPP mouse. Neurodegener Dis (2008) 5(2):65–71. doi:10.1159/000112834
190. Farlow M, Arnold SE, van Dyck CH, Aisen PS, Snider BJ, Porsteinsson AP, et al. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement (2012) 8(4):261–71. doi:10.1016/j.jalz.2011.09.224
191. Comery TA, Martone RL, Aschmies S, Atchison KP, Diamantidis G, Gong X, et al. Acute gamma-secretase inhibition improves contextual fear conditioning in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci (2005) 25(39):8898–902. doi:10.1523/JNEUROSCI.2693-05.2005
192. Martone RL, Zhou H, Atchison K, Comery T, Xu JZ, Huang X, et al. Begacestat (GSI-953): a novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein gamma-secretase for the treatment of Alzheimer’s disease. J Pharmacol Exp Ther (2009) 331(2):598–608. doi:10.1124/jpet.109.152975
193. Tong G, Castaneda L, Wang JS, Sverdlov O, Huang SP, Slemmon R, et al. Effects of single doses of avagacestat (BMS-708163) on cerebrospinal fluid Abeta levels in healthy young men. Clin Drug Investig (2012) 32(11):761–9. doi:10.1007/s40261-012-0006-4
194. Li T, Huang Y, Jin S, Ye L, Rong N, Yang X, et al. Gamma-secretase modulators do not induce Abeta-rebound and accumulation of beta-C-terminal fragment. J Neurochem (2012) 121(2):277–86. doi:10.1111/j.1471-4159.2011.07560.x
195. Mitani Y, Yarimizu J, Saita K, Uchino H, Akashiba H, Shitaka Y, et al. Differential effects between gamma-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J Neurosci (2012) 32(6):2037–50. doi:10.1523/JNEUROSCI.4264-11.2012
196. Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, et al. The mechanism of gamma-secretase dysfunction in familial Alzheimer disease. EMBO J (2012) 31(10):2261–74. doi:10.1038/emboj.2012.79
197. Cirrito JR, May PC, O’Dell MA, Taylor JW, Parsadanian M, Cramer JW, et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci (2003) 23(26):8844–53. doi:0270-6474/03/238844-10
198. Abramowski D, Wiederhold KH, Furrer U, Jaton AL, Neuenschwander A, Runser MJ, et al. Dynamics of Abeta turnover and deposition in different beta-amyloid precursor protein transgenic mouse models following gamma-secretase inhibition. J Pharmacol Exp Ther (2008) 327(2):411–24. doi:10.1124/jpet.108.140327
199. Garcia-Alloza M, Subramanian M, Thyssen D, Borrelli LA, Fauq A, Das P, et al. Existing plaques and neuritic abnormalities in APP:PS1 mice are not affected by administration of the gamma-secretase inhibitor LY-411575. Mol Neurodegener (2009) 4:19. doi:10.1186/1750-1326-4-19
200. Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med (2006) 12(7):856–61. doi:10.1038/nm1438
201. Fleisher AS, Raman R, Siemers ER, Becerra L, Clark CM, Dean RA, et al. Phase 2 safety trial targeting amyloid beta production with a gamma-secretase inhibitor in Alzheimer disease. Arch Neurol (2008) 65(8):1031–8. doi:10.1001/archneur.65.8.1031
202. Callaway E. Gene mutation defends against Alzheimer’s disease. Nature (2012) 487(7406):153. doi:10.1038/487153a
203. Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature (2012) 488(7409):96–9. doi:10.1038/nature11283
204. Mikulca JA, Nguyen V, Gajdosik DA, Teklu SG, Giunta EA, Lessa EA, et al. Potential novel targets for Alzheimer pharmacotherapy: II. Update on secretase inhibitors and related approaches. J Clin Pharm Ther (2014) 39(1):25–37. doi:10.1111/jcpt.12112
205. Menting KW, Claassen JA. beta-secretase inhibitor; a promising novel therapeutic drug in Alzheimer’s disease. Front Aging Neurosci (2014) 6:165. doi:10.3389/fnagi.2014.00165
206. Kokjohn TA, Roher AE. Amyloid precursor protein transgenic mouse models and Alzheimer’s disease: understanding the paradigms, limitations, and contributions. Alzheimers Dement (2009) 5(4):340–7. doi:10.1016/j.jalz.2009.03.002
207. DeMattos RB, Bales KR, Parsadanian M, O’Dell MA, Foss EM, Paul SM, et al. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer’s disease. J Neurochem (2002) 81(2):229–36. doi:10.1046/j.1471-4159.2002.00889.x
208. Cummings J, Gould H, Zhong K. Advances in designs for Alzheimer’s disease clinical trials. Am J Neurodegener Dis (2012) 1(3):205–16.doi:AJND1210001
209. Mulder SD, van der Flier WM, Verheijen JH, Mulder C, Scheltens P, Blankenstein MA, et al. BACE1 activity in cerebrospinal fluid and its relation to markers of AD pathology. J Alzheimers Dis (2010) 20(1):253–60. doi:10.3233/JAD-2010-1367
210. Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, Fox NC, et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol (2012) 11(3):241–9. doi:10.1016/S1474-4422(12)70015-7
211. Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K, et al. An effector-reduced anti-beta-amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci (2012) 32(28):9677–89. doi:10.1523/JNEUROSCI.4742-11.2012
212. Burstein AH, Zhao Q, Ross J, Styren S, Landen JW, Ma WW, et al. Safety and pharmacology of ponezumab (PF-04360365) after a single 10-minute intravenous infusion in subjects with mild to moderate Alzheimer disease. Clin Neuropharmacol (2013) 36(1):8–13. doi:10.1097/WNF.0b013e318279bcfa
213. Landen JW, Zhao Q, Cohen S, Borrie M, Woodward M, Billing CB Jr, et al. Safety and pharmacology of a single intravenous dose of ponezumab in subjects with mild-to-moderate Alzheimer disease: a phase I, randomized, placebo-controlled, double-blind, dose-escalation study. Clin Neuropharmacol (2013) 36(1):14–23. doi:10.1097/WNF.0b013e31827db49b
214. Miyoshi I, Fujimoto Y, Yamada M, Abe S, Zhao Q, Cronenberger C, et al. Safety and pharmacokinetics of PF-04360365 following a single-dose intravenous infusion in Japanese subjects with mild-to-moderate Alzheimer’s disease: a multicenter, randomized, double-blind, placebo-controlled, dose-escalation study. Int J Clin Pharmacol Ther (2013) 51(12):911–23. doi:10.5414/CP201816
215. Aisen PS, Gauthier S, Ferris SH, Saumier D, Haine D, Garceau D, et al. Tramiprosate in mild-to-moderate Alzheimer’s disease – a randomized, double-blind, placebo-controlled, multi-centre study (the Alphase Study). Arch Med Sci (2011) 7(1):102–11. doi:10.5114/aoms.2011.20612
Keywords: Alzheimer’s disease, amyloid cascade hypothesis, amyloid beta, tau, clinical trials, drugs
Citation: Ritter A and Cummings J (2015) Fluid biomarkers in clinical trials of Alzheimer’s disease therapeutics. Front. Neurol. 6:186. doi: 10.3389/fneur.2015.00186
Received: 20 April 2015; Accepted: 10 August 2015;
Published: 31 August 2015
Edited by:
Charlotte Elisabeth Teunissen, VU University Medical Center Amsterdam, NetherlandsReviewed by:
Ricardo Insausti, University of Castilla-La Mancha, SpainDouglas Galasko, University of California San Diego, USA
Copyright: © 2015 Ritter and Cummings. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aaron Ritter, Cleveland Clinic Lou Ruvo Center for Brain Health, 888 West Bonneville Avenue, Las Vegas, NV 89016, USA,cml0dGVyYUBjY2Yub3Jn