 Chiara La Morgia
Chiara La Morgia Michele Carbonelli3
Michele Carbonelli3 Piero Barboni
Piero Barboni Alfredo Arrigo Sadun
Alfredo Arrigo Sadun Valerio Carelli
Valerio Carelli- 1UOC Clinica Neurologica, IRCCS Istituto delle Scienze Neurologiche di Bologna, Ospedale Bellaria, Bologna, Italy
- 2Unità di Neurologia, Dipartimento di Scienze Biomediche e NeuroMotorie (DIBINEM), Università di Bologna, Bologna, Italy
- 3Studio Oculistico d’Azeglio, Bologna, Italy
- 4Istituto Scientifico San Raffaele, Milan, Italy
- 5Doheny Eye Institute, University of California Los Angeles, Los Angeles, CA, USA
Hereditary optic neuropathies are diseases affecting the optic nerve. The most common are mitochondrial hereditary optic neuropathies, i.e., the maternally inherited Leber’s hereditary optic neuropathy (LHON) and dominant optic atrophy (DOA). They both share a mitochondrial pathogenesis that leads to the selective loss of retinal ganglion cells and axons, in particular of the papillo-macular bundle. Typically, LHON is characterized by an acute/subacute loss of central vision associated with impairment of color vision and swelling of retinal nerve fibers followed by optic atrophy. DOA, instead, is characterized by a childhood-onset and slowly progressive loss of central vision, worsening over the years, leading to optic atrophy. The diagnostic workup includes neuro-ophthalmologic evaluation and genetic testing of the three most common mitochondrial DNA mutations affecting complex I (11778/ND4, 3460/ND1, and 14484/ND6) for LHON and sequencing of the nuclear gene OPA1 for DOA. Therapeutic strategies are still limited including agents that bypass the complex I defect and exert an antioxidant effect (idebenone). Further strategies are aimed at stimulating compensatory mitochondrial biogenesis. Gene therapy is also a promising avenue that still needs to be validated.
Introduction
Hereditary optic neuropathies are a heterogeneous group of diseases, which may be either an isolated optic neuropathy or optic neuropathy in the context of a more complex multi-systemic disease. The estimated minimum prevalence of hereditary optic neuropathies is about 1 in 10,000 (1).
Optic neuropathy, as part of a more complex spectrum of clinical symptoms, is common in hereditary neurodegenerative disorders with evidence of mitochondrial dysfunction [for a review see Ref. (2)] such as OPA1 “plus” syndrome (3), mitochondrial encephalomyopathy lactic acidosis and stroke-like syndrome (MELAS) (4), LHON/dystonia/MELAS/Leigh overlapping syndrome (5, 6), myoclonic epilepsy with ragged red fibers (MERRF) (7), POLG1-related syndrome (8), Friedreich ataxia (9), Charcot–Marie Tooth type 2A (10), Mohr–Tranebjerg syndrome (11), SPG7 syndrome (12), Wolfram syndrome (13), spinocerebellar ataxias (14), and DNMT1-related disorders (15).
The main non-syndromic hereditary optic neuropathies are Leber’s hereditary optic neuropathy (LHON) and dominant optic atrophy (DOA) and both are primary mitochondrial hereditary optic neuropathies (16). Other rare recessive or X-linked forms of optic neuropathy are also described, in most cases not yet resolved from the genetic point of view.
Genetic Basis of Isolated Optic Neuropathies
Leber’s hereditary optic neuropathy is a maternally inherited disorder and in 90–95% of cases it is due to one of three mitochondrial DNA (mtDNA) mutations (11778/ND4, 3460/ND1, and 14484/ND6) (17). Other rare mtDNA mutations have been reported in association with LHON (18). These rare mutations may need testing for, by complete sequence of mtDNA, if the clinical suspicion of LHON is high and a maternal inheritance is obvious.
Dominant optic atrophy, in about 70% of cases, is due to mutations in the OPA1 gene. Three other loci have been associated with autosomal DOA and designated as OPA4 (19), OPA5 (20), and OPA8 (21). Furthermore, heterozygous mutations in the OPA3 gene have been associated with DOA and cataract (22), as well as heterozygous mutations in the WSF1 gene have been reported in association with DOA and hearing loss (23). Both OPA3 and WSF1 genes are mostly associated with recessive mutations leading to Costeff (24) and Wolfram syndromes, respectively. Additional loci have been identified in X-linked (OPA2) (25) and recessive (OPA6 and OPA7) hereditary optic neuropathies, the latter recently associated with mutations in TMEM126A (26, 27).
The next sections will focus on LHON and DOA, the most frequent and prototypical hereditary optic neuropathies, for which an extensive body of studies is available.
Clinical Presentation of LHON/DOA and Clinical Workup
Leber’s hereditary optic neuropathy typically affects young adult males presenting with unilateral or bilateral subacute/acute painless loss of central vision. The clinical course typically involves a rapid worsening of visual acuity over a few weeks (nadir reached in about one to 6 months) and subsequent stabilization of visual loss, entering the chronic phase. Loss of vision can be very severe leading to legal blindness in most cases (17). In the acute phase, there is bilateral sequential vision loss, significant impairment of color vision, and usually maintenance of the pupillary light reflex. Fundus examination shows, in the acute phase, pseudoedema of the optic disk and retinal nerve fibers initially involving mainly the inferior and superior arcades with teleangectias (Figure 1A) and temporal or diffuse optic disk pallor in later stage of the disease. Visual fields reveal central or cecocentral scotoma, which progressively worsens over time (28). Optical coherence tomography (OCT) demonstrates an initial thickening of the temporal and inferior peripapillary fibers, which is followed by the thickening of the superior and, lastly, of the nasal fibers (Figure 1A). The temporal fibers are lost first and the nasal fibers are the most spared (29). Fluorescein angiography in the acute phase usually fails to show leakage of fluorescein (hence pseudo disk edema) and this is a useful marker for differentiating LHON from inflammatory optic neuropathies with true disk edema that causes fluorescein leakage (28). Another useful clinical sign, which distinguishes LHON from optic neuritis (the most important clinical differential diagnosis for LHON) is the usual absence of a relative afferent pupillary defect. The maintenance of the pupillary light response is partly due to the bilateral symmetry of the disease process and partly to the relative sparing of melanopsin retinal ganglion cells (mRGCs) in LHON, as revealed by post-mortem investigations (30). These cells are about 1% of the total RGCs and they contribute mainly to circadian photo-entrainment, but through the retinal projections to the olivary pretectal nucleus (OPN), they also regulate the pupillary light reflex. Clinical studies demonstrated the maintenance of the pupil responses in LHON patients (31, 32). The diagnostic workup in LHON also may include other neurophysiologic tests such as visual evoked potentials (VEPs) that reveal delayed latency and reduced amplitude of cortical responses and pattern electroretinograms (PERGs), which demonstrate reduced amplitude and delayed latency of responses (33).
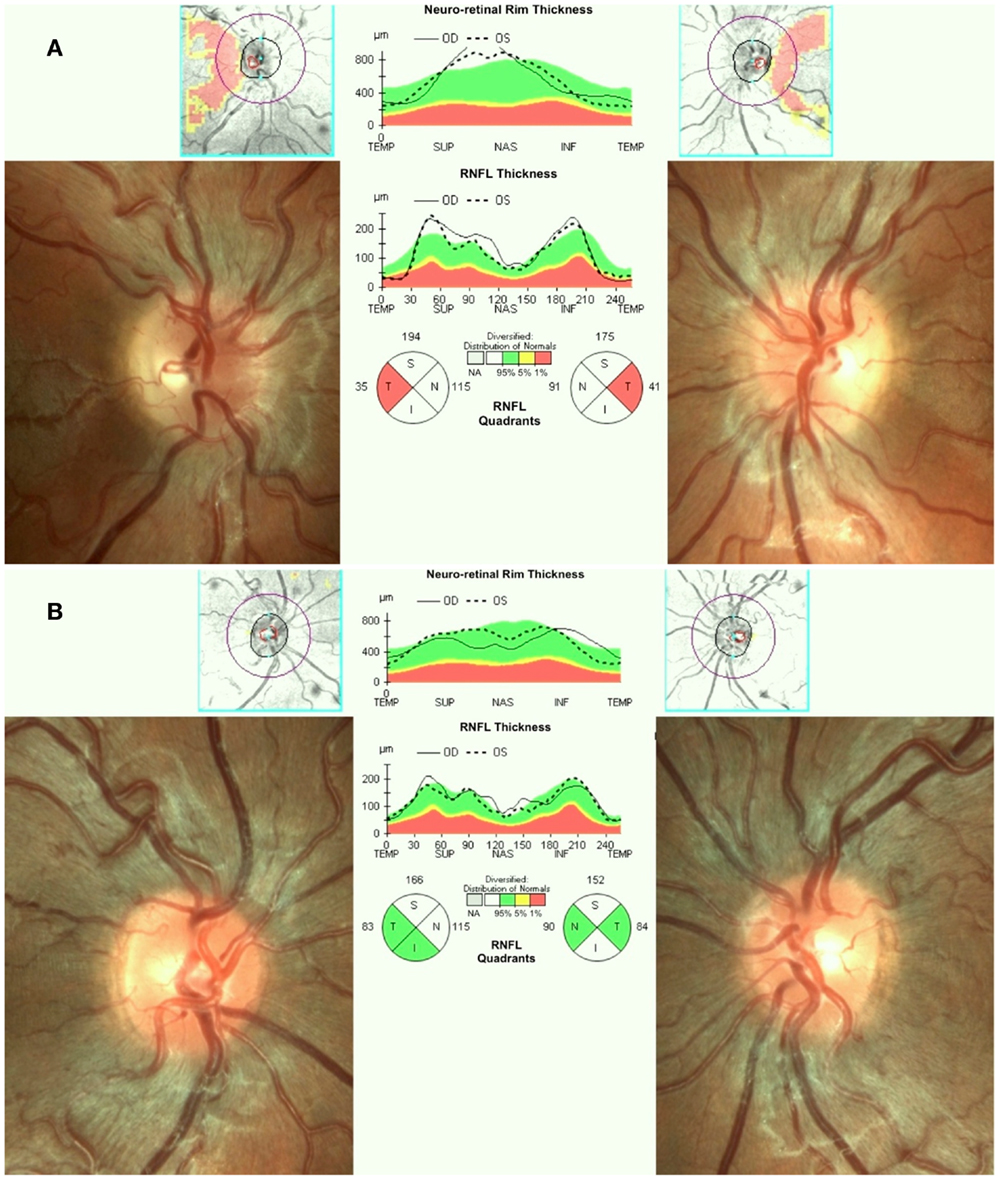
Figure 1. (A) Fundus and OCT in the acute-phase of LHON. Fundus exam shows moderate swelling of the retinal nerve fibers mainly involving the superior and inferior arcades around the optic disk associated with loss of fibers in the papillo-macular bundle (temporal pallor), retinal vessels tortuosity, and telangiectasias. Optical coherence tomography (OCT) shows retinal nerve fiber layer thinning of the temporal quadrant with thickening of all the other quadrants. (B) Fundus and OCT of a LHON carrier. Fundus examination reveals mild swelling of the superior and inferior retinal nerve fibers and microangiopathy. OCT demonstrates retinal nerve fiber layer thickening more evident in the supero-nasal (OD) and supero-inferior (OS) quadrants.
Some LHON patients may spontaneously recover vision during the clinical course (34). The rate of spontaneous recovery is variable for the three LHON mutations being 4–20% for 11778/ND4 and 3460/ND1 mutation, and 50% or more for the 14484/ND6 (34, 35). This recovery may occur after the nadir of the disease at about 6–8 months and up to 5 years after the onset (1).
The family history is very helpful since it often reveals the presence of other affected individuals along the maternal lineage, in prevalence males.
Leber’s hereditary optic neuropathy is due in the large majority of cases (90–95%) to the three classical LHON mutations, which are homoplasmic (i.e., all the mtDNA copies harbor the mutation) in most of the families. Despite the genetic risk of disease seems to be equal among all individuals along the maternal lineage, the penetrance is incomplete and the disease affects prevalently males (17). Genetic counseling implies in these patients that the offspring of a woman carrying the LHON mutation will harbor the mutation, whereas offspring of males are spared. The probability to become affected is roughly 10% in women and 50% in men carrying the 11778/ND4 LHON mutation (17). The reason for the lower penetrance in females is possibly related to the protective role exerted by estrogens on cellular metabolism (36). The risk of recurrence for individuals younger than 50 years of age and maternally related to the index case has been calculated by Harding and colleagues (37). A specific mtDNA background, haplogroup J, due to recurrence of non-synonymous variants affecting complex I and III subunit genes, has been associated with higher penetrance of the mutations 11778/ND4 and 14484/ND6 (38).
Other known environmental risk factors for LHON are smoking, alcohol, and the use of some antibiotics (i.e., macrolids, aminoglicosides, ethambutol, isoniazide, and linezolid) (39, 40). Clinical counseling should include avoiding these risk factors.
Though usually LHON presents as an isolated optic neuropathy, in a few cases, there may be the coexistence of additional signs and symptoms, such as myoclonus, MS-like features, epilepsy, heart conduction defects, peripheral neuropathy, dystonia, and myopathy (41).
Other ancillary tests that have been shown useful to complete the clinical characterization of LHON patients include brain and muscle magnetic resonance spectroscopy (42, 43) and lactic acid after standardized exercise (44).
Asymptomatic carriers of LHON mutation may also have swollen retinal nerve fibers that can be seen on fundus examination and by OCT (45, 46) (Figure 1B), abnormalities in color vision (47), and in neural conduction along the visual pathways (33, 48).
Dominant optic atrophy usually affects both males and females in young childhood, and in the large majority of cases, is due to mutations in the OPA1 gene (49, 50). There is incomplete penetrance and the classical clinical picture of DOA differs from LHON in that the former presents as a slowly progressive loss of vision with a smaller central or cecocentral scotoma (51). The clinical severity is very variable even within the same pedigree (52). As in LHON, OCT demonstrates preferential involvement of the papillo-macular bundle with a corresponding loss of fibers in the temporal sector of the optic disk and relative sparing of the nasal quadrant (53), which leads to temporal or diffuse optic atrophy. The main differential diagnosis for DOA is normal tension glaucoma, due to the frequent cupping of chronic DOA cases (54). Interestingly, OCT also demonstrates that optic disk size is reduced in DOA compared to controls and this might be explained by the role played by OPA1 in regulating developmental apoptosis and shaping optic nerve head morphology (55).
As for LHON, the clinical examination in DOA demonstrates the maintenance of the pupillary light reflex. This finding is corroborated by histological studies showing a relative sparing of mRGCs in DOA as well (30, 56).
VEPs may be normal or disclose a delayed latency and reduced amplitude of cortical responses (57).
Genetic counseling for DOA implies the rules of Mendelian autosomal dominant diseases and so the probability that offspring of an affected individual will inherit the mutation is 50% and this is equal for males and females. Prenatal genetic diagnosis is feasible, as well as pre-implantation diagnosis, thus providing the option to avoid transmission of the mutant OPA1 allele to offspring of an affected parent. As for LHON, DOA also may present a “plus” phenotype, which includes deafness, chronic external ophthalmoplegia, peripheral neuropathy, cerebellar atrophy, and mitochondrial myopathy (3, 58). In these cases, muscle biopsy histology and histoenzymology may disclose the hallmarks of mitochondrial myopathy with cytochrome c oxidase negative fibers, whereas molecular analysis will reveal the pathological accumulation of multiple mtDNA deletions (58). Moreover, these patients often present increased lactic acid levels after standardized exercise. The OPA1 mutations associated with the “plus” phenotype are in most cases missense mutations affecting the GTP-ase domain of the OPA1 protein (58), as opposed to the majority of mutations associated with non-syndromic DOA, mostly predicted to induce haploinsufficiency.
Follow-Up Management
Patients with hereditary optic neuropathies should be followed regularly during the disease course, particularly in the acute/subacute phase. The most important exams for the clinical follow-up of these patients are color vision, visual fields, visual acuity, and OCT.
Visual acuity is usually poor in LHON and some patients may end with bare light perception. DOA patients present a more variable clinical severity (52). Visual fields usually deteriorate from small to very large central scotoma in LHON and may remain small and stable in DOA over long periods of time (59). In LHON, visual fields may sometimes show reduction in size of the scotoma or its fenestration (60), both leading to an improvement in visual acuity (Figure 2). However, spontaneous visual recovery has never been described in DOA (61). Only one rare OPA1 mutation has been associated with a reversible LHON-like visual loss (62). A component of the visual improvement in LHON can be the adaptive phenomenon of eccentric fixation (63).
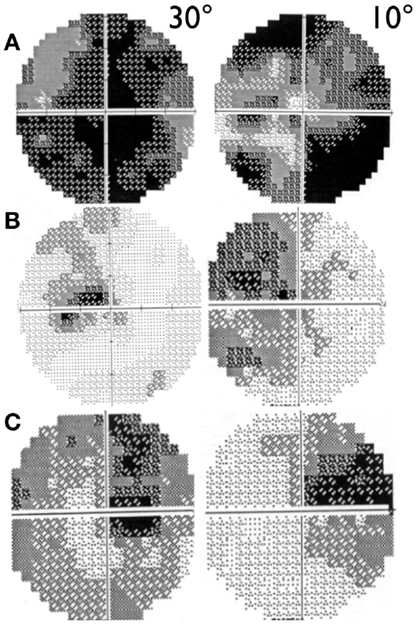
Figure 2. 30° and 10° visual field pattern of recovery in LHON. (A) Example of scotoma fenestration (OS) in a LHON patient carrying the 14484/ND6 mutation (B) Example of scotoma contraction (OS) in a LHON patient carrying the 11778/ND4 mutation and (C) Example of both scotoma fenestration and contraction (OD) in a LHON patient carrying the 11778/ND4 mutation.
Clinical Significance
Although diseases as LHON and DOA are rare, they constitute a terrible tragedy for the patients who go blind and for their families. Unlike most common causes of blindness, these hereditary optic neuropathies are not associated with old age. Generally, patients with LHON and DOA loose their vision before young adulthood and the disease tend to be bilaterally symmetrical. Hence, typical patients will loose their vision as a teenager or in their 20s and face a lifetime of blindness and disability. Parents and siblings are often part of the process of diagnosis and management.
Therapeutic Options
Therapeutic options for hereditary optic neuropathies are still limited (64) and up to now only one randomized double-blind placebo-controlled study with idebenone in LHON has been concluded (65).
Idebenone (a quinone analog of coenzyme Q10) has been reported to be effective in increasing the rate of recovery in LHON especially in patients with discordant visual acuity and if the patients are treated early in the disease course and for a long time (35, 65). These findings may suggest that there is a specific window of opportunity for increasing the rate of visual recovery with idebenone treatment. Coenzyme Q10 and idebenone are able to bypass the complex I defect caused by the LHON mutation and permit the transfer of electrons from NADH directly to complex III (66). A new double-blind placebo-controlled trial with idebenone in LHON is about to start with a longer follow-up and different design predefined primary end-points and dosages (Yu-Wai-Man, personal communication). Idebenone treatment has also been reported to ameliorate color vision in LHON (67).
Very preliminarily, encouraging results with idebenone treatment have been also reported in DOA patients (61) for whom spontaneous recovery has not been previously described. The rationale for the use of idebenone in DOA is that also in OPA1-related DOA, there is a complex I defect as in LHON (68).
More recently, other drugs are under investigations, such as the novel quinone molecule, alfa-tocotrienol EPI-743, which has shown promising results in an open-label trial on a limited group of LHON patients (69). These results need to be confirmed in larger controlled trial.
Other therapeutic strategies for LHON may include the use of drugs that might stimulate mitochondrial biogenesis, which has been demonstrated as a possible compensatory mechanism for LHON and other mitochondrial disorders (70). These include bezafibrate and rosiglitazone and novel molecules such as resveratrol and AICAR (5-aminoimidazole-4-carboxamide ribonucleoside) (71).
For a variety of reasons, gene therapy is a promising avenue for LHON treatment. The eye is easily accessible and provides a privileged space into which inject a viral vector. The most studied approach in LHON has been the AAV-mediated nuclear allotopic expression of mtDNA-encoded genes delivered to mitochondria (72, 73). This approach has been used in mouse models of LHON by the intravitreal delivery of an AAV vector carrying the wild-type ND4 (74, 75). This approach is now starting to be applied in LHON patients. However, it remains somehow controversial in terms of feasibility and efficiency (76). Other strategies include the direct delivery within mitochondria of the whole mtDNA or of an AAV vector containing the mtDNA-encoded ND4 subunit gene (77). These approaches are extremely promising but need to be validated.
For DOA, being a nuclear-encoded disease, gene therapy strategy should be more straightforward and feasible, as demonstrated by the recent encouraging results obtained with the gene therapy in Leber’s congenital amaurosis (78, 79). A proof of principle that genetically determined RGCs loss can be rescued by an AAV-based strategy with intravitreal delivery of the viral vector has been provided in an AIF mouse model, the so-called Harlequine mouse (80). There is issue of packaging, the 30 exons of the OPA1 gene into the AAV. Another problem to solve is which isoform of the OPA1 gene should be used as the most efficient to rescue the phenotype in RGCs. Currently, the specific role played by the eight OPA1 isoforms has not yet been systematically investigated. Another critical point is that the delivery of wild-type OPA1 to RGCs would likely complement haploinsufficiency mutations but more investigations are needed on the purported dominant negative mechanism of missense mutations. Nonetheless, gene therapy in DOA is a realistic objective, which will be pursued in the next future, providing some hope for treatment of DOA patients.
Conflict of Interest Statement
Dr. Alfredo Arrigo Sadun is the PI on unrestricted research grants from Edison Pharmaceuticals. Dr. Valerio Carelli received reimbursements of travel expenses for consulting activities from Edison Pharmaceuticals. All other co-authors have no conflict of interest.
Acknowledgments
Research on hereditary optic neuropathies was supported by Telethon Grants GGP06233 (to Valerio Carelli), GGP11182 (to Massimo Zeviani and Valerio Carelli), and GPP10005 (to Valerio Carelli), Mitocon Onlus (to Valerio Carelli); Research to Prevent Blindness (RPB), the International Foundation for Optic Nerve Diseases (IFOND), Struggling Within Leber’s, The Poincenot Family, the Eierman Foundation, and a National Eye Institute grant EY03040 (to Alfredo Arrigo Sadun).
References
1. Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies-disease mechanisms and therapeutic strategies. Prog Retin Eye Res (2011) 30:81–114. doi: 10.1016/j.preteyeres.2010.11.002
2. Carelli V, La Morgia C, Valentino ML, Barboni P, Ross-Cisneros FN, Sadun AA. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta (2009) 1787:518–28. doi:10.1016/j.bbabio.2009.02.024
3. Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissière A, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain (2008) 131:338–51. doi:10.1093/brain/awm298
4. Pulkes T, Eunson L, Patterson V, Siddiqui A, Wood NW, Nelson IP, et al. The mitochondrial DNA G13513A transition in ND5 is associated with a LHON/MELAS overlap syndrome and may be a frequent cause of MELAS. Ann Neurol (1999) 46:916–9. doi:10.1002/1531-8249(199912)46:6<916::AID-ANA16>3.0.CO;2-R
5. Bruyn GW, Went LN. A sex-linked heredo-degenerative neurological disorder, associated with Leber’s optic atrophy. I. Clinical studies. J Neurol Sci (1964) 1:59–80. doi:10.1016/0022-510X(64)90054-1
6. Novotny EJ Jr, Singh G, Wallace DC, Dorfman LJ, Louis A, Sogg RL, et al. Leber’s disease and dystonia: a mitochondrial disease. Neurology (1986) 36:1053–60. doi:10.1212/WNL.36.8.1053
7. Mancuso M, Orsucci D, Angelini C, Bertini E, Carelli V, Comi GP, et al. Phenotypic heterogeneity of the 8344A >G mtDNA “MERRF” mutation. Neurology (2013) 80:2049–54. doi:10.1212/WNL.0b013e318294b44c
8. Milone M, Wang J, Liewluck T, Chen LC, Leavitt JA, Wong LJ. Novel POLG splice site mutation and optic atrophy. Arch Neurol (2011) 68:806–11. doi:10.1001/archneurol.2011.124
9. Fortuna F, Barboni P, Liguori R, Valentino ML, Savini G, Gellera C, et al. Visual system involvement in patients with Friedreich’s ataxia. Brain (2009) 132:116–23. doi:10.1093/brain/awn269
10. Rouzier C, Bannwarth S, Chaussenot A, Chevrollier A, Verschueren A, Bonello-Palot N, et al. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy “plus” phenotype. Brain (2012) 135:23–34. doi:10.1093/brain/awr323
11. Ha AD, Parratt KL, Rendtorff ND, Lodahl M, Ng K, Rowe DB, et al. The phenotypic spectrum of dystonia in Mohr-Tranebjaerg syndrome. Mov Disord (2012) 27:1034–40. doi:10.1002/mds.25033
12. Klebe S, Depienne C, Gerber S, Challe G, Anheim M, Charles P, et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain (2012) 135:2980–93. doi:10.1093/brain/aws240
13. Rigoli L, Di Bella C. Wolfram syndrome 1 and Wolfram syndrome 2. Curr Opin Pediatr (2012) 24:512–7. doi:10.1097/MOP.0b013e328354ccdf
14. Stricker S, Oberwahrenbrock T, Zimmermann H, Schroeter J, Endres M, Brandt AU, et al. Temporal retinal nerve fiber loss in patients with spinocerebellar ataxia type 1. PLoS One (2011) 6:e23024. doi:10.1371/journal.pone.0023024
15. Moghadam KK, Pizza F, La Morgia C, Franceschini C, Tonon C, Lodi R, et al. Narcolepsy is a common phenotype in HSAN IE and ADCA-DN. Brain (2014) 37(Pt 6):1643–55. doi:10.1093/brain/awu069
16. Carelli V, La Morgia C, Sadun AA. Mitochondrial dysfunction in optic neuropathies: animal models and therapeutic options. Curr Opin Neurol (2013) 26:52–8. doi:10.1097/WCO.0b013e32835c5f0b
17. Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res (2004) 23:53–89. doi:10.1016/j.preteyeres.2003.10.003
18. Achilli A, Iommarini L, Olivieri A, Pala M, Hooshiar Kashani B, Reynier P, et al. Rare primary mitochondrial DNA mutations and probable synergistic variants in Leber’s hereditary optic neuropathy. PLoS One (2012) 7:e42242. doi:10.1371/journal.pone.0042242
19. Kerrison JB, Arnould VJ, Ferraz Sallum JM, Vagefi MR, Barmada MM, Li Y, et al. Genetic heterogeneity of dominant optic atrophy, Kjer type: identification of a second locus on chromosome 18q12.2-12.3. Arch Ophthalmol (1999) 117:805–10.
20. Barbet F, Hakiki S, Orssaud C, Gerber S, Perrault I, Hanein S, et al. A third locus for dominant optic atrophy on chromosome 22q. J Med Genet (2005) 42:e1. doi:10.1136/jmg.2004.025502
21. Carelli V, Schimpf S, Fuhrmann N, Valentino ML, Zanna C, Iommarini L, et al. A clinically complex form of dominant optic atrophy (OPA8) maps on chromosome 16. Hum Mol Genet (2011) 20:1893–905. doi:10.1093/hmg/ddr071
22. Reynier P, Amati-Bonneau P, Verny C, Olichon A, Simard G, Guichet A, et al. OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J Med Genet (2004) 41:e110. doi:10.1136/jmg.2003.016576
23. Rendtorff ND, Lodahl M, Boulahbel H, Johansen IR, Pandya A, Welch KO, et al. Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy and hearing impairment. Am J Med Genet A (2011) 155A:1298–313. doi:10.1002/ajmg.a.33970
24. Anikster Y, Kleta R, Shaag A, Gahl WA, Elpeleg O. Type III 3-methylglutaconic aciduria (optic atrophy plus syndrome, or Costeff optic atrophy syndrome): identification of the OPA3 gene and its founder mutation in Iraqi Jews. Am J Hum Genet (2001) 69:1218–24. doi:10.1086/324651
25. Assink JJ, Tijmes NT, ten Brink JB, Oostra RJ, Riemslag FC, de Jong PT, et al. A gene for X-linked optic atrophy is closely linked to the Xp11.4-Xp11.2 region of the X chromosome. Am J Hum Genet (1997) 61:934–9.
26. Barbet F, Gerber S, Hakiki S, Perrault I, Hanein S, Ducroq D, et al. A first locus for isolated autosomal recessive optic atrophy (ROA1) maps to chromosome 8q. Eur J Hum Genet (2003) 11:966–71. doi:10.1038/sj.ejhg.5201070
27. Hanein S, Perrault I, Roche O, Gerber S, Khadom N, Rio M, et al. TMEM126A, encoding a mitochondrial protein, is mutated in autosomal-recessive nonsyndromic optic atrophy. Am J Hum Genet (2009) 84:493–8. doi:10.1016/j.ajhg.2009.03.003
28. Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol (2005) 140:517–23. doi:10.1016/j.ajo.2005.03.017
29. Barboni P, Carbonelli M, Savini G, Ramos Cdo V, Carta A, Berezovsky A, et al. Natural history of Leber’s hereditary optic neuropathy: longitudinal analysis of the retinal nerve fiber layer by optical coherence tomography. Ophthalmology (2010) 117:623–7. doi:10.1016/j.ophtha.2009.07.026
30. La Morgia C, Ross-Cisneros FN, Sadun AA, Hannibal J, Munarini A, Mantovani V, et al. Melanopsin retinal ganglion cells are resistant to neurodegeneration in mitochondrial optic neuropathies. Brain (2010) 133:2426–38. doi:10.1093/brain/awq155
31. Moura AL, Nagy BV, La Morgia C, Barboni P, Oliveira AG, Salomão SR, et al. The pupil light reflex in Leber’s hereditary optic neuropathy: evidence for preservation of melanopsin-expressing retinal ganglion cells. Invest Ophthalmol Vis Sci (2013) 54:4471–7. doi:10.1167/iovs.12-11137
32. Kawasaki A, Collomb S, Léon L, Münch M. Pupil responses derived from outer and inner retinal photoreception are normal in patients with hereditary optic neuropathy. Exp Eye Res (2014) 120:161–6. doi:10.1016/j.exer.2013.11.005
33. Ziccardi L, Sadun F, De Negri AM, Barboni P, Savini G, Borrelli E, et al. Retinal function and neural conduction along the visual pathways in affected and unaffected carriers with Leber’s hereditary optic neuropathy. Invest Ophthalmol Vis Sci (2013) 54:6893–901. doi:10.1167/iovs.13-12894
34. Stone EM, Newman NJ, Miller NR, Johns DR, Lott MT, Wallace DC. Visual recovery in patients with Leber’s hereditary optic neuropathy and the 11778 mutation. J Clin Neuroophthalmol (1992) 12:10–4.
35. Carelli V, La Morgia C, Valentino ML, Rizzo G, Carbonelli M, De Negri AM, et al. Idebenone treatment in Leber’s hereditary optic neuropathy. Brain (2011) 134:e188. doi:10.1093/brain/awr180
36. Giordano C, Montopoli M, Perli E, Orlandi M, Fantin M, Ross-Cisneros FN, et al. Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy. Brain (2011) 134:220–34. doi:10.1093/brain/awq276
37. Harding AE, Sweeney MG, Govan GG, Riordan-Eva P. Pedigree analysis in Leber hereditary optic neuropathy families with a pathogenic mtDNA mutation. Am J Hum Genet (1995) 57:77–86.
38. Carelli V, Achilli A, Valentino ML, Rengo C, Semino O, Pala M, et al. Haplogroup effects and recombination of mitochondrial DNA: novel clues from the analysis of Leber hereditary optic neuropathy pedigrees. Am J Hum Genet (2006) 78:564–74. doi:10.1086/501236
39. Sadun AA, La Morgia C, Carelli V. Mitochondrial optic neuropathies: our travels from bench to bedside and back again. Clin Experiment Ophthalmol (2013) 41:702–12. doi:10.1111/ceo.12086
40. Kirkman MA, Yu-Wai-Man P, Korsten A, Leonhardt M, Dimitriadis K, De Coo IF, et al. Gene-environment interactions in Leber hereditary optic neuropathy. Brain (2009) 132:2317–26. doi:10.1093/brain/awp158
41. Nikoskelainen EK, Marttila RJ, Huoponen K, Juvonen V, Lamminen T, Sonninen P, et al. Leber’s “plus”: neurological abnormalities in patients with Leber’s hereditary optic neuropathy. J Neurol Neurosurg Psychiatry (1995) 59:160–4. doi:10.1136/jnnp.59.2.160
42. La Morgia C, Achilli A, Iommarini L, Barboni P, Pala M, Olivieri A, et al. Rare mtDNA variants in Leber hereditary optic neuropathy families with recurrence of myoclonus. Neurology (2008) 70:762–70. doi:10.1212/01.wnl.0000295505.74234.d0
43. Lodi R, Carelli V, Cortelli P, Iotti S, Valentino ML, Barboni P, et al. Phosphorus MR spectroscopy shows a tissue specific in vivo distribution of biochemical expression of the G3460A mutation in Leber’s hereditary optic neuropathy. J Neurol Neurosurg Psychiatry (2002) 72:805–7. doi:10.1136/jnnp.72.6.805
44. Montagna P, Plazzi G, Cortelli P, Carelli V, Lugaresi E, Barboni P, et al. Abnormal lactate after effort in healthy carriers of Leber’s hereditary optic neuropathy. J Neurol Neurosurg Psychiatry (1995) 58:640–1. doi:10.1136/jnnp.58.5.640
45. Sadun AA, Salomao SR, Berezovsky A, Sadun F, Denegri AM, Quiros PA, et al. Subclinical carriers and conversions in Leber hereditary optic neuropathy: a prospective psychophysical study. Trans Am Ophthalmol Soc (2006) 104:51–61.
46. Barboni P, Savini G, Feuer WJ, Budenz DL, Carbonelli M, Chicani F, et al. Retinal nerve fiber layer thickness variability in Leber hereditary optic neuropathy carriers. Eur J Ophthalmol (2012) 22:985–91. doi:10.5301/ejo.5000154
47. Ventura DF, Gualtieri M, Oliveira AG, Costa MF, Quiros P, Sadun F, et al. Male prevalence of acquired color vision defects in asymptomatic carriers of Leber’s hereditary optic neuropathy. Invest Ophthalmol Vis Sci (2007) 48:2362–70. doi:10.1167/iovs.06-0331
48. Sacai PY, Salomão SR, Carelli V, Pereira JM, Belfort R Jr, Sadun AA, et al. Visual evoked potentials findings in non-affected subjects from a large Brazilian pedigree of 11778 Leber’s hereditary optic neuropathy. Doc Ophthalmol (2010) 121:147–54. doi:10.1007/s10633-010-9241-2
49. Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet (2000) 26:207–10. doi:10.1038/79936
50. Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet (2000) 26:211–5. doi:10.1038/79944
51. Votruba M, Moore AT, Bhattacharya SS. Clinical features, molecular genetics, and pathophysiology of dominant optic atrophy. J Med Genet (1998) 35:793–800. doi:10.1136/jmg.35.10.793
52. Cohn AC, Toomes C, Potter C, Towns KV, Hewitt AW, Inglehearn CF, et al. Autosomal dominant optic atrophy: penetrance and expressivity in patients with OPA1 mutations. Am J Ophthalmol (2007) 143:656–62. doi:10.1016/j.ajo.2006.12.038
53. Barboni P, Savini G, Parisi V, Carbonelli M, La Morgia C, Maresca A, et al. Retinal nerve fiber layer thickness in dominant optic atrophy measurements by optical coherence tomography and correlation with age. Ophthalmology (2011) 118:2076–80. doi:10.1016/j.ophtha.2011.02.027
54. Votruba M, Thiselton D, Bhattacharya SS. Optic disc morphology of patients with OPA1 autosomal dominant optic atrophy. Br J Ophthalmol (2003) 87:48–53. doi:10.1136/bjo.87.1.48
55. Barboni P, Carbonelli M, Savini G, Foscarini B, Parisi V, Valentino ML, et al. OPA1 mutations associated with dominant optic atrophy influence optic nerve head size. Ophthalmology (2010) 117:1547–53. doi:10.1016/j.ophtha.2009.12.042
56. Perganta G, Barnard AR, Katti C, Vachtsevanos A, Douglas RH, MacLaren RE, et al. Non-image-forming light driven functions are preserved in a mouse model of autosomal dominant optic atrophy. PLoS One (2013) 8:e56350. doi:10.1371/journal.pone.0056350
57. Holder GE, Votruba M, Carter AC, Bhattacharya SS, Fitzke FW, Moore AT. Electrophysiological findings in dominant optic atrophy (DOA) linking to the OPA1 locus on chromosome 3q 28-qter. Doc Ophthalmol (1999) 95:217–28.
58. Yu-Wai-Man P, Griffiths PG, Gorman GS, Lourenco CM, Wright AF, Auer-Grumbach M, et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain (2010) 133:771–86. doi:10.1093/brain/awq007
59. Puomila A, Huoponen K, Mäntyjärvi M, Hämäläinen P, Paananen R, Sankila EM, et al. Dominant optic atrophy: correlation between clinical and molecular genetic studies. Acta Ophthalmol Scand (2005) 83:337–46. doi:10.1111/j.1600-0420.2005.00448.x
60. Nakamura M, Yamamoto M. Variable pattern of visual recovery of Leber’s hereditary optic neuropathy. Br J Ophthalmol (2000) 84:534–5. doi:10.1136/bjo.84.5.534
61. Barboni P, Valentino ML, La Morgia C, Carbonelli M, Savini G, De Negri A, et al. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain (2013) 136:e231. doi:10.1093/brain/aws280
62. Cornille K, Milea D, Amati-Bonneau P, Procaccio V, Zazoun L, Guillet V, et al. Reversible optic neuropathy with OPA1 exon 5b mutation. Ann Neurol (2008) 63:667–71. doi:10.1002/ana.21376
63. Altpeter EK, Blanke BR, Leo-Kottler B, Nguyen XN, Trauzettel-Klosinski S. Evaluation of fixation pattern and reading ability in patients with Leber hereditary optic neuropathy. J Neuroophthalmol (2013) 33:344–8. doi:10.1097/WNO.0b013e31829d1f5b
64. Pfeffer G, Horvath R, Klopstock T, Mootha VK, Suomalainen A, Koene S, et al. New treatments for mitochondrial disease-no time to drop our standards. Nat Rev Neurol (2013) 9:474–81. doi:10.1038/nrneurol.2013.129
65. Klopstock T, Yu-Wai-Man P, Dimitriadis K, Rouleau J, Heck S, Bailie M, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain (2011) 134:2677–86. doi:10.1093/brain/awr170
66. Geromel V, Darin N, Chrétien D, Bénit P, DeLonlay P, Rötig A, et al. Coenzyme Q(10) and idebenone in the therapy of respiratory chain diseases: rationale and comparative benefits. Mol Genet Metab (2002) 77:21–30. doi:10.1016/S1096-7192(02)00145-2
67. Rudolph G, Dimitriadis K, Büchner B, Heck S, Al-Tamami J, Seidensticker F, et al. Effects of idebenone on color vision in patients with leber hereditary optic neuropathy. J Neuroophthalmol (2013) 33:30–6. doi:10.1097/WNO.0b013e318272c643
68. Zanna C, Ghelli A, Porcelli AM, Karbowski M, Youle RJ, Schimpf S, et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain (2008) 131:352–67. doi:10.1093/brain/awm335
69. Sadun AA, Chicani CF, Ross-Cisneros FN, Barboni P, Thoolen M, Shrader WD, et al. Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch Neurol (2012) 69:331–8. doi:10.1001/archneurol.2011.2972
70. Giordano C, Iommarini L, Giordano L, Maresca A, Pisano A, Valentino ML, et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain (2014) 137:335–53. doi:10.1093/brain/awt343
71. Wenz T, Williams SL, Bacman SR, Moraes CT. Emerging therapeutic approaches to mitochondrial diseases. Dev Disabil Res Rev (2010) 16:219–29. doi:10.1002/ddrr.109
72. Guy J, Qi X, Pallotti F, Schon EA, Manfredi G, Carelli V, et al. Rescue of a mitochondrial deficiency causing Leber hereditary optic neuropathy. Ann Neurol (2002) 52:534–42. doi:10.1002/ana.10354
73. Bonnet C, Augustin S, Ellouze S, Bénit P, Bouaita A, Rustin P, et al. The optimized allotopic expression of ND1 or ND4 genes restores respiratory chain complex I activity in fibroblasts harboring mutations in these genes. Biochim Biophys Acta (2008) 1783:1707–17. doi:10.1016/j.bbamcr.2008.04.018
74. Ellouze S, Augustin S, Bouaita A, Bonnet C, Simonutti M, Forster V, et al. Optimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunction. Am J Hum Genet (2008) 83:373–87. doi:10.1016/j.ajhg.2008.08.013
75. Guy J, Qi X, Koilkonda RD, Arguello T, Chou TH, Ruggeri M, et al. Efficiency and safety of AAV-mediated gene delivery of the human ND4 complex I subunit in the mouse visual system. Invest Ophthalmol Vis Sci (2009) 50:4205–14. doi:10.1167/iovs.08-3214
76. Perales-Clemente E, Fernández-Silva P, Acín-Pérez R, Pérez-Martos A, Enríquez JA. Allotopic expression of mitochondrial-encoded genes in mammals: achieved goal, undemonstrated mechanism or impossible task? Nucleic Acids Res (2011) 39:225–34. doi:10.1093/nar/gkq769
77. Yu H, Koilkonda RD, Chou TH, Porciatti V, Ozdemir SS, Chiodo V, et al. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc Natl Acad Sci U S A (2012) 109:E1238–47. doi:10.1073/pnas.1119577109
78. Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr, Mingozzi F, Bennicelli J, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med (2008) 358:2240–8. doi:10.1056/NEJMoa0802315
79. Testa F, Maguire AM, Rossi S, Pierce EA, Melillo P, Marshall K, et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital amaurosis type 2. Ophthalmology (2013) 120:1283–91. doi:10.1016/j.ophtha.2012.11.048
Keywords: optic nerve, LHON, OPA1, DOA, hereditary, optic atrophy, leber, mitochondria
Citation: La Morgia C, Carbonelli M, Barboni P, Sadun AA and Carelli V (2014) Medical management of hereditary optic neuropathies. Front. Neurol. 5:141. doi: 10.3389/fneur.2014.00141
Received: 23 May 2014; Accepted: 16 July 2014;
Published online: 31 July 2014.
Edited by:
Mohammad Hosein Nowroozzadeh, Shiraz University of Medical Sciences, IranCopyright: © 2014 La Morgia, Carbonelli, Barboni, Sadun and Carelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara La Morgia, UOC Clinica Neurologica, IRCCS Istituto delle Scienze Neurologiche di Bologna, Ospedale Bellaria, Via Altura, 3 40139 Bologna, Italy; Unità di Neurologia, Dipartimento di Scienze Biomediche e NeuroMotorie (DIBINEM), Università di Bologna, Bologna, Italy e-mail:Y2hpYXJhbGFtb3JnaWFAZ21haWwuY29t;Y2hpYXJhLmxhbW9yZ2lhQHVuaWJvLml0