
 Sean Robbins
Sean Robbins
- Department of Anesthesiology, Center for Translational Research in Neurodegenerative Disease, University of Florida, Gainesville, FL, USA
Preclinical stroke models provide insights into mechanisms of cellular injury and potential therapeutic targets. Renewed efforts to standardize preclinical practices and adopt more rigorous approaches reflect the assumption that a better class of compounds will translate into clinical efficacy. While the need for novel therapeutics is clear, it is also critical that diagnostics be improved to allow for more rapid treatment upon hospital admission. Advances in imaging techniques have aided in the diagnosis of stroke, yet current limitations and expenses demonstrate the need for new and complementary approaches. Intracerebral hemorrhage (ICH) exhibits the highest mortality rate, displays unique pathology and requires specialized treatment strategies relative to other forms of stroke. The aggressive nature and severe consequences of ICH underscore the need for novel therapeutic approaches as well as accurate and expeditious diagnostic tools. The use of experimental models will continue to aid in addressing these important issues as the field attempts to translate basic science findings into the clinical setting. Several preclinical models of ICH have been developed and are widely used to recapitulate human pathology. Because each model has limitations, the burden lies with the investigator to clearly define the question being asked and select the model system that is most relevant to that question. It may also be necessary to optimize and refine pre-existing paradigms, or generate new paradigms, as the future success of translational research is dependent upon the ability to mimic human sequelae and assess clinically relevant outcome measures as means to evaluate therapeutic efficacy.
Introduction
Preclinical studies are the foundation for drug development and the primary basis upon which to define mechanism of action, yet the success of animal studies is immaterial if efficacy is lost or toxicity occurs in patient populations. The field of central nervous research, most notably stroke, has been plagued by promising preclinical data for compounds that disappoint when applied in the clinical setting. The blame for these translational discrepancies has been assigned, in large part, to poor study design in both research sectors. For example, inappropriate statistical analyses, underpowered studies, experimenter bias, lack of validation studies, lack of comorbid conditions, and inappropriate cohort selection are just a few of the attributed causes (STAIR, 1999; STAIR-II, 2001; O’Collins et al., 2006; Fisher et al., 2009).
Over the past decade, resurgence in methodological inquiry has prompted detailed investigations into experimental design in both the basic science (STAIR, 1999; Dirnagl, 2010) and clinical research (STAIR-II, 2001) environments. Out of these inquiries arose several important realizations regarding the manner in which preclinical studies should be conducted, with a renewed emphasis on various aspects that ultimately relate to whether experimental paradigms resemble the human condition. Several important points have also been raised regarding methods to improve clinical trial design, though these will not be discussed here. While many of the guidelines and recommendations for preclinical studies were framed within the context of ischemic stroke, most are directly applicable to hemorrhagic stroke, including intracerebral hemorrhage (ICH).
A thoughtful approach to conducting the most rigorous, clinically relevant preclinical studies inevitably centers upon whether the models under scrutiny recapitulate aspects of the clinical pathology that ultimately reduce the quality of daily living in stroke patients. As such, the future success of novel therapeutics necessarily hinges upon the relevance of the models in which they are tested and the outcomes by which efficacy is assessed. With this in mind, it is clear that translating preclinical successes to patient populations is not untenable; rather, it requires careful consideration of experimental paradigms and methodological practices (MacLellan et al., 2012). The pages that follow highlight several aspects of clinical ICH pathology and identified risk factors that have been or should be incorporated into experimental studies. Future directions are then discussed within the contexts of developing diagnostic tools and modeling risk factors. Together, these approaches should improve the overall quality and translational value of preclinical ICH investigations.
Clinical Aspects of Intracerebral Hemorrhage
Stroke currently accounts for 73.7 billion dollars in annual medical costs and is the fourth leading cause of death in the United States. Approximately 13% of strokes occur through some type of hemorrhage, resulting in blood leakage into the brain parenchyma or subarachnoid space and subsequent injury by virtue of excessive cerebral pressure and various mechanisms of cellular toxicity. Although it constitutes the minority of stroke cases, hemorrhagic stroke claims the most lives with 30–40% mortality, half of which occurs within 2 days of onset (Roger et al., 2012). Several unique circumstances may lead to hemorrhage of a cerebral artery, including β-amyloid deposition and associated microbleeds (Hartz et al., 2012; Smith et al., 2012), brain trauma (Eisenberg et al., 1990; Servadei et al., 2002), hemorrhagic transformation following embolic stroke (Rosell et al., 2008; England et al., 2010), or physiological abnormalities in vessel stability and integrity. In all cases, the common denominator is weakened arterial vasculature that eventually ruptures.
The primary goals of experimental research are to (1) elucidate the biological mechanisms that mediate injury progression, and (2) identify, develop, and test novel therapies that can be applied in the clinical setting to improve patient outcomes. These goals are achieved primarily through use of experimental models that recapitulate certain aspects of ICH pathology, which may include mimicking vascular abnormalities, hematoma expansion, and/or inflammatory responses. Although ICH can result from many causes, some of which remain unknown, several aspects of ICH pathology have been well-characterized and demonstrated to account for a great many cases within the clinical setting.
Vascular Abnormalities
The most common vascular abnormalities associated with hemorrhagic stroke are aneurysms and arteriovenous malformations (AVMs), each of which can lead to ICH (Farhat, 2011) or subarachnoid hemorrhage (SAH) depending upon the location of the vessel. Furthermore, the size and location of the affected vessel is directly related to the severity of the injury, with larger, deeper-seated vessels producing greater devastation upon rupture.
Although aneurysms and AVMs both involve swelling of weakened vessels, cerebral aneurysms are often characterized as “ballooning” vessels. Aneurysms typically manifest after the age of 40, and are generally asymptomatic (0.5–3% result in bleeding) despite the fact that they account for the majority of SAH cases. AVMs are characterized by tangled arterial vessels that bypass the brain, consequently diverting oxygenated blood directly into the venous circulation. These atypical vessels dilate and weaken over time, and eventually succumb to high arterial blood pressure. In contrast to aneurysms, which develop over time and are typically located within the deep cerebral vasculature, AVMs are congenital (but are not generally considered heritable) and can develop anywhere within the brain or on its surface (modified from strokeassociation.org).
Upon considering the vast spectrum of causes, including varying degrees of brain trauma, vascular abnormalities, β-amyloid depositions, and embolic occlusions, it is no surprise that hemorrhage produces a wide range of neuropathies. Indeed, hemorrhagic insult can manifest as motor disturbances, speech impairments, emotional impulsivity, and/or learning and memory dysfunction. Within the general context of stroke, the most recent data indicates that the incidence of ICH is approximately 10% compared to 3% for SAH (Roger et al., 2012). Moreover, ICH is particularly devastating since blood extravasation into the brain causes cellular toxicity in addition to the accumulation of intracranial pressure.
In the preclinical setting, these factors weigh heavily upon the experimental models employed, the outcome measures assessed and the treatment modalities tested (these issues will be discussed at length in subsequent sections). As clinicians refine their treatment strategies using experience and the best available imaging techniques, basic scientists continue to search for novel therapies using the most clinically relevant models and methodological approaches. In spite of these efforts, a stroke occurs approximately every 40 s (strokeassociation.org), and therefore the most effective means to combat stroke in the immediate sense may be through identification of risk factors, lifestyle alterations, and early diagnosis upon admission to the clinic.
Plaque Deposition and Microbleeds
Deposition of plaques within the vascular lumen can weaken vascular integrity, resulting in leaky vessels and eventual rupture. Cerebral amyloid angiopathy (CAA), characterized by the deposition of β-amyloid fragments within the cerebral arterial vasculature, has been linked to hemorrhage (Smith and Greenberg, 2009; Vasilevko et al., 2010). This pathology increases over time in the elderly population and is predominantly localized to the posterior lobar regions of the brain (Vinters and Gilbert, 1983). These amyloid deposits produce vascular pathology, including alterations in vessel wall thickness and lumen diameter, that are associated with impaired cognitive function (Zekry et al., 2003) either in the presence or absence of Alzheimer Disease (Vinters and Gilbert, 1983).
The degree of CAA correlates with ICH, with microaneurysms resulting from excessive plaque loads (Vonsattel et al., 1991). Indeed, microhemorrhages are directly related to cognitive deficits associated with the accumulation of plaques and have been demonstrated to predict subsequent hemorrhagic episodes (Greenberg et al., 2004). Interestingly, diffusion-weighted magnetic resonance imaging (MRI) has demonstrated the ability to detect hyperintensities that represent subacute infarctions within the cerebral cortex and subcortical white matter of advanced CAA patients (Kimberly et al., 2009). While it stands to reason that the degree of subacute infarction would correlate with hemorrhage burden, it will be interesting to determine whether these “silent” infarctions (Smith et al., 2012) can provide any information regarding disease etiology or progression (for a comprehensive review of CAA, see Viswanathan and Greenberg, 2011).
Risk Factors and Diagnostics
Of the many stroke risk factors identified to date, it is perhaps most helpful to consider those which can be controlled as we seek more effective strategies to prevent ICH. For example, high blood pressure is the leading cause of stroke, and lowering blood pressure reduced the risk of hypertension-related ICH and CAA-related ICH (Arima et al., 2010). Diabetes, smoking, and high blood cholesterol are also major risk factors since these conditions are often accompanied by high blood pressure and buildup of atherosclerotic plaques. In particular, diabetes and hyperglycemia are associated with increased risk for hemorrhagic transformation in patients with ischemic stroke (Poppe et al., 2009; Kruyt et al., 2010). Because the hallmark of ICH is pressure-induced rupturing of weakened vessels, lifestyle alterations including increased exercise and adopting a healthier diet are relatively simple, yet effective, means of preventing atherosclerosis and controlling blood pressure. High blood pressure often occurs in conjunction with diabetes, which is also a major risk factor for ICH.
In spite of these preventative strategies, there are other inherent risks and uncontrollable factors associated with age, sex, race, and others (for current comprehensive data concerning risk factors, see the most recent report from the American Heart Association; Roger et al., 2012). Because risk for stroke can be partly dependent upon genetic factors and age, it is imperative to improve diagnostics and treatment strategies in addition to adopting preventative measures. Importantly, ICH exhibits unique characteristics that render the brain more susceptible to injury, as reflected by the high mortality rate relative to other stroke sub-types. For this reason, it is critically important to develop effective tools that can quickly identify cerebral hemorrhage, as well as circumstances that signify a substantial risk for ICH in patients suffering from other types of stroke.
In the current clinical setting, time is of the essence due to the narrow therapeutic window for thrombolytic therapy and the high mortality rate associated with ICH. Several diagnostic approaches are employed to eliminate confounding causes of stroke symptoms and accurately identify the type of stroke prior to the initiation of any therapy. This latter point is critical since ischemic and hemorrhagic strokes require distinct treatment modalities, and incorrect diagnosis may not only be ineffective but may actually worsen the outcome (i.e., administration of tissue plasminogen activator in cases of ICH). The initial patient evaluation most often includes a physical assessment, analysis of patient history, brain imaging, and other laboratory tests to determine eligibility for treatment (Adams et al., 2007). Although advances in diffusion-weighted MRI technology, novel imaging techniques, analysis, and interpretation have increased the accuracy of differentiating between ischemic and hemorrhagic stroke (Fiebach et al., 2002), many acute care facilities are not equipped to perform this testing and must rely on other means of analysis. CT scans, among various other techniques, are reasonably effective but imperfect in detecting cerebral hemorrhage, and unfortunately none of the available imaging techniques provide a prognosis for spontaneous hemorrhagic transformations or complications following thrombolytic therapy.
Due to the complexity of diagnosing stroke, the narrow therapeutic window for treatment and the necessity of differentiating between stroke sub-types, another approach that has gained popularity is the potential use of blood-borne biomarkers as diagnostic tools and prognostic indicators. Because there are clear advantages to this approach in terms of time and feasibility, the identification of a molecular signature may prove valuable as both a diagnostic tool and a prognostic indicator.
Biomarkers
One area of great interest in terms of diagnostic value (and effectiveness of treatments) is the identification of biomarkers. The major goal is to identify one or more molecules that signify ICH, or indicate high risk for ICH, that are present in the systemic circulation of stroke patients upon hospital admission. This would allow for point-of-care analysis that is relatively quick, non-invasive and accurate, and therefore could be implemented into the triage system for acute patients to expedite treatment. An example, though still arguable, of the potential for biomarkers is the detection of specific β-amyloid fragments in the plasma of patients displaying CAA-related ICH (Hernandez-Guillamon et al., 2012), and the hope is to define the molecular signature for ICH as well as other stroke sub-types.
In the case of hemorrhagic stroke, several potential biomarkers have emerged that may be useful for diagnosis of ICH and prediction of hemorrhagic transformation following thrombolytic therapy (for review, see Foerch et al., 2009). Astroglial-specific proteins have been deemed as candidates for acute phase ICH, presumably due to more rapid injury and blood brain barrier (BBB) disruption relative to less injurious forms of stroke. For example, plasma glial fibrillary acidic protein (GFAP) has been reported to be elevated in ICH patients upon admission relative to those classified with ischemic stroke or stroke mimics (Foerch et al., 2012), and pretreatment levels of S100β were shown to predict hemorrhagic complications following thrombolysis (Foerch et al., 2007). Several reports support the use of GFAP as a biomarker based on reliable detection of acute phase ICH (Foerch et al., 2006), ability to differentiate from ischemic stroke within 6 h of stroke onset (Dvorak et al., 2009) and the presence of cleaved GFAP byproducts, more specifically the detectable caspase-cleaved GFAP fragments proximal to plaque-associated vessels (Mouser et al., 2006).
Importantly, several studies have also validated the use of antibodies generated against protein breakdown products and cleavage fragments that are produced following brain injury. Some examples include antigens generated in response to calpain- and caspase-mediated cell death (Pineda et al., 2007; Mondello et al., 2010) and GFAP breakdown products (Papa et al., 2011) that are elevated in plasma from patients that suffered a traumatic brain injury. Other biomarkers indicative of BBB injury have shown promise in predicting hemorrhage following thrombolysis, including matrix metalloproteinase 9 (MMP-9; Montaner et al., 2003) and cellular fibronectin (Castellanos et al., 2004). However, a cautionary interpretation of these and other biomarker data is necessary until results are rigorously validated in multi-institutional, double-blinded studies including patients with various forms of CNS pathology. This is perhaps the crux of the caveat concerning biomarkers, and further research into the acute molecular mechanisms of ICH pathology is likely necessary to advance the field in this regard.
Recent efforts aimed at identifying novel proteins that are not “pan-CNS injury” markers may yield promising results, though the field continues to lack a consensus molecular signature for ICH. For example, an ELISA-based method for visinin like protein 1 differentiated between healthy subjects and ischemic stroke patients (Stejskal et al., 2011), although it is not clear whether this protein is elevated specifically in response to (ischemic) stroke or CNS injury in general. Pentraxin 3 was associated with higher mortality in ischemic stroke patients (Ryu et al., 2012), yet elevations in this protein are also associated with several cardiovascular risk factors. Since pentraxin 3 may represent atherosclerotic plaque burden rather than ischemic stroke per se, differentiation from plaque-associated ICH and hemorrhagic transformation would not be possible. Another study demonstrated that increased levels of brain natriuretic peptide correlated with functional outcomes and mortality at 3–6 months following cardioembolic stroke but not other sub-types (Kingwell, 2012), lending support to the notion that specificity for individual sub-types is possible.
Based upon the accumulated evidence to date, however, it is conceivable that the use of a single biomarker will not provide adequate specificity to discriminate between ICH and other stroke sub-types. This point has been recognized and addressed by studies using biomarker panels, with the goal of enhancing specificity by obtaining complementary data in the form of multiple protein levels. One such example is a panel consisting of MMP-9, S100β, BNP, and D-Dimer that showed promise by discriminating between stroke and stroke mimics (Rustici et al., 2011). It is not clear at this point whether this approach will be fruitful in improving diagnosis and stroke differentiation, although it is certainly an area worth pursuing. Improvements in future study design and additional validation studies will be important in determining whether specific biomarkers or biomarker panels possess the sensitivity and specificity to be effective in the clinical setting (Whiteley et al., 2008).
Preclinical Models of Intracerebral Hemorrhage
The use of animal models has been of great benefit for identifying mechanisms of injury and novel therapeutics. In recent years, however, the failure of candidate therapeutics in clinical stroke trials has placed additional pressure on basic scientists to develop better preclinical models that more closely mimic human conditions. There are several well-accepted models that are commonly utilized to recapitulate hemorrhagic stroke (Figure 1). Although there is no perfect model, each is effective in reproducing certain aspects of the clinical pathology (Table 1). What follows is a detailed comparison of the various preclinical models, with special attention afforded to those aspects which relate to translating findings to the clinical setting.
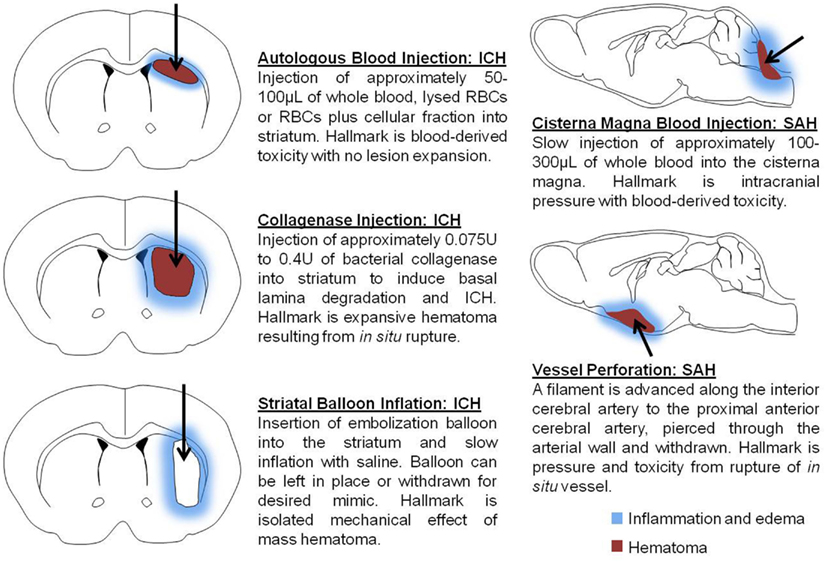
Figure 1. Schematic depicts models commonly used to mimic intracerebral hemorrhage (ICH) and subarachnoid hemorrhage (SAH). Autologous blood injection involves single- or double injection of autologous blood, or blood fractions, to generate a hematoma within the brain parenchyma. Collagenase injection is accomplished through the injection of recombinant bacterial collagenase to mimic blood extravasation following the rupture of cerebral arterial vasculature. Striatal balloon inflation is utilized to assess the mass hematoma effect on cellular injury and to evaluate the consequences of hematoma removal. Models of SAH produce blood accumulation in the subarachnoid space either by injection into the cistern magna or perforation of the anterior cerebral artery. Arrows represent injection route and demarcate the mass hematoma region.
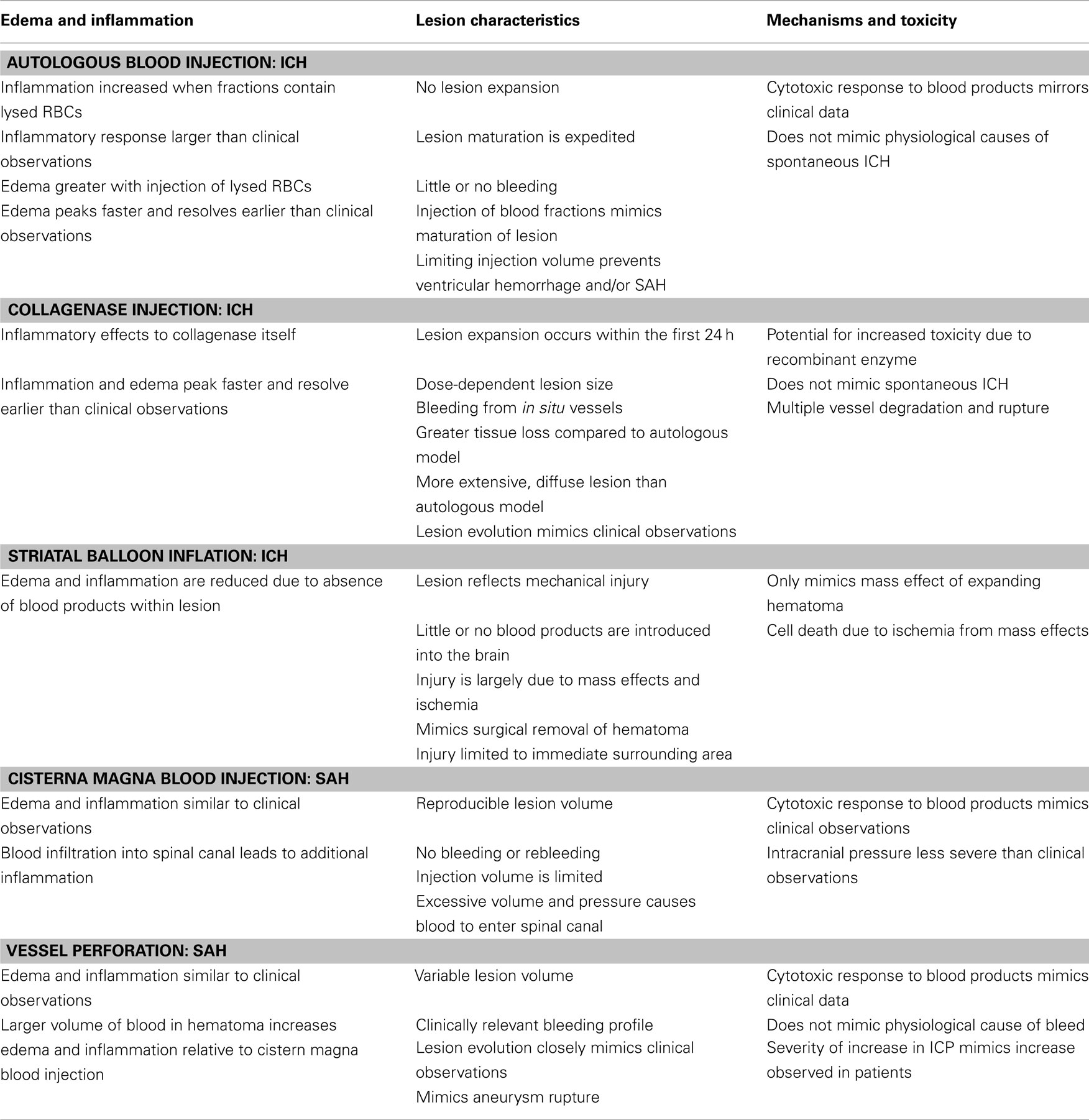
Table 1. Comparison of injury mechanisms and characteristics in experimental models of intracerebral and subarachnoid hemorrhage.
Collagenase Model
The collagenase model is one of the more common models of ICH utilized today. This model entails the injection of between 0.075 and 0.4 U of a bacterial collagenase into the brain, typically into the basal ganglia or striatum. The collagenases degrade the collagen found in the basal lamina of blood vessels, resulting in degraded vessels that become leaky and ultimately rupture (Rosenberg et al., 1990). The bleeding profile of experimental ICH begins with bleeding approximately 10 min after injection and progresses to hematoma between 4 and 24 h later. In this regard, the hematoma progression in the experimental condition mimics that which occurs in human ICH (Rosenberg et al., 1990), particularly concerning the expansion observed after initial formation and rebleeding (Wang and Tsirka, 2005).
The prescription of oral anticoagulant therapy is a common complication seen in the clinic since it impairs clotting and often exaggerates bleeding following ICH (Steiner et al., 2006; Foerch et al., 2008). Thus, the combined model of oral anticoagulant therapy and collagenase injection mimics this comorbidity for purposes of identifying ways to circumventing this clinical issue. The model itself involves the administration of some anticoagulant often prescribed to patients, e.g., warfarin, at therapeutic levels prior to the induction of ICH via collagenase injection.
Relevance to the clinical pathology
The collagenase model reproduces some of the pattern of bleeding from in situ vessels seen in the clinical presentation (Wang and Tsirka, 2005). The fact that the hemorrhage arises from in situ vessels patterns somewhat the spontaneous bleeding found in clinical ICH. However, because the bleeding is induced by an exogenous enzyme that degrades the vessels, this model is imperfect for investigations into the physiological causes of spontaneous ICH. An additional caveat is that the injection of collagenase leads to the breakdown of multiple vessels throughout the effected region, whereas clinical ICH often results from the rupture of a single vessel (MacLellan et al., 2008; Qureshi et al., 2009).
The hematoma progression in the collagenase model exhibits a profile similar to that observed in the clinical setting, with rebleeding and gradual expansion of the hematoma over the first 24 h (MacLellan et al., 2008; Liew et al., 2012). Administration of oral anticoagulant therapy in combination with ICH mimics the hemorrhagic response seen in patients that have been prescribed anticoagulants due to clotting impairments (Steiner et al., 2006). A caveat of the collagenase model is that the collagenase used to induce ICH is possibly toxic to other cell types in addition to the basal lamina, and thus may result in extraneous physiological changes that are not representative of the clinical pathology. Indeed, many believe that recombinant collagenase enzymes are responsible for heightened inflammation and cellular toxicity relative to the human condition, although this assertion has yet to be validated experimentally.
As such, while the collagenous model is useful for investigating mechanisms of neuronal and glial cell death and activation, findings should be interpreted with these limitations in mind (Xue and Del Bigio, 2003). In addition, the model has the potential to cause excessive damage and hemorrhage if the dose of collagenase is too large (Rosenberg et al., 1990). The inclusion of oral anticoagulant therapy into this ICH model translates quite well to the human condition despite the reliance upon artificial induction rather than spontaneous bleeding from an in situ vessel.
Autologous Blood Model
At present, the injection of autologous whole blood or blood fractions is one of the most common strategies used to recapitulate ICH and is one of the oldest models. This model entails collecting blood from a superficial artery, such as in the tail, and injecting it stereotactically into the brain (Nath et al., 1986). The most common target for injection is the striatum, as injections into the cortex can potentially precipitate SAH and further complicate the already intricate process of ICH (Xue and Del Bigio, 2000b). There are multiple ways to inject autologous blood into the brain; the injection can be composed of whole blood, lysed blood, or red blood cell (RBC) plus different cellular fractions (Xue and Del Bigio, 2000b; Hua et al., 2002). Administration of different cellular fractions allows for the investigation of the manner in which each cell type contributes to mechanisms of injury. Approximately 50–100 μL of whole blood is injected, typically at a rate of 5–10 μL per min. Use of a faster injection rate or larger volume tends to produce additional damage associated with the mechanical expansion and even intraventricular hemorrhage (IVH; Yang et al., 1994). Backflow of blood along the needle track can also lead to formation of IVH or SAH, which challenges reproducibility and translational value.
Relevance to the clinical pathology
Injection of whole blood into the brain translates to the clinic insofar as the physiological response seen; however, because the injection in this model is given as more or less a single bolus, there is little to no expansion of the hematoma as is observed with the collagenase model. The most significant factor is the failure of this model to produce bleeding from in situ vessels; with no bleeding from in situ vessels, there are no rebleeding events reminiscent of those observed in the clinic. One advantage of this model is the production of inflammation and toxicity that somewhat resembles clinical ICH (Xue and Del Bigio, 2000a), although it should be noted that the inflammation that occurs in both the collagenase model and the autologous blood model is significantly higher than that which has been detected in patients (Qureshi et al., 2009).
The act of injecting blood via a syringe at a fixed rate is often considerably slower and at lower pressures than bleeding from an in situ artery, necessarily limiting the translation of this model to the clinical setting. To address this issue, one strategy would be to inject the blood at a rate consistent with physiological arterial pressure to more closely mimic the mechanical force that produces ICH in patient populations (Nath et al., 1986). Autologous injection is also hindered by the limited blood volume that can be injected prior to excessive injury. Since the vast majority of clinical ICH results from vascular malformations (Qureshi et al., 2001), each of these injection models suffer from the limitation of no prior vascular pathology.
Injection of the different fractions of autologous blood leads to varied responses to the fraction injected (Huang et al., 2002; Zhao et al., 2009). Following ICH, RBCs begin to lyse soon after entering the brain parenchyma, releasing their intracellular contents. The injection of lysed blood is utilized to mimic the result of the breakdown of the blood after the initial hematoma formation, and simulate a more mature hematoma (Zhao et al., 2009). However, upon considering the inflammatory response, it should be noted that the injection of lysed RBCs leads to a marked increase in inflammation in the first 24 h after ICH. Injection of autologous blood represents a single bolus event of blood, and because there is no rebleeding events, the hematoma matures relatively fast and resolves faster than in the collagenase model. The collagenase model hematoma requires several weeks to mature, allows for a longer evaluation time and the maturation pertains more closely to the clinical model (MacLellan et al., 2008; Wang, 2010). The lesion size produced in the blood injection model can be variable due to backflow of the blood along the needle track or entrance of the blood into the ventricles or along the corpus callosum.
This result can be alleviated by employing the double injection model (Wang et al., 2008). The double injection model entails an initial injection of a small blood volume that coagulates along the needle track prior to a second, larger injection to introduce the hematoma. The coagulation of blood along the needle track alleviates the backflow of blood along the needle track and alleviates the need for incorporating heparin, as anticoagulant therapy has been shown to contribute to hematoma expansion (Flibotte et al., 2004), mortality (Cucchiara et al., 2008), and rebleeding events (Vermeer et al., 2002; Matute et al., 2012) in ICH patients. However, it should be noted that newer pharmacological agents that are currently under investigation may alleviate adverse outcomes associated with traditional therapies (for review, see Moussouttas, 2012).
Balloon Inflation Model
The balloon inflation model entails the insertion of an embolization balloon into the brain, most often in the same location used for the previous two models. The volume of the balloon and the length of inflation are easily reproduced. The resulting lesion is directly proportional to the size of the inflation, allowing for manipulation of injury severity. The most common volume injected into the balloon is approximately 300 μL, injected over the course of 1 min (Lopez Valdes et al., 2000). The balloon model is utilized to investigate the mass effect of the hematoma. In contrast to the injection ICH models, the balloon model produces injury that is purely mechanical and lacks introduction of blood or metabolites resulting from RBC lysis. Therefore, this model is valuable for studying hematoma mass effects in isolation since cell death results primarily from mechanisms initiated by ischemia alone (Lopez Valdes et al., 2000).
Relevance to the clinical pathology
The balloon model replicates the mass effects seen in the expansion of the hematoma in ICH, and is a valuable tool for investigating the effects of removal of the hematoma. This can be achieved simply by deflating and withdrawing the balloon from the skull. This is particularly relevant to clinical treatment strategies, as a significant portion of the patients presenting with ICH have some form of surgical intervention for hematoma removal (Lopez Valdes et al., 2000; Huang et al., 2002; Thai et al., 2006). However, since the balloon model is relatively limited in the scope of area affected, as there are no blood products introduced into the lesion, it is not possible to evaluate the blood-derived toxicity mechanisms involved in ICH. The absence of blood products in the lesion allows for investigation of the mechanisms of neuronal death in isolation. The solely mechanical nature of the injury in this model translates well to the clinical data of surgical intervention. However, findings should be interpreted considering the limitation that ICH injury represents a complex interplay of mass effects and deleterious cascades, many of which this model does not address.
Subarachnoid Hemorrhage Model
Currently, experimental modeling of SAH is achieved through either injection of blood into the cisternae magna or the perforation of intracranial arteries. Methodologically, the blood injection model is similar to the autologous blood injection model above with the exception of the specific location of injection. The perforation model is a modification of the intraluminal filament model used for middle cerebral artery occlusion in experimental ischemic stroke (Park et al., 2008). The perforation model entails the insertion of a filament to the proximal anterior cerebral artery (ACA), advancement through the wall of the ACA and subsequent withdrawal to induce SAH. A majority of SAH cases arise from the rupture of aneurysms, and while both models of SAH reproduce the accumulation of blood into the subarachnoid space, the perforation model more accurately mimics the hemorrhage itself (Park et al., 2008; Lee et al., 2009).
Relevance to the clinical pathology
The injection and perforation models are both effective means of investigating of the mechanisms of injury resulting from blood accumulation in the subarachnoid space. However, with the injection model, the blood often spreads into the spinal canal, a condition not observed in the clinic (Lee et al., 2009). Another caveat of the injection model is that the mechanism of hemorrhage induction does not mimic aneurysm rupture. Despite these issues, the injection model benefits from the fact that hemorrhage volume is controllable and highly reproducible experimentally. Between the two models, the perforation model most closely mimics the mechanisms and pattern of bleeding observed in the clinic. This method also resembles aneurysm rupture, with initial bleeding and rebleeding events leading to gradual evolution of the hematoma (Park et al., 2008), and is a more accurate mimic of the intracranial pressure associated with clinical ICH. However, a caveat that arises in the perforation model is the highly variable hemorrhage size observed experimentally. Therefore, although vessel perforation is effective in recapitulating some of the clinical observations, it can be problematic when evaluating experimental treatments due to difficulty in achieving reproducible hemorrhage volume (Lee et al., 2009).
Outcome Measures in Preclinical Models
In addition to the type of model employed, a major determinant of the future success of preclinical models, in terms of both experimental efficacy and translational value, is the evaluation of outcomes. Various measures have been used in the assessment of injury and therapeutic efficacy, encompassing elements of cellular injury, brain edema, and performance on tasks that require neurological and sensorimotor faculties. Recently there has been an added emphasis on multiple outcome measures that assess both histological and behavioral recovery, as the bench mark for efficacy appears to be shifting from histological to functional improvement.
Neurohistological Outcomes
Most recent studies have evaluated both functional and histological outcomes. Of the histological outcomes, the most commonly used have been hematoma volume, quantification of neuronal loss, and immunohistochemistry probing for various markers of inflammation and immune cell responsiveness (Xi et al., 2006; Kirkman et al., 2011). Certain patterns have emerged from these investigations. For example, inflammation and edema are greater in the collagenase and blood injection models when compared to the clinical data (Wasserman et al., 2008; Qureshi et al., 2009; Kirkman et al., 2011). The time course for edema and inflammation in experimental ICH is much faster than the onset and duration observed in patients with ICH (Xi et al., 1998; Xue and Del Bigio, 2000a, 2003; Huang et al., 2002). Additionally, MRI studies of the time course of infarct expansion reveal that only in the collagenase model does the lesion continue to mature for several weeks, while the blood injection model takes considerably less time to achieve complete hematoma expansion (MacLellan et al., 2010). These findings are particularly relevant since clinical data suggests that the hematoma takes several weeks to months to resolve without surgical intervention (Qureshi et al., 2009).
Between the two models, the collagenase model causes significantly greater loss of neurons compared to the autologous blood model (MacLellan et al., 2010). Hematoma expansion is a significant factor in human ICH and is present in most patients presenting with ICH. To date, the only model that appears to reproduce some clinical hematoma expansion is the collagenase model. However, the loss of tissue in the collagenase model is far greater than that seen in the autologous blood model. Additionally, damage caused by the needle is significant when using even the smallest needle (McCluskey et al., 2008), and therefore presents an additional caveat in the translation of injection models. Thus, the significant amount of damage caused by the needle can influence all other evaluated factors and must be taken into account when interpreting results from preclinical injection studies.
Secondary injury largely results from hematoma expansion. The contributing factors to this delayed injury phase include inflammation, cytotoxicity of blood products, oxidative stress, and infiltrating immune cells (Wang and Doré, 2007). In experimental ICH, edema, and inflammation are preceded by BBB breakdown, which leads to infiltration of peripheral immune cells and enhanced pro-inflammatory signaling (Aronowski and Zhao, 2011). The most common method of measuring edema is by determining the difference between the wet and dry weight of the brain, as weight increases proportionally with fluid extravasation. Edema peaks at approximately 3 days and resolves within several days (Xi et al., 2006). The time course of edema in the clinical setting is slightly different, peaking at approximately 14 days after ICH (Staykov et al., 2011). This temporal difference between the clinical data and the experimental data is due in part to differing amounts of white matter in rodents and humans (Adeoye et al., 2011).
The inflammatory response to ICH involves a complex interplay of resident immune cell activation, peripheral immune cell extravasation, and cellular signaling. The activation of resident microglia and astrocytes is accompanied by extravasation of neutrophils, peripheral macrophages, and RBCs into the brain parenchyma. Immune cells produce various cytokines and proteases (Power et al., 2003) that contribute to a pro-inflammatory microenvironment (Aronowski and Zhao, 2011). Although activated microglia and neutrophils play a key role in hematoma resolution, both cell types produce inflammatory mediators and reactive oxygen species (ROS) that may exacerbate injury following ICH. Elevations in oxidative stress and cytokine production occur within hours following ICH (Wang and Doré, 2007) and are generally considered to be indicators of inflammation. Oxidative stress results from ROS production by microglia, macrophages, and neutrophils, as well as the release of free iron from hemoglobin degradation (Aronowski and Zhao, 2011). TNFα and IL-1β are among the most common cytokines investigated, most likely due to their putative pro-inflammatory roles, although alterations in numerous other cytokines and chemokines have been documented (Wang and Doré, 2007).
The net effect of this immune cell response remains unknown, yet the involvement of these cells is considered by many to exacerbate ICH injury. At present, few studies have definitively shown more than an temporal association between activated immune cells, the release of inflammatory mediators and progressive injury. Nonetheless, immune cell activation is commonly utilized as an indicator of injury severity. This is generally accomplished through immunohistochemical quantification with antibodies generated against cell surface antigens selectively expressed by neutrophils (myeloperoxidase), microglia/macrophages (CD11b, ED1, and/or Iba1), and reactive astrocytes (GFAP). With regard to the detection of activated microglia/macrophages, cell surface antigen expression is assessed in conjunction with morphological analysis, as activated cells display an amoeboid morphology relative to the squamous or ramified phenotypes characteristic of resting cells. It should be noted that these cell surface antigens are differentially expressed on various immune cells depending upon the maturation and activation state, and there is currently no cell surface antigen that can be exploited for absolute identification of resident microglia relative to peripheral, extravasating monocytes/macrophages.
Because a multitude of cytokines, chemokines, and prostaglandins contribute to the overall inflammatory state of the cerebral microenvironment, it is likely that a combination of signals drives the neural sequelae. Therefore, future in vivo studies investigating these temporal response profiles in conjunction with mechanistic in vitro approaches may improve our general understanding of the ways in which this concerted signaling ultimately contributes to neural injury following ICH.
Functional Outcomes
While it is important to assess hematoma volume, expansion, and neuronal loss, most ICH patients have significant neurological and functional deficits that are often permanent (Cramer, 2008; Qureshi et al., 2009). Because the bench mark for preclinical efficacy involves restoration of function, behavioral outcomes are currently viewed as a critical component of determining the potential of novel therapies. The functional responses in experimental ICH mimic those seen in the clinic with some correlation to lesion size, location, and degree of deficit (Hua et al., 2002). The most common methods for evaluating functional outcomes involve a series of behavioral tests that are scored on a neurological scale. This scale allows for the calculation of a neurological deficit score (NDS) that can be used for relative comparisons of neurobehavioral impairment (Hua et al., 2002). Other testing modalities involve the assessment of ability to perform tasks that require motor coordination and/or cognition. For ICH, some of the most common behavioral tests currently used involve limb placement, balance, and circling. Functional recovery is most often examined relatively shortly after ICH, and most published studies have demonstrated increases in recovery of function in the behavioral tests and improvement in NDS. While this mimics the recovery of simple tasks in patients, it does not model long-term deficits (Hua et al., 2006).
One explanation for the quick recovery in the rodent model is the relatively small amount of white matter and resolution of edema (Fisher et al., 2009). Between some of the commonly used collagenase and autologous blood injection models, the time for neurological deficits to return to normal was longer in the collagenase model (MacLellan et al., 2008). However, behavioral tests that require more advanced sensorimotor functioning are preferable when attempting to mimic the human condition. Rodent studies have documented long-term behavioral deficits using common ICH models (≥1 month following ICH; Beray-Berthat et al., 2010), although these tests may not possess the sensitivity to detect differences in the severity of injury (MacLellan et al., 2006).
Despite their utility, behavioral tests, and NDS are poor predictors of hematoma size, with location having a more substantial impact on NDS. Importantly, none of the above tests address cognitive function in ICH. In fact, few experimental studies have rigorously examined the cognitive impact of ICH despite evidence that ICH patients experience cognitive deficits (Su et al., 2007; MacLellan et al., 2009). The paucity of reproducible cognitive assessments has been attributed, in part, to confounding interpretation of testing that involves mixed motor and cognitive components. However, mixed results within the literature are perhaps more problematic when attempting to resolve issues related to duration of deficit, type of deficit, and the degree of functional restoration that can be attributed to spontaneous recovery. These issues may be alleviated by future studies from multiple laboratories that have adopted standardized behavioral testing procedures. Indeed, a major drawback of behavioral testing in general, and particularly NDS scoring methods, is the subjective nature of assessment. Therefore, it will be imperative to adopt standardized testing procedures that employ more objective means of assessing functional outcomes.
Improving Translational Value
One of the major limitations of preclinical studies is the degree of external validity, which concerns whether results obtained from sample populations (and in this case, experimental models) can be predictive of those in patient populations. Because the experimental method is based heavily upon maximizing internal validity within a particular model or paradigm, attempts to control all variables other than that being investigated necessarily limit the generalizations that can be made regarding effects on heterogeneous populations.
Although this caveat is inherent and essentially unavoidable when attempting to address specific effects, the translational value of experimental studies can be improved by expanding the overall scope of investigations (Figure 2). Several reports have prescribed methods and strategies to improve the translational value of preclinical stroke models (STAIR, 1999; Fisher et al., 2009), including ICH (Kirkman et al., 2011; MacLellan et al., 2012). The following suggestions are specifically relevant to risk factors and diagnostic strategies that should be considered in future efforts to improve patient outcomes following ICH.

Figure 2. Diagram depicts the suggested strategy for improving translational studies using preclinical models. For testing novel therapeutics, initial experiments that do not show efficacy should be re-evaluated for methodological soundness, and in the absence of experimental flaws, these treatment strategies should be abandoned. Studies that demonstrate efficacy in single models, sexes, or species should be regarded as preliminary and subsequently validated with more rigorous, comparative studies. The suggested validation approach involves comparisons between sexes, distinct ICH models, aged animals, and comorbid conditions that are common in the clinical setting prior to testing in higher order species. Due to the heterogeneity of patient populations, mixed results from validation studies should be subjected to additional evaluations, when appropriate, for application to specific patient cohorts. Negative data should be reported in all instances to guide the field with future investigations.
Modeling Risk Factors
Several underlying risk factors have been identified for stroke, in general, and can be applied to ICH. However, in the case of hemorrhage, it will be of particular importance to identify factors that (1) may represent a unique or increased risk for ICH, and (2) can be incorporated into experimental models, thereby improving the likelihood of successful translation into the clinical setting. Although initial studies in young, healthy animals are preferred for initial screening of novel therapies, subsequent studies including comorbid conditions must be conducted to validate results prior to clinical application.
Hypertension and diabetes
In contrast to the many purported proximal causes of ICH injury, the incontrovertible physical cause of vascular rupture, arterial pressure, has not been modeled effectively at present. Because high blood pressure is one of the major risks for stroke (Roger et al., 2012) and is associated with hemorrhage following thrombolytic therapy (Butcher et al., 2010), future studies should incorporate strategies to mimic physiologically relevant blood pressure, as well as high blood pressure, and conduct comparative studies to assess outcomes and treatment efficacy.
The stroke-prone spontaneously hypertensive rat has been widely used to model high blood pressure, and demonstrates increased susceptibility to ICH (Wu et al., 2011). Although the phenotype may introduce additional caveats such as cardiac abnormalities and other unique features that contribute to stroke pathology (for review, see Bailey et al., 2011), modeling hemorrhage in this or other spontaneous models of hypertension-induced ICH (Iida et al., 2005) has some advantages. For example, hypertension is inherent in the species and/or induced through the diet, therefore there is no need to induce the phenotype with pharmaceutical agents that could confound interpretation of the results. Since cardiovascular disease is a systemic problem, these phenotypes are a closer representation of the comorbidity in patients compared to relatively healthy animals. The use of these models also allows for investigating interactions between novel treatments and currently prescribed medications, which could eliminate red herrings that show preclinical efficacy when administered alone but produce toxicity in combination with other pharmaceutical agents.
In light of potential caveats with hypertensive animals, another approach would be to employ the autologous injection model using a flow rate that more closely mimics physiological arterial pressure. However, it is conceivable that increased injection pressure could produce injury severity that exceeds that which is typically observed in clinical ICH. It is noteworthy that the autologous blood model lacks the hematoma expansion that is characteristically observed in patients, and therefore this approach may recapitulate the human pathology, similar to the collagenase model, while lacking the excessive inflammatory component of recombinant collagenase injection. Future investigations are needed to determine the feasibility and relevance of this approach.
Consistent with a role for exacerbating ICH, recent data showed that diabetic and hyperglycemic rats are more susceptible to hemorrhage and less responsive to thrombolytic therapy (Won et al., 2011; Fan et al., 2012). Similar effects have been reported in diabetic mouse models (Chen et al., 2011; Liu et al., 2011). At present, there are an increasing number of studies incorporating the diabetic comorbidity into preclinical ICH models. Continued investigations into this link will likely increase our understanding of the precipitating factors leading to cerebral hemorrhage.
Cerebral amyloid angiopathy
As in the patient population, amyloid deposition produces vascular pathology in animal models. The use of genetically modified mice will likely be important in gaining a better understanding of CAA progression, as shifting the production of different β-amyloid species influences the tissue localization and degree of deposition (Herzig et al., 2004). There are several mouse models that exhibit cerebral vascular pathology similar to that observed in CAA patients, including weakened cerebral vasculature, age-related plaque accumulation, microbleeding, and macrohemorrhage (Van Dorpe et al., 2000; Christie et al., 2001; Kimchi et al., 2001; Winkler et al., 2001). Transgenic studies have confirmed a role for the known risk factor epsilon4 apolipoprotein E allele (Fryer et al., 2003) and demonstrated efficacy in reducing plaque load and microhemorrhage by targeting ApoE interactions (Yang et al., 2011). These and other encouraging results (for review, see Viswanathan and Greenberg, 2011) highlight the potential of experimental models in advancing diagnosis and treatment of CAA-related ICH.
Biomarker Screening
A critically important issue for effective treatment is the ability to diagnose ICH accurately and expeditiously. Despite advancements in imaging techniques, the expenses associated with the most recent technologies and the requirement for preliminary evaluations will render many patients unable to benefit from imaging analyses. To increase the efficiency and scope of diagnostic testing, the most promising strategy may be the development of biomarker panels that can be used to screen patients for molecular signatures that define the type and/or severity of injury.
Because basic science research provides the foundation for mechanistic inquiry and benefits from greater experimental control relative to clinical studies, preclinical investigations should incorporate assessments of promising biomarkers in attempts to generate molecular pathological blueprints for the resulting sequelae. The use of panels, as opposed to single protein targets, is likely to be of greater benefit due to the multifactorial molecular cascades associated with various forms of neuropathology, including ICH. In the case of brain trauma, a major cause of ICH, such panels were developed (Mondello et al., 2011a) to assess a variety of brain-associated proteins that are elevated in plasma, including dendritic marker MAP-2, myelination marker MBP, neuronal marker NSE, gliosis marker GFAP, and neuronal ubiquitination marker UCH-L1 (Brophy et al., 2011; Mondello et al., 2011b). Animal studies should pursue these quantitative analyses with other brain- or vascular-associated proteins and continue the search for proteolytic cleavage products of brain antigens. In addition to identifying novel profiles for clinical screening, experimental investigations should incorporate assessments of biomarkers that have shown promise in the clinical setting, such as those mentioned, as means of validating the translational value of the models.
Rigorous methodological practices
Recommendations for good general practices in experimental ischemic stroke research methodology have been detailed by the Stroke Therapy Academic Industry Roundtable (STAIR, 1999; Fisher et al., 2009). These practices have also been endorsed in recent reviews by Kirkman et al. (2011) and MacLellan et al. (2012), and are consistent with the priorities described by the National Institute of Neurological Disorders and Stroke (NINDS, 2005) and the American Heart and Stroke Association. These prescriptions underscore the essential problem of experimental research, the quality of which ultimately hinges upon maximizing internal validity by evaluating the independent variable through experimental control of other confounding variables. In the laboratory, this method is critical to determine causality. In terms of external validity, however, it lacks the ability to assess other comorbid conditions that are important determinants of whether clinical efficacy can be achieved.
As a consequence, studies need to be expanded to include various groups that represent all of the conditions and comorbidities present within mixed patient populations. For example, sex differences need to be investigated by comparing male and female animals. In these investigations, attention should be paid to estrous cycle when evaluating mechanisms of injury and recovery. In treatment studies, physiological parameters need to be monitored to determine the mechanisms responsible for the observed effects, and whether the compounds produce adverse physiological responses (i.e., increased blood pressure) that would contraindicate use in specific patient populations. Due to the low body mass of rodents, acute changes in temperature and body weight should be monitored closely, as even slight changes can produce substantial alterations in physiology and behavior.
For assessment of efficacy, histological outcomes need to be measured in conjunction with behavioral batteries that include all aspects of clinical ICH pathology, including motor and cognitive disturbances, at both short- and long-term time points following ICH. When evaluating behavioral outcomes, it is also important to consider circadian rhythms. Because rodents exhibit a nocturnal active cycle, animals should be housed on a reverse light cycle to provide a more accurate measure of motor activity and cognition. Toxicity markers should be evaluated following treatment, and successful strategies should be validated in multiple models due to differences in the degree of hematoma expansion, neuroinflammation, and cellular injury (Xue and Del Bigio, 2003). Sample size calculations should be conducted to ensure adequate power to detect significant differences, and also to limit the potential for false positives that can result from inflated group sizes. When appropriate, animal exclusion should be reported, including the number of animals excluded, the cause for removal from the study, and the mortality rate resulting from surgical or other manipulations. Evaluations should also be conducted using all possible strategies to reduce experimenter bias, including random assignment to experimental groups, double-blinded outcome assessments, and a priori determinations of exclusion criteria.
As mentioned, the inclusion of phenotypes representative of prevalent comorbidities (diabetes, CAA) and risk factors (hypertension, age) are perhaps most crucial in improving the translational value of future studies involving ICH. The development of newer models that more closely mimic the underlying human pathology will also be of great benefit for future investigations. A recent study described a new transgenic mouse model in which animals develop AVMs that produced debilitating phenotypes, including severe motor disturbances and lethality (Milton et al., 2012). In this regard, the utilization of knockouts and transgenic animals, in conjunction with other recommendations, could provide substantial advancements in the field.
By combining genetically modified methods with clinically relevant phenotypes, we have recently demonstrated a link between prostaglandin receptor function and β-amyloid-mediated stroke injury (Zhen et al., 2011). Similar approaches will likely increase our knowledge of injury mechanisms, further address the selectivity of novel treatments and identify additional targets for therapeutic interventions. Investigations with aged animals following the completion of studies in young, healthy animals is one recommendation which, given the clinical scope of ICH, will be critical in translating effective preclinical results into clinical therapeutic efficacy. Importantly, this combined approach should maximize the potential of elucidating causal factors, such as the role of cerebral arterial β-amyloid deposition (Lee et al., 2012) in hemorrhage predisposition, and identifying biomarkers that can be applied to specific patient populations upon presentation in the clinic.
In conjunction with these approaches, future studies will benefit from the utilization of enhanced image technologies that allow for live, in vivo brain imaging. MRI was demonstrated to correlate with histological outcomes following ICH (Del Bigio et al., 1996). Preclinical models should attempt to incorporate this and other imaging techniques, as these methods would provide greater opportunity for correlating biomarkers with injury progression and/or treatment efficacy. Finally, validation studies in higher order species is necessary due to the limitations of rodent models. For example, autologous blood injection into the pig brain allows for more gradual hematoma development, greater reproducibility, and more closely mimics the human condition due to the greater abundance of white matter (Wagner et al., 1996). The International Classification of Functioning, Disability and Health (ICF) has also emphasized the importance of environmental factors that interact with biological processes to influence susceptibility and patient outcomes. These classifications incorporate various influences related to body function, biological structure, activities associated with daily living, access/utilization of various health care resources and other sociodemographic characteristics that are poorly modeled in rodents (Kucukdeveci et al., 2011; Quintas et al., 2012). Therefore, expanding the use of ICH models in higher order species will also provide more opportunities to assess factors that reflect psychosocial contributions to susceptibility and recovery.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (2RO1NS046400) and the NIH/National Center for Complementary and Alternative Medicine (1R21AT005246; R21 AT005085). Studies included within this manuscript were selected based on the quality of the work and the relevance to the topic, and this review was not meant to be all-inclusive.
References
Adams, H. P. Jr., Del Zoppo, G., Alberts, M. J., Bhatt, D. L., Brass, L., Furlan, A., Grubb, R. L., Higashida, R. T., Jauch, E. C., Kidwell, C., Lyden, P. D., Morgenstern, L. B., Qureshi, A. I., Rosenwasser, R. H., Scott, P. A., and Wijdicks, E. F. (2007). Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke 38, 1655–1711.
Adeoye, O., Clark, J. F., Kahatri, P., Wagner, K. R., Zuccarello, M., and Pyne-Geithman, G. J. (2011). Do current animal models of intracerebral hemorrhage mirror the human pathology? Transl. Stroke Res. 2, 17–25.
Arima, H., Tzourio, C., Anderson, C., Woodward, M., Bousser, M. G., Macmahon, S., Neal, B., and Chalmers, J. (2010). Effects of perindopril-based lowering of blood pressure on intracerebral hemorrhage related to amyloid angiopathy: the PROGRESS trial. Stroke 41, 394–396.
Aronowski, J., and Zhao, X. (2011). Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke 42, 1781–1786.
Bailey, E. L., Smith, C., Sudlow, C. L., and Wardlaw, J. M. (2011). Is the spontaneously hypertensive stroke prone rat a pertinent model of sub cortical ischemic stroke? A systematic review. Int. J. Stroke 6, 434–444.
Beray-Berthat, V., Delifer, C., Besson, V. C., Girgis, H., Coqueran, B., Plotkine, M., Marchand-Leroux, C., and Margaill, I. (2010). Long-term histological and behavioural characterisation of a collagenase-induced model of intracerebral haemorrhage in rats. J. Neurosci. Methods 191, 180–190.
Brophy, G. M., Mondello, S., Papa, L., Robicsek, S. A., Gabrielli, A., Tepas, J. III, Buki, A., Robertson, C., Tortella, F. C., Hayes, R. L., and Wang, K. K. (2011). Biokinetic analysis of ubiquitin C-terminal hydrolase-L1 (UCH-L1) in severe traumatic brain injury patient biofluids. J. Neurotrauma 28, 861–870.
Butcher, K., Christensen, S., Parsons, M., De Silva, D. A., Ebinger, M., Levi, C., Jeerakathil, T., Campbell, B. C., Barber, P. A., Bladin, C., Fink, J., Tress, B., Donnan, G. A., and Davis, S. M. (2010). Postthrombolysis blood pressure elevation is associated with hemorrhagic transformation. Stroke 41, 72–77.
Castellanos, M., Leira, R., Serena, J., Blanco, M., Pedraza, S., Castillo, J., and Davalos, A. (2004). Plasma cellular-fibronectin concentration predicts hemorrhagic transformation after thrombolytic therapy in acute ischemic stroke. Stroke 35, 1671–1676.
Chen, J., Cui, X., Zacharek, A., Cui, Y., Roberts, C., and Chopp, M. (2011). White matter damage and the effect of matrix metalloproteinases in type 2 diabetic mice after stroke. Stroke 42, 445–452.
Christie, R., Yamada, M., Moskowitz, M., and Hyman, B. (2001). Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am. J. Pathol. 158, 1065–1071.
Cramer, S. C. (2008). Repairing the human brain after stroke: I. Mechanisms of spontaneous recovery. Ann. Neurol. 63, 272–287.
Cucchiara, B., Messe, S., Sansing, L., Kasner, S., and Lyden, P. (2008). Hematoma growth in oral anticoagulant related intracerebral hemorrhage. Stroke 39, 2993–2996.
Del Bigio, M. R., Yan, H. J., Buist, R., and Peeling, J. (1996). Experimental intracerebral hemorrhage in rats. Magnetic resonance imaging and histopathological correlates. Stroke 27, 2312–2319; discussion 2319–2320.
Dirnagl, U. (2010). Standard operating procedures (SOP) in experimental stroke research: SOP for middle cerebral artery occlusion in the mouse. Nat. Precedings 19, 1–10.
Dvorak, F., Haberer, I., Sitzer, M., and Foerch, C. (2009). Characterisation of the diagnostic window of serum glial fibrillary acidic protein for the differentiation of intracerebral haemorrhage and ischaemic stroke. Cerebrovasc. Dis. 27, 37–41.
Eisenberg, H. M., Gary, H. E. Jr., Aldrich, E. F., Saydjari, C., Turner, B., Foulkes, M. A., Jane, J. A., Marmarou, A., Marshall, L. F., and Young, H. F. (1990). Initial CT findings in 753 patients with severe head injury. A report from the NIH Traumatic Coma Data Bank. J. Neurosurg. 73, 688–698.
England, T. J., Bath, P. M., Sare, G. M., Geeganage, C., Moulin, T., O’Neill, D., Woimant, F., Christensen, H., De Deyn, P., Leys, D., and Ringelstein, E. B. (2010). Asymptomatic hemorrhagic transformation of infarction and its relationship with functional outcome and stroke subtype: assessment from the Tinzaparin in Acute Ischaemic Stroke Trial. Stroke 41, 2834–2839.
Fan, X., Qiu, J., Yu, Z., Dai, H., Singhal, A. B., Lo, E. H., and Wang, X. (2012). A rat model of studying tissue-type plasminogen activator thrombolysis in ischemic stroke with diabetes. Stroke 43, 567–570.
Fiebach, J. B., Schellinger, P. D., Jansen, O., Meyer, M., Wilde, P., Bender, J., Schramm, P., Juttler, E., Oehler, J., Hartmann, M., Hahnel, S., Knauth, M., Hacke, W., and Sartor, K. (2002). CT and diffusion-weighted MR imaging in randomized order: diffusion-weighted imaging results in higher accuracy and lower interrater variability in the diagnosis of hyperacute ischemic stroke. Stroke 33, 2206–2210.
Fisher, M., Feuerstein, G., Howells, D. W., Hurn, P. D., Kent, T. A., Savitz, S. I., and Lo, E. H. (2009). Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke 40, 2244–2250.
Flibotte, J. J., Hagan, N., O’Donnell, J., Greenberg, S. M., and Rosand, J. (2004). Warfarin, hematoma expansion, and outcome of intracerebral hemorrhage. Neurology 63, 1059–1064.
Foerch, C., Arai, K., Jin, G., Park, K. P., Pallast, S., Van Leyen, K., and Lo, E. H. (2008). Experimental model of warfarin-associated intracerebral hemorrhage. Stroke 39, 3397–3404.
Foerch, C., Curdt, I., Yan, B., Dvorak, F., Hermans, M., Berkefeld, J., Raabe, A., Neumann-Haefelin, T., Steinmetz, H., and Sitzer, M. (2006). Serum glial fibrillary acidic protein as a biomarker for intracerebral haemorrhage in patients with acute stroke. J. Neurol. Neurosurg. Psychiatr. 77, 181–184.
Foerch, C., Montaner, J., Furie, K. L., Ning, M. M., and Lo, E. H. (2009). Invited article: searching for oracles? Blood biomarkers in acute stroke. Neurology 73, 393–399.
Foerch, C., Niessner, M., Back, T., Bauerle, M., De Marchis, G. M., Ferbert, A., Grehl, H., Hamann, G. F., Jacobs, A., Kastrup, A., Klimpe, S., Palm, F., Thomalla, G., Worthmann, H., and Sitzer, M. (2012). Diagnostic accuracy of plasma glial fibrillary acidic protein for differentiating intracerebral hemorrhage and cerebral ischemia in patients with symptoms of acute stroke. Clin. Chem. 58, 237–245.
Foerch, C., Wunderlich, M. T., Dvorak, F., Humpich, M., Kahles, T., Goertler, M., Alvarez-Sabin, J., Wallesch, C. W., Molina, C. A., Steinmetz, H., Sitzer, M., and Montaner, J. (2007). Elevated serum S100B levels indicate a higher risk of hemorrhagic transformation after thrombolytic therapy in acute stroke. Stroke 38, 2491–2495.
Fryer, J. D., Taylor, J. W., Demattos, R. B., Bales, K. R., Paul, S. M., Parsadanian, M., and Holtzman, D. M. (2003). Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J. Neurosci. 23, 7889–7896.
Greenberg, S. M., Eng, J. A., Ning, M., Smith, E. E., and Rosand, J. (2004). Hemorrhage burden predicts recurrent intracerebral hemorrhage after lobar hemorrhage. Stroke 35, 1415–1420.
Hartz, A. M., Bauer, B., Soldner, E. L., Wolf, A., Boy, S., Backhaus, R., Mihaljevic, I., Bogdahn, U., Klunemann, H. H., Schuierer, G., and Schlachetzki, F. (2012). Amyloid-beta contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke 43, 514–523.
Hernandez-Guillamon, M., Delgado, P., Penalba, A., Rodriguez-Luna, D., Molina, C. A., Rovira, A., Alvarez-Sabin, J., Boada, M., and Montaner, J. (2012). Plasma beta-amyloid levels in cerebral amyloid angiopathy-associated hemorrhagic stroke. Neurodegener. Dis. 10, 320–323.
Herzig, M. C., Winkler, D. T., Burgermeister, P., Pfeifer, M., Kohler, E., Schmidt, S. D., Danner, S., Abramowski, D., Sturchler-Pierrat, C., Burki, K., Van Duinen, S. G., Maat-Schieman, M. L., Staufenbiel, M., Mathews, P. M., and Jucker, M. (2004). Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 7, 954–960.
Hua, Y., Nakamura, T., Keep, R. F., Wu, J., Schallert, T., Hoff, J. T., and Xi, G. (2006). Long-term effects of experimental intracerebral hemorrhage: the role of iron. J. Neurosurg. 104, 305–312.
Hua, Y., Schallert, T., Keep, R. F., Wu, J., Hoff, J. T., and Xi, G. (2002). Behavioral tests after intracerebral hemorrhage in the rat. Stroke 33, 2478–2484.
Huang, F. P., Xi, G., Keep, R. F., Hua, Y., Nemoianu, A., and Hoff, J. T. (2002). Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J. Neurosurg. 96, 287–293.
Iida, S., Baumbach, G. L., Lavoie, J. L., Faraci, F. M., Sigmund, C. D., and Heistad, D. D. (2005). Spontaneous stroke in a genetic model of hypertension in mice. Stroke 36, 1253–1258.
Kimberly, W. T., Gilson, A., Rost, N. S., Rosand, J., Viswanathan, A., Smith, E. E., and Greenberg, S. M. (2009). Silent ischemic infarcts are associated with hemorrhage burden in cerebral amyloid angiopathy. Neurology 72, 1230–1235.
Kimchi, E. Y., Kajdasz, S., Bacskai, B. J., and Hyman, B. T. (2001). Analysis of cerebral amyloid angiopathy in a transgenic mouse model of Alzheimer disease using in vivo multiphoton microscopy. J. Neuropathol. Exp. Neurol. 60, 274–279.
Kingwell, K. (2012). Stroke: brain natriuretic peptide could be a biomarker for outcomes after cardioembolic stroke. Nat. Rev. Neurol. 8, 60.
Kirkman, M. A., Allan, S. M., and Parry-Jones, A. R. (2011). Experimental intracerebral hemorrhage: avoiding pitfalls in translational research. J. Cereb. Blood Flow Metab. 31, 2135–2151.
Kruyt, N. D., Biessels, G. J., Devries, J. H., and Roos, Y. B. (2010). Hyperglycemia in acute ischemic stroke: pathophysiology and clinical management. Nat. Rev. Neurol. 6, 145–155.
Kucukdeveci, A. A., Tennant, A., Grimby, G., and Franchignoni, F. (2011). Strategies for assessment and outcome measurement in physical and rehabilitation medicine: an educational review. J. Rehabil. Med. 43, 661–672.
Lee, J. H., Bacskai, B. J., and Ayata, C. (2012). Genetic animal models of cerebral vasculopathies. Prog. Mol. Biol. Transl. Sci. 105, 25–55.
Lee, J. Y., Sagher, O., Keep, R., Hua, Y., and Xi, G. (2009). Comparison of experimental rat models of early brain injury after subarachnoid hemorrhage. Neurosurgery 65, 331–343; discussion 343.
Liew, H. K., Hsu, C. W., Wang, M. J., Kuo, J. S., Li, T. Y., Peng, H. F., Wang, J. Y., and Pang, C. Y. (2012). Therapeutic benefit of urocortin in rats with intracerebral hemorrhage. J. Neurosurg. 116, 193–200.
Liu, J., Gao, B. B., Clermont, A. C., Blair, P., Chilcote, T. J., Sinha, S., Flaumenhaft, R., and Feener, E. P. (2011). Hyperglycemia-induced cerebral hematoma expansion is mediated by plasma kallikrein. Nat. Med. 17, 206–210.
Lopez Valdes, E., Hernandez Lain, A., Calandre, L., Grau, M., Cabello, A., and Gomez-Escalonilla, C. (2000). Time window for clinical effectiveness of mass evacuation in a rat balloon model mimicking an intraparenchymatous hematoma. J. Neurol. Sci. 174, 40–46.
MacLellan, C. L., Auriat, A. M., Mcgie, S. C., Yan, R. H., Huynh, H. D., De Butte, M. F., and Colbourne, F. (2006). Gauging recovery after hemorrhagic stroke in rats: implications for cytoprotection studies. J. Cereb. Blood Flow Metab. 26, 1031–1042.
MacLellan, C. L., Langdon, K. D., Churchill, K. P., Granter-Button, S., and Corbett, D. (2009). Assessing cognitive function after intracerebral hemorrhage in rats. Behav. Brain Res. 198, 321–328.
MacLellan, C. L., Paquette, R., and Colbourne, F. (2012). A critical appraisal of experimental intracerebral hemorrhage research. J. Cereb. Blood Flow Metab. 32, 612–627.
MacLellan, C. L., Silasi, G., Auriat, A. M., and Colbourne, F. (2010). Rodent models of intracerebral hemorrhage. Stroke 41, S95–S98.
MacLellan, C. L., Silasi, G., Poon, C. C., Edmundson, C. L., Buist, R., Peeling, J., and Colbourne, F. (2008). Intracerebral hemorrhage models in rat: comparing collagenase to blood infusion. J. Cereb. Blood Flow Metab. 28, 516–525.
Matute, M. C., Masjuan, J., Egido, J. A., Fuentes, B., Simal, P., Diaz-Otero, F., Reig, G., Diez-Tejedor, E., Gil-Nunez, A., Vivancos, J., and Alonso De Lecinana, M. (2012). Safety and outcomes following thrombolytic treatment in stroke patients who had received prior treatment with anticoagulants. Cerebrovasc. Dis. 33, 231–239.
McCluskey, L., Campbell, S., Anthony, D., and Allan, S. M. (2008). Inflammatory responses in the rat brain in response to different methods of intra-cerebral administration. J. Neuroimmunol. 194, 27–33.
Milton, I., Ouyang, D., Allen, C. J., Yanasak, N. E., Gossage, J. R., Alleyne, C. H. Jr., and Seki, T. (2012). Age-dependent lethality in novel transgenic mouse models of central nervous system arteriovenous malformations. Stroke 43, 1432–1435.
Mondello, S., Muller, U., Jeromin, A., Streeter, J., Hayes, R. L., and Wang, K. K. (2011a). Blood-based diagnostics of traumatic brain injuries. Expert Rev. Mol. Diagn. 11, 65–78.
Mondello, S., Papa, L., Buki, A., Bullock, M. R., Czeiter, E., Tortella, F. C., Wang, K. K., and Hayes, R. L. (2011b). Neuronal and glial markers are differently associated with computed tomography findings and outcome in patients with severe traumatic brain injury: a case control study. Crit. Care 15, R156.
Mondello, S., Robicsek, S. A., Gabrielli, A., Brophy, G. M., Papa, L., Tepas, J., Robertson, C., Buki, A., Scharf, D., Jixiang, M., Akinyi, L., Muller, U., Wang, K. K., and Hayes, R. L. (2010). AlphaII-spectrin breakdown products (SBDPs): diagnosis and outcome in severe traumatic brain injury patients. J. Neurotrauma 27, 1203–1213.
Montaner, J., Molina, C. A., Monasterio, J., Abilleira, S., Arenillas, J. F., Ribo, M., Quintana, M., and Alvarez-Sabin, J. (2003). Matrix metalloproteinase-9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation 107, 598–603.
Mouser, P. E., Head, E., Ha, K. H., and Rohn, T. T. (2006). Caspase-mediated cleavage of glial fibrillary acidic protein within degenerating astrocytes of the Alzheimer’s disease brain. Am. J. Pathol. 168, 936–946.
Moussouttas, M. (2012). Challenges and controversies in the medical management of primary and antithrombotic-related intracerebral hemorrhage. Ther. Adv. Neurol. Disord. 5, 43–56.
Nath, F. P., Jenkins, A., Mendelow, A. D., Graham, D. I., and Teasdale, G. M. (1986). Early hemodynamic changes in experimental intracerebral hemorrhage. J. Neurosurg. 65, 697–703.
NINDS. (2005). Priorities for clinical research in intracerebral hemorrhage: report from a National Institute of Neurological Disorders and Stroke workshop. Stroke 36, e23–e41.
O’Collins, V. E., Macleod, M. R., Donnan, G. A., Horky, L. L., Van Der Worp, B. H., and Howells, D. W. (2006). 1,026 experimental treatments in acute stroke. Ann. Neurol. 59, 467–477.
Papa, L., Lewis, L. M., Falk, J. L., Zhang, Z., Silvestri, S., Giordano, P., Brophy, G. M., Demery, J. A., Dixit, N. K., Ferguson, I., Liu, M. C., Mo, J., Akinyi, L., Schmid, K., Mondello, S., Robertson, C. S., Tortella, F. C., Hayes, R. L., and Wang, K. K. (2011). Elevated levels of serum glial fibrillary acidic protein breakdown products in mild and moderate traumatic brain injury are associated with intracranial lesions and neurosurgical intervention. Ann. Emerg. Med. doi:10.1016/j.annemergmed.2011.08.021. [Epub ahead of print].
Park, I. S., Meno, J. R., Witt, C. E., Suttle, T. K., Chowdhary, A., Nguyen, T. S., Ngai, A. C., and Britz, G. W. (2008). Subarachnoid hemorrhage model in the rat: modification of the endovascular filament model. J. Neurosci. Methods 172, 195–200.
Pineda, J. A., Lewis, S. B., Valadka, A. B., Papa, L., Hannay, H. J., Heaton, S. C., Demery, J. A., Liu, M. C., Aikman, J. M., Akle, V., Brophy, G. M., Tepas, J. J., Wang, K. K., Robertson, C. S., and Hayes, R. L. (2007). Clinical significance of alphaII-spectrin breakdown products in cerebrospinal fluid after severe traumatic brain injury. J. Neurotrauma 24, 354–366.
Poppe, A. Y., Majumdar, S. R., Jeerakathil, T., Ghali, W., Buchan, A. M., and Hill, M. D. (2009). Admission hyperglycemia predicts a worse outcome in stroke patients treated with intravenous thrombolysis. Diabetes Care 32, 617–622.
Power, C., Henry, S., Del Bigio, M. R., Larsen, P. H., Corbett, D., Imai, Y., Yong, V. W., and Peeling, J. (2003). Intracerebral hemorrhage induces macrophage activation and matrix metalloproteinases. Ann. Neurol. 53, 731–742.
Quintas, R., Cerniauskaite, M., Ajovalasit, D., Sattin, D., Boncoraglio, G., Parati, E. A., and Leonardi, M. (2012). Describing functioning, disability, and health with the international classification of functioning, disability, and health brief core set for stroke. Am. J. Phys. Med. Rehabil. 91, S14–S21.
Qureshi, A. I., Mendelow, A. D., and Hanley, D. F. (2009). Intracerebral haemorrhage. Lancet 373, 1632–1644.
Qureshi, A. I., Suri, M. F., Ling, G. S., Khan, J., Guterman, L. R., and Hopkins, L. N. (2001). Absence of early proinflammatory cytokine expression in experimental intracerebral hemorrhage. Neurosurgery 49, 416–420; discussion 421.
Roger, V. L., Go, A. S., Lloyd-Jones, D. M., Benjamin, E. J., Berry, J. D., Borden, W. B., Bravata, D. M., Dai, S., Ford, E. S., Fox, C. S., Fullerton, H. J., Gillespie, C., Hailpern, S. M., Heit, J. A., Howard, V. J., Kissela, B. M., Kittner, S. J., Lackland, D. T., Lichtman, J. H., Lisabeth, L. D., Makuc, D. M., Marcus, G. M., Marelli, A., Matchar, D. B., Moy, C. S., Mozaffarian, D., Mussolino, M. E., Nichol, G., Paynter, N. P., Soliman, E. Z., Sorlie, P. D., Sotoodehnia, N., Turan, T. N., Virani, S. S., Wong, N. D., Woo, D., and Turner, M. B. (2012). Heart disease and stroke statistics – 2012 update: a report from the American Heart Association. Circulation 125, e2–e220.
Rosell, A., Foerch, C., Murata, Y., and Lo, E. H. (2008). Mechanisms and markers for hemorrhagic transformation after stroke. Acta Neurochir. Suppl. 105, 173–178.
Rosenberg, G. A., Mun-Bryce, S., Wesley, M., and Kornfeld, M. (1990). Collagenase-induced intracerebral hemorrhage in rats. Stroke 21, 801–807.
Rustici, M. C., Chiappini, E., Salvadori, M., Sollai, S., Galli, L., and De Martino, M. (2011). Clinical usefulness of the semiquantitative procalcitonin test in the diagnosis of bacterial infections in a third level children’s hospital. Clin. Lab. 57, 497–506.
Ryu, W. S., Kim, C. K., Kim, B. J., Kim, C., Lee, S. H., and Yoon, B. W. (2012). Pentraxin 3: a novel and independent prognostic marker in ischemic stroke. Atherosclerosis 220, 581–586.
Servadei, F., Murray, G. D., Teasdale, G. M., Dearden, M., Iannotti, F., Lapierre, F., Maas, A. J., Karimi, A., Ohman, J., Persson, L., Stocchetti, N., Trojanowski, T., and Unterberg, A. (2002). Traumatic subarachnoid hemorrhage: demographic and clinical study of 750 patients from the European brain injury consortium survey of head injuries. Neurosurgery 50, 261–267; discussion 267–269.
Smith, E. E., and Greenberg, S. M. (2009). Beta-amyloid, blood vessels, and brain function. Stroke 40, 2601–2606.
Smith, E. E., Schneider, J. A., Wardlaw, J. M., and Greenberg, S. M. (2012). Cerebral microinfarcts: the invisible lesions. Lancet Neurol. 11, 272–282.
STAIR. (1999). Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 30, 2752–2758.
STAIR-II. (2001). Recommendations for clinical trial evaluation of acute stroke therapies. Stroke 32, 1598–1606.
Staykov, D., Wagner, I., Volbers, B., Hauer, E. M., Doerfler, A., Schwab, S., and Bardutzky, J. (2011). Natural course of perihemorrhagic edema after intracerebral hemorrhage. Stroke 42, 2625–2629.
Steiner, T., Rosand, J., and Diringer, M. (2006). Intracerebral hemorrhage associated with oral anticoagulant therapy: current practices and unresolved questions. Stroke 37, 256–262.
Stejskal, D., Sporova, L., Svestak, M., and Karpisek, M. (2011). Determination of serum visinin like protein-1 and its potential for the diagnosis of brain injury due to the stroke – a pilot study. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. Repub. 155, 263–268.
Su, C. Y., Chen, H. M., Kwan, A. L., Lin, Y. H., and Guo, N. W. (2007). Neuropsychological impairment after hemorrhagic stroke in basal ganglia. Arch. Clin. Neuropsychol. 22, 465–474.
Thai, Q. A., Pradilla, G., Legnani, F. G., Kretzer, R. M., Hsu, W., and Tamargo, R. J. (2006). Lysis of intracerebral hematoma with stereotactically implanted tissue plasminogen activator polymers in a rabbit model. J. Neurosurg. 105, 424–429.
Van Dorpe, J., Smeijers, L., Dewachter, I., Nuyens, D., Spittaels, K., Van Den Haute, C., Mercken, M., Moechars, D., Laenen, I., Kuiperi, C., Bruynseels, K., Tesseur, I., Loos, R., Vanderstichele, H., Checler, F., Sciot, R., and Van Leuven, F. (2000). Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the London mutant of human APP in neurons. Am. J. Pathol. 157, 1283–1298.
Vasilevko, V., Passos, G. F., Quiring, D., Head, E., Kim, R. C., Fisher, M., and Cribbs, D. H. (2010). Aging and cerebrovascular dysfunction: contribution of hypertension, cerebral amyloid angiopathy, and immunotherapy. Ann. N. Y. Acad. Sci. 1207, 58–70.
Vermeer, S. E., Algra, A., Franke, C. L., Koudstaal, P. J., and Rinkel, G. J. (2002). Long-term prognosis after recovery from primary intracerebral hemorrhage. Neurology 59, 205–209.
Vinters, H. V., and Gilbert, J. J. (1983). Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 14, 924–928.
Viswanathan, A., and Greenberg, S. M. (2011). Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 70, 871–880.
Vonsattel, J. P., Myers, R. H., Hedley-Whyte, E. T., Ropper, A. H., Bird, E. D., and Richardson, E. P. Jr. (1991). Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann. Neurol. 30, 637–649.
Wagner, K. R., Xi, G., Hua, Y., Kleinholz, M., De Courten-Myers, G. M., Myers, R. E., Broderick, J. P., and Brott, T. G. (1996). Lobar intracerebral hemorrhage model in pigs: rapid edema development in perihematomal white matter. Stroke 27, 490–497.
Wang, J. (2010). Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog. Neurobiol. 92, 463–477.
Wang, J., and Doré, S. (2007). Inflammation after intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 27, 894–908.
Wang, J., Fields, J., and Dore, S. (2008). The development of an improved preclinical mouse model of intracerebral hemorrhage using double infusion of autologous whole blood. Brain Res. 1222, 214–221.
Wang, J., and Tsirka, S. E. (2005). Tuftsin fragment 1-3 is beneficial when delivered after the induction of intracerebral hemorrhage. Stroke 36, 613–618.
Wasserman, J. K., Yang, H., and Schlichter, L. C. (2008). Glial responses, neuron death and lesion resolution after intracerebral hemorrhage in young vs. aged rats. Eur. J. Neurosci. 28, 1316–1328.
Whiteley, W., Tseng, M. C., and Sandercock, P. (2008). Blood biomarkers in the diagnosis of ischemic stroke: a systematic review. Stroke 39, 2902–2909.
Winkler, D. T., Bondolfi, L., Herzig, M. C., Jann, L., Calhoun, M. E., Wiederhold, K. H., Tolnay, M., Staufenbiel, M., and Jucker, M. (2001). Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J. Neurosci. 21, 1619–1627.
Won, S. J., Tang, X. N., Suh, S. W., Yenari, M. A., and Swanson, R. A. (2011). Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by Increasing superoxide production. Ann. Neurol. 70, 583–590.
Wu, G., Bao, X., Xi, G., Keep, R. F., Thompson, B. G., and Hua, Y. (2011). Brain injury after intracerebral hemorrhage in spontaneously hypertensive rats. J. Neurosurg. 114, 1805–1811.
Xi, G., Keep, R. F., and Hoff, J. T. (1998). Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J. Neurosurg. 89, 991–996.
Xi, G., Keep, R. F., and Hoff, J. T. (2006). Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 5, 53–63.
Xue, M., and Del Bigio, M. R. (2000a). Intracerebral injection of autologous whole blood in rats: time course of inflammation and cell death. Neurosci. Lett. 283, 230–232.
Xue, M., and Del Bigio, M. R. (2000b). Intracortical hemorrhage injury in rats: relationship between blood fractions and brain cell death. Stroke 31, 1721–1727.
Xue, M., and Del Bigio, M. R. (2003). Comparison of brain cell death and inflammatory reaction in three models of intracerebral hemorrhage in adult rats. J. Stroke Cerebrovasc. Dis. 12, 152–159.
Yang, G. Y., Betz, A. L., Chenevert, T. L., Brunberg, J. A., and Hoff, J. T. (1994). Experimental intracerebral hemorrhage: relationship between brain edema, blood flow, and blood-brain barrier permeability in rats. J. Neurosurg. 81, 93–102.
Yang, J., Ji, Y., Mehta, P., Bates, K. A., Sun, Y., and Wisniewski, T. (2011). Blocking the apolipoprotein E/amyloid-beta interaction reduces fibrillar vascular amyloid deposition and cerebral microhemorrhages in TgSwDI mice. J. Alzheimers Dis. 24, 269–285.
Zekry, D., Duyckaerts, C., Belmin, J., Geoffre, C., Moulias, R., and Hauw, J. J. (2003). Cerebral amyloid angiopathy in the elderly: vessel walls changes and relationship with dementia. Acta Neuropathol. 106, 367–373.
Zhao, X., Song, S., Sun, G., Strong, R., Zhang, J., Grotta, J. C., and Aronowski, J. (2009). Neuroprotective role of haptoglobin after intracerebral hemorrhage. J. Neurosci. 29, 15819–15827.
Zhen, G., Kim, Y. T., Li, R. C., Yocum, J., Kapoor, N., Langer, J., Dobrowolski, P., Maruyama, T., Narumiya, S., and Dore, S. (2011). PGE(2) EP1 receptor exacerbated neurotoxicity in a mouse model of cerebral ischemia and Alzheimer’s disease. Neurobiol. Aging. doi: 10.1016/j.neurobiolaging.2011.09.017. [Epub ahead of print].
Keywords: behavior, CNS, inflammation, ischemia, preclinical, rodent, stroke, therapy
Citation: Leonardo CC, Robbins S and Doré S (2012) Translating basic science research to clinical application: models and strategies for intracerebral hemorrhage. Front. Neur. 3:85. doi: 10.3389/fneur.2012.00085
Received: 28 February 2012; Paper pending published: 26 March 2012;
Accepted: 07 May 2012; Published online: 29 May 2012.
Edited by:
Steven M. Greenberg, Harvard Medical School, USAReviewed by:
Craig S. Anderson, University of Sydney, AustraliaRu-Lan Hsieh, Shin Kong Wu Ho-Su Memorial Hospital, Taiwan
Copyright: © 2012 Leonardo, Robbins and Doré. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Sylvain Doré, Department of Anesthesiology, Center for Translational Research in Neurodegenerative Disease, University of Florida College of Medicine, 1275 Center Drive, Gainesville, FL 32610, USA. e-mail:c2RvcmVAdWZsLmVkdQ==