
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 17 December 2010
Sec. Spinal Cord Medicine
Volume 1 - 2010 | https://doi.org/10.3389/fneur.2010.00149
 Angshuman Mukherjee
Angshuman Mukherjee Ambar Chakravarty*
Ambar Chakravarty*
Spasticity, a classical clinical manifestation of an upper motor neuron lesion, has been traditionally and physiologically defined as a velocity dependent increase in muscle tone caused by the increased excitability of the muscle stretch reflex. Clinically spasticity manifests as an increased resistance offered by muscles to passive stretching (lengthening) and is often associated with other commonly observed phenomenon like clasp-knife phenomenon, increased tendon reflexes, clonus, and flexor and extensor spasms. The key to the increased excitability of the muscle stretch reflex (muscle tone) is the abnormal activity of muscle spindles which have an intricate relation with the innervations of the extrafusal muscle fibers at the spinal level (feed-back and feed-forward circuits) which are under influence of the supraspinal pathways (inhibitory and facilitatory). The reflex hyperexcitability develops over variable period of time following the primary lesion (brain or spinal cord) and involves adaptation in spinal neuronal circuitries caudal to the lesion. It is highly likely that in humans, reduction of spinal inhibitory mechanisms (in particular that of disynaptic reciprocal inhibition) is involved. While simply speaking the increased muscle stretch reflex may be assumed to be due to an altered balance between the innervations of intra and extrafusal fibers in a muscle caused by loss of inhibitory supraspinal control, the delayed onset after lesion and the frequent reduction in reflex excitability over time, suggest plastic changes in the central nervous system following brain or spinal lesion. It seems highly likely that multiple mechanisms are operative in causation of human spasticity, many of which still remain to be fully elucidated. This will be apparent from the variable mechanisms of actions of anti-spasticity agents used in clinical practice.
Spasticity is a common phenomenon seen in neurologic disorders that result in loss of mobility and may produce pain due to muscle spasms. It is a state of sustained increase in tone of a muscle when it is passively lengthened.
In simple terms of clinical neurology, spasticity is defined as increased resistance to passive movement due to a lowered threshold of tonic and phasic stretch reflexes (Burke et al., 1972).
Physiologically spasticity is defined as a motor disorder characterized by a velocity dependent increase in the tonic stretch reflexes (muscle tone) with exaggerated tendon jerks, resulting from hyperexcitability of the stretch reflexes as one component of the upper motor neuron (UMN) syndrome (Lance, 1980). The velocity dependent increase in resistance to passive stretch often melts suddenly resulting in clasp-knife phenomenon. The definition of spasticity was further elaborated by addition of several features of spastic paresis to form a more comprehensive picture of UMN syndrome which are described below (Young, 1989; Delwaide and Gerard, 1993).
1. In a patient with spasticity, brisk tendon jerks sometimes accompanied by clonus and velocity dependent muscle hypertonia to stretch preferentially affecting certain muscle groups, are the effects of a combination of hyperexcitability of an afferent pathway to motor neurons and disturbed processing of other peripheral afferent pathways at the spinal cord level.
2. In spasticity, other positive symptoms or signs such as flexor (or extensor) spasm, clasp knife phenomenon. Babinski sign, exaggerated cutaneous withdrawal (flexor, pain) reflexes, autonomic hyperflexia, dystonia, and contractures may limit voluntary movement and cause discomfort.
3. In addition to the above features, several negative features are also included in spastic states such as paresis, lack of dexterity and fatigability.
In the pathophysiology of spasticity and spastic paretic syndrome there are two broad categories of inter-related influencing mechanisms namely:
1. Spinal mechanism concerning changes in the functioning of the spinal neurons and motor subsystems.
2. Supraspinal and suprasegmental mechanisms.
Before discussing spinal mechanisms of spasticity. “Motor control system” and “Motor functions of the spinal cord”, are summarized below.
This system has the following components
1. Cerebral cortex as a whole is essential for sending analytical and command motor signals for execution through:
a. Frontal motor area forming corticospinal (pyramidal) pathways.
b. Premotor and supplementary motor cortices which are important for programming, i.e., sequencing and modulation of all voluntary movements.
c. Prefrontal cortex projecting to premotor and supplementary motor areas and help by planning and initiation of willed activity.
d. Parietal cortical areas (5,7) which are important for guidance of movement.
e. Association areas acting through conscious (visual, tactile, auditory) or unconscious (proprioceptive) informations also guide motor system.
2. Subcortical centers – basal ganglia (striatum, pallidum, substantial nigra, subthalamic nucleus) and cerebellum are important for maintenance of tone, posture, and co-ordination of movement.
3. Brainstem is the major relay station which is active through its nuclei specially pons and medullary reticular nuclei, vestibular, and red nuclei on muscle stretch reflexes, posture, reflex, and repetitive movements.
4. Spinal cord – contains final common pathways for motor execution and active through its specialized neuronal circuits and motor subsystems. It involves:
a. Motor unit – consisting of a motor neuron and all the muscles it innervates, which is the functional module of motor control system. Alpha motoneurons are the final common pathway for the skeletal muscle activity.
b. Spinal cord reflexes – enhance the ability of motor control system for coordinated motor activity.
These include:
i. Cutaneous reflex – like withdrawal (flexor pain nociceptive) reflex.
ii. Muscle reflex – stretch reflex.
Motor functions are basically dependent on the following factors:
1. Muscle receptors and muscle stretch reflexes:
Muscle function is dependent on excitation of anterior horn motoneurons and continuous sensory feedback from each muscle to the spinal cord regarding its length and tension. Muscle spindles consisting of specialized intrafusal muscle fibers act as receptors to send information of muscle length or rate of change of length. Golgi tendon organs transmit information about tendon tension or rate of change of tension. Two types of sensory endings are found in receptor area of muscle spindle – primary (group Ia afferent fiber) and secondary (group II afferent fiber) (Figure 1) Golgi tendon organs send information through group Ib afferent fibers. Large alpha efferent fibers innervate extrafusal skeletal muscle fibers and small gamma efferent fibers innervate intrafusal (spindle) fibers.
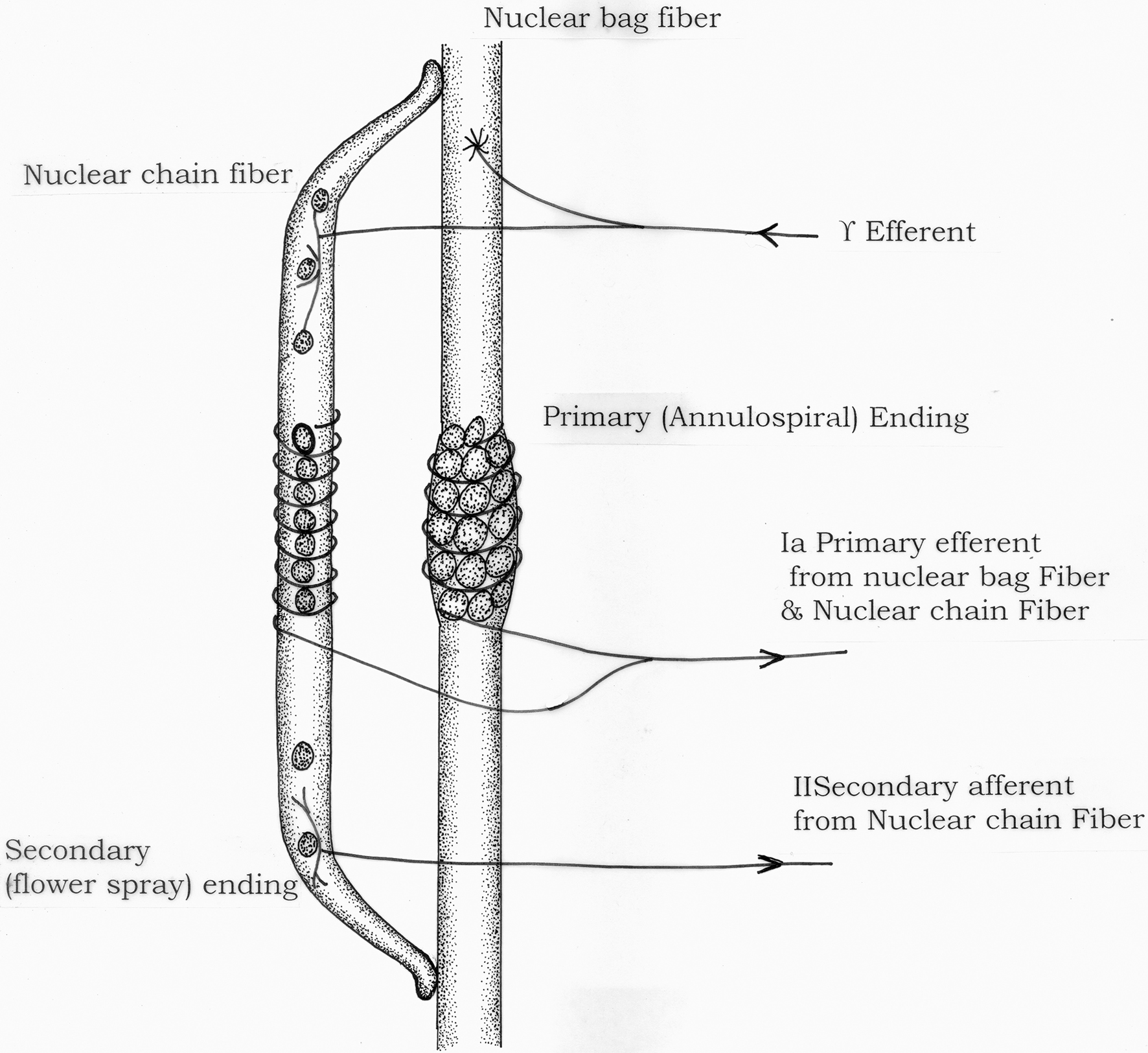
Figure 1. Diagrammatic representation of muscle spindle.
Muscle stretch reflex (myotatic reflex) is the function of the muscle spindle. Whenever a muscle is stretched, the excited spindles cause reflex contraction of the same muscle and also the synergistic muscles. “Dynamic stretch reflex” is caused by rapid stretch of the muscle and elicited through potent stimulation primarily by la afferent fibers from nuclear bag in spindle through monosynaptic pathway. Dynamic response is over within fraction of a second when a weaker static stretch reflex continues for a prolonged period. Static reflex is mediated by nuclear chain fires (mainly group II afferent and also some group la afferent fibers) acting through interneurons in the cord, i.e., polysynaptically.
Muscle tone is generated by muscle spindles by acting through the stretch reflex. Muscle tone is the constant muscular activity that is necessary as a background to actual movement in order to maintain the basic attitude of the body particularly against the force of gravity (Carpenter, 1984). As tone opposes movement and tends to keep muscles at preset lengths, it has to be changed in steps during a movement. Gamma fibers are ideally suited for this and whenever a command is sent to alpha motor fibers, gamma fibers are also excited. There occurs alpha–gamma co-activation to produce contraction of both extrafusal and intrafusal fibers according to the position and force commands from the brain to the spinal cord.
Clinical elicitation of stretch reflex is done in two ways :
a. Static – by passive stretching (tone testing).
b. Dynamic – by muscle and tendon jerks.
Clonus occurs when the dynamic stretch reflex is highly sensitized and facilitated. The dynamic response dies out within a fraction of a second to elicit a new cycle and in this way the muscle contraction (e.g., gastrocnemius) oscillates for a long period to produce clonus.
2. Interneurons: Most integrative functions in the spinal cord are mediated by interneurons. Interneurons which are involved in every segmental and stretch reflex pathways are excited or inhibited by several peripheral and descending fiber systems (Lundberg, 1979).
Interneuron systems involved in the stretch reflex arc and in the pathophysiology of spasticity are discussed below.
1. Renshaw cells and recurrent inhibition:
Renshaw cells are situated in lamina VII of ventral horn medial to motoneurons. Collateral from an alpha motoneuron axon excites Renshaw cell which in turn inhibits the same and also other motoneuron innervating the synergistic muscles. This alpha motoneuron-Renshaw cell – alpha motoneuron pathway of inhibition forms a negative feedback circuit to control motoneuron excitation and is called recurrent inhibition (Pompetano, 1984). In addition, Renshaw cells inhibit gamma motoneurons and la inhibitory interneurons (Jankowska and Roberts, 1972).
2. Reciprocal la inhibition:
Stretch of a muscle activates la afferent fires to produce monosynaptic excitation of homonymous alpha motoneurons. There occurs in addition disynaptic inhibition of alpha motoneurons innervating antagonist muscles (reciprocal inhibition). It is now established that la interneurons receive the same diverse excitatory and inhibitory inputs from segmental afferents (e.g., flexor afferents) and supraspinal descending tracts as are received by alpha motoneurons (Hultborn et al., 1976). These inputs excite alpha motoneurons to contract synergistic muscles and also excite la inhibitory interneurons to inhibit in turn alpha motoneurons to antagonistic muscles during stretch reflex activity.
Clinical electrophysiological studies by H – reflex demonstrating this phenomenon had been performed early by Misra and Pandey (1994) in Neurolathyrism – a pure motor spastic tropical paraparesis caused by ingestion of grass peas containing the neurotoxin beta – ODAP. These authors demonstrated increased motoneuron excitability, altered transmission in the premotoneuronal pathway and lack of reciprocal inhibition in the generation of spasticity in this condition. More recently, Crone et al. (2007) also demonstrated reduced reciprocal inhibition in neurolathyrism.
3. Inhibition from group II afferents:
In addition to established role of group II fibers in stretch reflex arc, these fibers from secondary spindle endings are known to produce flexion reflex by exciting flexor alpha motoneurons and inhibiting extensor motoneurons.
4. Non-reciprocal lb inhibition:
1b afferent fibers from Golgi tendon organs end on lb inhibitory interneurons which synapse with alpha motoneurons to both homonymous and heteronymous muscles. Like Renshaw cell and la inhibitory interneurons, lb interneurons also receive diverse segmental and supraspinal inputs. Therefore, lb inhibition is not a simple autogenic inhibitory safety mechanism to regulate muscle tension only. It is a part of complex system regulating muscle tension to control posture and movement.
5. Presynaptic inhibition:
The amplitude of EPSP generated in a motoneuron in response to la afferent stimulation diminishes if there occurs prior depolarization of this la afferent fiber through axo–axonic synapse with a specific interneuron. The specific interneurons involved in this process of presynaptic inhibition are also controlled by descending pathways. This permits automatic suppression of unimportant afferent informations (Schmidt, 1971).
6. Flexor reflex afferents
Nociceptive reflex or simply pain reflex produces contraction of flexor muscles of a limb (withdrawal) and crossed extensor reflex of opposite limb. This is mediated by polysynaptic connection between flexor reflex afferents (FRA), interneurons and motoneurons of extensor as well as flexor muscles.
1. Increased fusimotor drive: Exaggerated muscle stretch reflexes in spasticity was attributed to the increased sensitivity of the muscle spindles due to increased fusimotor activity some 20–30 years ago. The posterior root section for treatment of spasticity in cerebral palsy and diluted procaine injection near intramuscular nerve for treating hyperactive stretch reflex (Rushworth, 1960) were based on this theory. The local anesthetic injection was assumed to block small diameter fusimotor fibers but not larger diameter alpha motor axons. Later experiments using microneurography studies (Hagbarth, 1981) failed to demonstrate any change in the discharge of muscle spindle afferents in spastic patients making it unlikely that any significant changes in fusimotor drive exist. The hypothesis that increased fusimotor outflow is involved in the pathophysiology of spasticity has consequently been discredited.
2. Primary hyperexcitability of alpha motoneurons following spinal lesions – plateau potentials: Recent research has shown that several active membrane properties can shape the motoneuronal output (Rekling et al., 2000; Powers and Binder, 2001; Heckman et al., 2003; Hultborn et al., 2004; Heckmann et al., 2005). Voltage dependent, persistent inward Ca2+ and Na+ currents are of particular relevance, as they amplify and prolong the response of motoneurons to synaptic excitation. These inward currents can produce prolonged depolarizations (plateau potentials) when opposing outward currents are reduced or the Ca2+ channels are facilitated, e.g., by serotonergic and noradrenergic innervations of the motoneurons.
When a graded depolarizing current is introduced through an intracellular electrode into a motoneuron of a decerebrate cat, a critical threshold (plateau threshold) is reached. Above this threshold, further depolarization will trigger a regenerative activation of sustained inward current. In the decerebrate cat (with tonic descending serotonergic drive) the plateau potentials are easily evoked. However, following an acute spinal transaction they cannot be evoked unless the persistent inward current is specifically increased, e.g., by monoaminergic agonist. In a few cases, it was possible to demonstrate that plateau potentials can again be induced in the chronic spinal state without adding any neurotransmitter precursors or agonists (Feganel and Dumtrijevic, 1982). This suggested that plateau potentials, returning long after spinal injury, can play a role in the pathophysiology of spasticity. Little is known about the possible contribution of plateau potentials to the development of spasticity in humans because of the difficulty in demonstrating the existence of such intrinsic membrane properties in the intact organism.
3. Enhanced cutaneous reflexes: In spasticity, cutaneous reflexes (flexor or withdrawal) are enhanced. Dorsal horn neurons give rise to both long axons which form ascending tracts and short propriospinal axons to innervate motor neurons of cord. Rostral lesions in CNS disrupting descending reticulospinal tract (RST) or spinothalamic tract alter normal gating mechanisms in dorsal horn so that pain is experienced to rather innocuous stimuli. This mishandling of segmental inputs helped by failure of presynaptic inhibition (mediated through GABA-ergic synapses on primary afferents in substantia gelatinosa) results in hyperactivity in long tract neurons to be felt as pain as an associated feature in spasticity.
Similarly excitation of short propriospinal interneuron system in the cord produces hyperactive nociceptive reflexes. This system acts as an arousal system for motoneurons in absence of brainstem reticular system in a cord deprived of supraspinal influences (Burke and Ashby, 1972). The clinical signs of this phenomenon include Babinski’s response, triple flexion of leg and gross flexor, or sometimes extensor spasm which may be produced by simple and non-noxious cutaneous stimuli.
Current hypotheses stress more on alterations in inhibitory mechanisms in spinal neuronal circuitry than an excitatory processes although both may be inter-related in a patient with spasticity.
Figure 2 illustrates the spinal reflex circuits that could be involved in the development of spasticity. The monosynaptic Ia excitation which underlies the dynamic and tonic components of the stretch reflex may be inhibited by various spinal reflexes pathways.
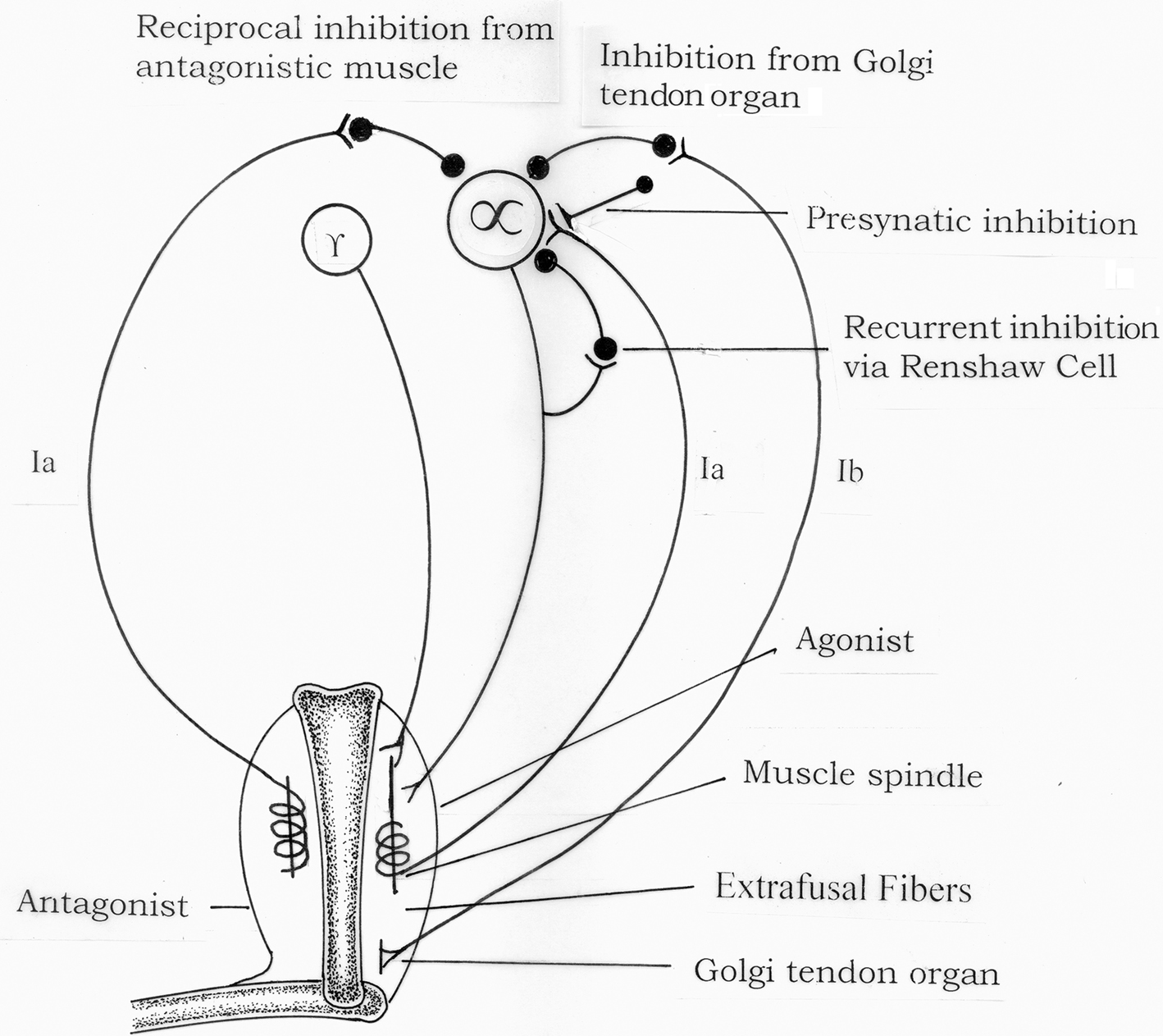
Figure 2. Spinal pathways which may be responsible for development of spasticity.
These include:
1. Presynaptic inhibition of Ia afferent terminals.
2. Disynaptic reciproval Ia inhibition from muscle spindle Ia afferents from the antagonist muscles.
3. Recurrent inhibition via motor axon collaterals and Renshaw cells.
4. Non-reciprocal Ib inhibition from Golgi tendon organs.
5. Inhibition from muscle spindle group II afferents (not shown in the Figure 3).
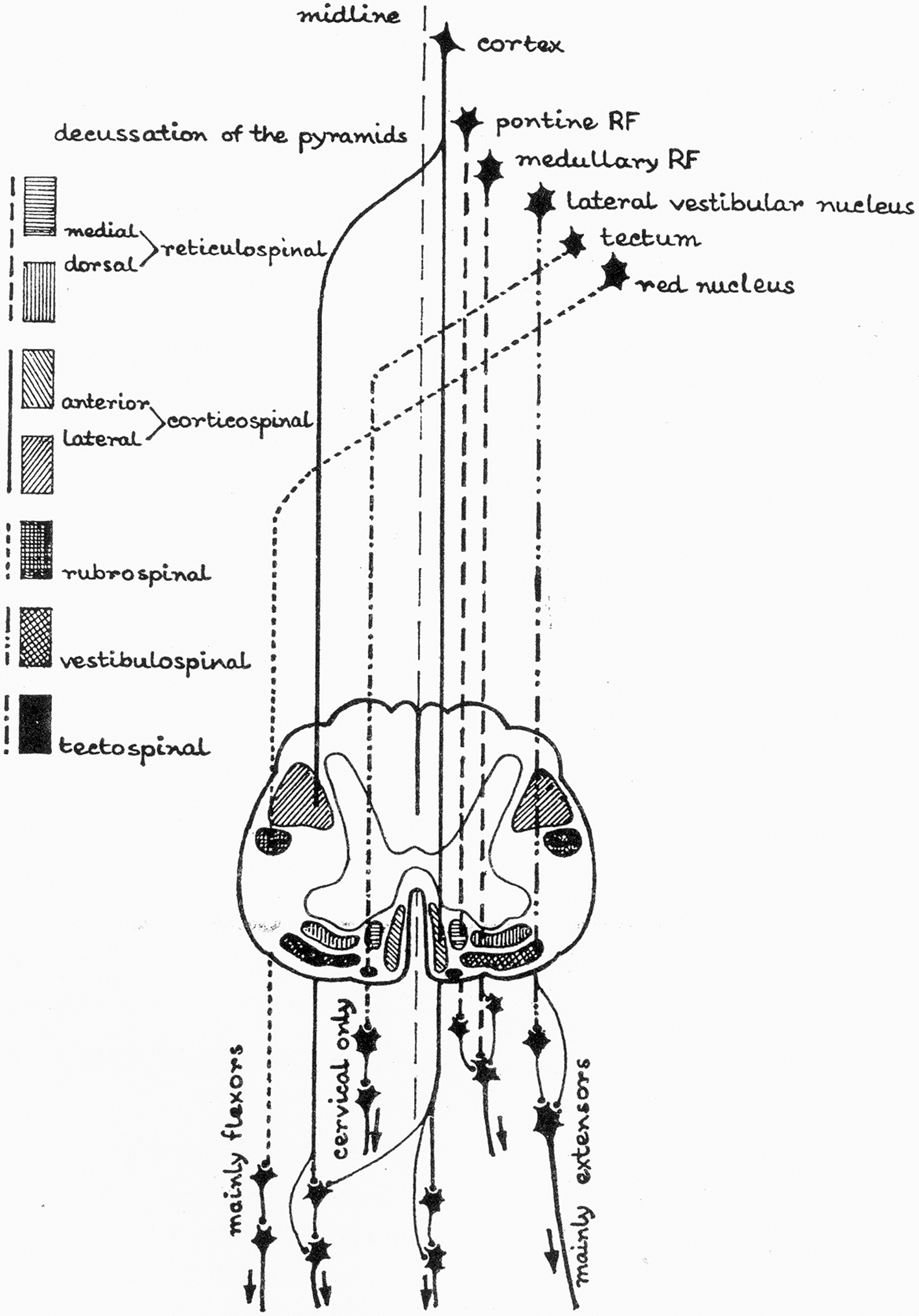
Figure 3. Supraspinal descending pathways in spinal cord (RF, reticular formation).
The changes in reflex transmission in these pathways may depend both on an altered supraspinal drive (if any remains) and on secondary changes at cellular level in the spinal cord below the lesion which may include:
1. Presynaptic inhibition of Ia afferent terminals:
As discussed before, the inhibition is through axo–axonic synapses which are GABA-ergic and on activation reduces the amount of transmitter released by Ia terminals on the motoneuron. If there occurs a reduction in the normally maintained tonic level of presynaptic inhibition, there will be increased response on alpha motoneurons by Ia input and spasticity may ensure.
A technique used to study presynaptic inhibition in human subjects was to vibrate the Achilles tendon and record the resulting depression of the soleus H-reflex (Burke and Ashby, 1972; Ashby et al., 1974). As this vibratory inhibition was subsequently found to be decreased in spastic patients, it became generally accepted that spasticity involved reduced presynaptic inhibition of Ia afferents (Ashby et al., 1974; Ashby and Verrier, 1975, 1976). However, later studies have cast doubts on this interpretation. Using a more optimal technique to evaluate presynaptic inhibition, Nielsen et al. (1995) demonstrated that presynaptic inhibition was reduced in spastic patients with multiple sclerosis. A similar finding was made for patients with spinal cord injury, but not for hemiplegic stroke patients (Paist et al., 1994). Presynaptic inhibition thus seems to be reduced in some spastic patients but not in all.
2. Disynaptic reciprocal Ia inhibition:
Reduced reciprocal inhibition is a strong candidate for playing a major role in the pathophysiology of spasticity (Crone and Nielsen, 1994; Crone et al., 2004, 2006). In spastic patients, reflex spread is common with reflex induced co-contraction of antagonist muscle groups, a failure of reciprocal inhibition. The Ia inhibitory interneurons are activated by descending motor fibers, damage to which could reduce this type of inhibition.
3. Recurrent inhibition:
Recurrent inhibition mediated by Renshaw cells have been studied by complex H-reflex techniques (Pierrot-Deseilligny and Bussel, 1975). In some patients with both supraspinal as well as traumatic spinal lesions increased recurrent inhibition may be seen, which obviously plays no role in development of spasticity (Katz and Pierrot-Deseilligny, 1982; Shefner et al., 1992). Only in patients with progressive paraparesis of ALS is a reduction found at rest and it is doubtful that this reduction contributes to the spasticity observed in these patients (Mazzochio and Rossi, 1989; Shefner et al., 1992). Changes in recurrent inhibition thus probably plays no major role in the pathophysiology of spasticity.
4. Non-reciprocal Ib inhibition:
This inhibition is caused by activation of Ib afferents coming from Golgi tendon organs and is mediated by segmental interneurons projecting to mononeurons of the same muscle. Ib inhibition may be demonstrated in human subjects by specialized H reflex studies (Raynor and Shefner, 1994). Whereas this inhibition is easily demonstrated in healthy subjects, there was failure to produce any inhibition on the paretic side in hemiplegic patients, along with a facilitatory effect in some subjects (Pierrot-Deseilligny et al., 1979). This observation suggests that alteration of Ib inhibition excitation plays a role in the pathophysiology of spasticity. However, reflex effects from Golgi tendon Ib afferents are unchanged after spinal cord lesions in humans.
At this stage it is important to take note of the fact that the anterior horn cell (AHC) or spinal motoneuron is the key nucleus in the operation of all spinal reflexes. Dysfunction of AHC leads to hypotonicity – as is evident in pure AHC affecting diseases like poliomyelitis, spinal muscular atrophy and the progressive muscular atrophic form of motor neuron disease (MND). Associated dysfunction of supraspinal pathways with some surviving spinal motoneurons might cause spastic weakness as commonly seen in the amyotrophic lateral sclerosis form of MND.
The importance of supraspinal and suprasegmental control of spinal reflexes was progressively understood since the role of muscle stretch reflex to generate muscle contraction was discovered by Liddell and Sherrington (1924), Delwaide and Oliver (1988) Descending influences control spinal reflexes by converging along with primary peripheral afferents on common interneuronal pool projecting to motoneurons. Imbalance of the descending inhibitory and facilitatory influences on muscle stretch reflexes is thought to be the cause of spasticity (Lundberg, 1975). These influences are discussed below.
There are five important descending tracts, of these, corticospinal tract originates from cerebral cortex. Other four come from closely neighboring parts in the brain stem and these are – Reticulospinal Vestibulospinal, Rubrospinal, and Tectospinal tracts. In human spastic paretic syndrome, the three important pathways are – corticospinal, reticulospinal, and vestibulospinal.
1. Corticospinal pathway – Isolated pyramidal lesions have not produced spasticity in conditions such as destruction of motor cortex (area 4), unilateral lesion in cerebral peduncle, lesions in basis pontis and medullary pyramid (Bucy et al., 1964; Brooks, 1986). Instead of spasticity these lesions produced weakness, hypotonia, and hyporeflexia. Pyramidal tract lesion alone is more responsible for weakness and loss of superficial reflexes such as abdominal reflexes rather than spasticity, hyper-reflexia and Babinski’s sign. Spasticity however may be caused in lesions of area 4 if the lesions include the premotor and supplementary motor areas. Fibers responsible for spasticity run with the pyramidal tract to end in the bulbar reticular formation (corticoreticular pathway). Lesions (vascular) in the anterior limb of internal capsule and not in the posterior limb produce spasticity as fibers from supplementary motor area pass through anterior limb. Large middle cerebral artery territory infarcts involving corticospinal and corticoreticular pathways produce spasticity (Gilman et al., 1973). Failure of isolated pyramidal lesion to produce spasticity does not however infer that this tract has no influence over muscle tone. Ipsilateral supplementary motor and premotor areas and contralateral motor cortex can take up some of the functions of pyramidal tract and prevent spasticity to develop.
2. Corticoreticular pathways and dorsal reticulospinal tract
Medullary reticular formation is active as a powerful inhibitory center to regulate muscle tone (stretch reflex) and the cortical motor areas control tone through this center. Lesions of premotor area (frontal cortex) or internal capsule reduces control over medullary center to produce hypertonicity.
Dorsal RST situated in the ventral part of the lateral funiculus of the spinal cord carries the inhibitory influence from the medullary center. This tract is non-monoaminergic, but unlike ventral (medial) RST, it inhibits FRA as well as stretch reflex arc. “Flexor spams” are release phenomenon of flexor reflexes due to damage to dorsal reticulospinal pathway (Fisher and Curry, 1965). Clasp-knife phenomenon is also a release phenomenon due to loss of inhibitory effects on FRA.
1. Vestibulospinal pathway: Vestibulospinal tract (VST) is a descending motor tract originating from lateral vestibular (Deiter’s) nucleus and is virtually uncrossed. The tract ends mostly on interneurons but also excites motor neurons monosynaptically. This excitatory pathway helps to maintain posture and to support against gravity and so control extensors rather than flexors. This pathway is important in maintaining decerebrate rigidity but has lesser role in human spasticity (Fries et al., 1993).
The cerebellum through its connections with the vestibular nuclei and reticular formation may indirectly modulate muscle stretch reflexes and tone.
2. Medial (ventral) RST – Through this tract reticular formation exerts facilitatory influence on spasticity. The tract has a diffuse origin being mainly from pontine tegmentum. Unlike dorsal RST, it is not affected by stimulation of motor cortex or internal capsule and not inhibitory to FRA. This pathway is more important than vestibulospinal system in maintaining spastic extensor tone (Schreiner et al., 1949; Shahani and Young, 1973).
The four descending pathways which are important in spastic paretic syndrome are arranged as follows in the spinal cord:
1. Lateral funiculus contains corticospinal tract (CST) and dorsal RST.
2. Anterior funiculus contains VST and medical RST (in close proximity with medial longitudinal fasciculus).
Muscle tone is maintained by a controlled balance on stretch reflex arc by inhibitory influence of CST and dorsal RST and facilitatory influence (on extensor tone) by medial RST and to a lesser extent in humans by VST.
1. In cortical and internal capsular lesions, the controlling drive on the inhibitory center in the medullary brain stem is lost and so in absence of inhibitory influence of dorsal RST originating from this center, facilitatory action of medial RST becomes unopposed. This results in spastic hemiplegia with antigravity posturing, but flexor spams are unusual.
2. Spinal lesions – (a) Incomplete (partial) myelopathy involving lateral funiculus (e.g., early multiple sclerosis) (Peterson et al., 1975) may affect CST only to produce paresis, hypotonia, hyporeflexia, and loss of cutaneous reflexes. If dorsal RST is involved in addition, unopposed medial RST activity then results in hyper-reflexia and spasticity (similar to cortical or capsular lesions), the latter being marked in antigravity muscles to produce paraplegia in extension. Extensor and flexor spasms may occur, the former being commoner.
(b) Severe myelopathy with involvement of all the four descending pathways produces less marked spasticity compared to isolated lateral cord lesion because of lack of unopposed excitatory influences of medial RST and VST. The latter factor is also responsible for lack of extensor hypertonia and in presence of release of flexor reflexes by dorsal RST lesion, helps to produce paraplegia in flexion. Paraplegia in flexion is also possible in partial myelopathy if FRA get stimulated by factors like pressure sores.
(c) Isolated dorsal RST involvement with CST sparing (proved pathologically and electrophysiologically) (Oppenheimer, 1978; Thompson et al., 1987) may explain marked spasticity and spasms but little weakness in many cases of spastic paraparesis. Only hyper-reflexia with normal tone is again a possibility in isolated anterior cord lesion.
3. Clinically spasticity may be of different types due to involvement of descending pathways. Depending on the predominant involvement of phasic (dynamic) or tonic (static) components of muscle stretch reflexes, the spasticity may be “phasic” and “tonic” Precollicular lesions in cat produce essentially phasic and decerebration at a lower level produces essentially tonic spasticity (Burke et al., 1972). Patients of chronic spinal cord injury who are ambulatory with minimum voluntary movement reveals more of “phasic” spasticity in the form of increased tendon jerks and clonus. Non-ambulatory patients with or without voluntary movement revels more of “tonic” spasticity on passive stretch at ankle and vibratory tonic reflex testing by noting tonic response of triceps surae on vibrating the Achilles tendon (Burke et al., 1972).
4. Neuroplasticity of the spinal cord in the form of receptor supersensitivity of neurons to a loss of synaptic input and sprouting of axon terminals are also responsible for hypertonicity in complete myelopathy with delayed reorganization after a variable period of spinal shock (Davis, 2000). This hypertonicity is not velocity dependent as in partial myelopathy and results from nearly continuous flexor spasms. Paraplegia in flexion may be associated with mass reflexes (exaggerated flexor spasms) in this condition.
5. Cerebellum and muscle tone: The cerebellum does not seem to have a direct effect on muscle tone determining spinal reflex pathways as there is no direct descending cerebello-spinal tract. The ascending spino-cerebellar tracts carry afferent impulses relating to joint and limb position and range and direction of movement. Such impulses are also carried by the posterior column tracts to the medullary centers and then onto the cerebellar cortex through the inferior cerebellar peduncles. The cerebral and cerebellar cortices are inter-connected by feed-back–feed-forward loops which project through the corticospinal and other descending extrapyramidal pathways to the spinal cord. The cerebellum mainly influences muscle tone through its connections with the vestibular and brain stem reticular nuclei. Pure cerebellar lesions classically produce hypotonia. But associated corticospinal tract involvement produces varying degrees of spasticity as seen in some forms of spino-cerebellar atrophies (SCA) and the spastic ataxia of Charlevoix–Saguenay encountered in French Canadian stock. On the other hand chronic cerebellar stimulation had been used to relieve spasticity in cerebral palsy (Ebner et al., 1982). Earlier experimental study demonstrated that cerebellar surface stimulation reduced the amplitude of the tonic and phasic stretch reflexes (Dimitrijevic, 1984). This modified the organization of the segmental reflexes producing a more normal reciprocal relationships of EMG activity in the agonist and antagonist.
How does UMN lesion cause spasticity and associated phenomena? The major problem is a loss of control of the spinal reflexes. Spinal reflex activity is normally tightly regulated and if inhibitory control is lost, the balance is tipped in favor of excitation, resulting in hyperexcitability of the spinal reflexes. The problem is made difficult by the fact that individual patients have lesions affecting different pathways to different extent and that the subsequent adaptations in the spinal networks, as a result to the primary lesion, may vary considerably. The different spinal mechanisms – plateau potentials, reciprocal inhibition and presynaptic inhibition – may have different roles in different patients. It is likely that spasticity is not caused by a single mechanism, but rather by an intricate chain of alterations in different inter-dependent networks.
The fact that there is a period of shock, followed by a transition period when reflexes return, but are not hyperactive suggests that this is not just simply a question of switching off supraspinal inhibition, or altering the balance. It implies that there must be some sort of rearrangement, a kind of neuronal plasticity, occurring within the spinal cord, and most probably at the cerebral level as well. One possibility is sprouting of afferent axons (Raisman, 1969; Benecke, 1985; Raineteau and Schwab, 2001; Bareyre et al., 2004). Afferent fibers might sprout, attach to previously inhibitory synapses, and convert them to excitatory synapses. Alternatively there could be development of denervation hypersensitivity due to upregulation of receptors (Sravraky, 1961).
Spasticity may also be explained by changes in mechanical properties of muscles and not only by hyper-reflexia (Dietz et al., 1981; Thilmann et al., 1991). The increased mechanical resistance may be caused by alterations in tendon compliance and physiological changes in muscle fibers which affect functional movement of leg occurring at low angular velocities. Contractures are extreme effects of mechanical resistance which can be prevented by early treatment of hypertonia with botulinum toxin (BTX) in spastic cerebral palsy.
In conclusion, it needs to be mentioned that the progress being made in the electrophysiologic analysis of spinal control mechanisms in spasticity and measurement of spasticity are helpful for greater understanding of pathophysiology of the condition. Newly used drugs have multiple sites of actions (Delwarde and Pennisi, 1994). On the whole it seems highly likely that more than one pathophysiologic abnormality contributes to development of spasticity (Sheean, 2001; Nielsen et al., 2007).
The use of BTX in treatment of spasticity has already been mentioned. When injected at or near the motor point of affected muscle BTX binds to the SV2 receptor on the presynaptic membrane allowing for entry of the toxin into the axon terminal. Once inside the axon, BTX light chains act to impede exocytosis of acetylcholine (ACH). This allows for fusion of neurotransmitter-containing intra-axonal vesicles with the presynaptic membrane, resulting in extrusion of ACH into the synaptic cleft. The reduced presynaptic outflow of ACH at the neuromuscular junction causes diminution of muscle contraction. BTX reduces the frequency and quantity but not the amplitude of miniature endplate potential (MEPP). The motor EPP is reduced below the muscle membrane threshold and the ability to generate muscle fiber action potentials and subsequent contraction is diminished (Ney and Joseph, 2007).
Of the orally active agents, Baclofen is a centrally acting GABA analog. It binds to GABA receptor at the presynaptic terminal and inhibits muscle stretch reflex. Baclofen can also be used intrathecally.
Dantrolene interferes with the release of calcium from the sarcoplasmic reticulum of the muscle.
Tizanidine is an imidazole derivative with agonist action on alpha-2-adrenergic receptors in the central nervous system. The exact mechanism of its action in reduction of human muscle tone is not known but a central action can be speculated.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ashby, P., and Verrier, M. (1975). Neurophysiological changes following spinal cord lesions in man. Can. J. Neurol. Sci. 2, 91–100.
Ashby, P., and Verrier, M. (1976). Neurophysiologic changes in hemiplegia. Possible explanation for the initial disparity between muscle tone and tendon reflexes. Neurology 26, 1145–1151.
Ashby, P., Verrier, M., and Lightfoot, E. (1974). Segmental reflex pathways in spinal shock and spinal spasticity in man. J. Neurol. Neurosurg. Psychiatr. 37, 1352–1360.
Bareyre, F. M., Kerschensteiner, M., Rainetear, O., Mettenleiter, T. C., Weinmann, O., and Schwab, M. E. (2004). The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat. Neurosci. 7, 269–277.
Benecke, R. (1985). “Basic neurophysiological mechanisms in spasticity,” in Treating Spasticity. Pharmacological Advances, ed. C. D. Marsden (London: Hans Huber Publishers), 11–17.
Bucy, P. C., Kephnger, J. E., and Siqucira, E. B. (1964). Destruction of the pyramidal tract in man. J. Neurosurg. 21, 385–398.
Burke, D., and Ashby, P. (1972). Are spinal ‘presynaptic’ inhibitory mechanisms suppressed in spascity? J. Neurol. Sci. 15, 321–326.
Burke, D., Knowles, L., Andrews, C. J., and Ashby, P. (1972). Spasticity decrebrate rigidity in the clasp knife phenomenon: an experimental study in the cat. Brain 95, 31–48.
Crone, C., Johnsen, L. L., Bieringsorensen, F., and Nielsen, J. B. (2006). Appearance of reciprocal facilitation of ankle extensors from ankle flexors in patients with stroke or spinal cord injury. Brain 126, 495–507.
Crone, C., and Nielsen, J. (1994). Central control of disynaptic reciprocal inhibition in humans. Acta Physiol. Scand. 152, 351–363.
Crone, C., Petersen, N. T., Gimenz-Roldan, S., Lungholt, B., Nyborg, K., and Neilsen, J. B. (2007). Reduced reciprocal inhibition is seen only in spastic limbs in patients with neurolathyrism. Exp. Brain Res. 181, 193–197.
Crone, C., Petersen, N. T., Nielsen, J. E., Hansen, N. L., and Nielsen, J. B. (2004). Reciprocal inhibition and corticospinal transmission in the arm and leg in patients with autosomal dominant pure spastic paraparesis (ADPSP). Brain 127, 2693–2702.
Davis, R. (2000). Cerebellar stimulation for cerebral palsy, spasticity, function and seizures. Arch. Med. Res. 31, 290–299.
Delwaide, P. J., and Gerard, P. (1993). “Reduction of non-reciprocal (lb) inhibition: a key factor for interpreting spastic muscle stiffness,” International Congress on Stroke Rehabilitation, Berlin.
Delwaide, P. J., and Oliver, E. (1988). Short-latency autogenic inhibition (lB) inhibition) in human spasticity. J. Neurol. Neurosurg. Psychiatr. 51, 1546–1550.
Delwarde, P. J., and Pennisi, G. (1994). Tizanidine and electrophysiologic analysis of spinal control mechanisms in human with spasticity. Neurology 44 (11 Suppl. 9), S21–S28.
Dietz, V., Quimern, J., and Berger, W. (1981). Electrophysiological studies of gait in spasticity and rigidity. Evidence that altered mechanical properties of muscle contribute to hypertonia. Brain 104, 431–449.
Dimitrijevic, M. R. (1984). “Neural control of chronic upper motor neuron syndromes,” in Electromyography in CNS Disorders, ed B. T. Shahani (Boston, London: Butterworths), 111–127.
Ebner, T. J., Bloedel, J. R., and Schwartz, A. B. (1982). The effects of cerebellar stimulation on the stretch reflex in the spastic monkey. Brain 105, 425–442.
Feganel, J., and Dumtrijevic, M. R. (1982). Study of propriospinal interneuron system in man. Cutaneous extenceptive conditioning of stretch reflexes. J. Neurol. Sci. 56, 155–172.
Fisher, C. M., and Curry, H. B. (1965). Pure motor hemiplegia of vascular origin. Arch. Neurol. 13, 30–44.
Fries, W., Danek, A., Schidumann, K., and Hamburger, C. (1993). Motor recovery following capsular stroke. Role of descending pathways from multiple motor areas. Brain 116, 369–382.
Gilman, S., Marco, L. A., and Ebel, H. C. (1973). Effects of medullary pyramidotomy in the monkey. II. Abnormalities of spindle afferent response. Brain 94, 515–530.
Hagbarth, K. E. (1981). “Fusimotor and stretch reflex functions studied in recordings from muscle spindle afferents in man,” in Muscle Receptors and Movement, Vol. 13. eds A. Taylor and A. Prochazka (New York: Oxford University Press), 109–115.
Heckmann, C. H., Gorassini, M. A., and Bennett, D. J. (2005). Persistent inward currents in motoneuron dendrites: implications for motor output. Muscle Nerve 31, 135–156.
Heckman, C. J., Lee, R. H., and Brownstone, R. M. (2003). Hyperexcitable dendrites in motoneurons and their neuromodulatory control during motor behavior. Trends Neurosci. 26, 688–695.
Hultborn, H., Brownstone, R. B., Toth, T. I., and Gossard, J. P. (2004). Key mechanisms for setting the input-output gain across the motoneuron pool. Prog. Brain Res. 143, 77–95.
Hultborn, H., Illert, M., and Santini, M. (1976). Convergence on interneurones mediating the reciprocal la inhibition of motoneurons. III. Effects from supraspinal pathways. Acta Physiol. Scand. 96, 368–391.
Jankowska, E., and Roberts, W. J. (1972). Synaptic actions of single interneurons mediating reciprocal la inhibition of motoneurons. J. Physiol. (Lond) 222, 623–642.
Katz, R., and Pierrot-Deseilligny, E. (1982). Recurrent inhibition of alphamotoneurons in patients with upper motor neuron lesions. Brain 105, 103–124.
Lance, J. W. (1980). “Symposium,” in Spasticity: Disordered Motor Control, eds R. G. Feldman, R. R. Young, and W. P. Koella (Chicago: Year Book Medical Pubs), 485–495.
Liddell, E. G. T., and Sherrington, C. S. (1924). Reflexes in response to stretch (myotatic reflexes). Proc. R. Soc. 96B, 212–242.
Lundberg, A. (1975). “Control of spinal mechanism from the brain,” in The Nervous System, Vol. 1, eds D. Tower and R. Brady (New York: Raven Press), 253–265.
Mazzochio, R., and Rossi, A. (1989). Recurrent inhibition in human spinal spasticity. Int. J. Neurol. Sci. 10, 337–347.
Misra, U. K., and Pandey, C. M. (1994). H reflex studies in neurolathyrism. Electroencephalogr. Clin. Neurophysiol. 93, 281–285.
Ney, J. P., and Joseph, K. R. (2007). Neurologic uses of Botulinum toxin type A. Neuropsychiatr. Dis. Treat. 6, 785–790.
Nielsen, J., Petersen, N., and Crone, C. (1995). Changes in transmission across synapses of la afferents in spastic patients. Brain 118, 995–1004.
Nielsen, J. B., Crone, C., and Hultborn, H. (2007). The spinal pathophysiology of spasticity – from a basic science point of view. Acta Physiol. 189, 171–180.
Oppenheimer, D. R. (1978). The cervical cord in multiple sclerosis. Neuropathol. Appl. Neurobiol. 4, 151–162.
Paist, M., Mazevet, D., Dietz, V., and Pierrot-Descilligny, E. (1994). A quantitative assessment of presynaptic inhibition of la afferents in spastics. Differences in hemiplegics and paraplegics. Brain 117, 1449–1455.
Peterson, B. W., Maunz, R. A., Pitts, N. G., and Mackel, R. G. (1975). Patterns of projection and branching of reticulospinal neurons. Exp. Brain Res. 23, 333–351.
Pierrot-Deseilligny, E., and Bussel, B. (1975). Evidence for recurrent inhibition by motoneuron in human subjects. Brain 88, 105–108.
Pierrot-Deseilligny, E., Katz, R., and Morin, C. (1979). Evidence of lb inhibition in human subjects. Brain Res. 166, 176–179.
Pompetano, O. (1984). “Recurrent inhibition,” in Handbook of the Spinal Cord, Vols. 2 and 3, ed. F. A. Davidoff (New York: Marcel Dekker), 461–557.
Powers, R. K., and Binder, M. D. (2001). Input-output functions of mammalian motoneurons. Rev. Physiol. Biochem. Pharmacol. 143, 137–263.
Raineteau, O., and Schwab, M. E. (2001). Plasticity of motor systems after incomplete spinal cord injury. Nat. Rev. Neurosci. 2, 263–273.
Raisman, G. (1969). Neuronal plasticity in the septal nuclei of the adult rat. Brain Res. 14, 25–48.
Raynor, E. M., and Shefner, J. M. (1994). Recurrent inhibition is decreased in patients with amyotrophic lateral sclerosis. Neurology 44, 2148–2150.
Rekling, J. C., Funk, G. D., Bayliss, D. A., Dong, X. W., and Feldman, J. L. (2000). Sunaptic control of motoneuronal excitability Phys. Rev. 80, 767–852.
Rushworth, G. (1960). Spasticity and rigidity: an experimental study and review. J. Neurol. Neurosurg. Psychiatr. 23, 99–118.
Schmidt, R. F. (1971). Presynaptic inhibition in the vertebrate central nervous system. Ergeb. Physiol. 63, 91–101.
Schreiner, L. M., Mandsley, D. B., and Magoum, H. W. (1949). Role of brain stem facilitatory systems in maintenance of spasticity. Neurophysiology 12, 207–216.
Shahani, B. T., and Young, R. R. (1973). “Human flexor spasm,” in New Developments in Electromyography and Clinical Neurophysiology, Vol. 3, ed J. E. Desmedt (Basal: S Karger AG), 734–743.
Shefner, J. M., Berman, S. A., Sarkarati, M., and Young, R. R. (1992). Recurrent inhibition is increased in patients with spinal cord injury. Neurology 42, 2162–2168.
Sravraky, G. W. (1961). Supersensitivity Following Lesions of the Nervous System. An Aspect of the Relativity of Nervous Integration. Toronto: University of Toronto Press, pp. 1–210.
Thilmann, A. F., Fellows, S. J., and Grams, E. (1991). The mechanism of spastic muscle hypertonus: variation in reflex gain over the time course of spasticity. Brain 114, 233–244.
Thompson, P. D., Day, B. L., and Rothwell, J. C. (1987). The interpretation of electromyographic responses to electrical stimulation of the motor cortex in diseases of the upper motor neuron. J. Neurol. Sci. 80, 91–110.
Keywords: spasticity, muscle tone, muscle spindle, spinal reflexes, supraspinal pathways
Citation: Mukherjee A and Chakravarty A (2010) Spasticity mechanisms – for the clinician. Front. Neur. 1:149. doi: 10.3389/fneur.2010.00149
Received: 05 August 2010;
Accepted: 13 November 2010;
Published online: 17 December 2010.
Edited by:
U. K. Misra, Sanjay Gandhi Post Graduate Institute of Medical Sciences, IndiaReviewed by:
U. K. Misra, Sanjay Gandhi Post Graduate Institute of Medical Sciences, IndiaCopyright: © 2010 Mukherjee and Chakravarty. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Ambar Chakravarty, Department of Neurology, Vivekananda Institute of Medical Sciences, 99 Sarat Bose Road, Kolkata 700026, India.e-mail:c2FzY2hha3JhQHlhaG9vLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.