 Amit Joharapurkar
Amit Joharapurkar Vrajesh Pandya
Vrajesh Pandya
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Nephrol. , 22 October 2024
Sec. Clinical Research in Nephrology
Volume 4 - 2024 | https://doi.org/10.3389/fneph.2024.1459425
Desidustat is a small molecule inhibitor of hypoxia-inducible factor-prolyl hydroxylase (HIF-PH) discovered and developed by Zydus Lifesciences for the treatment of anemia associated with chronic kidney disease (CKD). This review summarizes the preclinical and clinical profile of desidustat which led to its approval and clinical use in India.
Anemia (defined as hemoglobin [Hb]<11 g/dL in women and<12 g/dL in men) has a prevalence of <10% in stages I & II, about 20–40% in stage III, and 50–60% in stage IV of CKD. This prevalence even exceeds 70% in stage V due to the decline in estimated glomerular filtration rate (eGFR) (1). Global prevalence of CKD was estimated to be 11% to 14% in the first two decades of twenty-first century (2). Hemoglobin (Hb) and red blood cells (RBC) deficiency observed in CKD is caused due to a combination of factors like deficiency in iron utilization, relative erythropoietin (EPO) deficiency, and resistance to the actions of erythropoietin (3). Systemic inflammation, uremia, and vitamin deficiency also contribute to the progression of CKD-induced anemia. The severity of anemia has a direct correlation with progression of chronic kidney disease (decrease in eGFR) and morbidity due to cardiorenal diseases (4). Anemia is also observed in diabetic patients, independent of eGFR and albuminuria (3).
EPO is synthesized and secreted by the fibroblast-like interstitial peritubular cells of the kidneys, and the perisinusoidal cells of the liver may also secrete EPO in a minor fashion (5). In bone marrow, the hematopoietic stem cells (HSCs) differentiate into colony forming units-erythroid (CFU-Es) and then to erythroblasts, which form reticulocytes by enucleation. This process requires stimulation by EPO. Erythrocytes or RBC are formed from reticulocytes within one or two days, which circulate in the blood for 120 days, following which, they are killed by phagocytosis in the spleen and liver (6). EPO acts on specific EPO receptors (EPORs) which signal through Janus tyrosine kinase-2 (JAK2). JAK2 stimulates signal transduction and activator of transcription-5 (STAT-5), RAS−RAF−MAP kinase, and phosphoinositide-3 kinase/AKT kinase (protein kinase B). On the other hand, erythroid progenitors undergo apoptosis by the cluster of differentiation 95 (CD95), a membrane protein of the tumor necrosis factor (TNF) receptor family, which triggers apoptosis after binding to the CD95 ligand, which is produced by mature erythroblasts. Thus, a negative feedback cycle controls the production of mature erythrocytes in blood (7).
Apart from erythropoietin, which differentiates the erythrocyte precursors, iron is the next important contributor in efficient synthesis of hemoglobin and RBC synthesis by reticulocyte maturation is iron.
Iron from diet is absorbed in the duodenum and proximal jejunum, when it is in the ferrous (Fe2+) state or bound by a protein such as heme. Duodenal cytochrome B (Dcytb), on the brush border of the enterocytes converts the insoluble ferric (Fe3+) to absorbable ferrous (Fe2+) ions and divalent metal cation transporter 1 (DMT1) transports ferrous (Fe2+) iron in to the cell. The cellular iron is transported in the circulation by the transmembrane efflux protein ferroportin. Ferrous iron (Fe2+) is oxidized (by ceruloplasmin and hephaestin) into ferric iron (Fe3+) for binding to transferrin, which serves as the iron-carrier protein in plasma. Transferrin chelates iron to make it soluble, prevents the formation of reactive oxygen species, and facilitates its transport into cells.
Iron metabolism is monitored by the hepatic hormone hepcidin, which exclusively regulates the transporter ferroportin. Inflammatory states increase cytokine (e.g., IL-6) release, which stimulates hepcidin expression in the liver. Hepcidin causes decreased iron absorption through ferroportin degradation and decreases the release of iron from macrophages (8). In addition to hepcidin, levels of DMT1 and Dcytb are upregulated in the hypoxic environment of the intestinal mucosa by hypoxia-inducible factor-2 (HIF-2α) (9). High levels of iron, inflammatory cytokines, and oxygen lead to increased levels of hepcidin. Hepcidin binds to ferroportin, resulting in its internalization and degradation and effectively shunting cellular iron into ferritin stores and preventing its absorption into the blood. Thus, increased hepcidin traps body iron in macrophages, hepatocytes, and enterocytes. Thereby, hepcidin potentiates the excretion of iron through the sloughing of enterocytes (and their ferritin stores) into the feces and out of the body.
The treatment of CKD-induced anemia is done primarily using recombinant EPO (10). However, in end-stage renal disease and in nondialysis chronic kidney disease patients, treatment with EPO or ESA (erythropoiesis stimulating agents) showed inadequate efficacy and cardiovascular toxicity in terms of risk of cardiovascular hospitalization or stroke (11). The risks due to EPO treatment can be correlated with inflammatory markers and urinary protein/creatinine ratio (12). Earlier, the increase in Hb and blood volume or viscosity thereby was considered the reason for cardiovascular disease. However, it is now evident that elevation of Hb into the normal range in a spontaneous way is not detrimental, but rather beneficial for cardiovascular health (13). ESA treatment, either supraphysiological or after prolonged exposure, also results into ESA resistance which is associated with the disease progression, infection and inflammation, increased cytokine signaling, and defective iron metabolism (14). On the other hand, it has been demonstrated that improving ESA sensitivity reduces the cardiovascular toxicity of the erythropoietin preparations (15), and higher doses of EPO can induce morbidity and mortality (16). A sizable population of CKD patients require higher doses of EPO, which may culminate into EPO resistance and cardiovascular morbidities (17). Anti-EPO antibodies are generated in some CKD patients after treatment with ESA and it is related with EPO resistance, resulting in worsening of the anemia (18). EPO antibodies mediate pure red cell aplasia after treatment with recombinant EPO products.
In CKD patients, body iron regulation is disturbed, with increased iron loss, decreased iron absorption, and limited iron availability leading to iron deficiency. Intravenous (IV) iron therapy is frequently prescribed to replace lost iron, which eventually causes rise in hepcidin levels and traps iron in the tissue stores. Also, the increased iron in circulation imposes toxicity without effective erythropoiesis (19).
High altitude or hypoxia enhances RBC synthesis by stimulating EPO production by induction of hypoxia-inducible factor (HIF) (20). There are three isoforms of HIF (HIF-1, -2, and -3). HIF-1 mainly regulates the metabolic response, while HIF-2 stimulates erythropoiesis in response to hypoxia. HIF-3 is less closely related to HIF-1 and HIF-2, and its role is not yet fully understood. HIF consists of two subunits, namely α and β. HIF-α is hydroxylated at two proline residues (Pro402 and Pro564 for HIF-1α, and Pro405 and Pro531 for HIF-2α) by prolyl hydroxylase domain enzyme (PHD). Once hydroxylated, HIF-α is recognized by Hippel−Lindau tumor suppressor protein (pVHL), which causes its proteasomal degradation by E3 ubiquitin ligase. PHD enzymes require oxygen, iron, ascorbate, and the substrate 2-oxoglutarate (2- OG) for their activity. Oxygen deficiency (hypoxia) causes PHD inhibition, which activates HIF-α, while HIF-β is constitutively expressed.
There are three isoforms of PHD, namely, PHD1, PHD2, and PHD3 (21). The active sites of all isoforms of PHDs share a high-sequence homology. PHD1 shows more affinity toward HIF-2α than HIF-1α and hydroxylates it under normoxia condition. Deletion of the PHD1 gene induces hypoxia tolerance by shifting the tricarboxylic acid cycle to the glycolytic pathway (22). Tolerance to hypoxia is due to reduced oxidative stress and may not be due to angiogenesis, erythropoiesis, or vasodilation. PHD2 gene deletion accelerated RBC synthesis without a significant increase in EPO levels (23). Inactivation of PHD1 and PHD3 leads to erythrocytosis by activating the hepatic HIF-2α, whereas only PHD2 deficiency leads to erythrocytosis by activating the renal EPO pathway (21).
Interstitial fibroblasts of kidneys generate EPO (24). Fibrosis in the renal tissue causes EPO deficiency and anemia. HIF-2 stimulates EPO gene transcription by binding to hypoxia-responsive regulatory elements. HIF-2 induces efficient absorption as well as utilization of iron by stimulation of genes required for iron transport and metabolism like DMT1 and Dcytb. HIF indirectly stimulates efficient iron utilization through regulation of hepcidin (25). Inflammation upregulates hepcidin through the STAT-3 pathway or the BMP/SMAD1/5/8 pathway (19, 25). As already discussed, increased hepcidin causes degradation of ferroportin and causes entrapment of iron in tissue stores, resulting in a functional iron deficiency, which cannot be corrected using oral iron supplementation. As the iron deficiency worsens, iron regulatory proteins bind to HIF- 2α and further decrease erythropoiesis (26). This feedback mechanism makes the anemia in CKD patients difficult to treat using erythropoietin analogues or iron supplementation.
Activation of HIF signaling using PHD inhibition is a unique and rational approach to correct anemia associated with chronic diseases. Desidustat is a novel HIF-PHD inhibitor discovered and developed by Zydus Lifesciences for the treatment of anemia in patients with CKD patients on dialysis and also those who are not on dialysis. Desidustat got its first approval in India in 2022 (27). Currently, PHD inhibitors are approved in different countries. Roxadustat, the first clinically used PHD inhibitor, is approved in a number of countries, including China, Japan, Chile, South Korea, and Europe for the treatment of anemia in CKD in NDD and DD adult patients. However, it was not approved in USA for treatment of anemia of CKD (both NDD and DD patients), because a higher portion of NDD-CKD patients and DD-CKD patients experienced vascular access thrombosis compared with controls (28). Recently, vadadustat and daprodustat have been approved for clinical use in USA in addition to their previous approvals in Japan and China. In US, vadadustat and daprodustat are approved by FDA only for dialysis patients who have been receiving dialysis at least for four months. Few other HIF-PHD inhibitors, molidustat (Japan and China) and enarodustat (Japan) have also found their way to the market. These HIF-PHD inhibitors (Figure 1) have met their primary goal of correcting anemia in CKD patients, however, these molecules demonstrated differential profile in terms of efficacy and toxicity.
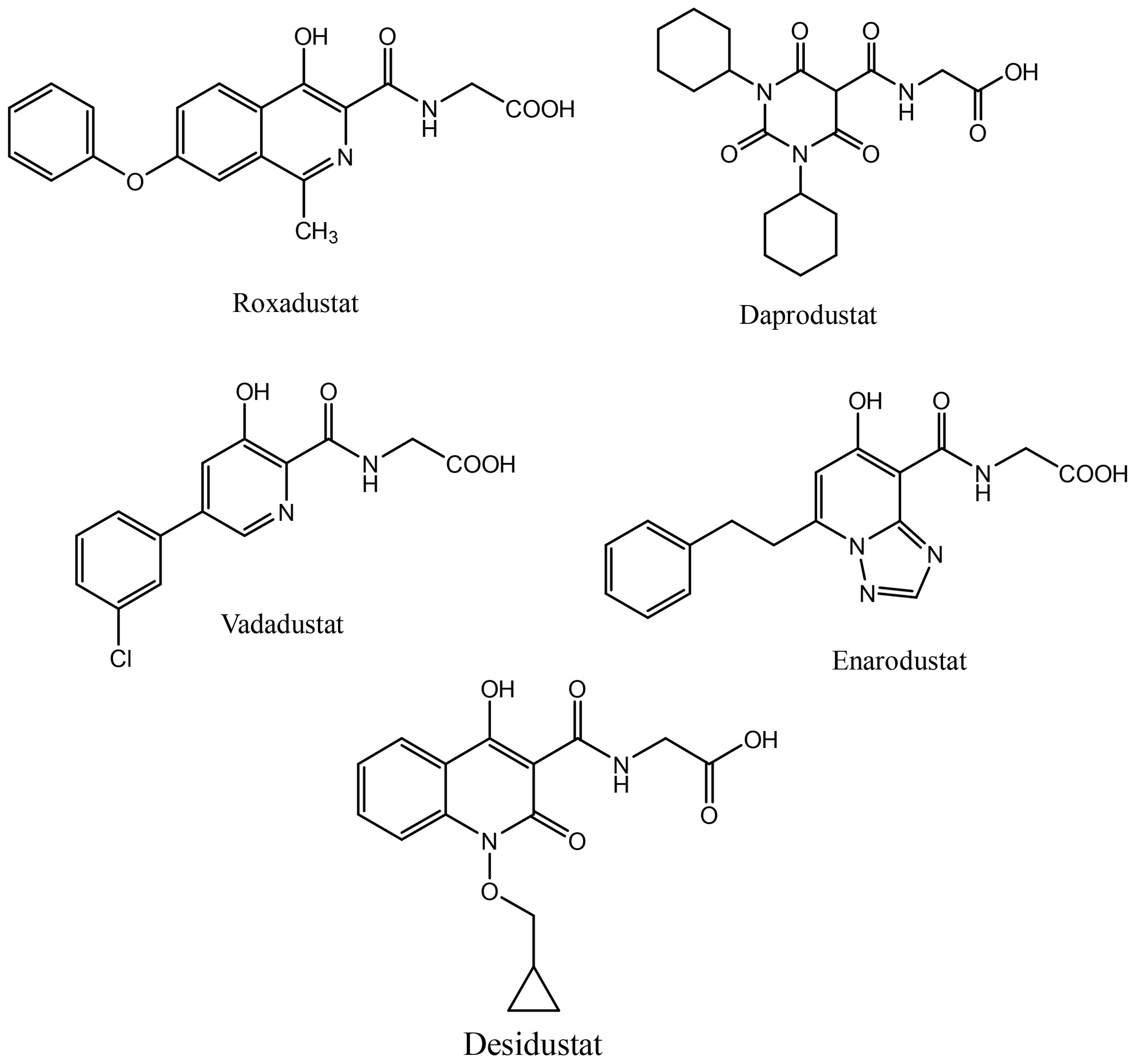
Figure 1. Selected PHD inhibitors.
Clinically approved HIF-PHD inhibitors are the functional inhibitors of 2-oxoglutarate, which is a substrate of PHD (29). Since the pathways controlled by 2-oxoglutarate cover a diverse range of physiological processes, inhibition of PHD may affect many genes that are unrelated to anemia, whose protein products exert important functions. The genes that are modulated by HIF but may be unrelated to erythropoiesis (or anemia) include those related to glucose metabolism, energy homeostasis, angiogenesis, migration and interaction of cells and many other metabolic pathways. The modulation of such genes by HIF-PHD inhibitors may cause the complications like cancer progression, retinal diseases, pulmonary hypertension, and cyst formation in polycystic kidney diseases in adults (30), due to the downstream pathways like induction of vascular endothelial growth factor, proliferation of endothelial colony-forming cells, or chloride-dependent fluid secretion in case of cysts (30, 31). However, these safety concerns were not found to be significant in the clinical trials of the HIF-PHD inhibitors so far.
The transient and frequent PHD inhibition caused by the HIF-PHD inhibitors may also enhance the production of reactive oxygen species since the switch of metabolism from aerobic to anaerobic glycolysis (32). HIF stabilization modulate osteogenic differentiation through the expression of RUNX2 gene, and HIF-PHD inhibitors may cause vascular calcification (30). Daprodustat, a HIF-PHD inhibitor caused vascular calcification in a mice model of CKD and hyperphosphatemia, which was due to stabilization of HIF (33). Clinical studies with a long follow-up period are needed to evaluate the possible risk of clinically relevant calcification in CKD patients with hyperphosphatemia.
HIF-PHD inhibitor treatment may also cause thromboembolism, roxadustat caused more thrombosis than ESAs and vadadustat, according to an analysis, and the lowest rates of thrombosis were seen with molidustat and ESAs, with the US FDA raising concern for thrombotic risk of certain HIF-PHD inhibitors (34). Thrombosis risk is increased by HIF-PHD inhibitor treatment because they can modulate the tissue factors involved in thrombosis or protein kinase B that contributes to platelet and monocyte activation (30). HIF is also partially responsible for the remodeling of heart leading to worsening of heart failure (35). The use of HIF-PHD inhibitor daprodustat was associated with an increase in the estimated hazard ratio (HR) for hospitalization for heart failure in patients with a history of heart failure in patients not undergoing dialysis (36). It is possible that administration of HIF-PHD inhibitors to patients with heart failure causes an imbalance between the energy demand and supply in heart, leading to cardiac hypertrophy.
Most of the phase 3 studies with HIF-PHD inhibitors were done with strict exclusion criteria for patients with comorbidities of cardiac metabolism. Hence, post-marketing trials would be needed to ensure the safety of HIF-PHD in clinical use.
The in vitro activity of desidustat was assessed in two HIF heterodimerization bioassays which measured stabilization of HIF in live cells and in real time (37). EC50 and Emax values were determined as a measure of relative potency and efficacy, respectively. Desidustat achieved maximum stabilization of both, HIF1α and HIF2α. In HIF1 and 2 bioassays, desidustat was found to be a HIF stabilizer (EC50 = 32.6 μM for HIF1α and 22.1 μM for HIF2α), while it showed the optimum efficacy (Emax = 119% in both HIF1α and HIF2α assays). In this assay, roxadustat showed similar activity as that of desidustat with 105% and 98.9% Emax at micromolar concentrations. However, daprodustat was comparatively weaker, since it could show only 37.9% and 51.8% Emax. Desidustat stabilizes HIF in liver as well as kidneys of rats, after single dose administration (37).
Desidustat caused a dose-related increase in serum erythropoietin when dosed from 5 mg/kg to 60 mg/kg oral dose, and also caused a dose-related increase in reticulocyte count. In these rats, the HIF stabilization caused by desidustat was also associated with transient increase in serum iron which was normalized within 48 days of dosing. After repeated dosing (alternate day for a week), the lowest dose of 5 mg/kg caused an increase of 0.9 g/dl in hemoglobin in these rats, which increased in a dose-related way up to the 60 mg/kg doses tested. These doses caused a dose-related increase in erythropoietin, but not in serum Vascular Endothelial Growth Factor (VEGF) (38).
The erythropoietic activity of roxadustat was determined in a rat model of anemia of CKD induced by five-sixth nephrectomy. The nephrectomized rats showed a significant loss in hemoglobin, RBC count and hematocrit, compared to the sham-treated animals. These nephrectomized animals were treated with desidustat on alternate day with the dose ranging from 05 mg/kg to 60 mg/kg by oral route, with darbepoietin (16 µg/kg, subcutaneous, once a week) as the comparator for 28 days. Starting from dose of 15 mg/kg, desidustat showed a significant and dose-related improvement in hemoglobin, RBC count and hematocrit, the serum iron was also increased but it normalized with 24 hours of last dose. The increases in the hemoglobin and hematocrit showed by desidustat at 15 mg/kg dose were comparable to those shown by darbepoietin. In addition, desidustat treatment caused a meaningful reduction in liver hepcidin after twenty-eight days of the treatment (38).
Chronic inflammatory diseases are often associated with anemia, and treatment with erythropoiesis stimulating agents (ESAs) are associated with potentially hazardous side effects and poor outcomes. In rodent models of inflammation, desidustat was assessed as an effective treatment of anemia of inflammation. In BALB/c mice, a single dose treatment of desidustat (15mg/kg, orally) attenuated the effect of lipopolysaccharide (LPS) - or turpentine oil-induced inflammation and increased serum erythropoietin (EPO), iron, and reticulocyte count, and decreased serum hepcidin levels, in a dose related manner (39).
The model of anemia of chronic disease (ACD) was established by administration of proteoglycan-polysaccharide (PG-PS). Desidustat can reverse the anemia caused by the PG-PS stimulus, which induces inflammation and functional iron deficiency. In female Lewis rats, treatment with desidustat (15 and 30 mg/kg, orally) markedly reduced PGPS-induced anemia and increased hemoglobin, red blood cell (RBC) and white blood cell (WBC) count, hematocrit, serum iron and spleen iron. These effects of desidustat were associated with reduction in hepcidin gene (HAMP) expression as well as reduction in serum hepcidin, and increased EPO expression in liver and kidneys. Desidustat increased the expression of duodenal cytochrome B (DcytB), ferroportin (FPN1) and divalent metal transporter 1 (DMT1) in duodenum, and FPN1 and monocyte chemoattractant protein-1 (MCP-1) in liver suggesting an overall influence on iron metabolism (39).
Cisplatin administration in mice causes the decrease in hemoglobin, hematocrit, and RBC count. Oral treatment with desidustat (15 mg/kg and 30 mg/kg, alternate day) for twenty-eight days caused a dose-related increase in all these three parameters (38).
Studies in SD rats and C57 mice (38–40) studies indicate that 15 mg/kg on alternate day dose is the efficacious dose for improvement in hemoglobin, in anemia induced by nephrectomy or inflammation. Desidustat reduces alternate compliment pathways through partial inhibition of factor B, which is useful in treatment of immune-mediated renal and hematological disorders (41). Desidustat prevents acute kidney injury in rats induced by ischemia (42). Treatment with desidustat reversed the decrease in serum erythropoietin caused by the ischemia injury and prevented the increase in serum creatinine, urea, and KIM-1-the known markers of kidney injury. Desidustat treatment caused a significant increase in serum erythropoietin levels, which was accompanied by reduction in serum IL-6 and IL-1β levels. Thus, desidustat treatment can improve erythropoietin by a combination of direct HIF-stabilizing effect and reduction in the proinflammatory cytokines (42). Adenine converts into 2, 8-dihydroxyadenine in the body and deposits as crystals in the proximal tubular epithelia of the kidney, which induces inflammation and subsequent tubulointerstitial fibrosis and anemia (43). Desidustat treatment effectively reduced the adenine-induced nephritis in mice as indicated by reduced creatinine and urea, and reduced the urinary albumin to creatinine ratio. This treatment caused a profound decrease in the inflammatory milieu induced by the adenine supplementation with decreased IL-1 β and IL-6 levels in both kidney and serum. Also, a significant reduction in liver hepcidin and renal myeloperoxidase indicated by HIF stabilization and reduction in oxidative stress associated fibrosis (42).
Though erythropoietin therapy is the mainstay of CKD-induced anemia, a significant CKD patients are refractory to erythropoietin (EPO) effects due to inflammation, deranged iron utilization, and generation of EPO antibodies (14). Effect of desidustat on EPO-refractory renal anemia was assessed in Sprague Dawley rats that were made anemic by cisplatin (5 mg/kg, IP, single dose) and turpentine oil (5 mL/kg, SC, once a week). These rats were given recombinant human EPO (rhEPO, 1 μg/kg) and desidustat (15 or 30 mg/kg) for eight weeks. Separately, rhEPO (1–5 μg/kg) was given to anemic rats to sustain the normal hemoglobin levels and desidustat (15 mg/kg) for eight weeks. In another experiment, the anemic rats were treated rhEPO (5 μg/kg) for two weeks and then desidustat (15 mg/kg) for the next two weeks. Dosing of rhEPO was thrice a week, and for desidustat, it was on alternate days. Desidustat inhibited EPO-resistance caused by rhEPO treatment, decreased hepcidin, IL-6, IL-1β, and increased iron and liver ferroportin. Desidustat reduced EPO requirement and anti-EPO antibodies. Desidustat also maintained normal hemoglobin levels after cessation of rhEPO treatment. Thus, novel prolyl hydroxylase inhibitor desidustat can treat EPO resistance via improved iron utilization and decreased inflammation (10).
Desidustat (15 mg/kg, oral) administration in rats is rapidly absorbed with Tmax of 1.0 h, with Cmax of 10.48µg/ml in the blood. The peak erythropoietin concentration was observed at 6 h after the dose, and the resultant increase in reticulocyte and hemoglobin-RBC indicated an alternate day dosing for an optimum efficacy (38, 40). The clearance of the compound is moderate, with t1/2 of 6.35 h. The major route of excretion was found to be the renal route, with 39% of unchanged drug excreted in the urine. However, the lack of clinically meaningful effect on systemic clearance of desidustat, even after 5/6th removal of kidneys or complete removal of the two kidneys in rats, suggest the importance of non-renal clearance mechanism on the overall disposition of desidustat (40).
In single-dose studies, desidustat was well-tolerated up to 300 mg/kg in mice, up to 800 mg/kg in rats, and up to 75 mg/kg in beagle dogs by the oral route. Based on one-month toxicity studies, 60 mg/kg in rats and 15 mg/kg in dogs was considered the maximum tolerated dose (MTD) and 5 mg/kg was considered the no observed adverse effect level (NOAEL) in the 28-day toxicity studies in dogs (44). The toxicity signs were mainly associated with accelerated pharmacology, related to the hemoglobin build-up in the blood, and it was reversible.
Phase I study was a randomized, double-blind, placebo-controlled trial, conducted in addition to a third part with open-label to evaluate the food/sex effect (44). Fifty six participants were enrolled in the first part, where a single dose of desidustat with strengths of 10, 25, 50, 100, 150, 200, and 300 mg was given to the volunteers. In the second part, every day single dose of 100, 150, 200, and 300 mg was given to thirty two participants. The gender and food effect were studied in twelve participants with 150 mg dose of desidustat.
The Cmax was 566.4, 6745, and 17858.3 ng/ml at 10 mg, 100 mg and 300 mg dose, with a dose-related pattern, and the AUC (h.ng/ml) for 24 hours was found to be 3597.2, 46,844.7, and 115,898.8, respectively for these doses. The tmax mean was found to be 1.25 to 3 h at this dose range, and the t1/2 mean range was 6.9 to 11.3 hours. Similar to the preclinical pharmacokinetic study, the excretion of drug was found in urine, with 27.5 to 41.1% drug excreted unchanged in the urine. The data indicated no accumulation of the drug at 100, 150, 200, and 300 mg doses.
Absorption of desidustat was delayed in presence of food (median tmax was 2 h as against 5 h in fed condition, with Cmax decreased by 2.39 fold in presence of food). However, the half-life of the drug was similar in fed or fasted conditions. The mean Cmax for male and female subjects were not different, though the extent of absorption in males was more than in females.
Desidustat showed a dose-related increase in mean serum EPO levels from 100 mg to 300 mg dose, with mean serum EPO Cmax values ranging from 6.6 mIU/L at 10 mg to 79.9 mIU/L at 300 mg dose level. The EPO level peak in placebo-treated subjects was 9.6 mIU/L, and in all subjects, the EPO levels returned to the baseline within 24 hours of the single dose administration. The serum EPO profile after multiple dosing followed a similar profile to single dosing, with a lag in time of approximately 6–8 h to EPO Cmax from pharmacokinetic tmax for desidustat. A higher number of subjects who received desidustat showed a hemoglobin increase of >0.5 g/dl at any timepoint compared to the placebo group. Also, the hemoglobin increase of >1.0 g/dl was seen more in subjects treated with desidustat, when compared to those that received placebo.
The drug-related adverse events (AEs) were of grade 1 severity in the single dose study and they included dizziness and headache were the most common symptoms. In the multiple dose study, the treatment-related AEs were all grade 1 in severity, with the exception of dizziness in two subjects, which was classified as grade 2 in severity (44).
Phase 2 trial of desidustat was a placebo-controlled, randomized, double-blind, parallel group and multicentric study (45), Desidustat doses were 100, 150, and 200 mg, administered alternate day for 6 weeks. The mean age of the patients was 48 years, with 57% females, and 43% male patients, and their mean baseline hemoglobin was 9.44 g/dL. At the end of the treatment, the mean increase in hemoglobin level in the treated patients was found to be 1.57, 2.22, and 2.92 g/dL in 100, 150, and 200 mg treatment arms, respectively, while the change in the placebo group was 0.46 g/dL. The response rate for hemoglobin rise (>1g/dL) was 66% in 100 mg, 75% in 150 mg, and 83% in 200 mg treatment arms, versus 23% in placebo arm, respectively. Total iron binding capacity (TIBC) was found to be increased in all treatment groups at the end of treatment. The transferrin saturation (TSAT) was decreased in 150 mg and 200 mg treatment arms. LDL levels were decreased in 150 mg treatment arm. Hepcidin levels were significantly decreased in all the desidustat treatment arms. The increase in the EPO levels was observed from 4 h to 24 hours post-dose in the first week as well as in the last week of the treatment.
A dose-related increase in Cmax and AUC0–t was observed in desidustat arms after single and multiple doses, and no accumulation was observed after repeated dosing for six weeks. The mean half-life of 6.56–13.79 h observed after first dose was found to be 6.20–8.70 h in the sixth week of the treatment (45).
Treatment emergent adverse events (TEAE) were lesser in the treatment arms than in the placebo arm. TEAEs were mainly abdominal pain, vomiting, and headache, and majority of TEAEs were mild. No patient was discontinued from the trial due to AE, and no deaths occurred during the trial. The number of patients experiencing AEs during the trial do not significantly differ (p > 0.05) in the treatment and placebo arms. No safety issues were observed in terms of laboratory values, vitals, and liver function tests (45).
In phase 3 study, desidustat was tested against darbepoetin. Patients of anemia (n=588) due to CKD without dialysis need, with baseline hemoglobin of 7.0-10.0 g/dL were randomized to receive either desidustat 100 mg (oral tablets thrice a week) or biosimilar darbepoetin (subcutaneous injection, 0.75 μg/kg once in two weeks) for 24 weeks (46).
Desidustat treatment caused a hemoglobin rise of 1.95 g/dL from the baseline, while the increase observed in darbepoietin group was 1.83 g/dL in the darbepoetin, confirming that desidustat treatment was noninferior to darbepoietin, with more responders in desidustat group for hemoglobin rise than in the darbepoietin group. Also, desidustat caused a significant decrease in hepcidin and low-density lipoprotein, and did not change the vascular endothelial growth factor after the treatment, at the tested dose (47). Majority of the AEs were mild, unrelated with the treatment, and resolved, and there were no treatment-related deaths. The treatment emergent adverse event hypertension had more incidents in the darbepoetin treated group (5.78%) compared to the desidustat group (1.70%).
In dialysis-dependent patients of CKD phase 3 study of desidustat was conducted across 38 centers in India (47). A total of 392 patients with clinical diagnosis of anemia due to CKD with dialysis need (Erythrocyte Stimulating Agent [ESA] naïve or prior ESA users) and with baseline hemoglobin levels of 8.0-11.0 g/dL (inclusive) were randomized in a 1:1 ratio to receive either desidustat oral tablets (thrice a week) or epoetin alfa subcutaneous injection for 24 weeks to maintain a hemoglobin level of 10-12 g/dL. The primary endpoint was to assess the change in the hemoglobin level between the desidustat and the epoetin alfa groups from the baseline to evaluation period week 16-24. The key secondary efficacy endpoint was the number of patients with hemoglobin response. The change in hemoglobin from the baseline to week 16-24 was 0.95 g/dL in the desidustat group and 0.80 g/dL in the epoetin alfa group, which met the prespecified noninferiority margin. The number of hemoglobin responders was significantly higher in the desidustat group (106 [59.22%]) when compared to the epoetin alfa group (89 [48.37%]) (p = 0.0382). The safety profile of the desidustat oral tablet was comparable with the epoetin alfa injection. There were no new risks or no increased risks seen with the use of desidustat compared to epoetin alfa. Thus, at the tested dose, desidustat profile is comparable in safety with epoetin alfa.
Most of the PHD inhibitors show nanomolar potency in PHD binding assays, though from translational mechanism perspective, HIF stabilization is a rate-limiting target engagement tool to assess the efficacy of any PHD inhibitor (37). Daprodustat (48) roxadustat (49), vadadustat (50), and desidustat (37) demonstrate HIF stabilization at micromolar potency in cell-based assays. A notable difference is the suboptimal effect (Emax) of daprodustat, which may have been translated in lesser EPO release caused by daprodustat in preclinical as well as clinical studies (51). On the other hand, the reticulocyte count, which is the hallmark of erythropoiesis, was found to be significantly increased with desidustat, as compared to vadadustat (52).
In CKD patients with anemia chronic inflammation and comorbidities are prominent. Desidustat has shown efficacy for improving hemoglobin at the same dose in anemia models of inflammation (PGPS-induced) or nephrectomy-induced CKD (38, 39, 42). PHD inhibitors have been reported to have toxicities related to accelerated pharmacology, arising out of elevated hemoglobin. However, no off-target toxicities observed following desidustat treatment (27, 44–47), as against more cancer-related death or tumor progression events and more esophageal or gastric erosion events in the daprodustat treatment (53). Reports indicated a higher portion of nondialysis-CKD patients and dialysis dependent-CKD patients treated with roxadustat experienced vascular access thrombosis compared with controls (28, 54). Vadadustat failed to meet noninferiority criteria in Major Adverse Cardiovascular Events (MACE) trial in the non-dialysis patient population, showed increased risk of thromboembolic events, driven by vascular access thrombosis in dialysis patients, and the risk of drug-induced liver injury (52). The roxadustat preclinical toxicity study indicated the side effects such as chronic or chronic active valve inflammation, increased production, myxomatous degeneration, and fibrinous thrombus in the heart; glandular mucosal erosion, ulcer, and inflammation in the stomach (54). Thus, appearance of off-target or severely increased thrombotic or gastrointestinal side effects in the trials were the major reasons for the decrease in the benefit/risk profile of the currently used drugs of PHD inhibitor class. Compared to other PHD inhibitors, desidustat treatment, either in nondialysis patients or in dialysis-dependent patients is noninferior to either epoetin alpha or darbepoietin (46, 47), without any serious side effect related to tumor progression, gastrointestinal trouble, or thrombosis. Significant enough, it showed a prominent decrease in hepcidin (45) levels.
The FDA review highlighted that the appearance of the thrombotic or hematological side effects of roxadustat could be related to the rise in the hemoglobin in the first eight weeks of trial, especially in the nondialysis patients (55). The increase in hemoglobin in the nondialysis patients, within eight weeks of treatment with roxadustat, was, indeed higher than the comparator ESA treatment (56), while it was like ESA in case of daprodustat (57) and desidustat (46). However, a major difference appears to be significantly higher efficacy in terms of hemoglobin rise by desidustat treatment, compared to daprodustat (36, 46). Thus, at tested doses and duration, desidustat seems to be effective with optimum increase in erythropoietin levels in preclinical and clinical settings, without sudden increase in hemoglobin within eight weeks of treatment in clinic, and it is devoid of any serious side effects, especially the off-target thrombotic or gastrointestinal side effects in up to phase 3 clinical trials (46, 47).
Most PHD inhibitors approved as on now, have a common pharmacophore containing glycinamide with carboxylic group at the terminal. Majority of these molecules have hydrophobic rings attached directly to central core or through spacer groups. Among these inhibitors, daprodustat differs in having sp3 rich cyclohexyl ring; however this has led to the formation of several polyoxygenated metabolites, which is mainly mediated by CYP2C8 and secondarily by CYP3A4 (57, 58). Vadadustat forms GSH-adduct during metabolism (52). GSH adduct formation can be associated with drug-induced liver, skin, and hematopoietic toxicity of many drugs leading to clinical toxicities (59). On other hand, Desidustat is comparatively a flat molecule and has relatively less hydrophobic sites for such metabolism (29). This is reflected in presence of minimally formed (<10%) metabolites in clinical studies.
Desidustat treatment is recommended once every alternate day, while vadadustat or daprodustat are prescribed for once daily administration. Desidustat administration does not induce any reactive or active metabolites and based on in vitro data, no clinically relevant drug-drug interactions can be predicted that are mediated by CYPs or transporter interactions. Additionally, no drug interaction is expected with phosphate binders and oral iron preparation (as no significant alteration in aqueous solubility of desidustat in the presence of phosphate binders or multivalent cations) (27). Table 1 shows the summary of the pharmacokinetic data of selected PHD inhibitors.
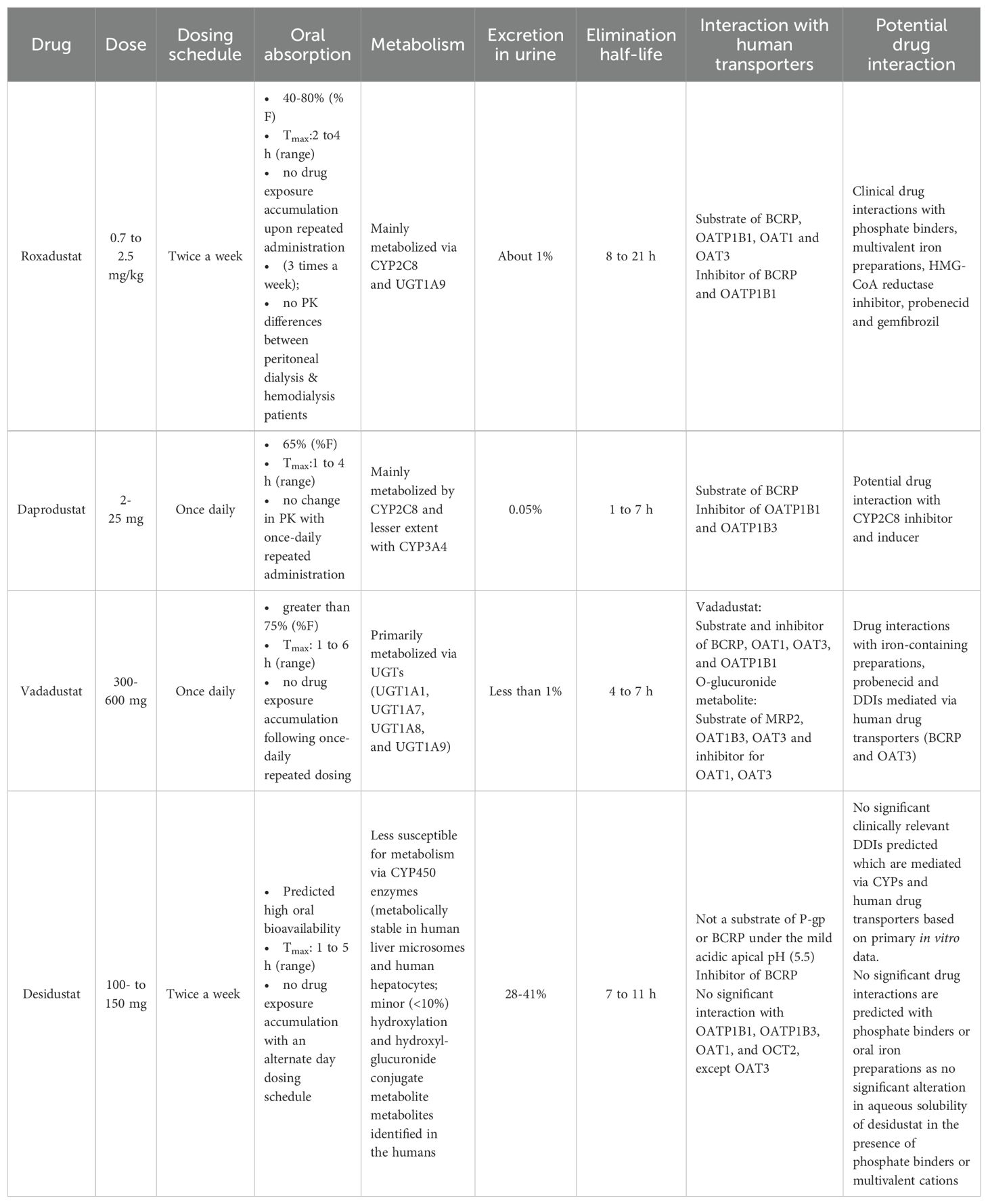
Table 1. Pharmacokinetics of selected PHD inhibitors.
Some anemic chronic kidney disease (CKD) patients are refractory to erythropoietin (EPO) effects due to inflammation, deranged iron utilization, and generation of EPO antibodies. Desidustat inhibited EPO-resistance caused by rhEPO treatment in preclinical studies, decreased hepcidin, IL-6, IL-1β, and increased iron and liver ferroportin (36). This treatment also reduced EPO requirement and anti-EPO antibodies and maintained normal hemoglobin levels after cessation of rhEPO treatment. A substantial and sustained improvement in hemoglobin levels was also observed in a clinical case of pure red cell aplasia (PRCA) emphasizing the crucial role of desidustat intervention in EPO-induced PRCA cases (59). Thus, desidustat can treat EPO resistance via improved iron utilization and decreased inflammation.
HIF-PHD inhibitors have demonstrated their therapeutic use in managing anemia in CKD patients, since the first approval and launch of roxadustat in China and Japan. This was followed by approvals of vadaustat and daprodustat in Japan, and of daprodustat and vadadustat in USA for the dialysis patients who were on dialysis for at least four months. An ideal HIF-PHD inhibitor should be devoid of the potential liabilities such as drug-induced liver injury, higher incidences of cancer, easophageal erosions, etc. CKD patients are usually treated with multiple drugs including statins, anti-hypertensives, anti-diabetics, clopidogrel, iron supplements or phosphate binders (60). Hence, it is important for an ideal HIF-PH inhibitor to demonstrate minimal potential for drug-drug interactions. These ideal characteristics of a HIF-PHD inhibitor with low risk/benefit ratio may provide a better therapeutic index (60–64). On this background, desidustat profile looks promising. Further long-term clinical and post-marketing studies may prove useful to realize the potential of desidustat.
AJ: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Writing – original draft, Writing – review & editing. VP: Formal analysis, Methodology, Writing – original draft, Writing – review & editing. HP: Formal analysis, Investigation, Methodology, Writing – review & editing. MJ: Conceptualization, Investigation, Methodology, Resources, Writing – review & editing. RD: Conceptualization, Formal analysis, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Authors AJ, VP, HP, MJ, and RD was/were employed by Zydus Lifesciences Limited.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. St Peter WL, Guo H, Kabadi S, Gilbertson DT, Peng Y, Pendergraft T, et al. Prevalence, treatment patterns, and healthcare resource utilization in Medicare and commercially insured non-dialysis-dependent chronic kidney disease patients with and without anemia in the United States. BMC Nephrol. (2018) 19:67. doi: 10.1186/s12882-018-0861-1
2. Stauffer ME, Fan T. Prevalence of anemia in chronic kidney disease in the United States. PloS One. (2014) 9:e84943. doi: 10.1371/journal.pone.0084943
3. Babitt JL, Lin HY. Mechanisms of anemia in CKD. J Am Soc Nephrol. (2012) 23:1631–4. doi: 10.1681/ASN.2011111078
4. Inker LA, Grams ME, Levey AS, Coresh J, Cirillo M, Collins JF, et al. Relationship of estimated GFR and albuminuria to concurrent laboratory abnormalities: an individual participant data meta- analysis in a global consortium. Am J Kidney Dis. (2019) 73:206–17. doi: 10.1053/j.ajkd.2018.08.013
5. Eckardt KU. Erythropoietin production in liver and kidneys. Curr Opin Nephrol Hypertens. (1996) 5:28–34. doi: 10.1097/00041552-199601000-00007
6. Willekens FL, Were JM, Groenen-Döpp YA, Roerdinkholder-Stoelwinder B, De Pauw B, Bosman GJ. Erythrocyte vesiculation: a self-protective mechanism? Br J Haematol. (2008) 141:549–56. doi: 10.1111/j.1365-2141.2008.07055.x
7. Richmond TD, Chohan M, Barber DL. Turning cells red: signal transduction mediated by erythropoietin. Trends Cell Biol. (2005) 15:146–55. doi: 10.1016/j.tcb.2005.01.007
8. Gupta J, Mitra N, Kanetsky PA, Devaney J, Wing MR, Reilly M, et al. Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin J Am Soc Nephrol. (2012) 7:1938–46. doi: 10.2215/CJN.03500412
9. Panwar B, Gutiérrez OM. Disorders of iron metabolism and anemia in chronic kidney disease. Semin Nephrol. (2016) 36:252–61. doi: 10.1016/j.semnephrol.2016.05.002
10. Joharapurkar AA, Patel VJ, Kshirsagar SG, Patel MS, Savsani HH, Kajavadara C, et al. Prolyl hydroxylase inhibitor desidustat improves anemia in erythropoietin hyporesponsive state. Curr Res Pharmacol Drug Discovery. (2022) 3:100102. doi: 10.1016/j.crphar.2022.100102
11. Skali H, Parving HH, Parfrey PS, Burdmann EA, Lewis EF, Ivanovich P, et al. TREAT Investigators: Stroke in patients with type 2 diabetes mellitus, chronic kidney disease, and anemia treated with darbepoetin alfa: the Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) experience. Circulation. (2011) 124:2903–8. doi: 10.1161/CIRCULATIONAHA.111.030411
12. McMurray JJ, Uno H, Jarolim P, Desai AS, de Zeeuw D, Eckardt KU, et al. Predictors of fatal and nonfatal cardiovascular events in patients with type 2 diabetes mellitus, chronic kidney disease, and anemia: an analysis of the Trial to Reduce cardiovascular Events with Aranesp (darbepoetin-alfa) Therapy (TREAT). Am Heart J. (2011) 162:748–755.e3. doi: 10.1016/j.ahj.2011.07.016
13. McCullough PA, Lepor NE. The deadly triangle of anemia, renal insufficiency, and cardiovascular disease: implications for prognosis and treatment. Rev Cardiovasc Med. (2005) 6:1–10. doi: 10.1016/j.carrev.2005.06.002
14. Mallick S, Rafiroiu A, Kanthety R, Iqbal S, Malik R, Rahman M. Factors predicting erythropoietin resistance among maintenance hemodialysis patients. Blood Purif. (2012) 33:238–44. doi: 10.1159/000335256
15. Raine AE. Hypertension, blood viscosity, and cardiovascular morbidity in renal failure: implications of erythropoietin therapy. Lancet. (1988) 1:97–100. doi: 10.1016/s0140-6736(88)90293-0
16. Besarab A, Bolton WK, Browne JK, Egrie JC, Nissenson AR, Okamoto DM, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med. (1998) 339:584–90. doi: 10.1056/NEJM199808273390903
17. Zhang Y, Thamer M, Stefanik K, Kaufman J, Cotter DJ. Epoetin requirements predict mortality in hemodialysis patients. Am J Kidney Dis. (2004) 44:866–76. doi: 10.1016/S0272-6386(04)01086-8
18. Macdougall IC, Casadevall N, Locatelli F, Combe C, London GM, Di Paolo S, et al. Incidence of erythropoietin antibody-mediated pure red cell aplasia: the prospective immunogenicity surveillance registry (PRIMS). Nephrol Dial Transplant. (2015) 30:451–60. doi: 10.1093/ndt/gfu297
19. Wish JB, Aronoff GR, Bacon BR, Brugnara C, Eckardt KU, Ganz T, et al. Positive iron balance in chronic kidney disease: how much is too much and how to tell? Am J Nephrol. (2018) 47:72–83. doi: 10.1159/000486968
20. Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, et al. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J. (2004) 381:761–7. doi: 10.1042/BJ20040620
21. Takeda K, Aguila HL, Parikh NS, Li X, Lamothe K, Duan LJ, et al. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood. (2008) 111:3229–35. doi: 10.1182/blood-2007-09-114561
22. Aragonés J, Schneider M, Van Geyte K, Fraisl P, Dresselaers T, Mazzone M, et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. (2008) 40:170–80. doi: 10.1038/ng.2007.62
23. Takeda K, Cowan A, Fong GH. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation. (2007) 116:774–81. doi: 10.1161/CIRCULATIONAHA.107.701516
24. Kobayashi H, Liu Q, Binns TC, Urrutia AA, Davidoff O, Kapitsinou PP, et al. Distinct subpopulations of FOXD1 stroma-derived cells regulate renal erythropoietin. J Clin Invest. (2016) 126:1926–38. doi: 10.1172/JCI83551
25. Zaritsky J, Young B, Wang HJ, Westerman M, Olbina G, Nemeth E, et al. Hepcidin–a potential novel biomarker for iron status in chronic kidney disease. Clin J Am Soc Nephrol. (2009) 4:1051–6. doi: 10.2215/CJN.05931108
26. Wilkinson N, Pantopoulos K. IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2α mRNA translation. Blood. (2013) 122:1658–68. doi: 10.1182/blood-2013-03-492454
27. Dhillon S. Desidustat: first approval. Drugs. (2022) 82:1207–12. doi: 10.1007/s40265-022-01744-w
28. Gul K, Zaman N, Azam SS. Roxadustat and its failure: A comparative dynamic study. J Mol Graph Model. (2023) 120:108422. doi: 10.1016/j.jmgm.2023.108422
29. Joharapurkar AA, Pandya VB, Patel VJ, Desai RC, Jain MR. Prolyl hydroxylase inhibitors: A breakthrough in the therapy of anemia associated with chronic diseases. J Med Chem. (2018) 61:6964–82. doi: 10.1021/acs.jmedchem.7b01686
30. Nakanishi T, Kuragano T. Growing concerns about using hypoxia-inducible factor prolyl hydroxylase inhibitors for the treatment of renal anemia. Clin Kidney J. (2024) 17:sfae051. doi: 10.1093/ckj/sfae051
31. Buchholz B, Eckardt KU. Role of oxygen and the HIF-pathway in polycystic kidney disease. Cell Signal. (2020) 69:109524. doi: 10.1016/j.cellsig.2020.109524
32. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
33. Tóth A, Csiki DM, Nagy B Jr, Balogh E, Lente G, Ababneh H, et al. Daprodustat accelerates high phosphate-induced calcification through the activation of HIF-1 signaling. Front Pharmacol. (2022) 13:798053. doi: 10.3389/fphar.2022.798053
34. Ku E, Del Vecchio L, Eckardt KU, Haase VH, Johansen KL, Nangaku M, et al. Novel anemia therapies in CKD: conclusions from a Kidney Disease: Improving Global Outcomes(KDIGO) Controversies Conference. Kidney Int. (2023) 104:655–80. doi: 10.1016/j.kint.2023.05.009
35. Sant’Ana PG, Tomasi LC, Murata GM, Vileigas DF, Mota GAF, Souza SLB, et al. Hypoxia- inducible factor 1-alpha and glucose metabolism during cardiac remodeling progression from hypertrophy to heart failure. Int J Mol Sci. (2023) 24:6201. doi: 10.3390/ijms24076201
36. Singh AK, Carroll K, McMurray JJV, Solomon S, Jha V, Johansen KL, et al. Daprodustat for the treatment of anemia in patients not undergoing dialysis. N Engl J Med. (2021) 385:2313–24. doi: 10.1056/NEJMoa2113380
37. Janssens LK, Stove CP. Sensing an oxygen sensor: development and application of activity-based assays directly monitoring HIF heterodimerization. Anal Chem. (2021) 93:14462–70. doi: 10.1021/acs.analchem.1c02923
38. Jain MR, Joharapurkar AA, Pandya V, Patel V, Joshi J, Kshirsagar S, et al. Pharmacological characterization of ZYAN1, a novel prolyl hydroxylase inhibitor for the treatment of anemia. Drug Res (Stuttg). (2016) 66:107–12. doi: 10.1055/s-0035-1554630
39. Jain M, Joharapurkar A, Patel V, Kshirsagar S, Sutariya B, Patel M, et al. Pharmacological inhibition of prolyl hydroxylase protects against inflammation-induced anemia via efficient erythropoiesis and hepcidin downregulation. Eur J Pharmacol. (2019) 843:113–20. doi: 10.1016/j.ejphar.2018.11.023
40. Patel H, Joharapurkar AA, Pandya VB, Patel VJ, Kshirsagar SG, Patel P, et al. Influence of acute and chronic kidney failure in rats on the disposition and pharmacokinetics of ZYAN1, a novel prolyl hydroxylase inhibitor, for the treatment of chronic kidney disease-induced anemia. Xenobiotica; fate foreign compounds Biol Syst. (2018) 48:37–44. doi: 10.1080/00498254.2016.1278287
41. Patel VJ, Joharapurkar AA, Kshirsagar SG, Patel MS, Savsani HH, Dodiya HS, et al. HIF stabilizer desidustat protects against complement-mediated diseases. Drug Res (Stuttg). (2024) 74:325–34. doi: 10.1055/a-2347-9919
42. Joharapurkar AA, Patel VJ, Kshirsagar SG, Patel MS, Savsani HH, Jain MR. Prolyl hydroxylase inhibitor desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines and oxidative stress. Drug Dev Res. (2021) 82:852–60. doi: 10.1002/ddr.21792
43. Yokozawa T, Oura H, Okada T. Metabolic effects of dietary purine in rats. J Nutr Sci Vitaminol (Tokyo). (1982) 28:519–26. doi: 10.3177/jnsv.28.519
44. Kansagra KA, Parmar D, Jani RH, Srinivas NR, Lickliter J, Patel HV, et al. Phase I clinical study of ZYAN1, A novel prolyl-hydroxylase (PHD) inhibitor to evaluate the safety, tolerability, and pharmacokinetics following oral administration in healthy volunteers. Clin Pharmacokinet. (2018) 57:87–102. doi: 10.1007/s40262-017-0551-3
45. Parmar DV, Kansagra KA, Patel JC, Joshi SN, Sharma NS, Shelat AD, et al. Outcomes of desidustat treatment in people with anemia and chronic kidney disease: A phase 2 study. Am J Nephrol. (2019) 49:470–8. doi: 10.1159/000500232
46. Agrawal D, Varade D, Shah H, Nazar A, Krishnan J, Shukla V, et al. Desidustat in anemia due to non-dialysis-dependent chronic kidney disease: A phase 3 study (DREAM-ND). Am J Nephrol. (2022) 53:352–60. doi: 10.1159/000523961
47. Gang S, Khetan P, Varade D, Chinta VR, Mavani S, Gupta U, et al. Desidustat in anemia due to dialysis-dependent chronic kidney disease: A phase 3 study (DREAM-D). Am J Nephrol. (2022) 53:343–51. doi: 10.1159/000523949
48. Ariazi JL, Duffy KJ, Adams DF, Fitch DM, Luo L, Pappalardi M, et al. Discovery and preclinical characterization of GSK1278863 (Daprodustat), a small molecule hypoxia inducible factor-prolyl hydroxylase inhibitor for anemia. J Pharmacol Exp Ther. (2017) 363:336–47. doi: 10.1124/jpet.117.242503
49. Del Balzo U, Signore PE, Walkinshaw G, Seeley TW, Brenner MC, Wang Q, et al. Nonclinical characterization of the hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat, a novel treatment of anemia of chronic kidney disease. J Pharmacol Exp Ther. (2020) 374:342–53. doi: 10.1124/jpet.120.265181
50. Zuk A, Si Z, Loi S, Bommegowda S, Hoivik D, Danthi S, et al. Preclinical characterization of vadadustat (AKB-6548), an oral small molecule hypoxia-inducible factor prolyl-4-hydroxylase inhibitor, for the potential treatment of renal anemia. J Pharmacol Exp Ther. (2022) 383:11–24. doi: 10.1124/jpet.122.001126
51. Caltabiano S, Cizman B, Burns O, Mahar KM, Johnson BM, Ramanjineyulu B, et al. Effect of renal function and dialysis modality on daprodustat and predominant metabolite exposure. Clin Kidney J. (2019) 12:693–701. doi: 10.1093/ckj/sfz013
52. Mitsubishi Tanabe Pharma Corporation. Vadadustat (VAFSEO tablets): Japanese prescribing information. 2020. Available online at: https://www.pmda.go.jp/PmdaSearch/iyakuDetail/ResultDataSetPDF/400315_39990D3F1020_1_01. (accessed July 21, 2020).
53. McCallum W, Weiner DE. HIF-PHIs for anemia management in CKD: potential and uncertainty ASCEND. Clin J Am Soc Nephrol. (2022) 17:1255–8. doi: 10.2215/CJN.02440222
54. Ugawa T, Ashizaki M, Murata A, Majikawa Y. Roxadustat (Evrenzo® tablet), a therapeutic drug for renal anemia: pharmacological characteristics and clinical evidence in Japan. Nihon Yakurigaku Zasshi. (2021) 156(3):187–97. doi: 10.1254/fpj.21001
55. US Food & Drug Administration. Cardiovascular and Renal Drugs Advisory Committee (2021). Available online at: https://www.fda.gov/advisory-committees/human-drug-advisory-committees/cardiovascular-and-renal-drugs-advisory-committee (accessed July 07, 2021).
56. Akizawa T, Iwasaki M, Yamaguchi Y, Majikawa Y, Reusch M. Phase 3, randomized, double-blind, active-comparator (Darbepoetin alfa) study of oral roxadustat in CKD patients with anemia on hemodialysis in Japan. J Am Soc Nephrol. (2020) 31:1628–39. doi: 10.1681/ASN.2019060623
57. Johnson BM, Stier BA, Caltabiano S. Effect of food and gemfibrozil on the pharmacokinetics of the novel prolyl hydroxylase inhibitor GSK1278863. Clin Pharmacol Drug Dev. (2014) 3:109–17. doi: 10.1002/cpdd.83
58. Dhillon S. Daprodustat: first approval. Drugs. (2020) 80:1491–7. doi: 10.1007/s40265-020-01384-y
59. Kashiv P, Malde S, Dubey S, Gupta S, Pawar T, Sejpal KN, et al. A case of erythropoietin (EPO)-induced pure red cell aplasia and its treatment efficacy with desidustat. Cureus. (2024) 16:e62022. doi: 10.7759/cureus.62022
60. Papotti B, Marchi C, Adorni MP, Potì F. Drug-drug interactions in polypharmacy patients: The impact of renal impairment. Curr Res Pharmacol Drug Discovery. (2021) 2:100020. doi: 10.1016/j.crphar.2021.100020
61. Palleria C, Di Paolo A, Giofrè C, Caglioti C, Leuzzi G, Siniscalchi A, et al. Pharmacokinetic drug-drug interaction and their implication in clinical management. J Res Med Sci. (2013) 18:601–10.
62. Patel H, Modi N, Chaudhari J, Patel P, Giri P, Patel H, et al. Nonclinical pharmacokinetic evaluation of desidustat: a novel prolyl hydroxylase inhibitor for the treatment of anemia. Eur J Drug Metab Pharmacokinet. (2022) 47:725–40. doi: 10.1007/s13318-022-00788-3
63. Thompson RA, Isin EM, Ogese MO, Mettetal JT, Williams DP. Reactive metabolites: current and emerging risk and hazard assessments. Chem Res Toxicol. (2016) 29:505–33. doi: 10.1021/acs.chemrestox.5b00410
Keywords: prolyl hydroxylase inhibitor, HIF, chronic kidney disease-induced anemia, hepcidin, hemoglobin
Citation: Joharapurkar A, Pandya V, Patel H, Jain M and Desai R (2024) Desidustat: a novel PHD inhibitor for the treatment of CKD-induced anemia. Front. Nephrol. 4:1459425. doi: 10.3389/fneph.2024.1459425
Received: 04 July 2024; Accepted: 02 October 2024;
Published: 22 October 2024.
Edited by:
Vikram Sabapathy, University of Virginia, United StatesReviewed by:
Rohit Upadhyay, Tulane University, United StatesCopyright © 2024 Joharapurkar, Pandya, Patel, Jain and Desai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amit Joharapurkar, YW1pdGpvaGFyYXB1cmthckB6eWR1c2xpZmUuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.