 Mythri Shankar
Mythri Shankar Manjusha Yadla
Manjusha Yadla
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Nephrol., 12 September 2024
Sec. Onconephrology
Volume 4 - 2024 | https://doi.org/10.3389/fneph.2024.1439288
This article is part of the Research TopicOnconephrology: Evolving Concepts and ChallengesView all 6 articles
Monoclonal gammopathy of renal significance (MGRS) is where kidney injury occurs due to the accumulation or effects of abnormal monoclonal proteins. These proteins, originating from non-cancerous or pre-cancerous plasma cells or B cells, deposit in specific areas of the kidney. Mechanisms contributing to MGRS include high levels of vascular endothelial growth factor secretion, autoantibodies targeting complement components, and targeting specific receptors leading to nephropathy. Kidney lesions in monoclonal gammopathy of renal significance (MGRS) are classified based on the presence of organized or nonorganized deposits, including fibrillar, microtubular, or crystal inclusions. Kidney biopsy is essential for confirming the diagnosis of MGRS by identifying monoclonal immunoglobulin deposits. Immunofluorescence helps determine the class of light and/or heavy chain involved in MGRS. The treatment approach is clone-directed and hence it depends on the presence of B cell clone or plasma cell clone or any detectable monoclonal protein. Chemotherapy targeting plasma cell or B cell malignancies and autologous hematopoietic cell transplantation may be used to manage MGRS. Kidney outcomes in MGRS patients strongly correlate with the hematologic response to chemotherapy.
Circulating monoclonal protein (M protein) is seen in 3% of individuals older than 50 years. This percentage increases to 5% after age 70 years (1, 2). Such individuals are diagnosed with a condition known as monoclonal gammopathy of undetermined significance (MGUS). MGUS is a condition where M-protein is below 30 g/l and a bone marrow examination shows less than 10% monoclonal plasma cells, along with the absence of damage to vital organs or events associated with multiple myeloma. It is important to note that MGUS is an asymptomatic condition. However, in some instances, monoclonal gammopathy can lead to severe organ damage, but it doesn’t meet the criteria for overt multiple myeloma (MM) or other malignant lymphoproliferative disorders such as B-cell non-Hodgkin lymphoma, which includes Waldenström’s macroglobulinemia (WM) or chronic lymphocytic leukemia (CLL) (3).
Hence, the International Kidney and Monoclonal Gammopathy Research Group (IKMG), in the year 2012, coined the term “monoclonal gammopathy of renal significance” (MGRS) to characterize a range of kidney disorders caused by the secretion of M-protein from a clone of B-cells, lymphoplasmacytic cells, or plasma cells that fail to satisfy current hematologic criteria for specific treatment. Recently, the IKMG expanded the definition of MGRS to encompass all B-cell or plasma cell proliferative disorders that generate a kidney-damaging M-protein, including conditions like smoldering multiple myeloma (MM), smoldering Waldenström’s macroglobulinemia (WM), and monoclonal B-cell lymphocytosis (MBL). Additionally, low-grade B-cell lymphomas and low-grade chronic lymphocytic leukemia (CLL) associated with kidney problems are now considered part of the updated MGRS definition. These clones, responsible for secreting M-proteins, can have a direct nephrotoxic effect and can also indirectly harm the kidneys through complement activation. Furthermore, these small B-cell or plasma cell clones can affect organs, such as the skin and peripheral nerves, leading to introduction of the concept of “monoclonal gammopathy of clinical significance” (MGCS) (4).
Anti-tumor therapy is currently not recommended for MGUS, smoldering myeloma, or asymptomatic WM (Table 1, Figure 1). The patients are closely monitored and treatment is considered only if they develop any symptoms or at the risk of developing any symptoms. However, this approach is not valid in MGRS as the M-protein may still cause kidney damage even if the tumor burden is not high. In such instances, the monoclonal gammopathy has a known significance due to the presence of kidney disease, which carries an increased risk of progressing to end-stage kidney disease (ESKD). Furthermore, there have been reported cases of recurrence after kidney transplantation (5–12).

Table 1. Definitions of B-cell and plasma cell proliferative disorders.
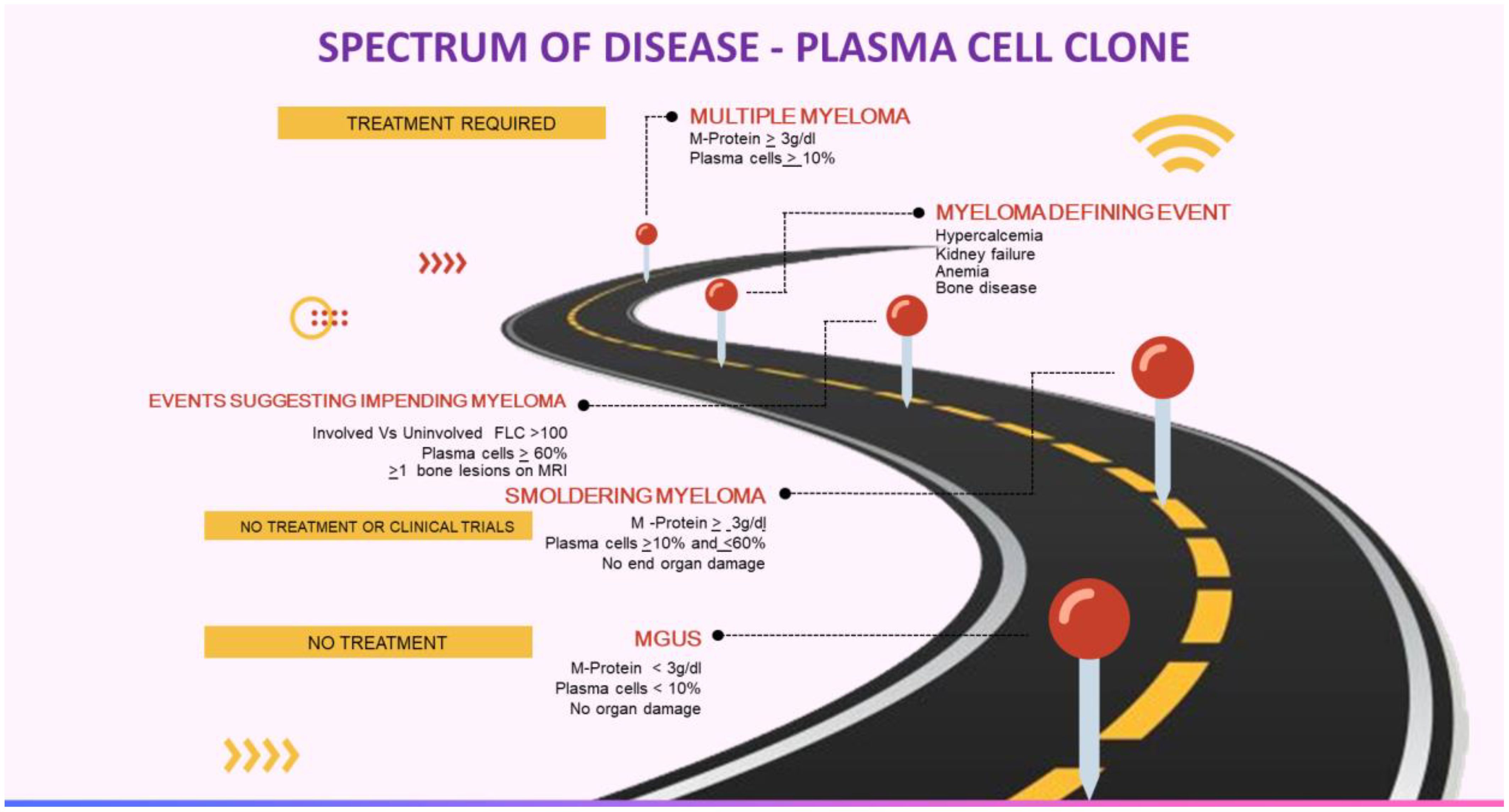
Figure 1. Multiple myeloma continuum. Courtesy: Mythri Shankar.
In MGRS, kidney damage primarily occurs due to the abnormal buildup or impact of monoclonal proteins in the kidney. These proteins, which may be light chains, heavy chains, or entire immunoglobulins, are produced by small, benign or pre-malignant clones of plasma cells or B cells. The specific locations within the kidney—such as the glomeruli, tubules, vessels, or interstitial areas—where these monoclonal proteins accumulate vary according to the unique biochemical characteristics of the involved pathogenic light and/or heavy chains.
Moreover, other mechanisms that contribute to the development of MGRS have also been identified, in addition to the deposition of monoclonal proteins within the kidney:
● vascular endothelial growth factor (VEGF) secretion in high amounts is implicated in patients with POEMS syndrome—characterized by polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes. This includes conditions like membranoproliferative glomerulonephritis-like lesions, thrombotic microangiopathy, and mesangiolysis with the formation of microcapillaries (13).
● Monoclonal proteins may function as C3 nephritic factor or autoantibodies targeting complement components such as complement factor H, factor I, and complement receptor 1 (CR1). This results in uncontrolled activation of the alternative complement pathway, causing C3 glomerulopathy linked with monoclonal gammopathy. Both processes facilitate the accumulation of complement factors such as C3 in the kidney, without substantial deposits of immunoglobulin (14).
● Circulating monoclonal immunoglobulin autoantibodies may target the phospholipase A2 receptor, leading to a type of membranous nephropathy that often recurs quickly after kidney transplantation (15). Additionally, there have been reports of monoclonal anti-glomerular basement membrane (GBM) disease caused by circulating monoclonal antibodies that attack type IV collagen (16).
The kidney lesions linked to MGRS can be classified based on the ultrastructural features of any deposits found in the kidney (17). These deposits can be categorized as either organized (i.e., having a substructure) or nonorganized (i.e., granular and lacking substructure). Occasionally, deposits may not be visible within the kidney.
Organized deposits in MGRS lesions are further classified as microtubular deposits, fibrillar deposits or crystal inclusions.
These include amyloidosis and fibrillary GN. In amyloidosis, the fibrillar deposits are Congo red positive, while in fibrillary glomerulonephritis, it is usually negative. Staining for the DnaJ heat shock protein family (Hsp40) member B9 (DNAJB9) helps differentiate between fibrillary glomerulonephritis (positive) and amyloid (negative) (18). However, recent reports suggest that fibrillary glomerulonephritis could be an independent entity unrelated to monoclonal gammopathy (19).
Lesions in MGRS with microtubular deposits are seen in conditions like monotypic immunotactoid glomerulopathy and monoclonal (type 1 and some type 2) cryoglobulinemia. Microtubules differ from fibrils because they are usually larger and feature hollow centers (20).
Lesions with crystal inclusions cover disorders like light chain proximal tubulopathy. It is a condition where the light chain crystals deposit in proximal tubular epithelial cells. Rarely, a non-crystalline deposit variant of proximal tubulopathy is also seen. Light chain crystalline podocytopathy features crystalline inclusions primarily within podocytes but also in other kidney cell types like proximal and distal tubular cells, endothelial cells, interstitial histiocytes, and mesangial cells (21). In crystal-storing histiocytosis, intracytoplasmic light chain crystalline inclusions are found within interstitial histiocytes, and sometimes in proximal tubular cells and podocytes (22). Cryocrystalglobulinemia is characterized by deposits made up of entire monoclonal immunoglobulins within glomerular endothelial cells or in the subendothelial space or in the vascular lumens or mesangial cells. Extrarenal deposits are also observed in all three conditions (23).
Nonorganized deposits in MGRS lesions include conditions classified under monoclonal immunoglobulin deposition diseases (MIDDs), such as light chain, heavy chain, or both light and heavy chain deposition disease, and monoclonal gammopathy-associated proliferative glomerulonephritis (24). This includes diseases involving deposits that are monoclonal immunoglobulin G (IgG), and less commonly, immunoglobulin M (IgM), immunoglobulin A (IgA) or light chain-only. Membranoproliferative glomerulonephritis is the most frequent type of injury observed in monoclonal gammopathy-associated proliferative glomerulonephritis, which encompasses conditions like proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) (25, 26) and C3 glomerulopathy with monoclonal gammopathy. In C3 glomerulopathy, the deposits mainly consist of C3 complement (27, 28). This category also includes rare cases of MGRS that histologically resemble polyclonal immunoglobulin-mediated kidney diseases, such as membranous nephropathy and anti-glomerular basement membrane (GBM) antibody (Goodpasture’s) disease, which show monotypic staining during immunofluorescence microscopy (15) (Figure 2).
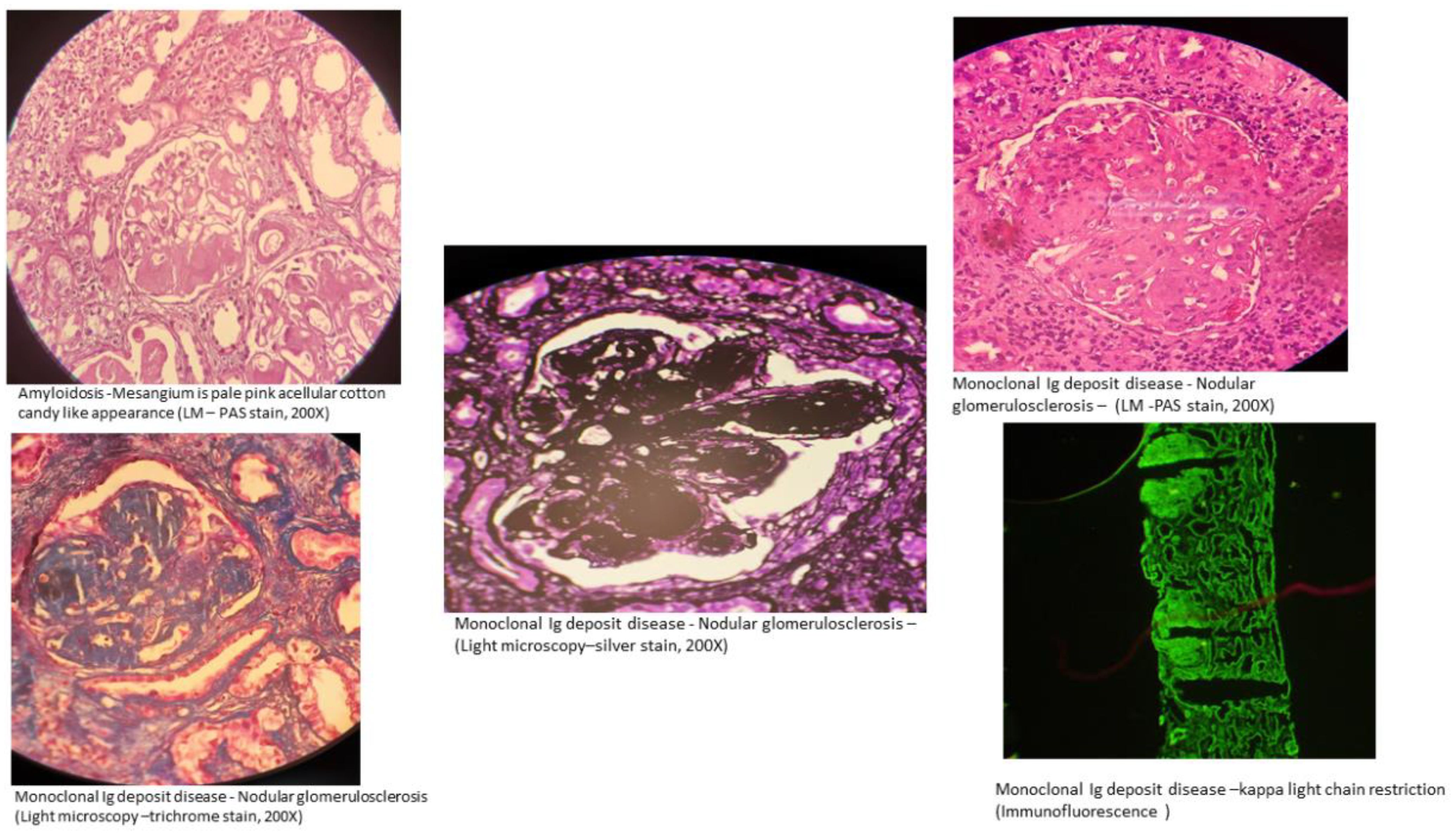
Figure 2. Kidney biopsy images of MGRS. Image Credit: Vinay .KS
MGRS lesions without any deposits encompass conditions such as thrombotic microangiopathy linked to monoclonal gammopathy and POEMS syndrome (13, 29).
Although it offers a structured approach to diagnosing kidney diseases associated with MGRS, it does not offer details regarding the clinical progression or prognosis of these various conditions.
Kidney disease in patients with MGRS may arise as a complication of already identified premalignant hematologic disorder or non-malignant hematologic disorder, such as MGUS or smoldering multiple myeloma, or it may be the first sign of a monoclonal gammopathy. Similar to the kidney diseases seen in multiple myeloma (30) and other malignant monoclonal gammopathies, those related to MGRS can manifest as acute or subacute kidney injury, chronic kidney disease, proteinuria and/or nephrotic syndrome, or electrolyte imbalances (31). The most common initial symptoms include impaired kidney function and proteinuria, possibly accompanied by hematuria. Additionally, MGRS can mimic kidney diseases typically not associated with monoclonal gammopathies, like membranous nephropathy and anti-glomerular basement membrane (GBM) antibody (Goodpasture’s) disease (15, 32).
The diagnosis of MGRS should be considered under the following circumstances:
Indicators associated with MGRS include proteinuria ≥1.5 g/day, hematuria, and an abnormal serum-free light chain ratio (33).
The suspicion should also arise in all patients with a premalignant or non-malignant monoclonal gammopathy with unexplained kidney function impairment and/or proteinuria. Additionally, patients presenting with unexplained kidney dysfunction and/or proteinuria, who upon further investigation with serum or urine protein electrophoresis, immunofixation, or a serum free light chain assay is found to have a monoclonal gammopathy should be suspected of MGRS. It is well established that urine-free light chain assay is not a clinically significant test at this point in time.
In most cases where MGRS is suspected, a kidney biopsy is performed unless there are reasons not to proceed. Confirmation of diagnosis of MGRS is by identifying monoclonal immunoglobulin deposits in the kidney via biopsy, which are characteristically restricted to a particular class of heavy chain or light chain as determined by immunofluorescence. The definitive method to demonstrate the nephrotoxic effects of monoclonal proteins is kidney biopsy, as the identification of monoclonal protein in serum or urine alone does not confirm its role in kidney disease. However, a kidney biopsy may be postponed under certain clinical situations:
In some cases of C3 glomerulonephritis, standard immunofluorescence might not reveal monoclonal immunoglobulin deposits. Here, protease digestion followed by paraffin immunofluorescence is the recommended method to unmask any hidden immunoglobulin deposits. Missing these “masked” monoclonal immunoglobulins can lead to a misdiagnosis, only identifying C3 glomerulonephritis instead of MGRS (33, 34). If no monoclonal immunoglobulins are detectable by paraffin immunofluorescence, the confirmation of circulating monoclonal protein through serum or urine protein electrophoresis, immunofixation, and/or serum free light chain assay is required to diagnose C3 glomerulopathy due to monoclonal gammopathy. Additional tests should include evaluating the alternative complement pathway, such as assessing C3 nephritic factor, C3 levels, C4 levels, C5b-9 complex and anti-complement factor H autoantibodies.
Interestingly, the majority (70 to 80 percent) of proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) may not demonstrate plasma cell or B cell clones in bone marrow examinations. In such cases, the monoclonal protein is found exclusively in the kidney and diagnosis is solely by kidney biopsy.
Patients presenting with albuminuria or nephrotic syndrome who have a confirmed diagnosis of immunoglobulin light chain (AL) amyloidosis from biopsies of non-kidney tissues may receive a presumptive diagnosis of renal AL amyloidosis without requiring a kidney biopsy.
Furthermore, patients diagnosed with monoclonal gammopathy displaying signs of Fanconi syndrome (e.g., subnephrotic-range proteinuria, aminoaciduria, hypophosphatemia, normoglycemic glycosuria, hypouricemia), can be presumptively diagnosed with light chain proximal tubulopathy.
Patients with monoclonal gammopathy can undergo kidney biopsy safely. A study involving 1993 patients who had either native or transplant kidney biopsies showed that the incidence of major hemorrhagic complications was comparable between patients with and without monoclonal gammopathy (35).
For patients diagnosed with MGRS, further assessments aim to characterize the clone to determine the appropriate treatment strategy.
For patients who either do not show a detectable clone in initial tests, or who possess an IgM monoclonal protein (suggesting a likely B cell or lymphoplasmacytic clone), additional diagnostic measures may include imaging such as computed tomography (CT) of the chest, abdomen, and pelvis, along with positron emission tomography (PET), if available, to identify any B cell clone (25, 36, 37) (Figure 3).
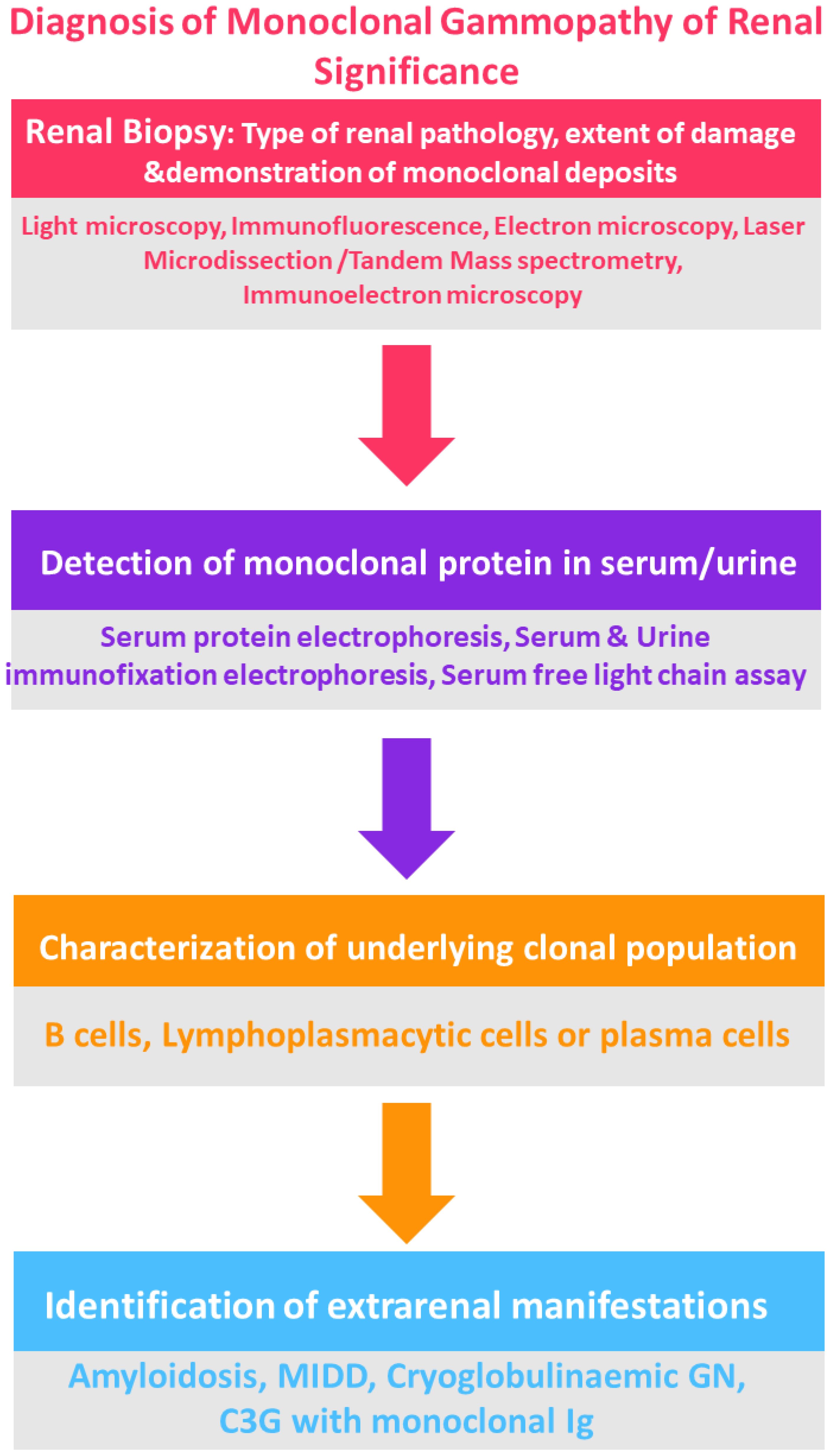
Figure 3. Flowchart for the diagnosis of MGRS. Courtesy: Mythri Shankar.
This includes serum protein electrophoresis and immunofixation, 24-hour urine protein electrophoresis and immunofixation, along with a serum free light chain assay. While the urine-free light chain assay is not recommended due to its lack of any added information, using these tests together improves the sensitivity for detecting monoclonal proteins, particularly in patients with small clones producing minimal protein levels. It is essential that any circulating monoclonal protein discovered matches the type present in kidney deposits. Identifying a serum or urine monoclonal protein is also critical for tracking the effectiveness of treatment.
This analysis should encompass immunohistochemistry and flow cytometry to evaluate the surface and intracellular markers of plasma cells and B cells. Staining for kappa and lambda light chains is essential to verify that the identified clone corresponds to the light chain restriction of the monoclonal deposits in the kidney. Additionally, cytogenetic and fluorescence in situ hybridization (FISH) analyses are becoming more common in guiding treatment decisions and may prove beneficial in specific scenarios.
For patients without a detectable clone from prior testing, or who possess an IgM monoclonal protein typically produced by B cells, additional evaluations may be necessary. This involves conducting imaging studies such as CT scans of the chest, abdomen, and pelvis, augmented with PET scans when possible, to identify potential B cell clones. Additionally, flow cytometry of peripheral blood lymphocytes is carried out to detect small, low-grade clones like those observed in chronic lymphocytic leukemia and monoclonal B cell lymphocytosis (MBL).
Patients diagnosed with a variant of MGRS that may be associated with extrarenal complications, including AL amyloidosis, monoclonal immunoglobulin deposition disease (MIDD), or monoclonal [type I] cryoglobulinemia, should be specifically assessed for these additional clinical manifestations.
The success in identifying a pathogenic clone in patients with MGRS varies depending on the specific disorder. For instance, in studies of patients with light chain deposition disease, bone marrow biopsy identified a plasma cell clone in 65 to nearly 100 percent of cases (11). In contrast, the detection rate of a clonal source in patients with PGNMID is much lower, ranging from 25 to 30 percent (25). When considering all major case series of MGRS patients, about 40 percent of cases do not have an identifiable clone (38).
The main goal in managing monoclonal gammopathy of renal significance (MGRS) is to maintain kidney function and inhibit the progression of any associated extrarenal manifestations. While most MGRS cases result from the deposition of monoclonal immunoglobulins in the kidneys, no treatments are currently available to stop this deposition or remove the already deposited materials (24) (Figure 4).
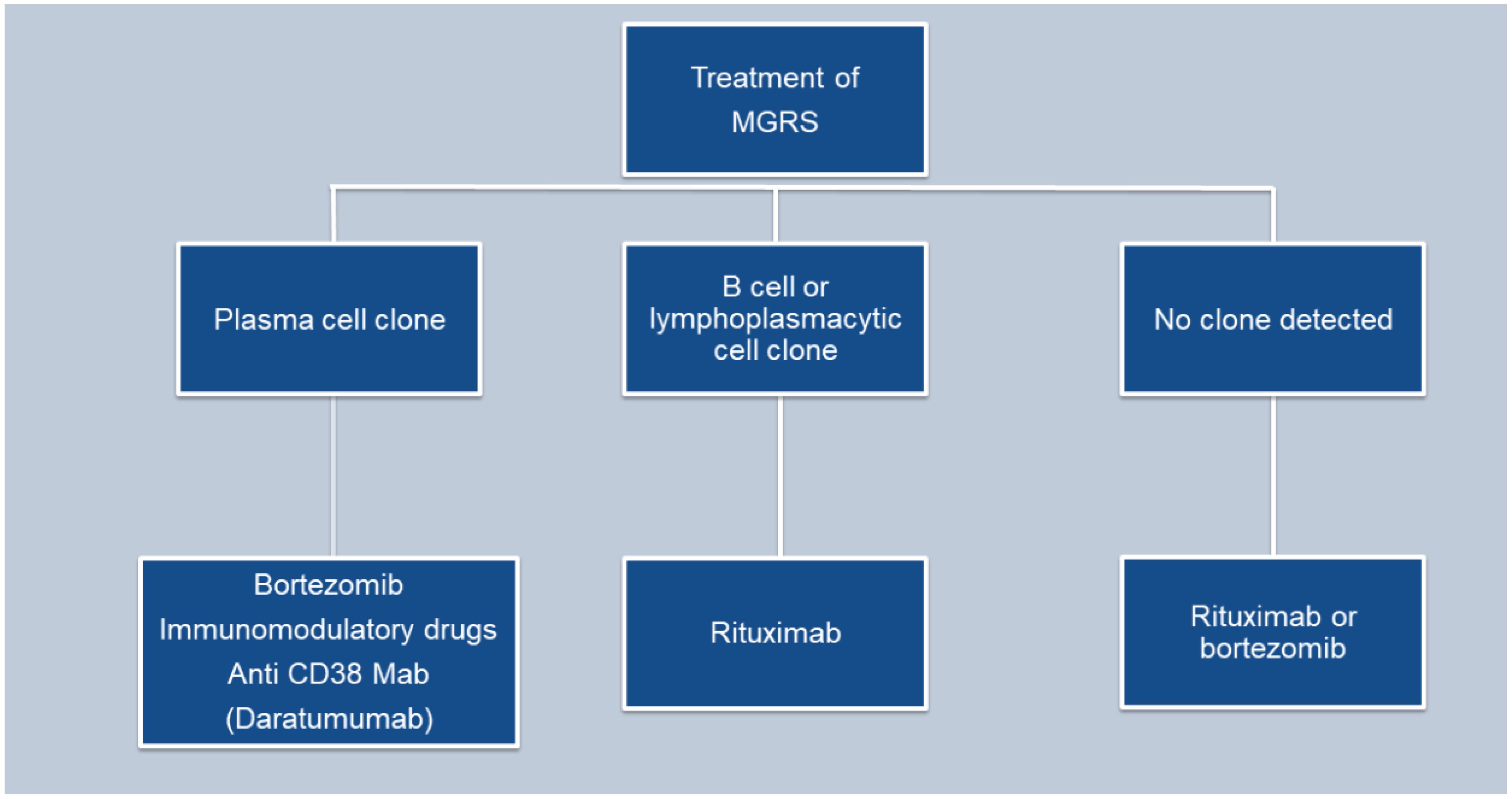
Figure 4. Flowchart depicting the treatment of MGRS.
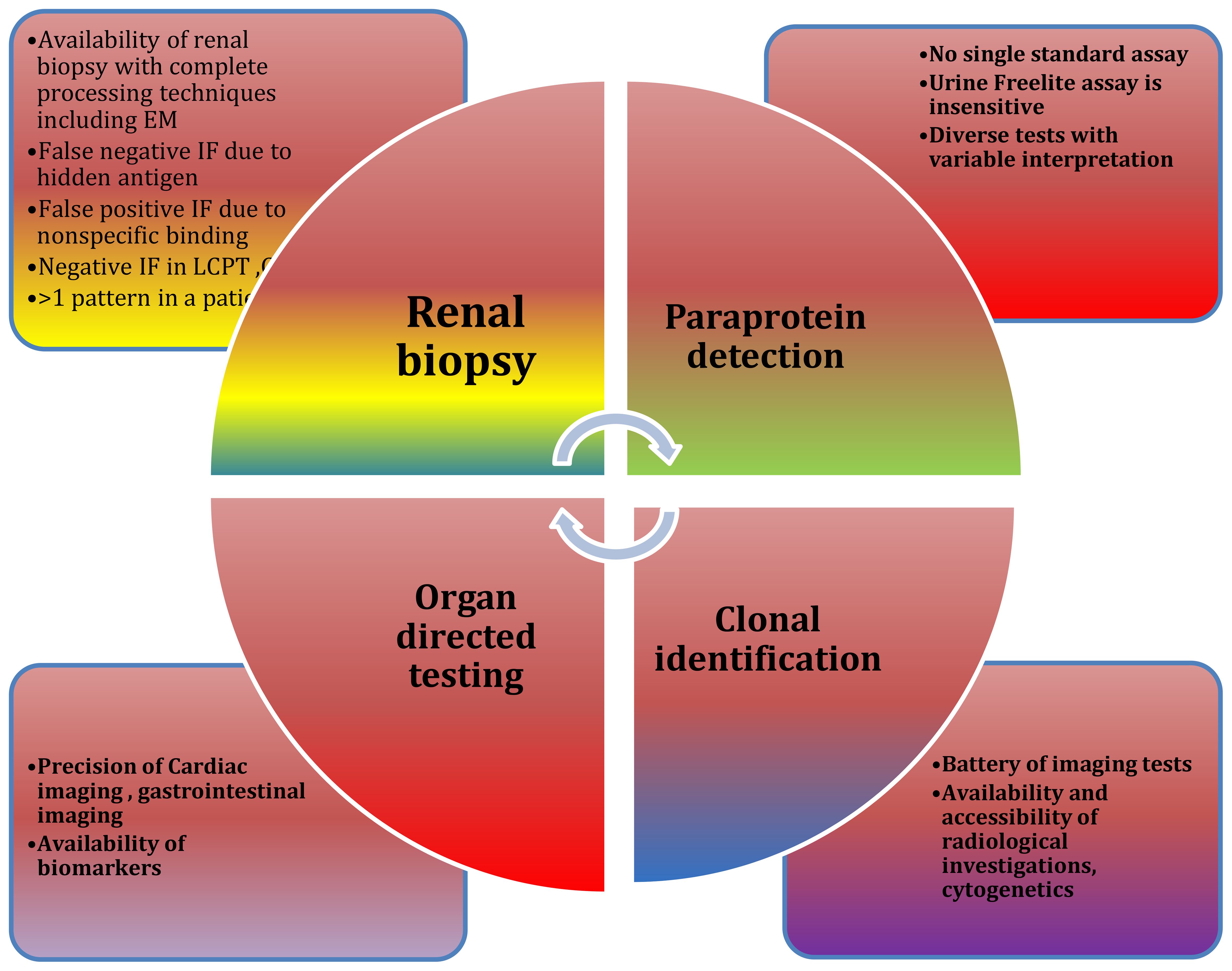
Figure 5. Diagnostic challenges: Challenges are at multiple levels while approaching MGRS.
The treatment strategy for MGRS largely hinges on the type of kidney injury, the characteristics of the clone (be it plasma cell, B cell, or lymphoplasmacytic) that is producing the nephrotoxic monoclonal immunoglobulin, and the ability to reverse or halt further kidney damage. Recent studies indicate that kidney outcomes in MGRS patients correlate strongly with the hematologic response to chemotherapy (39, 40).
Typically, the chemotherapeutic agents used to manage MGRS, target plasma cell or other B cell malignancies. These include proteasome inhibitors like bortezomib, carfilzomib, and ixazomib; monoclonal antibodies such as rituximab and daratumumab; alkylating agents including cyclophosphamide, bendamustine, and melphalan; immunomodulatory drugs like thalidomide, lenalidomide, and pomalidomide; and glucocorticoids such as prednisone and dexamethasone. In cases of conditions such as amyloidosis or monoclonal immunoglobulin deposition disease (MIDD), treatment strategies may include autologous hematopoietic cell transplantation. Preferably, medications that do not necessitate dosage alterations based on kidney function are used to reduce side effects, particularly cytopenias. Treatment decisions for MGRS should be made in association with a hematologist or an oncologist having expertise in using anti-myeloma and anti-lymphoma therapies.
Patients with PGNMID or C3 glomerulopathy with monoclonal gammopathy, are at an increased risk of progressive kidney disease. To prevent further kidney damage and decline in kidney function, treatment typically centers on eliminating the pathogenic clone responsible for the condition (24) (Tables 2, 3).
The treatment strategy for patients with PGNMID hinges on whether a B cell clone or plasma cell clone or detectable monoclonal protein is present in the urine or serum. The absence of randomized trials means no definitive guide for the optimal treatment approach exists. The support for a clone-directed treatment of PGNMID is largely based on observational studies and one small, uncontrolled trial (38, 41).
For patients with a detectable plasma cell clone, the treatment regimen mirrors that used for multiple myeloma (24). This typically includes a combination of bortezomib, cyclophosphamide and dexamethasone.
Daratumumab is considered an alternative, although data supporting its use are limited (41). Treatment may continue for up to six months if there is evidence of hematologic response and no toxicity.
In patients with an identifiable B cell clone, the treatment protocol mirrors that employed for Waldenström macroglobulinemia. The preferred treatment is rituximab (an anti-CD20 monoclonal antibody), either alone or combined with cyclophosphamide and dexamethasone, or bendamustine, given that most IgM-producing cells are CD20 positive (24).
In patients with PGNMID who do not have a detectable plasma or B cell clone, treatment is determined by the presence of monoclonal protein in the serum or urine. When kidney deposition of monoclonal immunoglobulin matches a detectable monoclonal protein of the same isotype in the serum or urine, it implies a causative relationship. Treatment for these patients involves chemotherapy targeted at eliminating the presumed clone responsible for producing the monoclonal protein. The specific chemotherapy regimen selected depends on the isotype of the monoclonal immunoglobulin identified in the serum or urine, given the absence of a detectable clone.
In patients with non-IgM monoclonal proteins (such as IgG or IgA) detected in the serum or urine and kidney, therapy strategy akin to that used for multiple myeloma is used. For IgM monoclonal protein, treatment is akin to Waldenstorm gammaglobulinemia.
With respect to AL Amyloidosis, at the beginning of the 21st century, patient classification was updated based on Mayo Clinic criteria into stages I, II, or III. This classification was determined by the levels of N-terminal prohormone of brain natriuretic peptide (NT-proBNP) and troponin T. Stage I patients had low levels of both markers (below 332 ng/L and 0.035 mg/L, respectively), stage II patients had high levels of one marker, and stage III patients had high levels of both markers (48, 49). Recently, immunoglobulin free light chain (FLC) levels have been added to these criteria, with minor adjustments to the cut-off points and the inclusion of FLC burden. The main objective of therapy is to achieve the best and most durable hematologic response, tailored to the often delicate condition of the patients. For those with severe cardiac disease, a rapid response is essential due to the direct myocardial toxicity of amyloidogenic light chains, making the quick suppression of FLC a crucial prognostic factor, especially in patients with stage III cardiac disease.
- For patients in stages I and II, the initial treatment should be melphalan combined with dexamethasone (M-Dex). Adding bortezomib to this regimen is expected to enhance hematologic and organ response rates. bortezomib should be introduced rapidly after 1 or 2 courses of M-Dex if there is no clonal response. For patients with advanced chronic kidney disease (CKD), cyclophosphamide is preferred over melphalan, and regimens such as cyclophosphamide-bortezomib-dexamethasone (CBD, also known as CyBorD or VCD) have shown effectiveness. Alternatively, thalidomide can replace bortezomib in the CTD regimen.
- Stage III cardiac patients face significant challenges due to poor median survival rates. Promising preliminary results have been observed with the CBD regimen, which appears to significantly reduce early mortality based on small case series. In young, carefully selected patients, cardiac transplantation might be considered, ideally after achieving hematologic remission.
- In selected patients, particularly those in stages I and II, high-dose melphalan and autologous stem cell transplantation (HDM/ASCT) should be considered if there is no severe renal insufficiency or other advanced organ failure (50).
Given the rarity of Monoclonal Immunoglobulin Deposition Disease (MIDD), controlled studies are lacking, and treatment approaches are based on expert consensus. Achieving an optimal hematologic response is crucial, similar to the treatment of AL amyloidosis, as it can lead to the regression of monoclonal immunoglobulin deposits if complete and sustained remission is achieved (51).
Small retrospective studies have shown that high-dose melphalan combined with autologous stem cell transplantation (HDM/ASCT) is an effective treatment, offering high hematologic response rates and low mortality related to the treatment. This is in contrast to AL amyloidosis, where patients often experience more systemic complications, leading to higher treatment-related mortality. Most data on HDM/ASCT predate the introduction of newer antimyeloma drugs. Initial results suggest that bortezomib-based treatments may yield similar hematologic response rates to HDM/ASCT, akin to treatments for multiple myeloma (52–55).
- For patients with CKD stages 1 to 3, the primary goal is to protect kidney function. A bortezomib-based regimen, such as cyclophosphamide, bortezomib, and dexamethasone (CBD), is recommended as the first line of treatment. HDM/ASCT should be considered for selected patients with good overall health and no significant extrarenal issues, particularly if only a partial hematologic response is achieved with the initial treatment.
- For patients with CKD stages 4 and 5, the chances of renal recovery are minimal. For those not eligible for kidney transplantation, the main goal is to preserve the function of other organs, especially the heart. A bortezomib-based regimen like CBD is recommended. If kidney transplantation is planned, the treatment goal is to ensure long-term function of the transplant, which requires an optimal clonal response. In such cases, HDM/ASCT should be considered after 3 to 4 cycles of a CBD-like regimen.
In MGRS-Amyloidosis, a poor prognosis was linked to high creatinine levels, elevated beta-2-microglobulin, and the need for hemodialysis at the time of diagnosis. In MGRS-Non Amyloidosis, the only factor associated with a higher risk of death was being over the age of 65 (56).
Diagnostic challenges at multiple levels are depicted in Figure 5. Emerging therapies for MGRS are show in Tables 4.
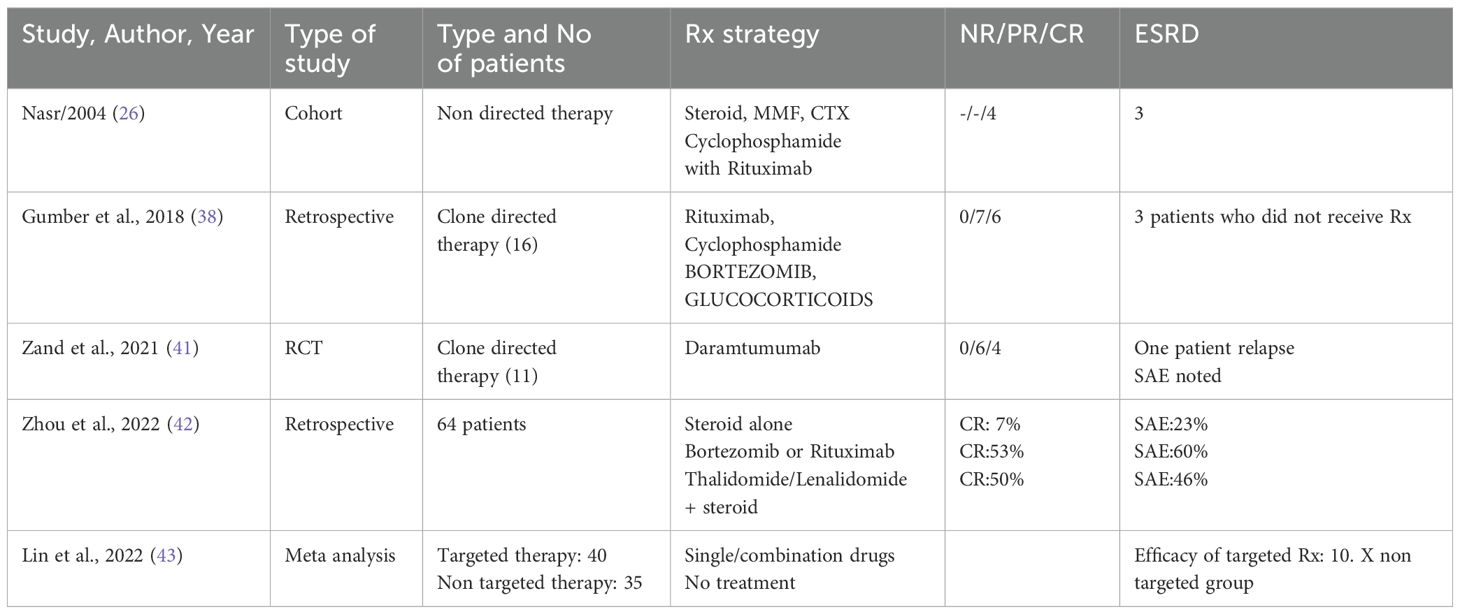
Table 2. Treatment of PGNMID.
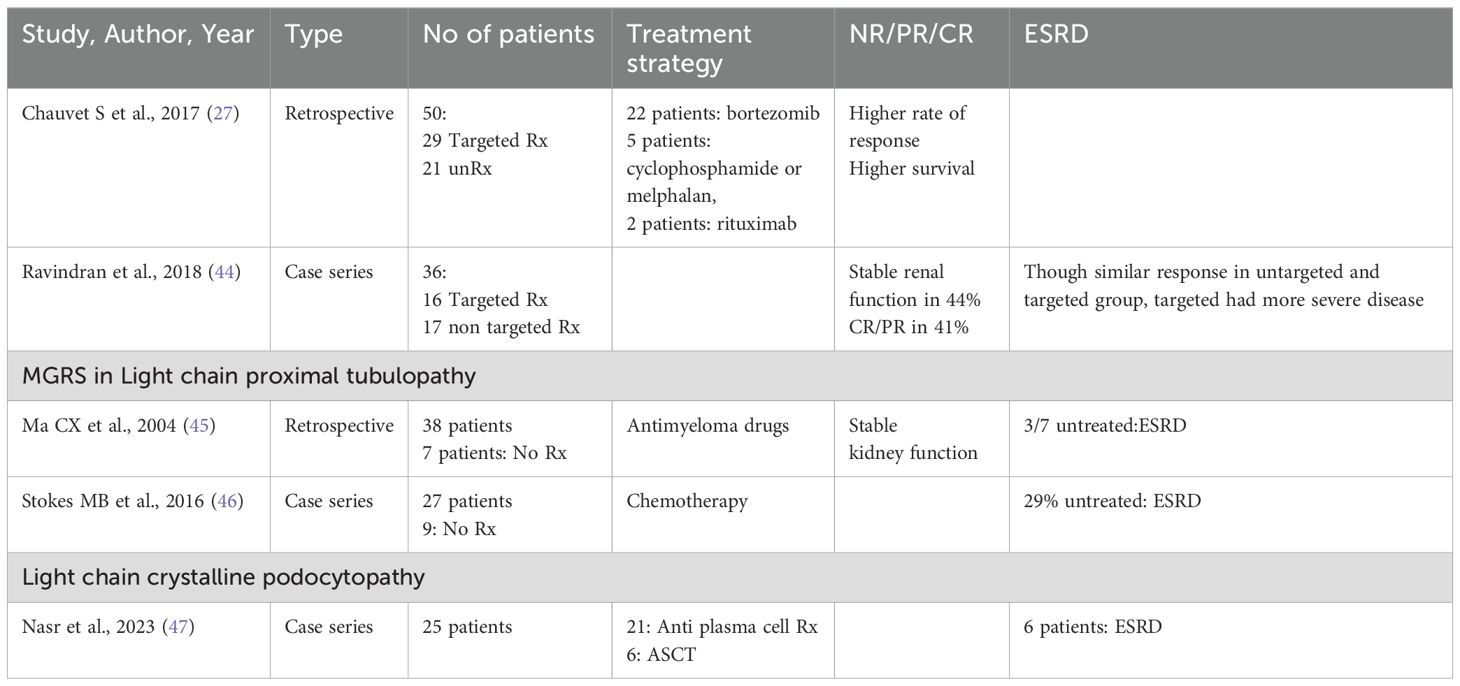
Table 3. Treatment of C3glomerulopathy.
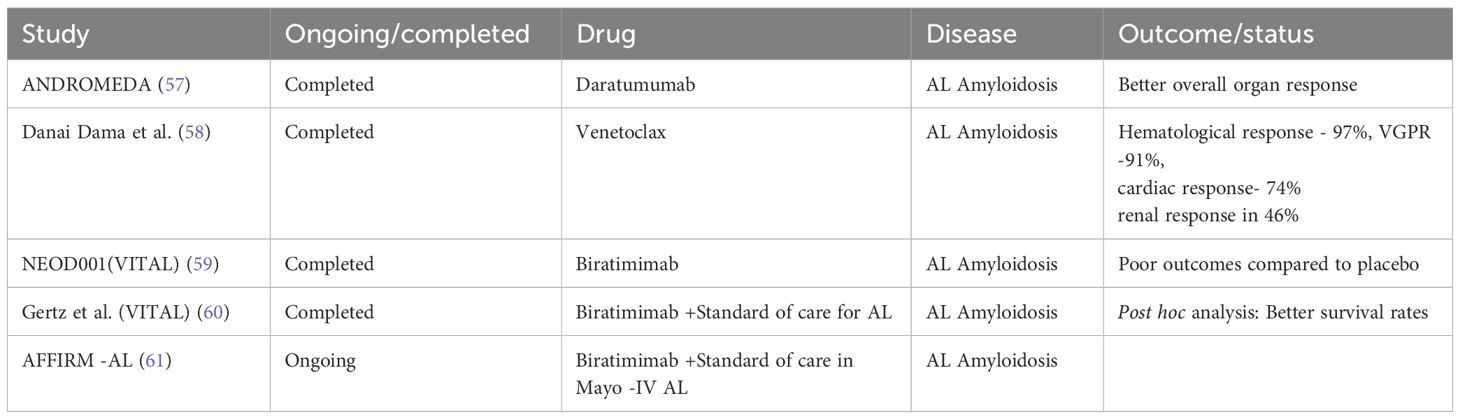
Table 4. Newer therapies in management of AL Amyloidosis appear to be promising, offering the advantages of improved organ response and the overall survival rate.
The following tests are conducted monthly to assess together the hematologic and kidney responses to treatment:
● Serum protein electrophoresis and immunofixation.
● 24-hour urine collection for total protein, protein electrophoresis, and immunofixation.
● Serum free light chain assay.
● Serum creatinine.
Evaluations can be conducted every two to three months for patients who have finished active treatment. Nevertheless, a substantial portion of MGRS patients, especially those with PGNMID, initially may not exhibit detectable circulating monoclonal proteins. In these cases, assessing a hematologic response isn’t feasible, and monitoring is often limited to serum creatinine levels and proteinuria quantification. Despite this, we should continue to track monoclonal protein levels as initially outlined since these proteins may become detectable later in the disease’s progression (62, 63).
Here is a conclusion for the document:
In summary, monoclonal gammopathy of renal significance (MGRS) encompasses a diverse array of kidney disorders driven by monoclonal proteins. The diagnostic and treatment approaches for MGRS must be tailored to the specific type of kidney damage and the characteristics of the pathogenic monoclonal proteins involved. Effective management relies on early detection, precise characterization of the monoclonal protein, and targeted therapy aimed at reducing the production of these proteins. Despite advances in treatment strategies, including the use of bortezomib-based regimens and autologous hematopoietic cell transplantation, the prognosis of MGRS remains variable. Continued research and clinical trials are essential to improve outcomes and develop more effective treatments for patients affected by this complex condition.
MS: Writing – original draft, Writing – review & editing. MY: Conceptualization, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rajkumar SV, Dimopoulos MA, Palumbo A. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. (2014) 15:548.
2. Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Offord JR, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. (2006) 354:1362–9.
3. van de Donk NW, Palumbo A, Johnsen HE, Engelhardt M, Gay F, Gregersen H, et al. The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: recommendations from the European Myeloma Network. Haematologica. (2014) 99:984–96.
4. Fermand JP, Bridoux F, Dispenzieri A, Jaccard A, Kyle RA, Leung N, et al. Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood. (2018) 132:1478–85.
5. Angioi A, Amer H, Fervenza FC, Sethi S. Recurrent light chain proximal tubulopathy in a kidney allograft. Am J Kidney Dis. (2016) 68:483–7.
6. Roth RM, Benson D, Hebert LA, Bissell MG, Satoskar AA, Nadasdy T, et al. Progressive renal light chain amyloidosis with the absence of detectable free monoclonal light chains after an autologous hematopoietic stem cell transplant for amyloid light chain amyloidosis. Arch Pathol Lab Med. (2013) 137:1304–8.
7. Miyazaki D, Yazaki M, Gono T, Kametani F, Tsuchiya A, Matsuda M, et al. AH amyloidosis associated with an immunoglobulin heavy chain variable region (VH1) fragment: a case report. Amyloid. (2008) 15:125–8.
8. Mallett A, Tang W, Hart G, McDonald SP, Hawley CM, Badve SV, et al. End-stage kidney disease due to fibrillary glomerulonephritis and immunotactoid glomerulopathy - outcomes in 66 consecutive ANZDATA registry cases. Am J Nephrol. (2015) 42:177–84.
9. Sathyan S, Khan FN, Ranga KV. A case of recurrent immunotactoid glomerulopathy in an allograft treated with rituximab. Transplant Proc. (2009) 41:3953–5.
10. Neel A, Perrin F, Decaux O, Dejoie T, Tessoulin B, Halliez M, et al. Long-term outcome of monoclonal (type 1) cryoglobulinemia. Am J Hematol. (2014) 89:156–61.
11. Sayed RH, Wechalekar AD, Gilbertson JA, Bass P, Mahmood S, Sachchithanantham S, et al. Natural history and outcome of light chain deposition disease. Blood. (2015) 126:2805–10. doi: 10.1182/blood-2015-07-658872
12. Nambirajan A, Bhowmik D, Singh G, Agarwal SK, Dinda AK. Monoclonal gammopathy of renal significance with light chain deposition disease diagnosed postrenal transplant: a diagnostic and therapeutic challenge. Transpl Int. (2015) 28:375–9.
13. Sanada S, Ookawara S, Karube H, Shindo T, Goto T, Nakamichi T, et al. Marked recovery of severe renal lesions in POEMS syndrome with high-dose melphalan therapy supported by autologous blood stem cell transplantation. Am J Kidney Dis. (2006) 47:672–9. doi: 10.1053/j.ajkd.2006.01.004
14. Jokiranta TS, Solomon A, Pangburn MK, Zipfel PF, Meri S. Nephritogenic lambda light chain dimer: a unique human miniautoantibody against complement factor H. J Immunol. (1999) 163:4590–6.
15. Debiec H, Hanoy M, Francois A, Guerrot D, Ferlicot S, Johanet C, et al. Recurrent membranous nephropathy in an allograft caused by IgG3κ targeting the PLA2 receptor. J Am Soc Nephrol. (2012) 23:1949–54. doi: 10.1681/ASN.2012060577
16. Tsuji T, Ohashi N, Sato T, Goto D, Nagata S, Matsuyama T, et al. Monoclonal immunoglobulin G1 κ-type atypical antiglomerular basement membrane disease accompanied by necrotizing glomerulonephritis. Clin Nephrol. (2020) 93:152–7. doi: 10.5414/CN109889
17. Leung N, Bridoux F, Batuman V, Chaidos A, Cockwell P, D’Agati VD, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol. (2019) 15:45–59. doi: 10.1038/s41581-018-0077-4
18. Alexander MP, Dasari S, Vrana JA, Riopel J, Valeri AM, Markowitz GS, et al. Congophilic fibrillary glomerulonephritis: A case series. Am J Kidney Dis. (2018) 72:325–36. doi: 10.1053/j.ajkd.2018.03.017
19. Andeen NK, Kung VL, Robertson J, Gurley SB, Avasare RS, Sitaraman S. Fibrillary glomerulonephritis, DNAJB9, and the unfolded protein response. Glomerular Dis. (2022) 2:164–75. doi: 10.1159/000525542
20. Terrier B, Karras A, Kahn JE, Le Guenno G, Marie I, Benarous L, et al. The spectrum of type I cryoglobulinemia vasculitis: new insights based on 64 cases. Med (Baltimore). (2013) 92:61–8. doi: 10.1097/MD.0b013e318288925c
21. Larsen CP, Bell JM, Harris AA, Messias NC, Wang YH, Walker PD. The morphologic spectrum and clinical significance of light chain proximal tubulopathy with and without crystal formation. Mod Pathol. (2011) 24:1462–9. doi: 10.1038/modpathol.2011.104
22. El Hamel C, Thierry A, Trouillas P, Bridoux F, Carrion C, Quellard N, et al. Crystal-storing histiocytosis with renal Fanconi syndrome: pathological and molecular characteristics compared with classical myeloma-associated Fanconi syndrome. Nephrol Dial Transplant. (2010) 25:2982–90. doi: 10.1093/ndt/gfq129
23. Leung N, Buadi FK, Song KW, Magil AB, Cornell LD. A case of bilateral renal arterial thrombosis associated with cryocrystalglobulinaemia. NDT Plus. (2010) 3:74–7. doi: 10.1093/ndtplus/sfp140
24. Sethi S, Rajkumar SV. Monoclonal gammopathy-associated proliferative glomerulonephritis. Mayo Clin Proc. (2013) 88:1284–93. doi: 10.1016/j.mayocp.2013.08.002
25. Bhutani G, Nasr SH, Said SM, Sethi S, Fervenza FC, Morice WG, et al. Hematologic characteristics of proliferative glomerulonephritides with nonorganized monoclonal immunoglobulin deposits. Mayo Clin Proc. (2015) 90:587–96. doi: 10.1016/j.mayocp.2015.01.024
26. Nasr SH, Markowitz GS, Stokes MB, Seshan SV, Valderrama E, Appel GB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. (2004) 65:85–96. doi: 10.1111/j.1523-1755.2004.00365.x
27. Chauvet S, Frémeaux-Bacchi V, Petitprez F, Karras A, Daniel L, Burtey S, et al. Treatment of B-cell disorder improves renal outcome of patients with monoclonal gammopathy-associated C3 glomerulopathy. Blood. (2017) 129:1437–47. doi: 10.1182/blood-2016-08-737163
28. Zand L, Kattah A, Fervenza FC, Smith RJ, Nasr SH, Zhang Y, et al. C3 glomerulonephritis associated with monoclonal gammopathy: a case series. Am J Kidney Dis. (2013) 62:506–14. doi: 10.1053/j.ajkd.2013.02.370
29. Martins M, Bridoux F, Goujon JM, Meuleman MS, Ribes D, Rondeau E, et al. Complement activation and thrombotic microangiopathy associated with monoclonal gammopathy: A national french case series. Am J Kidney Dis. (2022) 80:341–52. doi: 10.1053/j.ajkd.2021.12.014
30. Shankar M, Anandh U, Guditi S. Multiple facets of multiple myeloma in kidney biopsy: A multicenter retrospective study. Indian J Nephrol. (2024) 34:31–6. doi: 10.4103/ijn.ijn_362_22
31. Shankar M, Anandh U, Guditi S. PARAKID: Navigating the relation between paraproteins and kidney lesions: A multi-center retrospective observational study. Clin Nephrol. (2023) 100:269–74. doi: 10.5414/CN111123
32. Borza DB, Chedid MF, Colon S, Lager DJ, Leung N, Fervenza FC. Recurrent Goodpasture’s disease secondary to a monoclonal IgA1-kappa antibody autoreactive with the alpha1/alpha2 chains of type IV collagen. Am J Kidney Dis. (2005) 45:397–406. doi: 10.1053/j.ajkd.2004.09.029
33. Larsen CP, Ambuzs JM, Bonsib SM, Boils CL, Cossey LN, Messias NC, et al. Membranous-like glomerulopathy with masked IgG kappa deposits. Kidney Int. (2014) 86:154–61. doi: 10.1038/ki.2013.548
34. Larsen CP, Messias NC, Walker PD, Fidler ME, Cornell LD, Hernandez LH, et al. Membranoproliferative glomerulonephritis with masked monotypic immunoglobulin deposits. Kidney Int. (2015) 88:867–73. doi: 10.1038/ki.2015.195
35. Fish R, Pinney J, Jain P, Addison C, Jones C, Jayawardene S, et al. The incidence of major hemorrhagic complications after renal biopsies in patients with monoclonal gammopathies. Clin J Am Soc Nephrol. (2010) 5:1977–80. doi: 10.2215/CJN.00650110
36. Katzmann JA, Kyle RA, Benson J, Larson DR, Snyder MR, Lust JA, et al. Screening panels for detection of monoclonal gammopathies. Clin Chem. (2009) 55:1517–22. doi: 10.1373/clinchem.2009.126664
37. Palladini G, Russo P, Bosoni T, Verga L, Sarais G, Lavatelli F, et al. Identification of amyloidogenic light chains requires the combination of serum-free light chain assay with immunofixation of serum and urine. Clin Chem. (2009) 55:499–504. doi: 10.1373/clinchem.2008.117143
38. Gumber R, Cohen JB, Palmer MB, Kobrin SM, Vogl DT, Wasserstein AG, et al. A clone-directed approach may improve diagnosis and treatment of proliferative glomerulonephritis with monoclonal immunoglobulin deposits. Kidney Int. (2018) 94:199–205. doi: 10.1016/j.kint.2018.02.020
39. Cohen C, Royer B, Javaugue V, Szalat R, El Karoui K, Caulier A, et al. Bortezomib produces high hematological response rates with prolonged renal survival in monoclonal immunoglobulin deposition disease. Kidney Int. (2015) 88:1135–43. doi: 10.1038/ki.2015.201
40. Czarnecki PG, Lager DJ, Leung N, Dispenzieri A, Cosio FG, Fervenza FC. Long-term outcome of kidney transplantation in patients with fibrillary glomerulonephritis or monoclonal gammopathy with fibrillary deposits. Kidney Int. (2009) 75:420–7. doi: 10.1038/ki.2008.577
41. Zand L, Rajkumar SV, Leung N, Sethi S, El Ters M, Fervenza FC. Safety and efficacy of daratumumab in patients with proliferative GN with monoclonal immunoglobulin deposits. J Am Soc Nephrol. (2021) 32:1163–73. doi: 10.1681/ASN.2020101541
42. Zhou H, Li M, Zeng C, Chen Z, Zhang T, Cheng Z. Efficacy of immunomodulatory drugs in combination with dexamethasone in proliferative glomerulonephritis with monoclonal immunoglobulin deposits. Kidney Int Rep. (2022) 7(10):2166-175. doi: 10.1016/j.ekir.2022.07.009
43. Lin L, Chen N. A review on the diagnosis and treatment of proliferative glomerulonephritis with monoclonal immunoglobulin deposits. Int J Gen Med. (2022) 15:8577–82. doi: 10.2147/IJGM.S386733
44. Ravindran A, Fervenza FC, Smith RJH. Sethi S.C3glomerulopathy associated with monoclonal Ig is a distinct subtype. Kidney Int. (2018) 94:178.
45. Ma CX, Lacy MQ, Rompala JF, Dispenzieri A, Rajkumar SV, Greipp PR, et al. Aquired Fanconi syndrome is an indolent disorder in the absence of over multiple myeloma. Blood. (2004) 104:40.
46. Stokes MB, Valeri AM, Herlitz L, Khan AM, Siegel DS, Markowitz GS, et al. Light chain proximal tubulopathy: clinical and pathologic characteristics in the modern treatment era. J Am Soc Nephrol. (2016) 27:1555.
47. Nasr SH, Kudose S, Javaugue V, Harel S, Said SM, Pascal V, et al. Pathological characteristics of light chain crystalline podocytopathy. Kidney Int. (2023) 103:616.
48. Palladini G, Dispenzieri A, Gertz MA, Kumar S, Wechalekar A, Hawkins PN, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. (2012) 30:4541–9. doi: 10.1200/JCO.2011.37.7614
49. Dispenzieri A, Gertz MA, Kyle RA, Lacy MQ, Burritt MF, Therneau TM, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. (2004) 22:3751–7. doi: 10.1200/JCO.2004.03.029
50. Venner CP, Lane T, Foard D, Rannigan L, Gibbs SD, Pinney JH, et al. Cyclophosphamide, bortezomib, and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival. Blood. (2012) 119:4387–90. doi: 10.1182/blood-2011-10-388462
51. Mikhael JR, Schuster SR, Jimenez-Zepeda VH, Bello N, Spong J, Reeder CB, et al. Cyclophosphamide-bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyloidosis. Blood. (2012) 119:4391–4. doi: 10.1182/blood-2011-11-390930
52. Wechalekar AD, Goodman HJ, Lachmann HJ, Offer M, Hawkins PN, Gillmore JD. Safety and efficacy of risk-adapted cyclophosphamide, thalidomide, and dexamethasone in systemic AL amyloidosis. Blood. (2007) 109:457–64. doi: 10.1182/blood-2006-07-035352
53. Cibeira MT, Sanchorawala V, Seldin DC, Quillen K, Berk JL, Dember LM, et al. Outcome of AL amyloidosis after high-dose melphalan and autologous stem cell transplantation: long-term results in a series of 421 patients. Blood. (2011) 118:4346–52. doi: 10.1182/blood-2011-01-330738
54. Cordes S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Dingli D, et al. Ten-year survival after autologous stem cell transplantation for immunoglobulin light chain amyloidosis. Cancer. (2012) 118:6105–9. doi: 10.1002/cncr.27660
55. Kastritis E, Migkou M, Gavriatopoulou M, Zirogiannis P, Hadjikonstantinou V, Dimopoulos MA. Treatment of light chain deposition disease with bortezomib and dexamethasone. Haematologica. (2009) 94:300–2. doi: 10.3324/haematol.13548
56. Gozzetti A, Guarnieri A, Zamagni E, Zakharova E, Coriu D, Bittrich M, et al. Monoclonal gammopathy of renal significance (MGRS): Real-world data on outcomes and prognostic factors. Am J Hematol. (2022) 97:877–84. doi: 10.1002/ajh.26566
57. Kastritis E, Palladini G, Minnema MC, Wechalekar AD, Jaccard A, Lee HC, et al. Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med. (2021) 385:46–58.
58. Dima D, Hughes M, Orland M, Ullah F, Goel U, Anwer F, et al. Outcomes of venetoclax-based therapy in patients with t(11;14) light chain amyloidosis after failure of daratumumab-based therapy. Amyloid. (2024) 1–7.
59. Liedtke M, Merlini G, Landau H, Comenzo RL, Sanchorawala V, Weiss BM, et al. The VITAL amyloidosis study: A randomized, double-blind, placebo controlled, global, phase 3 study of NEOD001 in patients with AL amyloidosis and cardiac dysfunction. Blood. (2016) 128:5690.
60. Gertz MA, Cohen AD, Comenzo RL, Kastritis E, Landau HJ, Libby EN, et al. Birtamimab plus standard of care in light-chain amyloidosis: the phase 3 randomized placebo-controlled VITAL trial. Blood. (2023) 142:1208–18.
61. A Study to Evaluate the Efficacy and Safety of Birtamimab in Mayo Stage IV Patients With AL Amyloidosis (AFFIRM-AL) . Available online at: https://clinicaltrials.gov/study/NCT04973137 (April 14, 2024).
62. Kyle RA, Durie BG, Rajkumar SV, Landgren O, Blade J, Merlini G, et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. (2010) 24:1121–7.
Keywords: monoclonal, gammopathy, renal, significance, pathogenesis, treatment, kidney, plasma cells
Citation: Shankar M and Yadla M (2024) Unraveling monoclonal gammopathy of renal significance: a mini review on kidney complications and clinical insights. Front. Nephrol. 4:1439288. doi: 10.3389/fneph.2024.1439288
Received: 27 May 2024; Accepted: 16 August 2024;
Published: 12 September 2024.
Edited by:
Etienne Macedo, University of California, San Diego, United StatesReviewed by:
Arzu Velioglu, Marmara University, TürkiyeCopyright © 2024 Shankar and Yadla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manjusha Yadla, bWFuanV5YWRsYUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.