 Rita Kalot1,2
Rita Kalot1,2 Thomas M. Kitzler
Thomas M. Kitzler Elena Torban
Elena Torban- 1Department of Medicine and Department of Physiology, McGill University, Montreal, QC, Canada
- 2The Research Institute of the McGill University Health Center, Montreal, QC, Canada
- 3Department of Human Genetics, McGill University, Montreal, QC, Canada
- 4McGill University Health Center, Montreal, QC, Canada
Ciliopathies are a group of rare genetic disorders caused by defects to the structure or function of the primary cilium. They often affect multiple organs, leading to brain malformations, congenital heart defects, and anomalies of the retina or skeletal system. Kidney abnormalities are among the most frequent ciliopathic phenotypes manifesting as smaller, dysplastic, and cystic kidneys that are often accompanied by renal fibrosis. Many renal ciliopathies cause chronic kidney disease and often progress to end-stage renal disease, necessitating replacing therapies. There are more than 35 known ciliopathies; each is a rare hereditary condition, yet collectively they account for a significant proportion of chronic kidney disease worldwide. The primary cilium is a tiny microtubule-based organelle at the apex of almost all vertebrate cells. It serves as a “cellular antenna” surveying environment outside the cell and transducing this information inside the cell to trigger multiple signaling responses crucial for tissue morphogenesis and homeostasis. Hundreds of proteins and unique cellular mechanisms are involved in cilia formation. Recent evidence suggests that actin remodeling and regulation at the base of the primary cilium strongly impacts ciliogenesis. In this review, we provide an overview of the structure and function of the primary cilium, focusing on the role of actin cytoskeleton and its regulators in ciliogenesis. We then describe the key clinical, genetic, and molecular aspects of renal ciliopathies. We highlight what is known about actin regulation in the pathogenesis of these diseases with the aim to consider these recent molecular findings as potential therapeutic targets for renal ciliopathies.
Introduction
Ciliopathies are a heterogeneous group of rare genetic disorders that arise due to defects in the structure or function of the primary cilium. The severity of the disease presentation in ciliopathies is highly variable and depends on the nature of the ciliary dysfunction, which, in turn, is dependent on both the affected gene and the type of genetic variant (i.e., mutation) involved. Disease-causing variants in over 180 human genes give rise to more than 35 established human ciliopathies, including Bardet Biedl Syndrome (BBS), Nephronophthisis (NPHP), Polycystic Kidney Disease (PKD), Retinitis Pigmentosa (RP), Joubert Syndrome (JBTS), Orofacial-Digital Syndrome (OFD) and many others. Ciliopathies often affect multiple organs, resulting in a range of congenital defects such as cardiac malformation, retinal degeneration, or skeletal defects. Kidney abnormalities are among the most frequent ciliopathic phenotypes, including renal hypo-dysplasia, fluid-filled cysts, or renal fibrosis (1–4). Most renal ciliopathies cause chronic kidney disease and progress to end-stage renal disease (ESRD). Although each disease is an individually rare hereditary condition, collectively, ciliopathies contribute significantly to chronic kidney disease worldwide with an associated burden on families, society and the health system.
The primary cilium is a specialized microtubule-based organelle that extends from the surface of almost all vertebrate cells. Although primary cilium was observed in mammalian cells as early as 1898 (5), for almost a hundred years after its discovery, it was thought to be a vestigial organelle that lacked functional properties (6). The role of the primary cilium as a key cellular signaling hub crucial for tissue morphogenesis and homeostasis was only discovered in the 1990s (7, 8).
The primary cilium is nucleated at the basal body which anchors it to the apical plasma membrane. The ciliary microtubule core, known as the axoneme, is covered by a ciliary membrane, which contains thousands of signaling molecules, receptors, channels, and transporters. Therefore, the primary cilium acts as a “cellular antenna” that protrudes outside of the cell, senses the surrounding extracellular environment, and transduces the information inside the cell to trigger signaling pathways that govern important cellular responses (9). The primary cilium integrates different cellular components such as cytoskeletal actin and microtubule networks with unique cellular trafficking machinery and a repertoire of signaling molecules. Disruptions to these functions cause human ciliopathies including diseases with kidney involvement (10).
In this review, we will provide an overview of the structure and function of the primary cilium and describe the clinical, genetic and molecular aspects of the renal ciliopathies. We will emphasize the role of the actin cytoskeleton at the primary cilium and describe recent molecular findings that might be of therapeutic importance for renal ciliopathies, as summarized in Box 1.
Primary cilia and ciliogenesis
Motile and sensory cilia
Cilia have evolved to accomplish two major functions, motility and sensation, and, accordingly, are classified as motile and sensory (11). Motile cilia (or flagella) were the first organelles ever observed in mid-16th century, when Antony Van Leeuwenhoek used the simple light microscope he invented to visualize a tiny protozoan organism in rainwater that rapidly moved with the help of multiple miniature “legs” (12). These “legs” were later named “cilia” or “eyelashes” in Latin for the plural or “cilium” or an “eyelash” for a single structure (12). Motile cilia are found in a subset of tissues such as the bronchi of the lungs, cells lining brain ventricles and other organs (13), and are characterized by their ability to move in a coordinated manner to propel fluid or cells in the extracellular space (14). The axoneme of the motile cilium is composed of 9 microtubular duplets, which are the extensions of the 9 microtubular triplets that make up the basal body. In addition, motile cilia have an inner central pair of microtubules, an arrangement described as (9 + 2) and other proteins that enable motile cilia to beat coherently and directionally (15). Unlike motile cilia, a sensory, or primary, cilium is solitary (present in a single copy per cell), extending from the apical surface of almost all cells, including kidney or lung epithelia. The primary cilium lacks the central microtubule pair and the molecules associated with the ciliary motility. The primary cilium adopts a (9 + 0) arrangement and harbors an abundance of signaling molecules within its membrane enabling it to function as a signaling hub (16–18). Motile and primary cilia share a plethora of common structural proteins, however, many proteins, especially those required for ciliary motility or sensory function, are unique to each type (18).
The early stage of ciliogenesis
Cilia biogenesis, referred to as “ciliogenesis” hereafter, begins when a cell exits the cell cycle. Upon cell cycle exit in G0, the mother/daughter centriole pair, that serves as the microtubule organizing center (MTOC) during cell division, is liberated and the mother centriole undergoes maturation by acquiring morphologically and functionally distinct distal and subdistal appendages; it then ascends towards and docks beneath the apical surface, thereby becoming the basal body (19). The distal appendages are recruited to the basal body by centrosomal proteins and are required for anchoring the basal body to the apical cell membrane, while subdistal appendages help nucleate microtubules to build ciliary axoneme (20–23). Mother centriole maturation is governed by multiple proteins, many of which are linked to human ciliopathies. For example, mutations in two of the genes encoding distal appendage proteins, CEP164 and CEP83, cause nephronophthisis phenotypes, while the distortion of centrosomal proteins OFD1 and C2CD3 (both are also engaged in the distal appendage assembly) is linked to JBTS, OFD, and RP (9). Additionally, centriolar satellites, which are electron-dense structures that move around the basal body, maintain the centrosome and cilium by facilitating the transport of ciliary and centrosomal proteins (9).
Pathways of ciliogenesis
Depending on the cell type, two pathways of ciliogenesis, “intracellular” and “extracellular”, have been described (24, 25) (Figure 1). The intracellular pathway mostly occurs in non-polarized cells, although it was described in some epithelial cells as well (26). It begins with the formation of a large ciliary vesicle which encloses the mother centriole. The ciliary vesicle forms via trafficking of Golgi-derived cargo protein vesicles toward the distal appendages of the mother centriole and fusing with them. The ciliary vesicle grows and then fuses with the plasma membrane forming the ciliary membrane and the ciliary pocket. The latter is a membrane invagination that keeps the primary cilium submerged in the cytoplasm and is a site for ciliary protein trafficking and active endocytosis (24, 27, 28). On the other hand, polarized epithelial cells (such as kidney epithelium) utilize the extracellular pathway to form the primary cilium at the cellular apex. In this pathway, the primary cilium is formed only after the mother centriole has acquired its distal and subdistal appendages and docked directly to the plasma membrane. The core ciliogenic machinery is shared between the intracellular and extracellular pathways, however, some differences between the two routes exist (reviewed in (28)). For example, the exocyst complex is critical for ciliogenesis in the polarized kidney epithelial cells but is dispensable in the retinal pigmental epithelial RPE-1 cells employing intracellular ciliogenesis (29). Emerging evidence suggests that defects attributed to each specific ciliogenic pathway might contribute to different ciliopathy phenotypes or tissue-specific defects (28, 30, 31).
Box 1 Key points.
● The primary cilium is a specialized microtubule-based organelle that extends from the surface of almost all vertebrate cells and functions as a key signaling nexus for tissue morphogenesis and homeostasis.
● Defects to primary cilium formation and function cause a heterogeneous group of hereditary disorders known as ciliopathies. Ciliopathies that feature kidney abnormalities (renal ciliopathies) are among the most common.
● Recent evidence has revealed complex actin dynamics at the basal body that critically affect primary cilium formation.
● Many ciliopathy-associated proteins directly or indirectly regulate actin-cytoskeletal dynamics during cilium formation.
● Actin dynamics are disrupted across renal ciliopathy disorders, contributing to cystic transformation, fibrosis, and renal malformations due to dysregulated cilium-dependent developmental signaling processes.
● The actin-dependent mechanisms in renal ciliopathies are an emerging field that promises to uncover important mechanistic aspects of disease pathogenesis and discover new potential therapeutic targets.
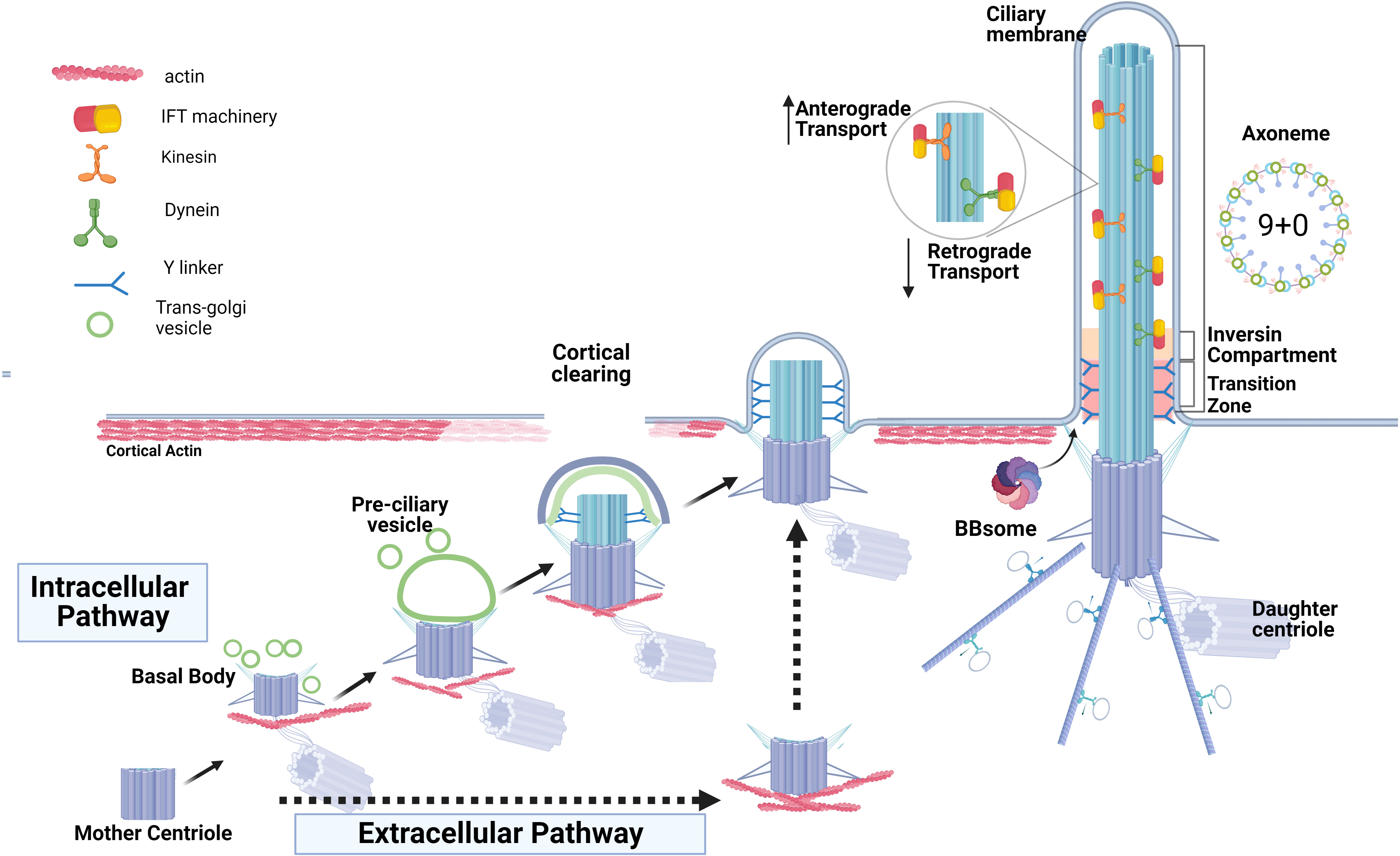
Figure 1 Two routes of ciliogenesis and the ciliary structures. The formation of the primary cilium by intracellular pathway commences when the mother centriole matures by acquiring distal and sub-distal appendages and transforming into the basal body. This is followed by the formation and enlargement of a pre-ciliary vesicle around the mother centriole via fusion of centriole’s appendages with the ciliary-protein cargo vesicles originating from trans-Golgi. The basal body ascends and then docks to the apical membrane and the ciliary vesicle fuses with the plasma membrane to form the ciliary membrane and the ciliary pocket. The extracellular route of ciliogenesis in the epithelial cells (intermittent black arrows) is characterized by direct docking of the basal body to the apical membrane and cilia elongation without the formation of a ciliary pocket. Basal body docking is preceded by clearing of cortical actin at the cell apex, an obligatory step permitting ciliogenesis. Docked basal body templates axoneme extension, and cilia growth via bi-directional anterograde and retrograde Intraflagella transport, IFT, is depicted. The inversin compartment and the transition zone immediately above the basal body form a diffusion barrier to control protein content within the ciliary shaft and ciliary membrane.
Initiation of ciliogenesis
To initiate ciliogenesis, the removal of the CP110-CEP97 protein complex from the mother centriole is required (32). This complex is localized at the distal end of centrioles and exerts control over centriole length by inhibiting centriolar microtubule extension (33–35). The elimination of CP110 is important for the transformation of the mother centriole into the basal body, as retention of CP110 at the mother centriole blocks cilium formation (36–38). Membrane remodeling occurs directly above the site of future basal body docking and is known as “cortical clearing”; it is an obligatory step that precedes docking in both intracellular and extracellular ciliogenesis pathways and is followed by basal body microtubule extension and assembly of the ciliary axoneme (39).
Primary cilia compartments
Transition zone
The transition zone (TZ) is a highly organized compartment at the base of the cilium, acting as a gate that controls protein entry to and exit from the ciliary compartment (40, 41). The TZ consists of transition fibers connecting the distal part of the basal body to the base of the ciliary membrane and Y-shaped structures (known as Y-links) (Figure 1). The Y-links serve as physical connectors between the microtubular doublet of axoneme and the ciliary membrane (40, 42). The precise mechanisms underlying the formation of the TZ and how it effectively carries out its regulatory functions are not well understood and remain areas of ongoing active investigation (43). Transition fibers act as docking sites for ciliary trafficking machinery (44, 45). In addition, disrupting the formation of transition fibers impedes basal body docking and blocks ciliogenesis at an early stage (21, 46). The TZ is essential to the function and integrity of the primary cilium, notably to regulate the downstream cellular pathways such as Sonic Hedgehog (SHH) and WNT signaling pathways (47).
Components of the transition zone
The transition zone is composed of a complex network of several protein compartments with at least 28 proteins associated with the TZ (9) (Figure 1). NPHP1, NPHP4 and NPHP8 form the nephrocystin module (NPHP1-4-8) involved in apical organization of renal epithelial cells (48). The nephrocystin module interacts with the CEP290 module (CEP290-NPHP5) at the basal body, as well as with the MKS module of the transition zone (43). The MKS module is critical to ciliogenesis and SHH signaling and is composed of several evolutionarily conserved proteins that are associated with severe ciliopathy phenotypes. INVS, NPHP3, NEK8, and ANKS6 localize to the inversin compartment, which forms distally to the transition zone along the axoneme (49). Disruption of any of these modules leads to the abnormal formation and function of the primary cilium. For example, the absence of CEP290 results in TZ malfunction that leads to alterations in cilia composition. Additionally, an increasing number of proteins are being recognized as reliant on an intact TZ for their precise localization within the cilium (47–50). Proximity ligation assays broadened and refined the protein-protein interaction network at TZ, confirming the majority of well-known TZ proteins and revealing novel prospective TZ protein candidates and potential linkages to other ciliary proteins excluded from the TZ (51–54).
Intraflagellar transport
The cilium is built via a unique, evolutionary-conserved cellular machinery known as intraflagellar transport or IFT, that is dedicated to the transport of ciliary proteins and lipid components toward and within the primary cilium (30, 55). IFT machinery is composed of anterograde transport complexes (IFT-B) powered by kinesin motors (56, 57) and retrograde transport complexes (IFT-A), which are mainly dynein motor-dependent (58, 59). Six proteins constitute the IFT-A group, while sixteen proteins were identified in the IFT-B group. Proteins of both IFT-B and IFT-A complexes are enriched with domains that enable extensive interactions with multiple partners, as observed in IFT-IFT and IFT-cargo interactions (60). Utilizing total internal reflection fluorescence microscopy and photobleaching techniques, it was shown that a solitary anterograde train pauses upon reaching the ciliary tip and subsequently divides into multiple retrograde trains (61). This observation implies a significant structural reconfiguration of the trains and a dynamic cross-talk between the IFT-A and IFT-B proteins, although the details of how the switch from anterograde to retrograde transport occurs are mainly unknown (62). Genetic variants in several IFT machinery components are associated with multi-systemic ciliopathies including BBS (63), JBTS (64), OFD1 (65), and NPHP (66) signifying the importance of this machinery in maintaining the cilium.
The Bardet-Biedl syndrome protein complex
The BBSome is a ciliary trafficking protein complex composed of eight BBS protein subunits (BBS1, 2, 4, 5, 7, 8, 9 and BBIP10) that has similarity to the coat protein trafficking systems (67) (68). Several additional proteins act as chaperonins and are required for BBSome assembly (69). The BBSome constituents are localized in the cilium proper, the basal body, and centriolar satellites. They regulate both ciliary cargo protein transport and signaling processes. The BBSome’s role is pivotal for the ciliary entry of many signaling molecules: e.g. SMO protein (a key part of the SHH signaling pathway) or several G-protein-coupled receptors which are absent from the cilium when the BBSome is malfunctional (70, 71). Likewise, somatostatin receptor type 3 is absent from the cilia of hippocampal neurons in Bbs2- or Bbs4-mutant mice (71). In addition, the BBSome maintains microtubule stability by preventing histone deacetylase HDAC6-mediated microtubule deacetylation and destabilization through its BBIP10 subunit (which binds to and sequesters the HDAC6) (68). More recent findings also highlight the BBSome’s involvement in ciliary protein exit thus regulating the equilibrium of ciliary receptor concentration and their localization on ciliary membrane (72, 73). Mutations in the genes encoding BBSome subunits and BBSome chaperonins cause pleiotropic Bardet-Biedl syndrome (72, 74, 75) (discussed below). Overall, proper cilia formation depends on the coordinated interplay between the different ciliary compartments and their prospective components.
The roles of actin in ciliogenesis
Hints of actin involvement in the formation of primary cilia
In the last decade, a particular role for the actin cytoskeleton in ciliogenesis emerged (10). For example, vesicle trafficking, basal body docking, centrosome positioning, and intraflagellar transport are all actin-dependent processes. The primary cilium is also rich in actin regulatory proteins, and genetic or pharmaceutical disruption of actin dynamics may negatively or positively affect ciliation in cells in vitro [reviewed in (10)] (Figure 2). Actin localization within the photoreceptor connecting cilium was initially observed by Chaitin et al. as early as 1984 (76). Mammalian photoreceptors consist of an inner segment, the cell body where proteins are synthesized, and the outer segment, where photo-sensing takes place. These segments are connected by the “connecting” cilium that uses IFT machinery to move proteins from the inner to the outer segment. Liu et al. demonstrated that actin bundles act as tracks, facilitating the transportation of the periciliary membrane complex to the basal body located at the base of the connecting cilium (77, 78). This study for the first time revealed a potentially important role of actin cytoskeleton in the regulation of primary cilium dynamics.
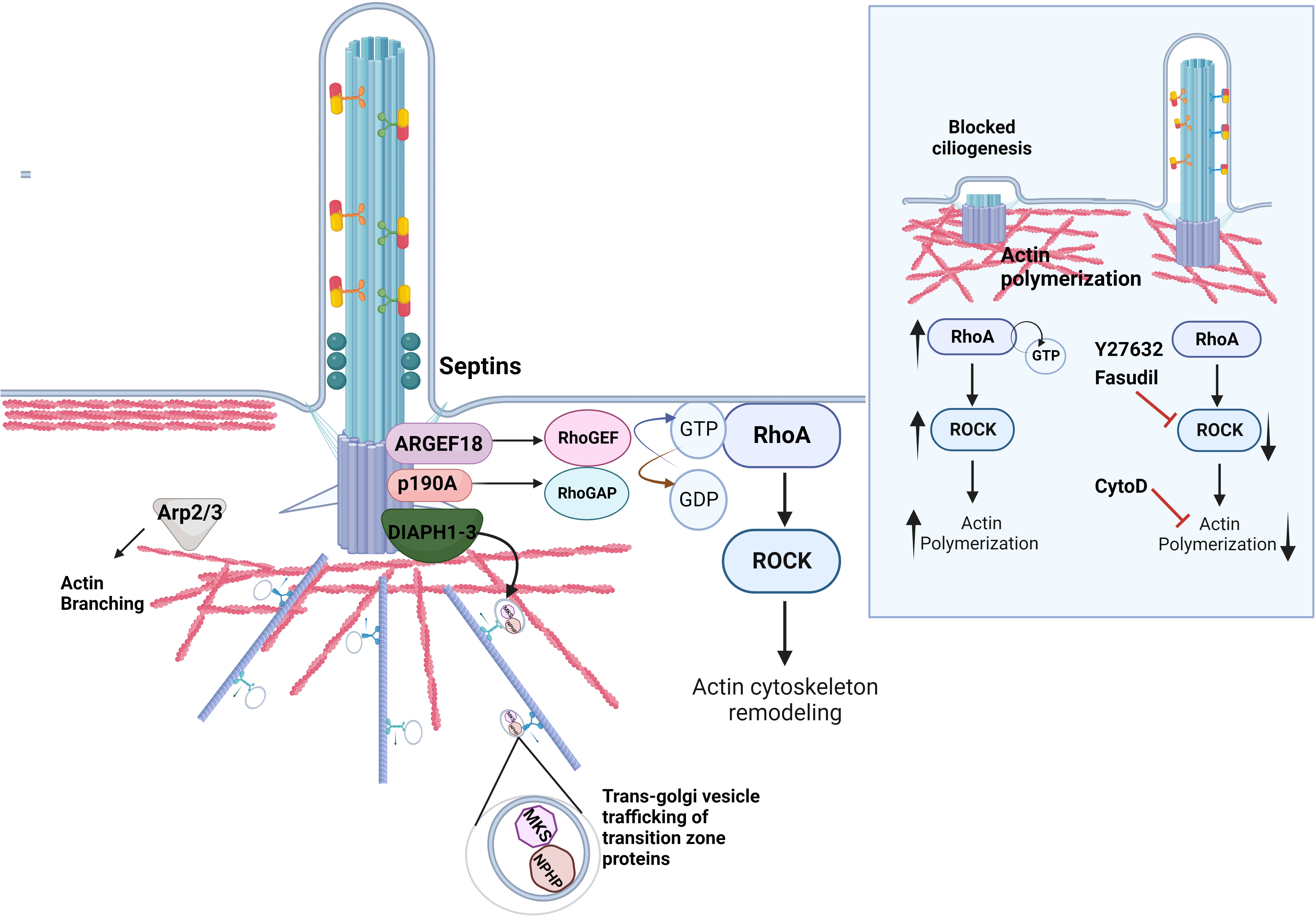
Figure 2 Regulation of actin dynamics in ciliogenesis. The actin cytoskeleton at the ciliary base contributes to cilia assembly and function. A set of actin regulatory proteins including DIAPH1-3 family of formins is important for the proper transport of ciliary proteins as well as actin nucleation and polymerization. The proteins of the family of Rho GTPases RhoA directly impact ciliogenesis by regulating actin dynamics at the ciliary base. RhoA cycles between an active GTP-bound and an inactive GDP-bound form with the help of RhoGEFs and RhoGAPs, respectively. Septins localize to the transition zone of the primary cilium, contribute to the diffusion barrier formation and influence the activity of the RhoA and its effector RhoA kinase, ROCK, at the basal body via ARHGEF18. p190A RhoGAP localizes to the basal body and inhibits excessive RhoA activation. Similarly, Arp2/3 complex mediates F-actin branching, which inhibits ciliogenesis. Inlet: Excessive RhoA activity due to mutations in the genes that regulate the Rho pathway lead to increased actin polymerization around the ciliary base, which hinders the transport of ciliary protein cargo and retains disassembly factors, inhibiting ciliation. The rescue of ciliogenesis can be achieved by utilizing the ROCK inhibitors Fasudil and Y2732, or the actin depolymerizing agent Cytochalasin D.
Genomic and proteomic screens defining the ciliome
To understand the regulatory mechanisms governing the formation of the primary cilium, several ciliogenesis modulator screens were carried out to identify key drivers and repressors influencing the formation and maintenance of primary cilia (79, 80) and to demarcate the ciliome, which represents the extensive repertoire of ciliary proteins currently counting over 1000 molecules (81). Independent studies such as Syscilia Gold standard (82), CiliaCarta (83), and Cildb (9, 84) were dedicated to defining the ciliome and identifying candidate ciliary genes using genomic, proteomic, transcriptomic, and evolutionary data, often integrating Bayesian statistics for predictive scoring of ciliary function. Moreover, recent research efforts provided valuable transcriptomic resources for curated primary ciliomes, establishing tissue and temporal specificity within various subgroups of differentially expressed genes associated with ciliary structures (85).
The involvement of actin in ciliogenesis was revealed in a comprehensive functional high-throughput RNA interference screen aiming to define genes involved in ciliogenesis (79). The screen evaluated a total of 7,784 therapeutically relevant human genes and revealed several molecules related to actin dynamics and vesicle trafficking required for ciliogenesis. Among these, gelsolin, an actin filament severing protein, emerged as a positive regulator, while the actin nucleating proteins of the Arp2/3 complex, that drive actin branching, were identified as negative regulators. Additionally, cilia-associated proteomics, combined with proximity-based biotinylation (Bio-ID) of the cilium-targeted APEX2 peroxidase enzyme, enabled Kohli et al. to detect a high stoichiometry of actin-binding proteins (86). Notably, actinin-1 & 4, tropomyosins-3 & 4, ezrin, gelsolin, cortactin and other actin regulatory proteins were found to be abundantly associated with cilia. Gupta et al. employed a BioID approach to systematically map the centrosome-cilium interface (53). This study generated a protein topology network of over 7,000 interactions with 58 bait proteins. The analysis of interactions and phenotypic profiles revealed protein modules involved in centriole duplication ciliogenesis and centriolar satellite biogenesis. Importantly, the study highlighted extensive novel interactions between centrosome-cilium interface proteins and actin regulators such as RhoA, ARHGAP21, and DIAPH3 (53). Additionally, Choksi et al. (87) undertook genome-wide expression profiling and large-scale functional studies in a zebrafish model to describe a comprehensive network of motile cilia genes controlled by Foxj1, the master regulator of cilia biogenesis. Their gene ontology analysis revealed a set of 78 cytoskeletal proteins, including the actin regulator ARHGEF18. Ultrastructural studies found actin within cilia proper where it participates in regulating ciliary length by “decapitating” the ciliary tip (87). In fact, localized actin is essential for ectocytosis (vesicle shedding), aiding G protein-coupled receptor recycling at the ciliary tip (88). Overall, these studies uncovered an intriguing phenomenon: actin polymerization and branching must be precisely regulated spatially and timely to allow the formation of primary cilium, its function and maintenance.
Actin-dependent mechanisms of ciliogenesis
Actin dynamics at early stages of ciliogenesis
Actin dynamics are critical for ciliogenesis during the early stages since actin governs directional transportation of the mother centriole (future basal body) to the appropriate cell cortex (89). Subsequent steps include the anchoring of the basal body to the underlying actin cytoskeleton through focal adhesion proteins, followed by localized actin clearing at the apical surface and microtubule nucleation (39, 90). Prior to the basal body docking, a reduction in cortical actin was shown to positively affect ciliary vesicle trafficking and to promote axoneme extension (39). Actin remodeling at the cell’s apex is particularly important in polarized epithelial cells, in which the basal body docks directly to the apical membrane to initiate ciliogenesis. Although actin is asymmetrically distributed in all cells, the polarized epithelium is characterized by a thick cortical actin layer, which is cleared as the basal body docking occurs (39). Several molecules including RAB19 and lysosomal membrane-tethering HOPS complex are implicated in cortical clearing in the polarized dog kidney Madine-Darby collecting duct epithelial cells (extracellular pathway) as well as in the retinal epithelial cells RPE-1, which utilize intracellular ciliogenesis route (39, 91). However, the precise sequence of events during cortical clearing or whether a specific complement of actin regulatory proteins exists in polarized vs non-polarized cells is still unknown. On the contrary, a thick actin network is required in multiciliated cells, in which thousands of basal bodies per cell find orderly integration within a dense cortical actin mesh, directly anchoring to the actin and microtubular cytoskeleton for proper positioning and organized planar alighment (13, 92).
During ciliogenesis, actin reorganization coincides with microtubule polymerization, stabilization, and modification (10). Actomyosin contraction and the asymmetry of the stable microtubule network within the cell generate the force required to push the centrosome to the apical surface. The crosstalk between actin and microtubule networks is tightly regulated by cytoskeleton-associated proteins, many of which play crucial roles in cilium formation and maintenance (10, 93) Furthermore, the transport of vesicle cargo powered by myosin II and myosin Va (actin regulatory protein) delivers essential ciliary membrane components including Arl13b (10), the proteins of exocyst complex, the BBSome and others to the base of the growing cilia. Myosin Va is involved at the earliest stages of ciliogenesis, where it transports preciliary vesicles through its association with Rab 8 and Rab11 small GTPases (94) that mark vesicle trafficking route to the basal body (10).
Cilia disassembly
Actin at the basal body is also important for cilia disassembly (95). Cilia disassembly occurs when the cell re-enters the cell cycle or during injury and epithelia-to-mesenchyme transition, e.g., in fibrosis or cancer (95–97). Three mitosis-regulating kinase families, Aurora A kinase (AURKA), Polo kinase (PLK1) and NimA-related kinase (NEK) and their associated proteins have been implicated in this process (98–100). Both AURKA and PLK1 phosphorylate and activate histone deacetylase 6 (HDAC6), which deacetylates axonemal tubulin, thereby destabilizing the microtubule-based ciliary core and promoting cilia resorption (98, 99). NEK family members phosphorylate other proteins, e.g. Kif24, that also favors tubulin depolymerization (100). Actin polymerization, especially actin branching, appears to hasten cilia disassembly (95). Although the detailed mechanisms remain unclear, it is believed that the centrosomal polymerized actin plays a role in retaining disassembly determinants AURKA, PLK1 and NEK at the base of the cilium leading to an enhanced cilia disassembly (95). Interestingly, translocation of Hippo pathway downstream effectors YAP and TAZ to the nucleus is cued by the actin polymerization cascade and results in increased expression of AURKA and PLK1 accompanied by suppression of ciliogenesis (101). This event appears to reinforce a feedback loop where excessive RhoA and actin polymerization trigger mechanisms of cilia disassembly. On the contrary, cytoplasmic sequestration of YAP/TAZ from the nucleus increased ciliogenesis and lowered AURKA and PLK1 expression levels, indicating that YAP/TAZ transcriptionally controls the expression of some disassembly factors (101).
Molecular determinants of actin regulation at the primary cilium
DIAPH family of formins
Multiple actin regulatory proteins control actin dynamics throughout the different stages of ciliogenesis [reviewed in (28, 95)] (Figure 2). Actin filament formation requires actin nucleation factors such as formins. The DIAPH family of formins, which includes DIAPH1, DIAPH2 and DIAPH3, features N-terminal GTPase-binding domains that interact with and are activated by small GTP-binding proteins of the Rho subfamily. Polander et al. showed that depleting DIAPH1 by RNA interference led to impaired ciliogenesis and reduced ciliary length. Reciprocally, targeting DIAPH1 to the basal body using centrin or PACT domains induced elongation of cilia and the formation of bulbous ciliary tips, suggesting that DIAPH1 likely acts in regulating ciliary vesicle trafficking. Similarly, DIAPH2 and DIAPH3 localize at the ciliary base and are implicated in the regulation of cilia maintenance by mediating post-Golgi and recycling endosomal vesicle trafficking (102). Depletion of DIAPH2 and DIAPH3 leads to decreased cilia length and accumulation of the ciliary structural protein IFT20 and small GTPase Rab11 at the cilium. On the contrary, both IFT20 and Rab11 levels increase upon targeting DIAPH2 and DIAPH3 to the ciliary base (103).
Rho family of GTPases and its regulators
The establishment of the actin network in ciliated cells critically relies on small GTPases that act as regulators of actin dynamics and are capable of orchestrating pathways for actin assembly and remodeling, microtubule organization, as well as contributing to activation of essential transcription factors that trigger cell differentiation (104, 105). The Rho family of GTPases counts more than 20 members (106); RhoA, Rac1, and Cdc42 are the most studied. Functioning as molecular switches, these GTPases cycle between their active, GTP-bound state, and their inactive, GDP-bound configuration. The cyclic transition depends on three distinct classes of molecules: 1) guanine nucleotide exchange factors (GEFs), responsible for catalyzing the exchange of GDP for GTP, thereby activating Rho-family proteins; 2) GTPase activating proteins (GAPs), which expedite the inherent GTP hydrolytic activity of the Rho-family, thereby tempering the signaling cascade and 3) Guanine nucleotide dissociation inhibitors (GDIs), that bind to Rho GTPases and stabilize them in their inactive states (106–108).
Using primary cultures of mouse multiciliated tracheal epithelial cells (mTEC), Pan et al. showed that RhoA plays a critical role in the development of a distinct apical actin network (109). The apical actin web is indispensable for the docking of basal bodies, their uniform tilting (planar polarization) and the subsequent elongation of the ciliary axoneme. Park et al. showed that the Rho GTPase activation by planar cell polarity proteins Dishevelled and Inturned is required for basal body docking to the apical plasma membrane of multiciliated Xenopus cells (110). It is plausible that the regulation of Rho activity for apical docking and planar polarization of basal bodies involves distinct RhoA regulatory proteins, a notion reinforced by observations that knockdown of ARHGEF11 triggers various embryonic anomalies indicative of impaired cilia-mediated fluid flow, likely due to deregulation of basal body planar alignment and, as a consequence, lack of uniform directional ciliary beating (110).
Employing an ENU mutagenesis strategy, Steward et al. generated a mouse model displaying renal hypoplasia associated with glomerular cysts, which stemmed from a point mutation in the ArhGAP35 gene that encodes p190A RHOGAP, one of the main mammalian GAPs (111). This loss-of-function mutation induced mis-localization of p190ARHOGAP away from the basal body, ultimately yielding escalated RhoA activity, increased actin polymerization at the ciliary base and a curtailed ciliogenesis seen as both diminished percentage of ciliated cells and shorter primary cilia in mouse proximal tubules as well as mouse embryonic fibroblasts (MEFs). Importantly, pharmacological inhibition of RhoA kinase (ROCK) activity rescued ciliary length in mutant MEFs (111). Building on these findings, Streets et al. screened several ARHGAPs and identified ARHGAP5, -29, and -35 as required for cell ciliation and ciliary length (112). After analyzing PKD1 homozygous cells it was found that p190A RHOGAP was absent at the basal body of these mutant cells, which also featured elevated RhoA activity at the basal body and shorter cilia. In conclusion, the lack of centrosomal PC1-ARHGAP35 interactions in the PKD1 mutant cells might contribute to cyst formation in ADPKD (112).
Branched filamentous actin
Branched F-actin, which is primarily distributed in the cell cortex, is nucleated by the ARP2/3 complex. Notably, silencing the key component of the ARP2/3 complex, ACTR3 (also known as ARP3) led to a marked increase in ciliary length and facilitated ciliogenesis in hTERT-RPE cells (79). Similarly, Coa et al. showed that overexpression of mir-129-3p not only stimulated ciliation in proliferating mammalian cells but the primary cilia were also elongated (38). These effects were caused by simultaneous downregulation of four positive regulators of branched F-actin: ABLIM1, ABLIM3, TOCA1, and ARP2. Furthermore, the modulation of Arp2/3 complex activity led to the accumulation of ciliary vesicles in the cilia proper, suggesting that Arp2/3 complex plays a role in IFT-B distribution within cilia, therefore highlighting its significance in regulating both IFT entry into the cilia and ciliary length (113).
Septins
Septins are the family of GTP-binding scaffold proteins that associate with the cell membrane and cytoskeleton (114). During ciliogenesis, septins participate in the formation of the diffusion barrier, partitioning the ciliary membrane from the rest of the apical plasma membrane. Septins also regulate RhoA activity and actin polymerization facilitating trafficking of ciliary cargo proteins (13). For example, SEPTIN9 enhances the activity of ARHGEF18, which in turn activates RhoA (115). In this context, activated RhoA is required for the assembly of the complete exocyst complex (116), which orchestrates transport of post-Golgi vesicles containing transition zone proteins of the NPHP and MKS complexes (115, 117).
Inhibition of actin polymerization pathway and ciliogenesis
In 2010, Kim et al. reported that the inhibition of actin assembly through drug intervention facilitates ciliogenesis in immortalized human retinal pigmented epithelial cells (hTERT-RPE) (79). This effect was attributed to the stabilization of the pericentrosomal preciliary compartment (PPC), a vesiculotubular structure responsible for storing transmembrane proteins needed for the initial phases of ciliogenesis. Actin depolymerization halts vesicle transport, causing vesicles to accumulate within the PPC. Furthermore, actin depolymerization at the base of the cilium leads to the formation of actin nodes that exhibit a marked affinity for delivering ciliary membrane cargo proteins to the ciliary vesicle. Thus, the reduction of actin polymerization facilitates the efficient delivery of cargo to the ciliary vesicle (79). On the contrary, actin polymerization, and especially actin branching, establishes a physical barrier to vesicle transport and ciliary membrane remodeling, inhibiting ciliogenesis (95) (Figure 2). Thus, actin dynamics in ciliogenesis constitute a remarkable phenomenon where the depolymerization of actin serves to promote both the assembly and elongation of cilia. For instance, the application of Cytochalasin D (CytoD), a small molecule that induces the depolymerization of filamentous (F)-actin, rapidly triggers both the formation of primary cilia and an excessive elongation of cilia likely through enhancing trafficking of essential ciliary proteins such as Arl13b and the dynamics of IFT machinery (79, 118). Remarkably, CytoD induces ciliogenesis even under conditions that typically trigger ciliary disassembly, such as serum stimulation of cultured cells (79). Interestingly, CytoD seems to also cause cytoplasmic retention of YAP and TAZ, thereby indirectly inhibiting transcription of ciliary disassembly factors (101). Ciliary elongation was also achieved by treating cells with a selective RhoA kinase inhibitor Y27632 (119). On the contrary, factors that induce branched actin assembly exert inhibitory effects on ciliogenesis (38, 120, 121). However, suppression of branched-actin regulators such as Arp2 via microRNA-mediated approaches (38) or treatment with a specific Arp2/3 inhibitor enhances trafficking of ciliary proteins (113).
In summary, actin polymerization affects different aspects of cilia formation and maintenance. The intricate interplay between actin dynamics and ciliogenesis relies on multiple actin regulatory proteins that orchestrate the balance between elongation and disassembly of the cilium, ultimately ensuring fine-tuning of cilia formation, maintenance, and function.
Renal ciliopathies: molecular mechanisms of disease and actin regulation
Preamble
The first indication that abnormalities of primary cilia can cause cystic kidney phenotypes, resembling human polycystic kidney disease (PKD), came from the studies of the Oak Ridge Polycystic Kidney (orpk) mouse (122, 123). Genetic analysis revealed that the orpk mouse harbored a hypomorphic allele in the Ift88 gene, which is a component of the anterograde IFT-B complex (122, 124). The residual IFT88 activity from the orpk allele was sufficient for the generation of shorter, functionally impaired, primary cilia causing dilatation of proximal tubules, glomerular cysts as well as polydactyly and hydrocephalus (124–126). The studies of the orpk mouse have established the key role of the primary cilium in cystic kidney disease and started a new chapter in the exploration of the cilia role in human disease. Since then, hundreds of genes involved in the generation and function of the primary cilium have been identified. While the quest for mapping all genes relevant to human health is far from complete, the emerging picture revealed different functional protein compartments that are essential for the cilia role in mammalian development (Figure 3). In this ongoing process, new evidence has emerged, linking several key ciliary proteins to the regulation of the actin cytoskeleton. Below, we describe what is currently known about the role of actin regulation in different renal ciliopathies. Exploring the link between actin regulation and ciliopathies may change our present understanding of disease mechanisms and foster the development of novel treatments.
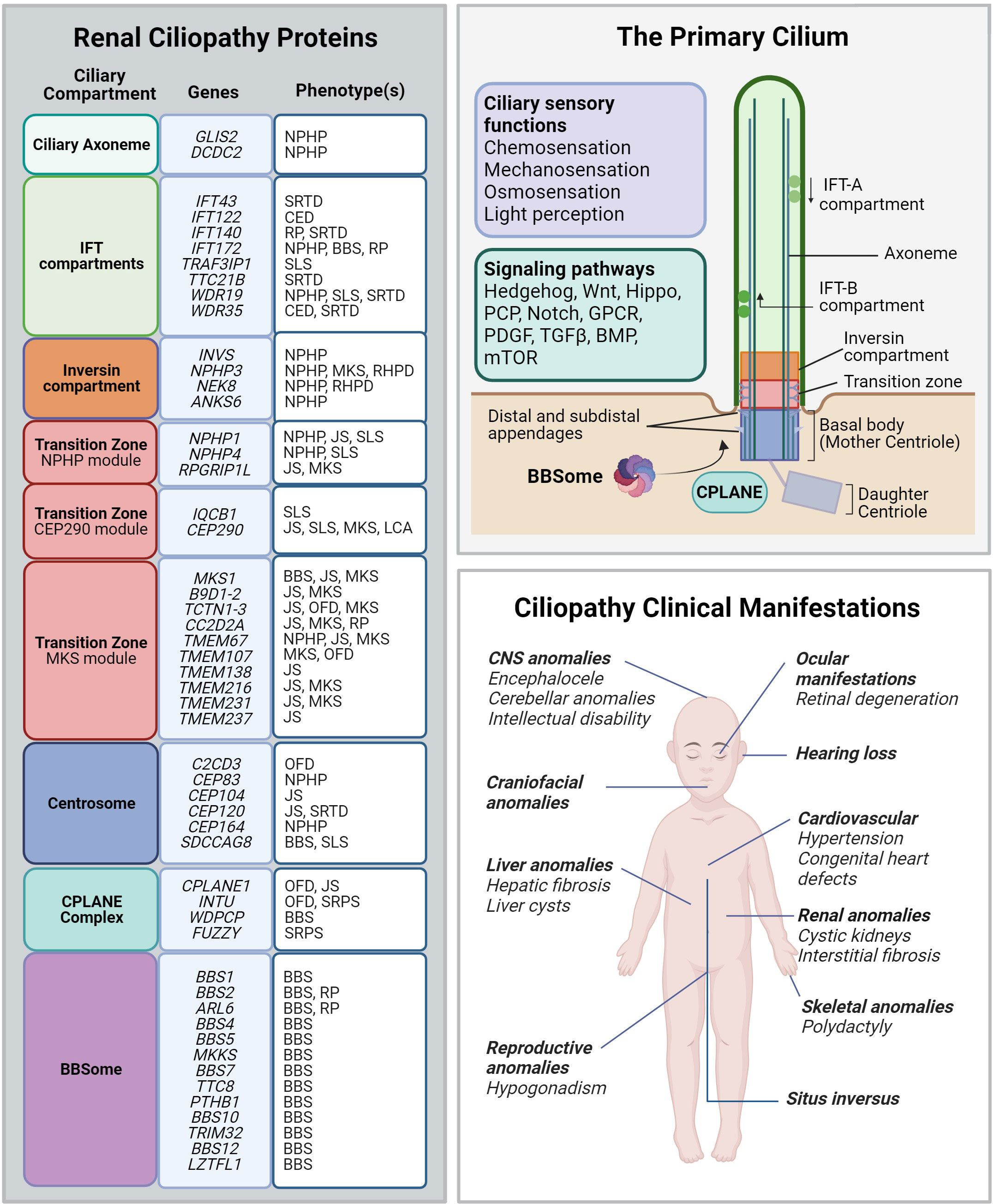
Figure 3 Renal ciliopathy gene-disease associations. Left panel: Genetic and phenotype information for renal ciliopathy gene-disease associations obtained from OMIM. Renal ciliopathies demonstrate tremendous genetic and phenotypic heterogeneity: pathogenic variants in many different genes can result in the same clinical ciliopathy phenotype, and different variants within the same gene can manifest in different phenotypes. The affected proteins in the same disease are often found to localize to the same ciliary compartment or converge on the same ciliary pathway. BBS, Bardet-Biedl syndrome; CED, Cranioectodermal dysplasia; JS, Joubert syndrome; LCA, Leber congenital amaurosis; NPHP, Nephronophthisis; OFD, Orofaciodigital syndrome; RHPD, Renal-hepatic-pancreatic dysplasia; RP, Retinitis pigmentosa; SLS, Senior-Løken syndrome. SRTD, Short-rib thoracic dysplasia. Upper right panel: Schematic depiction of the primary cilium and its protein compartments. The primary cilium regulates developmental programs from many sensory inputs (listed in the violet box) through multiple signaling pathways (listed in the green box). GPCR, G protein-coupled receptors; PCP, Planar cell polarity; PDGF, Platelet-derived growth factor; TGF-β, Transforming growth factor beta; BMP, Bone morphogenic protein; mTOR, Mammalian target of rapamycin. Lower right panel: Spectrum of the most common ciliopathic phenotypes discussed in the review.
Polycystic kidney disease
Autosomal-dominant polycystic kidney disease (ADPKD) is the most common cause of inherited kidney disease, with a reported incidence of 1:400 to 1:1,000 (127, 128). It primarily presents with renal cysts, liver cysts, and intracranial aneurysms (127). End-stage renal disease (ESRD) occurs in approximately 50% of patients by the age of 60 years. ADPKD is caused by heterozygous pathogenic variants in the genes PKD1 (ADPKD1, OMIM #173900) and PKD2 (ADPKD2, OMIM #613095), which comprise about 80% and 5-10% of total cases, respectively (127, 129, 130) (128, 131). The other cases of ADPKD either remain unsolved or are due to other genes [reviewed in (132, 133)].
Autosomal-recessive Polycystic Kidney Disease (ARPKD) occurs less frequently with a prevalence of 1 in 20,000 and is caused by biallelic variants in the PKHD1 gene (ARPKD, OMIM #263200) (132). It is characterized by enlarged, echogenic kidneys with fusiform dilatation of the collecting ducts. Most patients progress to ESRD, however, there is some clinical variability. Notably, patients frequently have liver disease with dilated biliary ducts, congenital hepatic fibrosis and portal hypertension. The most characteristic ARPKD presentation is described in neonates, consisting of a history of oligohydramnios, massively enlarged kidneys, and the characteristic “Potter Sequence” with pulmonary hypoplasia that leads to respiratory insufficiency and perinatal death in 30% of affected newborns (134).
PKD molecular mechanisms of disease
PKD1 and PKD2 encode the transmembrane proteins Polycystin1 (PC1) and Polycystin2 (PC2), respectively. PC1 and PC2 form a heteromeric complex at the primary cilium, cell-cell junctions and cell-extracellular matrix interface (127, 129, 130). PKHD1 encodes fibrocystin/polyductin, a ciliary membrane protein that is associated with the PC1/PC2 complex and localized to the primary cilium and basal body in renal tubular epithelial cells and bile duct cells (132, 135–137). The polycystin proteins are understood to function at the primary cilium and are important for maintaining the integrity of the renal epithelium. However, their precise cellular roles and the pathogenic mechanisms underlying cyst formation remain elusive (138). The polycystins are involved in mediating numerous cellular signaling pathways, including intracellular Ca2+, cAMP, mammalian target of rapamycin (mTOR), and others (127, 138). Initially, it was thought that polycystin complex at the primary cilium responded to external stimuli (e.g., fluid flow) to induce intracellular Ca2+ signaling (139), however, this role has been lately disputed (140). Moreover, its putative mechanosensory role at the primary cilium has also been contended (140). Others have suggested that ciliary-mediated proliferation pathways integral to cell division may be activated following loss of PC function, leading to unrestricted proliferation of renal tubular epithelial cells. The cystic growth can then produce a cascade of consequences including tubular obstruction, inflammation, ischemia, and metabolic disruption of renal tissue (127). On the cellular level, cystogenesis has been characterized by disruptions to apical-basal polarity, excessive WNT signaling or dysregulated planar cell polarity, aberrant extracellular matrix formation, and cell cycle regulation (141–143).
In ADPKD, it is hypothesized that a “second-hit” in the polycystin genes that reduces the PC function is required for cystogenesis in renal epithelial cells (127, 138, 144). Extensive research has shown that haploinsufficiency or complete loss of the polycystin protein function leads to a spectrum of cystic kidney phenotypes in a dosage-dependent manner (144, 145). Importantly, in vivo studies in mice show that ablation of the primary cilium in Pkd1-/- and Pkd2-/- mice suppresses cystogenesis and ameliorates the cystic phenotypes, suggesting that the polycystins may restrict cilium-originating signals required for cystogenesis (145). Although ciliary morphology is not grossly perturbed in PKD, as seen in other ciliopathy disorders, coordination of ciliary turnover and cell-cycle control may also be disrupted (146).
PKD proteins and actin regulation
Actin dynamics appear to play a role in PKD pathogenesis, as cystic cells are characterized by significant actin cytoskeletal disorganization and an accumulation of active RhoA at the basal body (112). Streets et al. showed that PC1 interacts with GTPase activating protein p190A RHOGAP, encoded by the ARHGAP35 gene. Biallelic loss of PKD1 results in the loss of p190A RHOGAP at the basal body and excessive centrosomal RhoA activity, implicating RhoA/ROCK signaling at the basal body in ADPKD pathogenesis (112). Cai and colleagues elucidated the essential role of the RhoA-YAP-c-Myc signaling axis in PKD cystogenesis (147). In Pkd1 mutant mouse tissues, aberrant activation of RhoA triggers translocation of the transcription factor YAP to the nucleus. This results in the upregulation of c-myc promoting cell proliferation and renal tubule dilatation in vivo. Crucially, these effects were reversed with the inhibition of RhoA signaling (147). However, more recently, the involvement of transcriptionally-active YAP in ADPKD pathogenesis was disputed (148).
NPHP and NPHP-related ciliopathies
Nephronophthisis (NPHP) is an autosomal-recessive renal ciliopathy that was initially described by Fanconi et al. in 1951 based on the disappearance of nephrons in the kidneys of affected children (149). Patients typically present with polydipsia, polyuria, microalbuminuria, and chronic tubulointerstitial nephritis; most patients progress to ESRD before the age of 30. Thus, NPHP is considered one of the most common hereditary causes of renal failure in the pediatric population and young adults (150). However, incidence estimates vary widely from 1 in 50,000 in Canada to 1 in 900,000 in the United States (151). In comparison to PKD, the kidneys are typically not enlarged and may be smaller in size. This explains the origin of the disease name, which derives from Greek and means “vanishing of the kidney”. Histological examination reveals a characteristic triad: loss of corticomedullary differentiation with occasional corticomedullary cysts, tubular atrophy with a thickening of the basement membrane, and interstitial fibrosis (150). Although cysts are found in about 70% of NPHP cases, they are smaller than in ADPKD and their presence is not necessary for an NPHP diagnosis (152). The first discovered and most common is nephronophthisis type 1 (NPHP1) (OMIM #256100), caused by biallelic variants in the NPHP1 gene (often due to a homozygous deletion of the entire gene). NPHP1 accounts for 20-50% of all genetically solved NPHP cases (153–155). About one-third of NPHP1 cases are syndromic, often presenting with Senior-Løken syndrome, SLS (retinal degeneration) and Joubert syndrome, JS (cerebellar and oculomotor anomalies) (156). Currently, more than 25 genes have been identified as genetic causes of NPHP (155, 157). However, the majority (70%) of NPHP cases remain unsolved (157).
NPHP molecular mechanisms of disease
Many NPHP proteins localize to specific compartments at the primary cilium or centrosome and are involved in ciliogenesis or ciliary functions (Figure 3). Some NPHP proteins are directly involved in forming the basal body as well as facilitating its migration and docking to the cell membrane (158). Interestingly, many NPHP proteins involved in ciliopathies with either kidney or eye involvement cluster within the transition zone modules (156). The inappropriate accumulation of non-ciliary proteins in the cilium proper due to aberrant ciliary gating is hypothesized to contribute to disrupted ciliary function in NPHP and other renal ciliopathies (159). Many ciliary-mediated signaling pathways, including Hippo, cAMP, mTOR pathway and others, are implicated in NPHP disease pathogenesis [reviewed in (154)]. NPHP phenotypes are further associated with upregulation of pro-inflammation and pro-fibrotic pathways culminating in renal interstitial fibrosis and inflammation (150, 160, 161). The complex interactions between NPHP proteins and their roles in supporting ciliogenesis and ciliary function play a role in the phenotypic heterogeneity observed in NPHP-related ciliopathies.
NPHP proteins and actin regulation
Nephrocystins interact with actin and microtubule structures to coordinate ciliary signaling in renal epithelial cells, which mediates cell adhesion and cell division of renal tubular cells (162–164). Nephrocystin 1 (NPHP1) localizes to the basal body of the primary cilium and the apical surface of renal epithelial cells (160). NPHP1 functionally interacts with nephrocystin 4 (NPHP4) and nephrocystin 8 (NPHP8), forming the NPHP1-4-8 module within the ciliary transition zone in both polarized renal epithelial cells and at the centrosome of dividing cells. The NPHP1-4-8 protein complex is involved in cortical actin cytoskeletal organization and regulation of cellular apicobasal polarity during tubule morphogenesis and tissue maintenance (48, 161, 164). NPHP1 also interacts with proteins at focal adhesion complexes (164), which, in addition to enabling cell-extracellular matrix contacts, facilitate anchoring the basal body to the actin cytoskeleton in multiciliated cells (165). Loss of either NPHP1 or NPHP4 in vitro results in disrupted tight junction formation, aberrant ciliary function and loss of normal apicobasal polarity (166). In multiciliated Xenopus laevis cells, NPHP4 is involved in subcortical actin organization through interactions with formin DAAM1 and the PCP effector, Inturned (167).
Pathogenic variants in INVS result in an infantile form of NPHP (OMIM #602088), featuring large cystic kidneys and situs inversus in some patients. INVS encodes inversin, which forms the “inversin compartment” and controls switching of the canonical and PCP signaling pathways, believed to be important in renal development and tissue maintenance (168). Inversin is involved in regulating the cortical actin cytoskeletal network during mitosis, and loss of its function leads to mitotic spindle misalignment, defective PCP signaling, disrupted mitotic cell rounding, and loss of epithelial organization and integrity (169). Together, this gives rise to defective tubulogenesis as the cells fail to preserve oriented cell divisions and incorporate longitudinally along the developing nephron (170).
Aberrant RhoA activity at the centrosome appears to play a significant role in NPHP pathogenesis through activation of its downstream target, ROCK, and by promoting abnormal actin formation and activation of the Hippo pathway (95). The human kidney proximal tubular epithelial cells with an NPHP1 knockdown and the renal tissue from Nphp1 knockout mice exhibit increased levels of GEF-H1, an important RhoA GTPase effector factor in mammalian renal epithelium that mediates RhoA activation (171). The NPHP phenotype could be rescued in these mice by a knockdown of GEF-H1, which acts upstream of RhoA, thus reducing the RhoA activity and preventing renal cystogenesis, interstitial fibrosis and inflammation (171).
Bardet-Biedl syndrome
Bardet-Biedl Syndrome (BBS) is a group of autosomal-recessive ciliopathies manifesting during early childhood. The diagnosis is based on the presence of several major diagnostic criteria (retinal dystrophy, obesity, intellectual disability, polydactyly, genital anomalies and renal malformations) but may also include other variable features affecting multiple organ systems (172). The prevalence ranges from 1 in 140,000 to 1 in 160,000 but has been reported more commonly on the island of Newfoundland, affecting 1 in 17,000 (173). Renal manifestations of BBS more closely resemble the fibrocystic phenotypes observed in NPHP, JS, and MKS, compared to PKD, and include dysplastic kidneys or nephronophthisis, occurring in about 40% of cases, and often leading to end-stage renal disease (174, 175). Renal disease in BBS is highly variable in presentation due to high genetic locus and allelic heterogeneity; however, the molecular basis behind this phenotypic variability remains poorly understood (175, 176). At least 26 different known genes have been identified as causes of BBS and likely contribute to the great variability in severity and clinical pleiotropy (177). Many of the BBS-causing genes encode the core BBS proteins forming the BBSome, a protein complex that facilitates ciliary vesicle trafficking toward and within primary cilium (Figure 3) (68). Disruption to any of the BBSome subunits can result in the clinical manifestations of BBS, providing strong evidence for the functional interdependency among all BBSome components.
BBS molecular mechanisms of disease
Defects to the BBSome can significantly alter the composition of the ciliary membrane leading to the loss, mis-localization or abnormal retention of membrane receptors and other molecules within cilium. For example, the photosensory protein rhodopsin is lost in the outer photoreceptor segment of Bbs2, Bbs4 and Bbs8 loss-of-function mutant mice, while other proteins inappropriately accumulate in the outer segment, leading to increased photoreceptor apoptosis and progressive retinal degeneration (178–180). Studies of the cells isolated from patients with BBS and pathogenic variants in BBS1, BBS5 and BBS10 demonstrate shorter cilia and abnormal SHH signaling; the latter likely arises due to inappropriate trapping of the key SHH component, Smoothened, within ciliary shaft (181). Downregulation of SHH signaling during development is linked to renal malformations and may contribute to cystogenesis in BBS and other renal ciliopathies (182). Suppression of Bbs transcripts in zebrafish leads to stabilization of β-catenin and inappropriate activation of canonical WNT pathway, that might contribute to the cystic renal phenotype in BBS (183). The BBSome is crucial for the localizations of G-protein-coupled receptors (GPCRs) at the primary cilium of certain neurons, and some of the phenotypic manifestations of BBS are thought to result from misplacement of specific GPCRs (71). Thus, like other syndromic ciliopathies, the pathogenic mechanisms of BBS are largely linked to defects in signaling pathways mediated by the primary cilium.
BBS proteins and actin regulation
Studies of Bbs mutant cells established a potential role of dysregulated actin polymerization in the pathogenesis of BBS (184). Defects in actin cytoskeletal regulation during development are implicated in renal malformations and renal cystogenesis in BBS. Renal medullary cells isolated from Bbs4 and Bbs6 deficient mice displayed a reduced percent of ciliated cells, shorter cilia, and abnormal actin fiber accumulation at the cell apex. Additionally, paucity of lamellipodia and filopodia was detected as well as loss of peripheral focal adhesions, the defects usually associated with decreased cell motility that was seen in the Bbs mutant cells. Inappropriate actin aggregation was attributed to significantly increased RhoA activity and dynamics of actin polymerization since treatment of mutant cells with selective RhoA kinase inhibitor Y27632 or actin polymerization inhibitor CytoD rescued actin aggregation phenotype. Importantly, these inhibitors also rescued ciliary length and increased percent of ciliated cells, pointing to the strong link between abnormal actin polymerization, increased RhoA activity and ciliary defects. Intriguingly, Bbs4 and Bbs6 proteins previously detected only at the basal body and in the cilium, also co-localized with the focal adhesions and governed their assembly. Thus, the results of Hernandez-Hernandez et al. support the hypothesis that BBS proteins regulate actin cytoskeletal arrangement through RhoA and focal adhesion dynamics, functions that are now recognized as integral to ciliogenesis (184).
Meckel-Gruber syndrome
Meckel-Gruber syndrome (MKS, OMIM #24900) is a perinatally lethal autosomal-recessive ciliopathy that presents with a constellation of severe developmental anomalies including neural tube defects, skeletal malformations, congenital heart defects, liver anomalies, and enlarged cystic dysplastic kidneys (152, 185). The worldwide incidence for MKS is 1 in 13,250 to 1 in 140,000 live births (186). It is the ciliopathy with the most severe presentation, often arising from biallelic full loss-of-function variants in ciliary genes that lead to total or significant loss of ciliary function (152). Several genes have been associated with MKS, including MKS1, TMEM67, CEP290, CC2D2A, and TMEM216, many of which encode proteins that localize to the transition zone and are essential for basal body docking (152, 187). Most MKS-causing genes are also associated with milder ciliopathy phenotypes, causing NPHP or JS, suggesting that these disease entities exist along an “allelic spectrum” and depend on the nature of the underlying genomic variant (188).
MKS molecular mechanisms of disease
Many of the MKS proteins converge on the same pathway during ciliogenesis, and their loss-of-function models have demonstrated a critical role in mediating basal body anchoring in association with actin cytoskeletal dynamics (187, 189–191). The transition zone protein TMEM67 (also known as Meckelin), encoded by TMEM67, plays a crucial role in ciliogenesis. Biallelic truncating pathogenic variants in TMEM67 (MKS3) commonly lead to Meckel syndrome (MKS3, OMIM #607361); however, “milder” TMEM67 allelic combinations (e.g., biallelic missense variants) lead to less severe phenotypic presentations such as JS or isolated NPHP (192, 193). TMEM67 and MKS1 interact together and are required to mediate basal body docking to the apical cell surface (189, 194).
MKS proteins and actin regulation
The involvement of MKS proteins in actin regulation is only beginning to emerge. The TMEM67 is suggested to promote actin-cytoskeletal reorganization required for basal body docking by regulating RhoA-ROCK signaling (190). Loss of TMEM67 or MKS1 leads to disrupted ciliogenesis and impaired epithelial morphogenesis (189). Similarly, loss of TMEM216 in human fibroblasts or its knockdown in zebrafish was shown to cause hyperactivation of RhoA, hypothesized to disrupt the required actin reorganization at the cell apex preventing normal basal body docking (187).
Skeletal ciliopathies with renal involvement
Approximately 10% of syndromic NPHP cases present with skeletal anomalies often found alongside liver, eye, and CNS anomalies (156). Short-rib thoracic dysplasia with and without polydactyly (SRTD) are a group of skeletal ciliopathy disorders with shared features of polydactyly, shortening of the long bones and severe rib and thoracic malformations (152, 195). The most common forms of SRTD are Jeune asphyxiating thoracic dysplasia (JATD), Ellis–van Creveld syndrome, Sensenbrenner syndrome, Mainzer-Saldino syndrome (MZSDS), and short-rib polydactyly syndromes (SRPS) (196). Polycystic kidneys are occasionally detected in patients with Oral-facial-digital syndromes (OFD), which are primarily characterized by dysmorphic craniofacial and oral anomalies and polydactyly (197). Ciliopathies with skeletal malformations often arise due to defects in genes encoding for the IFT-A and IFT-B components or the dynein motor arms of the primary cilium, leading to defects in ciliary function, including mediating SHH signaling (Figure 3) (152). Defects to the IFT-A complex of proteins (e.g., WDR35, IFT122, IFT43, IFT172, IFT140 and WDR19), can present with renal disease phenotypes such as chronic renal failure, NPHP, and renal cysts (63, 198–203) and, in some cases, isolated NPHP or NPHP-like nephropathy (63). Although the underlying mechanisms are not well characterized, several groups have posited that IFT proteins may be regulating planar cell polarity in the elongating renal tubules, although this supposition requires experimental confirmation. There are over 16 genes associated with OFD phenotypes, however, the X-linked OFD1 (OMIM # 300170) is the most common (~50% of all OFD cases) and causes polycystic kidney disease in half of affected patients (152, 204, 205). OFD1 protein localizes to the centrosome and is essential for recruiting the distal appendage proteins during ciliogenesis (206). Recent research uncovered the expanded role of OFD1 in microtubule organization and cell-cycle regulation as well actin filament branching (207, 208).
Skeletal ciliopathies caused by distortion of CPLANE function
A special subset of skeletal ciliopathies featuring renal malformations is caused by defects in the Ciliopathy and Planar Cell Polarity, CPLANE, genes (209). Originally discovered in Drosophila (210), where these genes are known as “Planar cell polarity effectors” and participate in PCP signaling, vertebrate homologs of inturned, fuzzy and fritz (WDPCP) regulate ciliogenesis. Their depletion in frogs led to significantly shorter cilia (211); homozygous mutations of Fuzzy, Inturned and Wdpcp in mice caused stunted sparse primary cilia and features of classical ciliopathies such as craniofacial defects, polydactyly, and dysplastic kidneys (212–214). CPLANE proteins interact with each other (215), localize to the basal body and control important aspects of vesicular trafficking to the basal body (209, 216). Comprehensive tandem affinity purification studies revealed interactions with various ciliary proteins including IFT43 and WDR35 (209) [both cause Sensenbrenner syndrome (201, 217)]. They also interact with and form a functional complex with two additional proteins, Joubert syndrome 17 encoded by the JBTS17 gene (also known as CPLANE1) (209) and a small GTPase effector RSG1 (209). Pathogenic variants in CPLANE1 cause JS and OFD, often presenting with renal involvement (218). Variants in FUZ, INTU, and WDPCP are associated with embryonically lethal cases of SRPS, OFD syndrome, and neural tube defects (209, 219, 220); renal hypoplasia was described in some of these patients.
CPLANE and actin regulation
Knockdown of Fuzzy or Inturned reduces the thickness of the actin cortical network in multiciliated Xenopus cells (211), implying that in this context, CPLANE proteins might function as positive regulators of actin polymerization. Similarly, mouse Wdpcp mutant cells display thinner actin-based stress fibers and minimal actin-based membrane protrusions; the latter affects cell motility (213). The effect of WDPCP on actin was traced to its interaction with actin regulatory protein Septin 2 (213). However, Drosophila CPLANE homologs function to restrict actin polymerization in the wing cells and their loss leads to an excessive actin cytoskeleton consistent with the negative actin regulatory function in the fly cells. In agreement with this, we recently observed both excessive RhoA activity and increased actin polymerization at the basal body of Fuzzy mutant cells (unpublished data). Importantly, ciliogenesis could be rescued with CytoD or ROCK inhibitors in vitro and ex vivo (Figure 4). Thus, the accumulated evidence suggests that CPLANE proteins affect ciliogenesis via differential effects on actin polymerization in various cells, and this may contribute to the ciliopathic phenotypes of skeletal/renal ciliopathies.
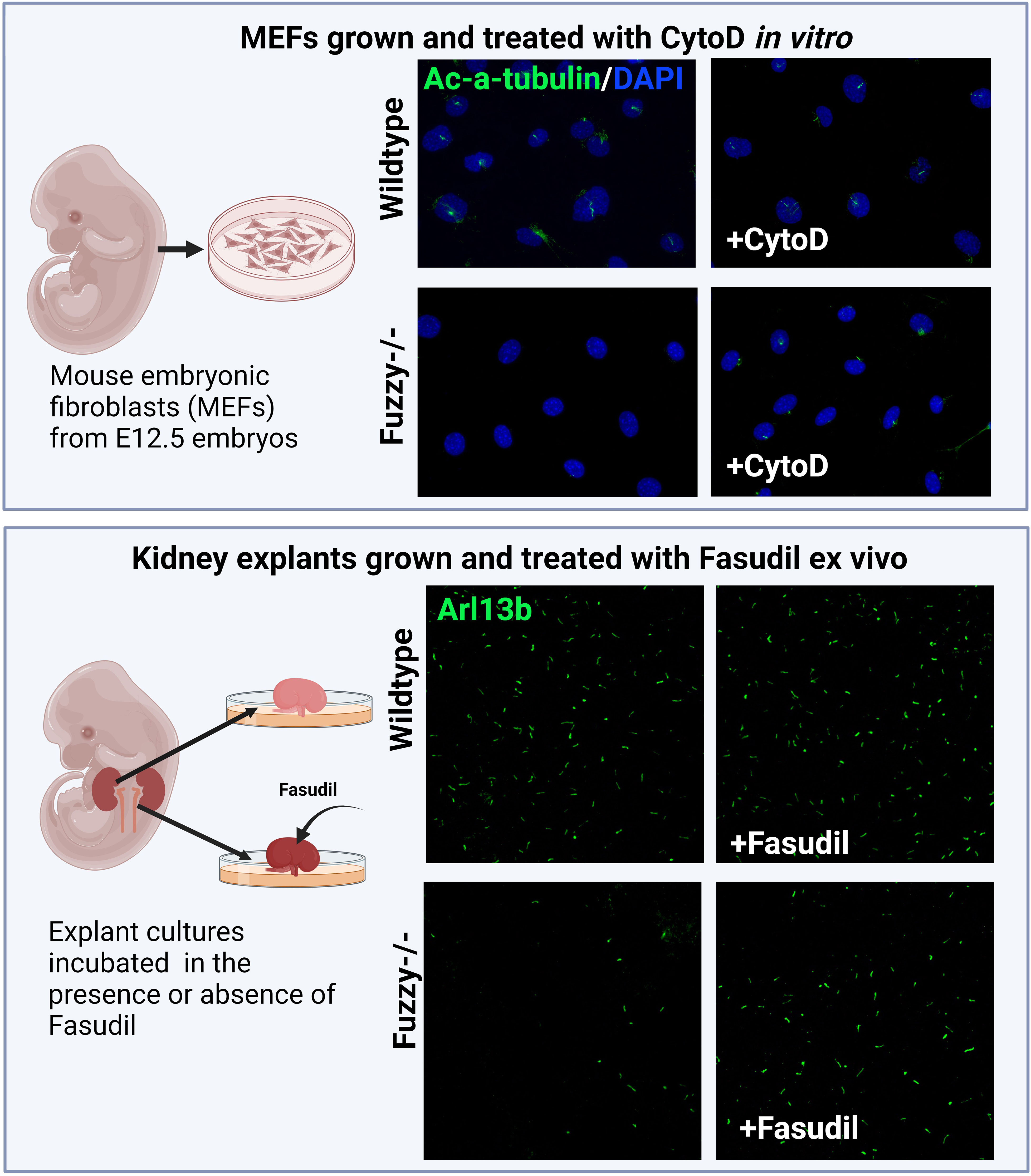
Figure 4 Inhibition of actin signaling pathway and/or polymerization rescues ciliogenesis in a CPLANE ciliopathy model. Upper panels: Schemata of generating mouse embryonic fibroblasts (MEFs) from E12.5 embryos. Wildtype MEFs are ciliated at ~ 80-85% after 24 hours of starvation in vitro whereas Fuzzy-/- MEFs lack cilia. Treatment of unciliated Fuzzy-/- MEFs with 0.5µM of CytoD leads to 60% ciliated mutant cells and no effect on the wildtype cells; cilia were visualized with anti-acetylated-α-tubulin antibody and 4′,6-diamidino-2-phenylindole to stain nuclei (unpublished data). Lower panels: Schematic depiction of an experiment in which embryonic kidney explants from E12.5 wildtype and Fuzzy-/- embryos were cultured ex vivo for 72 hours in the presence or absence of 2.5µM Fasudil followed by a whole mount tissue immunostaining with anti-Arl13b antibody to detect primary cilia. Treatment with Fasudil elongates ciliary length in mutant tissues with no effect on the wild-type tissues (unpublished data).
Actin-involvement in ciliopathy-associated diseases
Townes-Brock syndrome 1
The majority of the pathogenic mechanisms of renal ciliopathies described above are associated with increased centrosomal actin, however, Townes-Brock syndrome 1 (TBS1, OMIM #107480) features reduced actin polymerization. TBS1, an autosomal-dominant disorder, results from heterozygous loss-of-function variants in the SALL1 gene and features ciliopathy-spectrum phenotype with renal abnormalities often leading to ESRD (221). SALL1 encodes a transcription factor that interacts with CEP97 and CP110, two negative regulators of ciliogenesis, and removes them from the mother centriole, thereby favoring ciliogenesis. Loss of SALL1 function leads to defects in ciliogenesis and SHH signaling in patient-derived fibroblasts (221). Recently, Bosal-Basterra et al. reported that truncated SALL1 protein interacts with LUZP1, a leucine-zipper motif-containing protein that localizes around centrioles and binds to filamentous actin (222). Loss of LUZP1 reduces F-actin and increases ciliogenesis. Importantly, interactions between mutant SALL1 and LUZP1 target LUZP1 for degradation leading to the disruption of both ciliogenesis and SHH signaling through reduced F-actin polymerization, indicating that dysregulation of centrosomal actin cytoskeleton contributes to the pathogenesis of TBS (222).
Lowe syndrome
Lowe syndrome (OMIM # 309000) is an extremely rare and severe X-linked oculo-cerebro-renal disorder that results from pathogenic variants in the OCRL gene encoding inositol polyphosphate 5-phosphatase OCRL-1. The proximal tubular dysfunction leads to renal failure in the second or third decade (223). Some patients with OCRL variants present with Dent disease type 2, which is thought to be a milder presentation of classical Lowe syndrome (223). OCRL-1 is involved in membrane and endosomal trafficking. It also binds Rho GTPases to modify F-actin polymerization important for phagocytosis, cell migration, cell polarity, and ciliogenesis (223–225). In OCRL-1 mutant cells, actin polymerization is decreased and primary cilium formation and function are disrupted (226) due to defects in ciliary vesicle processing during ciliogenesis (225, 227).
Overall, the studies of renal ciliopathies described above indicate that many renal ciliopathy proteins localize to the basal body and participate in the complex interactions that organize centrosomal actin remodeling required for the successful formation of primary cilium. These emerging data have uncovered a frequent distortion of actin regulation and remodeling in pathogenesis of renal ciliopathies.
Primary cilium in renal cell carcinomas
Renal cell carcinomas (RCC) frequently present with cystic kidneys, prompting studies of primary cilia in the context of RCC (228, 229). Germline heterozygous pathogenic variants in the Von Hippel-Lindau (VHL) gene is the most common genetic cause of RCC, where renal cysts often precede tumorigenesis (230). VHL encodes a tumor suppressor protein VHL that localizes to the Par3-Par6-aPKC complex at the primary cilium to facilitate ciliogenesis by stabilizing and orienting microtubule formation (228, 231, 232). VHL-deficient cells are not ciliated and exhibit defects in cellular polarity and cell-cycle regulation, leading to uncontrolled cell proliferation (233, 234). Interestingly, VHL also acts as a E3-ubiquitin ligase, and in this capacity, targets the ciliary disassembly factor Aurora Kinase A (AURKA) for degradation (235). Moreover, loss of VHL function results in high levels of AURKA, affecting stability of the microtubular axoneme and leading to disassembly of the primary cilium followed by development of cystic kidneys and RCC (235). Other ciliary genes involved in RCC have been recently reviewed elsewhere (236). Tuberous sclerosis TSC is an autosomal-dominant multi-system disorder that results from pathogenic variants in the tumor suppressor genes, TSC1 and TSC2, leading to aberrant activation of the mTOR pathway (237) and often presenting with renal cysts associated with cilia disruption (238). Currently, the role of actin regulation at the primary cilium assembly and disassembly in renal cancers and associated cystic phenotypes have not been studied.
Potential therapies
Renal ciliopathies are a significant cause of chronic kidney disease and ESRD. Currently, the only long-term treatments for kidney manifestations are renal replacement therapy through hemodialysis and kidney transplantation. High frequency of ADPKD, that affects more than 12 million people worldwide, prioritized significant scientific and financial resources to develop various treatment options. However, despite a significant effort, Tolvaptan has so far been the only FDA-approved medication for ADPKD (239). On the other hand, there are currently no approved treatments for other renal ciliopathies. Recent clinical trials for BBS were limited to obesity and hyperphagia treatment by Setmelanotide (a selective melanocortin-4 receptor agonist) with no focus on treating kidney phenotypes (240).
Drugs that prevent cilia disassembly via axoneme stabilization through histone deacetylase inhibition have also been tested in several pre-clinical models (241–243). Histone deacetylase 6 (HDAC6) inhibitors such as ACY-1215, Tubacin, Trichostatin A (TSA), Valproic acid (VPA) and Nicotinamide have shown to ameliorate cystic kidney disease in in vitro and in vivo models, however, some of these drugs have uncertain safety profiles and are yet to be tested in clinical trials. Excessive cell proliferation in the VHL-dependent RCC cells was rescued by treatment with alisertib, an AURKA−specific chemical inhibitor (244), however, ciliary integrity in the treated RCC cells was not investigated. Other treatments that target cellular metabolism (e.g., basal body localized-AMPK agonists such as Metformin), inflammation, EGF signaling, mTOR inhibitors, HDAC inhibitors, CDK inhibitors, Hedgehog agonists, somatostatin analogues, in addition to gene therapy via microRNAs, are all extensively reviewed elsewhere (243).
Recent evidence on the role of the RhoA/ROCK pathway in ciliogenesis heightened interest in the studies of actin regulation in relation to ciliopathies. Actin polymerization inhibitors such as Cytochalasin D or selective ROCK inhibitors Y27632 have shown to rescue cilia defects to some degree in different ciliopathy disease models (39, 101, 112, 184) (Figure 4). Importantly, inhibition of RhoA signaling with Y27632 suppresses cystogenesis in both the 3D culture of Pkd1 mutant collecting duct cells and in Pkd1 mutant mouse kidneys in vivo (147). However, their use in humans is difficult to envision due to their toxicity. On the other hand, ROCK inhibitor Fasudil, that has a reduced toxicity and a more favorable safety profile, was clinically approved for use in humans as a systemic ROCK inhibitor to treat cerebral vasospasm in Japan and China (245). Although it is still not FDA-approved, Fasudil is being used in several clinical trials addressing cardiovascular disorders (246, 247), Parkinson’s disease, Amyotrophic Lateral Sclerosis, and other conditions (https://clinicaltrials.gov). Given the persistent deregulation of RhoA signaling and actin polymerization in many models of renal ciliopathies, including ADPKD, NPHP and BBS, it would be reasonable to test Fasudil in human trials to treat ciliopathies, e.g., ADPKD or NPHP. With the advent of new inhibitors of actin regulatory pathways that can be targeted to specific tissues and have acceptable safety profiles, these molecules can be harnessed for their potential use in treating ciliopathies postnatally.
Conclusions
The primary cilium is an enigmatic organelle central to the development and homeostasis of all human organs. Dysfunction of primary cilium perturbs numerous cellular processes, negatively affecting tissue morphogenesis and tissue maintenance causing a constellation of severe, frequently congenital, phenotypes in humans, known as ciliopathies. Pathogenic variants in over 180 genes have been identified to date as the cause of human ciliopathies, which often present as malformations of the kidney. However, analysis of cohorts of patients with specific ciliopathies shows that only a fraction of genes has been identified. Assembly of clinically well-characterized patient cohorts and the use of a comprehensive genome-wide molecular diagnostic testing are critical if we are to identify the hidden genetic contributors and will pave the way towards improved diagnostic criteria and elucidation of the molecular mechanisms which will allow personalized therapies.
Analysis of the mechanisms causing ciliopathies has revealed an unexpected key role of actin cytoskeletal dynamics in ciliogenesis and ciliary function. Moreover, it is becoming clear that disruption to these mechanisms plays a part in the pathogenesis of various classes of renal ciliopathies, including ADPKD, NPHP, or BBS. However, whether actin dysregulation is a driving force in the cellular phenotype or merely is a consequence of an upstream mechanism that alters actin dynamics remains unknown. Importantly, deregulation of basal body actin network affects both cilial assembly and disassembly. For example, loss of cilia in renal cancer appears to be driven by the heightened activity of the cilium disassembly, but it remains unknown whether actin deregulation at the base of the cilium contributes to the cilia loss in RCC. Regardless of the precise nature of the cause-and-effect relationship, persistent actin network disorganization in the cells of patients affected with ciliopathies may offer an opportunity to identify novel preventative and restorative treatments.
The cellular and molecular mechanisms underlying renal ciliopathies are currently under intense investigation. The expanding complexity of the genetic errors and phenotypic consequences and the basis for tissue specificity associated with specific genes promise to be an exciting journey.
Author contributions
RK: Conceptualization, Writing – original draft, Writing – review & editing. ZS: Conceptualization, Writing – original draft, Writing – review & editing. TK: Funding acquisition, Supervision, Writing – review & editing, Conceptualization. ET: Conceptualization, Funding acquisition, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank Sima Babayeva for producing immunofluorescence images for the Figures. RK is supported by the Fonds de recherche du Québec en Santé graduate award; ZS is supported by the Canadian Institute of Health Research (CIHR) graduate award. TK is supported through the Fonds de recherche du Québec en Santé (FRQS) Salary Grant and the SickKids New Investigator Research Grant (NI21-1159). ET is supported by the CIHR grant CIHR230929.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet (2006) 7:125–48. doi: 10.1146/annurev.genom.7.080505.115610
2. Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet (2010) 11:331–44. doi: 10.1038/nrg2774
3. Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med (2011) 364:1533–43. doi: 10.1056/NEJMra1010172
4. Scholey JM. Intraflagellar transport motors in cilia: moving along the cell's antenna. J Cell Biol (2008) 180:23–9. doi: 10.1083/jcb.200709133
5. Bloodgood RA. From central to rudimentary to primary: the history of an underappreciated organelle whose time has come. primary cilium Methods Cell Biol (2009) 94:3–52. doi: 10.1016/s0091-679x(08)94001-2
7. Bangs F, Anderson KV. Primary cilia and mammalian hedgehog signaling. Cold Spring Harbor Perspect Biol (2017) 9:a028175. doi: 10.1101/cshperspect.a028175
8. Margaria JP, Campa CC, De Santis MC, Hirsch E, Franco I. The PI3K/Akt/mTOR pathway in polycystic kidney disease: A complex interaction with polycystins and primary cilium. Cell signal (2020) 66:109468. doi: 10.1016/j.cellsig.2019.109468
9. Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol (2017) 18:533–47. doi: 10.1038/nrm.2017.60
10. Copeland J. Actin-based regulation of ciliogenesis–the long and the short of it. (Elsevier) (2020) 102:132–8. doi: 10.1016/j.semcdb.2019.12.005
11. Bloodgood RA. Sensory reception is an attribute of both primary cilia and motile cilia. J Cell Sci (2010) 123:505–9. doi: 10.1242/jcs.066308
12. Satir P. Landmarks in cilia research from Leeuwenhoek to us. Cell Motil Cytoskeleton (1995) 32:90–4. doi: 10.1002/cm.970320203
13. Mirvis M, Stearns T, James Nelson W. Cilium structure, assembly, and disassembly regulated by the cytoskeleton. Biochem J (2018) 475:2329–53. doi: 10.1042/BCJ20170453
14. Brooks ER, Wallingford JB. Multiciliated cells. Curr Biol (2014) 24:R973–82. doi: 10.1016/j.cub.2014.08.047
15. Mizuno N, Taschner M, Engel BD, Lorentzen E. Structural studies of ciliary components. J Mol Biol (2012) 422:163–80. doi: 10.1016/j.jmb.2012.05.040
16. Pedersen LB, Veland IR, Schrøder JM, Christensen ST. Assembly of primary cilia. Dev dyna: an Off Publ Am Assoc Anatom (2008) 237:1993–2006. doi: 10.1002/dvdy.21521
17. Christensen ST, Pedersen LB, Schneider L, Satir P. Sensory cilia and integration of signal transduction in human health and disease. Traffic (2007) 8:97–109. doi: 10.1111/j.1600-0854.2006.00516.x
18. Ishikawa H, Thompson J, Yates JR, Marshall WF. Proteomic analysis of mammalian primary cilia. Curr Biol (2012) 22:414–9. doi: 10.1016/j.cub.2012.01.031
19. Avasthi P, Marshall WF. Stages of ciliogenesis and regulation of ciliary length. Differentiation (2012) 83:S30–42. doi: 10.1016/j.diff.2011.11.015
20. Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell (2012) 150:533–48. doi: 10.1016/j.cell.2012.06.028
21. Schmidt KN, Kuhns S, Neuner A, Hub B, Zentgraf H, Pereira G. Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis. J Cell Biol (2012) 199:1083–101. doi: 10.1083/jcb.201202126
22. Failler M, Gee HY, Krug P, Joo K, Halbritter J, Belkacem L, et al. Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am J Hum Genet (2014) 94:905–14. doi: 10.1016/j.ajhg.2014.05.002
23. Li J, Lu D, Liu H, Williams BO, Overbeek PA, Lee B, et al. Sclt1 deficiency causes cystic kidney by activating ERK and STAT3 signaling. Hum Mol Genet (2017) 26:2949–60. doi: 10.1093/hmg/ddx183
24. Sorokin S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J Cell Biol (1962) 15:363–77. doi: 10.1083/jcb.15.2.363
25. Youson JH. Replication of basal bodies and ciliogenesis in a ciliated epithelium of the lamprey. Cell Tissue Res (1982) 223:255–66. doi: 10.1007/BF01258487
26. Ghossoub R, Molla-Herman A, Bastin P, Benmerah A. The ciliary pocket: a once-forgotten membrane domain at the base of cilia. Biol Cell (2011) 103:131–44. doi: 10.1042/BC20100128
27. Molla-Herman A, Ghossoub R, Blisnick T, Meunier A, Serres C, Silbermann F, et al. The ciliary pocket: an endocytic membrane domain at the base of primary and motile cilia. J Cell Sci (2010) 123:1785–95. doi: 10.1242/jcs.059519
28. Hoffman HK, Prekeris R. Roles of the actin cytoskeleton in ciliogenesis. J Cell Sci (2022) 135:jcs259030. doi: 10.1242/jcs.259030
29. Rivera-Molina FE, Xi Z, Reales E, Wang B, Toomre D. Exocyst complex mediates recycling of internal cilia. Curr Biol (2021) 31:5580–5589.e5585. doi: 10.1016/j.cub.2021.09.067
30. Bernabé-Rubio M, Alonso MA. Routes and machinery of primary cilium biogenesis. Cell Mol Life Sci (2017) 74:4077–95. doi: 10.1007/s00018-017-2570-5
31. Labat-de-Hoz L, Rubio-Ramos A, Casares-Arias J, Bernabé-Rubio M, Correas I, Alonso MA. A model for primary cilium biogenesis by polarized epithelial cells: role of the midbody remnant and associated specialized membranes. Front Cell Dev Biol (2021) 8:622918. doi: 10.3389/fcell.2020.622918
32. Spektor A, Tsang WY, Khoo D, Dynlacht BD. Cep97 and CP110 suppress a cilia assembly program. Cell (2007) 130:678–90. doi: 10.1016/j.cell.2007.06.027
33. Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD. CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev Cell (2002) 3:339–50. doi: 10.1016/S1534-5807(02)00258-7
34. Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof Y-D, Nigg EA. Plk4-induced centriole biogenesis in human cells. Dev Cell (2007) 13:190–202. doi: 10.1016/j.devcel.2007.07.002
35. Schmidt TI, Kleylein-Sohn J, Westendorf J, Le Clech M, Lavoie SB, Stierhof Y-D, et al. Control of centriole length by CPAP and CP110. Curr Biol (2009) 19:1005–11. doi: 10.1016/j.cub.2009.05.016
36. Kobayashi T, Dynlacht BD. Regulating the transition from centriole to basal body. J Cell Biol (2011) 193:435–44. doi: 10.1083/jcb.201101005
37. Seeley ES, Nachury MV. The perennial organelle: assembly and disassembly of the primary cilium. J Cell Sci (2010) 123:511–8. doi: 10.1242/jcs.061093
38. Cao J, Shen Y, Zhu L, Xu Y, Zhou Y, Wu Z, et al. miR-129-3p controls cilia assembly by regulating CP110 and actin dynamics. Nat Cell Biol (2012) 14:697–706. doi: 10.1038/ncb2512
39. Jewett CE, Soh AW, Lin CH, Lu Q, Lencer E, Westlake CJ, et al. RAB19 directs cortical remodeling and membrane growth for primary ciliogenesis. Dev Cell (2021) 56:325–340. e328. doi: 10.1016/j.devcel.2020.12.003
40. Reiter JF, Blacque OE, Leroux MR. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep (2012) 13:608–18. doi: 10.1038/embor.2012.73
41. Jensen VL, Leroux MR. Gates for soluble and membrane proteins, and two trafficking systems (IFT and LIFT), establish a dynamic ciliary signaling compartment. Curr Opin Cell Biol (2017) 47:83–91. doi: 10.1016/j.ceb.2017.03.012
42. Fisch C, Dupuis-Williams P. Ultrastructure of cilia and flagella–back to the future! Biol Cell (2011) 103:249–70. doi: 10.1042/BC20100139
43. Gonçalves J, Pelletier L. The ciliary transition zone: finding the pieces and assembling the gate. Mole Cells (2017) 40:243. doi: 10.14348/molcells.2017.0054
44. Deane JA, Cole DG, Seeley ES, Diener DR, Rosenbaum JL. Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr Biol (2001) 11:1586–90. doi: 10.1016/S0960-9822(01)00484-5
45. Nachury MV, Seeley ES, Jin H. Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu Rev Cell Dev Biol (2010) 26:59–87. doi: 10.1146/annurev.cellbio.042308.113337
46. Tanos BE, Yang H-J, Soni R, Wang W-J, Macaluso FP, Asara JM, et al. Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev (2013) 27:163–8. doi: 10.1101/gad.207043.112
47. Wheway G, Nazlamova L, Hancock JT. Signaling through the primary cilium. Front Cell Dev Biol (2018) 6:8. doi: 10.3389/fcell.2018.00008
48. Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell (2011) 145:513–28. doi: 10.1016/j.cell.2011.04.019
49. Gupta S, Ozimek-Kulik JE, Phillips JK. Nephronophthisis-pathobiology and molecular pathogenesis of a rare kidney genetic disease. Genes (2021) 12:1762. doi: 10.3390/genes12111762
50. Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet (2011) 43:776–84. doi: 10.1038/ng.891
51. Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol (2012) 14:61–72. doi: 10.1038/ncb2410
52. Remans K, Bürger M, Vetter IR, Wittinghofer A. C2 domains as protein-protein interaction modules in the ciliary transition zone. Cell Rep (2014) 8:1–9. doi: 10.1016/j.celrep.2014.05.049
53. Gupta GD, Coyaud É, Gonçalves J, Mojarad BA, Liu Y, Wu Q, et al. A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell (2015) 163:1484–99. doi: 10.1016/j.cell.2015.10.065
54. Dean S, Moreira-Leite F, Varga V, Gull K. Cilium transition zone proteome reveals compartmentalization and differential dynamics of ciliopathy complexes. Proc Natl Acad Sci (2016) 113:E5135–43. doi: 10.1073/pnas.1604258113
55. Bernabé-Rubio M, Andrés G, Casares-Arias J, Fernández-Barrera J, Rangel L, Reglero-Real N, et al. Novel role for the midbody in primary ciliogenesis by polarized epithelial cells. J Cell Biol (2016) 214:259–73. doi: 10.1083/jcb.201601020
56. Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL. Chlamydomonas kinesin-II–dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol (1998) 141:993–1008. doi: 10.1083/jcb.141.4.993
57. Kozminski KG, Beech PL, Rosenbaum JL. The Chlamydomonas kinesin-like protein FLA10 is involved in motility associated with the flagellar membrane. J Cell Biol (1995) 131:1517–27. doi: 10.1083/jcb.131.6.1517
58. Pazour GJ, Wilkerson CG, Witman GB. A dynein light chain is essential for the retrograde particle movement of intraflagellar transport (IFT). J Cell Biol (1998) 141:979–92. doi: 10.1083/jcb.141.4.979
59. Porter ME, Bower R, Knott JA, Byrd P, Dentler W. Cytoplasmic dynein heavy chain 1b is required for flagellar assembly in Chlamydomonas. Mol Biol Cell (1999) 10:693–712. doi: 10.1091/mbc.10.3.693
60. Taschner M, Bhogaraju S, Lorentzen E. Architecture and function of IFT complex proteins in ciliogenesis. Differentiation (2012) 83:S12–22. doi: 10.1016/j.diff.2011.11.001
61. Chien A, Shih SM, Bower R, Tritschler D, Porter ME, Yildiz A. Dynamics of the IFT machinery at the ciliary tip. Elife (2017) 6:e28606. doi: 10.7554/eLife.28606
62. Klena N, Pigino G. Structural biology of cilia and intraflagellar transport. Annu Rev Cell Dev Biol (2022) 38:103–23. doi: 10.1146/annurev-cellbio-120219-034238
63. Bujakowska KM, Zhang Q, Siemiatkowska AM, Liu Q, Place E, Falk MJ, et al. Mutations in IFT172 cause isolated retinal degeneration and Bardet–Biedl syndrome. Hum Mol Genet (2015) 24:230–42. doi: 10.1093/hmg/ddu441
64. Luo M, Lin Z, Zhu T, Jin M, Meng D, He R, et al. Disrupted intraflagellar transport due to IFT74 variants causes Joubert syndrome. Genet Med (2021) 23:1041–9. doi: 10.1038/s41436-021-01106-z
65. Thevenon J, Duplomb L, Phadke S, Eguether T, Saunier A, Avila M, et al. Autosomal recessive IFT57 hypomorphic mutation cause ciliary transport defect in unclassified oral-facial-digital syndrome with short stature and brachymesophalangia. Clin Genet (2016) 90:509–17. doi: 10.1111/cge.12785
66. Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet (2011) 43:189–96. doi: 10.1038/ng.756
67. Loktev AV, Zhang Q, Beck JS, Searby CC, Scheetz TE, Bazan JF, et al. A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev Cell (2008) 15:854–65. doi: 10.1016/j.devcel.2008.11.001
68. Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell (2007) 129:1201–13. doi: 10.1016/j.cell.2007.03.053
69. Zhang Q, Nishimura D, Vogel T, Shao J, Swiderski R, Yin T, et al. BBS7 is required for BBSome formation and its absence in mice results in Bardet-Biedl syndrome phenotypes and selective abnormalities in membrane protein trafficking. J Cell Sci (2013) 126:2372–80. doi: 10.1242/jcs.111740
70. Seo S, Zhang Q, Bugge K, Breslow DK, Searby CC, Nachury MV, et al. A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and Smoothened. PloS Genet (2011) 7:e1002358. doi: 10.1371/journal.pgen.1002358
71. Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. Bardet–Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci (2008) 105:4242–6. doi: 10.1073/pnas.0711027105
72. Lechtreck K-F, Johnson EC, Sakai T, Cochran D, Ballif BA, Rush J, et al. The Chlamydomonas reinhardtii BBSome is an IFT cargo required for export of specific signaling proteins from flagella. J Cell Biol (2009) 187:1117–32. doi: 10.1083/jcb.200909183
73. Ye F, Nager AR, Nachury MV. BBSome trains remove activated GPCRs from cilia by enabling passage through the transition zone. J Cell Biol (2018) 217:1847–68. doi: 10.1083/jcb.201709041
74. Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet–Biedl syndrome. Nature (2003) 425:628–33. doi: 10.1038/nature02030
75. Wingfield JL, Lechtreck K-F, Lorentzen E. Trafficking of ciliary membrane proteins by the intraflagellar transport/BBSome machinery. Essays Biochem (2018) 62:753–63. doi: 10.1042/EBC20180030
76. CHaitin MH, Schneider BG, Hall MO, Papermaster DS. Actin in the photoreceptor connecting cilium: immunocytochemical localization to the site of outer segment disk formation. J Cell Biol (1984) 99:239–47. doi: 10.1083/jcb.99.1.239
77. Liu X, Vansant G, Udovichenko IP, Wolfrum U, Williams DS. Myosin VIIa, the product of the Usher 1B syndrome gene, is concentrated in the connecting cilia of photoreceptor cells. Cell Motil Cytoskeleton (1997) 37:240–52. doi: 10.1002/(SICI)1097-0169(1997)37:3<240::AID-CM6>3.0.CO;2-A
78. Ge R, Cao M, Chen M, Liu M, Xie S. Cytoskeletal networks in primary cilia: Current knowledge and perspectives. J Cell Physiol (2022) 237:3975–83. doi: 10.1002/jcp.30865
79. Kim J, Lee JE, Heynen-Genel S, Suyama E, Ono K, Lee K, et al. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature (2010) 464:1048–51. doi: 10.1038/nature08895
80. Wheway G, Schmidts M, Mans DA, Szymanska K, Nguyen T-MT, Racher H, et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat Cell Biol (2015) 17:1074–87. doi: 10.1038/ncb3201
81. Heydeck W, Fievet L, Davis EE, Katsanis N. The complexity of the cilium: spatiotemporal diversity of an ancient organelle. Curr Opin Cell Biol (2018) 55:139–49. doi: 10.1016/j.ceb.2018.08.001
82. Vasquez SSV, van Dam J, Wheway G. An updated SYSCILIA gold standard (SCGSv2) of known ciliary genes, revealing the vast progress that has been made in the cilia research field. Mol Biol Cell (2021) 32:br13. doi: 10.1091/mbc.E21-05-0226
83. Van Dam TJ, Kennedy J, van der Lee R, De Vrieze E, Wunderlich KA, Rix S, et al. CiliaCarta: An integrated and validated compendium of ciliary genes. PloS One (2019) 14:e0216705. doi: 10.1371/journal.pone.0216705
84. Arnaiz O, Malinowska A, Klotz C, Sperling L, Dadlez M, Koll F, et al. Cildb: a knowledgebase for centrosomes and cilia. Database (2009) 2009:bap022. doi: 10.1093/database/bap022
85. Elliott KH, Balchand SK, Bonatto Paese CL, Chang C-F, Yang Y, Brown KM, et al. Identification of a heterogeneous and dynamic ciliome during embryonic development and cell differentiation. Development (2023) 150:dev201237. doi: 10.1242/dev.201237
86. Kohli P, Höhne M, Jüngst C, Bertsch S, Ebert LK, Schauss AC, et al. The ciliary membrane-associated proteome reveals actin-binding proteins as key components of cilia. EMBO Rep (2017) 18:1521–35. doi: 10.15252/embr.201643846
87. Choksi SP, Babu D, Lau D, Yu X, Roy S. Systematic discovery of novel ciliary genes through functional genomics in the zebrafish. Development (2014) 141:3410–9. doi: 10.1242/dev.108209
88. Corral-Serrano JC, Lamers IJ, van Reeuwijk J, Duijkers L, Hoogendoorn AD, Yildirim A, et al. PCARE and WASF3 regulate ciliary F-actin assembly that is required for the initiation of photoreceptor outer segment disk formation. Proc Natl Acad Sci (2020) 117:9922–31. doi: 10.1073/pnas.1903125117
89. Euteneuer U, Schliwa M. Evidence for an involvement of actin in the positioning and motility of centrosomes. J Cell Biol (1985) 101:96–103. doi: 10.1083/jcb.101.1.96
90. Francis SS, Sfakianos J, Lo B, Mellman I. A hierarchy of signals regulates entry of membrane proteins into the ciliary membrane domain in epithelial cells. J Cell Biol (2011) 193:219–33. doi: 10.1083/jcb.201009001
91. Hoffman HK, Prekeris R. HOPS-dependent lysosomal fusion controls Rab19 availability for ciliogenesis in polarized epithelial cells. J Cell Sci (2024) 137(5):jcs261047. doi: 10.1242/jcs.261047
92. Clare DK, Magescas J, Piolot T, Dumoux M, Vesque C, Pichard E, et al. Basal foot MTOC organizes pillar MTs required for coordination of beating cilia. Nat Commun (2014) 5:4888. doi: 10.1038/ncomms5888
93. Pitaval A, Senger F, Letort G, Gidrol X, Guyon L, Sillibourne J, et al. Microtubule stabilization drives 3D centrosome migration to initiate primary ciliogenesis. J Cell Biol (2017) 216:3713–28. doi: 10.1083/jcb.201610039
94. Lindsay AJ, Jollivet F, Horgan CP, Khan AR, Raposo G, McCaffrey MW, et al. Identification and characterization of multiple novel Rab–myosin Va interactions. Mol Biol Cell (2013) 24:3420–34. doi: 10.1091/mbc.e13-05-0236
95. Smith CEL, Lake AVR, Johnson CA. Primary cilia, ciliogenesis and the actin cytoskeleton: A little less resorption, A little more actin please. Front Cell Dev Biol (2020) 8:622822. doi: 10.3389/fcell.2020.622822
96. Rozycki M, Lodyga M, Lam J, Miranda MZ, Fatyol K, Speight P, et al. The fate of the primary cilium during myofibroblast transition. Mol Biol Cell (2014) 25:643–57. doi: 10.1091/mbc.E13-07-0429
97. Wang L, Dynlacht BD. The regulation of cilium assembly and disassembly in development and disease. Development (2018) 145(18):dev151407. doi: 10.1242/dev.151407
98. Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell (2007) 129:1351–63. doi: 10.1016/j.cell.2007.04.035
99. Lee KH, Johmura Y, Yu LR, Park JE, Gao Y, Bang JK, et al. Identification of a novel Wnt5a-CK1varepsilon-Dvl2-Plk1-mediated primary cilia disassembly pathway. EMBO J (2012) 31:3104–17. doi: 10.1038/emboj.2012.144
100. Kim S, Lee K, Choi JH, Ringstad N, Dynlacht BD. Nek2 activation of Kif24 ensures cilium disassembly during the cell cycle. Nat Commun (2015) 6:8087. doi: 10.1038/ncomms9087
101. Kim J, Jo H, Hong H, Kim MH, Kim JM, Lee JK, et al. Actin remodelling factors control ciliogenesis by regulating YAP/TAZ activity and vesicle trafficking. Nat Commun (2015) 6:6781. doi: 10.1038/ncomms7781
102. Palander O. Regulation of a Downstream Effector (DIAPH1, 2 and 3) of RhoA Signaling in Ciliogenesis and Maintenance of the Primary Cilia. Canada: University of Toronto (2020).
103. Palander O, Lam A, Collins RF, Moraes TJ, Trimble WS. Nonredundant roles of DIAPHs in primary ciliogenesis. J Biol Chem (2021) 296:100680. doi: 10.1016/j.jbc.2021.100680
104. Hall A. Rho GTPases and the actin cytoskeleton. Science (1998) 279:509–14. doi: 10.1126/science.279.5350.509
105. Van Aelst L, Symons M. Role of Rho family GTPases in epithelial morphogenesis. Genes Dev (2002) 16:1032–54. doi: 10.1101/gad.978802
106. Schmidt S, Debant A. Function and regulation of the Rho guanine nucleotide exchange factor Trio. Small GTPases (2014) 5:e983880. doi: 10.4161/sgtp.29769
107. Pedersen E, Brakebusch C. Rho GTPase function in development: how in vivo models change our view. Exp Cell Res (2012) 318:1779–87. doi: 10.1016/j.yexcr.2012.05.004
108. Gamblin SJ, Smerdon SJ. GTPase-activating proteins and their complexes. Curr Opin Struct Biol (1998) 8:195–201. doi: 10.1016/S0959-440X(98)80038-9
109. Pan J, You Y, Huang T, Brody SL. RhoA-mediated apical actin enrichment is required for ciliogenesis and promoted by Foxj1. J Cell Sci (2007) 120:1868–76. doi: 10.1242/jcs.005306
110. Park TJ, Mitchell BJ, Abitua PB, Kintner C, Wallingford JB. Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat Genet (2008) 40:871–9. doi: 10.1038/ng.104ng.104
111. Stewart K, Gaitan Y, Shafer ME, Aoudjit L, Hu D, Sharma R, et al. A point mutation in p190A RhoGAP affects ciliogenesis and leads to glomerulocystic kidney defects. PloS Genet (2016) 12:e1005785. doi: 10.1371/journal.pgen.1005785
112. Streets AJ, Prosseda PP, Ong AC. Polycystin-1 regulates ARHGAP35-dependent centrosomal RhoA activation and ROCK signaling. JCI Insight (2020) 5(16):e135385. doi: 10.1172/jci.insight.135385
113. Yeyati PL, Schiller R, Mali G, Kasioulis I, Kawamura A, Adams IR, et al. KDM3A coordinates actin dynamics with intraflagellar transport to regulate cilia stability. J Cell Biol (2017) 216:999–1013. doi: 10.1083/jcb.201607032
114. Mostowy S, Cossart P. Septins: the fourth component of the cytoskeleton. Nat Rev Mol Cell Biol (2012) 13:183–94. doi: 10.1038/nrm3284
115. Safavian D, Kim MS, Xie H, El-Zeiry M, Palander O, Dai L, et al. Septin-mediated RhoA activation engages the exocyst complex to recruit the cilium transition zone. J Cell Biol (2023) 222:e201911062. doi: 10.1083/jcb.201911062
116. Adamo JE, Rossi G, Brennwald P. The Rho GTPase Rho3 has a direct role in exocytosis that is distinct from its role in actin polarity. Mol Biol Cell (1999) 10:4121–33. doi: 10.1091/mbc.10.12.4121
117. Guo W, Tamanoi F, Novick P. Spatial regulation of the exocyst complex by Rho1 GTPase. Nat Cell Biol (2001) 3:353–60. doi: 10.1038/35070029
118. Fu W, Wang L, Kim S, Li J, Dynlacht BD. Role for the IFT-A complex in selective transport to the primary cilium. Cell Rep (2016) 17:1505–17. doi: 10.1016/j.celrep.2016.10.018
119. Kakiuchi A, Kohno T, Kakuki T, Kaneko Y, Konno T, Hosaka Y, et al. Rho-kinase and PKCα inhibition induces primary cilia elongation and alters the behavior of undifferentiated and differentiated temperature-sensitive mouse cochlear cells. J Histochem Cytochem (2019) 67:523–35. doi: 10.1369/0022155419841013
120. Bershteyn M, Atwood SX, Woo W-M, Li M, Oro AE. MIM and cortactin antagonism regulates ciliogenesis and hedgehog signaling. Dev Cell (2010) 19:270–83. doi: 10.1016/j.devcel.2010.07.009
121. Failler M, Giro-Perafita A, Owa M, Srivastava S, Yun C, Kahler DJ, et al. Whole-genome screen identifies diverse pathways that negatively regulate ciliogenesis. Mol Biol Cell (2021) 32:169–85. doi: 10.1091/mbc.E20-02-0111
122. Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol (2000) 151:709–18. doi: 10.1083/jcb.151.3.709
123. Yoder BK, Tousson A, Millican L, Wu JH, Bugg CE, Schafer JA, et al. Polaris, a protein disrupted in orpk mutant mice, is required for assembly of renal cilium. Am J Physiology-Renal Physiol (2002) 282:F541–52. doi: 10.1152/ajprenal.00273.2001
124. Moyer JH, Lee-Tischler MJ, Kwon H-Y, Schrick JJ, Avner ED, Sweeney WE, et al. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science (1994) 264:1329–33. doi: 10.1126/science.8191288
125. Cano DA, Murcia NS, Pazour GJ, Hebrok M. orpk mouse model of polycystic kidney disease reveals essentialrole of primary cilia in pancreatic tissue organization. Development (2004) 131:3457–67. doi: 10.1242/dev.01189
126. Lehman JM, Michaud EJ, Schoeb TR, Aydin-Son Y, Miller M, Yoder BK. The oak ridge polycystic kidney mouse: modeling ciliopathies of mice and men. Dev Dyn (2008) 237:1960–71. doi: 10.1002/dvdy.21515
127. The European Polycystic Kidney Disease, C. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell (1994) 77:881–94. doi: 10.1016/0092-8674(94)90137-6
128. Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers (2018) 4:50. doi: 10.1038/s41572-018-0047-y
129. Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millán JL, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet (1995) 10:151–60. doi: 10.1038/ng0695-151
130. Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science (1996) 272:1339–42. doi: 10.1126/science.272.5266.1339
131. Cornec-Le Gall E, Torres VE, Harris PC. Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J Am Soc Nephrol (2018) 29:13–23. doi: 10.1681/asn.2017050483
132. Bergmann C, Von Bothmer J, Ortiz Brüchle N, Venghaus A, Frank V, Fehrenbach H, et al. Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol (2011) 22:2047–56. doi: 10.1681/ASN.2010101080
133. Lanktree MB, Haghighi A, di Bari I, Song X, Pei Y. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin J Am Soc Nephrol (2021) 16:790–9. doi: 10.2215/CJN.02320220
134. Hartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics (2014) 134:e833–845. doi: 10.1542/peds.2013-3646
135. Ward CJ, Yuan D, Masyuk TV, Wang X, Punyashthiti R, Whelan S, et al. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum Mol Genet (2003) 12:2703–10. doi: 10.1093/hmg/ddg274
136. Zhang M-Z, Mai W, Li C, Cho S-y, Hao C, Moeckel G, et al. PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc Natl Acad Sci U.S.A. (2004) 101:2311–6. doi: 10.1073/pnas.0400073101
137. Fedeles SV, Gallagher A-R, Somlo S. Polycystin-1: a master regulator of intersecting cystic pathways. Trends Mol Med (2014) 20:251–60. doi: 10.1016/j.molmed.2014.01.004
138. Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest (2014) 124:2315–24. doi: 10.1172/JCI72272
139. Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet (2003) 33:129–37. doi: 10.1038/ng1076
140. Delling M, DeCaen PG, Doerner JF, Febvay S, Clapham DE. Primary cilia are specialized calcium signaling organelles. Nature (2013) 504:311–4. doi: 10.1038/nature12833
141. Luyten A, Su X, Gondela S, Chen Y, Rompani S, Takakura A, et al. Aberrant regulation of planar cell polarity in polycystic kidney disease. J Am Soc Nephrol JASN (2010) 21:1521–32. doi: 10.1681/ASN.2010010127
142. Drummond IA. Polycystins, focal adhesions and extracellular matrix interactions. Biochim Biophys Acta (2011) 1812:1322–6. doi: 10.1016/j.bbadis.2011.03.003
143. Fischer E, Legue E, Doyen A, Nato F, Nicolas JF, Torres V, et al. Defective planar cell polarity in polycystic kidney disease. Nat Genet (2006) 38:21–3. doi: 10.1038/ng1701
144. Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan H-F, Gainullin VG, et al. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest (2012) 122:4257–73. doi: 10.1172/JCI64313
145. Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet (2013) 45:1004–12. doi: 10.1038/ng.2715
146. Gerakopoulos V, Ngo P, Tsiokas L. Loss of polycystins suppresses deciliation via the activation of the centrosomal integrity pathway. Life Sci Alliance (2020) 3:e202000750. doi: 10.26508/lsa.202000750
147. Cai J, Song X, Wang W, Watnick T, Pei Y, Qian F, et al. A RhoA–YAP–c-Myc signaling axis promotes the development of polycystic kidney disease. Genes Dev (2018) 32:781–93. doi: 10.1101/gad.315127.118
148. Formica C, Kunnen S, Dauwerse JG, Mullick AE, Dijkstra KL, Scharpfenecker M, et al. Reducing YAP expression in Pkd1 mutant mice does not improve the cystic phenotype. J Cell Mol Med (2020) 24:8876–82. doi: 10.1111/jcmm.15512
149. Fanconi G, Hanhart E, von Albertini A, Uhlinger E, Dolivo G, Prader A. Familial, juvenile nephronophthisis (idiopathic parenchymal contracted kidney). Helv. Paediatr Acta (1951) 6:1–49.
150. Wolf MTF, Hildebrandt F. Nephronophthisis. Pediatr Nephrol (2011) 26:181–94. doi: 10.1007/s00467-010-1585-z
151. Simms RJ, Hynes AM, Eley L, Sayer JA. Nephronophthisis: a genetically diverse ciliopathy. Int J Nephrol (2011) 2011:527137. doi: 10.4061/2011/527137
152. Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harbor Perspect Biol (2017) 9(3):a028191. doi: 10.1101/cshperspect.a028191
153. Hildebrandt F, Otto E, Rensing C, Nothwang HG, Vollmer M, Adolphs J, et al. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet (1997) 17:149–53. doi: 10.1038/ng1097-149
154. Srivastava S, Molinari E, Raman S, Sayer JA. Many genes—One disease? Genetics of nephronophthisis (NPHP) and NPHP-associated disorders. Front Pediatr (2018) 5:287. doi: 10.3389/fped.2017.00287
155. Barroso-Gil M, Olinger E, Sayer JA. Molecular genetics of renal ciliopathies. Biochem Soc Trans (2021) 49:1205–20. doi: 10.1042/BST20200791
156. Petzold F, Billot K, Chen X, Henry C, Filhol E, Martin Y, et al. The genetic landscape and clinical spectrum of nephronophthisis and related ciliopathies. Kidney Int (2023) 104:378–87. doi: 10.1016/j.kint.2023.05.007
157. Luo F, Tao YH. Nephronophthisis: A review of genotype–phenotype correlation. Nephrol (Carlton) (2018) 23:904–11. doi: 10.1111/nep.13393
158. Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol (2011) 192:1023–41. doi: 10.1083/jcb.201012116
159. McConnachie DJ, Stow JL, Mallett AJ. Ciliopathies and the kidney: A review. Am J Kidney Dis (2021) 77:410–9. doi: 10.1053/j.ajkd.2020.08.012
160. Salomon R, Saunier S, Niaudet P. Nephronophthisis. Pediatr Nephrol (2009) 24:2333–44. doi: 10.1007/s00467-008-0840-z
161. Hurd TW, Hildebrandt F. Mechanisms of nephronophthisis and related ciliopathies. Nephron Exp Nephrol (2010) 118:e9–e14. doi: 10.1159/000320888
162. Benzing T, Gerke P, Höpker K, Hildebrandt F, Kim E, Walz G. Nephrocystin interacts with Pyk2, p130Cas, and tensin and triggers phosphorylation of Pyk2. Proc Natl Acad Sci U.S.A. (2001) 98:9784–9. doi: 10.1073/pnas.171269898
163. Donaldson JC, Dise RS, Ritchie MD, Hanks SK. Nephrocystin-conserved domains involved in targeting to epithelial cell-cell junctions, interaction with filamins, and establishing cell polarity*. J Biol Chem (2002) 277:29028–35. doi: 10.1074/jbc.M111697200
164. Mollet G, Silbermann F, Delous M, Salomon R, Antignac C, Saunier S. Characterization of the nephrocystin/nephrocystin-4 complex and subcellular localization of nephrocystin-4 to primary cilia and centrosomes. Hum Mol Genet (2005) 14:645–56. doi: 10.1093/hmg/ddi061
165. Antoniades I, Stylianou P, Skourides PA. Making the connection: ciliary adhesion complexes anchor basal bodies to the actin cytoskeleton. Dev Cell (2014) 28:70–80. doi: 10.1016/j.devcel.2013.12.003
166. Delous M, Hellman NE, Gaudé H-M, Silbermann F, Le Bivic A, Salomon R, et al. Nephrocystin-1 and nephrocystin-4 are required for epithelial morphogenesis and associate with PALS1/PATJ and Par6. Hum Mol Genet (2009) 18:4711–23. doi: 10.1093/hmg/ddp434
167. Yasunaga T, Hoff S, Schell C, Helmstädter M, Kretz O, Kuechlin S, et al. The polarity protein Inturned links NPHP4 to Daam1 to control the subapical actin network in multiciliated cells. J Cell Biol (2015) 211:963–73. doi: 10.1083/jcb.201502043
168. Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Krönig C, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet (2005) 37:537–43. doi: 10.1038/ng1552
169. Werner ME, Ward HH, Phillips CL, Miller C, Gattone VH, Bacallao RL. Inversin modulates the cortical actin network during mitosis. Am J Physiol Cell Physiol (2013) 305:C36–47. doi: 10.1152/ajpcell.00279.2012
170. Lienkamp S, Ganner A, Walz G. Inversin, Wnt signaling and primary cilia. Differentiation (2012) 83:S49–55. doi: 10.1016/j.diff.2011.11.012
171. Hu Q, Lai J, Chen H, Cai Y, Yue Z, Lin H, et al. Reducing GEF-H1 expression inhibits renal cyst formation, inflammation, and fibrosis via rhoA signaling in nephronophthisis. Int J Mol Sci (2023) 24:3504. doi: 10.3390/ijms24043504
172. Forsyth R, Gunay-Aygun M. Bardet-Biedl Syndrome Overview. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews®. Seattle: University of Washington (1993).
173. Moore SJ, Green JS, Fan Y, Bhogal AK, Dicks E, Fernandez BA, et al. Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study. Am J Med Genet A (2005) 132a:352–60. doi: 10.1002/ajmg.a.30406
174. O'Dea D, Parfrey PS, Harnett JD, Hefferton D, Cramer BC, Green J. The importance of renal impairment in the natural history of bardet-biedl syndrome. Am J Kidney Dis (1996) 27:776–83. doi: 10.1016/S0272-6386(96)90513-2
175. Marchese E, Ruoppolo M, Perna A, Capasso G, Zacchia M. Exploring key challenges of understanding the pathogenesis of kidney disease in bardet–biedl syndrome. Kidney Int Rep (2020) 5:1403–15. doi: 10.1016/j.ekir.2020.06.017
176. Putoux A, Attie-Bitach T, Martinovic J, Gubler M-C. Phenotypic variability of Bardet-Biedl syndrome: focusing on the kidney. Pediatr Nephrol (2012) 27:7–15. doi: 10.1007/s00467-010-1751-3
177. Tian X, Zhao H, Zhou J. Organization, functions, and mechanisms of the BBSome in development, ciliopathies, and beyond. eLife (2023) 12:e87623. doi: 10.7554/eLife.87623
178. Abd-El-Barr MM, Sykoudis K, Andrabi S, Eichers ER, Pennesi ME, Tan PL, et al. Impaired photoreceptor protein transport and synaptic transmission in a mouse model of bardet-biedl syndrome. Vision Res (2007) 47:3394–407. doi: 10.1016/j.visres.2007.09.016
179. Datta P, Allamargot C, Hudson JS, Andersen EK, Bhattarai S, Drack AV, et al. Accumulation of non-outer segment proteins in the outer segment underlies photoreceptor degeneration in Bardet–Biedl syndrome. Proc Natl Acad Sci U.S.A. (2015) 112:E4400–9. doi: 10.1073/pnas.1510111112
180. Mayer SK, Thomas J, Helms M, Kothapalli A, Cherascu I, Salesevic A, et al. Progressive retinal degeneration of rods and cones in a Bardet-Biedl syndrome type 10 mouse model. Dis Model Mech (2022) 15:dmm049473. doi: 10.1242/dmm.049473
181. Hey CAB, Larsen LJ, Tümer Z, Brøndum-Nielsen K, Grønskov K, Hjortshøj TD, et al. BBS Proteins Affect Ciliogenesis and Are Essential for Hedgehog Signaling, but Not for Formation of iPSC-Derived RPE-65 Expressing RPE-Like Cells. Int J Mol Sci (2021) 22:1345. doi: 10.3390/ijms22031345
182. Tran PV, Talbott GC, Turbe-Doan A, Jacobs DT, Schonfeld MP, Silva LM, et al. Downregulating hedgehog signaling reduces renal cystogenic potential of mouse models. J Am Soc Nephrol (2014) 25:2201. doi: 10.1681/ASN.2013070735
183. Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet (2007) 39:1350–60. doi: 10.1038/ng.2007.12
184. Hernandez-Hernandez V, Pravincumar P, Diaz-Font A, May-Simera H, Jenkins D, Knight M, et al. Bardet–Biedl syndrome proteins control the cilia length through regulation of actin polymerization. Hum Mol Genet (2013) 22:3858–68. doi: 10.1093/hmg/ddt241
185. Alexiev BA, Lin X, Sun C-C, Brenner DS. Meckel-gruber syndrome: pathologic manifestations, minimal diagnostic criteria, and differential diagnosis. Arch Pathol Lab Med (2006) 130:1236–8. doi: 10.5858/2006-130-1236-MS
186. Parelkar SV, Kapadnis SP, Sanghvi BV, Joshi PB, Mundada D, Oak SN. Meckel-Gruber syndrome: A rare and lethal anomaly with review of literature. J Pediatr Neurosci (2013) 8:154–7. doi: 10.4103/1817-1745.117855
187. Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, Brancati F, et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet (2010) 42:619–25. doi: 10.1038/ng.594
188. Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol (2011) 26:1039–56. doi: 10.1007/s00467-010-1731-7
189. Dawe HR, Smith UM, Cullinane AR, Gerrelli D, Cox P, Badano JL, et al. The Meckel–Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet (2007) 16:173–86. doi: 10.1093/hmg/ddl459
190. Dawe HR, Adams M, Wheway G, Szymanska K, Logan CV, Noegel AA, et al. Nesprin-2 interacts with meckelin and mediates ciliogenesis via remodelling of the actin cytoskeleton. J Cell Sci (2009) 122:2716–26. doi: 10.1242/jcs.043794
191. Huang L, Szymanska K, Jensen VL, Janecke AR, Innes AM, Davis EE, et al. TMEM237 is mutated in individuals with a joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet (2011) 89:713–30. doi: 10.1016/j.ajhg.2011.11.005
192. Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, Paruchuri Y, et al. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J Med Genet (2009) 46:663–70. doi: 10.1136/jmg.2009.066613
193. Van De Weghe JC, Gomez A, Doherty D. The joubert–meckel–nephronophthisis spectrum of ciliopathies. Annu Rev Genomics Hum Genet (2022) 23:301–29. doi: 10.1146/annurev-genom-121321-093528
194. Dawe HR, Farr H, Gull K. Centriole/basal body morphogenesis and migration during ciliogenesis in animal cells. J Cell Sci (2007) 120:7–15. doi: 10.1242/jcs.03305
195. Handa A, Voss U, Hammarsjö A, Grigelioniene G, Nishimura G. Skeletal ciliopathies: a pattern recognition approach. Jpn J Radiol (2020) 38:193–206. doi: 10.1007/s11604-020-00920-w
196. Huber C, Cormier-Daire V. Ciliary disorder of the skeleton. Am J Med Genet Part C: Semin Med Genet (2012) 160C:165–74. doi: 10.1002/ajmg.c.31336
197. Bruel A-L, Franco B, Duffourd Y, Thevenon J, Jego L, Lopez E, et al. 15 years of research on Oral-Facial-Digital syndromes: from 1 to 16 causal genes. J Med Genet (2017) 54:371–80. doi: 10.1136/jmedgenet-2016-104436
198. Walczak-Sztulpa J, Eggenschwiler J, Osborn D, Brown DA, Emma F, Klingenberg C, et al. Cranioectodermal dysplasia, sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet (2010) 86:949–56. doi: 10.1016/j.ajhg.2010.04.012
199. Mill P, Lockhart PJ, Fitzpatrick E, Mountford HS, Hall EA, Reijns MAM, et al. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am J Hum Genet (2011) 88:508–15. doi: 10.1016/j.ajhg.2011.03.015
200. Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet (2011) 89:634–43. doi: 10.1016/j.ajhg.2011.10.001
201. Arts HH, Bongers EMHF, Mans DA, Beersum S, Oud MM, Bolat E, et al. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J Med Genet (2011) 48:390–5. doi: 10.1136/jmg.2011.088864
202. Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, et al. Defects in the IFT-B component IFT172 cause jeune and mainzer-saldino syndromes in humans. Am J Hum Genet (2013) 93:915–25. doi: 10.1016/j.ajhg.2013.09.012
203. Senum SR, Li YM, Benson KA, Joli G, Olinger E, Lavu S, et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am J Hum Genet (2022) 109:136–56. doi: 10.1016/j.ajhg.2021.11.016
204. Chetty-John S, Piwnica-Worms K, Bryant J, Bernardini I, Fischer RE, Heller T, et al. Fibrocystic disease of liver and pancreas; under-recognized features of the X-linked ciliopathy oral-facial-digital syndrome type 1 (OFD1). Am J Med Genet (2010) Part A 152A:2640–5. doi: 10.1002/ajmg.a.33666
205. Franco B, Thauvin-Robinet C. Update on oral-facial-digital syndromes (OFDS). Cilia (2016) 5:12. doi: 10.1186/s13630-016-0034-4
206. Singla V, Romaguera-Ros M, Garcia-Verdugo JM, Reiter JF. Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev Cell (2010) 18:410–24. doi: 10.1016/j.devcel.2009.12.022
207. Alfieri M, Iaconis D, Tammaro R, Perone L, Calì G, Nitsch L, et al. The centrosomal/basal body protein OFD1 is required for microtubule organization and cell cycle progression. Tissue Cell (2020) 64:101369. doi: 10.1016/j.tice.2020.101369
208. Cao M, Zou X, Li C, Lin Z, Wang N, Zou Z, et al. An actin filament branching surveillance system regulates cell cycle progression, cytokinesis and primary ciliogenesis. Nat Commun (2023) 14:1687. doi: 10.1038/s41467-023-37340-z
209. Toriyama M, Lee C, Taylor SP, Duran I, Cohn DH, Bruel AL, et al. The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nat Genet (2016) 48:648–56. doi: 10.1038/ng.3558
210. Gubb D, Garcia-Bellido A. A genetic analysis of the determination of cuticular polarity during development in Drosophila melanogaster. J Embryol Exp Morphol (1982) 68:37–57.
211. Park TJ, Haigo SL, Wallingford JB. Ciliogenesis defects in embryos lacking inturned or fuzzy function are associated with failure of planar cell polarity and Hedgehog signaling. Nat Genet (2006) 38:303–11. doi: 10.1038/ng1753
212. Gray RS, Abitua PB, Wlodarczyk BJ, Szabo-Rogers HL, Blanchard O, Lee I, et al. The planar cell polarity effector Fuz is essential for targeted membrane trafficking, ciliogenesis and mouse embryonic development. Nat Cell Biol (2009) 11:1225–32. doi: 10.1038/ncb1966
213. Cui C, Chatterjee B, Lozito TP, Zhang Z, Francis RJ, Yagi H, et al. Wdpcp, a PCP protein required for ciliogenesis, regulates directional cell migration and cell polarity by direct modulation of the actin cytoskeleton. PloS Biol (2013) 11:e1001720. doi: 10.1371/journal.pbio.1001720
214. Zeng H, Hoover AN, Liu A. PCP effector gene Inturned is an important regulator of cilia formation and embryonic development in mammals. Dev Biol (2010) 339:418–28. doi: 10.1016/j.ydbio.2010.01.003S0012-1606(10)00010-2
215. Langousis G, Cavadini S, Boegholm N, Lorentzen E, Kempf G, Matthias P. Structure of the ciliogenesis-associated CPLANE complex. Sci Adv (2022) 8:eabn0832. doi: 10.1126/sciadv.abn0832
216. Zilber Y, Babayeva S, Seo JH, Liu JJ, Mootin S, Torban E. The PCP effector Fuzzy controls cilial assembly and signaling by recruiting Rab8 and Dishevelled to the primary cilium. Mol Biol Cell (2013) 24:555–65. doi: 10.1091/mbc.E12-06-0437
217. Gilissen C, Arts HH, Hoischen A, Spruijt L, Mans DA, Arts P, et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet (2010) 87:418–23. doi: 10.1016/j.ajhg.2010.08.004
218. Srour M, Schwartzentruber J, Hamdan FF, Ospina LH, Patry L, Labuda D, et al. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am J Hum Genet (2012) 90:693–700. doi: 10.1016/j.ajhg.2012.02.011
219. Zhang W, Taylor SP, Ennis HA, Forlenza KN, Duran I, Li B, et al. Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies. Hum Mutat (2018) 39:152–66. doi: 10.1002/humu.23362
220. Seo JH, Zilber Y, Babayeva S, Liu J, Kyriakopoulos P, De Marco P, et al. Mutations in the planar cell polarity gene, Fuzzy, are associated with neural tube defects in humans. Hum Mol Genet (2011) 20:4324–33. doi: 10.1093/hmg/ddr359
221. Bozal-Basterra L, Martín-Ruíz I, Pirone L, Liang Y, Sigurðsson JO, Gonzalez-Santamarta M, et al. Truncated SALL1 impedes primary cilia function in townes-brocks syndrome. Am J Hum Genet (2018) 102:249–65. doi: 10.1016/j.ajhg.2017.12.017
222. Bozal-Basterra L, Gonzalez-Santamarta M, Muratore V, Bermejo-Arteagabeitia A, Da Fonseca C, Barroso-Gomila O, et al. LUZP1, a novel regulator of primary cilia and the actin cytoskeleton, is a contributing factor in Townes-Brocks Syndrome. eLife (2020) 9:e55957. doi: 10.7554/eLife.55957
223. Bökenkamp A, Ludwig M. The oculocerebrorenal syndrome of Lowe: an update. Pediatr Nephrol (2016) 31:2201–12. doi: 10.1007/s00467-016-3343-3
224. Mehta ZB, Pietka G, Lowe M. The cellular and physiological functions of the lowe syndrome protein OCRL1. Traffic (Copenhagen Denmark) (2014) 15:471–87. doi: 10.1111/tra.12160
225. Madhivanan K, Ramadesikan S, Aguilar RC. Role of ocrl1 in primary cilia assembly. In Int Rev Cell Mol Biology (Elsevier) (2015) pp:331–47. doi: 10.1016/bs.ircmb.2015.02.003
226. Luo N, West CC, Murga-Zamalloa CA, Sun L, Anderson RM, Wells CD, et al. OCRL localizes to the primary cilium: a new role for cilia in Lowe syndrome. Hum Mol Genet (2012) 21:3333–44. doi: 10.1093/hmg/dds163
227. Mukherjee D, Sen A, Aguilar RC. RhoGTPase-binding proteins, the exocyst complex and polarized vesicle trafficking. Small GTPases (2014) 5:e28453. doi: 10.4161/sgtp.28453
228. Schraml P, Frew IJ, Thoma CR, Boysen G, Struckmann K, Krek W, et al. Sporadic clear cell renal cell carcinoma but not the papillary type is characterized by severely reduced frequency of primary cilia. Mod Pathol (2009) 22:31–6. doi: 10.1038/modpathol.2008.132
229. Basten SG, Willekers S, Vermaat JS, Slaats GG, Voest EE, van Diest PJ, et al. Reduced cilia frequencies in human renal cell carcinomas versus neighboring parenchymal tissue. Cilia (2013) 2:2. doi: 10.1186/2046-2530-2-2
230. Tekin B, Erickson LA, Gupta S. von Hippel-Lindau disease-related neoplasia with an emphasis on renal manifestations. Semin Diagn Pathol (2023) S0740-2570(23)00096–5. doi: 10.1053/j.semdp.2023.11.003
231. Schermer B, Ghenoiu C, Bartram M, Muller RU, Kotsis F, Hohne M, et al. The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J Cell Biol (2006) 175:547–54. doi: 10.1083/jcb.200605092
232. Hergovich A, Lisztwan J, Barry R, Ballschmieter P, Krek W. Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat Cell Biol (2003) 5:64–70. doi: 10.1038/ncb899
233. Kuehn EW, Walz G, Benzing T. Von hippel-lindau: a tumor suppressor links microtubules to ciliogenesis and cancer development. Cancer Res (2007) 67:4537–40. doi: 10.1158/0008-5472.CAN-07-0391
234. Lolkema MP, Mehra N, Jorna AS, van Beest M, Giles RH, Voest EE. The von Hippel-Lindau tumor suppressor protein influences microtubule dynamics at the cell periphery. Exp Cell Res (2004) 301:139–46. doi: 10.1016/j.yexcr.2004.07.016
235. Chowdhury P, Perera D, Powell RT, Talley T, Tripathi DN, Park YS, et al. Therapeutically actionable signaling node to rescue AURKA driven loss of primary cilia in VHL-deficient cells. Sci Rep (2021) 11:10461. doi: 10.1038/s41598-021-89933-7
236. Adamiok-Ostrowska A, Piekielko-Witkowska A. Ciliary genes in renal cystic diseases. Cells (2020) 9(4):907. doi: 10.3390/cells9040907
237. Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers (2016) 2:16035. doi: 10.1038/nrdp.2016.35
238. Bissler JJ, Christopher Kingswood J. Renal manifestation of tuberous sclerosis complex. Am J Med Genet C Semin Med Genet (2018) 178:338–47. doi: 10.1002/ajmg.c.31654
239. Zhou JX, Torres VE. Autosomal dominant polycystic kidney disease therapies on the horizon. Adv Kidney Dis Health (2023) 30:245–60. doi: 10.1053/j.akdh.2023.01.003
240. Haqq AM, Chung WK, Dollfus H, Haws RM, Martos-Moreno GÁ, Poitou C, et al. Efficacy and safety of setmelanotide, a melanocortin-4 receptor agonist, in patients with Bardet-Biedl syndrome and Alström syndrome: a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial with an open-label period. Lancet Diabetes Endocrinol (2022) 10:859–68. doi: 10.1016/S2213-8587(22)00277-7
241. Cebotaru L, Liu Q, Yanda MK, Boinot C, Outeda P, Huso DL, et al. Inhibition of histone deacetylase 6 activity reduces cyst growth in polycystic kidney disease. Kidney Int (2016) 90:90–9. doi: 10.1016/j.kint.2016.01.026
242. Cao Y, Semanchik N, Lee SH, Somlo S, Barbano PE, Coifman R, et al. Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc Natl Acad Sci (2009) 106:21819–24. doi: 10.1073/pnas.0911987106
243. Devlin L, Dhondurao Sudhindar P, Sayer JA. Renal Ciliopathies: Promising drug targets and prospects for clinical trials. Expert Opin Ther Targets (2023) 27(4-5):325–46. doi: 10.1080/14728222.2023.2218616
244. Ding XF, Zhou J, Chen G, Wu YL. VHL loss predicts response to Aurora kinase A inhibitor in renal cell carcinoma cells. Mol Med Rep (2018) 18:1206–10. doi: 10.3892/mmr.2018.9038
245. Feng Y, LoGrasso PV, Defert O, Li R. Rho kinase (ROCK) inhibitors and their therapeutic potential. J medicinal Chem (2016) 59:2269–300. doi: 10.1021/acs.jmedchem.5b00683
246. Fukumoto Y, Yamada N, Matsubara H, Mizoguchi M, Uchino K, Yao A, et al. Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension–A pilot efficacy trial–. Circ J (2013) 77:2619–25. doi: 10.1253/circj.CJ-13-0443
Keywords: renal ciliopathies, genetics, primary cilium, ciliogenesis, actin regulation
Citation: Kalot R, Sentell Z, Kitzler TM and Torban E (2024) Primary cilia and actin regulatory pathways in renal ciliopathies. Front. Nephrol. 3:1331847. doi: 10.3389/fneph.2023.1331847
Received: 01 November 2023; Accepted: 20 December 2023;
Published: 16 January 2024.
Edited by:
Rachel Katherine Miller, University of Texas Health Science Center at Houston, United StatesReviewed by:
Pamela Tran, University of Kansas Medical CenterRuhee Dere, Baylor College of Medicine, United States
Copyright © 2024 Kalot, Sentell, Kitzler and Torban. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Torban, ZWxlbmEudG9yYmFuQG1jZ2lsbC5jYQ==