
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Comput. Neurosci. , 29 August 2012
Volume 6 - 2012 | https://doi.org/10.3389/fncom.2012.00061
This article is part of the Research Topic Biophysically based Computational Models of Astrocyte ~ Neuron Coupling and their Functional Significance View all 11 articles
 Mark R. Witcher* and Thomas L. Ellis
Mark R. Witcher* and Thomas L. Ellis
Epilepsy is a common chronic neurologic disorder affecting approximately 1% of the world population. More than one-third of all epilepsy patients have incompletely controlled seizures or debilitating medication side effects in spite of optimal medical management. Medically refractory epilepsy is associated with excess injury and mortality, psychosocial dysfunction, and significant cognitive impairment. Effective treatment options for these patients can be limited. The cellular mechanisms underlying seizure activity are incompletely understood, though we here describe multiple lines of evidence supporting the likely contribution of astroglia to epilepsy, with focus on individual astrocytes and their network functions. Of the emerging therapeutic modalities for epilepsy, one of the most intriguing is the field of neuromodulation. Neuromodulatory treatment, which consists of administering electrical pulses to neural tissue to modulate its activity leading to a beneficial effect, may be an option for these patients. Current modalities consist of vagal nerve stimulation, open and closed-loop stimulation, and transcranial magnetic stimulation. Due to their unique properties, we here present astrocytes as likely important targets for the developing field of neuromodulation in the treatment of epilepsy.
Epilepsy is a common chronic neurologic disorder affecting 0.5–1% of the population with an estimated lifetime risk greater than 4% (Hauser et al., 1993; Hesdorffer et al., 2011). More than one-third of all epilepsy patients have incompletely controlled seizures or debilitating medication side effects in spite of optimal medical management (Sander, 1993; Kwan and Brodie, 2000; Sillanpaa and Schmidt, 2006). Medically refractory epilepsy is associated with excess injury and mortality, psychosocial dysfunction, and significant cognitive impairment (Brodie and Dichter, 1996). Treatment options for these patients include new antiepileptic drugs (AEDs), which may lead to seizure freedom in a small percentage of patients (Fisher, 1993; Engel, 2012) and resective surgery which is associated with long term seizure freedom in 60–80% of patients (Engel et al., 2003; Lee et al., 2005b; Engel, 2012). The cellular mechanisms underlying seizure activity are incompletely understood, though multiple lines of evidence support the contribution of astrocytic cells, both individually and in networks. Many properties of astrocytes also make them important targets for the developing field of neuromodulation in the treatment of epilepsy.
Historically, astroglia were thought to provide only metabolic and physical support for neurons. They serve as the primary source of energy for neurons (Brown et al., 2004) and serve to control ionic homeostasis and neuronal excitability by buffering potassium (Kofuji and Newman, 2004). It is now clear, however, that astroglia are directly involved in neuronal signaling, even locally at synapses (Barres, 1991; Bergles et al., 1997; Volterra et al., 2002; Hatton and Parpura, 2004; Lin and Bergles, 2004; Allen and Barres, 2005; Volterra and Meldolesi, 2005). Astroglia synthesize and recycle glutamate (Hertz and Zielke, 2004) and respond to synaptic release of neurotransmitters with both calcium waves and release of gliotransmitters that can further influence synaptic activity (Cornell-Bell et al., 1990a; Grosche et al., 1999; Schipke and Kettenmann, 2004; Pascual et al., 2005; Perea and Araque, 2005; Zorec et al., 2012), with important implications in the epileptic brain (Carmignoto and Haydon, 2012). Perisynaptic astroglial processes may detect spill out of glutamate and other substances from active synapses (Rusakov and Kullmann, 1998; Diamond, 2005), and respond structurally by extending and modifying their processes (Cornell-Bell et al., 1990b; Hirrlinger et al., 2004; Witcher et al., 2007, 2010). Variation in synapse strength and the degree to which substances escape the perimeter might determine whether astroglial processes grow toward and ensheath parts of some synapses and avoid or retract from others (Cornell-Bell et al., 1990b; Hatton and Parpura, 2004; Witcher et al., 2007). Astroglia also secrete substances that are critical to the formation and function of synapses during development (Mauch et al., 2001; Ullian et al., 2001, 2004; Christopherson et al., 2005; Goritz et al., 2005) and contain contact-mediated factors that influence synapse maturation (Mazzanti and Haydon, 2003; Murai et al., 2003; Hama et al., 2004).
Astrocytes also function as modulators of neurotransmitters. It is commonly accepted that astrocytes play a large role in glutamate uptake, mostly through the GLT-1 transporter (Danbolt, 2001), also known as the excitatory amino acid transporter-2 (EAAT2) transporter with assistance by additional uptake via the EAAT1 transporter (Shigeri et al., 2004). Astrocytes, through the regulation of extracellular glutamate diffusion, also appear to modulate intersynaptic crosstalk (Oliet et al., 2001, 2006; Theodosis et al., 2006) and modulate synaptic function (Piet et al., 2004). Astrocytic transporter placement may only be contributory to these effects, however, as glutamine synthetase plays a key role in the metabolism of uptaken glutamate (Magistretti, 2006). While glutamine synthetase has been shown to be upregulated in reactive astrocytes (Eddleston and Mucke, 1993), it appears to be reduced in the epileptic hippocampus (Eid et al., 2004; van der Hel et al., 2005), indicating potential changes in the uptake potential of astrocytic transporters. Glutamate transporters have similarly been shown to be reduced in the epileptic hippocampus (Proper et al., 2002). Similarly, GABA release from interneurons may be proportionately increased by intracellular astrocytic calcium through activation of the kainic subtype of neuronal ionotropic glutamate receptors (Liu et al., 2004). Disruption of this cycle could lead to a decrease in GABA release, and thus decreased inhibition of neurons. GABA and glutamate imbalance is believed to potentially be a key mechanism in pathologies such as epilepsy. Thus, astroglia likely modulate function of synapses individually and within synaptic networks.
Multiple pathologically induced changes in astrocytic function have been described. For example, neuronal calcium homeostasis may be modulated by growth factors such as NGF, known to be released by astrocytes in response to excitotoxic injection, providing a means of protecting cultured neurons against excitotoxicity (Eddleston and Mucke, 1993). Extracellular levels of calcium, as well as the propagation of multicellular calcium waves may be tied intricately to mechanically induced stimulation in astrocytes (Ostrow and Sachs, 2005). Under pathological conditions, mechanical stimulation could be provided in the injured CNS by reactive gliosis, swelling, mass effects, or tissue hypertrophy (Ostrow and Sachs, 2005). Astrocytic dysregulation of the vascular system has also been implicated in the epileptic brain. For example, altered calcium signaling by astrocytes can result in changes in local vascular tone (Zonta et al., 2003; Gomez-Gonzalo et al., 2011). Additionally, the upregulation or induction of multiple inflammatory factors within perivascular astrocytes, including IL-1β, complement components, and plasminogen activator in the epileptic brain may also have dramatic effects on the vascular system leading to disruption of the blood brain barrier (as reviewed in Aronica et al., 2012). Dysfunctional astrocytic calcium signaling may also underlay the development of a recurrent excitatory loop sustaining ictal discharge (Gomez-Gonzalo et al., 2010). Astrocytic dysfunction can also result in neuronal oxidative stress (Takuma et al., 2004), as failed support of dopaminergic neurons has been detected after astrocytes were experimentally deprived of glutathione (Drukarch et al., 1997).
Pathological dysfunction has also been observed in the gap-junctional coupling of astrocytes. Along these lines, increases have been noted in the expression of connexin-43 in experimental excitotoxic injuries (Haupt et al., 2007) as well as from epileptic patient specimens (Naus et al., 1991). Upregulation may result in increased intercellular signaling through calcium waves (Scemes and Giaume, 2006), an increased potential for astrocytic glutamate release (Parpura et al., 2004), and may play a role in the potassium buffering capacity of astrocytes (Wallraff et al., 2006). Through gap junctions, astrocytes can exert neurotrophic and neuroprotective influences (Naus et al., 2001; Takuma et al., 2004). Conversely, gap junction communication can be reversed in pathological conditions such as hypoxia (Martinez and Saez, 2000), potentially resulting in increased neuronal injury (Ozog et al., 2002).
It is therefore clear that astroglial cells function to support the microenvironments of neuronal cells and to modulate neural networks. Given their unique structural interactions with multiple neuronal contacts simultaneously (Figure 1; originally published in Witcher et al., 2007; and Movie S1 in Supplementary Material), it is evident that astrocytes can readily integrate into a functional network. Astroglial cells may even function as separate, parallel networks in roles not currently understood. It is highly likely that astroglial networks have a fundamental role in disease states addressed through neurosurgical approaches. Specifically, astrocytes play a vital role in the epileptic brain, the treatment algorithm of which often ends in neurosurgical resection, transection, or disconnection of cellular networks. Similarly, multiple neurosurgical entities currently depend on the modulation of cerebral networks through introduction and manipulation of neuromodulatory devices such as deep brain stimulators. These techniques have analogous implications for astrocytes and the astroglial network. These disease processes include dystonia, movement disorders such Parkinson’s disease and psychological disorders dependent on appropriate modulation of network function such as Obsessive-Compulsive Disorder, depression, and others. Through a better understanding and manipulation of the astrocytic networks of the human brain, we hypothesize that new targets and modulatory therapies can be developed in the treatment of epilepsy.
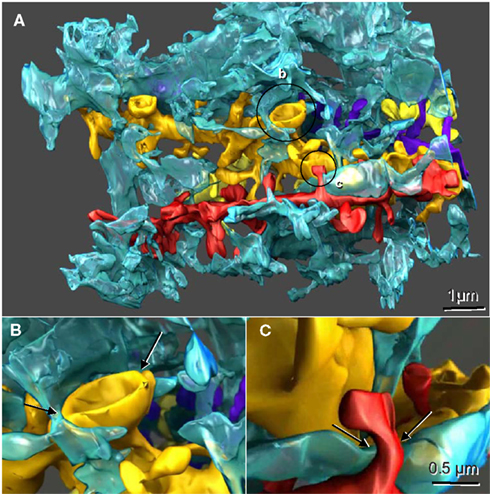
Figure 1. Reconstructed dendrites, synapses, and associated astroglial processes from rodent hippocampus. (A) Three-dimensional reconstruction of a single astroglial process (blue) interdigitating among many dendrites, four of which are reconstructed here (gold, yellow, red, and purple). Axonal boutons are not displayed. (B) Approximately 50% of the Axon-Spine Interface (ASI) of a mushroom spine was apposed by astroglia (arrows). (C) Only the neck of this thin dendritic spine was apposed by astroglia (arrows). Scale bar in (C) is for (B) and (C). Figure reproduced with permission from John Wiley and Sons.
One of the most consistently reported findings associated with seizure activity, whether a single event caused experimentally by focal application of convulsants (Jiang et al., 1998) or by repeated epileptic seizures in vivo (Scheibel and Scheibel, 1977), is hippocampal neuronal loss, subsequent deafferentation of dendritic partners, and loss of dendritic spines. Hippocampal alterations in mesial temporal lobe epilepsy (MTLE) include neuronal loss in the hippocampus, gliosis, and reorganization of subsets of neurons in the hippocampus (Sommer, 1880; Spencer, 2002). Lesions of CA1, in particular, are associated with the classically described Ammon’s Horn sclerosis (Mathern et al., 1997; Duvernoy, 2005), and greater loss of spines is associated with a greater degree of pathology. Other findings consistently reported in epileptic tissue include denuding of dendritic segments, as well as the formation of varicosities along dendritic shafts (Multani et al., 1994). The degree of synaptic loss can vary along individual dendrites (Scheibel and Scheibel, 1977). As reviewed by Fiala et al. (2002), these pathologies are also consistent with other neurodegenerative disorders, many of which cause deafferentation of dendrites. This observation led to the hypothesis that deafferentation may be the cause of dendritic spine pathology associated with disorders including epilepsy (Swann et al., 2000; Fiala et al., 2002). Structural evidence including the presence of giant spines, tortuous spines, an appearance of axon-free post synaptic densities (PSD) in dendritic spines, and aberrant synaptic partners (Raisman and Field, 1973; Baloyannis and Kim, 1979; Chen and Hillman, 1982) could support this hypothesis.
While recent attention has focused on changes in glial function in neurological diseases such as epilepsy (Tian et al., 2005; Binder and Steinhauser, 2006; Lee et al., 2006b; Matute et al., 2006), associated changes in perisynaptic astrocytic structure have been less of a focus, though changes in the amount of synaptic astrocytic contact would also be expected in the epileptic hippocampus. Important findings established or implicated in this form of epilepsy include increases in extracellular glutamate (During and Spencer, 1993), decreases in glutamate metabolism (Malthankar-Phatak et al., 2006), decreases in glutamate-stimulated GABA release (During et al., 1995), increased GABA transporter expression (Lee et al., 2006b), and increased lactate levels (Cavus et al., 2005), all of which can be associated with astrocytic dysfunction. While multiple studies have addressed the morphological changes of reactive astrocytes (Krishnan et al., 1994; van Paesschen et al., 1997; Mitchell et al., 1999; Briellmann et al., 2002) ultrastructural alterations remain understudied. Of primary importance is evidence which contradictorily supports both increased and decreased association between synaptic partners and astrocytic processes.
Multiple lines of evidence would support the possibility of increased perisynaptic astroglial apposition in the epileptic brain. The first is that in the epileptic hippocampus, microdialysis studies indicate that extracellular glutamate levels rise prior to and during ictal onset, peaking at levels associated with potential neurotoxicity (During and Spencer, 1993). Previous work has shown that astrocytes are responsible for the removal of extracellular glutamate, and that astrocytic presence can be neuroprotective (Rosenberg and Aizenman, 1989). Increased presence of perisynaptic astrocytic processes could therefore potentially serve as a mechanism by which to lower glutamate concentrations to baseline levels. It has also been shown that astrocytic processes are attracted to the neurotransmitter glutamate (Cornell-Bell et al., 1990b). The source of extracellular glutamate is likely synaptic in nature. It is therefore plausible that astrocytic processes would be drawn to these remaining synapses.
Further supporting increased apposition are the findings that astroglial contact stabilizes larger synapses (Witcher et al., 2007) and increases the efficacy of synaptic transmission (Pfrieger and Barres, 1997). The effects of increasing the amount of apposition at individual synapses is currently unknown, but previous research suggests that the amount of surround is not correlated to synapse size (Ventura and Harris, 1999) and that any amount of synaptic contact may be significant (Witcher et al., 2007). As synaptic loss is a known consequence of epilepsy, astrocytic apposition could function to stabilize remaining synapses.
Finally, previously reported increases in individual neurotransmitter receptor could support increased apposition, since MTLE is associated with an increase in glutamate receptors in the epileptic hippocampus. Human hippocampal astrocytes, similar to neurons, contain a wide variety of glutamate receptors, including AMPA receptors (Seifert et al., 2004; Matute et al., 2006), which are increased in CA1 in human epileptic tissue (Brines et al., 1997). Given the established concomitant decrease in neuronal density in epileptic hippocampi, it is plausible that this glutamate receptor increase is in the astrocytic component of the neuropil.
In contrast to the preceding evidence, other findings support the likelihood that perisynaptic astrocytic apposition could instead be decreased in epilepsy. One line of evidence lies in the morphological changes which take place subsequent to neuronal deafferentation. Due to the decrease in hippocampal neuronal density and concomitant dendritic pathologies consistently reported in both experimental and human epilepsy (Scheibel and Scheibel, 1977; Belichenko and Dahlstrom, 1995; Drakew et al., 1996; Thompson et al., 1996), it has been hypothesized that epilepsy serves as a model of neuronal deafferentation (Swann et al., 2000; Fiala et al., 2002). In the hippocampus, Schaffer collateral axons provide excitatory input from CA3 pyramidal neurons to CA1 pyramidal neurons via apical dendrites in CA1 stratum radiatum. In MTLE, where neuronal loss is prominent in both the hippocampal subfields of CA3 and CA1, loss of CA3 pyramidal neurons results in deafferentation of CA1 pyramidal neurons. As reported from other brain regions, astrocytic withdrawal can result in increased signaling between postsynaptic and presynaptic partners (Oliet et al., 2001), as well as increased signaling between adjacent synapses (Piet et al., 2004). Similarly, astrocytic withdrawal has been shown to regulate synaptic connectivity in the arcuate nucleus (Fernandez-Galaz et al., 1997) and similar effects have been implicated in the hippocampal dentate hilus (Luquin et al., 1993; Klintsova et al., 1995). Therefore, decrease in astrocyte surround at the synapse could be a compensatory mechanism useful for increasing axonal input to deafferented dendrites.
Modifications in the pattern of glutamate transporter expression in the sclerotic human hippocampus provide further support for astrocytic withdrawal from synapses. As many as five types of glutamate transporters have been described as present, and are designated as EAAT1–5, as reviewed by Danbolt (2001). The major glutamate transporters associated with hippocampal astrocytes include EAAT1 (GLAST), EAAT2 (GLT), and EAAT3 (EAAC) while the other EAAT subtypes are typically associated only with neurons (Danbolt, 2001). In addition to the astrocytic distribution of EAAT2, a presynaptic neuronal distribution was also shown in a subpopulation of excitatory hippocampal terminals, including the CA1 (Chen et al., 2002, 2004). In the CA1 subfield, approximately 60% of astrocytic membranes contain EAAT2 transporter proteins compared to approximately 30% of neurons (Chen et al., 2004). EAAT2 localizes in the cellular membrane of astrocytes (Danbolt, 2001), while in neurons it appears to label in the membrane as well as the cytoplasm (Chen et al., 2002, 2004). EAAT3 transporters, by contrast, appear to have a cytoplasmic distribution in both astrocytes and neurons (Danbolt, 2001). The distribution of EAAT1, however, is strictly astrocytic and localized almost exclusively to cytoplasmic membranes (Danbolt, 2001).
The EAAT1 transporter concentration does not vary between the soma and processes of astrocytes, but varies relative to neighboring structures, where concentration is increased along membranes apposing neuropil, and decreased in membrane apposing cellular somata, pial surfaces, or capillary endothelium (Danbolt, 2001). The EAAT1 transporter is therefore a reliable marker of astrocytic membrane in the neuropil, and can be particularly useful in the distal processes of astrocyte where GFAP filaments do not extend (Bushong et al., 2002, 2004). While the EAAT2 transporter is associated with the majority of glutamate uptake from the hippocampus, and a causative relationship has been shown between its knockout and the development of lethal, spontaneous seizures (Tanaka et al., 1997), its expression in neurons makes it non-specific to astrocytic membranes.
In the sclerotic human hippocampus, expression of the EAAT2 transporters are significantly decreased in CA1, and EAAT1 levels show a similar trend (Proper et al., 2002). Paradoxically, a well-known consequence of sclerosis is the hypertrophy of astrocytes, demonstrated repeatedly through expansion of the GFAP protein unique to astrocytic soma and perisomatic processes. This protein, however, is absent in small, distal astrocytic processes, such as those that appose synaptic membranes (Bushong et al., 2002). As the EAAT1 and EAAT2 transporters are localized in all portions of the astrocytic membrane regardless of distance from the soma, they serve as indicators of the astrocytic processes distal to the appearance of GFAP. While the decrease in EAAT2 is significant in TLE (Proper et al., 2002), the trend toward a concomitant decrease in astrocyte-specific EAAT1 suggests that the decrease is likely contributed to by astrocytic changes. Decrease in these transporters suggests a decrease in non-GFAP containing perisynaptic astrocytic processes, thereby supporting a potential withdrawal of these processes.
Using three-dimensional unbiased brick analysis, multiple interesting findings were recently shown from the human epileptic hippocampus (Witcher et al., 2010). Specifically, it was shown that synaptic loss was indeed associated with the process of epileptogenesis, and that synaptic density decreased as gliosis increased. It was also shown that the morphology of remaining synapses was altered, and in the severe epileptic state, normal-appearing neuronal spines were replaced by abnormal giant spines which likely have unique physiological properties. Remaining synapses did not show increased numbers of vesicles, refuting the idea that high extracellular glutamate (During and Spencer, 1993; Cavus et al., 2005) is likely based on decreased uptake of the neurotransmitter. Synapses remaining in the epileptic hippocampus were not restricted from intersynaptic communication (Witcher et al., 2010). Despite these changes, however, it was clear that the apposition of perisynaptic astroglia supported larger synapses (Witcher et al., 2007, 2010). Therefore, while the astroglia and synapses show fundamental changes in the pathologic brain, it is clear that positive benefits arise from the relationship.
Neurosurgical management of epilepsy is useful in patients who are refractory to optimized medical treatment (Engel et al., 2003; Lee et al., 2005b). Current modalities are dominated by resection or disconnection of epileptic cellular networks. Descriptions of modern methods and their psychosocial implications occupy a vast literature and include lesionectomies, anterior temporal lobectomy, amygdalohippocampectomy, extratemporal resection, and corpus callosotomy (Feindel et al., 2009; Wilson and Engel, 2010). There is also a developing role for stereotactic radiosurgery (Quigg et al., 2012). In spite of improvements in surgical technique, approximately 4% of patients will suffer death or permanent neurologic disability (ILAE, 1997). Moreover, more than one-third of patients will not be candidates for surgical resection (Kwan and Brodie, 2000). For patients who are not candidates for resective surgery, there are limited options. Neuromodulatory treatment, which consists of administering electrical pulses to neural tissue to modulate its activity leading to a beneficial effect (Witcher and Ellis, 2011), can be effective for these patients.
The interest in neuromodulation for neurological disorders is driven by a desire to discover less invasive surgical treatments, as well as new treatments for patients whose medical conditions remain refractory to existing modalities (Witcher and Ellis, 2011). Interestingly, the unique characteristics of astrocytes make them interesting targets in the developing field of neuromodulation. Specifically, the use of these technologies requires modulation of large neural networks, and likely involve modulation of or through astrocytes or astroglial networks. These mechanisms likely involve reactive astrocytosis, network manipulation, and modulation of the release of gliotransmitters.
Astrocytic processes are ubiquitous between cells and around excitatory synapses throughout the CNS (Witcher et al., 2007, 2010). This proximity of astrocytic processes to synapses allows synaptic placement of glutamate transporters at sites of glutamate release and also the ability to limit or delimit interactions between neighboring synapses (Witcher et al., 2007, 2010). Astrocytes have also been shown to express metabotropic receptors for many neurotransmitters, including glutamate, GABA, norepinephrine, and acetylcholine (Tritsch and Bergles, 2007). Astrocytic responses to the activation of these receptors implies sensing of neuronal function and results in oscillations or repetitive spikes in Ca2+, which likely has influence over neuronal network function (Di Castro et al., 2011; Takata et al., 2011). Gliotransmitters, namely ATP, D-serine, and glutamate are release in response to neuronal and astrocytic stimulation which also could cause neuronal network effects (Santello and Volterra, 2009; Halassa and Haydon, 2010). Mechanisms eliciting neuronal responses have been studied in both culture and slice models and include stimulation of metabotropic receptors, photolysis of caged IP3 or infused caged Ca2+, and repetitive depolarization of the astrocyte membrane presynaptic (Araque et al., 1998; Parri et al., 2001; Fiacco and McCarthy, 2004; Fellin et al., 2004; Jourdain et al., 2007).
Vagal nerve stimulation (VNS) is one example of neuromodulation that was developed in the 1980s, and which is now routinely available (Ben-Menachem, 2002). VNS, as an adjunct to medical management, may yield up to a 50% reduction in seizure frequency (VNSSG, 1995) although most of these patients will not be seizure free. Deep brain stimulation (DBS) is another example of neuromodulation. Given the significant experience and success of DBS for movement disorders (Krack et al., 2003) combined with its reversibility, programmability, and low risk of morbidity, there has been a resurgence of interest in using DBS devices for treating medically refractory epilepsy. Responsive neurostimulation (RNS) is a technology that detects seizure activity at a previously defined focus and applies an electrical stimulus to the site of seizure onset to terminate the seizure. Lastly, transcranial magnetic stimulation (TMS) is a nearly 25-year-old technology initially introduced as a means to non-invasively investigate corticospinal circuits. Currently, TMS is used primarily in clinical neurophysiology. Importantly, TMS can be used to evaluate and manipulate excitatory and inhibitory intracortical circuits with poststimulatory effect, allowing for a developing use in epileptic neuromodulation. A growing body of literature supports the involvement of astrocytes in the realization of therapeutic goals for each of these modalities, and will be reviewed below.
The vagal nerve has a complex anatomical arrangement which projects to the autonomic and reticular structures and well as limbic and thalamic neurons. Stimulation of the vagus-nerve and its bilateral multisynaptic targets has become a common technology for the treatment of epilepsy. Over 50,000 patients have been treated with the technology, and current reports indicate an approximately 50% efficacy in seizure reduction, rivaling the efficacy of antiepileptic treatment, and often decreasing dependence on them (Labar, 2002). Efficacy has also been shown to increase over time (Vonck et al., 1999). The low side effect profile of VNS (Morris and Mueller, 1999) has also proven to be advantageous for users.
The mechanism of efficacy remains unknown, though certain structures within the brain appear to be affected by VNS. As evidenced by studies using positron-emission technology (PET), the thalamus is consistently affected by VNS stimulation, and blood flow to the cerebellum and cerebral structures is consistently altered (Ko et al., 1996; Henry et al., 1998, 1999; Ben-Menachem, 2002). Thalamic involvement has also been supported through SPECT (Van et al., 2000; Vonck et al., 2000) and functional MRI (Narayanan et al., 2002; Liu et al., 2003) analysis.
Studies of VNS have been reported from multiple vertebrate models including rodents (McLachlan, 1993), canines (Zabara, 1992), and lower primates (Lockard et al., 1990). In the rodent penicillin/pentylenetetrazol model, interictal spike frequency was reduced by 33% (McLachlan, 1993), the effect of which was later found to be greatest in continuous stimulation and reduced in a time-dependent fashion after stimulation (Takaya et al., 1996). Later tests showed that cortical excitability in rats can be modulated through VNS (De Herdt et al., 2010). Canine strychnine and pentylenetetrazol models show similar efficacy with lasting reduction in motor seizures and tremors (Zabara, 1992). In the alumina gel monkey model, seizures were eliminated in half of test animals during stimulation periods with some persistence into post-stimulation period (Lockard et al., 1990). Clinical trials have indicated seizure reduction at both low and high stimulation paradigms, with significantly greater reduction in the high stimulation group (Handforth et al., 1998) and overall efficacy showed a mean seizure reduction of approximately 35–45% (Morris and Mueller, 1999).
Astrocytic involvement in the regulation of the vagal nerve nuclei supports their importance in the efficacy of VNS. McDougal et al. (2011) recently demonstrated the activation of astrocytes within the nucleus of the solitary tract (NST) when afferent stimulation of the vagal nerve was applied. Using confocal, live-cell calcium imaging of brainstem slices, they showed that afferent activation of the vagal nerve resulted in increases in astrocytic intracellular calcium concentrations as well as in neurons. They then showed that the effect on astrocytes was blocked by the AMPA receptor antagonism and was unaffected by antagonism of NMDA and metabotropic glutamate receptors. This activation was dependent on extracellular Ca2+ influx through AMPA receptors. This Ca2+ influx was further amplified by calcium-induced calcium release via the ryanodine receptor. Selective staining verified the presence of the AMPAR subunit GluR1 on astrocytes. Taken together, they concluded that NST astrocytes may be active participants in the regulation of vagal activity (McDougal et al., 2011). This supports previous work which concluded that neurons in the NST are regulated via astrocytic glutamate signaling under pathologic and potentially physiologic conditions (Hermann et al., 2009).
Neuromodulation through the direct implantation of chronic stimulating electrodes has become a standard of treatment in many neurological disorders. DBS lead implantation within the anterior nucleus of the thalamus (ANT), as well as other central nervous system (CNS) targets – including the caudate nucleus, centromedian nucleus of the thalamus, cerebellum, hippocampus, and subthalamic nucleus – results in seizure reduction in selected patients (Shandra and Godlevsky, 1990; Vercueil et al., 1998; Bragin et al., 2002; Lee et al., 2006a). In these studies, stimulation was delivered in an open-loop fashion, that is, in a pre-defined manner, independent of the momentary physiological activity of the brain. The exact mechanism of action of DBS in reducing seizure activity is, however, unknown. It is known that stereotactic lesions of the ANT in humans can result in reduced seizure frequency (Mullan et al., 1967). DBS may interfere with synchronized oscillations by neurotransmitter release (Lee et al., 2005a). Other evidence suggests that the most likely mechanism may involve stimulation-induced modulation of pathologic neural networks (McIntyre et al., 2004). High frequency DBS appears to reproduce the clinical effect of ablative procedures (Benabid et al., 1987). Moreover, at high frequencies, DBS may abolish cortical epileptiform activity (Lado et al., 2003). A microthalamotomy effect has been postulated based on the observation that some patients obtain reduction in seizure frequency prior to activation of the pulse generator (Andrade et al., 2006; Lim et al., 2007).
Although the precise mechanism by which DBS reduces seizure activity is unclear, inhibition of neurons immediately adjacent to the area of applied current is likely involved. A “reversible functional lesion” may be generated in structures integral to initiating or sustaining epileptic activity (Boon et al., 2007). The applied current may inhibit neurons with a pathologically lowered threshold of activation. Alternatively, DBS may act on neuronal network projections to nearby or remote CNS structures originating from the area of stimulation. This might take place through either activation of inhibitory projections or through the inhibition of excitatory projections.
As reviewed recently by Vedam-Mai et al. (2012), high frequency stimulation shows effect on astrocytic activity which has important implications in the role of astroglia in this modality. Astrocytes can be directly depolarized by stimulation (Kang et al., 1998) and have the potential to modulate local and distant neural networks using clinically relevant stimulation paradigms via the release of gliotransmitters including ATP and glutamate (Bekar et al., 2008; Tawfik et al., 2010).
Another important mechanism implicating astrocytes in clinical efficacy is reactive astrocytosis. Reactive astrocytosis is a well described phenomena of astrocytes at stimulator implant sites, and is defined as astroglial hypertrophy and upregulation of GFAP and other astrocytic proteins (Pekny and Nilsson, 2005). This finding was described initially in cats (Stock et al., 1979) and has since been described in multiple species including rats (Kraev et al., 2009), non-human primates (Griffith and Humphrey, 2006), and humans (Moss et al., 2004; Sun et al., 2008a; DiLorenzo et al., 2010; Vedam-Mai et al., 2011). In these series, reactive elements including multinucleated giant cells and macrophages were common findings. In human studies, common elements included thin glial rims surrounding the electrode tract; lymphocytes and monocytes have also been described near the electrode (Moss et al., 2004; DiLorenzo et al., 2010). The volume of the glial surround, which could greatly impact electrode function, is, however, not known (Moss et al., 2004). Investigation in rats, however, indicates regional variability in astroglial reactivity to implanted electrodes (Hirshler et al., 2010).
The effects on network activity by reactive astrocytosis then becomes an important focus of their overall effect. Reactive astrocytes display marked functional changes which could include direct neurotrophic effects though modified energetics or neurotrophic factor release, enhanced glutamate uptake, reorganization of metabolic pathways, and modulation of synaptic transmission (Liberto et al., 2004; Sofroniew, 2005; Escartin and Bonvento, 2008). Recently, a model using selective virus-induced reactive astrocytosis in rat hippocampal area CA1 demonstrated that astrocytosis resulted in specific deficits in inhibitory synaptic transmission, and caused disruptions in functional regulation of circuits resulting in enhanced excitability of the local network (Ortinski et al., 2010). Specifically, a reduction was found in elicited monosynaptic inhibitory responses, which led to a reduction in basal inhibitory neurotransmission without affecting intrinsic neuronal properties. This resulted from an alteration in the astrocytic glutamate/glutamine cycle which resulted in reduced synaptic GABA availability (Ortinski et al., 2010). Thus, a growing body of evidence supports that reactive astrocytosis at the electrode site could readily alter network effects of targeted neural circuits.
In contrast to open-loop stimulation, contingent or closed-loop stimulation is designed to suppress epileptiform activity by stimulating a defined epileptogenic target directly in response to detection of abnormal EEG activity. This form of closed-loop, responsive neural stimulation (RNS), has preliminarily been shown safe and efficacious (Sun et al., 2008b), and is currently being evaluated in a randomized trial to assess safety and efficacy in epileptic patients. While its experimental and clinical trial background are beyond the scope of this review, its’ similarities to DBS, notably contacting electrodes, pulse delivery, and network neuromodulatory effects imply a likelihood of astrocytic involvement analogous to open-loop stimulation.
Transcranial magnetic stimulation of cortical tissues was initially reported by Barker et al. (1985) and quickly found acceptance as a research vehicle for neurophysiologists. TMS was initially applied to the study of the motor system (Barker et al., 1985) and has since expanded to include investigations in psychiatric conditions (Pascual-Leone et al., 1996), and migraine headache (Lipton and Pearlman, 2010). Importantly, it has also become a viable option for the treatment of drug resistant epilepsy. TMS exerts its effects through repetitive non-invasive stimulation in which a pulsed magnetic field creates current flow in the brain which can temporarily excite or inhibit target areas (Hallett, 2000).
The basis of TMS as a therapeutic neuromodulatory is derived from the lasting effects from the application of a train of transcranial stimuli. Theoretically, the lasting effects of TMS can be used to modulate activity in focal areas of cortex (Fregni and Pascual-Leone, 2007). The induced effect depends on the nature of the stimulation; that is, the frequency, the timing, the focus, and the intensity of the repetitive stimulation (Kimiskidis, 2010). While some paradigms have been studied using animal models, the numbers of basic studies particular to epilepsy are somewhat limited.
Early study within the mouse hippocampal-entorhinal cortex slice model indicated that repetitive direct (i.e., non-transcranial) stimulation at 1 Hz can depress the generation of ictal activity in a 4-aminopyridine model (Barbarosie and Avoli, 1997), in a frequency-dependent manner (D’Arcangelo et al., 2005). This frequency dependence has been replicated in TMS. Low-frequency TMS stimulation shows the tendency to lower seizure activity (Akamatsu et al., 2001; Godlevsky et al., 2006; Rotenberg et al., 2008). High frequency stimulation has been shown to potentially have both protective and inductive effects dependent on the chronicity of treatment and potentially other, unexplored, factors (Jennum and Klitgaard, 1996; Ebert and Ziemann, 1999).
Similar results have been identified in human studies. High frequency TMS has been shown to enhance cortical excitability at high intensities (Berardelli et al., 1998), while low-frequency TMS has been shown to reduce cortical excitability (Cincotta et al., 2003) as well as decreased strength of neuronal signaling (Muellbacher et al., 2000). As detailed by Kimiskidis (2010), the clinical effects are theoretically similar to long term potentiation (LTP) and long term depression (LTD) elicited by high- and low-frequency electrical stimulation, respectively. It is therefore possible that TMS at lower frequencies may exert its effect through the initiation of LTD, while at higher frequencies, the proconvulsant effect may be initiated through the induction of an LTP-type effect (Ziemann, 2005).
Direct evidence for astrocytic involvement in the neuromodulatory therapy is limited. Early work in a murine model found that high frequency TMS had a dramatic effect in the upregulation of astroglial gene expression (Fujiki and Steward, 1997) Following multiple high frequency trains (25 Hz), GFAP mRNA levels were significantly increased in the hippocampal dentate gyrus to levels similar to that following electroconvulsive seizures, indicating induction of an astrocytic reactive response (Fujiki and Steward, 1997). Indirectly, the analogous effects to LTD and LTP have important implications for astrocyte involvement, as the important contributions of astrocytes and gliotransmitters to synaptic plasticity have been described in multiple neuronal circuits (Yang et al., 2003; Witcher et al., 2007; Henneberger et al., 2010; Ben Menachem-Zidon et al., 2011; Bonansco et al., 2011; Navarrete et al., 2012).
In spite of optimal medical management, many patients with epilepsy remain medically refractory and suffer from debilitating seizures. Some of these patients may benefit from neuromodulatory treatment. As the evidence above indicates, it is very likely that modulation of astroglial function is important to the efficacy of neuromodulation. Additional studies are needed to identify the appropriate patient populations for neuromodulation, optimal targets, optimal stimulation modalities, and paradigms. It is also critical that the cellular and network mechanisms underlying the effects of these treatments must be better elucidated. Further studies are needed to determine the contribution of neural and glial components of the nervous system, and future modalities must be developed which optimize both. Understanding these relationships may enable future technologies, perhaps even nanotechnologies, to flourish in the developing field of therapeutic neuromodulation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Computational_Neuroscience/10.3389/fncom.2012.00061/abstract
Movie S1. Reconstructed dendrites, synapses, and associated astroglial processes from rodent hippocampus. Note the spatial relationship of a single astrocytic process (blue) interdigitating among multiple unique dendrites (gold, yellow, red, and purple). Axonal boutons are not displayed. These spatial relationships likely allow for the interactions of astrocytes with neurons, the regulation of neuronal communication, and form the basis of the astroglial-neuronal network. Figure acknowledgment to Cosmocyte, Inc. (Savage, MD, USA) for production in rendering and display.
Akamatsu, N., Fueta, Y., Endo, Y., Matsunaga, K., Uozumi, T., and Tsuji, S. (2001). Decreased susceptibility to pentylenetetrazol-induced seizures after low-frequency transcranial magnetic stimulation in rats. Neurosci. Lett. 310, 153–156.
Allen, N., and Barres, B. (2005). Signaling between glia and neurons, focus on synaptic plasticity. Curr. Opin. Neurobiol. 15, 542–548.
Andrade, D., Zumsteg, D., Hamani, C., Hodaie, M., Sarkissian, S., Lozano, A., and Wennberg, R. (2006). Long-term follow-up of patients with thalamic deep brain stimulation for epilepsy. Neurology 66, 1571–1573.
Araque, A., Parpura, V., Sanzgiri, R., and Haydon, P. (1998). Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur. J. Neurosci. 10, 2129–2142.
Aronica, E., Ravizza, T., Zurolo, E., and Vezzani, A. (2012). Astrocyte immune responses in epilepsy. Glia 60, 1258–1268.
Baloyannis, S., and Kim, S. (1979). Experimental modification of cerebellar development in tissue culture, X-irradiation induces granular degeneration and unattached Purkinje cell dendritic spines. Neurosci. Lett. 12, 283–288.
Barbarosie, M., and Avoli, M. (1997). CA3-driven hippocampal-entorhinal loop controls rather than sustains in vitro limbic seizures. J. Neurosci. 17, 9308–9314.
Barker, A., Jalinous, R., and Freeston, I. (1985). Non-invasive magnetic stimulation of human motor cortex. Lancet 1, 1106–1107.
Bekar, L., Libionka, W., Tian, G., Xu, Q., Torres, A., Wang, X., Lovatt, D., Williams, E., Takano, T., Schnermann, J., Bakos, R., and Nedergaard, M. (2008). Adenosine is crucial for deep brain stimulation-mediated attenuation of tremor. Nat. Med. 14, 75–80.
Belichenko, P., and Dahlstrom, A. (1995). Studies on the 3-dimensional architecture of dendritic spines and varicosities in human cortex by confocal laser scanning microscopy and Lucifer yellow microinjections. J. Neurosci. Methods 57, 55–61.
Ben Menachem-Zidon, O., Avital, A., Ben-Menahem, Y., Goshenm, I., Kreisel, T., Shmueli, E., Segal, M., Ben Hur, T., and Yirmiya, R. (2011). Astrocytes support hippocampal-dependent memory and long-term potentiation via interleukin-1 signaling. Brain Behav. Immun. 25, 1008–16.
Benabid, A., Pollak, P., Louveau, A., Henry, S., and de Rougemont, J. (1987). Combined (thalamotomy and stimulation) stereotactic surgery of the VIM thalamic nucleus for bilateral Parkinson disease. Appl. Neurophysiol. 50, 344–346.
Ben-Menachem, E. (2002). Vagus-nerve stimulation for the treatment of epilepsy. Lancet Neurol. 1, 477–482.
Berardelli, A., Inghilleri, M., Rothwell, J., Romeo, S., Currà, A., Gilio, F., Modugno, N., and Manfredi, M. (1998). Facilitation of muscle evoked responses after repetitive cortical stimulation in man. Exp. Brain Res. 122, 79–84.
Bergles, D., Dzubay, J., and Jahr, C. (1997). Glutamate transporter currents in Bergmann glial cells follow the time course of extrasynaptic glutamate. Proc. Natl. Acad. Sci. U.S.A. 94, 14821–14825.
Binder, D., and Steinhauser, C. (2006). Functional changes in astroglial cells in epilepsy. Glia 54, 358–368.
Bonansco, C., Couve, A., Perea, G., Ferradas, C., Roncagliolo, M., and Fuenzalida, M. (2011). Glutamate released spontaneously from astrocytes sets the threshold for synaptic plasticity. Eur. J. Neurosci. 33, 1483–92.
Boon, P., Vonck, K., De Herdt, V., Van Dycke, A., Goethals, M., Goossens, L., Van Zandijcke, M., De Smedt, T., Dewaele, I., Achten, R., Wadman, W., Dewaele, F., Caemaert, J., and Van Roost, D. (2007). Deep brain stimulation in patients with refractory temporal lobe epilepsy. Epilepsia 48, 1551–1560.
Bragin, A., Wilson, C., and Engel, J. Jr. (2002). Rate of interictal events and spontaneous seizures in epileptic rats after electrical stimulation of hippocampus and its afferents. Epilepsia 43 Suppl 5, 81–85.
Briellmann, R., Kalnins, R., Berkovic, S., and Jackson, G. (2002). Hippocampal pathology in refractory temporal lobe epilepsy, T2-weighted signal change reflects dentate gliosis. Neurology 58, 265–271.
Brines, M. L., Sundaresan, S., Spencer, D. D., and de Lanerolle, N. C. (1997). Quantitative autoradiographic analysis of ionotropic glutamate receptor subtypes in human temporal lobe epilepsy, up-regulation in reorganized epileptogenic hippocampus. Eur. J. Neurosci. 9, 2035–2044.
Brown, A. M., Tekkok, S. B., and Ransom, B. R. (2004). “Glial-neuronal interactions and brain energy metabolism,” in Glial-Neuronal Signaling, eds G. Hatton and V. Parpura (Boston, MA: Kluwer Academic Publishers), 279–296.
Bushong, E. A., Martone, M. E., and Ellisman, M. H. (2004). Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int. J. Dev. Neurosci. 22, 73–86.
Bushong, E. A., Martone, M. E., Jones, Y. Z., and Ellisman, M. H. (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22, 183–192.
Carmignoto, G., and Haydon, P. G. (2012). Astrocyte calcium signaling and epilepsy. Glia 60, 1227–1233.
Cavus, I., Kasoff, W. S., Cassaday, M. P., Jacob, R., Gueorguieva, R., Sherwin, R. S., Krystal, J. H., Spencer, D. D., and Abi-Saab, W. M. (2005). Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann. Neurol. 57, 226–235.
Chen, S., and Hillman, D. E. (1982). Plasticity of the parallel fiber-Purkinje cell synapse by spine takeover and new synapse formation in the adult rat. Brain Res. 240, 205–220.
Chen, W., Aoki, C., Mahadomrongkul, V., Gruber, C. E., Wang, G. J., Blitzblau, R., Irwin, N., and Rosenberg, P. A. (2002). Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J. Neurosci. 22, 2142–2152.
Chen, W., Mahadomrongkul, V., Berger, U. V., Bassan, M., DeSilva, T., Tanaka, K., Irwin, N., Aoki, C., and Rosenberg, P. A. (2004). The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J. Neurosci. 24, 1136–1148.
Christopherson, K. S., Ullian, E. M., Stokes, C. C., Mullowney, C. E., Hell, J. W., Agah, A., Lawler, J., Mosher, D. F., Bornstein, P., and Barres, B. A. (2005). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433.
Cincotta, M., Borgheresi, A., Gambetti, C., Balestrieri, F., Rossi, L., Zaccara, G., Ulivelli, M., Rossi, S., Civardi, C., and Cantello, R. (2003). Suprathreshold 0.3 Hz repetitive TMS prolongs the cortical silent period, potential implications for therapeutic trials in epilepsy. Clin. Neurophysiol. 114, 1827–1833.
Cornell-Bell, A. H., Finkbeiner, S. M., Cooper, M. S., and Smith, S. J. (1990a). Glutamate induces calcium waves in cultured astrocytes, Long-range glial signaling. Science 247, 470–473.
Cornell-Bell, A. H., Thomas, P. G., and Smith, S. J. (1990b). The excitatory neurotransmitter glutamate causes filopodia formation in cultured hippocampal astrocytes. Glia 3, 322–334.
D’Arcangelo, G., Panuccio, G., Tancredi, V., and Avoli, M. (2005). Repetitive low-frequency stimulation reduces epileptiform synchronization in limbic neuronal networks. Neurobiol. Dis. 19, 119–128.
De Herdt, V., De Waele, J., Raedt, R., Wyckhuys, T., El Tahry, R., Vonck, K., Wadman, W., and Boon, P. (2010). Modulation of seizure threshold by vagus nerve stimulation in an animal model for motor seizures. Acta Neurol. Scand. 121, 271–276.
Di Castro, M. A., Chuquet, J., Liaudet, N., Bhaukaurally, K., Santello, M., Bouvier, D., Tiret, P., and Volterra, A. (2011). Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat. Neurosci. 14, 1276–84.
Diamond, J. S. (2005). Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes, transmitter uptake gets faster during development. J. Neurosci. 25, 2906–2916.
DiLorenzo, D., Jankovic, J., Simpson, R., Takei, H., and Powell, S. (2010). Long-term deep brain stimulation for essential tremor, 12-year clinicopathologic follow-up. Mov. Disord. 25, 232–238.
Drakew, A., Muller, M., Gahwiler, B. H., Thompson, S. M., and Frotscher, M. (1996). Spine loss in experimental epilepsy, quantitative light and electron microscopic analysis of intracellularly stained CA3 pyramidal cells in hippocampal slice cultures. Neuroscience 70, 31–45.
Drukarch, B., Schepens, E., Jongenelen, C. A., Stoof, J. C., and Langeveld, C. H. (1997). Astrocyte-mediated enhancement of neuronal survival is abolished by glutathione deficiency. Brain Res. 770, 123–130.
During, M. J., Ryder, K. M., and Spencer, D. D. (1995). Hippocampal GABA transporter function in temporal-lobe epilepsy. Nature 376, 174–177.
During, M. J., and Spencer, D. D. (1993). Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 341, 1607–1610.
Duvernoy, H. M. (2005). The Human Hippocampus, Functional Anatomy, Vascularization and Serial Sections with MRI. Berlin: Springer.
Ebert, U., and Ziemann, U. (1999). Altered seizure susceptibility after high-frequency transcranial magnetic stimulation in rats. Neurosci. Lett. 273, 155–158.
Eddleston, M., and Mucke, L. (1993). Molecular profile of reactive astrocytes – implications for their role in neurologic disease. Neuroscience 54, 15–36.
Eid, T., Thomas, M. J., Spencer, D. D., Runden-Pran, E., Lai, J. C. K., Malthankar, G. V., Kim, J. H., Danbolt, N. C., Ottersen, O. P., and de Lanerolle, N. C. (2004). Loss of glutamine synthetase in the human epileptogenic hippocampus, possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363, 28–37.
Engel, J. Jr., Wiebe, S., French, J., Sperling, M., Williamson, P., Spencer, D., Gumnit, R., Zahn, C., Westbrook, E., and Enos, B. (2003). Practice parameter, temporal lobe and localized neocortical resections for epilepsy. Epilepsia 44, 741–751.
Engel, J. (2012). Early surgical therapy for drug-resistant temporal lobe epilepsy: a randomized trial. JAMA 307, 922–930.
Escartin, C., and Bonvento, G. (2008). Targeted activation of astrocytes, a potential neuroprotective strategy. Mol. Neurobiol. 38, 231–241.
Feindel, W., Leblanc, R., and de Almeida, A. N. (2009). Epilepsy surgery, historical highlights 1909–2009. Epilepsia 50(Suppl 3), 131–151.
Fellin, T., Pascual, O., Gobbo, S., Pozzan, T., Haydon, P., and Carmignoto, G. (2004). Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43, 729–743.
Fernandez-Galaz, M. C., Morschl, E., Chowen, J. A., Torres-Aleman, I., Naftolin, F., and Garcia-Segura, L. M. (1997). Role of astroglia and insulin-like growth factor-I in gonadal hormone-dependent synaptic plasticity. Brain Res. Bull. 44, 525–531.
Fiacco, T., and McCarthy, K. (2004). Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J. Neurosci. 24, 722–732.
Fiala, J. C., Spacek, J., and Harris, K. M. (2002). Dendritic spine pathology, cause or consequence of neurological disorders? Brain Res. Brain Res. Rev. 39, 29–54.
Fregni, F., and Pascual-Leone, A. (2007). Technology insight, noninvasive brain stimulation in neurology perspectives on the therapeutic potential of rTMS and tDCS. Nat. Clin. Pract. Neurol. 3, 383–393.
Fujiki, M., and Steward, O. (1997). High frequency transcranial magnetic stimulation mimics the effects of ECS in upregulating astroglial gene expression in the murine CNS. Brain Res. Mol. Brain Res. 44, 301–308.
Godlevsky, L. S., Kobolev, E. V., van Luijtelaar, E. L., Coenen, A. M., Stepanenko, K. I., and Smirnov, I. V. (2006). Influence of transcranial magnetic stimulation on spike-wave discharges in a genetic model of absence epilepsy. Indian J. Exp. Biol. 44, 949–954.
Gomez-Gonzalo, M., Losi, G., Brondi, M., Uva, L., Sulis-Sato, S., De, C. M., Ratto, G. M., and Carmignoto, G. (2011). Ictal but not interictal epileptic discharges activate astrocyte endfeet and elicit cerebral arteriole responses. Front. Cell Neurosci. 5:8. doi:10.3389/fncel.2011.00008
Gomez-Gonzalo, M., Losi, G., Chiavegato, A., Zonta, M., Cammarota, M., Brondi, M., Vetri, F., Uva, L., Pozzan, T., de Curtis, M., Ratto, G. M., and Carmignoto, G. (2010). An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biol. 8, e1000352. doi:10.1371/journal.pbio.1000352
Goritz, C., Mauch, D. H., and Pfrieger, F. W. (2005). Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol. Cell. Neurosci. 29, 190–201.
Griffith, R., and Humphrey, D. (2006). Long-term gliosis around chronically implanted platinum electrodes in the Rhesus macaque motor cortex. Neurosci. Lett. 406, 81–6.
Grosche, J., Matyash, V., Moller, T., Verkhratsky, A., Reichenbach, A., and Kettenmann, H. (1999). Microdomains for neuron-glia interactions, parallel fiber signaling to Bergmann glial cells. Nat. Neurosci. 2, 139–143.
Halassa, M., and Haydon, P. (2010). Integrated brain circuits, astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol. 72, 335–55.
Hama, H., Hara, C., Yamaguchi, K., and Miyawaki, A. (2004). PKC signaling mediates global enhancement of excitatory synaptogenesis in neurons triggered by local contact with astrocytes. Neuron 41, 405–415.
Handforth, A., DeGiorgio, C. M., Schachter, S. C., Uthman, B. M., Naritoku, D. K., Tecoma, E. S., Henry, T. R., Collins, S. D., Vaughn, B. V., Gilmartin, R. C., Labar, D. R., Morris, G. L. III, Salinsky, M. C., Osorio, I., Ristanovic, R. K., Labiner, D. M., Jones, J. C., Murphy, J. V., Ney, G. C., and Wheless, J. W. (1998). Vagus nerve stimulation therapy for partial-onset seizures, a randomized active-control trial. Neurology 51, 48–55.
Hatton, G. I., and Parpura, V. (2004). Glial: Neuronal Signaling. Boston: Kluwer Academic Publishers.
Haupt, C., Witte, O. W., and Frahm, C. (2007). Up-regulation of Connexin43 in the glial scar following photothrombotic ischemic injury. Mol. Cell. Neurosci. 35, 89–99.
Hauser, W. A., Annegers, J. F., and Kurland, L. T. (1993). Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota, 1935–1984. Epilepsia 34, 453–468.
Henneberger, C., Papouin, T., Oliet, S., and Rusakov, D. (2010). Long-term potentiation depends on release of D-serine from astrocytes. Nature 463, 232–236.
Henry, T. R., Bakay, R. A., Votaw, J. R., Pennell, P. B., Epstein, C. M., Faber, T. L., Grafton, S. T., and Hoffman, J. M. (1998). Brain blood flow alterations induced by therapeutic vagus nerve stimulation in partial epilepsy, I. Acute effects at high and low levels of stimulation. Epilepsia 39, 983–990.
Henry, T. R., Votaw, J. R., Pennell, P. B., Epstein, C. M., Bakay, R. A., Faber, T. L., Grafton, S. T., and Hoffman, J. M. (1999). Acute blood flow changes and efficacy of vagus nerve stimulation in partial epilepsy. Neurology 52, 1166–1173.
Hermann, G., van Meter, M., Rood, J., and Rogers, R. (2009). Proteinase-activated receptors in the nucleus of the solitary tract, evidence for glial-neural interactions in autonomic control of the stomach. J. Neurosci. 29, 9292–9300.
Hertz, L., and Zielke, H. R. (2004). Astrocytic control of glutamatergic activity, astrocytes as stars of the show. Trends Neurosci. 27, 735–743.
Hesdorffer, D. C., Logroscino, G., Benn, E. K. T., Katri, N., Cascino, G., and Hauser, W. A. (2011). Estimating risk for developing epilepsy. Neurology 76, 23–27.
Hirrlinger, J., Hulsmann, S., and Kirchhoff, F. (2004). Astroglial processes show spontaneous motility at active synaptic terminals in situ. Eur. J. Neurosci. 20, 2235–2239.
Hirshler, Y., Polat, U., and Biegon, A. (2010). Intracranial electrode implantation produces regional neuroinflammation and memory deficits in rats. Exp. Neurol. 222, 42–50.
ILAE. (1997). A global survey on epilepsy surgery, 1980–1990, a report by the commission on neurosurgery of epilepsy, the international league against epilepsy. Epilepsia 38, 249–255.
Jennum, P., and Klitgaard, H. (1996). Repetitive transcranial magnetic stimulations of the rat. Effect of acute and chronic stimulations on pentylenetetrazole-induced clonic seizures. Epilepsy Res. 23, 115–122.
Jiang, M., Lee, C. L., Smith, K. L., and Swann, J. W. (1998). Spine loss and other persistent alterations of hippocampal pyramidal cell dendrites in a model of early-onset epilepsy. J. Neurosci. 18, 8356–8368.
Jourdain, P., Bergersen, L., Bhaukaurally, K., Bezzi, P., Santello, M., Domercq, M., Matute, C., Tonello, F., Gundersen, V., and Volterra, A. (2007). Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci. 10, 331–339.
Kang, J., Jiang, L., Goldman, S., and Nedergaard, M. (1998). Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1, 683–692.
Kimiskidis, V. K. (2010). Transcranial magnetic stimulation for drug-resistant epilepsies, rationale and clinical experience. Eur. Neurol. 63, 205–210.
Klintsova, A., Levy, W. B., and Desmond, N. L. (1995). Astrocytic volume fluctuates in the hippocampal CA1 region across the estrous cycle. Brain Res. 690, 269–274.
Ko, D., Heck, C., Grafton, S., Apuzzo, M. L., Couldwell, W. T., Chen, T., Day, J. D., Zelman, V., Smith, T., and DeGiorgio, C. M. (1996). Vagus nerve stimulation activates central nervous system structures in epileptic patients during PET H2(15) O blood flow imaging. Neurosurgery 39, 426–430.
Kofuji, P., and Newman, E. A. (2004). Potassium buffering in the central nervous system. Neuroscience 129, 1045–1056.
Krack, P., Batir, A., Van Blercom, N., Chabardes, S., Fraix, V., Ardouin, C., Koudsie, A., Limousin, P. D., Benazzouz, A., LeBas, J. F., Benabid, A. L., and Pollak, P. (2003). Five-year follow-up of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N. Engl. J. Med. 349, 1925–1934.
Kraev, I., Godukhin, O., Patrushev, I., Davies, H., Popov, V., and Stewart, M. (2009). Partial kindling induces neurogenesis, activates astrocytes and alters synaptic morphology in the dentate gyrus of freely moving adult rats. Neuroscience 162, 254–267.
Krishnan, B., Armstrong, D. L., Grossman, R. G., Zhu, Z. Q., Rutecki, P. A., and Mizrahi, E. M. (1994). Glial cell nuclear hypertrophy in complex partial seizures. J. Neuropathol. Exp. Neurol. 53, 502–507.
Kwan, P., and Brodie, M. J. (2000). Early identification of refractory epilepsy. N. Engl. J. Med. 342, 314–319.
Labar, D. R. (2002). Antiepileptic drug use during the first 12 months of vagus nerve stimulation therapy, a registry study. Neurology 59, S38–S43.
Lado, F. A., Velisek, L., and Moshe, S. L. (2003). The effect of electrical stimulation of the subthalamic nucleus on seizures is frequency dependent. Epilepsia 44, 157–164.
Lee, K. H., Hitti, F. L., Shalinsky, M. H., Kim, U., Leiter, J. C., and Roberts, D. W. (2005a). Abolition of spindle oscillations and 3-Hz absence seizurelike activity in the thalamus by using high-frequency stimulation, potential mechanism of action. J. Neurosurg. 103, 538–545.
Lee, S. K., Lee, S. Y., Kim, K. K., Hong, K. S., Lee, D. S., and Chung, C. K. (2005b). Surgical outcome and prognostic factors of cryptogenic neocortical epilepsy. Ann. Neurol. 58, 525–532.
Lee, K. J., Jang, K. S., and Shon, Y. M. (2006a). Chronic deep brain stimulation of subthalamic and anterior thalamic nuclei for controlling refractory partial epilepsy. Acta Neurochir. Suppl. 99, 87–91.
Lee, T. S., Bjornsen, L. P., Paz, C., Kim, J. H., Spencer, S. S., Spencer, D. D., Eid, T., and de Lanerolle, N. C. (2006b). GAT1 and GAT3 expression are differently localized in the human epileptogenic hippocampus. Acta Neuropathol. (Berl.) 111, 351–363.
Liberto, C., Albrecht, P., Herx, L., Yong, V., and Levison, S. (2004). Pro-regenerative properties of cytokine-activated astrocytes. J. Neurochem. 89, 1092–1100.
Lim, S. N., Lee, S. T., Tsai, Y. T., Chen, I. A., Tu, P. H., Chen, J. L., Chang, H. W., Su, Y. C., and Wu, T. (2007). Electrical stimulation of the anterior nucleus of the thalamus for intractable epilepsy, a long-term follow-up study. Epilepsia 48, 342–347.
Lin, S. C., and Bergles, D. E. (2004). Synaptic signaling between neurons and glia. Glia 47, 290–298.
Lipton, R. B., and Pearlman, S. H. (2010). Transcranial magnetic simulation in the treatment of migraine. Neurotherapeutics 7, 204–212.
Liu, Q. S., Xu, Q., Arcuino, G., Kang, J., and Nedergaard, M. (2004) Astrocyte-mediated activation of neuronal kainate receptors. Proc. Natl. Acad. Sci. U.S.A. 101, 3172–3177.
Liu, W. C., Mosier, K., Kalnin, A. J., and Marks, D. (2003). BOLD fMRI activation induced by vagus nerve stimulation in seizure patients. J. Neurol. Neurosurg. Psychiatr. 74, 811–813.
Lockard, J. S., Congdon, W. C., and DuCharme, L. L. (1990). Feasibility and safety of vagal stimulation in monkey model. Epilepsia 31(Suppl 2), S20–S26.
Luquin, S., Naftolin, F., and Garcia-Segura, L. M. (1993). Natural fluctuation and gonadal hormone regulation of astrocyte immunoreactivity in dentate gyrus. J. Neurobiol. 24, 913–924.
Magistretti, P. J. (2006). Neuron-glia metabolic coupling and plasticity. J. Exp. Biol. 209, 2304–2311.
Malthankar-Phatak, G. H., de Lanerolle, N., Eid, T., Spencer, D. D., Behar, K. L., Spencer, S. S., Kim, J. H., and Lai, J. C. (2006). Differential glutamate dehydrogenase (GDH) activity profile in patients with temporal lobe epilepsy. Epilepsia 47, 1292–1299.
Martinez, A. D., and Saez, J. C. (2000). Regulation of astrocyte gap junctions by hypoxia-reoxygenation. Brain Res. Brain Res. Rev. 32, 250–258.
Mathern, G. W., Babb, T. L., and Armstrong, D. (1997). “Hippocampal sclerosis,” in Epilepsy, A Comprehensive Textbook, eds J. Engel Jr. and T. A. Pedley (Philadelphia: Lippincott-Raven Publishers), 133–154.
Matute, C., Domercq, M., and Sanchez-Gomez, M. V. (2006). Glutamate-mediated glial injury, mechanisms and clinical importance. Glia 53, 212–224.
Mauch, D. H., Nagler, K., Schumacher, S., Goritz, C., Muller, E. C., Otto, A., and Pfrieger, F. W. (2001). CNS synaptogenesis promoted by glia-derived cholesterol. Science 294, 1354–1357.
Mazzanti, M., and Haydon, P. G. (2003). Astrocytes selectively enhance N-type calcium current in hippocampal neurons. Glia 41, 128–136.
McDougal, D. H., Hermann, G. E., and Rogers, R. C. (2011). Vagal afferent stimulation activates astrocytes in the nucleus of the solitary tract via AMPA receptors, evidence of an atypical neural-glial interaction in the brainstem. J. Neurosci. 31, 14037–14045.
McIntyre, C. C., Savasta, M., Kerkerian-Le, G. L., and Vitek, J. L. (2004). Uncovering the mechanism(s) of action of deep brain stimulation, activation, inhibition, or both. Clin. Neurophysiol. 115, 1239–1248.
McLachlan, R. S. (1993). Suppression of interictal spikes and seizures by stimulation of the vagus nerve. Epilepsia 34, 918–923.
Mitchell, L. A., Jackson, G. D., Kalnins, R. M., Saling, M. M., Fitt, G. J., Ashpole, R. D., and Berkovic, S. F. (1999). Anterior temporal abnormality in temporal lobe epilepsy, a quantitative MRI and histopathologic study. Neurology 52, 327–336.
Morris, G. L. III, and Mueller, W. M. (1999). Long-term treatment with vagus nerve stimulation in patients with refractory epilepsy. The vagus nerve stimulation study group E01–E05. Neurology 53, 1731–1735.
Moss, J., Ryder, T., Aziz, T., Graeber, M., and Bain, P. (2004). Electron microscopy of tissue adherent to explanted electrodes in dystonia and Parkinson’s disease. Brain 127, 2755–2763.
Muellbacher, W., Ziemann, U., Boroojerdi, B., and Hallett, M. (2000). Effects of low-frequency transcranial magnetic stimulation on motor excitability and basic motor behavior. Clin. Neurophysiol. 111, 1002–1007.
Mullan, S., Vailati, G., Karasick, J., and Mailis, M. (1967). Thalamic lesions for the control of epilepsy. A study of nine cases. Arch. Neurol. 16, 277–285.
Multani, P., Myers, R. H., Blume, H. W., Schomer, D. L., and Sotrel, A. (1994). Neocortical dendritic pathology in human partial epilepsy, a quantitative Golgi study. Epilepsia 35, 728–736.
Murai, K. K., Nguyen, L. N., Irie, F., Yamaguchi, Y., and Pasquale, E. B. (2003). Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat. Neurosci. 6, 153–160.
Narayanan, J. T., Watts, R., Haddad, N., Labar, D. R., Li, P. M., and Filippi, C. G. (2002). Cerebral activation during vagus nerve stimulation, a functional MR study. Epilepsia 43, 1509–1514.
Naus, C. C., Bechberger, J. F., and Paul, D. L. (1991). Gap junction gene expression in human seizure disorder. Exp. Neurol. 111, 198–203.
Naus, C. C., Ozog, M. A., Bechberger, J. F., and Nakase, T. (2001). A neuroprotective role for gap junctions. Cell Commun. Adhes. 8, 325–328.
Navarrete, M., Perea, G., de Sevilla, D., Gómez-Gonzalo, M., Núñez, A., Martín, E., and Araque, A. (2012). Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol. 10, e1001259. doi:10.1371/journal.pbio.1001259
Oliet, S. H., Panatier, A., and Piet, R. (2006). Functional neuronal-glial anatomical remodelling in the hypothalamus. Novartis Found. Symp. 276, 238–248.
Oliet, S. H., Piet, R., and Poulain, D. A. (2001). Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292, 923–926.
Ortinski, P., Dong, J., Mungenast, A., Yue, C., Takano, H., Watson, D., Haydon, P., and Coulter, D. (2010). Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat. Neurosci. 13, 584–591.
Ostrow, L. W., and Sachs, F. (2005). Mechanosensation and endothelin in astrocytes – hypothetical roles in CNS pathophysiology. Brain Res. Brain Res. Rev. 48, 488–508.
Ozog, M. A., Siushansian, R., and Naus, C. C. (2002). Blocked gap junctional coupling increases glutamate-induced neurotoxicity in neuron-astrocyte co-cultures. J. Neuropathol. Exp. Neurol. 61, 132–141.
Parpura, V., Scemes, E., and Spray, D. C. (2004). Mechanisms of glutamate release from astrocytes, gap junction “hemichannels,” purinergic receptors and exocytotic release. Neurochem. Int. 45, 259–264.
Parri, H., Gould, T., and Crunelli, V. (2001). Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat. Neurosci. 4, 803–812.
Pascual, O., Casper, K. B., Kubera, C., Zhang, J., Revilla-Sanchez, R., Sul, J. Y., Takano, H., Moss, S. J., McCarthy, K., and Haydon, P. G. (2005). Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116.
Pascual-Leone, A., Rubio, B., Pallardo, F., and Catalá, M. D. (1996). Rapid-rate transcranial magnetic stimulation of left dorsolateral prefrontal cortex in drug-resistant depression. Lancet 348, 233–237.
Perea, G., and Araque, A. (2005). Synaptic regulation of the astrocyte calcium signal. J. Neural Transm. 112, 127–135.
Pfrieger, F. W., and Barres, B. A. (1997). Synaptic efficacy enhanced by glial cells in vitro. Science 277, 1684–1687.
Piet, R., Vargova, L., Sykova, E., Poulain, D. A., and Oliet, S. H. (2004). Physiological contribution of the astrocytic environment of neurons to intersynaptic crosstalk. Proc. Natl. Acad. Sci. U.S.A. 101, 2151–2155.
Proper, E. A., Hoogland, G., Kappen, S. M., Jansen, G. H., Rensen, M. G., Schrama, L. H., van Veelen, C. W., van Rijen, P. C., van Nieuwenhuizen, O., Gispen, W. H., and de Graan, P. N. (2002). Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 125, 32–43.
Quigg, M., Rolston, J., and Barbaro, N. M. (2012). Radiosurgery for epilepsy, clinical experience and potential antiepileptic mechanisms. Epilepsia 53, 7–15.
Raisman, G., and Field, P. M. (1973). A quantitative investigation of the development of collateral reinnervation after partial deafferentation of the septal nuclei. Brain Res. 50, 241–264.
Rosenberg, P. A., and Aizenman, E. (1989). Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci. Lett. 103, 162–168.
Rotenberg, A., Muller, P., Birnbaum, D., Harrington, M., Riviello, J. J., Pascual-Leone, A., and Jensen, F. E. (2008). Seizure suppression by EEG-guided repetitive transcranial magnetic stimulation in the rat. Clin. Neurophysiol. 119, 2697–2702.
Rusakov, D. A., and Kullmann, D. M. (1998). Extrasynaptic glutamate diffusion in the hippocampus, ultrastructural constraints, uptake, and receptor activation. J. Neurosci. 18, 3158–3170.
Sander, J. W. (1993). Some aspects of prognosis in the epilepsies, a review. Epilepsia 34, 1007–1016.
Santello, M., and Volterra, A. (2009). Synaptic modulation by astrocytes via Ca2+-dependent glutamate release. Neuroscience 158, 253–259.
Scemes, E., and Giaume, C. (2006). Astrocyte calcium waves, what they are and what they do. Glia 54, 716–725.
Scheibel, A. B., and Scheibel, M. E. (1977). “Specific postnatal threats to brain development, dendritic changes,” in Proceeding of the Brain, Fetal and Infant, Current research on Normal and Abnormal Development, Conference, held 12-14-1976 ed. S. R. Berenberg (The Hague: Martinus Nijhoff Medical Division), 302–315.
Schipke, C. G., and Kettenmann, H. (2004). Astrocyte responses to neuronal activity. Glia 47, 226–232.
Seifert, G., Huttmann, K., Schramm, J., and Steinhauser, C. (2004). Enhanced relative expression of glutamate receptor 1 flip AMPA receptor subunits in hippocampal astrocytes of epilepsy patients with Ammon’s horn sclerosis. J. Neurosci. 24, 1996–2003.
Shandra, A. A., and Godlevsky, L. S. (1990). Antiepileptic effects of cerebellar nucleus dentatus electrical stimulation under different conditions of brain epileptisation. Indian J. Exp. Biol. 28, 158–161.
Shigeri, Y., Seal, R. P., and Shimamoto, K. (2004). Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res. Brain Res. Rev. 45, 250–265.
Sillanpaa, M., and Schmidt, D. (2006). Natural history of treated childhood-onset epilepsy, prospective, long-term population-based study. Brain 129, 617–624.
Sofroniew, M. (2005). Reactive astrocytes in neural repair and protection. Neuroscientist 11, 400–407.
Sommer, W. (1880). Erkrankung des ammonshornsalsaetiologisches moment der epilepsie. Arch. Psychiatr. Nervenkr. 10, 631–675.
Spencer, S. S. (2002). When should temporal-lobe epilepsy be treated surgically? Lancet Neurol. 1, 375–382.
Stock, G., Sturm, V., Schmitt, H., and Schlor, K. (1979). The influence of chronic deep brain stimulation on excitability and morphology of the stimulated tissue. Acta Neurochir. (Wien.) 47, 123–129.
Sun, D., Yu, H., Spooner, J., Tatsas, A., Davis, T., Abel, T., Kao, C., and Konrad, P. (2008a). Postmortem analysis following 71 months of deep brain stimulation of the subthalamic nucleus for Parkinson disease. J. Neurosurg. 109, 325–329.
Sun, F. T., Morrell, M. J., and Wharen, R. E. Jr. (2008b). Responsive cortical stimulation for the treatment of epilepsy. Neurotherapeutics 5, 68–74.
Swann, J. W., Al Noori, S., Jiang, M., and Lee, C. L. (2000). Spine loss and other dendritic abnormalities in epilepsy. Hippocampus 10, 617–625.
Takata, N., Mishima, T., Hisatsune, C., Nagai, T., Ebisui, E., Mikoshiba, K., and Hirase, H. (2011). Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J. Neurosci. 31, 18155–18165.
Takaya, M., Terry, W. J., and Naritoku, D. K. (1996). Vagus nerve stimulation induces a sustained anticonvulsant effect. Epilepsia 37, 1111–1116.
Takuma, K., Baba, A., and Matsuda, T. (2004). Astrocyte apoptosis, implications for neuroprotection. Prog. Neurobiol. 72, 111–127.
Tanaka, K., Watase, K., Manabe, T., Yamada, K., Watanabe, M., Takahashi, K., Iwama, H., Nishikawa, T., Ichihara, N., Kikuchi, T., Okuyama, S., Kawashima, N., Hori, S., Takimoto, M., and Wada, K. (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702.
Tawfik, V., Chang, S., Hitti, F., Roberts, D., Leiter, J., Jovanovic, S., and Lee, K. (2010). Deep brain stimulation results in local glutamate and adenosine release, investigation into the role of astrocytes. Neurosurgery 67, 367–375.
Theodosis, D. T., Trailin, A., and Poulain, D. A. (2006) Remodeling of astrocytes, a prerequisite for synapse turnover in the adult brain? Insights from the oxytocin system of the hypothalamus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R1175–R1182.
Thompson, S. M., Fortunato, C., McKinney, R. A., Muller, M., and Gahwiler, B. H. (1996). Mechanisms underlying the neuropathological consequences of epileptic activity in the rat hippocampus in vitro. J. Comp. Neurol. 372, 515–528.
Tian, G. F., Azmi, H., Takano, T., Xu, Q., Peng, W., Lin, J., Oberheim, N., Lou, N., Wang, X., Zielke, H. R., Kang, J., and Nedergaard, M. (2005). An astrocytic basis of epilepsy. Nat. Med. 11, 973–981.
Tritsch, N. X., and Bergles, D. E. (2007). Defining the role of astrocytes in neuromodulation. Neuron 54, 497–500.
Ullian, E. M., Christopherson, K. S., and Barres, B. A. (2004). Role for glia in synaptogenesis. Glia 47, 209–216.
Ullian, E. M., Sapperstein, S. K., Christopherson, K. S., and Barres, B. A. (2001). Control of synapse number by glia. Science 291, 657–661.
Van, L. K., Vonck, K., and Boon, P. (2000). Vagus nerve stimulation in refractory epilepsy, SPECT activation study. J. Nucl. Med. 41, 1145–1154.
van der Hel, W. S., Notenboom, R. G., Bos, I. W., van Rijen, P. C., van Veelen, C. W., and de Graan, P. N. (2005). Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology 64, 326–333.
van Paesschen, W., Revesz, T., Duncan, J. S., King, M. D., and Connelly, A. (1997). Quantitative neuropathology and quantitative magnetic resonance imaging of the hippocampus in temporal lobe epilepsy. Ann. Neurol. 42, 756–766.
Vedam-Mai, V., Krock, N., Ullman, M., Foote, K., Shain, W., Smith, K., Yachnis, A., Steindler, D., Reynolds, B., Merritt, S., Pagan, F., Marjama-Lyons, J., Hogarth, P., Resnick, A., Zeilman, P., and Okun, M. (2011). The national DBS brain tissue network pilot study, need for more tissue and more standardization. Cell Tissue Bank. 12, 219–31.
Vedam-Mai, V., van Battum, E., Kamphuis, W., Feenstra, M., Denys, D., Reynolds, B., Okun, M., and Hol, E. (2012). Deep brain stimulation and the role of astrocytes. Mol. Psychiatry 17, 124–31.
Ventura, R., and Harris, K. M. (1999). Three-dimensional relationships between hippocampal synapses and astrocytes. J. Neurosci. 19, 6897–6906.
Vercueil, L., Benazzouz, A., Deransart, C., Bressand, K., Marescaux, C., Depaulis, A., and Benabid, A. L. (1998). High-frequency stimulation of the subthalamic nucleus suppresses absence seizures in the rat, comparison with neurotoxic lesions. Epilepsy Res. 31, 39–46.
VNSSG. (1995). A randomized controlled trial of chronic vagus nerve stimulation for treatment of medically intractable seizures. The Vagus Nerve Stimulation Study Group. Neurology 45, 224–230.
Volterra, A., Magistretti, P. J., and Haydon, P. G. (2002). The Tripartite Synapse. Oxford: Oxford University Press.
Volterra, A., and Meldolesi, J. (2005). Astrocytes, from brain glue to communication elements, the revolution continues. Nat. Rev. Neurosci. 6, 626–640.
Vonck, K., Boon, P., D’Have, M., Vandekerckhove, T., O’Connor, S., and De Reuck, J. (1999). Long-term results of vagus nerve stimulation in refractory epilepsy. Seizure 8, 328–334.
Vonck, K., Boon, P., Van Laere, K., D’Havé, M., Vandekerckhove, T., O’Connor, S., Brans, B., Dierckx, R., and De Reuck, J. (2000). Acute single photon emission computed tomographic study of vagus nerve stimulation in refractory epilepsy. Epilepsia 41, 601–609.
Wallraff, A., Kohling, R., Heinemann, U., Theis, M., Willecke, K., and Steinhauser, C. (2006). The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. 26, 5438–5447.
Wilson, S., and Engel, J. Jr. (2010). Diverse perspectives on developments in epilepsy surgery. Seizure 19, 659–668.
Witcher, M., Park, Y., Lee, M., Sharma, S., Harris, K., and Kirov, S. (2010). Three-dimensional relationships between perisynaptic astroglia and human hippocampal synapses. Glia 58, 572–87.
Witcher, M., and Ellis, T. (2011). “Neuromodulatory treatment of medically refractory epilepsy,” in Novel Treatment of Epilepsy, ed. H. Foyaca-Sibat (Rijeka: Croatia), 287–310.
Witcher, M. R., Kirov, S. A., and Harris, K. M. (2007). Plasticity of perisynaptic astroglia during synaptogenesis in the mature rat hippocampus. Glia 55, 13–23.
Yang, Y., Ge, W., Chen, Y., Zhang, Z., Shen, W., Wu, C., Poo, M., and Duan, S. (2003). Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc. Natl. Acad. Sci. U.S.A. 100, 15194–15199.
Zabara, J. (1992). Inhibition of experimental seizures in canines by repetitive vagal stimulation. Epilepsia 33, 1005–1012.
Ziemann, U. (2005). “Evaluation of epilepsy and anticonvulsants,” in Magnetic Stimulation in Clinical Neurophysiology, 2nd Edn, eds M. Hallett and A. Chokroverty (Philadelphia: Elsevier), 253–270.
Zonta, M., Angulo, M. C., Gobbo, S., Rosengarten, B., Hossmann, K. A., Pozzan, T., and Carmignoto, G. (2003). Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 6, 43–50.
Keywords: astrocyte, tripartite synapse, neuromodulation, epilepsy, deep brain stimulation, vagal nerve stimulation
Citation: Witcher MR and Ellis TL (2012) Astroglial networks and implications for therapeutic neuromodulation of epilepsy. Front. Comput. Neurosci. 6:61. doi: 10.3389/fncom.2012.00061
Received: 30 April 2012; Paper pending published: 25 June 2012;
Accepted: 30 July 2012; Published online: 29 August 2012.
Edited by:
Liam J. McDaid, University of Ulster, UKCopyright: © 2012 Witcher and Ellis. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Mark R. Witcher, Department of Neurosurgery, Wake Forest University, Medical Center Boulevard, Winston-Salem, NC 27157, USA. e-mail:bXdpdGNoZXJAd2FrZWhlYWx0aC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.