 Xi Kuang
Xi Kuang Shuang Chen
Shuang Chen Qingmei Ye
Qingmei Ye- 1Hainan Health Vocational College, Haikou, China
- 2Department of Neurology, Hubei Provincial Hospital of Integrated Chinese and Western Medicine, Hubei University of Chinese Medicine, Wuhan, China
- 3Hainan General Hospital and Hainan Affiliated Hospital of Hainan Medical University, Haikou, China
Protein lactylation is a new form of post-translational modification that has recently been proposed. Lactoyl groups, derived mainly from the glycolytic product lactate, have been linked to protein lactylation in brain tissue, which has been shown to correlate with increased neuronal excitability. Ischemic stroke may promote neuronal glycolysis, leading to lactate accumulation in brain tissue. This accumulation of lactate accumulation may heighten neuronal excitability by upregulating protein lactylation levels, potentially triggering post-stroke epilepsy. Although current clinical treatments for seizures have advanced significantly, approximately 30% of patients with epilepsy remain unresponsive to medication, and the prevalence of epilepsy continues to rise. This study explores the mechanisms of epilepsy-associated neuronal death mediated by lactate metabolism and protein lactylation. This study also examines the potential for histone deacetylase inhibitors to alleviate seizures by modifying lactylation levels, thereby offering fresh perspectives for future research into the pathogenesis and clinical treatment of epilepsy.
Highlights
• The lactate metabolism and transport carried out in neurons and glial cells may influence the development of epilepsy by regulating protein lactylation levels.
• Lactate may mediate epilepsy-related neuronal loss by promoting HMGB1 lactylation.
• Ischemic stroke may promote HMGB1 lactylation by activating HIF-1, which can promote a shift in the mode of cellular energy acquisition from oxidative phosphorylation to glycolysis, thereby inducing post-stroke epilepsy.
• Histone deacetylases may affect protein lactylation by regulating the transcriptional activity of HIF-1.
• Histone deacetylase inhibitors may combat post-stroke epilepsy by modulating hypoxia-induced protein lactylation.
1 Introduction
Epilepsy, as a common chronic disease of the nervous system, has affected more than 70 million people of all ages worldwide, with the number continuing to rise annually (Eugen et al., 2019). The factors inducing seizures are diverse and multifaceted. Ischemic stroke is one of the common causes of epilepsy. Post-ischemic stroke epilepsy accounts for approximately 9% of all epilepsy cases, a figure slightly lower than that caused by cerebral hemorrhage. However, the growing number of individuals affected by ischemic stroke each year highlights its significant impact on public health (Lanqing et al., 2021; Carolina et al., 2021). The hypoxic environment caused by ischemic stroke often forces brain tissue to obtain energy through glycolysis, thereby producing a large amount of lactate.
Lactate in the brain may affect the progression of various neuropsychiatric diseases through its roles in learning, memory, and emotional regulation. This process may be related to lactate-mediated post-translational modifications of proteins (Hagihara et al., 2021). Lactylation (La), a novel type of post-translational protein modification, was identified by Zhang et al. (2019). With the ongoing research into protein lactylation modification in recent years, HAGIHARA H et al. found that lysine lactylation (Kla) may be prevalent in neurons and glial cells in the brain.
Furthermore, the level of lysine lactylation is regulated by lactic acid levels, neural excitation, and social defeat stress (Hagihara et al., 2021). It suggests that increased neuronal excitability may be closely related to the level of protein lactylation in neurons.
Abnormal excitation of neurons is a key driver of epileptogenesis. Ischemia/reperfusion injury (I/R) can induce epileptogenesis by mediating abnormal excitation of neurons (Paudel et al., 2020; Esih et al., 2021; Endres et al., 2022). It can be speculated that ischemia-induced stroke may lead to increased glucose metabolism in the brain, resulting in the production of large quantities of lactate, which can be transformed into acetyl groups to participate in the acetylation of proteins related to neuronal excitation and thus cause post-stroke epilepsy (Hagihara et al., 2021; He et al., 2023). That is, protein lactylation may be a potential regulatory target for post-ischemic stroke epilepsy. While anti-epileptic drugs can help most patients control their seizures, approximately 30% of individuals with epilepsy do not respond to current clinical medications (Sun et al., 2021). This review mainly focuses on the impact of lactate metabolism and protein acetylation on epilepsy and changes in acetylation modifications controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs). Furthermore, based on the mechanisms of action of HDAC inhibitors (HDACIs) in regulating acetylation modifications, we discuss the possibility that they could provide a new strategy for treating post-stroke epilepsy.
2 The lactate mediates epilepsy by regulating the functions of neurons and glial cells
Seizures are usually localized in the hippocampus, with temporal lobe epilepsy (TLE) being the most common (Sun et al., 2021). Previous studies have suggested that neuronal hyperexcitability in epilepsy is primarily mediated by neuronal death and neuroinflammation, which is mediated by astrogliosis and microglial activation, and the relationship between neuronal death and neuroinflammation may be complex (Haenisch et al., 2015; Roh et al., 2023). Seizures are usually accompanied by significant glial cell proliferation and loss of neurons, and glial cell proliferation-mediated neuroinflammation often leads to neuronal death (Feng et al., 2019). Protein lactylation modifications have been found in the hippocampus and prefrontal cortex and are strongly associated with neuronal excitability (Hagihara et al., 2021).
Currently, there is insufficient evidence to confirm that protein lactylation directly induces epilepsy by regulating neuronal excitability. However, lactate, as the donor of lactide, can serve as an energy source for metabolic activities in neurons and glial cells during ischemia and hypoxia, potentially influencing the progression of post-stroke epilepsy.
2.1 Glial cells mediate epilepsy by regulating the release of neurotransmitters
Studies on status epilepticus suggest that astrocyte and microglial activity changes, which influence critical homeostatic processes—such as synaptogenesis, extracellular ion concentrations, and excitation-inhibition balance—may occur early in epilepsy development. Inhibiting glial cell activity has been shown to reduce susceptibility to epilepsy (Shen et al., 2023). Neuroglia, such as astrocytes, microglia, and oligodendrocytes, may influence neuronal excitability by regulating the production of glutamate, adenosine triphosphate (ATP), and γ-aminobutyric acid (GABA) in the central nervous system, thereby modulating the progression of epilepsy (Shen et al., 2023).
Glutamate is the primary excitatory neurotransmitter in the central nervous system and can induce seizures by increasing synaptic transmission in the hippocampal region. Although the death of neurons caused by cerebral infarction can reduce the synthesis of the neurotransmitter glutamate, previous studies have confirmed that the level of glutamate in the blood and cerebrospinal fluid of ischemic stroke patients is related to the severity of infarction and neurological dysfunction (Nicolo et al., 2019). Research has shown that stroke can mediate neuroinflammatory damage by activating astrocytes’ TGF-β pathway, and the TGF-β signaling in astrocytes may have a pro-epileptic effect (Korotkov et al., 2020). Therefore, astrocytes may induce post-stroke epilepsy by mediating stroke-related glutamate excitotoxicity. Astrocytes primarily take up excessive glutamate from the synaptic cleft through two types of glutamate transporters, such as glutamate–aspartate transporter (GLAST) and glutamate transporter-1 (GLT1). The glutamate, which is transported into the cell by GLAST or GLT1, can be converted into glutamine by glutamine synthetase, which is a major precursor for the biosynthesis of the inhibitory neurotransmitter GABA (Sun et al., 2021; Peterson et al., 2021). Glutamate released by neurons is taken up by astrocytes and converted to glutamine to alleviate the excitotoxicity of glutamate on neurons. Glutamine serves as a source of energy for both glial cells and neurons (Nagy et al., 2018). Recent studies have shown that reducing the expression of nascent proteins and the activity of inward rectifying K+ channel subtype 4.1 (Kir4.1), respectively, leads to a decrease in the expression of GLAST and GLT1 in hippocampal astrocytes, which results in increased extracellular glutamate levels and decreased GABA release, leading to an increased risk of epileptogenesis (Sun et al., 2021; Centonze et al., 2023). Since knocking down nascent protein does not affect the expression of GABA transporter proteins in astrocytes, the reduction in GABA release may be attributed to reduced GABA synthesis (Sun et al., 2021). It has been shown that inhibiting the conversion of putrescine to spermine in astrocytes would lead to more remaining putrescine being available for the synthesis of GABA, thereby effectively reducing epileptic activity (Kovács et al., 2021).
Previous research suggests that compared to GLAST, GLT1 is the main transporter for the uptake of glutamate by astrocytes (Peterson et al., 2021). In the intrahippocampal kainic acid model of TLE, GLT1 regulates the frequency of seizures and the total time spent in seizures by mediating the uptake of most glutamate in the dorsal forebrain (Peterson et al., 2021). GLT1 mitigates ischemic stroke-induced neuroexcitotoxicity by increasing glutamate reuptake in astrocytes, thereby reducing neuronal cell death, which is an important cause of post-stroke epilepsy (Wang et al., 2022). Post-translational modifications of GLT-1, including palmitoylation, ubiquitination, nitrosylation, and succinylation, can regulate the distribution of GLT-1 and the rate at which it transports glutamate (Peterson et al., 2021). Although it has not yet been confirmed that palmitoylation can alter the activity of GLT1, based on reports linking protein palmitoylation to neuronal excitability, GLT1 lactylation may affect the excitability of neurons by regulating the concentration of glutamate between synapses (Hagihara et al., 2021).
Previous studies have shown that calcium ions mediate the release of glutamate and ATP from neurotransmitter-activated astrocytes to promote neuronal firing patterns (Shen et al., 2023; Chen et al., 2023). However, other research suggests that the majority of the neurotransmitter glutamate in the brain is synthesized and released by neuronal cells, with astrocytes playing a role in regulating the concentration of glutamate between synapses through reuptake. Until the concept of “glutamatergic astrocytes” was proposed by DeCeglia et al., the ability of astrocytes to transmit information like neurons was unknown. Glutamatergic astrocytes are located mainly in the hippocampus and express the vesicular glutamate transporter protein 1 (vGLUT1), which specifically loads glutamate into synaptic vesicles and promotes its release into the synaptic gap (de Ceglia et al., 2023). Although vGLUT1-mediated glutamate release may promote the occurrence of epilepsy, it does not prove that the glutamate transported by vGLUT1 is synthesized by astrocytes rather than acquired by astrocytes from the synaptic cleft. Glutamate can not only directly induce epilepsy by mediating the excitotoxicity of neurons but also induce epilepsy related to brain tissue damage by altering the function of inhibitory interneurons. Previous studies have confirmed that brain tissue damage can lead to an increase in synaptic inputs generated by pyramidal neurons, thereby driving an increase in the excitability of surviving hilar interneurons, which in turn leads to the occurrence of post-traumatic epilepsy (Butler et al., 2017). This may be closely related to the glutamatergic synapses formed between pyramidal cells and interneurons in the hippocampus, as a previous study has shown that pyramidal cells may regulate the transition between inhibitory interneurons and disinhibitory interneurons through the glutamatergic synapses formed with interneurons (Tzilivaki et al., 2023).
During status epilepticus, neurons, astrocytes, and microglia release ATP, activating the purinergic receptor P2Y1 on astrocytes and further exacerbating neuronal excitotoxicity. Meanwhile, ATP released from damaged neurons can activate NLRP3 inflammatory vesicles by binding to the ATP-gated ion channel P2X7 in microglia, mediating inflammatory injury in brain tissue (Liu et al., 2022). Glial cells and neurons can sustainably increase neuronal excitotoxicity by releasing excitatory neurotransmitters. However, adenosine kinase synthesized by astrocytes converts ATP to adenosine, alleviating neuronal excitotoxicity by activating presynaptic A1 adenosine receptors, thereby blocking this self-sustaining cycle of neuronal activity (Shen et al., 2023; Boison and Steinhäuser, 2018). Meanwhile, as brain-resident immune cells, microglia catalyze the conversion of ATP to adenosine by expressing CD39, an extracellular ATP/ADP hydrolase encoded by Entpd1, which can reduce seizures (Hu et al., 2023). Although activated microglia may promote the occurrence of epilepsy by releasing glutamate and inflammatory cytokines (such as interleukin-1β, interleukin-6, and tumor necrosis factor-α), inhibiting glutamate uptake, and reducing GABAergic transmission (Shen et al., 2023), the above-mentioned astrocytes and microglia can play a dual role in promoting and inhibiting epilepsy, which may depend on different stages of epileptogenesis.
Previous studies have shown that oligodendrocytes, like astrocytes and microglia, regulate the release and reuptake of neurotransmitters between neuronal synapses, thereby mediating epileptogenesis and development. Oligodendrocytes have been shown to be involved in the regulation of glutamate levels in the brain (Shen et al., 2023).
2.2 Lactate interference with glial cell function
Glucose is the main energy source for the brain, and interfering with the glucose metabolic process may affect brain function. Lactate produced by glycolysis may mediate glial cell activation and neuronal functional impairment by promoting histone acetylation (Pan et al., 2022; Wei et al., 2023). The increase in histone H3K18 acetylation levels in the hippocampus can directly activate the nuclear factor kappa-B (NF-κB) signaling pathway by stimulating the promoters of Rat transcription factor (Rela) and NF-κB, which can enhance the production and release of pro-inflammatory factors and exacerbate the inflammatory damage associated with epilepsy (Wei et al., 2023; Cai and Lin, 2022). This pathological change may be amplified by a positive feedback loop between glycolysis/H4K12la/ pyruvate kinase isozyme type M2 (PKM2). As a key enzyme in the glycolytic pathway, PKM2 can ensure that the cell’s energy metabolism switches from oxidative phosphorylation to glycolysis. The H3K18la mainly mediates the pro-inflammatory activation of microglia through the NF-κB signaling pathway. Microglia activation can exacerbate neuronal functional damage by promoting the formation of NLRP3 inflammasomes and Apoptosis-Associated Speck-Like Protein Containing a Caspase Recruitment Domain (ASC) specks (Pan et al., 2022).
Microglia cells have significantly higher levels of H4K12la at the promoters of hypoxia-inducible factor-1α (HIF-1α), PKM2, and lactate dehydrogenase (LDH), which leads to increased expression of these glycolysis-related proteins and further upregulates H3K18 and H4K12 acetylation (Pan et al., 2022; Wei et al., 2023). While there was no significant difference in H4K12la between the transgenic AD model mice and wild-type mice in the affected areas of their brains, the research suggests that the metabolism of astrocytes and neurons, like microglia, is regulated by lactate (Pan et al., 2022; Barros et al., 2023). Previous studies have suggested that cognitive impairment may be linked to the expression levels and activities of key enzymes in glycolysis, such as LDH, as well as glucose transporter protein 1 (GLUT1) and GLUT3, which are the primary cellular channels for glucose uptake in neurons and astrocytes, respectively (Yang et al., 2024; Wu et al., 2023). It is, therefore, possible that the glucose uptake of astrocytes and neurons may affect brain function by regulating lactate production. Based on the evidence of crosstalk between microglia, astrocytes, and neurons, the functions of starry glial and neuronal cells may also be influenced by protein acetylation, as in microglia (Mayorga-Weber et al., 2022; Mason, 2017). The lactate/H4K12la/PKM2 feedback loop in microglia might continuously promote the acetylation of histones in neurons and astrocytes by providing lactate. Furthermore, as GLUT1 on astrocytes transports more glucose into the cell, aerobic glycolysis gradually replaces mitochondrial oxidative phosphorylation as these cells’ main glucose metabolic pathway. The glucose taken up by neurons via GLUT3 not only participates in oxidative energy supply but also is metabolized by the pentose phosphate pathway to eventually produce glutathione to maintain redox balance (Mayorga-Weber et al., 2022) (Figure 1).
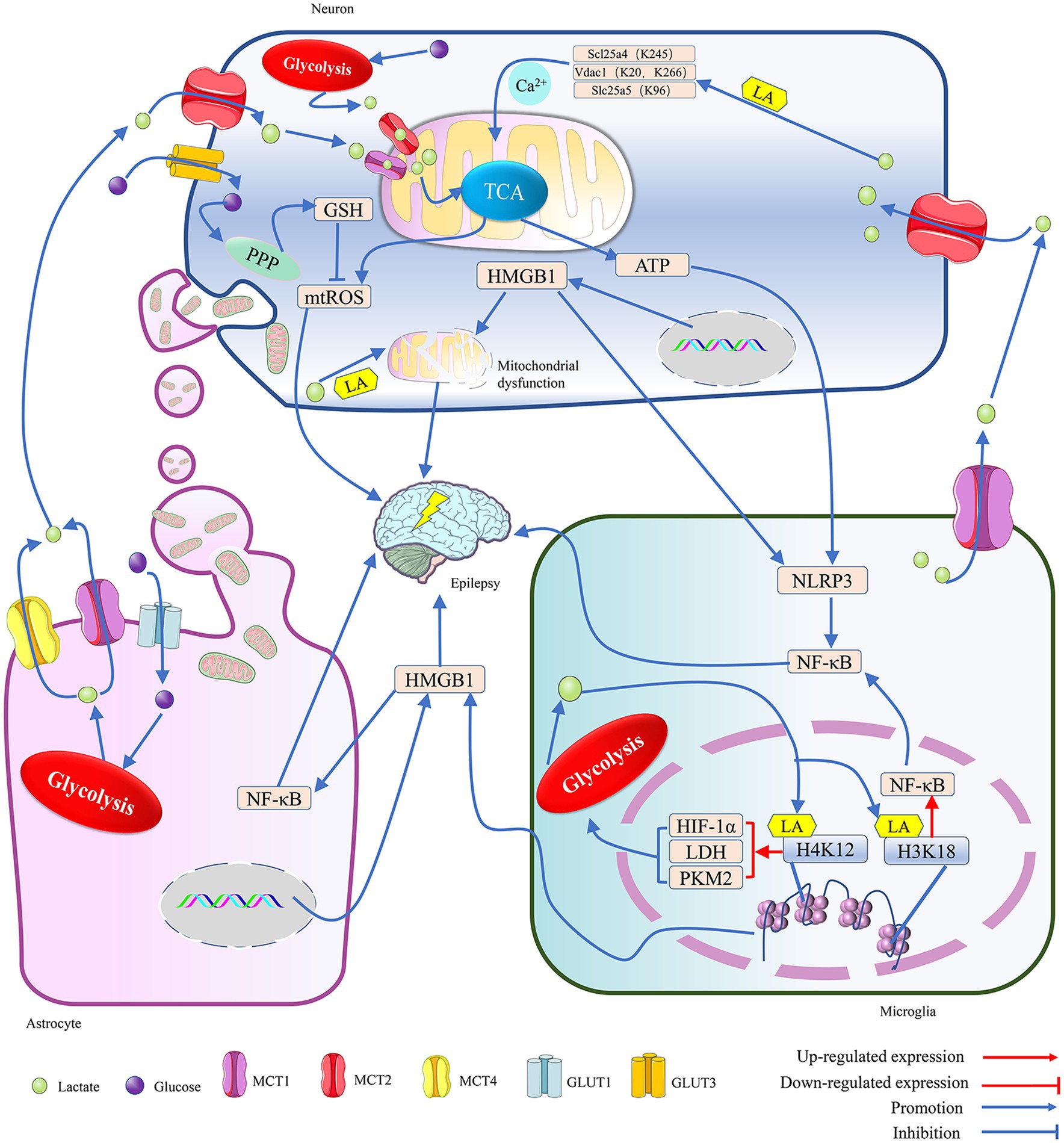
Figure 1. The metabolic crosstalk between neurons and glia involving lactate is implicated in epilepsy. 1. Both glucose and lactate can serve as energy substrates for neurons. Glucose is primarily taken up by neurons via GLUT3, while lactate, mainly provided by astrocytes and microglia, is mostly taken up by neurons via MCT2 during situations of intense stimulation of neural function. In a hypoxic environment, within neurons, the lactate generated from glucose through glycolysis and the lactate transferred into the cell by MCT2 are spread on the mitochondrial membrane by MCT1 and MCT2. There, they penetrate the TCA cycle, and the TCA contributes to the production of a significant amount of ATP and ROS through the promotion of oxidative phosphorylation. The ATP not only supports neuronal excitation but also activates the NLRP3/NF-κB pathway in microglia, leading to epilepsy. The mtROS may cause epilepsy by inducing oxidative stress in neurons, but PPP can reduce it by synthesizing glutathione, an antioxidant. Additionally, lactate can lead to epilepsy by enhancing the lactylation levels of proteins related to mitochondrial function, such as inducing Ca2+ overload-mediated mitochondrial apoptosis by promoting lactylation of key proteins of the Ca2+ signaling pathway, Scl25a4 (K245), Slc25a5 (K96) and Vdac1 (K20, K266). The transfer of HMGB1 from the nucleus to the cytoplasm can help coordinate homophilin lactylation, leading to epileptogenesis by mediating neuronal mitochondrial dysfunction. However, astrocytes maintain normal neuronal physiological activity and reduce epileptogenesis by providing healthy mitochondria to neurons. 2. In astrocytes, glucose taken up by the GLUT1 channel is converted into lactate, which is primarily exported by the MCT1/4 and used as an energy source by neurons. Both microglia and astrocytes can secrete HMGB1, which is a damage-associated molecular pattern and continues to induce the activation of glial cells and the inflammatory damage of neurons, leading to the occurrence of epilepsy. Meanwhile, HMGB1 secreted by microglia can mediate epileptogenesis through activation of the NF-κB pathway in astrocytes. 3. In a hypoxic environment, the lactate generated by glycolysis in microglia not only supports neuronal energy metabolism but also facilitates the lactylation of histones H3K18 and H4K12, which enhances the expression of NF-κB and glycolysis-related genes. NF-κB can trigger the onset of epilepsy, and the enzymes, such as LDH, HIF-1α, and PKM2, can further promote the production of lactate, which then helps in the lactylation of histones, ultimately intensifying the symptoms of epilepsy. Adenosine triphosphate (ATP); glucose transporter 1/3 (GLUT1/3); glutathione (GSH); high-mobility group box 1 (HMGB1); hypoxia-inducible factor 1 alpha (HIF-1α); K (Lysine); monocarboxylate transporter (MCT); mitochondria reactive oxygen species (mtROS); NLR family pyrin domain containing 3 (NLRP3); nuclear factor kappa-B (NF-κB); pentose phosphate pathway (PPP); pyruvate kinase isozymes M2 (PKM2); and tricarboxylic acid cycle (TCA).
Cerebral ischemia can induce the proliferation of astrocytes and stimulate their glycolytic metabolism (Lv et al., 2015), leading to the release of lactate, which can then enter neuron cells through monocarboxylic acid transporter (MCT) to cause an increase in neuronal activity (Barros et al., 2023; Yang et al., 2024; Wu et al., 2023). With the release of lactate from astrocytes, the negative feedback on glycolysis gradually weakens, allowing astrocytes to continuously produce lactate for energy transport to the neuron cells (Barros et al., 2023). Additionally, it has been demonstrated that neurons can uptake lactate released by microglia via MCT2 (Mayorga-Weber et al., 2022). Consequently, microglia and astrocytes may maintain neuron energy metabolism by continuously supplying lactate. This lactate may affect the physiological functions of neurons by promoting histone lactylation in hippocampal tissue (Nagy et al., 2018; Wei et al., 2023) (Figure 1).
MCT1-4 show cell-type-specific distribution. MCT1/4 is primarily located on the membranes of astrocytes, with MCT4 being unique to astrocytes. MCT2 is expressed exclusively by neurons, and microglia mainly rely on MCT1 for lactate transport (Yang et al., 2024; Jády et al., 2016). Lactate/pyruvate is transported between cells via MCTs. The imported pyruvate may enter the mitochondria and preferentially serve as a substrate for the pyruvate dehydrogenase system (PDHc) to provide energy for the cells, while the imported lactate is first oxidized by LDH to produce NADH, which then follows a specific shuttle mechanism to enter the mitochondria. When the shuttling of NADH is impeded, it may prevent the oxidation of lactate (Nagy et al., 2018) as a product of glycolysis; lactate/pyruvate can be used by the mitochondria for oxidative phosphorylation. Astrocytes can release lactate through MCT1/4, which may then enter neuronal cells via MCT2. Finally, the lactate enters the mitochondria through the MCT1/2 channels in the neuron’s cytoplasm (Yang et al., 2024; Wu et al., 2023; Yang et al., 2022), promoting mitochondrial energy metabolism and reactive oxygen species (ROS) production. The mitochondria ROS (mtROS) can induce dysfunction of neuronal mitochondria and damage the structure and function of synaptic elements (Jia et al., 2021). Moreover, the uptake of dysfunctional mitochondria released from glial cells by neurons can also lead to abnormalities in neural discharge (Zhang et al., 2023). Thus, lactate metabolism and mitochondrial function in glial cells can affect neuronal discharge frequency. It has been shown that an increase in the number of mitochondria and a disturbance in their dynamics in the hippocampus can lead to excessive excitation of hippocampal neurons and prolonged epileptic duration in mice (Bebensee et al., 2017; Lee et al., 2022) (Figure 1).
Activated microglia can cause neuronal damage by releasing dysfunctional mitochondria, whereas astrocytes can support neuronal survival by transferring functional mitochondria to them (Jia et al., 2023). Astrocytes might regulate neuronal activity by providing “powerhouses” (mitochondria) and the energy substrate lactate, thereby affecting anxiety-like behavior and cognitive deficits caused by seizures (Wu et al., 2023; Jia et al., 2023). Neurons can take up glucose through GLUT3 and break it down via glycolysis to generate energy, but their reliance on astrocytes for energy metabolism may help them avoid apoptosis induced by high glycolytic rates (Wu et al., 2023). Thus, the regulation of neuronal activity by astrocytes is closely related to mitochondrial function and lactate metabolism. Mitochondria have been shown to transfer between neurons and glial cells, and exogenous mitochondria can cross the blood–brain barrier to reduce ROS release induced by epilepsy, thereby reducing hippocampal neuron loss and glial activation (Jia et al., 2023). Numerous studies have confirmed that mitochondrial dysfunction can lead to increased lactate production through enhanced anaerobic glycolysis (Barros et al., 2023; Yang et al., 2024; Wu et al., 2023), and the lactate may mediate the occurrence and development of epilepsy by promoting protein lactylation modifications (Figure 1).
2.3 The impact of lactate metabolism on neuronal cell death
Previous studies have suggested that neuronal loss associated with programmed death plays an important role in the development of epilepsy (Liang et al., 2023).
2.3.1 NETosis
Neuroinflammation is one of the main causes of neuron loss (Li et al., 2021). Neuroinflammation is characterized not only by the proliferation of glial cells but also by disruption of the blood–brain barrier and migration of peripheral immune cells into the brain. Neutrophils infiltrate damaged brain tissue immediately after ischemic injury, and they can sustain neuronal damage around the infarct by releasing neutrophil extracellular trapping networks (NETs) (Li et al., 2021; Bernis et al., 2023; Byun et al., 2023). NETosis is a form of suicidal death for neutrophils, characterized by the formation and release of NETs, which primarily consist of double-stranded DNA, histones, and granule proteins (Li et al., 2021). During chronic inflammatory damage in the central nervous system, inhibiting the activity of key enzymes in the glycolytic pathway can block the activation of NETosis by reducing lactate levels (Awasthi et al., 2019; Ye et al., 2022; Wang et al., 2018). NETosis may exacerbate neuroinflammation by releasing inflammatory factors to induce neuronal loss in brain tissues after ischemic infarction. The hypoxic environment created by ischemic stroke can induce the occurrence of glycolysis, leading to the production of large amounts of lactate. The lactate may trigger NETosis and contribute to the development of post-stroke epilepsy by enhancing the lactylation of histones in neutrophils that infiltrate the ischemic focus.
2.3.2 Cuproptosis
Recent research has indicated that neuronal cuproptosis may be an important factor in the initiation and progression of TLE (Yang et al., 2023). Cuproptosis is a mitochondrial proteotoxic stress-dependent mode of regulated cell death (RCD), where the accumulation of copper in cells is crucial (Chen et al., 2023). Ferredoxin 1 (FDX1), a key protein in cuproptosis, can trigger mitochondrial protein lipidization, leading to mitochondrial protein toxicity stress. This form of stress is distinct from copper-induced mitochondrial oxidative stress and the iron-induced oxidative cell death associated with ferroptosis. As mitochondria are the primary targets in cuproptosis, the process includes the disintegration of the mitochondrial membrane and the functional loss of enzymes critical to the tricarboxylic acid cycle (Liu et al., 2022). When oxidative phosphorylation is inhibited, glycolysis is activated as the main energy source for cells (Barros et al., 2023). It seems that the occurrence of cuproptosis may help enhance the level of lactate. In turn, when aerobic glycolysis replaces the aerobic oxidation of sugars as the main energy source for cells, cuproptosis is likely to be suppressed (Xiong et al., 2023). It has been demonstrated that overexpression of FDX1 induces cuproptosis in tumor cells by catalyzing the lipoylation of PDH and α-ketoglutarate dehydrogenase (Schulz et al., 2023). However, HIF-1α can block FDX1-induced cuproptosis by indirectly inhibiting PDH (Chen et al., 2023). PDH promotes the entry of pyruvate into mitochondria and initiates the tricarboxylic acid cycle. Inhibition of PDH activity leads to mitochondrial energy depletion, which activates AMPK to promote cuproptosis (Xue et al., 2023). Copper overload may induce cuproptosis by mediating mitochondrial respiratory chain damage by over-activating the energy sensor AMPK. Cuproptosis can promote the release of the pro-inflammatory mediator High-Mobility Group Box 1 (HMGB1) (Liu et al., 2022; Xue et al., 2023) (Figure 2). It appears that cuproptosis-induced mitochondrial dysfunction may inhibit the exacerbation of cuproptosis by promoting the production of lactate, which has been shown to activate HIF-1α. HIF-1α may antagonize the effects of FDX1-mediated lipid acylation of PDH by inhibiting the activity of PDH to inhibit cuproptosis (Jiang et al., 2021).
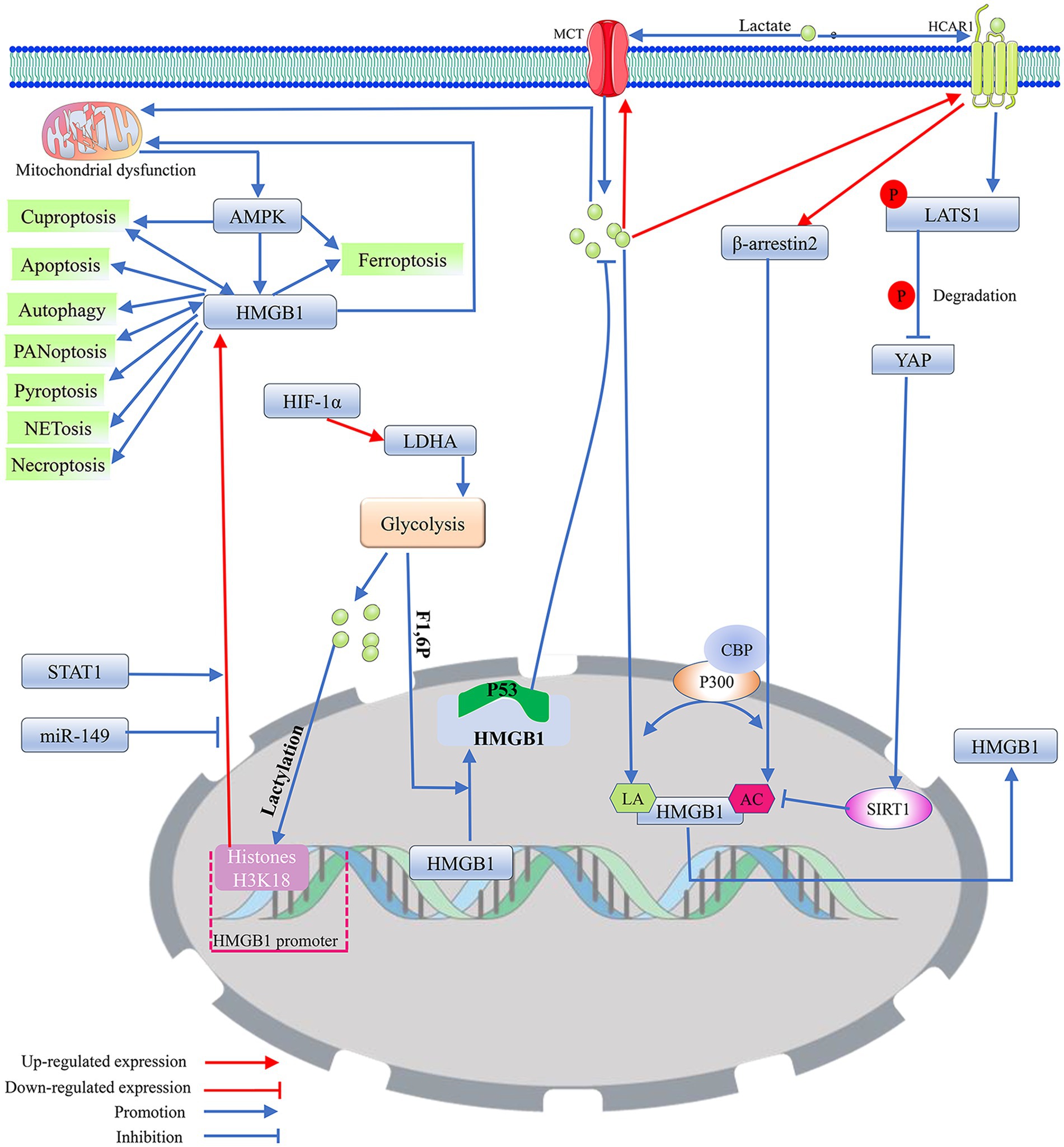
Figure 2. The potential regulatory mechanism of lactate on the biological activity of HMGB1. 1. As a DNA-binding protein, HMGB1 is usually located within the cell nucleus, but when it separates from the DNA strand and migrates out of the cell nucleus, its properties that enhance inflammation and immune responses are activated. Mitochondrial dysfunction promotes the nuclear translocation of HMGB1 through the activation of AMPK, thereby inducing HMGB1 mediated programmed cell death. Among them, cuproptosis and PANoptosis can further promote the nuclear translocation of HMGB1. In addition, miR-149 and STAT1 can affect HMGB1-mediated programmed cell death and mitochondrial dysfunction by regulating the expression level of HMGB1. 2. Exogenous lactate can upregulate β-arresin2 expression by binding to HCAR1, thereby promoting P300/CBP recruitment to the nucleus. P300/CBP promotes HMGB1 secretion and activation by catalyzing HMGB1 lactylation/acetylation. Meanwhile, the binding of exogenous lactate to HCAR1 can upregulate the level of LATS1 phosphorylation, which in turn promotes the phosphorylation and degradation of YAP. The degradation of YAP could promote the acetylation of HMGB1 by reducing SIRT1 activity. In addition, exogenous lactate can be taken up by cells via MCT, thereby mediating mitochondrial dysfunction by promoting protein lactylation, and mitochondrial dysfunction can promote HMGB1 activation. Elevated intracellular lactate levels upregulate the expression of MCT1 and HCAR1. 3. Transcription factor HIF-1α can promote glycolysis by enhancing the expression of LDHA. The lactate produced by glycolysis can not only directly promote HMGB1 lactylation but also enhance the expression of HMGB1 by upregulating the lactylation level of histone H3K18 in its promoter region. F1,6P, as an intermediate product of glycolysis, can bind to the K43/K44 sites of the HMGB1 protein to promote the separation of HMGB1 from DNA in the cell nucleus. The free form of HMGB1 can block P53 degradation by binding to the oncogenic factor P53, thereby reducing the production of lactate. Acetylation (AC); adenosine 5′-monophosphate-activated protein kinase (AMPK); CREB-binding protein (CBP); fructose-1,6-bisphosphate (F1,6P); hypoxia-inducible factor 1 (HIF-1); high-mobility group box 1 (HMGB1); hydroxy-carboxylic acid receptor 1 (HCAR1); lactylation (LA); large tumor suppressor kinase 1 (LATS1); lactate dehydrogenase A (LDHA); monocarboxylate transporter (MCT); signal transducer and activator of transcription 1 (STAT1); silencing regulatory protein 1 (SIRT1); and yes-associated protein (YAP).
2.3.3 Ferroptosis and disulfidptosis
Mitochondria, as the center of cellular energy metabolism, are not only the main target of cuproptosis but also the main site of iron utilization. The accumulation of excessive iron (Fe2+) in mitochondria, which is a key factor in triggering ferroptosis, can lead to mitochondrial functional impairment, which exacerbates oxidative stress-induced cellular damage through the release of ROS, destructive lipids, mitochondrial DNA, and proteins (Moos et al., 2023). Ferroptosis is a form of programmed cell death caused by lipid peroxidation associated with energy metabolism (Zhao et al., 2020). The onset and progression of ferroptosis may involve disruption of mitochondrial function, which often induces the Warburg effect (Yang et al., 2023). Elevated lactate concentration in cells may regulate iron ion metabolism by promoting lactylation modification of proteins, thereby promoting cellular ferroptosis (Zhang et al., 2023). Nevertheless, exogenous lactate can assist cells in replenishing their energy resources by activating HCAR1 or MCT1, increasing ATP production. With the enhancement of ATP levels, the potency of AMPK is gradually reduced, resulting in the inhibition of ferroptosis or cuproptosis (Xue et al., 2023) (Figure 2). Mitochondrial dysfunction is not the sole factor inducing the Warburg effect in neurons. In HT22 cells, the activation of small-conductance calcium-activated K+ (SK) channels may initiate aerobic glycolysis as the main energy source for the cell and reduce the generation of ROS during the process of mitochondrial energy metabolism to inhibit ROS-induced cellular oxidative stress, thereby preventing the cell from entering ferroptosis (Krabbendam et al., 2020).
With the continuous deepening of research on iron metabolism and ferroptosis in recent years, inhibiting ferroptosis in neuronal cells is considered a potential new strategy for treating epilepsy (Moos et al., 2023; Chen et al., 2020). It has been shown that the neuronal excitotoxicity inducer glutamate can promote ferroptosis by releasing mitochondrial ROS (Krabbendam et al., 2020). Cerebral infarction-induced cerebral ischemia and hypoxia promote the onset of glycolysis and lactate production in neuronal cells by upregulating the expression of LDHA (Yao and Li, 2023). Elevated lactate levels promote lactylation modification of lymphocyte cytoplasmic protein 1 in neuronal cells of Middle cerebral artery occlusion (MCAO) rats, thereby exacerbating cerebral infarction-induced neuronal loss, which is an important cause of post-stroke epilepsy (Zhang et al., 2023).
The neuronal solute carrier family 7 member 11 (SLC7A11)/GPX4 pathway may play a role in reducing the occurrence of epilepsy by inhibiting ferroptosis (Liang et al., 2023; Yuan et al., 2021). SLC7A11, a transport protein that facilitates the uptake of cystine and the efflux of glutamate, is instrumental in activating GPX4. This activation occurs through its role in cystine uptake, essential for the synthesis of glutathione, a key antioxidant that GPX4 uses to prevent lipid peroxidation and, consequently, ferroptosis in neuronal cells (Yang et al., 2023). However, the accumulation of disulfides, such as cystine, in the cell causes disulfide bond stress, which mediates the formation of disulfide bonds between actin cytoskeletal proteins and the breakdown of the actin filament (F-actin) network, ultimately leading to disulfidptosis (Liu et al., 2023). Therefore, the intake of cystine mediated by SLC7A11 can suppress neuronal ferroptosis while also triggering neuronal disulfidptosis. The efficiency of SLC7A11 in transporting cystine is affected by the level of glutamate in the cell. Although HIF-1α promotes the uptake of cysteine mediated by SLC7A11 by upregulating the expression of SLC1A1, lactate may not be involved in the resistance to ferroptosis driven by the glutamate metabolism of SLC7A11 (Yang et al., 2023).
2.3.4 Apoptosis
Recent research has shown that neuronal autophagy and apoptosis related to epilepsy are mediated by the mTOR signaling pathway (Liu et al., 2022), and these two forms of programmed cell death may exist in a complex interplay in the pathological changes leading to neuronal loss (Arab et al., 2023). Caspase-3 is one of the key effector molecules in executing cellular apoptosis. Activated caspase-3 and DNA-binding protein HMGB1 can rapidly transfer to mitochondria and degrade mitochondrial proteins, thereby mediating the loss of hippocampal CA1 and GABAergic interneurons to maintain sustained epilepsy (Kim et al., 2021). It is evident that HMGB1, like caspase-3, can mediate neuronal apoptosis-associated epilepsy by affecting mitochondrial structure and function. Recent studies have suggested that HMGB1 activation is regulated by lactylation modifications and that this process is closely linked to cellular lactate production (Yao and Li, 2023). Lactate promotes mitochondrial hyperfission by mediating an increase in lactylation of the mitochondrial Fission 1 protein (Fis1) lysine 20 (Fis1 K20la), which can contribute to mitochondrial hyperfission by inducing ATP depletion, mitochondrial ROS overproduction, and mitochondrial damage-mediated apoptosis. Conversely, activation of PDH downregulates lactylation of Fis1 K20 by decreasing the level of lactate produced by glycolysis, thereby alleviating the apoptosis-inducing effect of mitochondrial damage (An et al., 2023). Meanwhile, inhibiting H3 histone lactylation at H3K9 and H3K14 sites can induce apoptosis (Xu et al., 2023). It follows that lactate may mediate mitochondrial function-related apoptosis by regulating lactylation modifications of histones and non-histone proteins.
2.3.5 Pyroptosis
Pyroptosis, as an inflammatory mode of programmed cell death, can mediate the expression and release of caspase/Gasdermin D (GSDMD)-associated inflammatory factors through the activation of inflammasomes (Xia et al., 2021). The Nucleotide-binding oligomerization domain-like receptor family card domain-containing protein (NLRP3) inflammasome is an important activator of pyroptosis related to epilepsy, and it has been demonstrated that STAT3 can promote the expression of NLRP3 by catalyzing the acetylation of histone H3K9 on the NLRP3 promoter, thereby activating the NLRP3/caspase-1 pathway and exacerbating the damage to neurons in epileptic mice (Jiang et al., 2021). Epilepsy-related neuronal pyroptosis is initiated by the accumulation of ROS caused by mitochondrial impairment in neurons (Xu et al., 2023). Mitochondrial dysfunction often forces cells to obtain energy through glycolysis. A study on brain ischemia found that lactate upregulates HMGB1 expression by promoting histone lactylation of the HMGB1 promoter, which may lead to neuronal pyroptosis (Yao and Li, 2023). It is closely related to the activation of the NLRP3 inflammasome by HMGB1 (Liu et al., 2022). Thus, lactate may affect the occurrence of epilepsy by regulating neuronal apoptosis.
2.3.6 Autophagy
Autophagy is a process by which cells degrade their own excess or aging organelles and misfolded proteins (Yang et al., 2020). Lactate and pyruvate can activate mitochondrial autophagy and autophagy in primary neurons and astrocytes by lowering intracellular hydrogen ion concentration at non-toxic concentrations, which facilitates the restoration of mitochondrial function for the protection of these cells from apoptosis and necrosis (Fedotova et al., 2021). It has been shown that a lack of neuronal autophagy contributes to epilepsy and that seizures may be further exacerbated by triggering autophagy dysfunction, thus creating a vicious cycle (Ali et al., 2023). Lactate not only protects neurons by regulating mitochondrial autophagy within the cells but also maintains neuronal activity by regulating mitochondrial autophagy in glial cells (Zhu et al., 2022). Moreover, lactate may further promote cytoplasmic acidification by inducing a shift in cellular energy metabolism from oxidative phosphorylation to glycolysis, thereby sustaining mitochondrial autophagy (Komilova et al., 2021). With the exacerbation of neuronal autophagy, the autophagy articulation protein Sequestosome 1 is heavily depleted, which can inhibit the activation of the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)/mTOR pathway. Decreased mTOR activity impedes the downregulation of the transcription factor Hif-1α, resulting in increased expression of key glycolytic enzymes LDHA and hexokinase-2 (HK2) (Song et al., 2021). In addition, SIRT1, located in the nucleus and cytoplasm, directly or indirectly promotes the formation of autophagic vesicles and improves mitochondrial function, thus blocking epileptogenesis and development (Ali et al., 2023). Because both SIRT1 and lactate can mediate autophagy in neurons to control epileptogenesis, it is likely that there is an association between SIRT1 and lactate metabolism. A recent study has confirmed that lactate can improve long-term cognitive impairment in newborns repeatedly exposed to sevoflurane by activating SIRT1-mediated hippocampal neurogenesis and synaptic remodeling (Qiu et al., 2023).
2.3.7 Necroptosis
Necroptosis, which is a novel pattern of cell death associated with inflammation, is considered one of the complex mechanisms of neuronal death after status epilepticus (Roh et al., 2023). The accumulation of ROS due to mitochondrial damage can promote necroptosis of neurons in the hippocampus of acute epilepsy patients through activation of the RIPK1/RIPK3/MLKL pathway, and mitochondrial enzyme SIRT3 and the indole-derived small molecule NecroX-7 can alleviate seizures by suppressing the production of mitochondrial ROS (Roh et al., 2023; Song et al., 2021). This shows that mitochondria are important organelles that mediate epilepsy-related neuronal necroptosis. The glycolytic product lactate may regulate the activity of HIF-1α to block the release of mitochondrial ROS mediated by PDH, thereby regulating the necroptosis induced by RIPK (Chen et al., 2023; Jiang et al., 2021; Icard et al., 2023; Wong Fok Lung et al., 2020; Weindel et al., 2022; Luo et al., 2022). Furthermore, necroptosis is activated in hippocampal astrocytes and microglia to mediate neuroinflammation 4 h after status epilepticus induction (Wu et al., 2021).
2.3.8 PANoptosis
PANoptosis, which is an inflammatory programmed cell death driven by PANoptosome, has key features of pyroptosis, ferroptosis, apoptosis, and necroptosis (Lin et al., 2022; González-Rodríguez and Fernández-López, 2023). PANoptosis may be present in brain tissue I/R and is a new way of causing neuronal loss (González-Rodríguez and Fernández-López, 2023). Mitochondrial dysfunction likely affects PANoptosis of neurons, as it does most programmed forms of cell death (She et al., 2023). OGD/R induces PANoptosis of neurons by inducing mitochondrial fission and dysfunction (Zeng et al., 2023). It may be related to the hypoxia-induced enhancement of glycolytic metabolism, as several enzymes involved in glycolytic metabolism have been shown to directly regulate mitochondrial functions (Liu et al., 2023). Thus, mitochondrial oxidative stress and leakage of mitochondrial contents induced by multiple factors are important triggers of PANoptosis (Zeng et al., 2023; Liu et al., 2023; Bi et al., 2022). Numerous studies have demonstrated that the structural proteins of the PANoptosome are able to induce epilepsy-associated neuronal loss by mediating crosstalk in the neuronal programmed death pathway (Xia et al., 2023; Shi et al., 2023). PANoptosis may possess characteristics of various programmed cell deaths, including but not limited to pyroptosis, apoptosis, and necroptosis (Lin et al., 2022; González-Rodríguez and Fernández-López, 2023). Although it cannot currently be confirmed that PANoptosis includes cuproptosis, the evidence suggesting that (Fe-S) cluster damage can lead to cuproptosis, ferroptosis, and PANoptosis implies that these three programmed cell death pathways may be closely related to (Fe-S) cluster-mediated mitochondrial dysfunction (Chen et al., 2023; Lin et al., 2022). Therefore, PANoptosis, which possesses characteristics of all these programmed cell death types, may also be regulated by lactate metabolism, such as pyroptosis, apoptosis, and necroptosis.
2.4 Mitochondrial dysfunction intervention in the context of epilepsy-related neuronal programmed death
Accumulating evidence indicates that the occurrence and development of epilepsy are closely related to mitochondrial dysfunction (Moos et al., 2023). Damaged mitochondria can mediate many forms of neuronal programmed cell death by releasing mitochondrial contents, such as ROS, mitochondrial DNA, and mitochondria-associated proteins, leading to epilepsy-associated neuronal cell loss (Jia et al., 2023). Mitochondria can play an anticonvulsant and anti-neuroinflammatory role by synthesizing D-fructose 1,6-diphosphate, a glycolysis intermediate that inhibits sugar metabolism (Jia et al., 2023). It has been demonstrated that artificial mitochondrial transplantation may inhibit the loss of hippocampal neurons and the activation of glial cells by ameliorating the metabolic imbalance induced by mitochondrial dysfunction, thereby reducing hippocampal damage after seizures and ameliorating epilepsy-associated psychiatric and cognitive disorders (Jia et al., 2023). Therapeutic interventions aimed at improving mitochondrial function in the central nervous system may effectively alleviate neurological damage and cognitive impairments related to epilepsy. A recent study has demonstrated that honokiol attenuates mitochondrial dysfunction and its associated oxidative stress by regulating mitochondrial ROS homeostasis and that it may stabilize mitochondrial function through activation of mitochondrial DNA transcription mediated by the glutamate receptor N-methyl-D-aspartate receptor/AMPK/peroxisome proliferator-activated receptor γ-coactivator-1α/SIRT3 pathway (Wang et al., 2022). Overexpression of SIRT3 in neuronal cells is protective against ischemia–reperfusion injury in the mouse spinal cord (Gu et al., 2023). Meanwhile, the ketogenic diet-induced overexpression of SIRT1 can maintain the normal physiological function of neurons by promoting neuronal autophagy to eliminate protein aggregates and damaged mitochondria (Dyńka et al., 2022).
Brain injury and energy deficiency promote the transport of energy substrates, such as ketones, lactate, and acetoacetate, into cells by upregulating the number of MCT channels on the neuronal membrane, thereby ensuring normal neuronal function. Astrocytes may reduce glucose availability and frequency of epileptic seizures by producing ketones and shuttling them via MCT channels to neurons (Dyńka et al., 2022). However, some other studies suggest that transporting these energy substances and mitochondria may increase neuronal excitability, thereby triggering epilepsy. The energy substances and mitochondria transferred from glial cells to neurons may lead to neuronal excitation, an important factor in inducing epilepsy (Nagy et al., 2018; Wei et al., 2023; Jia et al., 2023). The increased synthesis of lactate caused by cerebral ischemia promotes mitochondrial apoptotic pathway-mediated neuronal death, which may induce the occurrence of post-stroke epilepsy by upregulating the lactylation levels of key proteins in the calcium signaling pathway, such as Scl25a4 (K245), Slc25a5 (K96), and Vdac1 (K20, K266) (Yao et al., 2023). However, when lactate and ketones replace glucose as the main energy source for neurons, glucose can enter the pentose phosphate pathway and produce a large amount of nicotinamide adenine dinucleotide phosphate, which can offset the oxidative effect of ROS released during the mitochondrial utilization of lactate for production (Mayorga-Weber et al., 2022). During status epilepticus, HMGB1, a non-histone DNA-binding protein, can be transferred from the cell nucleus to the mitochondria, thereby disrupting the energy metabolism balance of the mitochondria (Kim et al., 2021). HMGB1 in microglia can promote the release of inflammatory factors such as IL-1β and IL-18 by activating the NLRP3/NF-κB/MAPKs pathway, leading to the activation of the NLRP3 inflammasome in hippocampal neurons, which can subsequently lead to pyroptosis and apoptosis (Liu et al., 2022; Shen et al., 2018). The neuronal injury promotes the release of neurally mediated ATP, which reactivates the NLRP3/NF-κB/MAPKs pathway via binding to the purinergic receptor P2X ligand-gated ion channel 7 in microglia without HMGB1. HMGB1 may be a central element in the tandem of neuronal death and glial cell activation in the hippocampus, and HMGB1 can amplify the effects of neuroinflammatory injury by promoting the interaction between neuronal death and glial cell activation (Liu et al., 2022; Shen et al., 2018) (Figure 1).
2.5 Regulation of neuronal programmed death by HMGB1
2.5.1 HMGB1 and mitochondria
HMGB1 nucleus-to-cytoplasm translocation has been shown to maintain status epilepticus and drug-resistant temporal lobe epilepsy by mediating oxidative stress in brain tissue in neurons and glial cells (Pauletti et al., 2019). The function and activity of HMGB1 are determined not only by its subcellular localization but also by its oxidation–reduction status and post-translational modifications (Chen et al., 2023). Mitochondrial damage-mediated oxidative modification of HMGB1 promotes cytoplasmic accumulation and extracellular secretion of HMGB1. HMGB1 secreted into the extracellular compartment initiates the expression of NF-κB-mediated inflammatory factors (e.g., TNF-α and IL-1β) through activation of Toll-like receptor 4 (TLR4) on the cell surface, ultimately leading to inflammatory tissue damage (Zhao et al., 2023). Brain tissue ischemia induces HMGB1 translocation from the cell nucleus to the mitochondria and extracellular region in neurons and astrocytes, leading to neuronal apoptosis associated with epileptic status (Wang et al., 2020; Dai et al., 2023). During this process, the transposition of HMGB1 triggers the destruction of mitochondria, and the released contents of mitochondrial lysis further intensify HMGB1-mediated neuroinflammatory damage (Wang et al., 2020; Dai et al., 2023; Zhang et al., 2020) (Figure 2).
2.5.2 HMGB1 and various types of programmed cell death
Mitochondrial dysfunction can induce epileptogenesis by mediating multiple programmed cell deaths in neurons, and HMGB1 may intervene in epilepsy-associated multiple programmed cell deaths by modulating mitochondrial function. It has been demonstrated that HMGB1 released into the extracellular space can act as a damage-associated molecular pattern protein to mediate a variety of cellular programmed deaths (Chen et al., 2023; Tang et al., 2023). HMGB1, which is secreted extracellularly, may drive the disease process in epilepsy by inducing neuroinflammation and neuronal loss during the silent phase following the initial triggering event (Rosciszewski et al., 2019). HMGB1 transferred from the nucleus to the cytoplasm may activate the Myeloid differentiation factor 88 (MyD88)/NF-κB axis by binding to TLR4. The MyD88/NF-κB axis can induce neurological deficits by promoting neuronal apoptosis, autophagy, and the release of inflammatory factors, such as IL-1β and TNF-α (Lei et al., 2022; Fu et al., 2023; Shang et al., 2020) (Figure 3). STAT1 may upregulate the expression level of HMGB1 by binding to the HMGB1 promoter, thereby affecting the biological effects mediated by the TLR4/MyD88/NF-κB pathway (Fu et al., 2023). In the MCAO model, impaired neurons induce NETosis by releasing large amounts of HMGB1, which induces NETosis and can even be extruded as part of the NET. The extruded HMGB1 may further induce NETosis and platelet activation, which is one of the important reasons for the elevation of HMGB1, thereby exacerbating neuronal inflammatory damage (Kim and Lee, 2020). miR-149 may reduce the occurrence of epilepsy by inhibiting HMGB1-mediated neuroinflammatory injury (Kwak et al., 2023; Parsons et al., 2022; Wang et al., 2022). Cuproptosis and PANoptosis may further induce the release of inflammatory factors and programmed cell death by promoting the release of HMGB1 (Liu et al., 2022; Kwak et al., 2023). In addition, ATP depletion and calcium ions can upregulate the level of HMGB1 phosphorylation through activation of AMPK, which can promote the translocation and activation of HMGB1 (Liu et al., 2022). It cannot be excluded that HMGB1 may induce various forms of programmed death in neurons by regulating mitochondrial function (Figure 2).
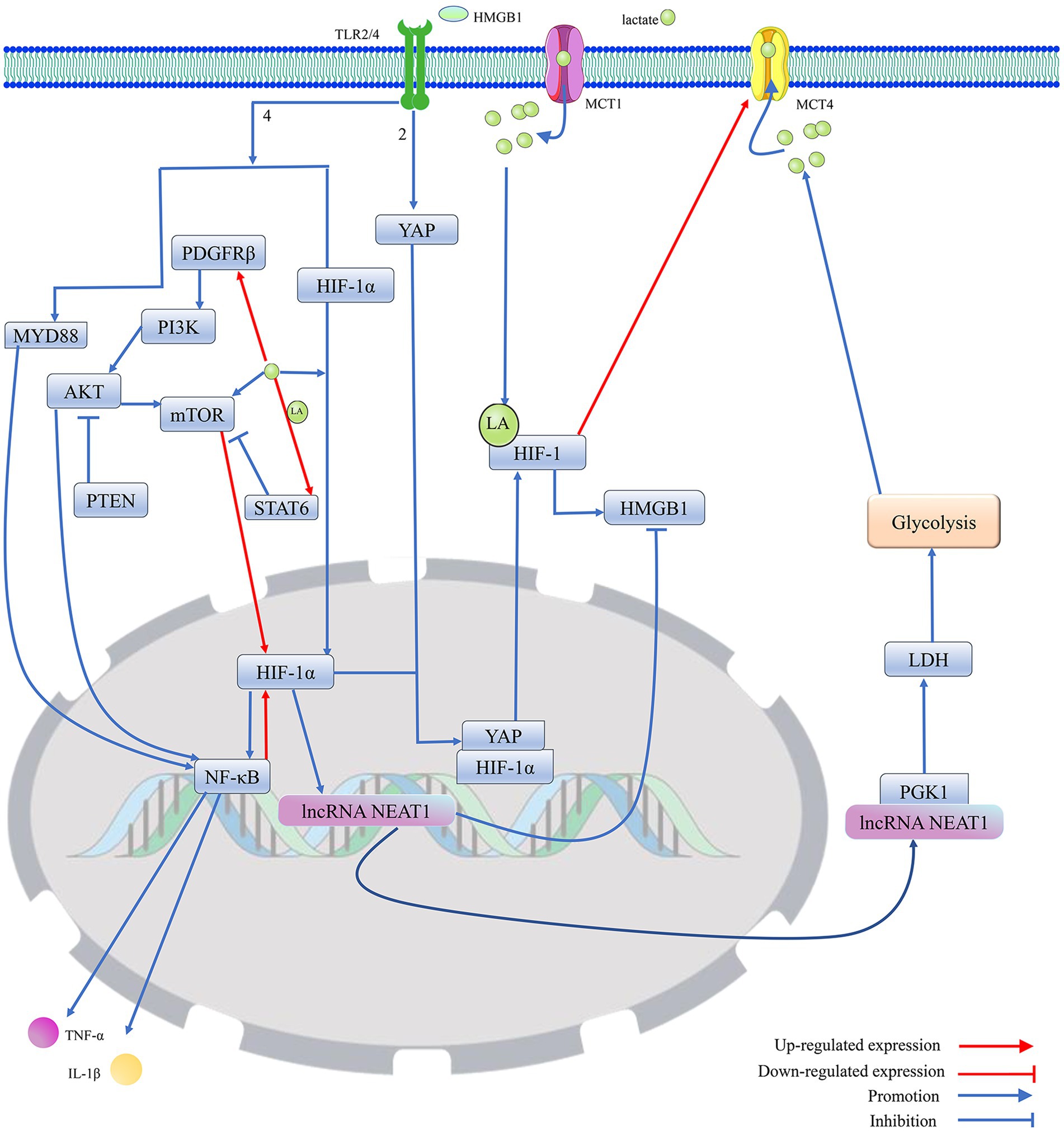
Figure 3. Protein lactonylation modification is an important influence in the process that is HIF-1-mediated HMGB1 activity. 1. Lactate, as a metabolite of cellular adaptation to hypoxic environments, is mainly produced by glycolysis. MCT1 and MCT4 are mainly responsible for the cellular uptake and release of lactate. Elevated intracellular lactate levels not only promote the translocation of HIF-1α from the cytoplasm to the nucleus to regulate gene expression but also stabilize the structure and enhance the biological activity of HIF-1 by promoting HIF-1 lactylation. Furthermore, lactate can promote the expression of HIF-1α by directly or indirectly enhancing the activation of mTOR. However, lactate can upregulate STAT6 expression by promoting histone lactylation, thereby blocking PDGFRβ/PI3K/AKT/mTOR-mediated HIF-1 expression. 2. Activated HIF-1 promotes the secretion and activation of HMGB1, which is secreted into the extracellular compartment and promotes the migration of YAP from the cytoplasm to the nucleus by binding to TLR2, thereby promoting the activation of HIF-1. Meanwhile, HMGB1, by binding to TLR4, not only directly promotes the migration of HIF-1 to the nucleus to exert transcriptional regulation but also activates the MYD88/NF-κB pathway to enhance the expression of HIF-1. However, HIF-1α can reduce the expression of HMGB1 by upregulating the transcript level of lncRNA NEAT1, thereby blocking the excessive activation of HIF-1 induced by HMGB1. The lncRNA NEAT1 can stabilize the structure of PGK1 by binding to PGK1, which mediates the glycolysis/lactate/HIF-1α pathway by increasing LDH activity, but HIF-1 can promote lactate efflux by upregulating the expression of MCT4, which blocks the excessive activation of HIF-1 induced by lactate. Furthermore, PTEN could block HIF-1 expression, which is mediated by the PDGFRβ/PI3K/AKT/NF-κB pathway, by inhibiting AKT, thereby reducing HMGB1-induced release of cellular inflammatory factors. Protein kinase B (AKT); hypoxia-inducible factor 1 (HIF-1); high-mobility group box. 1 (HMGB1); interleukin 1β (IL-1β); lactylation (LA); lactate dehydrogenase (LDH); monocarboxylate transporter (MCT); mechanistic target of rapamycin (mTOR); myeloid differentiation factor 88 (MYD88); nuclear enriched transcript 1 (NEAT1); platelet-derived growth factor receptor β (PDGFRβ); phosphoglycerate kinase 1 (PGK1); phosphatidylinositol 3-kinase (PI3K); phosphatase and tensin homolog (PTEN); signal transducer and activator of transcription 6 (STAT6); toll-like receptor (TLR); tumor necrosis factor-α (TNF-α); and yes-associated protein (YAP).
Neuroglia may be involved in HMGB1-mediated neuronal programmed cell death (Rosciszewski et al., 2019). HMGB1 induces NF-κB pathway activation in glial cells by activating signaling pathways involving TLR2, TLR4, and the receptor for advanced glycation end products (RAGE). Since activation of the NLRP3/NF-κB/MAPKs pathway in microglia is an important mechanism for HMGB1-induced hippocampal neuronal pyroptosis and apoptosis (Liu et al., 2022; Shen et al., 2018), the limited activation of NF-κB in astrocytes can lead to partial suppression of neuron loss in the absence of microglia, thereby producing a beneficial effect on controlling the occurrence of epileptic seizures (Cai and Lin, 2022; Rosciszewski et al., 2019). The damaged neurons can further promote the activation of microglia by secreting HMGB1 (Kitaoka et al., 2023). HMGB1 secreted by neurons can also directly act on astrocytes, thereby activating NF-κB p65-mediated inflammatory cascade effects (Kaya et al., 2023). It can be speculated that the intervention of glial cells may exacerbate the neuronal death effect induced by HMGB1.
2.5.3 The impact of lactate metabolism on the function of HMGB1
Research has shown that the increased protein lactylation level of hippocampal neurons is accompanied by upregulation of the HMGB1 expression level (Xie et al., 2023). Extracellular lactate can regulate cell death and immune cell polarization by binding to HCAR1 and transport protein MCT on the membrane (Zhai et al., 2022). Lactate primarily relies on the acetylase P300/CBP to introduce the lactide moiety into the lysine residue of HMGB1, thus directly participating in the lactylation of HMGB1. Meanwhile, exogenous lactate specifically increases the expression of β-arrestin2 by binding to HCAR1, which can catalyze the lactylation/acetylation of HMGB1 by facilitating the recruitment of P300/CBP to the nucleus, thereby mediating the secretion of HMGB1. Exogenous lactate promotes the phosphorylation and degradation of Yes-Associated Protein (YAP) by binding to HCAR. YAP can preserve SIRT1 expression and activation, and lactate-mediated degradation of YAP promotes HMGB1 acetylation by reducing SIRT1 activity. Lactate can induce the lactylation of HMGB1 in macrophages and promote its release from these cells (Yang et al., 2022). Fructose-1,6-bisphosphate, an intermediate product of glycolysis, serves as a novel HMGB1 ligand that promotes the segregation of HMGB1 from DNA in the nucleus by directly binding to K43/K44 on the HMGB1 protein. The free HMGB1 prefers to bind to P53 to form the HMGB1-P53 complex, which effectively prevents the degradation of P53, thus blocking glycolysis and removing damaged mitochondria (He et al., 2023; Li et al., 2023). Based on numerous reports that HMGB1 is an important gene in the regulation of glucose metabolism and mitochondria can regulate the activation of HMGB1, it is suggested that HMGB1 may be an important signaling factor related to mitochondrial dysfunction associated with protein lactylation modification-induced cell death (Li et al., 2023; Du et al., 2022; Liu et al., 2022) (Figure 2).
3 Regulating the level of protein lactylation may be a measure to intervene in the occurrence and development of epilepsy
Numerous studies have confirmed that histone acetylation is involved in regulating various cellular physiological activities (Table 1). Mitochondrial dysfunction often promotes the preferential catabolism of glucose by aerobic glycolysis to cause the accumulation of lactate, which can regulate cellular gene expression in the form of epigenetic modifications through regulating histone lactylation (He et al., 2023). Indeed, there exists a mutually promoting relationship between protein lactylation and mitochondrial dysfunction; mitochondrial dysfunction is a core element in the pathogenesis of epilepsy (Moos et al., 2023), and the disease process may be affected by protein lactylation.
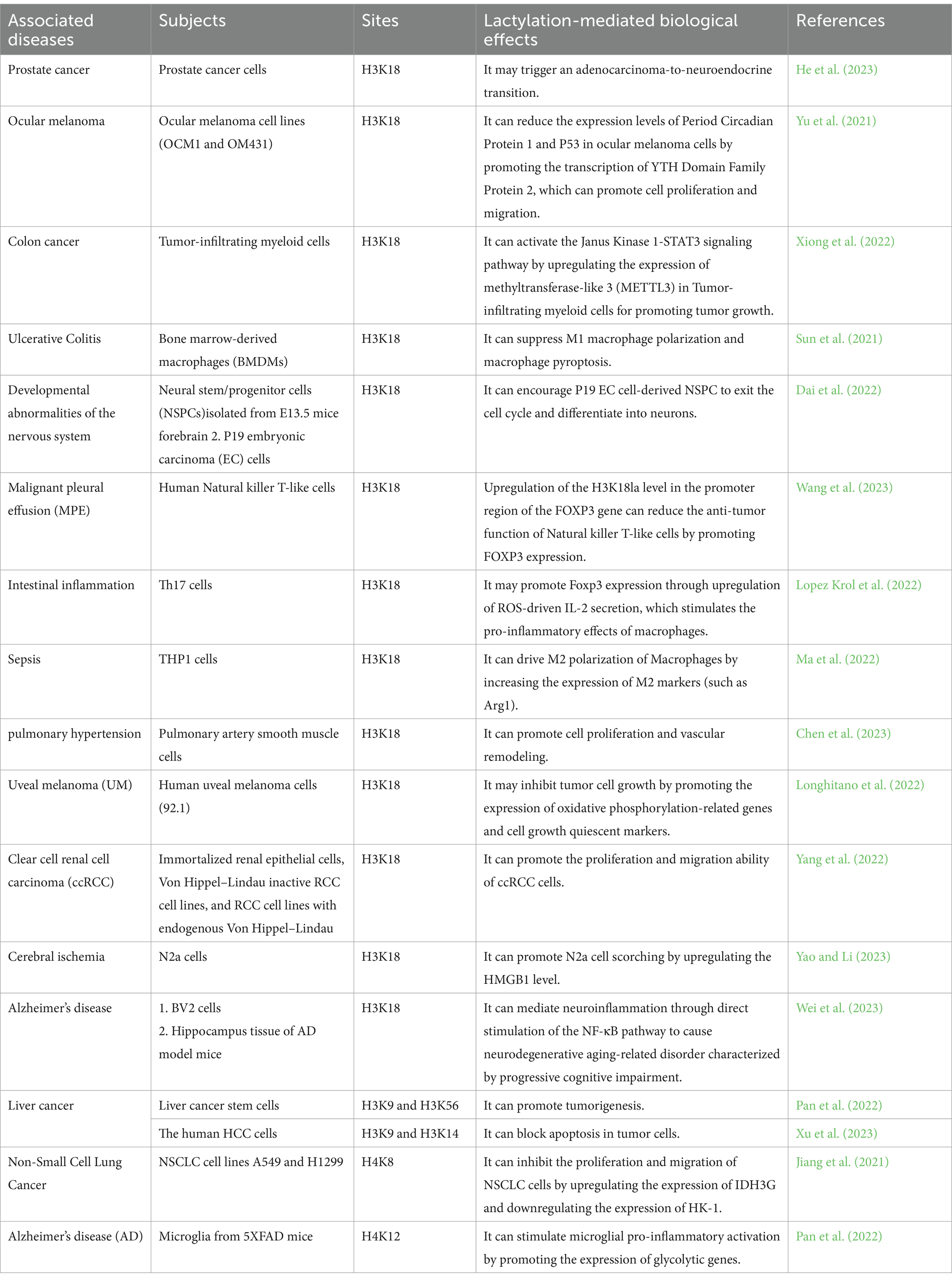
Table 1. The common sites of histone lactylation.
3.1 Mechanisms involved in influencing the level of protein lactylation
Since lactate can be catalyzed to produce lactyl coenzyme A, which has been confirmed as a direct precursor of the lactyl group, factors that affect lactate synthesis and transport may be involved in the regulation of protein lactylation modification (Yang et al., 2022; Dong et al., 2022) (Figure 4).
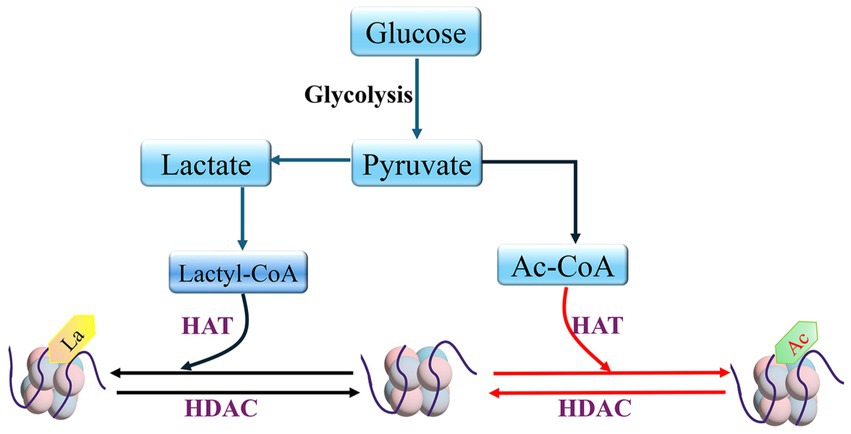
Figure 4. HDAC and HAT mediate histone lactylation and histone acetylation Lactyl coenzyme A (Lactyl-CoA); Acetyl-CoA (Ac-CoA).
3.1.1 The impact of proteins related to glucose metabolism on lactate production
Lactate is mainly derived from cellular glycolytic metabolism, and GLUT transfers glucose from the extracellular environment into the cell, thus providing the raw material for lactate production (Fan et al., 2023). HK2 catalyzes the conversion of glucose to glucose-6-phosphate, thereby driving the process of generating lactate (Rho et al., 2023). However, HK1 has been shown to be downregulated by lactate expression, thereby inhibiting the glycolytic pathway (Jiang et al., 2021). Phosphofructokinase-1 and phosphoglycerate kinase 1 (PGK1) progressively convert glucose-6-phosphoglycerate to 3-phosphoglycerate. Fructose-2,6-bisphosphatase 3 is a constitutive activator of phosphofructokinase-1, a key enzyme in glycolysis, and downregulation of fructose-2,6-bisphosphatase 3 and phosphofructokinase-1 expression in astrocytes reduces the production of lactate (Lv et al., 2015).
Knockdown of PGK1 inhibits the M1 polarization of microglia, thereby preventing the alleviation of neuroinflammation by disrupting glycolytic metabolism in the MCAO rat model (Cao et al., 2023). The enzyme 3-phosphoglycerate is transformed into pyruvate by pyruvate kinase, which has two structural isoforms: PKM1 and PKM2. PKM1 consistently functions as a high-activity tetramer, while PKM2 can exist either as a low-activity dimer or a high-activity tetramer. The low-activity dimer form of PKM2 promotes the conversion of pyruvate to lactate, playing a significant role in driving glycolysis. In contrast, the high-activity forms of both PKM2 and PKM1 facilitate the tricarboxylic acid cycle. A recent study has shown that an increase in the expression of PKM2 and a higher PKM2/PKM1 ratio promote glycolysis (Zhang et al., 2017). In the nervous system, the knockout of PKM2 can result in the compensatory overexpression of PKM1, which may promote the transformation of the energy metabolism of microglial cells from glycolysis to oxidative phosphorylation (Pan et al., 2022). PDH and LDH are key enzymes in determining the fate of pyruvate, an intermediate product of glycolysis, and PDH promotes the conversion of pyruvate to acetyl-coenzyme A, while LDH can shift the metabolism of pyruvate toward the production of lactate (Yang et al., 2022; Jia et al., 2021; Icard et al., 2023). LDH contains two subtypes, LDHA and LDHB. LDHA prefers to convert pyruvate into lactate, while LDHB tends to convert lactate back into pyruvate. However, downregulation of both LDHA and LDHB inhibits glycolysis and reduces lactate production (Yang et al., 2022). Pyruvate Dehydrogenase Phosphatase 1 (PDP1) and PDK control the rate of pyruvate transformation into acetyl-CoA by regulating the activity of PDH. PDK inhibits PDH activity by promoting the phosphorylation of serine 293 on PDH, while PDP1 normally catalyzes PDH dephosphorylation to activate PDH (Chen et al., 2023; Karagiota et al., 2023; Kantarci et al., 2020). Pyruvate transport to the mitochondria is normally blocked by the upregulation of PDK, leading to the production of lactate (Kantarci et al., 2020). In addition, lncRNA Nuclear Enriched Transcript 1 (NEAT1) can increase the activity of PGK1 and inhibit the degradation of PGK1, thereby promoting glycolysis (Liang et al., 2022) (Figures 3, 5).
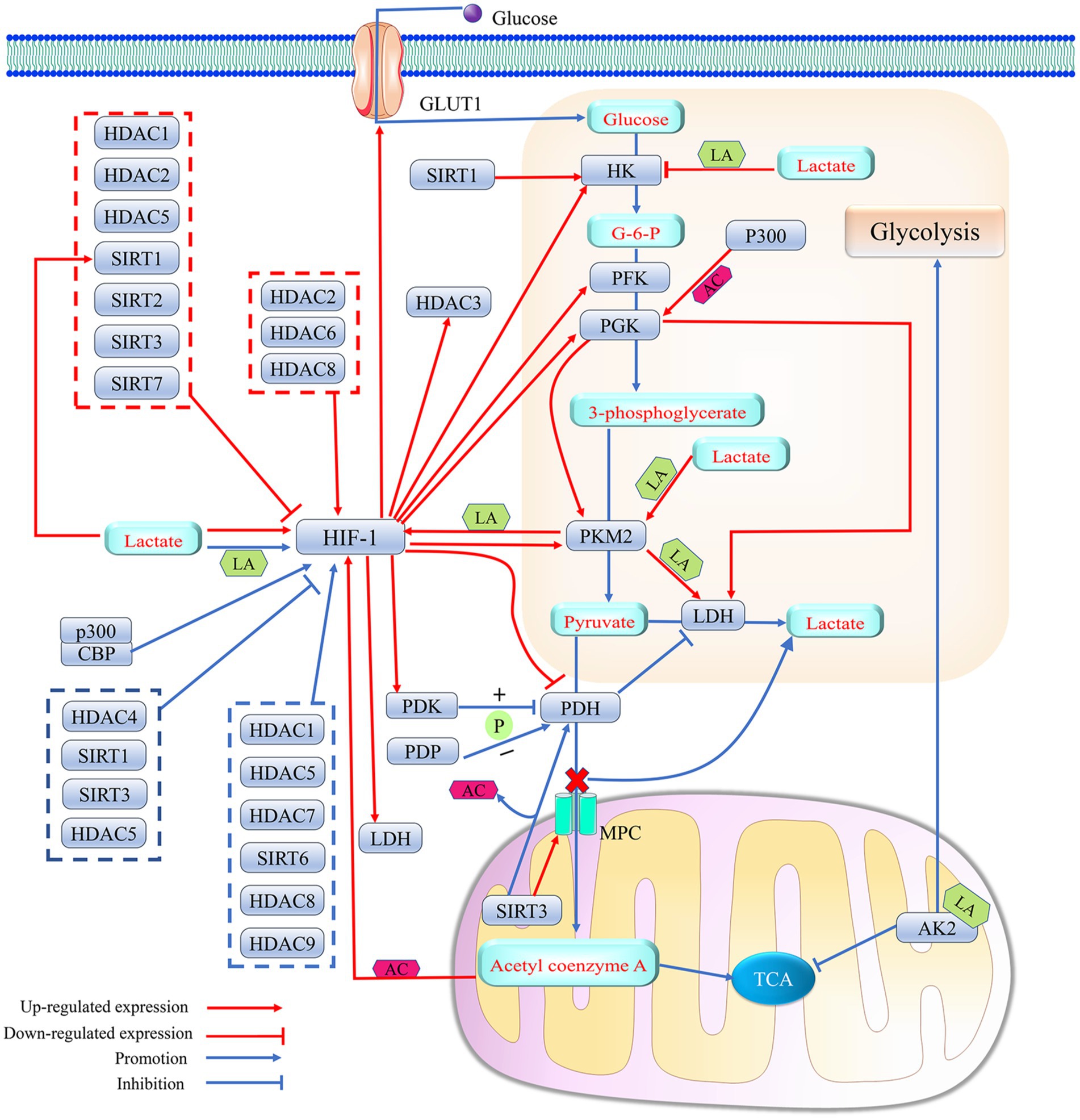
Figure 5. Histone deacetylase affects lactate metabolism by regulating HIF-1-mediated metabolic reprogramming effects. 1. HIF-1, as a transcription factor, enhances lactate production by regulating the expression levels of several key enzymes in glycolysis (HK, PGK, PFK, LDH, PDH and PKM2) and the glucose transport protein GLUT1. PDH promotes the entry of pyruvate into mitochondria via MPC and its conversion to acetyl coenzyme A, which initiates TCA by inhibiting the activity of LDH. HIF-1 catalyzes the phosphorylation of PDH by promoting the expression of PDK, thereby attenuating the activity of PDH. Elevated lactate levels in cells can promote lactylation modification of HIF-1α, which can elevate the transcription factor activity of HIF-1 to further enhance the efficiency of the glycolytic response. Histone acetylation or lactylation of the promoter of HIF-1 can promote HIF-1 expression. The level of promoter histone acetylation of HIF-1 is affected by the concentration of acetyl coenzyme A in cells, and the degree of HIF-1 lactylation can be regulated with PKM2. Meanwhile, the expression levels of both PKM2 and LDH can be upregulated by their promoter histone lactylation. A positive feedback loop consisting of lactate/PKM2/HIF-1/LDH further promotes protein lactylation. In addition, lactylation of the mitochondrial protein AK2 further promotes glycolysis and inhibits TCA. However, the positive feedback regulation of HIF-1α by lactate is not uncontrolled, and high lactate concentrations may block HK expression by upregulating histone lactylation levels in the HK promoter, thereby slowing down the onset of cellular glycolysis. Meanwhile, both PDP-mediated PDH dephosphorylation and SIRT3-catalyzed PDH deacetylation were able to produce less lactate by increasing PDH activity. 2. Histone acetylase P300 upregulates the expression level of PGK1, a key enzyme in glycolysis, by catalyzing its promoter histone acetylation. PGK1 promotes glycolysis by upregulating the expression of LDH and PKM2. Histone acetylase P300/CBP elevates the transcriptional activity of HIF-1 by promoting HIF-1 acetylation. 3. HDACs can intervene in lactate metabolism by regulating HIF-1 activity and/or expression levels. SIRT1 can even promote lactate production by directly upregulating the expression level of HK, while SIRT3 can help more pyruvate enter the mitochondria rather than converting it into lactate by promoting the expression of MPC. HIF-1 can act as a transcription factor to promote HDAC3 expression. Acetylation (AC); adenylate kinase 2 (AK2); glucose-6-phosphate (G-6-P); glucose transporter 1 (GLUT1); hexokinase (HK); histone acetylase P300; histone deacetylase (HDAC); hypoxia-inducible factor 1 (HIF-1); lactic dehydrogenase (LDH); lactylation (LA); mitochondrial pyruvate carrier (MPC); phosphorylation (P); pyruvate dehydrogenase kinase (PDK); pyruvate dehydrogenase (PDH); pyruvate dehydrogenase phosphatase (PDP); phosphoglycerate kinase (PGK); pyruvate kinase isozymes M2 (PKM2); phosphofructokinase-1 (PFK); silencing regulatory protein (SIRT); and tricarboxylic acid cycle (TCA).
Previous studies have confirmed possible interactions between key enzymes of glycolytic metabolism. PGK1 accelerates lactate synthesis in microglia by upregulating the expression of PKM2 and LDHA (Cao et al., 2023). The dimer PKM2 can enter the nucleus to form a complex with prolyl hydroxylase 3, thereby promoting the expression of HIF-1α and its target genes (LDH, GLUT1, and PDK1) (Awasthi et al., 2019). Simultaneously, PKM2 may promote the generation of lactate by promoting glycolysis, which can increase the transcript levels of HIF-1α, PKM2, and LDHA through upregulation of H4K12la levels on the HIF-1α, PKM2, and LDHA promoter, thus promoting the further production of lactic acid (Pan et al., 2022). In addition, the knockdown of mitochondrial pyruvate carrier protein 1 promotes the conversion of pyruvate to lactate by blocking the entry of pyruvate into mitochondria (Gao et al., 2023). In turn, lactate reduces the transcription of HK1 by upregulating histone lactylation in the HK-1 promoter, which can inhibit glycolysis (Jiang et al., 2021).
Furthermore, lactate can indirectly regulate its own production by affecting the activity and transcriptional levels of key enzymes in the tricarboxylic acid cycle, such as ATP citrate lyase 2 and IDH3G (Jiang et al., 2021; Yang et al., 2023). Adenylate kinase 2, an ATP-metabolizing enzyme located in the mitochondria, also plays a role in this regulatory network. A specific variant, adenylate kinase 2 K28Ia, promotes glycolysis (Yang et al., 2023) (Figure 5).
3.1.2 Effect of HIF-1 on lactate metabolism
HIF-1, which contains both HIF-1α and HIF-1β subunits, is often activated under hypoxic conditions. HIF-1α, which is the main subunit responsible for functional regulation, often helps cells adapt to hypoxia by regulating the expression of some genes (Liang et al., 2023). HIF-1 affects lactate supply by regulating the expression of glycolysis-related proteins, such as GLUT1, HK2, PFK, PGK1, PKM, LDHA, and PDK (Chen et al., 2023; Jiang et al., 2021; Chen et al., 2023; Eschricht et al., 2015; Hu et al., 2019). There may be positive feedback between lactate and HIF-1 (Jiang et al., 2021; Hu et al., 2019). HIF-1 not only enhances cellular lactate production by promoting the expression of LDHA but also promotes lactate excretion through the upregulation of MCT4, which can maintain the continuity of glycolysis by avoiding the competitive inhibition of LDHA mediated by lactate excess (Huang et al., 2022). Additionally, HIF-1α induces the shift of energy metabolism from oxidative phosphorylation to glycolysis by downregulating the expression of PDH (Lu et al., 2022). HIF-1 can upregulate the expression of PDK and LDHA (Karagiota et al., 2023). PDK prevents pyruvate from being converted to acetyl-coenzyme A by catalyzing the phosphorylation of PDH, which may allow LDHA to convert pyruvate to lactate (Zhang et al., 2019; Chen et al., 2023; Huang et al., 2022). Knockdown of HCAR1 can block lactate-mediated cellular physiological activities by downregulating the transcriptional level of MCT (Wagner et al., 2015). HIF-1 and lactate have been shown to regulate lactate-mediated cellular physiological activities by upregulating MCT and HCAR1 expression (Longhitano et al., 2022; Huang et al., 2022). It is thus clear that HIF-1 can intervene in protein-lactylation-mediated biological effects by regulating the expression of functional proteins related to lactate metabolism and transport (Figure 5).
3.2 The impact of HIF-1 on epilepsy
Ischemia/hypoxia-induced elevation of HIF-1α expression in brain tissue can mediate cognitive deficits associated with hippocampal neuronal loss by inducing mitochondrial structural damage and upregulating pyroptosis-associated effectors, TNF-α and IL-1β (Zhao et al., 2022). HIF-1α/meme oxygenase 1 may promote the development of epilepsy by inducing ferroptosis of hippocampal neurons and reducing the activity of antioxidant enzymes in hippocampal tissue (Liang et al., 2023). Numerous studies have suggested that cellular ferroptosis may be mediated by mitochondrial dysfunction and HMGB1 (Yang et al., 2023; Chen et al., 2023; Tang et al., 2023; Ge et al., 2023; Yuan et al., 2022). It can be inferred that HIF-1α may be involved in HMGB1-mediated mitochondrial dysfunction and programmed cell death associated with epilepsy.
3.2.1 HIF-1α and HMGB1
HIF-1α inhibits the expression of HMGB1 by upregulating the transcription level of lncRNA NEAT1, thereby reducing cell apoptosis and inflammatory damage (Luo et al., 2022). Indeed, HIF-1α, which may act as an upstream regulator of HMGB1, can affect the biological effects of HMGB1 by regulating its transcription level. Meanwhile, HIF-1α promotes the production of lactate from glycolysis by upregulating the expression level of LDHA, which exacerbates HMGB1-mediated ischemic neuronal pyroptosis by upregulating the histone H3K18la in the HMGB1 promoter (Yao and Li, 2023). Cell death usually favors the release of HMGB1 into the extracellular compartment. Extracellular HMGB1 can promote the expression of NF-κB and HIF-1α via binding to the TLR4 receptor (Peng et al., 2021). Lactate can enhance the translation level of NF-κB by promoting the translocation of the transcription factor HIF-1α into the nucleus (Peng et al., 2021). NF-κB located in the nucleus not only promotes the expression of inflammatory cytokines (IL-1β and TNF-α) but also enhances HMGB1 activity by elevating HIF-1α expression (Peng et al., 2021; Song et al., 2020). Thus, HIF-1α can regulate the function of HMGB1 through positive feedback. HMGB1 acts as a ligand for the cell surface receptor TLR2 to trigger YAP and HIF-1α nuclear translocation and activate the Hippo-YAP/HIF-1α pathway. Activated YAP not only promotes the expression of HIF-1α but also maintains HIF-1α stability by binding to HIF-1α in the nucleus, thereby promoting the expression of target genes of HIF-1α. Conversely, knockdown of HIF-1α may inhibit HMGB1 by reducing YAP nuclear translocation. It further confirms that HIF-1α can affect cell death mediated by HMGB1 (Zhang et al., 2019) (Figure 3).
3.2.2 HIF-1α and mitochondria
HIF-1α shifts energy production from oxidative phosphorylation to glycolysis by promoting the transcription of related genes, which reduces ROS production to reverse the mitochondrial dysfunction induced by oxidative stress (Li et al., 2019). Although HIF-1α does not stabilize mitochondrial function by promoting the transcription of mitochondrial DNA as SIRT3 does (Wang et al., 2022), HIF-1α, as a protein regulating iron homeostasis, decreases the partial pressure of oxygen, translocates to the mitochondria, and acts directly on the mitochondrial respiratory chain complex in hypoxia, thereby blocking the sustained effects of mitochondrial ROS on mitochondrial function and ultimately achieving multi-modal neuroprotection (Fouché et al., 2023). In addition, HIF-1α may exert cytoprotective effects by mediating mitochondrial autophagy (Lestón Pinilla et al., 2021; Fu et al., 2020). However, intermittent hypoxia induces cellular mitochondrial dysfunction via activation of HIF-1 (Moulin et al., 2022). In turn, ROS and lactate, which are generated with mitochondrial dysfunction, activate mTOR and HIF-1α-related signaling pathways (Tauffenberger et al., 2019; Ip et al., 2017), and the oxidative stress caused by mitochondrial dysfunction may protect the structure and function of HIF-1α by inhibiting its proteasomal degradation (Wu et al., 2023). This shows that there is an interactive relationship between HIF-1 and mitochondrial dysfunction and that the effect of HIF-1 on mitochondria is influenced by the extracellular environment and tissue variability (Figure 3). HIF-1, regulating glycolytic metabolism, can affect the level of protein lactylation by regulating lactate production. The protein lactylation may be one of the effective mechanisms by which HIF-1 regulates HMGBI, a core factor that mediates programmed death in multiple cells. Although the mechanisms of HIF-1-mediated neuronal loss, which is strongly associated with mitochondrial dysfunction-induced epilepsy, have not yet been clarified, it cannot be denied that HIF-1 may regulate the level of HMGBI lactylation to mediate programmed neuronal death or induce mitochondrial dysfunction to mediate the occurrence and development of epilepsy. It is speculated that HIF-1 may be an important target for regulating epilepsy-associated protein lactylation (Figure 6).
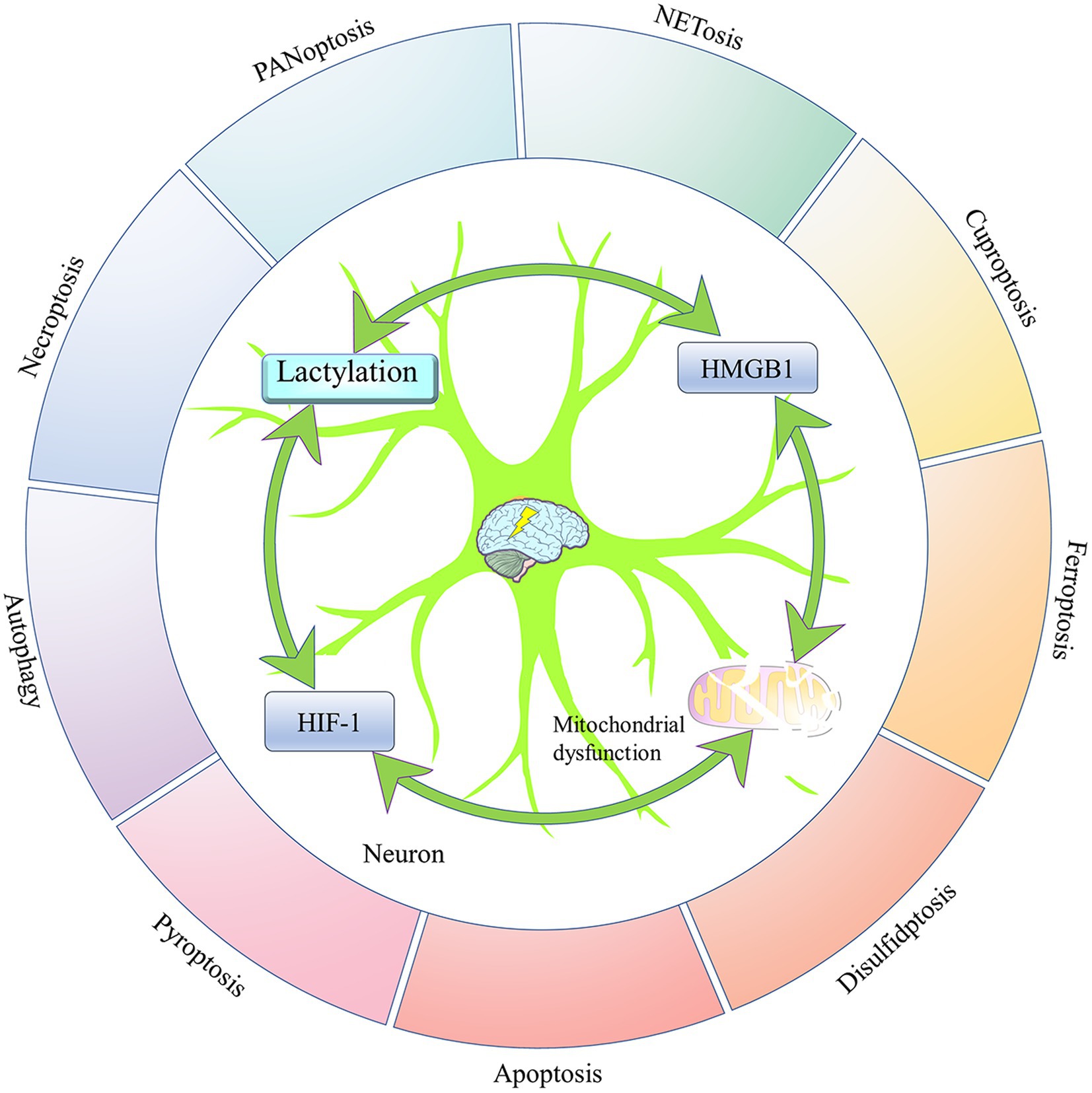
Figure 6. The four elements mediating epilepsy-related neuronal death. Neuronal death is one of the important pathological changes in epileptogenesis and development. Lactylation modification, HIF-1, and mitochondrial dysfunction all intervene in epilepsy-associated neuronal death by regulating the activity of HMGB1, which is a central factor in the regulation of multiple programmed cell deaths. In addition, there is an interactive relationship between lactylation modification, HIF-1, and mitochondrial dysfunction. Hypoxia-inducible factor 1 (HIF-1); and high-mobility group box 1 (HMGB1).
3.3 Regulation of protein lactylation by HATs and HDACs
3.3.1 Protein lactylation and delactylation catalyzed by HATs and HDACs
Previous studies have suggested that the acetyl group for protein acetylation is mainly supplied by acetyl coenzyme A and that the lactyl group for protein lactylation is mainly supplied by lactate (Dong et al., 2022). Acetyl coenzyme A and lactate in the body are mainly converted from pyruvate, an intermediate product of glycolytic metabolism, and both lactate modification and acetylation modification of proteins can be affected by glycolysis. Furthermore, it has been demonstrated that the mechanism of histone modification by lactate is similar to that of acetyl coenzyme A and that both protein lactylation and protein acetylation are regulated by acetylases and deacetylases (Moreno-Yruela et al., 2022) (Figure 4). Acetyltransferase P300, a newly identified lactonylated writer protein, was shown to promote gene expression by upregulating the levels of H3K9 acetylation, H4K12 acetylation, H4K16 acetylation, H4K12la, and H3K18la at the transcriptional start sites of pluripotency genes (Fan et al., 2023; Lin et al., 2022). P300 belongs to the nuclear A-type HAT. It has been demonstrated that the loss of P300 activity is associated with neuronal survival and long-term memory and that P300 may intervene in neuronal death by catalyzing histone acetylation or lactylation (Lu et al., 2015). P300 promotes PGK1 expression by catalyzing H3K27 acetylation in the PGK1 promoter region. PGK1 exacerbates ischemic stroke-induced brain injury by promoting microglia M1 polarization, inflammation, and glycolysis (Cao et al., 2023). Indeed, the regulation of protein lactylation by P300 can depend not only on the direct catalysis of lactide writing but also on the regulation of lactate production.
HDACs consist of HDAC1-11 and SIRT1-7 with deacetylase activity. A recent study revealed that HDACs, like P300, regulate protein lactylation and acetylation. Class I HDAC1, 2, and 3 are the most efficient lysine delactylases in vitro, mainly catalyzing the delactylation of H3K18 and H4K5. However, SIRT1-3, in vitro experiments, were shown to have a slight delactylation activity, and their main regulatory sites are in H3K18 and H3K5 (Fan et al., 2023; Moreno-Yruela et al., 2022). Furthermore, the knockdown of HDAC1-3 resulted in an increase in H4K5la but had little effect on pan-Kla and H3K18la. The differences in histone lactylation sites regulated by HDAC1-3 between in vitro and in vivo experiments suggest that the specificity of HDAC action sites may be affected by certain cofactors (Fan et al., 2023). However, some studies have suggested that HDAC1-3 predominantly regulates the level of H3K18la both in vitro and in vivo and that MS-275, a selective inhibitor of HDAC1-3, promotes gene expression by upregulating the levels of H3K14la and H3K18la (Dai et al., 2022). H3K18la can be widely distributed in the cells and tissues of humans and mice. Importantly, it has been discovered that H3K18la not only accumulates at promoters but also enriches in a tissue-specific manner at active enhancers, sharing a genomic distribution pattern with H3K27 acetylation (Galle et al., 2022). Mitochondrial dysfunction can promote pan-lysine lactylation and H3K18 lactylation without affecting pan-lysine acetylation and H3K27 acetylation (He et al., 2023). Lactylation and acetylation of histones may cooperate to execute the transcriptional regulation of genes. The acetylation of histones can competitively inhibit histone lactylation (Rho et al., 2023). However, HDAC inhibitors may activate neuronal transcriptional programs by mediating chemical modification of multiple forms of histone lysines, such as H3K9 acetylation, H3K9 crotonylation, H3K18 crotonylation, and H3K18la. There may be collaborative, rather than competitive, relationships between histone acetylation, butyrylation, and lactic acidification in neuronal cells (Dai et al., 2022). HDACs can not only regulate histone lactylation levels but also regulate lactylation modifications of non-histone proteins. SIRT1 is a potential non-histone delactylase in mammals (Sun et al., 2022). Meanwhile, HDACs affect the level of protein lactylation by regulating glucose metabolism. For example, SIRT1 promotes lactate production by positively regulating the expression of HK-2, a key enzyme in glycolysis (Chen et al., 2022) (Figure 5), while SIRT6 has been shown to be an inhibitor of glycolysis (Sun et al., 2021) (Figure 1). SIRT3 promotes the conversion of pyruvate to acetyl coenzyme A by catalyzing the deacetylation of PDH, thereby reducing lactate production and downregulating the lactylation of mitochondrial proteins (An et al., 2023).
3.3.2 The role of HATs and HDACs in the regulation of HIF-1
HDACs regulate HIF-1α activity through various mechanisms. HDACs can regulate the activity of HIF-1α by affecting its expression level, degradation rate, and post-translational modifications (Table 2). Various factors may influence this process, such as tissue specificity and external environmental pressures (Geng et al., 2011).
Table 2. The histone deacetylases regulating transcription factor HIF-1α.
It was shown that HDAC1 and HDAC2 downregulate the expression of fructose-1,6-bisphosphatase 1 by catalyzing the deacetylation of histone H3K27 in the enhancer of 1,6-bisphosphatase. Fructokinase 1,6-bisphosphate, a rate-limiting enzyme in gluconeogenesis, reduces the onset of glycolysis and decreases lactate production by inhibiting the expression of HIF-1α, thereby inhibiting histone lactylation (Yang et al., 2017; Fan et al., 2020). Meanwhile, HDAC2 promotes HIF-1α/6-phosphofructo-2-kinase (PFKFB3) axis-mediated neuroinflammatory injury by increasing the expression of HIF-1α (Liu et al., 2022). However, not all HDACs can act as upstream regulators of HIF-1, and there are currently few reports on the regulation of HIF-1 function by HDAC3, HDAC10, HDAC11, and SIRT5. It has even been suggested that HIF-1α may promote HDAC3 expression, serving as an upstream regulator of HDAC3 (Wang et al., 2021) (Figure 5). However, in epileptic hippocampal neurons, HDAC5 promotes HIF-1α enrichment at the PFKFB3 promoter by upregulating HIF-1α expression, thereby contributing to PFKFB3-induced neuronal apoptosis, oxidative stress, and inflammation (Pan et al., 2021).
Under hypoxic conditions, HDAC7-u enhances the transcriptional activity of HIF-1α, promoting the transcription of HIF-1α target genes. This effect is achieved by increasing HIF-1α’s transcriptional activity rather than altering its transcriptional level (Shakespear et al., 2013). HDAC9 enhances the transcriptional activity of HIF-1 by catalyzing HIF-1 deacetylation, thereby mediating ischemic stroke-induced neuronal Ferroptosis in cortical layers (Sanguigno et al., 2023).
SIRT1 stabilizes Von Hippel-Lindau activity by deacetylating Von Hippel-Lindau, effectively reversing its acetylation. This modification enhances the stability and function of the Von Hippel-Lindau protein. Under normoxic conditions, hydroxylated HIF-1α accelerates HIF-1α degradation by binding to von Hippel–Lindau ubiquitin ligase complexes (Li et al., 2022; Sailhamer et al., 2010). Activation of HIF-1α under hypoxic conditions induces a shift in energy metabolism from oxidative phosphorylation to glycolysis, which facilitates cellular adaptation to the hypoxic environment to protect neuronal viability (Kaitsuka et al., 2020; Zhu et al., 2022). The knockdown or inhibition of the activities of SIRT2 and SIRT3 has been shown to increase the expression of HIF-1α, which in turn activates cellular glycolysis to ensure the energy supply for neurons in the ischemic core (Kaitsuka et al., 2020; Zhu et al., 2022). The glycolysis in neurons depletes NAD+, which leads to the loss of biological activity of the NAD+-dependent deacetylase SIRT1, thereby maintaining the stability of HIF-1α structure and function (Li et al., 2022). Blocking the negative regulatory effect of HDACs on HIF-1α activity may help brain tissue adapt to the hypoxic environment to a certain extent, but elevated HIF-1α activity may increase the risk of inflammatory injury (Li et al., 2022; Kaitsuka et al., 2020; Zhu et al., 2022). Furthermore, SIRT6 independently upregulated HIF-1α expression to promote glycolysis (You et al., 2022).
It was shown that P300 may intervene in the functioning of the central nervous system by regulating the activity of key enzymes of glycolysis (Cao et al., 2023; Lu et al., 2015) and that P300/CBP may enhance the expression of HIF-1α target genes by promoting lysine acetylation of the transcription factor HIF-1α (Wang et al., 2024). It can be speculated that P300 may indirectly regulate protein lactylation modification by affecting the lactate production of glycolysis by modulating the transcriptional activity of HIF-1α.
In summary, HAT and HDAC regulate protein lactylation in vivo. This regulation depends not only on the catalytic activities of these enzymes but also on the availability of the lactate donor, lactide. These enzymes also indirectly regulate the expression of proteins related to glycolytic metabolism, which are induced by the transcription factor HIF-1α. Additionally, HAT and HDAC directly influence the activity of key glycolytic enzymes, further impacting lactate production. These mechanisms highlight the complex interplay between histone modification and metabolic control in cellular processes.
3.4 Regulation of HIF-1 by other substances
Research indicates that the stability and activation of HIF-1 are not only regulated by HATs and HDACs but also closely related to oxygen levels, cellular iron content, and the activation of other signaling pathways (Liang et al., 2023).
The autophagy-related factor P62 may enhance the expression of glycolysis-related proteins by upregulating HIF-1α expression (Calvo-Garrido et al., 2019). In turn, the products and key enzymes of glucose metabolism can adjust the biological activity and expression level of HIF-1. In microglia, lactate not only upregulates the transcriptional level of HIF-1α by promoting the histone H4K12la of the HIF-1α promoter (Pan et al., 2022; Jiang et al., 2021) but also enhances the production of lactate by directly promoting the lactylation of HIF-1α (Luo et al., 2022).
Accumulating lactate may promote the expression of the transcription factor STAT6 by upregulating histone lactylation in the STAT6 promoter region (Lu et al., 2022). Furthermore, STAT6 may reduce HIF-1α expression by inhibiting the mTOR-mediated glycolipid metabolism regulation signaling pathway (Lu et al., 2022; Park et al., 2019) (Figure 3).
Meanwhile, PDP1 can drive the acetylation of histone H3 in the promoter of HIF-1 target genes and promote gene expression by promoting the production of the glucose metabolism intermediate product acetyl coenzyme A. The expression of the HIF-1 target gene PDK1 can counteract the effects of PDP1 and diminish the biological impact of HIF-1 (Karagiota et al., 2023) (Figure 5). The epigenetically important regulators HAT and HDAC have been shown to be major regulators of lactylation modification, and a large number of studies have demonstrated that HAT and HDAC are critical for the progression of epilepsy (Wang et al., 2022). The regulation of HIF-1 by HAT and HDAC may be a potential pathway for their intervention in epilepsy-associated protein lactylation.
4 The impact of HDAC inhibitors on lactylation and epilepsy
Although the state of protein acetylation depends on the balance between HAT and HDAC, previous studies have shown that the imbalance between HAT and HDAC favors the latter, so the clinical development of HDAC inhibitors is focused on restoring this balance (Bezu et al., 2019). Currently, some HDAC inhibitors have been applied and studied as antiepileptic drugs (Wang et al., 2022). The protein lactylation induced by lactate is regulated by HAT and HDAC as protein acetylation (Yang et al., 2023). Suberoylanilide hydroxamic acid and trichostatin A, which are HDAC inhibitors, may reduce the rate of lactate production from glycolysis by promoting acetylation of key enzymes of glycolytic metabolism, thereby reducing protein lactylation modification (Wu et al., 2023; Zhang et al., 2022). It is undeniable that the antiepileptic effect of HDAC inhibitors may be partially mediated by the modulation of protein lactylation modifications.
4.1 The impact of HDAC inhibitors on neuronal loss and glial activation
The common HDAC inhibitors can be divided into four groups based on their structure: butyric acid derivatives (e.g., valproic acid), hydroxamic acid derivatives (e.g., suberoylanilide hydroxamic acid), benzamides (e.g., entestat) and cyclic tetrapeptides (e.g., romidepsin) (Bezu et al., 2019). Sodium butyrate can alleviate neurological damage caused by cerebral hypoxia-ischemia. The ability of trichostatin A to stimulate neurogenesis in the subgranular zone of the hippocampus may facilitate recovery from hypoxic–ischemic injury in the neonatal brain (Zalewska et al., 2020).
The conventional antiepileptic drug valproate may inhibit the activation of the NF-κb signaling pathway by promoting the acetylation of STAT1 and p65, thereby impeding microglia activation and attenuating I/R-induced neuroinflammation and brain damage (Dai et al., 2021). Suberoylanilide hydroxamic acid has been shown to prevent astrocyte and microglia activation, which can alleviate ischemia-induced neuroinflammation by inhibiting the deacetylation activity of HDAC1/2 (Dai et al., 2021), while the class IIa HDAC inhibitor MC1568 attenuates ischemic stroke-induced ferroptosis of cortical neurons by blocking HDAC9 transcription (Sanguigno et al., 2023). Furthermore, HDAC inhibitors significantly inhibit the immunological activation and aggregation induced by cerebral hemorrhage. Scriptaid has been proven to alleviate the inflammatory injury of cortical neurons caused by traumatic brain injury or cerebral hemorrhage by promoting the polarization of microglia/macrophages toward the protective M2 phenotype (Dai et al., 2021). Cerebral hemorrhage is highly susceptible to epilepsy, and the class I HDAC inhibitor entinostat protects neurons by inhibiting microglia activation, thereby ameliorating cerebral hemorrhage-induced neuroinflammatory damage (Bonsack and Sukumari-Ramesh, 2021). The class I HDAC inhibitor CI-994 and the HDAC1-specific inhibitor parthenolide may enhance synaptic plasticity in hippocampal neurons by inhibiting astrocyte and microglia immunoreactivity, thereby ameliorating epilepsy-induced cognitive impairment (Jia et al., 2023; Dai et al., 2021; Burns et al., 2022). RGFP966, the HDAC8-specific inhibitor WK2-16, and the HDAC3-specific inhibitor BG45 have been shown to reduce neuroinflammation-mediated hippocampal neuronal loss by inhibiting the proliferative activation of glial cells, thereby maintaining the morphological and functional stability of the cerebral cortex (Dai et al., 2021; Wang et al., 2023). Additionally, FK228, also known as romidepsin, can exert a neuroprotective effect by inhibiting NETosis-mediated neural inflammation and promoting the regeneration of neurons (Thakur et al., 2024). HDAC inhibitors may reduce the occurrence of post-stroke epilepsy by alleviating neuronal loss and neuroinflammation induced by ischemic brain injury.
4.2 The biological activity of HIF-1 is regulated by HDAC inhibitors
HIF-1α not only affects ischemia-induced hippocampal neuronal loss by regulating lactate metabolism but is also closely associated with epilepsy-related hippocampal neuronal death and oxidative stress (Liang et al., 2023; Zhao et al., 2022). Valproate, which has been shown to inhibit class I and class II HDACs, may reduce HIF-1α-mediated hippocampal neuronal loss by lowering the protein level of HIF-1α, thereby achieving antiepileptic effects (Luo et al., 2013; Simeone et al., 2017). Sodium butyrate not only enhances ubiquitination of HIF-1α Lys532 by catalyzing its acetylation to target HIF-1α for proteasomal degradation (Naia et al., 2017), but also upregulates the expression level of HIF-1α by inhibiting HDAC2 (Bashir and Olaniyi, 2023). Moreover, butyrate sodium combined with curcumin, which is a less toxic natural pan-HDAC inhibitor derived from food, prevents PI3K/Akt axis-mediated inhibition of HIF-1α activity through inhibition of HDAC1 (Islam et al., 2023). The development of food-borne HDAC inhibitors may be beneficial for epilepsy control. The ketogenic diet can increase the production of β-hydroxybutyrate, which may alleviate seizures associated with neuroinflammation by promoting the acetylation of histone H3K9 and H3K14, similar to HDAC inhibitors (Simeone et al., 2017).
Suberoylanilide hydroxamic acid, vorinostat, which is the first acetylation-modifying drug approved by the US Food and Drug Administration, mainly inhibits the activity of HDAC1 and HDAC2. It has been demonstrated that vorinostat inhibits the activity of HDACs, including HDAC4, to promote HIF-1α acetylation, which can inhibit the biological activity of HIF-1α and promote HIF-1α degradation (Geng et al., 2011; Sailhamer et al., 2010). Meanwhile, vorinostat promotes intracellular lactate efflux by upregulating the expression of MCT1 and MCT4, and lactate released into the extracellular space enhances the inhibitory effect of vorinostat on HDAC activity (Radoul et al., 2019). In addition, N-hydroxy-7-(2-naphthylthio) heptanamide, a novel synthetic HDAC inhibitor, was shown to inhibit HIF-1α expression to a greater extent than Vorinostat by in vitro and in vivo experiments in breast cancer (Park et al., 2011). Panobinostat (LBH589), a novel pan-HDAC inhibitor, has been shown to potently inhibit the viability of HIF-1α (Ni and Ni, 2021). The pan-histone deacetylase inhibitor PCI-24781 induced cellular autophagy by upregulating the concentration of HIF-1α (Bhalla et al., 2013).
In summary, as HIF-1α can affect the level of protein lactylation by regulating the expression of key enzymes of glycolysis and lactate transporter proteins, HDAC inhibitors may control ischemic stroke-induced protein lactylation by regulating the activity of HIF-1α, thereby alleviating abnormal excitation of neurons (Hagihara et al., 2021; Wu et al., 2023; Zhang et al., 2022). It may be one of the potential mechanisms by which HDAC inhibitors could help prevent post-ischemic stroke epilepsy.
5 Discussion
Stroke is one of the leading causes of death worldwide. Ischemic strokes account for 87% of strokes in humans (González-Rodríguez and Fernández-López, 2023). The accumulation of lactate caused by ischemic stroke may mediate neuroexcitotoxicity and neuronal apoptosis through the upregulation of protein lactylation levels, which can lead to post-stroke epilepsy (Hagihara et al., 2021; Wang et al., 2022; Zhang et al., 2023). Reperfusion injury in ischemic foci of the brain is mainly mediated by mitochondrial dysfunction, which can be induced through over-activated glycolysis (She et al., 2023; Liu et al., 2023). Although reperfusion of blood improves the oxygen supply to the ischemic lesion, it does not ensure that the mode of energy metabolism in the ischemic lesion and its surrounding tissues is switched from glycolysis to oxidative phosphorylation, implying that reperfusion may not completely block lactate production. Although there is no evidence that neurons initiate the “Warburg effect,” also known as “aerobic glycolysis,” as tumor cells do (Lin et al., 2022), peripheral immune cells differentiated from bone marrow hematopoietic stem cells have been observed to display the “Warburg effect,” such as macrophages and neutrophils (Chen et al., 2022). The “Warburg effect” of tumor cells is primarily for the rapid proliferation of tumor cells (Lin et al., 2022), while the “Warburg effect” of peripheral immune cells promotes an inflammatory cascade by supporting the expression of pro-inflammatory cytokines during the activation of immune cells (Zhang et al., 2019). Brain injury typically involves the disruption of the blood–brain barrier, and peripheral blood immune cells can easily migrate across the damaged barrier into the brain, where they work together with activated glial cells to exacerbate neuroinflammatory damage (Li et al., 2021; Bernis et al., 2023). After the restoration of blood supply to brain tissue, peripheral immune cells may continue to produce lactate and release it extracellularly, relying on the Warburg effect, and the lactate may be taken up by neurons via the MCT to promote neuronal protein lactylation, which may increase the risk of post-stroke epilepsy by mediating neuronal excitation (Hagihara et al., 2021; Zhang et al., 2019; Mayorga-Weber et al., 2022; Tröscher et al., 2021).
Furthermore, the enhancement of neuronal excitability further upregulates protein lactylation levels by promoting the conversion of glucose to lactate in the brain (Li and Freeman, 2015). However, as the lactate from aerobic glycolysis increases in peripheral immune cells infiltrating brain tissue, the level of protein lactylation is significantly upregulated, which can induce a shift of activated peripheral immune cells to an anti-inflammatory state for avoiding an unlimited extent of inflammatory damage to brain tissue (Zhang et al., 2019). It is evident that protein lactylation modifications mediate different biological effects in different cells.
Although protein lactylation alleviates ischemic stroke-induced neuroinflammation by affecting the activity of peripheral immune cells migrating to the ischemic lesion, the protein lactylation-mediated neuronal loss and glial cell activation should not be underestimated (Pan et al., 2022; Barros et al., 2023; Yang et al., 2024; Wu et al., 2023; Yao et al., 2023). The modification of protein lactylation in neurons and glia is associated not only with the facilitation of glycolysis but also with the cellular transport of lactate. If both lactate production and intracellular transformation of lactate in neurons can be controlled, post-stroke epilepsy would be better prevented. Since HIF-1 can act as a transcription factor for key enzymes of the glycolytic pathway and lactate transporter proteins, inhibiting the transcriptional activity of HIF-1 may be a potential measure to combat post-stroke epilepsy (Longhitano et al., 2022; Karagiota et al., 2023; Huang et al., 2022). HDAC may affect the expression of HIF-1-related target genes by directly or indirectly regulating HIF-1 transcription, degradation, and bioactivity, thereby influencing cellular glucose uptake, mitochondrial function, and lactate metabolism (Table 2). However, research has confirmed that lactate increases the activity of SIRT1, which is a potential protein lactylation and acetylation modification regulator in mammals (Sun et al., 2022; He et al., 2023), and the ketogenic diet may exert an anti-epileptic effect by inhibiting hypoxia-enhanced glycolysis (Sun et al., 2021; Hou et al., 2017). Thus, inhibition of lactate production and lactate-mediated protein lactylation may be the key to reducing post-stroke epilepsy rather than inhibition of HDAC enzyme activity, and it has been demonstrated that the HDAC inhibitors sodium butyrate and trichostatin A control lactate production by modulating the activities of pyruvate kinase and LDH (Rodrigues et al., 2015).
HMGB1 is a core factor mediating programmed cell death in various cells, and the nucleus-to-cytoplasm translocation of HMGB1 in neurons and glial cells may be an important trigger for status epilepticus (Pauletti et al., 2019; Chen et al., 2023; Tang et al., 2023). It has been demonstrated that N-(2′-hydroxyphenyl)-2-propylvaleramide may promote HMGB1 acetylation through inhibition of HDAC1, thereby inducing translocation of HMGB1 from the nucleus to the cytoplasm. Meanwhile, N-(2′-hydroxyphenyl)-2-propylvaleramide also induced HMGB1 secretion by promoting ROS synthesis (Sixto-López et al., 2020). The HDAC8-specific inhibitors, such as PCI-34051 and PCI-48012, and Scriptaid, all promote HMGB1 acetylation, which triggers HMGB1-mediated biological effects by inhibiting HDAC activity (Lopez et al., 2015; Chi et al., 2017). However, the mechanism of HDAC inhibitors regulating the biological effects of HMGB1 includes promoting the HMGB1 acetylation and regulating HMGB1 expression. Chidamide, a novel oral selective HDAC inhibitor, can downregulate the expression of HMGB1 (Liu et al., 2021). The novel HDAC inhibitor HFY-4A induces apoptosis by upregulating HMGB1 expression (Yin et al., 2023). The HDAC inhibitor pemetrexed, combined with sildenafil, may upregulate the expression of HMGB1 and promote the extracellular release of HMGB1 by inhibiting HDAC6, HDAC2, HDAC4, and HDAC9 (Booth et al., 2017). In addition, HDAC inhibitors promote the formation of the HMGB1-P53 complex by inhibiting SIRT1 activity, thereby reducing lactate generated from glycolysis (He et al., 2023; Li et al., 2023; Ma et al., 2020; Wang et al., 2023). HDAC inhibitors may regulate the activity of HMGB1 in various ways, thereby indirectly modulating protein lactylation and cell death.
HDAC can affect lactate production by regulating the activity of the transcription factor HIF-1 or key enzymes of glycolysis. Conversely, lactate can inhibit HDAC activity like conventional HDAC inhibitors (Figure 5). Lactate has been shown to induce histone H3 and H4 hyperacetylation, which may promote the expression of some genes by inhibiting class I and II HDAC (Wagner et al., 2015). In CD8+ T cells, lactate promotes protein acetylation by inhibiting HDAC activity, thereby inhibiting apoptosis (Feng et al., 2022). Lactate promotes the immunosuppressive effect of tumors by blocking the formation of a transcriptional repression complex between NF-κB and HDAC3 (Chang et al., 2021). Lactate can enhance HDAC6 activity by inhibiting HDAC11 activity, and HDAC6 upregulates IL-10 transcript levels and blocks the inflammatory effects of immune cells by promoting histone H3 acetylation in myeloid-derived suppressor cells and macrophages (Heim et al., 2020). Existing studies have confirmed that lactate can exert neuroprotective effects by regulating the expression levels of HDAC2/3/5 (Ding et al., 2020; Puri et al., 2019; Karnib et al., 2019; Genders et al., 2019). Lactate can alleviate inflammatory damage and protect neurological functions by modulating the function of HDACs.
There is a feedback regulation of HDAC expression by HIF-1α. HIF-1α in a hypoxic environment can enhance the deacetylation activity of HDAC2 (Wang et al., 2022). Meanwhile, HIF-1α can downregulate the transcript level of HDAC4 by binding to the promoter of HDAC4 (Pan and Zhao, 2021). The hypoxic environment drives HIF-1α to bind directly to the HDAC3 promoter, which reduces histone acetylation by upregulating HDAC3 expression (Wang et al., 2021; Yuan et al., 2022). However, the regulatory effects of HIF-1α on HDACs, like lactate, were not consistent. It has been shown that HIF-1α impairs the anti-inflammatory effect of Treg cells on pro-inflammatory macrophages by promoting the expression of SIRT2 in Treg cells, thereby contributing to ischemia- and hypoxia-induced neuroinflammatory injury (Shu et al., 2019). Both lactate and HIF-1α can act as HDAC inhibitors to regulate protein acetylation modifications. Based on the report of lactate mediating protein phosphorylation (Maschari et al., 2022), it is speculated that lactate not only induces protein lactylation but also alters protein function by regulating the levels of protein phosphorylation and acetylation.
Recent studies have suggested that histone acetylation and lactonylation may compete (Rho et al., 2023; Moreno-Yruela et al., 2022). The competition between the lactoyl and the acetyl groups for the epigenetic modifications of histone lysine is related not only to the activity of HDAC but also to the concentrations of lactic acid and acetyl coenzyme A. Pyruvate can be converted to lactate or acetyl coenzyme A by different enzymes, and the type of histone modification can be altered by regulating the activity of these enzymes (Dai et al., 2022). It has been shown that proteins tend to undergo acetylation rather than lactylation in the physiological state, while in hypoxia, histone lactylation can replace histone acetylation to regulate cellular function (Dai et al., 2022). However, it remains to be further explored whether ischemic stroke can mediate protein lactylation, replacing protein acetylation, which may alter cellular function by promoting glycolysis. Revealing this competitive relationship between lactylation and acetylation may facilitate the design of new therapeutic strategies for epilepsy after ischemic stroke. In addition, lactate and its mediated modification of protein lactylation can affect neuronal and glial cell function and the functional recovery of ischemic foci by modulating vascular endothelial cell function (Fan et al., 2023).
In summary, the modulation of protein lactylation levels by regulating lactate production and/or lactate membrane transport may be a potential strategy to combat epilepsy after ischemic stroke.
6 Conclusion
Ischemic brain injury results in lactate accumulation within the central nervous system, contributing to metabolic dysregulation. Lactate may induce post-stroke epilepsy by promoting protein lactylation in brain tissue. HAT and HDAC have been shown to modulate cellular function by catalyzing protein lactylation and delactylation, thereby mediating neuronal loss and glial cell activation, which are important factors contributing to epilepsy-associated neuronal hyperexcitability. Although the specific mechanism through which protein lactylation influences post-stroke epilepsy has not yet been validated, conducting research in this area may provide a new theoretical basis for using HDAC inhibitors in the clinical prevention and treatment of post-stroke epilepsy.
Author contributions
XK: Writing – original draft. SC: Writing – review & editing. QY: Conceptualization, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by The Natural Science Foundation of Hainan Province, China (Grant number: 817307).
Acknowledgments
We are grateful for the support from the Natural Science Foundation of Hainan Province, China (grant number: 817307) and Hainan Province Clinical Medical Center.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ali, N. H., Al-Kuraishy, H. M., Al-Gareeb, A. I., Alnaaim, S. A., Alexiou, A., Papadakis, M., et al. (2023). Autophagy and autophagy signaling in epilepsy: possible role of autophagy activator. Mol. Med. 29:142. doi: 10.1186/s10020-023-00742-2
An, S., Yao, Y., Hu, H., Wu, J., Li, J., Li, L., et al. (2023). PDHA1 hyperacetylation-mediated lactate overproduction promotes sepsis-induced acute kidney injury via Fis1 lactylation. Cell Death Dis. 14:457. doi: 10.1038/s41419-023-05952-4
Arab, H. H., Eid, A. H., Yahia, R., Alsufyani, S. E., Ashour, A. M., El-Sheikh, A. A. K., et al. (2023). Targeting autophagy, apoptosis, and SIRT1/Nrf2 Axis with Topiramate underlies its neuroprotective effect against cadmium-evoked cognitive deficits in rats. Pharmaceuticals (Basel) 16:1214. doi: 10.3390/ph16091214
Awasthi, D., Nagarkoti, S., Sadaf, S., Chandra, T., Kumar, S., and Dikshit, M. (2019). Glycolysis dependent lactate formation in neutrophils: a metabolic link between NOX-dependent and independent NETosis. Biochim. Biophys. Acta Mol. Basis Dis. 1865:165542. doi: 10.1016/j.bbadis.2019.165542
Barros, L. F., Ruminot, I., Sotelo-Hitschfeld, T., Lerchundi, R., and Fernández-Moncada, I. (2023). Metabolic recruitment in brain tissue. Annu. Rev. Physiol. 85, 115–135. doi: 10.1146/annurev-physiol-021422-091035
Bashir, A. M., and Olaniyi, K. S. (2023). Butyrate alleviates renal inflammation and fibrosis in a rat model of polycystic ovarian syndrome by suppression of SDF-1. BMC Pharmacol. Toxicol. 24:48. doi: 10.1186/s40360-023-00692-9
Bebensee, D. F., Can, K., and Müller, M. (2017). Increased mitochondrial mass and cytosolic redox imbalance in hippocampal astrocytes of a mouse model of Rett syndrome: subcellular changes revealed by ratiometric imaging of JC-1 and roGFP1 fluorescence. Oxidative Med. Cell. Longev. 2017:3064016. doi: 10.1155/2017/3064016
Bernis, M. E., Zweyer, M., Maes, E., Schleehuber, Y., and Sabir, H. (2023). Neutrophil extracellular traps release following hypoxic-ischemic brain injury in newborn rats treated with therapeutic hypothermia. Int. J. Mol. Sci. 24:3598. doi: 10.3390/ijms24043598
Bezu, L., Chuang, A. W., Liu, P., Kroemer, G., and Kepp, O. (2019). Immunological effects of epigenetic modifiers. Cancers 11:1911. doi: 10.3390/cancers11121911
Bhalla, S., Evens, A. M., Prachand, S., Schumacker, P. T., and Gordon, L. I. (2013). Paradoxical regulation of hypoxia inducible factor-1α (HIF-1α) by histone deacetylase inhibitor in diffuse large B-cell lymphoma. PLoS One 8:e81333. doi: 10.1371/journal.pone.0081333
Bi, Y., Xu, H., Wang, X., Zhu, H., Ge, J., Ren, J., et al. (2022). FUNDC1 protects against doxorubicin-induced cardiomyocyte PANoptosis through stabilizing mtDNA via interaction with TUFM. Cell Death Dis. 13:1020. doi: 10.1038/s41419-022-05460-x
Boison, D., and Steinhäuser, C. (2018). Epilepsy and astrocyte energy metabolism. Glia 66, 1235–1243. doi: 10.1002/glia.23247
Bonsack, F., and Sukumari-Ramesh, S. (2021). Entinostat improves acute neurological outcomes and attenuates hematoma volume after intracerebral hemorrhage. Brain Res. 1752:147222. doi: 10.1016/j.brainres.2020.147222
Booth, L., Roberts, J. L., Poklepovic, A., and Dent, P. (2017). [pemetrexed + sildenafil], via autophagy-dependent HDAC downregulation, enhances the immunotherapy response of NSCLC cells. Cancer Biol. Ther. 18, 705–714. doi: 10.1080/15384047.2017.1362511
Burns, A. M., Farinelli-Scharly, M., Hugues-Ascery, S., Sanchez-Mut, J. V., Santoni, G., and Gräff, J. (2022). The HDAC inhibitor CI-994 acts as a molecular memory aid by facilitating synaptic and intracellular communication after learning. Proc. Natl. Acad. Sci. USA 119:e2116797119. doi: 10.1073/pnas.2116797119
Butler, C. R., Boychuk, J. A., and Smith, B. N. (2017). Brain injury-induced synaptic reorganization in hilar inhibitory neurons is differentially suppressed by rapamycin. eNeuro 4:ENEURO.0134-17.2017. doi: 10.1523/ENEURO.0134-17.2017
Byun, D. J., Lee, J., Yu, J. W., and Hyun, Y. M. (2023). NLRP3 exacerbate NETosis-associated neuroinflammation in an LPS-induced inflamed brain. Immune Netw. 23:e27. doi: 10.4110/in.2023.23.e27
Cai, M., and Lin, W. (2022). The function of NF-kappa B during epilepsy, a potential therapeutic target. Front. Neurosci. 16:851394. doi: 10.3389/fnins.2022.851394
Calvo-Garrido, J., Maffezzini, C., Schober, F. A., Clemente, P., Uhlin, E., Kele, M., et al. (2019). SQSTM1/p62-directed metabolic reprogramming is essential for normal neurodifferentiation. Stem Cell Rep. 12, 696–711. doi: 10.1016/j.stemcr.2019.01.023
Cao, W., Feng, Z., Zhu, D., Li, S., Du, M., Ye, S., et al. (2023). The role of PGK1 in promoting ischemia/reperfusion injury-induced microglial M1 polarization and inflammation by regulating glycolysis. NeuroMolecular Med. 25, 301–311. doi: 10.1007/s12017-023-08736-3
Carolina, F., Nico, D., Barbara, E., Ansgar, F., Philip, S., Natalie, S., et al. (2021). Seizures after ischemic stroke: a matched multicenter study. Ann. Neurol. 90, 808–820. doi: 10.1002/ana.26212
Centonze, E., Marte, A., Albini, M., Rocchi, A., Cesca, F., Chiacchiaretta, M., et al. (2023). Neuron-restrictive silencer factor/repressor element 1-silencing transcription factor (NRSF/REST) controls spatial K(+) buffering in primary cortical astrocytes. J. Neurochem. 165, 701–721. doi: 10.1111/jnc.15755
Chang, H., Xu, Q., Li, J., Li, M., Zhang, Z., Ma, H., et al. (2021). Lactate secreted by PKM2 upregulation promotes Galectin-9-mediated immunosuppression via inhibiting NF-κB pathway in HNSCC. Cell Death Dis. 12:725. doi: 10.1038/s41419-021-03990-4
Chen, S., Chen, Y., Zhang, Y., Kuang, X., Liu, Y., Guo, M., et al. (2020). Iron metabolism and ferroptosis in epilepsy. Front. Neurosci. 14:601193. doi: 10.3389/fnins.2020.601193
Chen, W. J., Huang, S. Y., Chen, Y. W., Liu, Y. F., and Huang, R. S. (2023). Dietary folate deficiency promotes lactate metabolic disorders to sensitize lung cancer metastasis through MTOR-signaling-mediated druggable oncotargets. Nutrients 15:1514. doi: 10.3390/nu15061514
Chen, L., Huang, L., Gu, Y., Cang, W., Sun, P., and Xiang, Y. (2022). Lactate-lactylation hands between metabolic reprogramming and immunosuppression. Int. J. Mol. Sci. 23:11943. doi: 10.3390/ijms231911943
Chen, Y. H., Jin, S. Y., Yang, J. M., and Gao, T. M. (2023). The memory orchestra: contribution of astrocytes. Neurosci. Bull. 39, 409–424. doi: 10.1007/s12264-023-01024-x
Chen, C., Wei, M., Wang, C., Sun, D., Liu, P., Zhong, X., et al. (2020). The histone deacetylase HDAC1 activates HIF1α/VEGFA signal pathway in colorectal cancer. Gene 754:144851. doi: 10.1016/j.gene.2020.144851
Chen, M., Xia, Z., and Deng, J. (2023). Human umbilical cord mesenchymal stem cell-derived extracellular vesicles carrying miR-655-3p inhibit the development of esophageal cancer by regulating the expression of HIF-1α via a LMO4/HDAC2-dependent mechanism. Cell Biol. Toxicol. 39, 1319–1339. doi: 10.1007/s10565-022-09759-5
Chen, Y., Yang, H., Chen, S., Lu, Z., Li, B., Jiang, T., et al. (2022). SIRT1 regulated hexokinase-2 promoting glycolysis is involved in hydroquinone-enhanced malignant progression in human lymphoblastoid TK6 cells. Ecotoxicol. Environ. Saf. 241:113757. doi: 10.1016/j.ecoenv.2022.113757
Chen, J., Zhang, M., Liu, Y., Zhao, S., Wang, Y., Wang, M., et al. (2023). Histone lactylation driven by mROS-mediated glycolytic shift promotes hypoxic pulmonary hypertension. J. Mol. Cell Biol. 14:mjac073. doi: 10.1093/jmcb/mjac073
Chen, R., Zou, J., Kang, R., and Tang, D. (2023). The redox protein HMGB1 in cell death and cancer. Antioxid. Redox Signal. :null. doi: 10.1089/ars.2023.0007
Chi, J. H., Seo, G. S., Cheon, J. H., and Lee, S. H. (2017). Isoliquiritigenin inhibits TNF-α-induced release of high-mobility group box 1 through activation of HDAC in human intestinal epithelial HT-29 cells. Eur. J. Pharmacol. 796, 101–109. doi: 10.1016/j.ejphar.2016.12.026
Dai, S. K., Liu, P. P., Li, X., Jiao, L. F., Teng, Z. Q., and Liu, C. M. (2022). Dynamic profiling and functional interpretation of histone lysine crotonylation and lactylation during neural development. Development 149:dev200049. doi: 10.1242/dev.200049
Dai, X., Lv, X., Thompson, E. W., and Ostrikov, K. K. (2022). Histone lactylation: epigenetic mark of a glycolytic switch. Trends Genet. 38, 124–127. doi: 10.1016/j.tig.2021.09.009
Dai, S. J., Shao, Y. Y., Zheng, Y., Sun, J. Y., Li, Z. S., Shi, J. Y., et al. (2023). Inflachromene attenuates seizure severity in mouse epilepsy models via inhibiting HMGB1 translocation. Acta Pharmacol. Sin. 44, 1737–1747. doi: 10.1038/s41401-023-01087-6
Dai, Y., Wei, T., Shen, Z., Bei, Y., Lin, H., and Dai, H. (2021). Classical HDACs in the regulation of neuroinflammation. Neurochem. Int. 150:105182. doi: 10.1016/j.neuint.2021.105182
de Ceglia, R., Ledonne, A., Litvin, D. G., Lind, B. L., Carriero, G., Latagliata, E. C., et al. (2023). Specialized astrocytes mediate glutamatergic gliotransmission in the CNS. Nature 622, 120–129. doi: 10.1038/s41586-023-06502-w
Ding, Y., Xue, Q., Liu, S., Hu, K., Wang, D., Wang, T., et al. (2020). Identification of parthenolide dimers as activators of pyruvate kinase M2 in xenografts of glioblastoma multiforme in vivo. J. Med. Chem. 63, 1597–1611. doi: 10.1021/acs.jmedchem.9b01328
Dong, H., Zhang, J., Zhang, H., Han, Y., Lu, C., Chen, C., et al. (2022). YiaC and CobB regulate lysine lactylation in Escherichia coli. Nat. Commun. 13:6628. doi: 10.1038/s41467-022-34399-y
Du, J., Liu, J., Huang, X., Li, Y., Song, D., Li, Q., et al. (2022). Catalpol ameliorates neurotoxicity in N2a/APP695swe cells and APP/PS1 transgenic mice. Neurotox. Res. 40, 961–972. doi: 10.1007/s12640-022-00524-4
Dyńka, D., Kowalcze, K., and Paziewska, A. (2022). The role of ketogenic diet in the treatment of neurological diseases. Nutrients 14:5003. doi: 10.3390/nu14235003
Endres, M., Moro, M. A., Nolte, C. H., Dames, C., Buckwalter, M. S., and Meisel, A. (2022). Immune pathways in etiology, acute phase, and chronic sequelae of ischemic stroke. Circ. Res. 130, 1167–1186. doi: 10.1161/CIRCRESAHA.121.319994
Eschricht, S., Jarr, K. U., Kuhn, C., Lehmann, L., Kreusser, M., Katus, H. A., et al. (2015). Heat-shock-protein 90 protects from downregulation of HIF-1α in calcineurin-induced myocardial hypertrophy. J. Mol. Cell. Cardiol. 85, 117–126. doi: 10.1016/j.yjmcc.2015.05.018
Esih, K., Goričar, K., Soltirovska-Šalamon, A., Dolžan, V., and Rener-Primec, Z. (2021). Genetic polymorphisms, gene-gene interactions, and neurologic sequelae at two years follow-up in newborns with hypoxic-ischemic encephalopathy treated with hypothermia. Antioxidants (Basel) 10:1495. doi: 10.3390/antiox10091495
Eugen, T., Patrick, K., ByungIn, L., and Amitabh, D. (2019). Epilepsy in Asia: disease burden, management barriers, and challenges. Epilepsia 60, 7–21. doi: 10.1111/epi.14458
Fan, M., Yang, K., Wang, X., Chen, L., Gill, P. S., Ha, T., et al. (2023). Lactate promotes endothelial-to-mesenchymal transition via Snail1 lactylation after myocardial infarction. Sci. Adv. 9:eadc9465. doi: 10.1126/sciadv.adc9465
Fan, H., Yang, F., Xiao, Z., Luo, H., Chen, H., Chen, Z., et al. (2023). Lactylation: novel epigenetic regulatory and therapeutic opportunities. Am. J. Physiol. Endocrinol. Metab. 324, E330–E338. doi: 10.1152/ajpendo.00159.2022
Fan, Z., Zheng, W., Li, H., Wu, W., Liu, X., Sun, Z., et al. (2020). LOXL2 upregulates hypoxia-inducible factor-1α signaling through snail-FBP1 axis in hepatocellular carcinoma cells. Oncol. Rep. 43, 1641–1649. doi: 10.3892/or.2020.7541
Fedotova, E. I., Dolgacheva, L. P., Abramov, A. Y., and Berezhnov, A. V. (2021). Lactate and pyruvate activate autophagy and mitophagy that protect cells in toxic model of Parkinson's disease. Mol. Neurobiol. 59, 177–190. doi: 10.1007/s12035-021-02583-8
Feng, Q., Liu, Z., Yu, X., Huang, T., Chen, J., Wang, J., et al. (2022). Lactate increases stemness of CD8 + T cells to augment anti-tumor immunity. Nat. Commun. 13:4981. doi: 10.1038/s41467-022-32521-8
Feng, L., Murugan, M., Bosco, D. B., Liu, Y., Peng, J., Worrell, G. A., et al. (2019). Microglial proliferation and monocyte infiltration contribute to microgliosis following status epilepticus. Glia 67, 1434–1448. doi: 10.1002/glia.23616
Fouché, B., Turner, S., Gorham, R., Stephenson, E. J., Gutbier, S., Elson, J. L., et al. (2023). A novel mitochondria-targeting iron chelator neuroprotects multimodally via HIF-1 modulation against a mitochondrial toxin in a dopaminergic cell model of Parkinson's disease. Mol. Neurobiol. 60, 749–767. doi: 10.1007/s12035-022-03107-8
Fu, Z. J., Wang, Z. Y., Xu, L., Chen, X. H., Li, X. X., Liao, W. T., et al. (2020). HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 36:101671. doi: 10.1016/j.redox.2020.101671
Fu, Y., Xiang, Y., Wang, Y., Liu, Z., Yang, D., Zha, J., et al. (2023). The STAT1/HMGB1/NF-κB pathway in chronic inflammation and kidney injury after cisplatin exposure. Theranostics 13, 2757–2773. doi: 10.7150/thno.81406
Galle, E., Wong, C. W., Ghosh, A., Desgeorges, T., Melrose, K., Hinte, L. C., et al. (2022). H3K18 lactylation marks tissue-specific active enhancers. Genome Biol. 23:207. doi: 10.1186/s13059-022-02775-y
Gao, R., Li, Y., Xu, Z., Zhang, F., Xu, J., Hu, Y., et al. (2023). Mitochondrial pyruvate carrier 1 regulates fatty acid synthase lactylation and mediates treatment of nonalcoholic fatty liver disease. Hepatology 78, 1800–1815. doi: 10.1097/HEP.0000000000000279
Ge, C., Peng, Y., Li, J., Wang, L., Zhu, X., Wang, N., et al. (2023). Hydroxysafflor yellow a alleviates acute myocardial ischemia/reperfusion injury in mice by inhibiting ferroptosis via the activation of the HIF-1α/SLC7A11/GPX4 signaling pathway. Nutrients 15:3411. doi: 10.3390/nu15153411
Genders, A. J., Martin, S. D., McGee, S. L., and Bishop, D. J. (2019). A physiological drop in pH decreases mitochondrial respiration, and HDAC and Akt signaling, in L6 myocytes. Am. J. Physiol. Cell Physiol. 316, C404–C414. doi: 10.1152/ajpcell.00214.2018
Geng, H., Harvey, C. T., Pittsenbarger, J., Liu, Q., Beer, T. M., Xue, C., et al. (2011). HDAC4 protein regulates HIF1α protein lysine acetylation and cancer cell response to hypoxia. J. Biol. Chem. 286, 38095–38102. doi: 10.1074/jbc.M111.257055
Gong, Y., Tang, N., Liu, P., Sun, Y., Lu, S., Liu, W., et al. (2021). Newcastle disease virus degrades SIRT3 via PINK1-PRKN-dependent mitophagy to reprogram energy metabolism in infected cells. Autophagy 18, 1503–1521. doi: 10.1080/15548627.2021.1990515
González-Rodríguez, P., and Fernández-López, A. (2023). PANoptosis: new insights in regulated cell death in ischemia/reperfusion models. Neural Regen. Res. 18, 342–343. doi: 10.4103/1673-5374.343910
Gu, C., Kong, F., Zeng, J., Geng, X., Sun, Y., and Chen, X. (2023). Remote ischemic preconditioning protects against spinal cord ischemia-reperfusion injury in mice by activating NMDAR/AMPK/PGC-1α/SIRT3 signaling. Cell Biosci. 13:57. doi: 10.1186/s13578-023-00999-4
Haenisch, S., Zhao, Y., Chhibber, A., Kaiboriboon, K., Do, L. V., Vogelgesang, S., et al. (2015). SOX11 identified by target gene evaluation of miRNAs differentially expressed in focal and non-focal brain tissue of therapy-resistant epilepsy patients. Neurobiol. Dis. 77, 127–140. doi: 10.1016/j.nbd.2015.02.025
Hagihara, H., Shoji, H., Otabi, H., Toyoda, A., Katoh, K., Namihira, M., et al. (2021). Protein lactylation induced by neural excitation. Cell Rep. 37:109820. doi: 10.1016/j.celrep.2021.109820
He, D. L., Fan, Y. G., and Wang, Z. Y. (2023). Energy crisis links to autophagy and ferroptosis in Alzheimer's disease: current evidence and future avenues. Curr. Neuropharmacol. 21, 67–86. doi: 10.2174/1570159X20666220817140737
He, Y., Ji, Z., Gong, Y., Fan, L., Xu, P., Chen, X., et al. (2023). Numb/Parkin-directed mitochondrial fitness governs cancer cell fate via metabolic regulation of histone lactylation. Cell Rep. 42:112033. doi: 10.1016/j.celrep.2023.112033
Heim, C. E., Bosch, M. E., Yamada, K. J., Aldrich, A. L., Chaudhari, S. S., Klinkebiel, D., et al. (2020). Lactate production by Staphylococcus aureus biofilm inhibits HDAC11 to reprogramme the host immune response during persistent infection. Nat. Microbiol. 5, 1271–1284. doi: 10.1038/s41564-020-0756-3
Hou, K. L., Lin, S. K., Chao, L. H., Hsiang-Hua Lai, E., Chang, C. C., Shun, C. T., et al. (2017). Sirtuin 6 suppresses hypoxia-induced inflammatory response in human osteoblasts via inhibition of reactive oxygen species production and glycolysis-a therapeutic implication in inflammatory bone resorption. Biofactors 43, 170–180. doi: 10.1002/biof.1320
Hu, Y., Yao, Y., Qi, H., Yang, J., Zhang, C., Zhang, A., et al. (2023). Microglia sense and suppress epileptic neuronal hyperexcitability. Pharmacol. Res. 195:106881. doi: 10.1016/j.phrs.2023.106881
Hu, L., Zeng, Z., Xia, Q., Liu, Z., Feng, X., Chen, J., et al. (2019). Metformin attenuates hepatoma cell proliferation by decreasing glycolytic flux through the HIF-1α/PFKFB3/PFK1 pathway. Life Sci. 239:116966. doi: 10.1016/j.lfs.2019.116966
Huang, X., Zhao, L., and Peng, R. (2022). Hypoxia-inducible factor 1 and mitochondria: an intimate connection. Biomol. Ther. 13:50. doi: 10.3390/biom13010050
Hubbi, M. E., Hu, H., Kshitiz, G. D. M., and Semenza, G. L. (2013). Sirtuin-7 inhibits the activity of hypoxia-inducible factors. J. Biol. Chem. 288, 20768–20775. doi: 10.1074/jbc.M113.476903
Icard, P., Alifano, M., and Simula, L. (2023). The potential for citrate to reinforce epigenetic therapy by promoting apoptosis. Trends Endocrinol. Metab. 34, 586–589. doi: 10.1016/j.tem.2023.07.002
Ip, W. K. E., Hoshi, N., Shouval, D. S., Snapper, S., and Medzhitov, R. (2017). Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513–519. doi: 10.1126/science.aal3535
Islam, R., Dash, D., and Singh, R. (2023). An antioxidant ameliorates allergic airway inflammation by inhibiting HDAC 1 via HIF-1α/VEGF axis suppression in mice. Sci. Rep. 13:9637. doi: 10.1038/s41598-023-36678-0
Jády, A. G., Nagy, Á. M., Kőhidi, T., Ferenczi, S., Tretter, L., and Madarász, E. (2016). Differentiation-dependent energy production and metabolite utilization: a comparative study on neural stem cells, neurons, and astrocytes. Stem Cells Dev. 25, 995–1005. doi: 10.1089/scd.2015.0388
Jia, L., Liao, M., Mou, A., Zheng, Q., Yang, W., Yu, Z., et al. (2021). Rheb-regulated mitochondrial pyruvate metabolism of Schwann cells linked to axon stability. Dev. Cell 56, 2980–94.e6. doi: 10.1016/j.devcel.2021.09.013
Jia, X., Wang, Q., Ji, J., Lu, W., Liu, Z., Tian, H., et al. (2023). Mitochondrial transplantation ameliorates hippocampal damage following status epilepticus. Animal Model Exp. Med. 6, 41–50. doi: 10.1002/ame2.12310
Jiang, J., Huang, D., Jiang, Y., Hou, J., Tian, M., Li, J., et al. (2021). Lactate modulates cellular metabolism through histone lactylation-mediated gene expression in non-small cell lung cancer. Front. Oncol. 11:647559. doi: 10.3389/fonc.2021.647559
Jiang, Q., Tang, G., Zhong, X. M., Ding, D. R., Wang, H., and Li, J. N. (2021). Role of Stat3 in NLRP3/caspase-1-mediated hippocampal neuronal pyroptosis in epileptic mice. Synapse 75:e22221. doi: 10.1002/syn.22221
Jin, Y., Zhang, Z., Yu, Q., Zeng, Z., Song, H., Huang, X., et al. (2021). Positive reciprocal feedback of lncRNA ZEB1-AS1 and HIF-1 α contributes to hypoxia-promoted tumorigenesis and metastasis of pancreatic cancer. Front. Oncol. 11:761979. doi: 10.3389/fonc.2021.761979
Kaitsuka, T., Matsushita, M., and Matsushita, N. (2020). SIRT2 inhibition activates hypoxia-inducible factor 1α signaling and mediates neuronal survival. Biochem. Biophys. Res. Commun. 529, 957–962. doi: 10.1016/j.bbrc.2020.06.159
Kantarci, H., Gou, Y., and Riley, B. B. (2020). The Warburg effect and lactate signaling augment Fgf-MAPK to promote sensory-neural development in the otic vesicle. eLife 9:9. doi: 10.7554/eLife.56301
Karagiota, A., Kanoura, A., Paraskeva, E., Simos, G., and Chachami, G. (2023). Pyruvate dehydrogenase phosphatase 1 (PDP1) stimulates HIF activity by supporting histone acetylation under hypoxia. FEBS J. 290, 2165–2179. doi: 10.1111/febs.16694
Karnib, N., El-Ghandour, R., El Hayek, L., Nasrallah, P., Khalifeh, M., Barmo, N., et al. (2019). Lactate is an antidepressant that mediates resilience to stress by modulating the hippocampal levels and activity of histone deacetylases. Neuropsychopharmacology 44, 1152–1162. doi: 10.1038/s41386-019-0313-z
Kaya, Z., Belder, N., Sever-Bahcekapili, M., Donmez-Demir, B., Erdener, Ş. E., Bozbeyoglu, N., et al. (2023). Vesicular HMGB1 release from neurons stressed with spreading depolarization enables confined inflammatory signaling to astrocytes. J. Neuroinflammation 20:295. doi: 10.1186/s12974-023-02977-6
Kim, J. Y., Cho, H., Yoo, J., Kim, G. W., Jeon, Y. H., Lee, S. W., et al. (2023). HDAC8 deacetylates HIF-1α and enhances its protein stability to promote tumor growth and migration in melanoma. Cancers (Basel) 15:1123. doi: 10.3390/cancers15041123
Kim, S. W., and Lee, J. K. (2020). Role of HMGB1 in the interplay between NETosis and thrombosis in ischemic stroke: a review. Cells 9:1794. doi: 10.3390/cells9081794
Kim, J. E., Park, H., Kim, T. H., and Kang, T. C. (2021). LONP1 regulates mitochondrial accumulations of HMGB1 and Caspase-3 in CA1 and PV neurons following status epilepticus. Int. J. Mol. Sci. 22:2275. doi: 10.3390/ijms22052275
Kitaoka, S., Tomohiro, A., Ukeshima, S., Liu, K., Wake, H., Kimura, S. H., et al. (2023). Repeated social defeat stress induces HMGB1 nuclear export in prefrontal neurons, leading to social avoidance in mice. Cells 12:1789. doi: 10.3390/cells12131789
Komilova, N. R., Angelova, P. R., Berezhnov, A. V., Stelmashchuk, O. A., Mirkhodjaev, U. Z., Houlden, H., et al. (2021). Metabolically induced intracellular pH changes activate mitophagy, autophagy, and cell protection in familial forms of Parkinson's disease. FEBS J. 289, 699–711. doi: 10.1111/febs.16198
Korotkov, A., DWM, B., Banchaewa, L., Pustjens, B., van Scheppingen, J., Anink, J. J., et al. (2020). microRNA-132 is overexpressed in glia in temporal lobe epilepsy and reduces the expression of pro-epileptogenic factors in human cultured astrocytes. Glia 68, 60–75. doi: 10.1002/glia.23700
Kovács, Z., Skatchkov, S. N., Veh, R. W., Szabó, Z., Németh, K., Szabó, P. T., et al. (2021). Critical role of astrocytic polyamine and GABA metabolism in epileptogenesis. Front. Cell. Neurosci. 15:787319. doi: 10.3389/fncel.2021.787319
Krabbendam, I. E., Honrath, B., Dilberger, B., Iannetti, E. F., Branicky, R. S., Meyer, T., et al. (2020). SK channel-mediated metabolic escape to glycolysis inhibits ferroptosis and supports stress resistance in C. elegans. Cell Death Dis. 11:263. doi: 10.1038/s41419-020-2458-4
Kwak, M. S., Choi, S., Kim, J., Lee, H., Park, I. H., Oh, J., et al. (2023). SARS-CoV-2 infection induces HMGB1 secretion through post-translational modification and PANoptosis. Immune Netw. 23:e26. doi: 10.4110/in.2023.23.e26
Lanqing, Z., Jinwei, L., Reetta, K., Jukka, J., and Chuansheng, Z. (2021). Impact of drug treatment and drug interactions in post-stroke epilepsy. Pharmacol. Ther. 233:108030. doi: 10.1016/j.pharmthera.2021.108030
Lee, D. S., Kim, T. H., Park, H., and Kim, J. E. (2022). CDDO-me attenuates clasmatodendrosis in CA1 astrocyte by inhibiting HSP25-AKT mediated DRP1-S637 phosphorylation in chronic epilepsy rats. Int. J. Mol. Sci. 23:4569. doi: 10.3390/ijms23094569
Lei, C., Li, Y., Zhu, X., Li, H., and Chang, X. (2022). HMGB1/TLR4 induces autophagy and promotes neuroinflammation after intracerebral hemorrhage. Brain Res. 1792:148003. doi: 10.1016/j.brainres.2022.148003
Lestón Pinilla, L., Ugun-Klusek, A., Rutella, S., and De Girolamo, L. A. (2021). Hypoxia signaling in Parkinson's disease: there is use in asking "what HIF?". Biology (Basel) 10:723. doi: 10.3390/biology10080723
Li, B., and Freeman, R. D. (2015). Neurometabolic coupling between neural activity, glucose, and lactate in activated visual cortex. J. Neurochem. 135, 742–754. doi: 10.1111/jnc.13143
Li, Y., Fu, Y., Zhang, Y., Duan, B., Zhao, Y., Shang, M., et al. (2023). Nuclear fructose-1,6-bisphosphate inhibits tumor growth and sensitizes chemotherapy by targeting HMGB1. Adv. Sci. (Weinh) 10:e2203528. doi: 10.1002/advs.202203528
Li, Y., Liu, X. T., Zhang, P. L., Li, Y. C., Sun, M. R., Wang, Y. T., et al. (2022). Hydroxysafflor yellow a blocks HIF-1 α induction of NOX2 and protects ZO-1 protein in cerebral microvascular endothelium. Antioxidants (Basel) 11:728. doi: 10.3390/antiox11040728
Li, C., Xing, Y., Zhang, Y., Hua, Y., Hu, J., and Bai, Y. (2021). Neutrophil extracellular traps exacerbate ischemic brain damage. Mol. Neurobiol. 59, 643–656. doi: 10.1007/s12035-021-02635-z
Li, H. S., Zhou, Y. N., Li, L., Li, S. F., Long, D., Chen, X. L., et al. (2019). HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 25:101109. doi: 10.1016/j.redox.2019.101109
Liang, J., Liu, C., Xu, D., Xie, K., and Li, A. (2022). LncRNA NEAT1 facilitates glioma progression via stabilizing PGK1. J. Transl. Med. 20:80. doi: 10.1186/s12967-022-03273-2
Liang, P., Zhang, X., Zhang, Y., Wu, Y., Song, Y., Wang, X., et al. (2023). Neurotoxic A1 astrocytes promote neuronal ferroptosis via CXCL10/CXCR3 axis in epilepsy. Free Radic. Biol. Med. 195, 329–342. doi: 10.1016/j.freeradbiomed.2023.01.002
Liang, Z., Zheng, Z., Guo, Q., Tian, M., Yang, J., Liu, X., et al. (2023). The role of HIF-1α/HO-1 pathway in hippocampal neuronal ferroptosis in epilepsy. iScience 26:108098. doi: 10.1016/j.isci.2023.108098
Lin, J. F., Hu, P. S., Wang, Y. Y., Tan, Y. T., Yu, K., Liao, K., et al. (2022). Phosphorylated NFS1 weakens oxaliplatin-based chemosensitivity of colorectal cancer by preventing PANoptosis. Signal Transduct. Target. Ther. 7:54. doi: 10.1038/s41392-022-00889-0
Lin, J., Ji, Z., Di, Z., Zhang, Y., Yan, C., and Zeng, S. (2022). Overexpression of Tfap2a in mouse oocytes impaired spindle and chromosome organization. Int. J. Mol. Sci. 23:14376. doi: 10.3390/ijms232214376
Lin, J., Liu, G., Chen, L., Kwok, H. F., and Lin, Y. (2022). Targeting lactate-related cell cycle activities for cancer therapy. Semin. Cancer Biol. 86, 1231–1243. doi: 10.1016/j.semcancer.2022.10.009
Liu, J., Du, J., Li, Y., Wang, F., Song, D., Lin, J., et al. (2022). Catalpol induces apoptosis in breast cancer in vitro and in vivo: involvement of mitochondria apoptosis pathway and post-translational modifications. Toxicol. Appl. Pharmacol. 454:116215. doi: 10.1016/j.taap.2022.116215
Liu, L. X., Heng, J. H., Deng, D. X., Zhao, H., Zheng, Z. Y., Liao, L. D., et al. (2023). Sulconazole induces PANoptosis by triggering oxidative stress and inhibiting glycolysis to increase radiosensitivity in esophageal cancer. Mol. Cell. Proteomics 22:100551. doi: 10.1016/j.mcpro.2023.100551
Liu, Y. F., Hu, R., Zhang, L. F., Fan, Y., Xiao, J. F., and Liao, X. Z. (2022). Effects of dexmedetomidine on cognitive dysfunction and neuroinflammation via the HDAC2/HIF-1α/PFKFB3 axis in a murine model of postoperative cognitive dysfunction. J. Biochem. Mol. Toxicol. 36:e23044. doi: 10.1002/jbt.23044
Liu, J., Liu, Y., Wang, Y., Kang, R., and Tang, D. (2022). HMGB1 is a mediator of cuproptosis-related sterile inflammation. Front. Cell Dev. Biol. 10:996307. doi: 10.3389/fcell.2022.996307
Liu, L., Liu, Z., Zeng, C., Xu, Y., He, L., Fang, Q., et al. (2022). Dynorphin/KOR inhibits neuronal autophagy by activating mTOR signaling pathway to prevent acute seizure epilepsy. Cell Biol. Int. 46, 1814–1824. doi: 10.1002/cbin.11874
Liu, X., Nie, L., Zhang, Y., Yan, Y., Wang, C., Colic, M., et al. (2023). Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell Biol. 25, 404–414. doi: 10.1038/s41556-023-01091-2
Liu, C., She, Y., Huang, J., Liu, Y., Li, W., Zhang, C., et al. (2022). HMGB1-NLRP3-P2X7R pathway participates in PM 2.5 -induced hippocampal neuron impairment by regulating microglia activation. Ecotoxicol. Environ. Saf. 239:113664. doi: 10.1016/j.ecoenv.2022.113664
Liu, L., Zhang, J., Zhang, X., Cheng, P., Liu, L., Huang, Q., et al. (2021). HMGB1: an important regulator of myeloid differentiation and acute myeloid leukemia as well as a promising therapeutic target. J. Mol. Med. (Berl) 99, 107–118. doi: 10.1007/s00109-020-01998-5
Longhitano, L., Giallongo, S., Orlando, L., Broggi, G., Longo, A., Russo, A., et al. (2022). Lactate rewrites the metabolic reprogramming of uveal melanoma cells and induces quiescence phenotype. Int. J. Mol. Sci. 24:24. doi: 10.3390/ijms24010024
Lopez, G., Bill, K. L., Bid, H. K., Braggio, D., Constantino, D., Prudner, B., et al. (2015). HDAC8, a potential therapeutic target for the treatment of malignant peripheral nerve sheath tumors (MPNST). PLoS One 10:e0133302. doi: 10.1371/journal.pone.0133302
Lopez Krol, A., Nehring, H. P., Krause, F. F., Wempe, A., Raifer, H., Nist, A., et al. (2022). Lactate induces metabolic and epigenetic reprogramming of pro-inflammatory Th17 cells. EMBO Rep. 23:e54685. doi: 10.15252/embr.202254685
Lu, J., Fu, S., Dai, J., Hu, J., Li, S., Ji, H., et al. (2022). Integrated metabolism and epigenetic modifications in the macrophages of mice in responses to cold stress. J Zhejiang Univ Sci B 23, 461–480. doi: 10.1631/jzus.B2101091
Lu, X., Wang, L., Yu, C., Yu, D., and Yu, G. (2015). Histone acetylation modifiers in the pathogenesis of Alzheimer's disease. Front. Cell. Neurosci. 9:226. doi: 10.3389/fncel.2015.00226
Luo, H. M., Du, M. H., Lin, Z. L., Zhang, L., Ma, L., Wang, H., et al. (2013). Valproic acid treatment inhibits hypoxia-inducible factor 1α accumulation and protects against burn-induced gut barrier dysfunction in a rodent model. PLoS One 8:e77523. doi: 10.1371/journal.pone.0077523
Luo, M., Liu, Z., Hu, Z., and He, Q. (2022). Quercetin improves contrast-induced acute kidney injury through the HIF-1α/lncRNA NEAT1/HMGB1 pathway. Pharm. Biol. 60, 889–898. doi: 10.1080/13880209.2022.2058558
Luo, Y., Yang, Z., Yu, Y., and Zhang, P. (2022). HIF1α lactylation enhances KIAA1199 transcription to promote angiogenesis and vasculogenic mimicry in prostate cancer. Int. J. Biol. Macromol. 222, 2225–2243. doi: 10.1016/j.ijbiomac.2022.10.014
Lv, Y., Zhang, B., Zhai, C., Qiu, J., Zhang, Y., Yao, W., et al. (2015). PFKFB3-mediated glycolysis is involved in reactive astrocyte proliferation after oxygen-glucose deprivation/reperfusion and is regulated by Cdh1. Neurochem. Int. 91, 26–33. doi: 10.1016/j.neuint.2015.10.006
Ma, W., Ao, S., Zhou, J., Li, J., Liang, X., Yang, X., et al. (2022). Methylsulfonylmethane protects against lethal dose MRSA-induced sepsis through promoting M2 macrophage polarization. Mol. Immunol. 146, 69–77. doi: 10.1016/j.molimm.2022.04.001
Ma, S., Sun, L., Wu, W., Wu, J., Sun, Z., and Ren, J. (2020). USP22 protects against myocardial ischemia-reperfusion injury via the SIRT1-p53/SLC7A11-dependent inhibition of ferroptosis-induced cardiomyocyte death. Front. Physiol. 11:551318. doi: 10.3389/fphys.2020.551318
Maschari, D., Saxena, G., Law, T. D., Walsh, E., Campbell, M. C., and Consitt, L. A. (2022). Lactate-induced lactylation in skeletal muscle is associated with insulin resistance in humans. Front. Physiol. 13:951390. doi: 10.3389/fphys.2022.951390
Mason, S. (2017). Lactate shuttles in neuroenergetics-homeostasis, allostasis and beyond. Front. Neurosci. 11:43. doi: 10.3389/fnins.2017.00043
Mayorga-Weber, G., Rivera, F. J., and Castro, M. A. (2022). Neuron-glia (mis)interactions in brain energy metabolism during aging. J. Neurosci. Res. 100, 835–854. doi: 10.1002/jnr.25015
Moos, W. H., Faller, D. V., Glavas, I. P., Kanara, I., Kodukula, K., Pernokas, J., et al. (2023). Epilepsy: mitochondrial connections to the 'Sacred' disease. Mitochondrion 72, 84–101. doi: 10.1016/j.mito.2023.08.002
Moreno-Yruela, C., Zhang, D., Wei, W., Bæk, M., Liu, W., Gao, J., et al. (2022). Class I histone deacetylases (HDAC1-3) are histone lysine delactylases. Sci. Adv. 8:eabi6696. doi: 10.1126/sciadv.abi6696
Moulin, S., Thomas, A., Wagner, S., Arzt, M., Dubouchaud, H., Lamarche, F., et al. (2022). Intermittent hypoxia-induced cardiomyocyte death is mediated by HIF-1 dependent MAM disruption. Antioxidants (Basel) 11:1462. doi: 10.3390/antiox11081462
Nagy, A. M., Fekete, R., Horvath, G., Koncsos, G., Kriston, C., Sebestyen, A., et al. (2018). Versatility of microglial bioenergetic machinery under starving conditions. Biochim. Biophys. Acta Bioenerg. 1859, 201–214. doi: 10.1016/j.bbabio.2017.12.002
Naia, L., Cunha-Oliveira, T., Rodrigues, J., Rosenstock, T. R., Oliveira, A., Ribeiro, M., et al. (2017). Histone deacetylase inhibitors protect against pyruvate dehydrogenase dysfunction in Huntington's disease. J. Neurosci. 37, 2776–2794. doi: 10.1523/JNEUROSCI.2006-14.2016
Ni, J., and Ni, A. (2021). Histone deacetylase inhibitor induced pVHL-independent degradation of HIF-1α and hierarchical quality control of pVHL via chaperone system. PLoS One 16:e0248019. doi: 10.1371/journal.pone.0248019
Nicolo, J. P., O'Brien, T. J., and Kwan, P. (2019). Role of cerebral glutamate in post-stroke epileptogenesis. Neuroimage Clin. 24:102069. doi: 10.1016/j.nicl.2019.102069
Pan, L., Feng, F., Wu, J., Fan, S., Han, J., Wang, S., et al. (2022). Demethylzeylasteral targets lactate by inhibiting histone lactylation to suppress the tumorigenicity of liver cancer stem cells. Pharmacol. Res. 181:106270. doi: 10.1016/j.phrs.2022.106270
Pan, R. Y., He, L., Zhang, J., Liu, X., Liao, Y., Gao, J., et al. (2022). Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer's disease. Cell Metab. 34, 634–48.e6. doi: 10.1016/j.cmet.2022.02.013
Pan, W., Song, X., Hu, Q., and Zhang, Y. (2021). miR-485 inhibits histone deacetylase HDAC5, HIF1α and PFKFB3 expression to alleviate epilepsy in cellular and rodent models. Aging (Albany NY) 13, 14416–14432. doi: 10.18632/aging.203058
Pan, J., and Zhao, L. (2021). Long non-coding RNA histone deacetylase 4 antisense RNA 1 (HDAC4-AS1) inhibits HDAC4 expression in human ARPE-19 cells with hypoxic stress. Bioengineered 12, 2228–2237. doi: 10.1080/21655979.2021.1933821
Park, S. J., Kim, H., Kim, S. H., Joe, E. H., and Jou, I. (2019). Epigenetic downregulation of STAT6 increases HIF-1α expression via mTOR/S6K/S6, leading to enhanced hypoxic viability of glioma cells. Acta Neuropathol. Commun. 7:149. doi: 10.1186/s40478-019-0798-z
Park, K. C., Kim, S. W., Park, J. H., Song, E. H., Yang, J. W., Chung, H. J., et al. (2011). Potential anti-cancer activity of N-hydroxy-7-(2-naphthylthio) heptanomide (HNHA), a histone deacetylase inhibitor, against breast cancer both in vitro and in vivo. Cancer Sci. 102, 343–350. doi: 10.1111/j.1349-7006.2010.01798.x
Parsons, A. L. M., Bucknor, E. M. V., Castroflorio, E., Soares, T. R., Oliver, P. L., and Rial, D. (2022). The interconnected mechanisms of oxidative stress and neuroinflammation in epilepsy. Antioxidants (Basel) 11:157. doi: 10.3390/antiox11010157
Paudel, Y. N., Angelopoulou, E., Akyuz, E., Piperi, C., Othman, I., and Shaikh, M. F. (2020). Role of innate immune receptor TLR4 and its endogenous ligands in epileptogenesis. Pharmacol. Res. 160:105172. doi: 10.1016/j.phrs.2020.105172
Pauletti, A., Terrone, G., Shekh-Ahmad, T., Salamone, A., Ravizza, T., Rizzi, M., et al. (2019). Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain 142:e39. doi: 10.1093/brain/awz130
Peng, T., Du, S. Y., Son, M., and Diamond, B. (2021). HIF-1α is a negative regulator of interferon regulatory factors: implications for interferon production by hypoxic monocytes. Proc. Natl. Acad. Sci. USA 118:e2106017118. doi: 10.1073/pnas.2106017118
Peterson, A. R., Garcia, T. A., Cullion, K., Tiwari-Woodruff, S. K., Pedapati, E. V., and Binder, D. K. (2021). Targeted overexpression of glutamate transporter-1 reduces seizures and attenuates pathological changes in a mouse model of epilepsy. Neurobiol. Dis. 157:105443. doi: 10.1016/j.nbd.2021.105443
Puri, B. K., Kingston, M. C., and Monro, J. A. (2019). Fructose-associated hepatotoxicity indexed by the lactate dehydrogenase isoenzyme LDH-5. Med. Hypotheses 124, 40–41. doi: 10.1016/j.mehy.2019.02.019
Qiu, L. L., Tan, X. X., Yang, J. J., Ji, M. H., Zhang, H., Zhao, C., et al. (2023). Lactate improves Long-term cognitive impairment induced by repeated neonatal sevoflurane exposures through SIRT1-mediated regulation of adult hippocampal neurogenesis and synaptic plasticity in male mice. Mol. Neurobiol. 60, 5273–5291. doi: 10.1007/s12035-023-03413-9
Radoul, M., Najac, C., Viswanath, P., Mukherjee, J., Kelly, M., Gillespie, A. M., et al. (2019). HDAC inhibition in glioblastoma monitored by hyperpolarized 13 C MRSI. NMR Biomed. 32:e4044. doi: 10.1002/nbm.4044
Rho, H., Terry, A. R., Chronis, C., and Hay, N. (2023). Hexokinase 2-mediated gene expression via histone lactylation is required for hepatic stellate cell activation and liver fibrosis. Cell Metab. 35, 1406–23.e8. doi: 10.1016/j.cmet.2023.06.013
Rodrigues, M. F., Carvalho, É., Pezzuto, P., Rumjanek, F. D., and Amoêdo, N. D. (2015). Reciprocal modulation of histone deacetylase inhibitors sodium butyrate and trichostatin a on the energy metabolism of breast cancer cells. J. Cell. Biochem. 116, 797–808. doi: 10.1002/jcb.25036
Roh, Y., Lee, S. B., Kim, M., Kim, M. H., Kim, H. J., and Cho, K. O. (2023). Alleviation of hippocampal necroptosis and neuroinflammation by NecroX-7 treatment after acute seizures. Front. Pharmacol. 14:1187819. doi: 10.3389/fphar.2023.1187819
Rosciszewski, G., Cadena, V., Auzmendi, J., Cieri, M. B., Lukin, J., Rossi, A. R., et al. (2019). Detrimental effects of HMGB-1 require microglial-astroglial interaction: implications for the status epilepticus -induced neuroinflammation. Front. Cell. Neurosci. 13:380. doi: 10.3389/fncel.2019.00380
Sailhamer, E. A., Li, Y., Smith, E. J., Liu, B., Shuja, F., Soupir, C. P., et al. (2010). Hypoxic "second hit" in leukocytes from trauma patients: modulation of the immune response by histone deacetylase inhibition. Cytokine 49, 303–311. doi: 10.1016/j.cyto.2009.11.013
Sanguigno, L., Guida, N., Anzilotti, S., Cuomo, O., Mascolo, L., Serani, A., et al. (2023). Stroke by inducing HDAC9-dependent deacetylation of HIF-1 and Sp1, promotes TfR1 transcription and GPX4 reduction, thus determining ferroptotic neuronal death. Int. J. Biol. Sci. 19, 2695–2710. doi: 10.7150/ijbs.80735
Schulz, V., Basu, S., Freibert, S. A., Webert, H., Boss, L., Mühlenhoff, U., et al. (2023). Functional spectrum and specificity of mitochondrial ferredoxins FDX1 and FDX2. Nat. Chem. Biol. 19, 206–217. doi: 10.1038/s41589-022-01159-4
Shakespear, M. R., Hohenhaus, D. M., Kelly, G. M., Kamal, N. A., Gupta, P., Labzin, L. I., et al. (2013). Histone deacetylase 7 promotes toll-like receptor 4-dependent proinflammatory gene expression in macrophages. J. Biol. Chem. 288, 25362–25374. doi: 10.1074/jbc.M113.496281
Shang, L., Wang, L., Shi, X., Wang, N., Zhao, L., Wang, J., et al. (2020). HMGB1 was negatively regulated by HSF1 and mediated the TLR4/MyD88/NF-κB signal pathway in asthma. Life Sci. 241:117120. doi: 10.1016/j.lfs.2019.117120
She, R., Liu, D., Liao, J., Wang, G., Ge, J., and Mei, Z. (2023). Mitochondrial dysfunctions induce PANoptosis and ferroptosis in cerebral ischemia/reperfusion injury: from pathology to therapeutic potential. Front. Cell. Neurosci. 17:1191629. doi: 10.3389/fncel.2023.1191629
Shen, K., Mao, Q., Yin, X., Zhang, C., Jin, Y., Deng, A., et al. (2018). NLRP3 inflammasome activation leads to epileptic neuronal apoptosis. Curr. Neurovasc. Res. 15, 276–281. doi: 10.2174/1567202616666181122165540
Shen, W., Pristov, J. B., Nobili, P., and Nikolić, L. (2023). Can glial cells save neurons in epilepsy? Neural Regen. Res. 18, 1417–1422. doi: 10.4103/1673-5374.360281
Shi, C., Cao, P., Wang, Y., Zhang, Q., Zhang, D., Wang, Y., et al. (2023). PANoptosis: a cell death characterized by pyroptosis, apoptosis, and necroptosis. J. Inflamm. Res. 16, 1523–1532. doi: 10.2147/JIR.S403819
Shu, L., Xu, C. Q., Yan, Z. Y., Yan, Y., Jiang, S. Z., and Wang, Y. R. (2019). Post-stroke microglia induce Sirtuin2 expression to suppress the anti-inflammatory function of infiltrating regulatory T cells. Inflammation 42, 1968–1979. doi: 10.1007/s10753-019-01057-3
Simeone, T. A., Simeone, K. A., and Rho, J. M. (2017). Ketone bodies as anti-seizure agents. Neurochem. Res. 42, 2011–2018. doi: 10.1007/s11064-017-2253-5
Sixto-López, Y., Rosales-Hernández, M. C., Contis-Montes de Oca, A., Fragoso-Morales, L. G., Mendieta-Wejebe, J. E., Correa-Basurto, A. M., et al. (2020). N -(2'-Hydroxyphenyl)-2-Propylpentanamide (HO-AAVPA) inhibits HDAC1 and increases the translocation of HMGB1 levels in human cervical Cancer cells. Int. J. Mol. Sci. 21:5873. doi: 10.3390/ijms21165873
Song, S., Ding, Y., Dai, G. L., Zhang, Y., Xu, M. T., Shen, J. R., et al. (2021). Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharmacol. Sin. 42, 230–241. doi: 10.1038/s41401-020-0490-7
Song, Y., Zou, X., Zhang, D., Liu, S., Duan, Z., and Liu, L. (2020). Self-enforcing HMGB1/NF-κB/HIF-1α feedback loop promotes cisplatin resistance in hepatocellular carcinoma cells. J. Cancer 11, 3893–3902. doi: 10.7150/jca.42944
Sun, Y., Chen, Y., Xu, Y., Zhang, Y., Lu, M., Li, M., et al. (2022). Genetic encoding of ε- N -L-lactyllysine for detecting delactylase activity in living cells. Chem. Commun. (Camb.) 58, 8544–8547. doi: 10.1039/D2CC02643K
Sun, D., Tan, Z. B., Sun, X. D., Liu, Z. P., Chen, W. B., Milibari, L., et al. (2021). Hippocampal astrocytic neogenin regulating glutamate uptake, a critical pathway for preventing epileptic response. Proc. Natl. Acad. Sci. USA 118:e2022921118. doi: 10.1073/pnas.2022921118
Sun, S., Xu, X., Liang, L., Wang, X., Bai, X., Zhu, L., et al. (2021). Lactic acid-producing probiotic Saccharomyces cerevisiae attenuates ulcerative colitis via suppressing macrophage pyroptosis and modulating gut microbiota. Front. Immunol. 12:777665. doi: 10.3389/fimmu.2021.777665
Sun, L., Zhang, H., and Gao, P. (2021). Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 13, 877–919. doi: 10.1007/s13238-021-00846-7
Tang, D., Kang, R., Zeh, H. J., and Lotze, M. T. (2023). The multifunctional protein HMGB1: 50 years of discovery. Nat. Rev. Immunol. 23, 824–841. doi: 10.1038/s41577-023-00894-6
Tauffenberger, A., Fiumelli, H., Almustafa, S., and Magistretti, P. J. (2019). Lactate and pyruvate promote oxidative stress resistance through hormetic ROS signaling. Cell Death Dis. 10:653. doi: 10.1038/s41419-019-1877-6
Thakur, V., Gonzalez, M. A., Parada, M., Martinez, R. D., and Chattopadhyay, M. (2024). Role of histone deacetylase inhibitor in diabetic painful neuropathy. Mol. Neurobiol. 61, 2283–2296. doi: 10.1007/s12035-023-03701-4
Tong, Y., Kai, J., Wang, S., Yu, Y., Xie, S., Zheng, H., et al. (2021). VHL regulates the sensitivity of clear cell renal cell carcinoma to SIRT4-mediated metabolic stress via HIF-1α/HO-1 pathway. Cell Death Dis. 12:621. doi: 10.1038/s41419-021-03901-7
Tröscher, A. R., Gruber, J., Wagner, J. N., Böhm, V., Wahl, A. S., and von Oertzen, T. J. (2021). Inflammation mediated epileptogenesis as possible mechanism underlying ischemic post-stroke epilepsy. Front. Aging Neurosci. 13:781174. doi: 10.3389/fnagi.2021.781174
Tzilivaki, A., Tukker, J. J., Maier, N., Poirazi, P., Sammons, R. P., and Schmitz, D. (2023). Hippocampal GABAergic interneurons and memory. Neuron 111, 3154–3175. doi: 10.1016/j.neuron.2023.06.016
Ubaid, S., Pandey, S., Akhtar, M. S., Rumman, M., Singh, B., and Mahdi, A. A. (2022). SIRT1 mediates neuroprotective and neurorescue effects of camel α-lactalbumin and oleic acid complex on rotenone-induced Parkinson's disease. ACS Chem. Neurosci. 13, 1263–1272. doi: 10.1021/acschemneuro.1c00876
Wagner, W., Ciszewski, W. M., and Kania, K. D. (2015). L- and D-lactate enhance DNA repair and modulate the resistance of cervical carcinoma cells to anticancer drugs via histone deacetylase inhibition and hydroxycarboxylic acid receptor 1 activation. Cell Commun. Signal 13:36. doi: 10.1186/s12964-015-0114-x
Wang, D., Cao, L., Zhou, X., Wang, G., Ma, Y., Hao, X., et al. (2022). Mitigation of honokiol on fluoride-induced mitochondrial oxidative stress, mitochondrial dysfunction, and cognitive deficits through activating AMPK/PGC-1α/Sirt3. J. Hazard. Mater. 437:129381. doi: 10.1016/j.jhazmat.2022.129381
Wang, Y., Du, J., Lu, S., Li, X., Chen, Y., Yuan, C., et al. (2022). PKCα phosphorylation of GLT-1 at Ser562/563 induces glutamate excitotoxicity in ischemia in mice. Signal Transduct. Target. Ther. 7:82. doi: 10.1038/s41392-022-00897-0
Wang, L., Li, B., Bo, X., Yi, X., Xiao, X., and Zheng, Q. (2022). Hypoxia-induced LncRNA DACT3-AS1 upregulates PKM2 to promote metastasis in hepatocellular carcinoma through the HDAC2/FOXA3 pathway. Exp. Mol. Med. 54, 848–860. doi: 10.1038/s12276-022-00767-3
Wang, B., Liu, S., Fan, B., Xu, X., Chen, Y., Lu, R., et al. (2018). PKM2 is involved in neuropathic pain by regulating ERK and STAT3 activation in rat spinal cord. J. Headache Pain 19:7. doi: 10.1186/s10194-018-0836-4
Wang, D., Liu, K., Fukuyasu, Y., Teshigawara, K., Fu, L., Wake, H., et al. (2020). HMGB1 translocation in neurons after ischemic insult: subcellular localization in mitochondria and peroxisomes. Cells 9:643. doi: 10.3390/cells9030643
Wang, J., Liu, R., Wang, Y., Mo, H., Niu, Y., Xu, Q., et al. (2021). Repression of the miR-627-5p by histone deacetylase 3 contributes to hypoxia-induced hepatocellular carcinoma progression. J. Cancer 12, 5320–5330. doi: 10.7150/jca.58697
Wang, N., Peng, Y. J., Su, X., Prabhakar, N. R., and Nanduri, J. (2021). Histone deacetylase 5 is an early epigenetic regulator of intermittent hypoxia induced sympathetic nerve activation and blood pressure. Front. Physiol. 12:688322. doi: 10.3389/fphys.2021.688322
Wang, C., Shen, D., Hu, Y., Chen, J., Liu, J., Huang, Y., et al. (2023). Selective targeting of class I HDAC reduces microglial inflammation in the entorhinal cortex of young APP/PS1 mice. Int. J. Mol. Sci. 24:4805. doi: 10.3390/ijms24054805
Wang, Z., Shu, W., Zhao, R., Liu, Y., and Wang, H. (2023). Sodium butyrate induces ferroptosis in endometrial cancer cells via the RBM3/SLC7A11 axis. Apoptosis 28, 1168–1183. doi: 10.1007/s10495-023-01850-4
Wang, X., Simayi, A., Fu, J., Zhao, X., and Xu, G. (2022). Resveratrol mediates the miR-149/HMGB1 axis and regulates the ferroptosis pathway to protect myocardium in endotoxemia mice. Am. J. Physiol. Endocrinol. Metab. 323, E21–E32. doi: 10.1152/ajpendo.00227.2021
Wang, N., Su, X., Sams, D., Prabhakar, N. R., and Nanduri, J. (2024). P300/CBP regulates HIF-1-dependent sympathetic activation and hypertension by intermittent hypoxia. Am. J. Respir. Cell Mol. Biol. 70, 110–118. doi: 10.1165/rcmb.2022-0481OC
Wang, H., Sugimoto, K., Lu, H., Yang, W. Y., Liu, J. Y., Yang, H. Y., et al. (2021). HDAC1-mediated deacetylation of HIF1α prevents atherosclerosis progression by promoting miR-224-3p-mediated inhibition of FOSL2. Mol. Ther. Nucleic Acids 23, 577–591. doi: 10.1016/j.omtn.2020.10.044
Wang, Q., Wei, S., Li, L., Qiu, J., Zhou, S., Shi, C., et al. (2020). TGR5 deficiency aggravates hepatic ischemic/reperfusion injury via inhibiting SIRT3/FOXO3/HIF-1ɑ pathway. Cell Death Discov. 6:116. doi: 10.1038/s41420-020-00347-2
Wang, J., Yun, F., Sui, J., Liang, W., Shen, D., and Zhang, Q. (2022). HAT- and HDAC-targeted protein acetylation in the occurrence and treatment of epilepsy. Biomedicines 11:88. doi: 10.3390/biomedicines11010088
Wang, Z. H., Zhang, P., Peng, W. B., Ye, L. L., Xiang, X., Wei, X. S., et al. (2023). Altered phenotypic and metabolic characteristics of FOXP3 + CD3 + CD56 + natural killer T (NKT)-like cells in human malignant pleural effusion. Onco Targets Ther 12:2160558. doi: 10.1080/2162402X.2022.2160558
Wei, L., Yang, X., Wang, J., Wang, Z., Wang, Q., Ding, Y., et al. (2023). H3K18 lactylation of senescent microglia potentiates brain aging and Alzheimer's disease through the NFκB signaling pathway. J. Neuroinflammation 20:208. doi: 10.1186/s12974-023-02879-7
Weindel, C. G., Martinez, E. L., Zhao, X., Mabry, C. J., Bell, S. L., Vail, K. J., et al. (2022). Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell 185, 3214–31.e23. doi: 10.1016/j.cell.2022.06.038
Wong Fok Lung, T., Monk, I. R., Acker, K. P., Mu, A., Wang, N., Riquelme, S. A., et al. (2020). Staphylococcus aureus small colony variants impair host immunity by activating host cell glycolysis and inducing necroptosis. Nat. Microbiol. 5, 141–153. doi: 10.1038/s41564-019-0597-0
Wu, Z., Deshpande, T., Henning, L., Bedner, P., Seifert, G., and Steinhäuser, C. (2021). Cell death of hippocampal CA1 astrocytes during early epileptogenesis. Epilepsia 62, 1569–1583. doi: 10.1111/epi.16910
Wu, Y., He, Y., Liu, C., Ehle, C., Iyer-Bierhoff, A., Liu, B., et al. (2023). Histone deacetylase inhibitor (SAHA) reduces mortality in an endotoxemia mouse model by suppressing glycolysis. Int. J. Mol. Sci. 24:12448. doi: 10.3390/ijms241512448
Wu, A., Lee, D., and Xiong, W. C. (2023). Lactate metabolism, signaling, and function in brain development, synaptic plasticity, angiogenesis, and neurodegenerative diseases. Int. J. Mol. Sci. 24:13398. doi: 10.3390/ijms241713398
Wu, K., Li, B., Ma, Y., Tu, T., Lin, Q., Zhu, J., et al. (2023). Nicotinamide mononucleotide attenuates HIF-1α activation and fibrosis in hypoxic adipose tissue via NAD + /SIRT1 axis. Front. Endocrinol. (Lausanne) 14:1099134. doi: 10.3389/fendo.2023.1099134
Wu, H., Zhao, X., Hochrein, S. M., Eckstein, M., Gubert, G. F., Knöpper, K., et al. (2023). Mitochondrial dysfunction promotes the transition of precursor to terminally exhausted T cells through HIF-1α-mediated glycolytic reprogramming. Nat. Commun. 14:6858. doi: 10.1038/s41467-023-42634-3
Xia, C., Ou, S., Yang, Y., Zhang, W., Wu, W., Chen, Q., et al. (2023). ELP2-NLRP3-GSDMD/GSDME-mediated pyroptosis is induced by TNF-α in MC3T3-E1 cells during osteogenic differentiation. J. Cell. Mol. Med. 27, 4093–4106. doi: 10.1111/jcmm.17994
Xia, S., Yang, P., Li, F., Yu, Q., Kuang, W., Zhu, Y., et al. (2021). Chaihu-Longgu-Muli decoction exerts an antiepileptic effect in rats by improving pyroptosis in hippocampal neurons. J. Ethnopharmacol. 270:113794. doi: 10.1016/j.jep.2021.113794
Xie, J., Hong, S., Zhang, X., Li, Y., and Xie, R. (2023). Inhibition of glycolysis prevents behavioural changes in mice with MK801-induced SCZ model by alleviating lactate accumulation and lactylation. Brain Res. 1812:148409. doi: 10.1016/j.brainres.2023.148409
Xiong, J., He, J., Zhu, J., Pan, J., Liao, W., Ye, H., et al. (2022). Lactylation-driven METTL3-mediated RNA m 6 a modification promotes immunosuppression of tumor-infiltrating myeloid cells. Mol. Cell 82, 1660–77.e10. doi: 10.1016/j.molcel.2022.02.033
Xiong, C., Ling, H., Hao, Q., and Zhou, X. (2023). Cuproptosis: p53-regulated metabolic cell death? Cell Death Differ. 30, 876–884. doi: 10.1038/s41418-023-01125-0
Xu, H., Li, L., Wang, S., Wang, Z., Qu, L., Wang, C., et al. (2023). Royal jelly acid suppresses hepatocellular carcinoma tumorigenicity by inhibiting H3 histone lactylation at H3K9la and H3K14la sites. Phytomedicine 118:154940. doi: 10.1016/j.phymed.2023.154940
Xu, X. X., Shi, R. X., Fu, Y., Wang, J. L., Tong, X., Zhang, S. Q., et al. (2023). Neuronal nitric oxide synthase/reactive oxygen species pathway is involved in apoptosis and pyroptosis in epilepsy. Neural Regen. Res. 18, 1277–1285. doi: 10.4103/1673-5374.357906
Xue, Q., Kang, R., Klionsky, D. J., Tang, D., Liu, J., and Chen, X. (2023). Copper metabolism in cell death and autophagy. Autophagy 19, 2175–2195. doi: 10.1080/15548627.2023.2200554
Yang, K., Fan, M., Wang, X., Xu, J., Wang, Y., Tu, F., et al. (2022). Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 29, 133–146. doi: 10.1038/s41418-021-00841-9
Yang, J., Jin, X., Yan, Y., Shao, Y., Pan, Y., Roberts, L. R., et al. (2017). Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci. Rep. 7:43864. doi: 10.1038/srep43864
Yang, J., Luo, L., Zhao, C., Li, X., Wang, Z., Zeng, Z., et al. (2022). A positive feedback loop between inactive VHL-triggered histone lactylation and PDGFRβ signaling drives clear cell renal cell carcinoma progression. Int. J. Biol. Sci. 18, 3470–3483. doi: 10.7150/ijbs.73398
Yang, C., Pan, R. Y., Guan, F., and Yuan, Z. (2024). Lactate metabolism in neurodegenerative diseases. Neural Regen. Res. 19, 69–74. doi: 10.4103/1673-5374.374142
Yang, P., Qin, Y., Zhu, Y., Li, F., Xia, S. S., Zhou, B., et al. (2020). Chaihu-Longgu-Muli decoction relieves epileptic symptoms by improving autophagy in hippocampal neurons. J. Ethnopharmacol. 259:112990. doi: 10.1016/j.jep.2020.112990
Yang, Z., Su, W., Wei, X., Qu, S., Zhao, D., Zhou, J., et al. (2023). HIF-1α drives resistance to ferroptosis in solid tumors by promoting lactate production and activating SLC1A1. Cell Rep. 42:112945. doi: 10.1016/j.celrep.2023.112945
Yang, C., Wang, M., Chang, M., Yuan, M., Zhang, W., Tan, J., et al. (2023). Heterostructural nanoadjuvant CuSe/CoSe 2 for potentiating ferroptosis and photoimmunotherapy through intratumoral blocked lactate efflux. J. Am. Chem. Soc. 145, 7205–7217. doi: 10.1021/jacs.2c12772
Yang, Z., Yan, C., Ma, J., Peng, P., Ren, X., Cai, S., et al. (2023). Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat. Metab. 5, 61–79. doi: 10.1038/s42255-022-00710-w
Yang, D., Yin, J., Shan, L., Yi, X., Zhang, W., and Ding, Y. (2022). Identification of lysine-lactylated substrates in gastric cancer cells. iScience 25:104630. doi: 10.1016/j.isci.2022.104630
Yang, X., Zhang, X., Shen, K., Wang, Z., Liu, G., Huang, K., et al. (2023). Cuproptosis-related genes signature and validation of differential expression and the potential targeting drugs in temporal lobe epilepsy. Front. Pharmacol. 14:1033859. doi: 10.3389/fphar.2023.1033859
Yang, H., Zou, X., Yang, S., Zhang, A., Li, N., and Ma, Z. (2023). Identification of lactylation related model to predict prognostic, tumor infiltrating immunocytes and response of immunotherapy in gastric cancer. Front. Immunol. 14:1149989. doi: 10.3389/fimmu.2023.1149989
Yao, Y., Bade, R., Li, G., Zhang, A., Zhao, H., Fan, L., et al. (2023). Global-scale profiling of differential expressed lysine-lactylated proteins in the cerebral endothelium of cerebral ischemia-reperfusion injury rats. Cell. Mol. Neurobiol. 43, 1989–2004. doi: 10.1007/s10571-022-01277-6
Yao, X., and Li, C. (2023). Lactate dehydrogenase a mediated histone lactylation induced the pyroptosis through targeting HMGB1. Metab. Brain Dis. 38, 1543–1553. doi: 10.1007/s11011-023-01195-6
Ye, L., Jiang, Y., and Zhang, M. (2022). Crosstalk between glucose metabolism, lactate production and immune response modulation. Cytokine Growth Factor Rev. 68, 81–92. doi: 10.1016/j.cytogfr.2022.11.001
Yin, Y., Guan, X., Li, G., Chen, C., Duan, Y., and Yu, Z. (2023). The HDAC inhibitor HFY-4A improves TUSC2 transcription to induce immunogenic cell death in breast cancer. Toxicol. Appl. Pharmacol. 478:116698. doi: 10.1016/j.taap.2023.116698
You, Q., Wang, J., Yu, Y., Li, F., Meng, L., Chen, M., et al. (2022). The histone deacetylase SIRT6 promotes glycolysis through the HIF-1α/HK2 signaling axis and induces erlotinib resistance in non-small cell lung cancer. Apoptosis 27, 883–898. doi: 10.1007/s10495-022-01751-y
Yu, J., Chai, P., Xie, M., Ge, S., Ruan, J., Fan, X., et al. (2021). Histone lactylation drives oncogenesis by facilitating m 6 a reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 22:85. doi: 10.1186/s13059-021-02308-z
Yuan, J., Mo, Y., Zhang, Y., Zhang, Y., and Zhang, Q. (2022). Nickel nanoparticles induce epithelial-mesenchymal transition in human bronchial epithelial cells via the HIF-1α/HDAC3 pathway. Nanotoxicology 16, 695–712. doi: 10.1080/17435390.2022.2142169
Yuan, S., Wei, C., Liu, G., Zhang, L., Li, J., Li, L., et al. (2022). Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1α/SLC7A11 pathway. Cell Prolif. 55:e13158. doi: 10.1111/cpr.13158
Yuan, Y., Zhai, Y., Chen, J., Xu, X., and Wang, H. (2021). Kaempferol ameliorates oxygen-glucose deprivation/reoxygenation-induced neuronal ferroptosis by activating Nrf2/SLC7A11/GPX4 axis. Biomol. Ther. 11:923. doi: 10.3390/biom11070923
Zalewska, T., Jaworska, J., Sypecka, J., and Ziemka-Nalecz, M. (2020). Impact of a histone deacetylase inhibitor-trichostatin a on neurogenesis after hypoxia-ischemia in immature rats. Int. J. Mol. Sci. 21:3808. doi: 10.3390/ijms21113808
Zeng, Z., You, M., Fan, C., Rong, R., Li, H., and Xia, X. (2023). Pathologically high intraocular pressure induces mitochondrial dysfunction through Drp1 and leads to retinal ganglion cell PANoptosis in glaucoma. Redox Biol. 62:102687. doi: 10.1016/j.redox.2023.102687
Zhai, M., Cui, S., Li, L., Cheng, C., Zhang, Z., Liu, J., et al. (2022). Mechanical force modulates alveolar bone marrow mesenchymal cells characteristics for bone remodeling during orthodontic tooth movement through lactate production. Cells 11:3724. doi: 10.3390/cells11233724
Zhang, D., Du, J., Yu, M., and Suo, L. (2023). Urine-derived stem cells-extracellular vesicles ameliorate diabetic osteoporosis through HDAC4/HIF-1α/VEGFA axis by delivering microRNA-26a-5p. Cell Biol. Toxicol. 39, 2243–2257. doi: 10.1007/s10565-022-09713-5
Zhang, J., Gong, L., Zhu, H., Sun, W., Tian, J., Zhang, Y., et al. (2023). RICH2 decreases the mitochondrial number and affects mitochondrial localization in diffuse low-grade glioma-related epilepsy. Neurobiol. Dis. 188:106344. doi: 10.1016/j.nbd.2023.106344
Zhang, S., Hu, L., Jiang, J., Li, H., Wu, Q., Ooi, K., et al. (2020). HMGB1/RAGE axis mediates stress-induced RVLM neuroinflammation in mice via impairing mitophagy flux in microglia. J. Neuroinflammation 17:15. doi: 10.1186/s12974-019-1673-3
Zhang, F., Shao, C., Wu, Y., Zhao, W., Jing, X., Fang, C., et al. (2022). A dynamic fluorescence probed glycolysis suppression process in HeLa cells treated with trichostatin A. Analyst 147, 5082–5090. doi: 10.1039/D2AN01114J
Zhang, L., Shi, H., Chen, H., Gong, A., Liu, Y., Song, L., et al. (2019). Dedifferentiation process driven by radiotherapy-induced HMGB1/TLR2/YAP/HIF-1α signaling enhances pancreatic cancer stemness. Cell Death Dis. 10:724. doi: 10.1038/s41419-019-1956-8
Zhang, D., Tang, Z., Huang, H., Zhou, G., Cui, C., Weng, Y., et al. (2019). Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. doi: 10.1038/s41586-019-1678-1
Zhang, L., Wang, X., Che, W., Zhou, S., and Feng, Y. (2023). METTL3 silenced inhibited the ferroptosis development via regulating the TFRC levels in the intracerebral hemorrhage progression. Brain Res. 1811:148373. doi: 10.1016/j.brainres.2023.148373
Zhang, H., Wang, D., Li, M., Plecitá-Hlavatá, L., D'Alessandro, A., Tauber, J., et al. (2017). Metabolic and proliferative state of vascular adventitial fibroblasts in pulmonary hypertension is regulated through a MicroRNA-124/PTBP1 (polypyrimidine tract binding protein 1)/pyruvate kinase muscle axis. Circulation 136, 2468–2485. doi: 10.1161/CIRCULATIONAHA.117.028069
Zhang, W., Xu, L., Yu, Z., Zhang, M., Liu, J., and Zhou, J. (2023). Inhibition of the glycolysis prevents the cerebral infarction progression through decreasing the lactylation levels of LCP1. Mol. Biotechnol. 65, 1336–1345. doi: 10.1007/s12033-022-00643-5
Zhao, Y., Li, M., Yao, X., Fei, Y., Lin, Z., Li, Z., et al. (2020). HCAR1/MCT1 regulates tumor ferroptosis through the lactate-mediated AMPK-SCD1 activity and its therapeutic implications. Cell Rep. 33:108487. doi: 10.1016/j.celrep.2020.108487
Zhao, L., Song, Y., Zhang, Y., Liu, H., Shen, Y., Fan, Y., et al. (2022). HIF-1α/BNIP3L induced cognitive deficits in a mouse model of sepsis-associated encephalopathy. Front. Immunol. 13:1095427. doi: 10.3389/fimmu.2022.1095427
Zhao, S., Zhou, L., Wang, Q., Cao, J. H., Chen, Y., Wang, W., et al. (2023). Elevated branched-chain amino acid promotes atherosclerosis progression by enhancing mitochondrial-to-nuclear H 2 O 2 -disulfide HMGB1 in macrophages. Redox Biol. 62:102696. doi: 10.1016/j.redox.2023.102696
Zhou, W., Yang, J., Saren, G., Zhao, H., Cao, K., Fu, S., et al. (2020). HDAC6-specific inhibitor suppresses Th17 cell function via the HIF-1α pathway in acute lung allograft rejection in mice. Theranostics 10, 6790–6805. doi: 10.7150/thno.44961
Zhu, S., Donovan, E. L., Makosa, D., Mehta-D'souza, P., Jopkiewicz, A., Batushansky, A., et al. (2022). Sirt3 promotes chondrogenesis, chondrocyte mitochondrial respiration and the development of high-fat diet-induced osteoarthritis in mice. J. Bone Miner. Res. 37, 2531–2547. doi: 10.1002/jbmr.4721
Keywords: protein lactylation, histone deacetylase, epilepsy, high-mobility group box 1, hypoxia-inducible factor-1α, lactate
Citation: Kuang X, Chen S and Ye Q (2025) The lactate metabolism and protein lactylation in epilepsy. Front. Cell. Neurosci. 18:1464169. doi: 10.3389/fncel.2024.1464169
Edited by:
Eleonora Vannini, National Research Council (CNR), ItalyReviewed by:
Swati Banerjee, The University of Texas Health Science Center at San Antonio, United StatesKristina Lippmann, University Hospital Leipzig, Germany
Copyright © 2025 Kuang, Chen and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingmei Ye, c2VhOTgwMDYxNkAxMjYuY29t
†These authors have contributed equally to this work