 Moyan Wang1†
Moyan Wang1† Xin Chen1†
Xin Chen1† Shuangyang Li1†
Shuangyang Li1† Lingxue Wang1
Lingxue Wang1 Hongmei Tang1
Hongmei Tang1 Yuting Pu1
Yuting Pu1 Dechou Zhang1,2
Dechou Zhang1,2 Bangjiang Fang1,3*
Bangjiang Fang1,3* Xue Bai1,2*
Xue Bai1,2*- 1Department of Neurology, National Traditional Chinese Medicine Clinical Research Base, The Affiliated Traditional Chinese Medicine Hospital, Southwest Medical University, Luzhou, China
- 2Institute of Integrated Chinese and Western Medicine, Southwest Medical University, Luzhou, China
- 3Department of Emergency, Longhua Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, China
Intracerebral hemorrhage (ICH) is a severe condition that devastatingly harms human health and poses a financial burden on families and society. Bcl-2 Associated X-protein (Bax) and B-cell lymphoma 2 (Bcl-2) are two classic apoptotic markers post-ICH. Beclin 1 offers a competitive architecture with that of Bax, both playing a vital role in autophagy. However, the interaction between Beclin 1 and Bcl-2/Bax has not been conjunctively analyzed. This review aims to examine the crosstalk between autophagy and apoptosis in ICH by focusing on the interaction and balance of Beclin 1, Bax, and Bcl-2. We also explored the therapeutic potential of Western conventional medicine and traditional Chinese medicine (TCM) in ICH via controlling the crosstalk between autophagy and apoptosis.
1 Introduction
Stroke, always a magnet for medical researchers, causes permanent brain damage, serious disability or even death (Diener and Hankey, 2020). Hemorrhagic stroke is more fatal than ischemic stroke, and the former is subdivided into intracerebral hemorrhage (ICH) and subarachnoid hemorrhage (SAH) (Feigin et al., 2009). The incidence and prognosis of ICH vary with races and socioeconomic statuses. Asians suffer from a higher susceptibility to ICH than other ethnic groups (Johnson et al., 2016). Within the past four decades, the incidence of ICH has sharply declined in high-income countries, but almost doubled in low-and middle-income countries (Feigin et al., 2009). These fluctuations become more evident within the past two decades (Flaherty et al., 2007; Khellaf et al., 2010; Islam et al., 2008; Huhtakangas et al., 2011). A decreasing trend may be attributed to the rapid boom of medical resources, while a rising trend to the wide use of anticoagulant medications like warfarin (Lovelock et al., 2007).
Generally, ICH results in primary and secondary injuries. The primary injury happens when cerebral blood vessels abruptly rupture, leading to hematoma that exerts mechanical damages on adjacent tissues and a swift increase in intracranial pressure (Zhao et al., 2018). Red blood cell fragments and blood flow back into the brain cause secondary excitotoxic and cytotoxic brain injuries, like neuronal apoptosis, inflammatory response, oxidative stress, mitochondrial dysfunction and blood–brain barrier (BBB) destruction (Zhou et al., 2014; Chen et al., 2018). Regional ICH in the thalamus, cortex, brainstem, and areas beneath the cerebellar tentorium brings with severe motor, sense and language dysfunctions. Moreover, ICH patient also demonstrate nonspecific symptoms, such as headache, nausea, and meningeal irritation, all detrimental to the quality of life (Qureshi et al., 2001).
Massive apoptotic cells in the brain at post-ICH have been observed in in vivo models (Duan et al., 2020; Qureshi et al., 2001; Figure 1). Consistently, a great number of apoptotic cells are found around the site of hematoma in patients with spontaneous ICH (Qureshi et al., 2003). Ubiquitinated proteins and p62 accumulate in neurons from nearly all regions of the autophagy-defective brain, generating inclusion bodies age-dependently growing in size and quantity (Lee et al., 2010). Neurodegenerative diseases progress with an accumulation of autophagic vacuoles (Fleming et al., 2011). Cathepsin D is a lysosomal enzyme for regulating the accumulation of autophagic vacuoles (Araki et al., 2006). Produced in neurons and astrocytes rapidly following the brain hemorrhage, Cathepsin D shows the maximal activity at 1 week, and keeps continuous hyperactivity for 4 weeks or longer (He et al., 2008). Upregulation of autophagy-related proteins Beclin 1 and LC3 and downregulation of anti-apoptotic protein Bcl-2 at post-ICH prove that autophagy is activated by ICH (Shen et al., 2016). Some studies indicate that autophagy promotes cell survival (Carloni et al., 2008), whereas other research suggests that inhibiting autophagy may enhance survival probability (Puyal et al., 2009; Wang and Zhang, 2017). A long-term inhibition on autophagy is harmful to cell viability, while a short-term suppression may be acceptable (Smith et al., 2011). Mice lacking Atg7 in the central nervous system (CNS) present behavioral problems and die 28 weeks after birth, suggesting the health-threatening effects of autophagy deprivation (Komatsu et al., 2006). On the contrary, toxic proteins can be degraded by autophagy to favor cell survival and maintenance. Clinical studies have yet to explore the treatment of cerebral hemorrhage through the modulation of autophagy; however, acupuncture has been shown to concurrently reduce both autophagy and apoptosis (Yao and Zou, 2022). Consequently, autophagy represents a promising avenue for future research aimed at mitigating damage following cerebral hemorrhage (Chen et al., 2024).
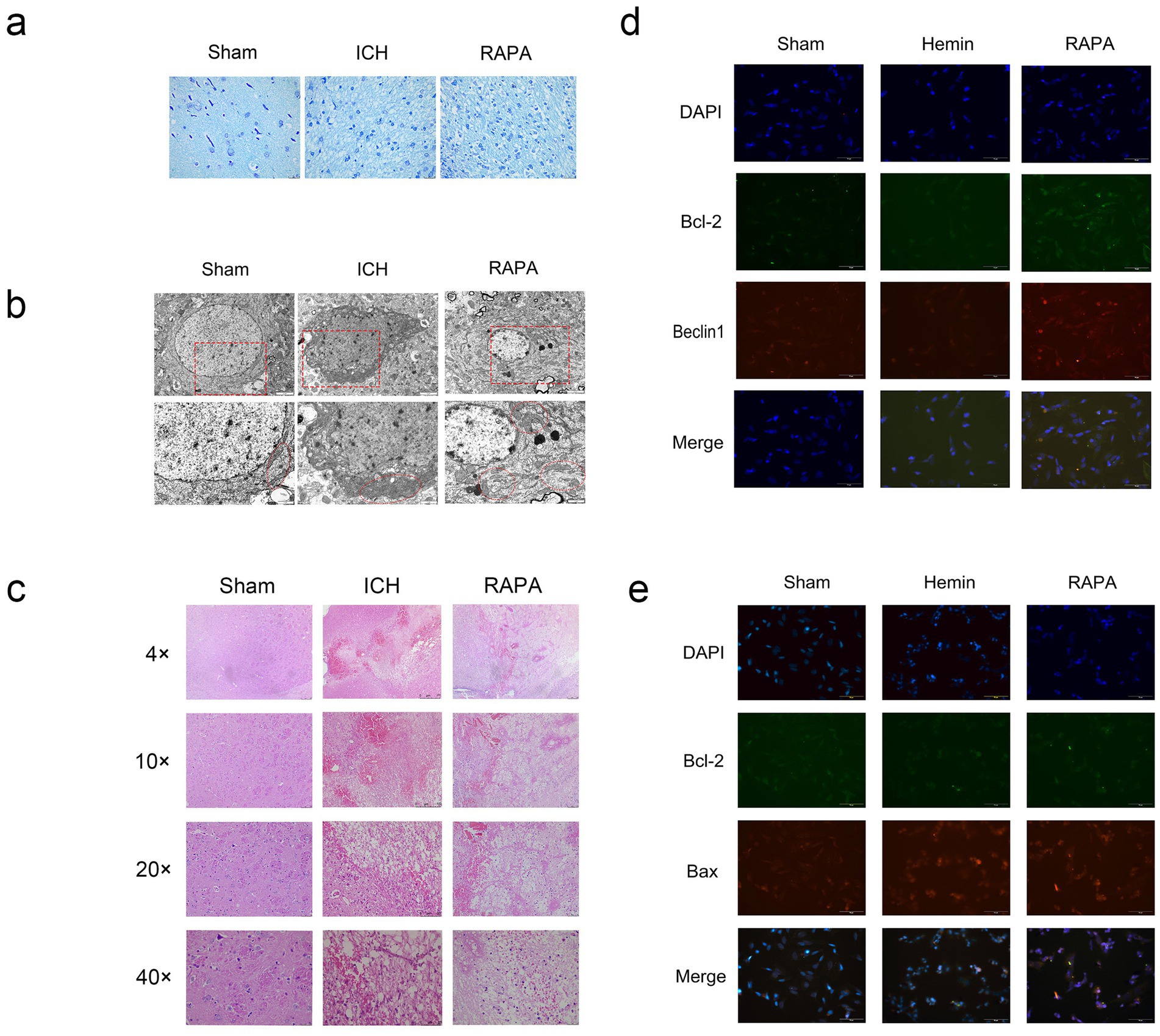
Figure 1. Rat intracerebral hemorrhage model induced by collagenase: the sham group was untreated, the Ich group was a simulated intracerebral hemorrhage group, and the Rapa group was treated with rapamycin. Sh-Sy5Y human neuroblastoma cells induced by hemin iron. The sham group was untreated, the hemin group was a simulated intracerebral hemorrhage group, and the Rapa group was treated with rapamycin. (a) Nissl staining (400×). (b) Electron microscopy (8,000×, 15,000×) (c) Hematoxylin–eosin staining (40×, 100×, 200×, 400×). (d) In vitro immunofluorescence Beclin1, Bcl-2 double staining results (400×). (e) In vitro immunofluorescence Beclin1, Bax double staining results (400×).
2 Critical roles of autophagy and apoptosis in neurological outcomes of ICH
Expression levels of Bcl-2 and Bax peak at 12 h and 1 day following ICH (Figure 2), respectively (Xuefei et al., 2011). At 6 h of ICH, blood osmosis and mechanical injury are examined in the brain sections, while nerve cell edema is absent and TUNEL-positive cells remain in a low proportion. Moreover, expression level of Bcl-2 decreases sharply in the brain sections after ICH, but increases to the baseline at 12 h and peaks at 3 days (Xiu'e and Liangqun, 2010). Besides apoptosis, autophagy involving Bcl-2 and Bax is speculated to participate in the acute phase of ICH, and the Bcl-2/Bax ratio can be used to infer changes in autophagy. Consistently, fluctuations in LC-3II/LC-3I surrounding hematomas and an increase within 6 h of ICH are indicative of autophagy in rats (Huaxian et al., 2003).
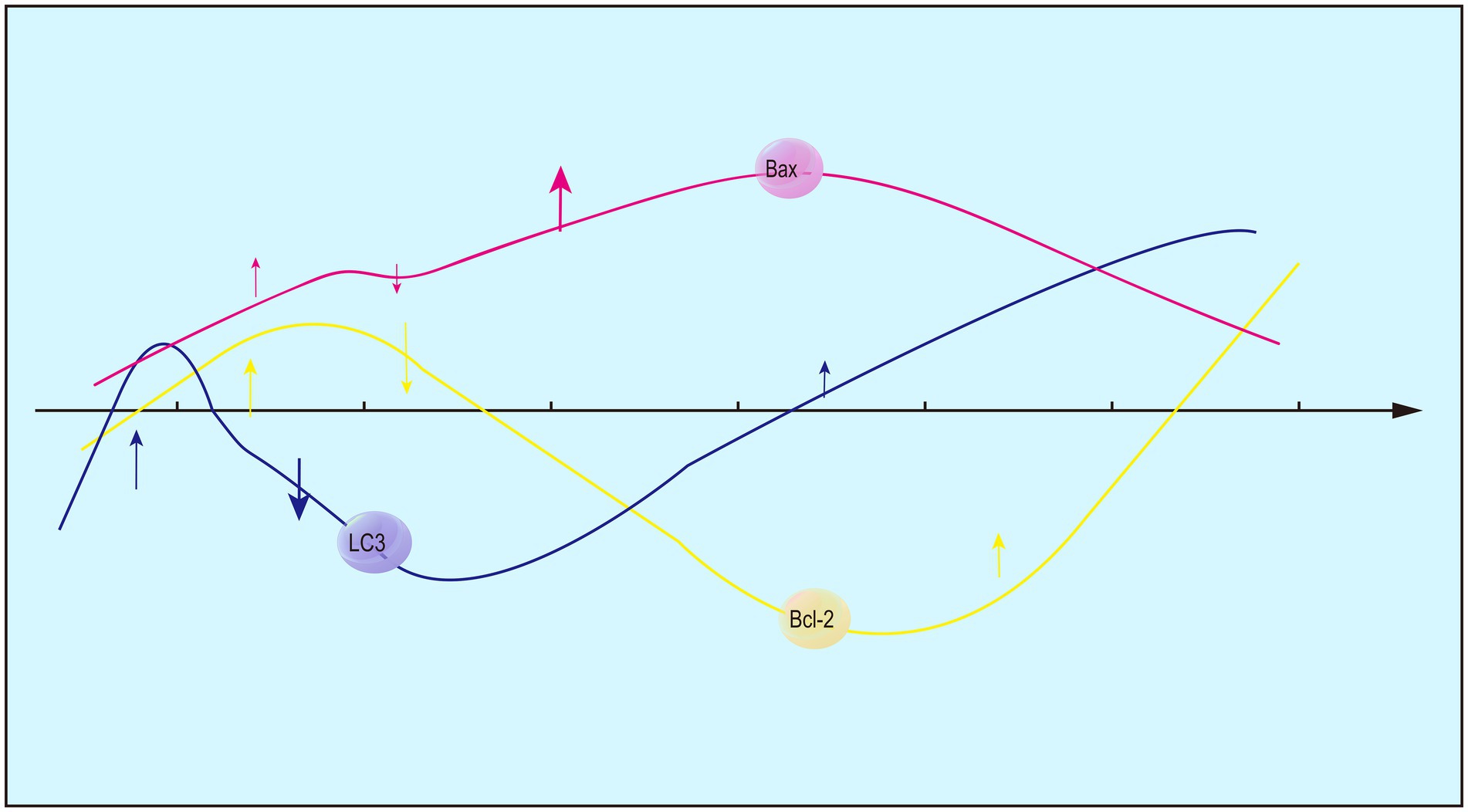
Figure 2. Changing trends in the expression of Bcl-2, Bax and LC3-II/LC3-I after ICH.
Investigator (Lee et al., 2015) utilized Western Blot analysis to determine that Bcl-2 expression peaked at 12 h following cerebral hemorrhage, while Bax expression peaked at 1 day. Pathological findings (Yanling et al., 2004) indicated that 6 h post-intracerebral hemorrhage (ICH), there was evidence of blood infiltration and mechanical damage, but no neuronal edema was observed. The number of TUNEL-positive cells was one-eighth of the peak value. However, in comparison to the sham group, Bcl-2 expression significantly decreased, only to rise above the average level of the saline group after 12 h, reaching its maximum at 3 days. This suggests that, in the acute phase, there are cellular responses beyond apoptosis, and it is plausible that Bcl-2 plays a role in the regulation of autophagy in conjunction with Bax. The Bcl-2/Bax ratio not only serves as an indicator of apoptosis but may also influence autophagy through alterations in this ratio, as evidenced by findings from a separate study (Duan et al., 2017). Furthermore, the autophagy marker LC-3/LC-3I was observed in rats with intracerebral hemorrhage (ICH) at 6 h post-injury, showing a significant recovery between 72 h and 7 days. Additionally, the study indicated that the observed autophagy may be indicative of excessive autophagic activity. By comparing autophagy agonists and autophagy inhibitors, it was demonstrated that excessive autophagy within 6 h led to an increase in the cleavage of Caspase-12, while inhibition of autophagy led to a reduction in damage.
Autophagy and apoptosis are normal physiological activities both involved in self-repair. To curb a pathological state, efforts can be made for increasing the possibility of autophagy without stimulating apoptosis. Not only rapid increases in expression levels of Bax and Bcl-2 following ICH respond to self-repair or self-clearance mechanism. Autophagy-induced apoptosis provides various effects. Inhibiting autophagy lowers apoptosis in rats with transient ischemic attack (Huang et al., 2020). On the contrary, an excessive autophagy during cardiac arrest and resuscitation causes neuronal apoptosis and worsens brain damage (Youle and Narendra, 2011). These diametrically opposed results may be attributed to the dual functions of autophagy that can actively promote apoptosis by removing damaged cells, maintaining tissue homeostasis, and eradicating harmful cells. Removal of damaged mitochondria during autophagic processes also prevents cell apoptosis caused by mitochondrial dysfunction (Green and Llambi, 2015; Ebrahimi-Fakhari et al., 2012). Autophagy and apoptosis have a blurred boundary, and it is challenging to quantify what a level of autophage can contribute to apoptosis.
3 Regulators of autophagy and apoptosis
Multiple factors and signaling pathways regulate both autophagy and apoptosis, and their upstream factors may be involved in the crosstalk between autophagy and apoptosis (Figure 3). For instance, downregulation of autophagy factor Atg5 inhibits both autophagy and apoptosis (Dong et al., 2019). Atg6 serves as a switch that connects apoptosis and autophagy, and its downregulation increases the susceptibility to TRAIL-induced cell death, suppresses apoptosis, and promotes autophagy (Cho et al., 2009). P53 is either activated or inhibited in autophagy, and also an initiator of apoptosis. DRAM links autophagy to p53, and loss of Atg7 exacerbates p53-dependent apoptosis (Crighton et al., 2006; He et al., 2013). Besides, p53 alters the Bcl-2/Bax ratio and the state of Bax overweighing Bcl-2, thereafter inducing apoptosis (Yan et al., 2021). The binding ability of p53 to Bax and Bad is enhanced by phosphorylation of Bcl-2 at Ser70, a process necessary for its anti-apoptotic function (Maiuri et al., 2007). Also, phosphorylation of Bcl-2 blocks the interaction between Bcl-2 and pro-apoptotic protein BH3, resulting in the apoptotic cell death when the autophagy activated by phosphorylated Bcl-2 no longer supports cell survival (Wei et al., 2008). AMPK breaks the balance between autophagy and apoptosis, and tilts it to the former. Additionally, phosphorylation of AMPK activates autophagic cell death (Liang et al., 2007; Kang et al., 2011). Caspase-dependent cell death is triggered by the cleavage of Beclin 1 and Atg7. Caspase-8 inhibits cell death and thus alleviates the severity of illness (Shimizu et al., 2004; Yu et al., 2004; Mizushima and Komatsu, 2011).
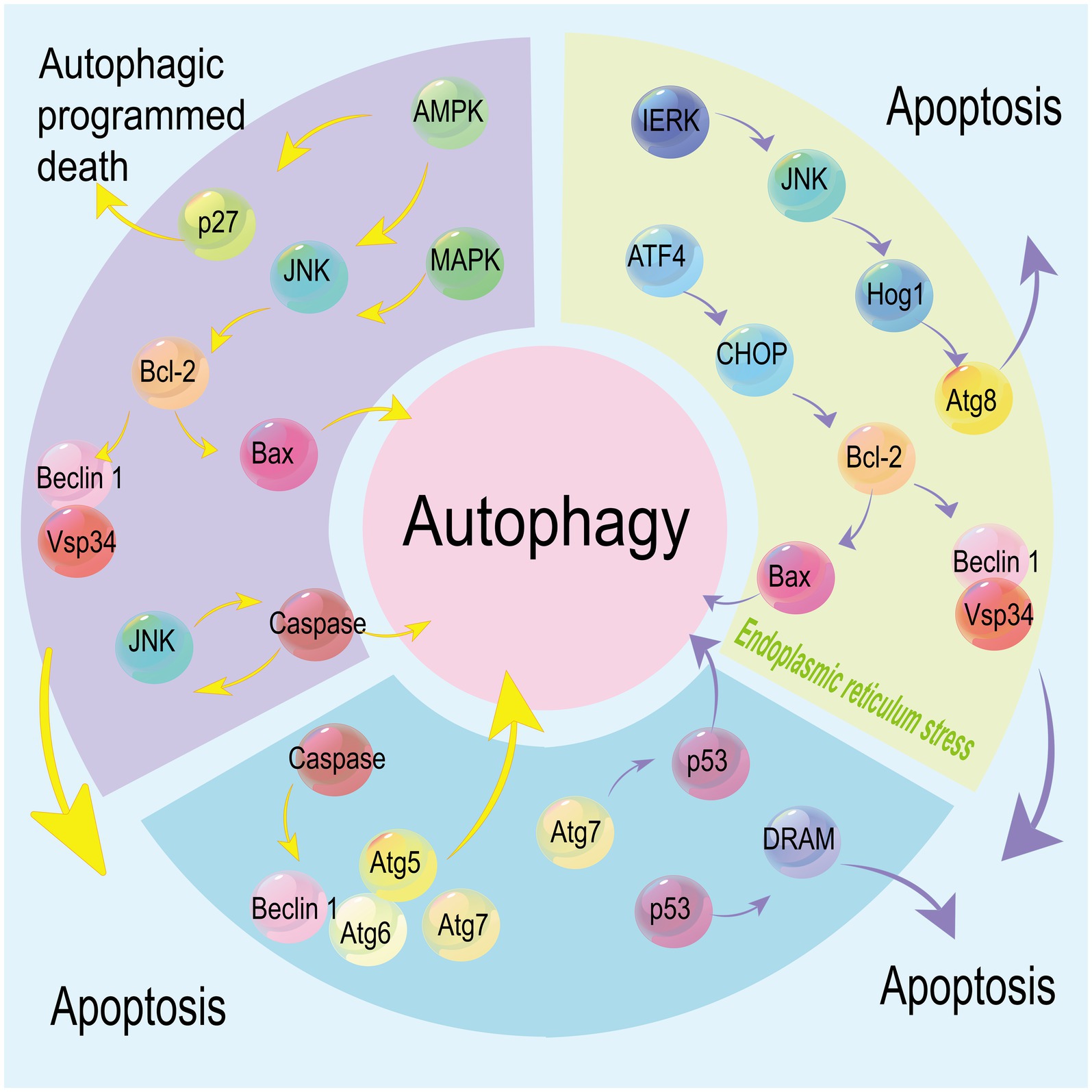
Figure 3. Multiple factors and signaling pathways mediate both autophagy and apoptosis. Under endoplasmic reticulum (ER) stress, autophagy regulated by the IERK/JNK/Hog1 axis via stabilizing Atg8 co-exists with apoptosis regulated by the ATF4/CHOP axis. p53 is involved in the regulation of autophagy and apoptosis mediated by DRAM and Atg7, respectively. Caspases mediate the cleavage of Beclin 1, Atg6, Atg5 and Atg7, linking apoptosis with autophagy signaling pathways. JNK is activated by MAPK and AMPK to trigger caspase-induced cell death, while phosphorylated AMPK also induces autophagy by stabilizing p27. Downstream Bcl-2 is involved in apoptosis and autophagy via mediating Bax and Beclin 1.
The MAPK8/JNK1/ERK signaling pathway or phosphorylation of DAPK stimulates autophagy by breaking the balance between Bcl-2 and Beclin 1 (Zalckvar et al., 2009; Fang et al., 2019). Besides autophagy, ER stress is another event triggering apoptosis. Stimulated by the unfolded protein response, ER membrane-resident stress sensor IRE1 interacts with TRAF2 and then activates JNK to initiate cell apoptosis. ER stress-induced autophagy can be activated by the IRE1/JNK/Hog1 and ATF4-associated signaling pathway through enhancing the ATG8/LC3 stability (White, 2016). Regulated by the ATF4/CHOP axis, an extreme ER stress stimulates cell apoptosis through downregulating Bcl-2 and upregulating Bax (Maiuri et al., 2007). The dynamic interaction of LC3B with Cav-1 and Fas controls cigarette smoking-induced apoptosis (Chen et al., 2010).
4 A subtle balance between apoptosis and autophagy
4.1 Bcl-2/Bax/Beclin 1
Members of the Bcl-2 family regulate cell death, and are involved in both apoptosis and autophagy (Gross et al., 1999). Functionally, a part of Bcl-2 family members is anti-apoptotic, and the remaining are pro-apoptotic. The antagonism between two camps determines the threshold of cell fate, serving as a common phenomenon in human beings. Members of the Bcl-2 family initiate or prevent the release of apoptotic factors via regulating the mitochondrial integrity by forming homodimers and heterodimers (Uren et al., 2017; Westphal et al., 2014). They also participate in the non-apoptotic cell death dependent on autophagy genes after knocking out the apoptotic genes BAX/BAK (Shimizu et al., 2004).
Through aggregation or segregation, the Bcl-2 family regulates cell apoptosis (Figure 4). The four conserved BH domains determine biological functions of the three subfamilies of Bcl-2. Generally, there is a homologous sequence shared by all the four BH domains of most anti-apoptotic members in the Bcl-2 family, while homologous sequences are only observed in the BH3 domain of pro-apoptotic members (Cohen et al., 2020). BH3-only proteins interact with the BAX N-terminal trigger site, and stimulate the translocation of cytoplasmic Bax to the mitochondrial outer membrane (MOM), leading to the altered mitochondrial outer membrane permeabilization (MOMP) and mitochondrial dysfunction. The formation of heterodimers by anti-apoptotic and pro-apoptotic Bcl-2 family members prevents Bax-induced MOMP alteration and mitochondrial dysfunction through trapping the exposed BH3-domains of pro-apoptotic members in the surface groove. The inhibited release of soluble pro-apoptotic factors (e.g., cytochrome c) from the intermembrane space ultimately prevents caspase-mediated apoptosis (Garner et al., 2019; Subburaj et al., 2015). In addition, BH3-only proteins directly or indirectly activate Bax/Bak by binding to Bcl-2 and inactivate it (Westphal et al., 2014; O'Neill et al., 2016). Bax and Bak cause apoptosis by generating pores or binding to BH3-only proteins (Uren et al., 2017; Chen et al., 2005). Cleavage products of the anti-apoptotic factors Bcl-2, Bcl-xl, and Mcl-1 produced by Caspase-9 effectors also cause apoptosis (Mizushima and Komatsu, 2011).
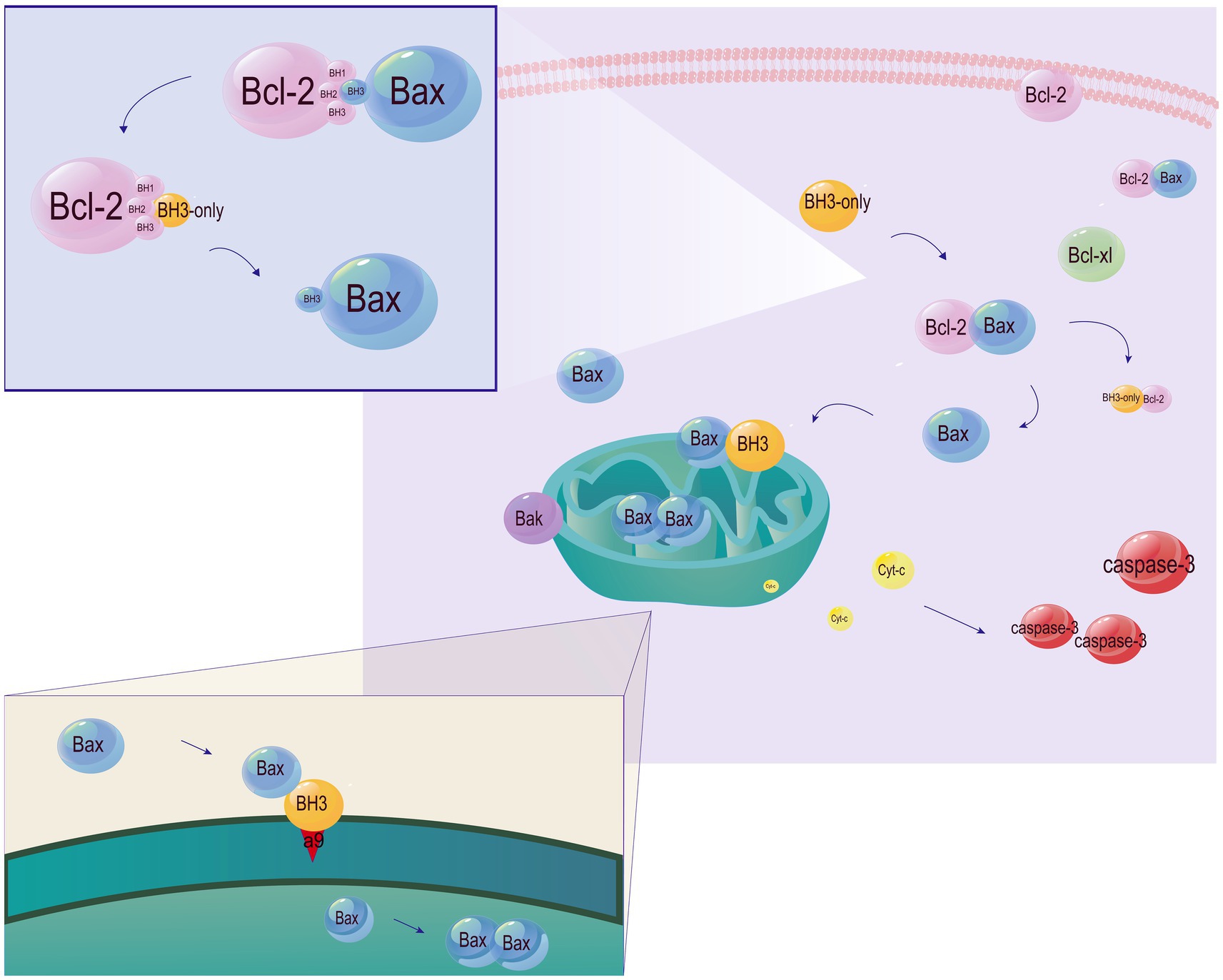
Figure 4. Involvement of the Bcl-2 family members in apoptosis. Under physiological conditions, Bak and Bax are localized in the outer mitochondrial membrane and mitochondria, respectively. Translocation of Bax to the cytoplasm increases the sensitivity to apoptosis. Bcl-2 and Bcl-xL are localized in the membrane and cytosol, respectively. Both of them control the translocation of Bax/Bak. BH3-only proteins are responsible for transmitting cell death signals to other members of the Bcl-2 family. Relying on the BH3 domain, BH3-only proteins bind to anti-apoptotic members of the Bcl-2 family and inactive them, thus indirectly activating Bax/Bak. Moreover, α9 released by BH3-only proteins from a hydrophobic groove directly activates Bax by inserting into the MOM. Bax monomers then form dimer units, self-assemble into oligomers and generate pores in the MOM, leading to the caspase cascade by the release of cytochrome C.
Beclin 1, a mammalian positive regulator of autophagy in the Bcl-2 family, contains a BH3 domain, a central coiled-coil domain and an evolutionarily conserved domain. Serving as an orthologue of yeast Atg6, Beclin 1 is considered to mediate autophagy and restore the autophagy activity having been destroyed by Atg6 (Kihara et al., 2001). During the development of Caenorhabditis elegans, loss of Beclin-1 increases caspase-dependent apoptosis and the number of apoptotic bodies (Takacs-Vellai et al., 2005). In as early as 2001, the Beclin 1 homolog of yeast Vps30 was found to assemble two complexes of Vps15 and Vps34 (Salminen et al., 2013). Beclin 1 interacts with Vps34 through the central coiled-coil domain and evolutionarily conserved domains, and the latter forms phosphatidylinositol-3-phosphate (PI3P) by phosphorylating phosphatidylinositol (Feng et al., 2007; Konishi et al., 2012). In early-stage phagosomes, Beclin 1, Rac1 and PI3P binding protein containing the FYVE domain assemble into the class III PI3K complex. Induction of the PI3K inhibitor 3-Methyladenine (3-MA) or knockout of Beclin 1 identically slows down the process of phagocytosis, indicating that phagocytosis is dependent on Beclin 1 and the activity of PI3K (Funderburk et al., 2010). The Beclin 1-Vps34 complex can also invite other partners to form new complexes. Specifically, interacting partners of the Beclin 1-Vps34 complex include Atg14L, UVRAG and Rubicon; while unstable or transient binding partners compose of Bif-1, Ambra 1, VMP1, nPIST, Rab5, ICP 34.5, and inositol 1,4,5-triphosphate receptor (IP3R). These important complexes are involved in all stages of autophagy to mediate the formation or maturation of autophagosomes, endocytic transport and autophagy signaling pathway. Endogenous Bcl-2 is able to interfere with the binding of Beclin 1 to Atg14L, UVRAG or Rubicon (Feng et al., 2007).
The BH3 domain of Beclin 1 binds to members of the Bcl-2 family. As a result, the apoptosis regulator Bcl-2 also mediates autophagy by interacting with Beclin 1 (Figure 4). Anti-apoptotic proteins encoded by cells and viruses effectively bind to the BH3 domain (He and Levine, 2010). The binding of Bcl-2 to Beclin 1 is enhanced in nutrient-rich conditions where autophagy is inhibited, and decreases in nutrient-deficient conditions where autophagy is stimulated. In addition, Bcl-2 impairs the interaction between Beclin 1 and Vps34 and reduces Beclin 1-associated Vps34 kinase activity, thereby potentially sequestering Beclin 1 away from the autophagy-inducing class III PI3K “core complex” (Pattingre et al., 2005). Loss of Beclin 1 suppresses autophagy, leading to cell death under nutrient deprivation and other stress conditions. The Bcl-2-Beclin 1 complex is critical for stimulus-induced autophagy in mammalian cells. Phosphorylated Bcl-2 inhibits autophagy by directly interacting with the BH3 domain of the ER-targeted Beclin 1. Approaches through phosphorylating Bcl-2 or binding to it interrupt its interaction with Beclin 1, thereby promoting autophagy (He et al., 2012; Danial and Korsmeyer, 2004). Members of the Bcl-2 family also influence other cellular processes like glucose homeostasis, cell cycle progression, calcineurin signaling, and transcriptional inhibition of p53 (Shimizu et al., 2004). Stimulated by starvation, phosphorylation of Bcl-2 at Thr69, Ser70, and Ser87 dislocates it from Beclin 1 and then induces autophagy (Wei et al., 2008). Replacement of these sites with alanine on Bcl-2 is reported to reduce the autophagy and glucose homeostasis in mouse skeletal muscles after exercise (Pattingre et al., 2009).
Bax also serves as a regulator of Beclin 1 (Figure 5). Bax/caspase induction cleaves Beclin 1 and inhibits autophagy. Pan-caspase inhibition stabilizes Beclin 1 and rescues Bax-mediated autophagy inhibition in the presence of Bax, suggesting the role of Beclin 1 as a caspase substrate (Yu et al., 2004). Overexpression of Bax does not increase the anti-apoptotic ability of cells, but inhibits autophagy through downregulating Beclin 1 via caspase cleavage. On the contrary, in cells equipping a strong anti-apoptotic capacity, overexpression of Bcl-xL or induction of caspase inhibitor stimulates autophagy by breaking the Bcl-xL-Beclin 1 interaction via Bax or Bcl-2 (Yong et al., 2010). Collectively, an orientation towards Bcl-2 blocks the interaction between Bcl-2 and Beclin 1 and thereafter increases autophagy.
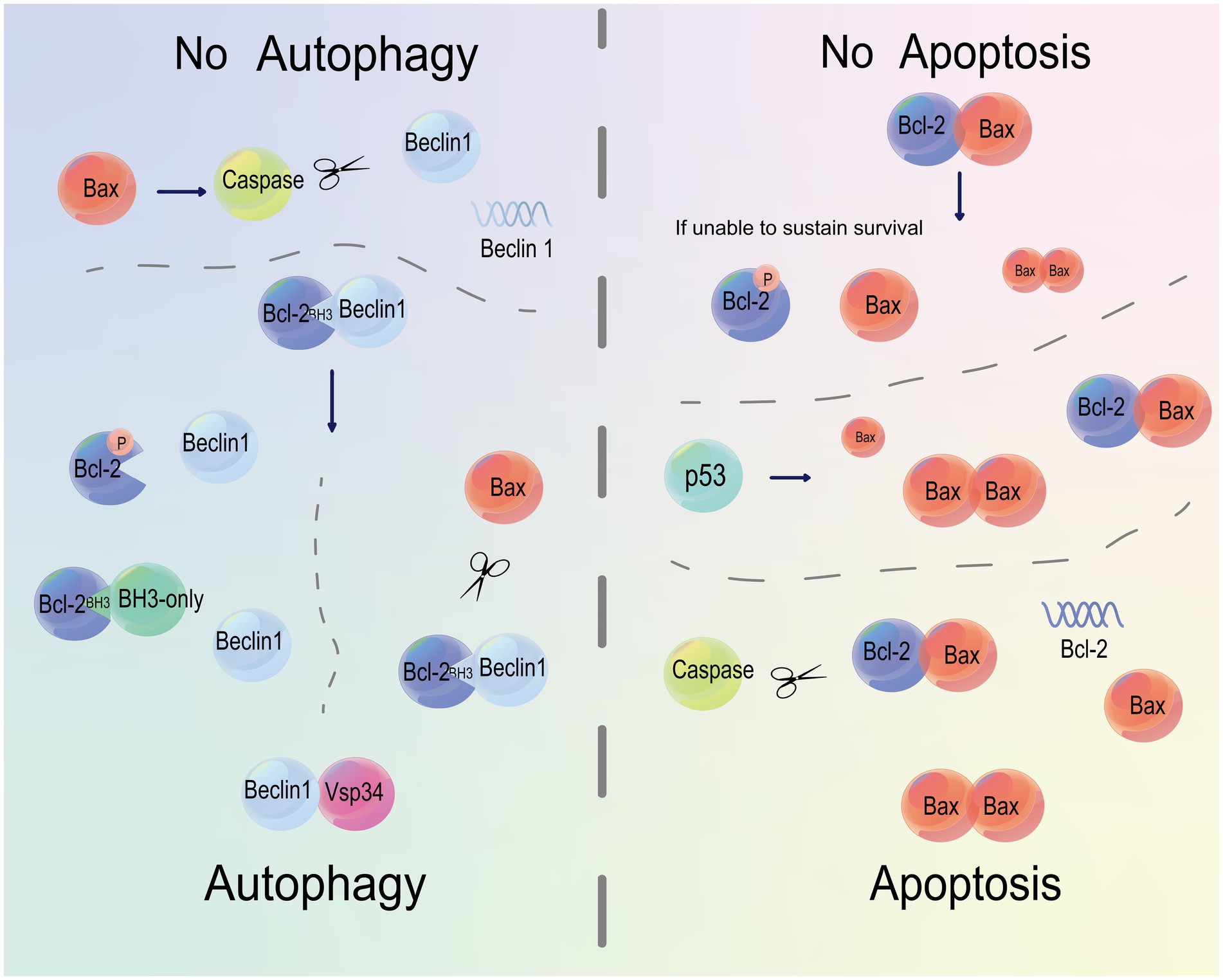
Figure 5. Bcl-2 and Bax regulate both apoptosis and autophagy. The binding of Bcl-2 to Beclin 1 or caspase cleavage of Beclin 1 inhibits autophagy. Phosphorylation of Bcl-2 or occupation on its BH3 domain activates autophagy by triggering the binding of Beclin 1 to Vsp34. Moreover, Bax can cleave Bcl-2 and Beclin 1. The binding of Bax to Bcl-2 prevents apoptosis. A decline in phosphorylated Bcl-2 by Bax cleavage below a threshold to maintain survival eventually causes apoptosis. p53 disrupts the balance between Bcl-2 and Bax, and orients this balance to the latter, leading to apoptosis. Caspases serve as regulators of apoptosis by cleaving Bcl-2.
4.2 Bcl-2/Bax/Beclin 1 regulate apoptosis and autophagy
Bcl-2 deficiency is not the only determinator for apoptosis, which also requires for the participation of Bax (Wall et al., 1999). The Bcl-2/Bax ratio is a predictive marker for cell sensitivity to apoptosis (Weidong and Zhengtang, 2007). Mathematical modeling of apoptosis shows that the ratio of pro-apoptotic and anti-apoptotic members of the Bcl-2 family serves as a hub to control apoptosis by directly determining the opening degree of mitochondrial ion channels of the outer membrane. Bcl-2 induces apoptosis by Bax homodimerization, but counters apoptosis by Bax heterodimerization. It is reported that Bax homodimerization/heterodimerization predicts cell fate. Cells containing 50% of Bax in heterodimerization of Bcl-2 and Bcl-xL are resistant to apoptosis, while those containing 80% of Bax in homodimerization are sensitive (Selvakumaran et al., 1994). The anti-apoptotic capacity of the Bcl-2 family largely relies on the expression of Bcl-2 and the formation of homodimers/heterodimers with Bcl-2 (Figure 4). Homodimerization of Bax triggers cell death, while heterodimerization of Bcl-2 to Bax favors cell viability. Generally, the Bcl-2/Bax ratio is intrinsic under normal physiological conditions, but altered by external stimuli like the p53-dependent cell death signaling (Crawford et al., 1998; Oltvai and Korsmeyer, 1994).
Overexpression of Bcl-2/Bcl-xL leads to autophagy, while overexpression of BH3-only proteins fails to induce non-apoptotic cell death in cells with loss of Bax/Bak. Hence, the balance between Bcl-2/Bcl-xL and Bax/Bak determines the status of autophagy (Luo and Rubinsztein, 2010). Caspases not only act as final executioners of apoptosis, but also participate in maintaining the above balance. Bax-induced caspase activation results in Beclin 1 cleavage and then inactivates autophagy. Pan-caspase inhibition remarkably stabilizes Beclin 1 and rescues from Bax-induced autophagy inhibition, indicating that Beclin 1 is the substrate of caspase. Autophagosomes are reduced following Bax-dependent cleavage of Beclin 1 or binding of Bcl-xL to Beclin 1. It is found that co-transfection of Bcl-xL and Bax normalizes the number of autophagosomes. Overexpression of Bax causes different outcomes of autophagy based on the anti-apoptotic state. Bax either reduces autophagy flux in cells without an increased anti-apoptotic ability by downregulating Beclin 1 via caspase cleavage, or induces autophagy in cells presenting an increased anti-apoptotic ability by overexpressing Bcl-xL or treatment of caspase inhibitors (Yu et al., 2004).
4.3 Balance among Bcl-2, Bax, and Beclin 1
Phosphorylation of Bcl-2 dissociates the Beclin 1-Bcl-2 complex and thus triggers autophagy. Bcl-2 also binds to Beclin 1 and breaks the Beclin 1-Vps34 complex, although it is not usually required for Bcl-2/Bcl-xL-induced suppression of autophagy. Compared with the Beclin 1-Vps34 complex, a multiprotein complex involving the Bcl-2/Bcl-xL, Beclin 1, and Vps34 inhibits autophagy by reducing the Class III PI3K activity of Vps34 (Pattingre et al., 2005). Loss of Bcl-2 results in cell death by inducing an excessive activity of autophagy, indicating that Bcl-2 functions as an interceptor to prevent autophagy-induced processes necessary for apoptosis (Yee et al., 2009). In cell apoptosis, Bcl-2 functions as a suppressor by preventing Bax homodimerization. During autophagy, Bax functions conversely to Bcl-2. Bax inhibits autophagy by promoting caspase-3-induced dissociation of Beclin 1 to break the interaction between Vps34 and Beclin 1. Besides, Bax stimulates autophagic flux influenced by the mitochondrial perturbation, and inhibits Bax-induced autophagy to further suppresses apoptosis. It is indicated that mitochondrial autophagy decides the cell fate (Maiuri et al., 2007). Members in the BH3-only subgroup of the Bcl-2 family, such as Bad, induce autophagy by acting on the BH3 domain and BH3 domain-binding groove of Bcl-2/Bcl-xL to impair the interaction between Bcl-2/Bcl-xL and Beclin 1 (Maiuri et al., 2007). Directly or indirectly, BH3-only proteins induce apoptosis through controlling the mitochondrial permeability of pro-apoptotic Bcl-2 family members (Oltersdorf et al., 2005). After a deep mining of the mechanism of apoptosis, BH3 mimetics of ABT-737 and its oral bioavailable analog Navitoclax have been designed and developed to stimulate cell apoptosis by targeting anti-apoptotic factors like Bcl-2, Bcl-xL and Bcl-w (Lee et al., 2015). Considering their architectures, we believed that BH3 mimetics are also promising mediators of cell autophagy.
4.4 Interaction between apoptosis and autophagy in the neurological damage of ICH
Autophagy is an initiator and competitor of apoptosis, as well as a collaborator to push cells into the abyss of death. Only Beclin 1 in the Bcl-2 family regulates autophagy, and is regulated by Bcl-2 and Bax. Apoptosis and autophagy hold differing sequences in cellular component degradation. During the process of apoptosis, the cytoskeleton is degraded in the early stage, and the organelles still exist in the late stage. In contrast, degradation of organelles occurs in the early stage of autophagy and the cytoskeleton remains in the late stage (Levine and Yuan, 2005). After triggering the apoptotic pathway, cells that are no longer needed commit suicide by shrinking and condensing the cytoplasm, organelles and chromatin. Eventually, cytoskeleton and nuclear proteins simultaneously undergo apoptosis (Slee et al., 2001). Macroautophagy is the major type of autophagy, in which portions of the cytoplasm are sequestered within autophagosomes and delivered to lysosomes for bulk degradation (Klionsky and Emr, 2000; Giricz et al., 2012). Lysosomes formed by autophagic vacuoles or autophagosomes during autophagy are responsible for engulfing the wrapped cytoplasm, organelles or proteins to synthesize essential molecules required for energy metabolism (Chen et al., 2010).
Autophagy, responded by a short-term stimulation, initiates to remove garbage organelles and particularly apoptosis-sensitive mitochondria. Through preventing the aggregation of lethal proteins and selectively removing pro-apoptotic proteins in the cytoplasm (e.g., caspase activators), autophagy shoulders a mission to prevent cell death. Therefore, autophagy, as a protective measure of self-rescue, reduces the sensitivity to apoptosis via eliminating harmful cellular components (Mariño et al., 2014; Bursch, 2001). However, a long-term stimulation uncovers the preface of programmed cell death (Gutierrez et al., 2014). Autophagy-dependent cell death, caused by excessive autophagy, is strictly defined as cell death caused by an autophagic flux. Presence of autophagosomes in dying cells, which may be probably caused by the blockage of autophagic flux (e.g., inhibition of caspase-10), is inconclusive of autophagic cell death (Mariño et al., 2014). Occasionally, blockage of the autophagosome-lysosome fusion increases autophagic flux, but inhibits autophagy. Current findings suggested that the positive expression of the autophagy substrate p62 is a reliable marker for autophagy-dependent cell death (Fricker et al., 2018). Instead of mutually exclusive, apoptosis and autophagy can both occur in the same type of cells. Initially stimulated by deprivation of growth factors, toxic drugs or hypoxia, autophagy usually mounts its stage prior to apoptosis, as an adaptive strategy. Once autophagy is powerless to counteract the long-term intolerable stress, apoptosis immediately joins to fight against unwanted cells (Yousefi et al., 2006; Kroemer et al., 2010). During their collaboration, autophagy assists to remove damaged components that are resistant to apoptosis (Lamparska-Przybysz et al., 2005).
Recovery of neurological function is closely linked with autophagy and apoptosis. Neuronal apoptosis in isolated brain regions results in the dysfunction of neural circuits, eventually leading to a selective loss of vulnerable neurons (Brown, 2021; Baron et al., 2014). Notably, neuronal apoptosis in the hematoma at post-ICH induces peripheral neuron damage, further aggravating neurological deficits. Through removing neuronal debris and reducing protein aggregation, autophagy exerts a protective role to promote neuron regeneration in patients with neurodegenerative diseases (Li et al., 2020; Martini-Stoica et al., 2016). Controversially, ICH rats overexpressing miR-146a present recovery of neurological function but a low profile of autophagy (Huan et al., 2020). The role of autophagy in ICH remains to be further analyzed.
5 ICH-associated autophagy and apoptosis
Autophagy and apoptosis of multiple types of brain cells, such as neurons, endothelial cells and microglia following ICH, pose vital influences on the prognosis. The formation of hematoma following ICH causes primary mechanical damages to surrounding tissues, and secondary hypoxic–ischemic toxicity further aggravates the mechanical damage.
Owing to reduced blood perfusion and hypoxia post-ICH, an ischemic penumbra is visualized around the ischemic core, with neurons are functionally impaired with lower activities, but not yet infarcted. Preserving tissue in the ischemic penumbra has therefore become the top priority (Back, 1998). Caspase-3 activation is initially observed in the ischemic penumbra after 3 h of ischemia–reperfusion injury (IRI), followed by the detection of its positive expression in the ischemic core at 24 h. On the contrary, the activity of caspase-3 remains low after the occlusion in the whole brain (Manabat et al., 2003). Suppression on apoptosis using caspase inhibitors offers a narrow treatment window, presenting efficacy on transient cerebral ischemia rather than the global cerebral ischemia (Li et al., 2000). Apoptosis is therefore believed as a rapid response in the local ischemic areas experiencing re-perfusion. The proportion of apoptotic cells is theoretically higher in the hypoperfused penumbra than in the ischemic core. Rapamycin is an autophagy activator that rescues hippocampal neuron death caused by global cerebral ischemia, suggesting that autophagy can prevent non-apoptotic cell death under certain conditions (Hwang et al., 2017). Autophagy abolition alone is sufficient to drive protein aggregation, leading to neuronal death and neurodegenerative diseases.
GFP-LC3 is an imaging marker on the autophagosomal membrane. Under physiological conditions, autophagy is barely detected in the brain (Mizushima et al., 2004). It is only detectable in the cortex and hippocampus following brain injuries (Weis et al., 2014). Following the stimulation from ischemia, autophagy increases in the early stage, particularly in cortical neurons, and gradually decreases alongside the weakening of lysosomes and aggravation of ischemic injuries (Liu et al., 2019). Just the increase in autophagosomes may result in an ineffective accumulation or even neurotoxicity. Ischemic injuries may be treated through interventions to keep a dynamic balance between autophagosomes and lysosomes (Menzies et al., 2017). In the brain, neurons are responsible for autophagosome biogenesis and trafficking. Due to their extreme sensitivity to autophagy, neurons also die with excessive autophagy (Benito-Cuesta et al., 2017).
Initially, apoptosis mainly occurs in neuronal cells following stroke (Manabat et al., 2003). After induction of excitotoxicity, p53, as a regulator of programmed cell death, triggers neuronal damages in sympathetic neurons after mitosis (Sakhi et al., 1994). Neurons connect and communicate with each other through synapses. When a neuron fails in response to a synaptic input or output, the connected neuron dies. In cases of ICH, neuronal death in the ischemic area results in the demise of neurons in the surrounding (known as the selective neuron loss) and remote regions (secondary neurodegeneration) (Brown, 2021; Baron et al., 2014). Neuronal apoptosis in the ischemic cerebral area may involve surrounding neurons, which is usually seen in ischemic stroke and Alzheimer’s disease.
Microglia are a type of immune cells that polarize in response to various brain injuries, including non-regional damages to neurons. Stimulated by excitotoxicity, sustained ischemia or inflammation, neurons induce the phagocytosis of microglia through releasing “find-me” and “eat-me” signals or binding to opsonins, like complement components C1q and C3b (Brown, 2021). Erythrolysis following ruptures of cerebral blood vessels triggers autophagy of microglia, and the released inflammatory factors further attack neurons (Yang et al., 2015). Based on this mechanism, autophagy inhibitors or Beclin 1 knockout contributes to rescuing transneuronal degeneration of thalamic neurons (Xing et al., 2012). Inhibition of autophagy by 3-MA or wort can also shrink infarct size and protect from oxygen glucose deprivation (OGD)-induced astrocyte damages in rats.
The BBB is composed of endothelial cells, also known as supporting cells, alongside with microglia and pericytes. The BBB maintains homeostasis of the central nervous system (CNS). Post-ICH, the integrity of endothelial cells is impaired, which increases the permeability of BBB and thus induces cerebral edema, leukocyte infiltration, and invasion of neurotoxic and active compounds. Eventually, neuronal loss occurs following the disruption of CNS homeostasis. Endothelial cells are targets of various initiators for apoptosis, including overexpression of Fas ligand, TGF-β and p53, deposition of amyloid-β peptide (Aβ) fragments, ATP depletion, increased Bax/Bcl-2 ratio, and caspase cascade (Rizzo and Leaver, 2010). There is a link between vascular endothelial cell apoptosis and neuronal death, and the vascular integrity reflects the degree of neuronal damage (Nagasawa and Kogure, 1989). Neuronal autophagy alleviates endothelial cell damage and hypoxia-induced BBB disruption via inhibiting membranous CLDN5 and CAV1-mediated transcytosis (Yang et al., 2021). Changes in BBB permeability influence the tight junctions between endothelial cells, and the overexpression of tight junction proteins favors to protect BBB permeability. It is reported that overexpression of exosomal miR-124-3p derived by microglia upregulates tight junction proteins, thereafter inducing autophagy but inhibiting apoptosis of endothelial cells (Zhao et al., 2022). Similarly, serum-derived exosomes suppress cell apoptosis in the striatum of rats with cerebral IRI through upregulating Bcl-2/Bax, ZO-1 and claudin-5 and downregulating caspase-3 in the cortex. Differing from the role of exosomal miR-124-3p in inhibiting apoptosis, serum-derived exosomes inhibit autophagy by reducing the LC3B-II/LC3B-I ratio and autophagy flux, thus inhibiting apoptosis (Huang et al., 2022). Astrocyte-derived exosomes consistently protect neurons by suppressing autophagy (Pei et al., 2019). Nevertheless, an excessive autophagy increases BBB permeability via degrading occludin (Kim et al., 2020).
Great efforts have been made for illustrating the transition between autophagy and apoptosis in neurons, microglia and endothelial cells. The distinct expression profiles of autophagic and apoptotic markers at certain time points of ICH models greatly reflect the dual role of autophagy. The autophagy and apoptosis varying across cell types require further in vivo experimental mining.
6 Autophagy and apoptosis in the treatment of ICH
6.1 ICH treatment by Western medicine
Post-ICH hematoma expansion is a risk factor for a poor prognosis, and hematoma volume is considered as a predictor of 30-day mortality and functional disability in ICH patients (Ruiz-Sandoval et al., 2007). As a common approach to treat ICH, surgical removal of hematoma lowers the compression on surrounding tissues and intracranial pressure. For high-risk individuals with large hematomas, either surgery or medication achieves a poor outcome (Gregson et al., 2012).
Tight blood pressure control is the major approach to prevent ICH. An early intensive blood pressure lowering has been recommended to limit hematoma expansion and treat ICH (Li et al., 2020). However, clinical benefits of an early intensive blood pressure lowering after acute ICH remain controversial (Moullaali et al., 2022). Medications are optional to ICH patients with surgical contraindications, including antihypertensive drugs, hemostatic drugs, antioxidants, and anti-inflammatory drugs (Greenberg et al., 2022).
6.1.1 Apoptosis-modulating drugs
Recombinant activated factor VII (rFVIIa) is a natural directly stopping bleeding to treat ICH. rFVIIa inhibit cell apoptosis by enhancing the Bcl-2/Bax ratio in rats with ICH (Ma et al., 2018). Clinical evidence has validated the efficacy of rFVIIa within 4 h of ICH in suppressing hematoma expansion, lowering mortality and improving the prognosis (Mayer et al., 2005).
Edaravone is applied to alleviate ICH-induced cerebral edema, neurological deficits and oxidative stress (Nakamura et al., 2008). It protects against neurological deficits in rats with ICH through downregulating caspase-3 and Bax, and upregulating Bcl-2 (Zhang et al., 2016).
Levetiracetam, widely used to treat epilepsy, exerts the function of protecting neurons post-ICH and preventing secondary injuries (Imai et al., 2020). Animal experiments have shown that levetiracetam inhibits inflammatory response and apoptosis in rats and mice with ICH by regulating the JAK2-STAT3 signaling pathway (Xiong et al., 2020).
Dexamethasone is able to inhibit cell apoptosis by modulating Bcl-2/Bax ratio and downregulating caspase-3, thus protecting against neuronal damage (Lee et al., 2015).
6.1.2 Autophagy-targeted drugs
Ebselen, an autophagy-targeted drug with anti-oxidative properties, alleviates silent brain infarction (SBI) following middle cerebral artery stroke by inhibiting expression levels of LC3-II and Beclin-1 (Li et al., 2015).
Prior to ischemic events, melatonin, a neuroprotective substance, can be used to prevent SBI through inhibiting ER stress-induced autophagy (Feng et al., 2017).
6.1.3 Dual-agents regulating apoptosis and autophagy
Cerebroprotein hydrolysate is a peptidergic drug that uniquely repairs nerve cells. It facilitates the recovery of secondary thalamic damage and neurological function after ICH by downregulating caspase-3 and LC3-II, and enhancing the Bcl-2/Bax ratio to inhibit both apoptosis and autophagy in the ipsilateral thalamus (Xing et al., 2014).
Interleukin 33 (IL-33) is reported to reduce brain water content and thus improves post-ICH neurological function. It executes its neuroprotective role through inhibiting inflammatory responses (downregulation of IL-1β and TNF-α), apoptosis (upregulation of Bcl-2 and downregulation of caspase-3) and autophagy (downregulation of LC3-II and Beclin-1) (Gao et al., 2017).
Prevention of SBI is the key strategy to improve the prognosis of ICH. Neuronal excitotoxicity, as an initiator of ICH-induced SBI, can be reduced by α-asarone (ASA) via suppressing mitochondria-mediated apoptosis and neuronal autophagy (Gao et al., 2022).
Lumbrokinase is featured by an antithrombotic activity. It inhibits apoptosis and autophagy by downregulating caspase-12 and NF-κB after cerebral infarction, and also suppresses inflammatory responses via reducing the NLRP3 inflammasome (Wang et al., 2022).
Atorvastatin exerts neuroprotective effects on ICH rats by fighting against apoptosis via downregulating calpain I, CHOP and GRP78 (Chen et al., 2022). Stimulation on autophagy is another mechanism underlying the function of atorvastatin in protecting subarachnoid hemorrhage (SAH) (Chen et al., 2020). Lovastatin inhibits apoptosis and regulates autophagy by silencing the AMPK/mTOR signaling pathway after ICH (Deng et al., 2023).
Minocycline is used to alleviate cerebral edema, maintain the integrity of BBB and improve the prognosis of ICH (Wang et al., 2019). Application of minocycline to ICH/SAH animal models achieves an anti-inflammatory outcome through inhibiting autophagy, apoptosis and release of inflammatory factors and promoting differentiation into M2 microglia (Wu et al., 2016; Dai et al., 2019).
6.2 ICH treatment by TCM
6.2.1 Apoptosis-associated TCM components
Baicalein is a flavone originally isolated from the traditional Chinese medicine (TCM) Scutellaria baicalensis. Intraperitoneal injection of baicalein reduces BBB permeability and inhibits cell apoptosis in ICH rats via inhibiting TLR4-mediated MAPK and NF-κB signaling pathways (Wei et al., 2017). Besides, baicalein suppresses apoptosis by inhibiting the Bax/Bcl-2 ratio (Wang et al., 2015).
Curcumin, a beta-diketone constituent extracted from the rhizome of Curcuma longa, effectively protects neuroinflammation and neuronal apoptosis by blocking M1 polarization of microglia following ICH (Wang et al., 2022).
Matrine is an alkaloid extracted from the dried roots, plants and fruits of the genus Sophora. It offers an inhibitory effect on inflammatory response, oxidative stress and apoptosis by inactivating the PI3K/Akt-mediated NF-κB signaling pathway and activating the Keap1/Nrf2-HO-1 signaling pathway (Liu et al., 2016).
Oxymatrine is also an alkaloid extracted from the root of Sophora flavescens. Similarly, it suppresses neuroinflammation, oxidative stress and neuronal apoptosis through downregulating TLR-4, NF-κB, TNF-α, IL-1β, and IL-6 (Huang et al., 2012).
Tetramethylpyrazine, also known as ligustrazine, is a chemical compound with neuroprotective properties. It relieves cerebral edema and inhibits the apoptosis of neurons in the surrounding tissues of hematoma via upregulating p-Akt and Bcl-2/Bax ratio and downregulating caspase-3 (Shao et al., 2018).
Ursolic acid is a pentacyclic triterpene acid widely presenting in loquat leaves, Ligustrum lucidum and Prunella vulgaris L. Treatment with ursolic acid reduces BBB permeability and brain water content in SAH rats through suppressing inflammatory factors, oxidative stress around hematomas and apoptosis (Zhang et al., 2014).
Ginsenoside Rb1, a triterpenoid saponin extracted from ginseng, favors the recovery of SAH rats by reducing cerebral edema, protecting BBB disruption and inhibiting p53-induced cell apoptosis (Li et al., 2010).
Ligustilide is a lactone compound extracted from Angelica sinensis and Ligusticum wallichii. Through downregulating p53 and caspase-3, ligustilide reduces the number of apoptotic cells in the surrounding area of SAH (Chen et al., 2011).
Triterpenoid saponins isolated from the Panax pseudoginseng greatly relieve cerebral edema and inhibit cell apoptosis (Xu et al., 2014).
Puerarin, a phytoestrogen found in the root of Pueraria, suppresses apoptosis and oxidative stress in surrounding brain regions of hematomas via inhibiting the PI3K/Akt-mediated inflammatory response and activating the SIRT signaling pathway (Zeng et al., 2021).
6.2.2 Autophagy-associated TCM components
Luteolin is a natural product found in a variety of edible TCM herbals, including chrysanthemum, Lonicera japonica and purple perilla. The neuroprotective function of luteolin post-ICH is dependent on the activated p62/Keap1/Nrf2 signaling pathway to stimulate autophagy and nuclear translocation of Nrf2. Moreover, it enhances autophagy and antioxidant property in both in vitro and in vivo ICH models, thus protecting against mitochondrial dysfunction in neurons (Tan et al., 2019).
Ginkgetin is a natural non-toxic biflavonoid extracted from the dried leaves of Ginkgo biloba. It protects cerebral microbleeds and neurological deficits by mediating autophagy via the JAK/STAT and MAPK signaling pathways (Adnan et al., 2020).
Artesunate is a semi-synthetic derivative of artemisinin. In the ICH rat model, artesunate increases autophagy to treat cerebral infarction via downregulating p-mTOR and upregulating Beclin-1 and Mcl-1 in the ischemic cerebral cortex and primary ischemic hippocampal neurons of rats (Shao et al., 2018).
Hydroxysafflor yellow A, the most effective water-soluble component of safflower, is usually adopted to treat cerebral infarction. It protects the brain after cerebral infarction by downregulating HIF-1, BNIP3, and Notch1 to inhibit cell autophagy (Zhang et al., 2022).
Total phenolics extracted from the lotus plumule protect neurological function after cerebral infarction and reduce cerebral necrosis and infarction by inhibiting autophagy via the PI3K/Akt signaling pathway (Qiao et al., 2023).
6.2.3 Apoptosis-associated TCM compound preparations
A combination of biologically active components of Ginkgo biloba greatly promotes the endogenous regression of hematoma by inhibiting neuronal apoptosis post-ICH through suppressing the LR4/NF-κB-dependent inflammatory response (Hu et al., 2011).
Didang Decoction (DDD) is a classic TCM compound preparation composed of rhubarb (Dahuang), peach seed (Taoren), leech (Shuizhi), and gadfly (Mengchong). Its neuroprotective effect in alleviating ICH-induced brain damages and cell apoptosis relies on the inhibition on the GPR78-IRE1/PERK signaling pathway (Huang et al., 2018).
Liangxue Tongyu Formulation, also commonly used to treat ICH, accelerates cell proliferation by activating the PI3K signaling pathway and inhibits glutamate-induced apoptosis of PC12 cells (Li et al., 2018).
6.2.4 Autophagy-associated TCM
Radix astragali-safflower is a classic herbal pair to treat cerebrovascular diseases. It effectively improves neurological function after cerebral infarction and prevents SBI by mediating autophagy through upregulating LC3II/LC3I, Beclin1, and p62 (Chen et al., 2022).
Tongqiao Huoxue Decoction, consisting of Radix Paeoniae Rubra, Ligusticum wallichii, red jujubes, safflower, Zingiberis Rhizoma Recens, Fistular onion stalk and Moschus, pharmacologically promotes blood circulation and dissipates stagnation. It alleviates cerebral ischemic injury by inhibiting autophagy through regulating the PI3K/Akt/mTOR signaling pathway (Hua et al., 2022).
Taohong Siwu Decoction exerts neuroprotective effects on cerebral infarction through upregulating LC3-II/LC3-I, Beclin1, Parkin, and PINK1, thereafter enhancing mitophagy and reducing the NLRP3 inflammasome (Ji et al., 2022).
6.2.5 Dual-regulation of TCM on autophagy and apoptosis
Resveratrol is a natural polyphenol extracted from TCM herbals of Veratrum, Polygonum multiflorum and Polygonum cuspidatum. Resveratrol reduces cerebral edema post-SAH via regulating both autophagy and apoptosis by the Akt and mTOR signaling pathways (Guo et al., 2018). Through mediating the AMPK/SIRT1 signaling pathway, resveratrol inhibits the activation of microglia and the release of inflammatory cytokines in a SAH model by regulating autophagy (Li and Han, 2018).
Acupuncture is a traditional TCM technique to stimulate hematoma absorption and improve neurological deficits after cerebral hemorrhage (Li et al., 2023). Through downregulating p-mTOR and p-S6K1, acupuncture triggers autophagy and inhibits the mTOR signaling pathway to alleviate neurological deficits in rats at post-ICH (Liu et al., 2022). Scalp acupuncture is a micro-system acupuncture technique that inhibits neuronal apoptosis by upregulating Beclin1, Parkin, PINK1 and NIX protein levels, and downregulating caspase-9 (Liu et al., 2021). Electroacupuncture at the acupoints of GV20 (Baihui)-GB7 (Quyan) enhances mitophagy and inhibits apoptosis after ICH, suggesting the therapeutic efficacy of electroacupuncture on ICH by balancing mitophagy and apoptosis (Guan et al., 2021).
7 Discussion
Apoptosis and autophagic cell death are not mutually exclusive; they can occur simultaneously within tissues or even within individual cells. Numerous studies have shed light on the interplay between autophagy and apoptosis at the cellular level. It has been suggested that autophagy may precede apoptosis, acting as an adaptive response to imminent cell death, particularly under conditions such as growth factor deprivation, exposure to toxic agents, hypoxia, and other forms of cellular damage. The lysosome, a crucial organelle in the autophagic process, also plays a regulatory role in apoptosis. Proteases released from lysosomal compartments can initiate apoptotic signaling, and lysosomes are involved in the trafficking of death receptors to the cell surface, influencing cellular sensitivity to external ligands (Bursch, 2001). Both autophagy and apoptosis are essential mechanisms for cellular self-preservation. When cellular injury occurs, apoptosis helps eliminate irreparably damaged cells, minimizing unnecessary energy expenditure. In contrast, autophagy temporarily boosts the energy supply required for cellular functions, mitigating damage and primarily acting as a protective strategy. However, the autophagic degradation of anti-apoptotic proteins can enhance apoptotic activity (Cao et al., 2021). From this perspective, the key factor in autophagy lies in the nature of the autophagic proteins involved. Additionally, autophagy can exacerbate cell death: sustained autophagy due to chronic stress—termed excessive autophagy—can lead to increased apoptosis and even trigger autophagic cell death. Thus, autophagic death and apoptosis are interconnected processes that often occur concurrently (Liu et al., 2023).
While autophagy may seem detrimental following cerebral hemorrhage, macrophages can have a beneficial role in this context, an area that warrants further investigation. Autophagy is also entangled with ferroptosis. Ferroptosis is a new type of non-apoptotic death. After cerebral hemorrhage, iron ions are deposited in the blood. Excessive free iron will produce hydroxyl radicals, leading to cell apoptosis and damage to the blood–brain barrier (BBB) (Wei et al., 2022). Activation of autophagy can promote ferroptosis (Liu et al., 2023). Multiple autophagy factors are involved in ferroptosis, such as NCOA4-promoted ferritinophagy, RAB7A-dependent lipophagy, BECN1-mediated system Xc inhibition, STAT3-induced changes in lysosomal membrane permeability, and HSP90-related chaperones (Xie et al., 2023). Depletion of core components of the autophagic machinery inhibits ferroptotic cell death, such as ATG5, ATG7 and BECN1/Vps30/Atg6 (beclin 1) (Liu et al., 2023). Concurrently, a deficiency in autophagy intensifies the production of reactive oxygen species (ROS) and apoptosis triggered by iron overload, indicating that autophagy plays a protective role in cellular responses to iron overload and oxidative stress (Sung et al., 2023). However, excessive iron overload-induced mitochondrial damage can induce further mitophagy, providing an additional source of iron for lipid peroxidation (Yu et al., 2022). Recent studies have demonstrated that the activation of macrophages can mitigate hematoma formation following cerebral hemorrhage and enhance neurological function. Nonetheless, the release of heme and iron subsequent to erythrocyte rupture may exacerbate ferroptosis by promoting autophagy (Fu et al., 2023).
8 Conclusion
Autophagy and apoptosis conjure up a complicated interplay after ICH. Autophagy serves as an initiator and competitor of apoptosis, and also accompanies apoptosis-induced cell death. In the Bcl-2 family, the Bcl-2/Bax ratio not only reflects a state of cell apoptosis, but also cooperates with Beclin 1 to regulate autophagy. ICH-induced apoptosis and autophagy struggle to maintain a dynamic balance. More evidences are needed to illustrate their interactions post-ICH.
Author contributions
MW: Writing – original draft. XC: Writing – original draft. SL: Writing – original draft. LW: Writing – review & editing. HT: Methodology, Writing – review & editing. YP: Data curation, Writing – review & editing. DZ: Data curation, Writing – review & editing. BF: Supervision, Writing – review & editing. XB: Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (ZYYCXTD-C-202207), Sichuan Science and Technology Program (2022YFS0613), Luzhou Science and Technology Program (2020-SYF-30), and Program of Southwest Medical University (2023ZYYJ01, 2023ZYYJ09; 2020XYLH-018,029).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2024.1445919/full#supplementary-material
References
Adnan, M., Rasul, A., Hussain, G., Shah, M. A., Zahoor, M. K., Anwar, H., et al. (2020). Ginkgetin: a natural biflavone with versatile pharmacological activities. Food Chem. Toxicol. 145:111642. doi: 10.1016/j.fct.2020.111642
Araki, T., Hayashi, M., and Saruta, T. (2006). Anion-exchange blocker enhances cytoplasmic vacuole formation and cell death in serum-deprived mouse kidney epithelial cells in mice. Cell Biol. Int. 30, 93–100. doi: 10.1016/j.cellbi.2005.10.020
Back, T. (1998). Pathophysiology of the ischemic penumbra--revision of a concept. Cell. Mol. Neurobiol. 18, 621–638. doi: 10.1023/A:1020629818207
Baron, J. C., Yamauchi, H., Fujioka, M., and Endres, M. (2014). Selective neuronal loss in ischemic stroke and cerebrovascular disease. J. Cereb. Blood Flow Metab. 34, 2–18. doi: 10.1038/jcbfm.2013.188
Benito-Cuesta, I., Diez, H., Ordoñez, L., and Wandosell, F. (2017). Assessment of autophagy in neurons and brain tissue. Cells 6:25. doi: 10.3390/cells6030025
Brown, G. C. (2021). Neuronal loss after stroke due to microglial phagocytosis of stressed neurons. Int. J. Mol. Sci. 22:13442. doi: 10.3390/ijms222413442
Bursch, W. (2001). The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 8, 569–581. doi: 10.1038/sj.cdd.4400852
Cao, W., Li, J., Yang, K., and Cao, D. (2021). An overview of autophagy: mechanism, regulation and research progress. Bull. Cancer 108, 304–322. doi: 10.1016/j.bulcan.2020.11.004
Carloni, S., Buonocore, G., and Balduini, W. (2008). Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol. Dis. 32, 329–339. doi: 10.1016/j.nbd.2008.07.022
Chen, Z. H., Lam, H. C., Jin, Y., Kim, H. P., Cao, J., Lee, S. J., et al. (2010). Autophagy protein microtubule-associated protein 1 light chain-3B (Lc3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc. Natl. Acad. Sci. USA 107, 18880–18885. doi: 10.1073/pnas.1005574107
Chen, Y., Lei, L., Wang, K., Liang, R., Qiao, Y., Feng, Z., et al. (2022). Huangqi-Honghua combination prevents cerebral infarction with Qi deficiency and blood stasis syndrome in rats by the autophagy pathway. Evid. Based Complement. Alternat. Med. 2022, 1–12. doi: 10.1155/2022/9496926
Chen, D., Sui, L., Chen, C., Liu, S., Sun, X., and Guan, J. (2022). Atorvastatin suppresses NLRP3 inflammasome activation in intracerebral hemorrhage via TLR4-and MyD88-dependent pathways. Aging 14, 462–476. doi: 10.18632/aging.203824
Chen, Y., Tang, W., Huang, X., An, Y., Li, J., Yuan, S., et al. (2024). Mitophagy in intracerebral hemorrhage: a new target for therapeutic intervention. Neural Regen. Res. 19, 316–323. doi: 10.4103/1673-5374.379019
Chen, D., Tang, J., Khatibi, N. H., Zhu, M., Li, Y., Wang, C., et al. (2011). Treatment with Z-ligustilide, a component of Angelica sinensis, reduces brain injury after a subarachnoid hemorrhage in rats. J. Pharmacol. Exp. Ther. 337, 663–672. doi: 10.1124/jpet.110.177055
Chen, L., Willis, S. N., Wei, A., Smith, B. J., Fletcher, J. I., Hinds, M. G., et al. (2005). Differential targeting of prosurvival Bcl-2 proteins by their Bh3-only ligands allows complementary apoptotic function. Mol. Cell 17, 393–403. doi: 10.1016/j.molcel.2004.12.030
Chen, J.-H., Wu, T., Xia, W.-Y., Shi, Z.-H., Zhang, C.-L., Chen, L., et al. (2020). An early neuroprotective effect of atorvastatin against subarachnoid hemorrhage. Neural Regen. Res. 15, 1947–1954. doi: 10.4103/1673-5374.280326
Chen, S., Zhao, L., Sherchan, P., Ding, Y., Yu, J., Nowrangi, D., et al. (2018). Activation of melanocortin receptor 4 with Ro27-3225 attenuates neuroinflammation through AMPK/JNK/p38 MAPK pathway after intracerebral hemorrhage in mice. J. Neuroinflammation 15:106. doi: 10.1186/s12974-018-1140-6
Cho, D. H., Jo, Y. K., Hwang, J. J., Lee, Y. M., Roh, S. A., and Kim, J. C. (2009). Caspase-mediated cleavage of Atg 6/Beclin-1 links apoptosis to autophagy in HeLa cells. Cancer Lett. 274, 95–100. doi: 10.1016/j.canlet.2008.09.004
Cohen, D. T., Wales, T. E., McHenry, M. W., Engen, J. R., and Walensky, L. D. (2020). Site-dependent cysteine Lipidation potentiates the activation of Proapoptotic Bax. Cell Rep. 30, 3229–39.e6. doi: 10.1016/j.celrep.2020.02.057
Crawford, R. A., Caldwell, C., Iles, R. K., Lowe, D., Shepherd, J. H., and Chard, T. (1998). Prognostic significance of the bcl-2 apoptotic family of proteins in primary and recurrent cervical cancer. Br. J. Cancer 78, 210–214. doi: 10.1038/bjc.1998.466
Crighton, D., Wilkinson, S., O'Prey, J., Syed, N., Smith, P., Harrison, P. R., et al. (2006). DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121–134. doi: 10.1016/j.cell.2006.05.034
Dai, S., Hua, Y., Keep, R. F., Novakovic, N., Fei, Z., and Xi, G. (2019). Minocycline attenuates brain injury and iron overload after intracerebral hemorrhage in aged female rats. Neurobiol. Dis. 126, 76–84. doi: 10.1016/j.nbd.2018.06.001
Danial, N. N., and Korsmeyer, S. J. (2004). Cell death: critical control points. Cell 116, 205–219. doi: 10.1016/S0092-8674(04)00046-7
Deng, X., Yang, J., Qing, R., Yuan, H., Yue, P., and Tian, S. (2023). Suppressive role of lovastatin in intracerebral hemorrhage through repression of autophagy. Metab. Brain Dis. 38, 361–372. doi: 10.1007/s11011-022-01101-6
Diener, H. C., and Hankey, G. J. (2020). Primary and secondary prevention of ischemic stroke and cerebral hemorrhage: Jacc focus seminar. J. Am. Coll. Cardiol. 75, 1804–1818. doi: 10.1016/j.jacc.2019.12.072
Dong, Y., Chen, H., Gao, J., Liu, Y., Li, J., and Wang, J. (2019). Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J. Mol. Cell. Cardiol. 136, 27–41. doi: 10.1016/j.yjmcc.2019.09.001
Duan, S., Wang, F., Cao, J., and Wang, C. (2020). Exosomes derived from Microrna-146a-5p-enriched bone marrow mesenchymal stem cells alleviate intracerebral hemorrhage by inhibiting neuronal apoptosis and microglial M1 polarization. Drug Des. Devel. Ther. 14, 3143–3158. doi: 10.2147/DDDT.S255828
Duan, X. C., Wang, W., Feng, D. X., Yin, J., Zuo, G., Chen, D. D., et al. (2017). Roles of autophagy and endoplasmic reticulum stress in intracerebral hemorrhage-induced secondary brain injury in rats. CNS Neurosci. Ther. 23, 554–566. doi: 10.1111/cns.12703
Ebrahimi-Fakhari, D., Mclean, P. J., and Unni, V. K. (2012). Alpha-synuclein's degradation in vivo: opening a new (cranial) window on the roles of degradation pathways in Parkinson disease. Autophagy 8, 281–283. doi: 10.4161/auto.8.2.18938
Fang, L., Yulin, Q., and Yaodan, Y. (2019). Iprimma affects T lymphocytes and Bcl-2 mrna expression in transplanted tumor tissues of lung cancer mice by inhibiting Tgf-β 1/Erk signal pathway. Chin. J. Cancer Biother. 26, 1203–1208. doi: 10.3872/j.issn.1007-385x.2019.11.004
Feigin, V. L., Lawes, C. M., Bennett, D. A., Barker-Collo, S. L., and Parag, V. (2009). Worldwide stroke incidence and early case fatality reported in 56 population-based studies: a systematic review. Lancet Neurol. 8, 355–369. doi: 10.1016/S1474-4422(09)70025-0
Feng, W., Huang, S., Wu, H., and Zhang, M. (2007). Molecular basis of Bcl-xL's target recognition versatility revealed by the structure of Bcl-xL in complex with the Bh3 domain of Beclin-1. J. Mol. Biol. 372, 223–235. doi: 10.1016/j.jmb.2007.06.069
Feng, D., Wang, B., Wang, L., Abraham, N., Tao, K., Huang, L., et al. (2017). Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via Perk and Ire1 signalings. J. Pineal. Res. 62. doi: 10.1111/jpi.12395
Flaherty, M. L., Kissela, B., Woo, D., Kleindorfer, D., Alwell, K., Sekar, P., et al. (2007). The increasing incidence of anticoagulant-associated intracerebral hemorrhage. Neurology 68, 116–121. doi: 10.1212/01.wnl.0000250340.05202.8b
Fleming, A., Noda, T., Yoshimori, T., and Rubinsztein, D. C. (2011). Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 7, 9–17. doi: 10.1038/nchembio.500
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Fu, K., Xu, W., Lenahan, C., Mo, Y., Wen, J., Deng, T., et al. (2023). Autophagy regulates inflammation in intracerebral hemorrhage: enemy or friend? Front. Cell Neurosci. 16:1036313. doi: 10.3389/fncel.2022.1036313
Funderburk, S. F., Wang, Q. J., and Yue, Z. (2010). The Beclin 1–VPS34 complex – at the crossroads of autophagy and beyond. Trends Cell Biol. 20, 355–362. doi: 10.1016/j.tcb.2010.03.002
Gao, X., Li, R., Luo, L., Zhang, D., Liu, Q., Zhang, J., et al. (2022). Alpha-asarone ameliorates neurological deterioration of intracerebral hemorrhagic rats by alleviating secondary brain injury via anti-excitotoxicity pathways. Phytomedicine 105:154363. doi: 10.1016/j.phymed.2022.154363
Gao, Y., Ma, L., Luo, C.-L., Wang, T., Zhang, M.-Y., Shen, X., et al. (2017). Il-33 exerts neuroprotective effect in mice intracerebral hemorrhage model through suppressing inflammation/apoptotic/Autophagic pathway. Mol. Neurobiol. 54, 3879–3892. doi: 10.1007/s12035-016-9947-6
Garner, T. P., Amgalan, D., Reyna, D. E., Li, S., Kitsis, R. N., and Gavathiotis, E. (2019). Small-molecule allosteric inhibitors of Bax. Nat. Chem. Biol. 15, 322–330. doi: 10.1038/s41589-018-0223-0
Giricz, Z., Mentzer, R. M. Jr., and Gottlieb, R. A. (2012). Autophagy, myocardial protection, and the metabolic syndrome. J. Cardiovasc. Pharmacol. 60, 125–132. doi: 10.1097/FJC.0b013e318256ce10
Green, D. R., and Llambi, F. (2015). Cell death signaling. Cold Spring Harb. Perspect. Biol. 7:a006080. doi: 10.1101/cshperspect.a006080
Greenberg, S. M., Ziai, W. C., Cordonnier, C., Dowlatshahi, D., Francis, B., Goldstein, J. N., et al. (2022). 2022 Guideline for the Management of Patients with Spontaneous Intracerebral Hemorrhage: a guideline from the American Heart Association/American Stroke Association. Stroke, 2022 53, e282–e361. doi: 10.1161/STR.0000000000000407
Gregson, B. A., Broderick, J. P., Auer, L. M., Batjer, H., Chen, X. C., Juvela, S., et al. (2012). Individual patient data subgroup meta-analysis of surgery for spontaneous supratentorial intracerebral hemorrhage. Stroke 43, 1496–1504. doi: 10.1161/STROKEAHA.111.640284
Gross, A., Mcdonnell, J. M., and Korsmeyer, S. J. (1999). Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 13, 1899–1911. doi: 10.1101/gad.13.15.1899
Guan, R., Li, Z., Dai, X., Zou, W., Yu, X., Liu, H., et al. (2021). Electroacupuncture at Gv20-Gb7 regulates mitophagy to protect against neurological deficits following intracerebral hemorrhage via inhibition of apoptosis. Mol. Med. Rep. 24:492. doi: 10.3892/mmr.2021.12131
Guo, D., Xie, J., Zhao, J., Huang, T., Guo, X., and Song, J. (2018). Resveratrol protects early brain injury after subarachnoid hemorrhage by activating autophagy and inhibiting apoptosis mediated by the Akt/mtor pathway. Neuroreport 29, 368–379. doi: 10.1097/WNR.0000000000000975
Gutierrez, E., Richardson, D. R., and Jansson, P. J. (2014). The anticancer agent di-2-pyridylketone 4, 4-dimethyl-3-thiosemicarbazone (Dp44mT) overcomes prosurvival autophagy by two mechanisms: persistent induction of autophagosome synthesis and impairment of lysosomal integrity. J. Biol. Chem. 289, 33568–33589. doi: 10.1074/jbc.M114.599480
He, C., Bassik, M. C., Moresi, V., Sun, K., Wei, Y., Zou, Z., et al. (2012). Exercise-induced Bcl2-regulated autophagy is required for muscle glucose homeostasis. Nature 481, 511–515. doi: 10.1038/nature10758
He, C., and Levine, B. (2010). The Beclin 1 interactome. Curr. Opin. Cell Biol. 22, 140–149. doi: 10.1016/j.ceb.2010.01.001
He, Y., Wan, S., Hua, Y., Keep, R. F., and Xi, G. (2008). Autophagy after experimental intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 28, 897–905. doi: 10.1038/sj.jcbfm.9600578
He, C., Zhu, H., Li, H., Zou, M. H., and Xie, Z. (2013). Dissociation of Bcl-2-Beclin 1 complex by activated Ampk enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 62, 1270–1281. doi: 10.2337/db12-0533
Hu, Y.-Y., Huang, M., Dong, X.-Q., Xu, Q.-P., Yu, W.-H., and Zhang, Z.-Y. (2011). Ginkgolide B reduces neuronal cell apoptosis in the hemorrhagic rat brain: possible involvement of toll-like receptor 4/nuclear factor-kappa B pathway. J. Ethnopharmacol. 137, 1462–1468. doi: 10.1016/j.jep.2011.08.034
Hua, Y., Zhai, Y., Wang, G., Wang, N., Wu, Q., Huang, Q., et al. (2022). Tong-Qiao-Huo-Xue decoction activates Pi3K/Akt/mtor pathway to reduce Bmecs autophagy after cerebral ischemia/reperfusion injury. J. Ethnopharmacol. 298:115585. doi: 10.1016/j.jep.2022.115585
Huan, S., Jin, J., Shi, C. X., Li, T., Dai, Z., and Fu, X.-J. (2020). Overexpression of mi R-146a inhibits the apoptosis of hippocampal neurons of rats with cerebral hemorrhage by regulating autophagy. Hum. Exp. Toxicol. 39, 1178–1189. doi: 10.1177/0960327120907131
Huang, Y., Gao, X., Zhou, X., Xie, B., Zhang, Y., Zhu, J., et al. (2020). Mitophagy in the Hippocampus is excessive activated after cardiac arrest and cardiopulmonary resuscitation. Neurochem. Res. 45, 322–330. doi: 10.1007/s11064-019-02916-z
Huang, M., Hu, Y. Y., Dong, X. Q., Xu, Q. P., Yu, W. H., and Zhang, Z. Y. (2012). The protective role of oxymatrine on neuronal cell apoptosis in the hemorrhagic rat brain. J. Ethnopharmacol. 143, 228–235. doi: 10.1016/j.jep.2012.06.028
Huang, Q., Lan, T., Lu, J., Zhang, H., Zhang, D., Lou, T., et al. (2018). DiDang Tang inhibits endoplasmic reticulum stress-mediated apoptosis induced by oxygen glucose deprivation and intracerebral hemorrhage through blockade of the Grp78-Ire1/Perk pathways. Front. Pharmacol. 9:1423. doi: 10.3389/fphar.2018.01423
Huang, L. Y., Song, J. X., Cai, H., Wang, P. P., Yin, Q. L., Zhang, Y. D., et al. (2022). Healthy serum-derived exosomes improve neurological outcomes and protect blood-brain barrier by inhibiting endothelial cell apoptosis and reversing autophagy-mediated tight junction protein reduction in rat stroke model. Front. Cell. Neurosci. 16:841544. doi: 10.3389/fncel.2022.841544
Huaxian, Z., Xingqun, L., Tao, T., Qinghua, L., and Xialing, L. (2003). Characteristics of Bcl-2, Bax protein expression and intervention in brain tissue of experimental cerebral hemorrhage in Chinese medicine. J. Cent. South Univ. (Med. Sci.). 28:229. doi: 10.3321/j.issn:1672-7347.2003.03.009
Huhtakangas, J., Tetri, S., Juvela, S., Saloheimo, P., Bode, M. K., and Hillbom, M. (2011). Effect of increased warfarin use on warfarin-related cerebral hemorrhage: a longitudinal population-based study. Stroke 42, 2431–2435. doi: 10.1161/STROKEAHA.111.615260
Hwang, J. Y., Gertner, M., Pontarelli, F., Court-Vazquez, B., Bennett, M. V. L., Ofengeim, D., et al. (2017). Global ischemia induces lysosomal-mediated degradation of mtor and activation of autophagy in hippocampal neurons destined to die. Cell Death Differ. 24, 317–329. doi: 10.1038/cdd.2016.140
Imai, T., Sugiyama, T., Iwata, S., Nakamura, S., Shimazawa, M., and Hara, H. (2020). Levetiracetam, an antiepileptic drug has neuroprotective effects on intracranial hemorrhage injury. Neuroscience 431, 25–33. doi: 10.1016/j.neuroscience.2020.01.036
Islam, M. S., Anderson, C. S., Hankey, G. J., Hardie, K., Carter, K., Broadhurst, R., et al. (2008). Trends in incidence and outcome of stroke in Perth, Western Australia during 1989 to 2001: the Perth community stroke study. Stroke 39, 776–782. doi: 10.1161/STROKEAHA.107.493643
Ji, Z.-J., Shi, Y., Li, X., Hou, R., Yang, Y., Liu, Z.-Q., et al. (2022). Neuroprotective effect of Taohong Siwu decoction on cerebral ischemia/reperfusion injury via Mitophagy-Nlrp3 Inflammasome pathway. Front. Pharmacol. 13:910217. doi: 10.3389/fphar.2022.910217
Johnson, C. O., Nguyen, M., Roth, G. A., Nichols, E., Alam, T., Abate, D., et al. (2016). Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the global burden of disease study 2019, 439–458. doi: 10.1016/S1474-4422(19)30034-1
Kang, R., Zeh, H. J., Lotze, M. T., and Tang, D. (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580. doi: 10.1038/cdd.2010.191
Khellaf, M., Quantin, C., D'athis, P., Fassa, M., Jooste, V., Hervieu, M., et al. (2010). Age-period-cohort analysis of stroke incidence in Dijon from 1985 to 2005. Stroke 41, 2762–2767. doi: 10.1161/STROKEAHA.110.592147
Kihara, A., Kabeya, Y., Ohsumi, Y., and Yoshimori, T. (2001). Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2, 330–335. doi: 10.1093/embo-reports/kve061
Kim, K. A., Kim, D., Kim, J. H., Shin, Y. J., Kim, E. S., Akram, M., et al. (2020). Autophagy-mediated occludin degradation contributes to blood-brain barrier disruption during ischemia in bEnd.3 brain endothelial cells and rat ischemic stroke models. Fluids Barr. CNS 17:21. doi: 10.1186/s12987-020-00182-8
Klionsky, D. J., and Emr, S. D. (2000). Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721. doi: 10.1126/science.290.5497.1717
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J. I., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Konishi, A., Arakawa, S., Yue, Z., and Shimizu, S. (2012). Involvement of Beclin 1 in engulfment of apoptotic cells. J. Biol. Chem. 287, 13919–13929. doi: 10.1074/jbc.M112.348375
Kroemer, G., Mariño, G., and Levine, B. (2010). Autophagy and the integrated stress response. Mol. Cell 40, 280–293. doi: 10.1016/j.molcel.2010.09.023
Lamparska-Przybysz, M., Gajkowska, B., and Motyl, T. (2005). Cathepsins and bid are involved in the molecular switch between apoptosis and autophagy in breast cancer Mcf-7 cells exposed to camptothecin. J. Physiol. Pharmacol. 56, 159–179. doi: 10.4161/auto.2149
Lee, I. N., Cheng, W. C., Chung, C. Y., Lee, M. H., Lin, M. H. C., Kuo, C. H., et al. (2015). Dexamethasone reduces brain cell apoptosis and inhibits inflammatory response in rats with intracerebral hemorrhage. J. Neurosci. Res. 93, 178–188. doi: 10.1002/jnr.23454
Lee, J. H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related Ps1 mutations. Cell 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008
Levine, B., and Yuan, J. (2005). Autophagy in cell death: an innocent convict? J. Clin. Invest. 115, 2679–2688. doi: 10.1172/JCI26390
Li, H., Colbourne, F., Sun, P., Zhao, Z., Buchan, A. M., and Iadecola, C. (2000). Caspase inhibitors reduce neuronal injury after focal but not global cerebral ischemia in rats. Stroke 31, 176–182. doi: 10.1161/01.STR.31.1.176
Li, Z., and Han, X. (2018). Resveratrol alleviates early brain injury following subarachnoid hemorrhage: possible involvement of the Ampk/Sirt 1/autophagy signaling pathway. Biol. Chem. 399, 1339–1350. doi: 10.1515/hsz-2018-0269
Li, X., Huang, X., Tang, Y., Zhao, F., Cao, Y., Yin, L., et al. (2018). Assessing the pharmacological and therapeutic efficacy of traditional Chinese medicine Liangxue Tongyu prescription for intracerebral hemorrhagic stroke in neurological disease models. Front. Pharmacol. 9:1169. doi: 10.3389/fphar.2018.01169
Li, R., Li, D., Wu, C., Ye, L., Wu, Y., Yuan, Y., et al. (2020). Nerve growth factor activates autophagy in Schwann cells to enhance myelin debris clearance and to expedite nerve regeneration. Theranostics 10, 1649–1677. doi: 10.7150/thno.40919
Li, Y., Tang, J., Khatibi, N. H., Zhu, M., Chen, D., Zheng, W., et al. (2010). Ginsenoside Rbeta 1 reduces neurologic damage, is anti-apoptotic, and down-regulates p53 and Bax in subarachnoid hemorrhage. Curr. Neurovasc. Res. 7, 85–94. doi: 10.2174/156720210791184952
Li, L., Wang, X., Guo, J., Chen, Y., and Wang, Z. (2023). Effect of acupuncture in the acute phase of intracerebral hemorrhage on the prognosis and serum Bdnf: a randomized controlled trial. Front. Neurosci. 17:1167620. doi: 10.3389/fnins.2023.1167620
Li, Q., Warren, A. D., Qureshi, A. I., Morotti, A., Falcone, G. J., Sheth, K. N., et al. (2020). Ultra-early blood pressure reduction attenuates hematoma growth and improves outcome in intracerebral hemorrhage. Ann. Neurol. 88, 388–395. doi: 10.1002/ana.25793
Li, Y., Zhang, J., Chen, L., Xing, S., Li, J., Zhang, Y., et al. (2015). Ebselen reduces autophagic activation and cell death in the ipsilateral thalamus following focal cerebral infarction. Neurosci. Lett. 600, 206–212. doi: 10.1016/j.neulet.2015.06.024
Liang, J., Shao, S. H., Xu, Z. X., Hennessy, B., Ding, Z., Larrea, M., et al. (2007). The energy sensing Lkb1-Ampk pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 9, 218–224. doi: 10.1038/ncb1537
Liu, J., Liu, Y., Wang, Y., Li, C., Xie, Y., Dj, K., et al. (2023). Tmem 164 is a new determinant of autophagy-dependent ferroptosis. Autophagy 19, 945–956. doi: 10.1080/15548627.2022.2111635
Liu, Y., Xue, X., Zhang, H., Che, X., Luo, J., Wang, P., et al. (2019). Neuronal-targeted Tfeb rescues dysfunction of the autophagy-lysosomal pathway and alleviates ischemic injury in permanent cerebral ischemia. Autophagy 15, 493–509. doi: 10.1080/15548627.2018.1531196
Liu, S., Yao, S., Yang, H., Liu, S. J., and Wang, Y. J. (2023). Autophagy: regulator of cell death. Cell Death Dis. 14:648. doi: 10.1038/s41419-023-06154-8
Liu, P., Yu, X., Dai, X., Zou, W., Yu, X., Niu, M., et al. (2021). Scalp acupuncture attenuates brain damage after intracerebral hemorrhage through enhanced Mitophagy and reduced apoptosis in rats. Front. Aging Neurosci. 13:718631. doi: 10.3389/fnagi.2021.718631
Liu, H., Zhang, B., Li, X.-W., Du, J., Feng, P.-P., and Cheng, C. (2022). Acupuncture inhibits mammalian target of rapamycin, promotes autophagy and attenuates neurological deficits in a rat model of hemorrhagic stroke. Acupunct. Med. 40, 59–67. doi: 10.1177/09645284211028873
Liu, X., Zhang, X., Ma, K., Zhang, R., Hou, P., Sun, B., et al. (2016). Matrine alleviates early brain injury after experimental subarachnoid hemorrhage in rats: Possible involvement of Pi3K/Akt-mediated Nf-κB inhibition and Keap1/Nrf2-dependent ho-1 inductionn. Cell. Mol. Biol. 62, 38–44. doi: 10.14715/cmb/2016.62.11.7
Lovelock, C. E., Molyneux, A. J., and Rothwell, P. M. (2007). Change in incidence and aetiology of intracerebral haemorrhage in Oxfordshire, UK, between 1981 and 2006: a population-based study. Lancet Neurol. 6, 487–493. doi: 10.1016/S1474-4422(07)70107-2
Luo, S., and Rubinsztein, D. C. (2010). Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 17, 268–277. doi: 10.1038/cdd.2009.121
Ma, Y.-M., Zheng, J.-X., Xu, X., Shan, Y.-S., Gao, Z.-W., and Chen, W.-G. (2018). Effects of recombinant activated coagulation factor vii on apoptosis and expressions of Bcl-2 and Bax in rats with intracerebral hemorrhage. Eur. Rev. Med. Pharmacol. Sci. 22, 5264–5269. doi: 10.26355/eurrev_201808_15725
Maiuri, M. C., Criollo, A., Tasdemir, E., Vicencio, J. M., Tajeddine, N., Hickman, J. A., et al. (2007). Bh3-only proteins and Bh3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy 3, 374–376. doi: 10.4161/auto.4237
Maiuri, M. C., le Toumelin, G., Criollo, A., Rain, J. C., Gautier, F., Juin, P., et al. (2007). Functional and physical interaction between Bcl-X(L) and a Bh3-like domain in Beclin-1. EMBO J. 26, 2527–2539. doi: 10.1038/sj.emboj.7601689
Manabat, C., Han, B. H., Wendland, M., Derugin, N., Fox, C. K., Choi, J., et al. (2003). Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke 34, 207–213. doi: 10.1161/01.STR.0000047101.87575.3C
Mariño, G., Niso-Santano, M., Baehrecke, E. H., and Kroemer, G. (2014). Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 15, 81–94. doi: 10.1038/nrm3735
Martini-Stoica, H., Xu, Y., Ballabio, A., and Zheng, H. (2016). The autophagy-lysosomal pathway in neurodegeneration: a Tfeb perspective. Trends Neurosci. 39, 221–234. doi: 10.1016/j.tins.2016.02.002
Mayer, S. A., Brun, N. C., Begtrup, K., Broderick, J., Davis, S., Diringer, M. N., et al. (2005). Recombinant activated factor vii for acute intracerebral hemorrhage. N. Engl. J. Med. 352, 777–785. doi: 10.1056/NEJMoa042991
Menzies, F. M., Fleming, A., Caricasole, A., Bento, C. F., Andrews, S. P., Ashkenazi, A., et al. (2017). Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034. doi: 10.1016/j.neuron.2017.01.022
Mizushima, N., and Komatsu, M. (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. doi: 10.1016/j.cell.2011.10.026
Mizushima, N., Yamamoto, A., Matsui, M., Yoshimori, T., and Ohsumi, Y. (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–1111. doi: 10.1091/mbc.e03-09-0704
Moullaali, T. J., Wang, X., Sandset, E. C., Woodhouse, L. J., Law, Z. K., Arima, H., et al. (2022). Early lowering of blood pressure after acute intracerebral haemorrhage: a systematic review and meta-analysis of individual patient data. J. Neurol. Neurosurg. Psychiatry 93, 6–13. doi: 10.1136/jnnp-2021-327195
Nagasawa, H., and Kogure, K. (1989). Correlation between cerebral blood flow and histologic changes in a new rat model of middle cerebral artery occlusion. Stroke 20, 1037–1043. doi: 10.1161/01.STR.20.8.1037
Nakamura, T., Kuroda, Y., Yamashita, S., Zhang, X., Miyamoto, O., Tamiya, T., et al. (2008). Edaravone attenuates brain edema and neurologic deficits in a rat model of acute intracerebral hemorrhage. Stroke 39, 463–469. doi: 10.1161/STROKEAHA.107.486654
Oltersdorf, T., Elmore, S. W., Shoemaker, A. R., Armstrong, R. C., Augeri, D. J., Belli, B. A., et al. (2005). An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681. doi: 10.1038/nature03579
Oltvai, Z. N., and Korsmeyer, S. J. (1994). Checkpoints of dueling dimers foil death wishes. Cell 79, 189–192. doi: 10.1016/0092-8674(94)90188-0
O'Neill, K. L., Huang, K., Zhang, J., Chen, Y., and Luo, X. (2016). Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 30, 973–988. doi: 10.1101/gad.276725.115
Pattingre, S., Bauvy, C., Carpentier, S., Levade, T., Levine, B., and Codogno, P. (2009). Role of Jnk1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 284, 2719–2728. doi: 10.1074/jbc.M805920200
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., et al. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939. doi: 10.1016/j.cell.2005.07.002
Pei, X., Li, Y., Zhu, L., and Zhou, Z. (2019). Astrocyte-derived exosomes suppress autophagy and ameliorate neuronal damage in experimental ischemic stroke. Exp. Cell Res. 382:111474. doi: 10.1016/j.yexcr.2019.06.019
Puyal, J., Vaslin, A., Mottier, V., and Clarke, P. G. H. (2009). Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann. Neurol. 66, 378–389. doi: 10.1002/ana.21714
Qiao, W., Zang, Z., Li, D., Shao, S., Li, Q., and Liu, Z. (2023). Liensinine ameliorates ischemia-reperfusion-induced brain injury by inhibiting autophagy via Pi3K/Akt signaling. Funct. Integr. Genomics 23:140. doi: 10.1007/s10142-023-01063-7
Qureshi, A. I., Ling, G. S., Khan, J., Suri, M. F. K., Miskolczi, L., Guterman, L. R., et al. (2001). Quantitative analysis of injured, necrotic, and apoptotic cells in a new experimental model of intracerebral hemorrhage. Crit. Care Med. 29, 152–157. doi: 10.1097/00003246-200101000-00030
Qureshi, A. I., Suri, M. F., Ostrow, P. T., Kim, S. H., Ali, Z., Shatla, A. A., et al. (2003). Apoptosis as a form of cell death in intracerebral hemorrhage. Neurosurgery 52, 1041–1048.discussion: 7–8. doi: 10.1227/01.NEU.0000057694.96978.BC
Qureshi, A. I., Tuhrim, S., Broderick, J. P., Batjer, H. H., Hondo, H., and Hanley, D. F. (2001). Spontaneous intracerebral hemorrhage. N. Engl. J. Med. 344, 1450–1460. doi: 10.1056/NEJM200105103441907
Rizzo, M. T., and Leaver, H. A. (2010). Brain endothelial cell death: modes, signaling pathways, and relevance to neural development, homeostasis, and disease. Mol. Neurobiol. 42, 52–63. doi: 10.1007/s12035-010-8132-6
Ruiz-Sandoval, J.́. L., Chiquete, E., Romero-Vargas, S., Padilla-Martínez, J. J., and González-Cornejo, S. (2007). Grading scale for prediction of outcome in primary intracerebral hemorrhages. Stroke 38, 1641–1644. doi: 10.1161/STROKEAHA.106.478222
Sakhi, S., Bruce, A., Sun, N., Tocco, G., Baudry, M., and Schreiber, S. S. (1994). p53 induction is associated with neuronal damage in the central nervous system. Proc. Natl. Acad. Sci. USA 91, 7525–7529. doi: 10.1073/pnas.91.16.7525
Salminen, A., Kaarniranta, K., Kauppinen, A., Ojala, J., Haapasalo, A., Soininen, H., et al. (2013). Impaired autophagy and app processing in Alzheimer's disease: the potential role of Beclin 1 interactome. Prog. Neurobiol. 106-107, 33–54. doi: 10.1016/j.pneurobio.2013.06.002
Selvakumaran, M., Lin, H. K., Miyashita, T., Wang, H. G., Krajewski, S., Reed, J. C., et al. (1994). Immediate early up-regulation of bax expression by p 53 but not Tgf beta 1: a paradigm for distinct apoptotic pathways. Oncogene 9, 1791–1798
Shao, M., Shen, Y., Sun, H., Meng, D., Huo, W., and Qi, X. (2018). Protectiveness of Artesunate given prior ischemic cerebral infarction is mediated by increased autophagy. Front. Neurol. 9:634. doi: 10.3389/fneur.2018.00634
Shao, Z., Wu, P., Wang, X., Jin, M., Liu, S., Ma, X., et al. (2018). Tetramethylpyrazine protects against early brain injury and inhibits the Perk/Akt pathway in a rat model of subarachnoid hemorrhage. Neurochem. Res. 43, 1650–1659. doi: 10.1007/s11064-018-2581-0
Shen, X., Ma, L., Dong, W., Wu, Q., Gao, Y., Luo, C., et al. (2016). Autophagy regulates intracerebral hemorrhage induced neural damage via apoptosis and Nf-κB pathway. Neurochem. Int. 96, 100–112. doi: 10.1016/j.neuint.2016.03.004
Shimizu, S., Kanaseki, T., Mizushima, N., Mizuta, T., Arakawa-Kobayashi, S., Thompson, C. B., et al. (2004). Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 6, 1221–1228. doi: 10.1038/ncb1192
Slee, E. A., Adrain, C., and Martin, S. J. (2001). Executioner caspase-3, −6, and −7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 276, 7320–7326. doi: 10.1074/jbc.M008363200
Smith, C. M., Chen, Y., Sullivan, M. L., Kochanek, P. M., and Clark, R. S. B. (2011). Autophagy in acute brain injury: feast, famine, or folly? Neurobiol. Dis. 43, 52–59. doi: 10.1016/j.nbd.2010.09.014
Subburaj, Y., Cosentino, K., Axmann, M., Pedrueza-Villalmanzo, E., Hermann, E., Bleicken, S., et al. (2015). Bax monomers form dimer units in the membrane that further self-assemble into multiple oligomeric species. Nat. Commun. 6:8042. doi: 10.1038/ncomms9042
Sung, H. K., Murugathasan, M., Abdul-Sater, A. A., and Sweeney, G. (2023). Autophagy deficiency exacerbates iron overload induced reactive oxygen species production and apoptotic cell death in skeletal muscle cells. Cell Death Dis. 14:252. doi: 10.1038/s41419-022-05484-3
Takacs-Vellai, K., Vellai, T., Puoti, A., Passannante, M., Wicky, C., Streit, A., et al. (2005). Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr. Biol. 15, 1513–1517. doi: 10.1016/j.cub.2005.07.035
Tan, X., Yang, Y., Xu, J., Zhang, P., Deng, R., Mao, Y., et al. (2019). Luteolin exerts neuroprotection via modulation of the p62/Keap1/Nrf2 pathway in intracerebral hemorrhage. Front. Pharmacol. 10:1551. doi: 10.3389/fphar.2019.01551
Uren, R. T., Iyer, S., and Kluck, R. M. (2017). Pore formation by dimeric Bak and Bax: an unusual pore? Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 372:20160218. doi: 10.1098/rstb.2016.0218
Wall, N. R., Mohammad, R. M., and Al-Katib, A. M. (1999). Bax: Bcl-2 ratio modulation by bryostatin 1 and novel antitubulin agents is important for susceptibility to drug induced apoptosis in the human early pre-B acute lymphoblastic leukemia cell line, Reh. Leukemia Res. 23, 881–888. doi: 10.1016/S0145-2126(99)00108-3
Wang, G., Li, Z., Li, S., Ren, J., Suresh, V., Xu, D., et al. (2019). Minocycline preserves the integrity and permeability of Bbb by altering the activity of Dkk1-Wnt signaling in Ich model. Neuroscience 415, 135–146. doi: 10.1016/j.neuroscience.2019.06.038
Wang, Y.-H., Liao, J.-M., Chen, K.-M., Su, H.-H., Liu, P.-H., Chen, Y.-H., et al. (2022). Lumbrokinase regulates endoplasmic reticulum stress to improve neurological deficits in ischemic stroke. Neuropharmacology 221:109277. doi: 10.1016/j.neuropharm.2022.109277
Wang, F., Xia, J.-J., Shen, L.-J., Jiang, T.-T., Li, W.-L., You, D.-L., et al. (2022). Curcumin attenuates intracerebral hemorrhage-induced neuronal apoptosis and neuroinflammation by suppressing Jak1/Stat1 pathway. Biochem. Cell Biol. 100, 236–245. doi: 10.1139/bcb-2021-0423
Wang, C., Xie, G., Zhou, C., Zhang, X., Li, T., Xu, J., et al. (2015). Baincalein alleviates early brain injury after experimental subarachnoid hemorrhage in rats: possible involvement of Tlr4/Nf-κB-mediated inflammatory pathway. Brain Res. 1594, 245–255. doi: 10.1016/j.brainres.2014.10.014
Wang, J.-P., and Zhang, M.-Y. (2017). Role for target of rapamycin (mtor) signal pathway in regulating neuronal injury after intracerebral hemorrhage. Cell. Physiol. Biochem. 41, 145–153. doi: 10.1159/000455983
Wei, Y., Pattingre, S., Sinha, S., Bassik, M., and Levine, B. (2008). Jnk1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30, 678–688. doi: 10.1016/j.molcel.2008.06.001
Wei, Y., Sinha, S., and Levine, B. (2008). Dual role of Jnk1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 4, 949–951. doi: 10.4161/auto.6788
Wei, Y., Song, X., Gao, Y., Gao, Y., Li, Y., and Gu, L. (2022). Iron toxicity in intracerebral hemorrhage: Physiopathological and therapeutic implications. Brain Res. Bull. 178, 144–154. doi: 10.1016/j.brainresbull.2021.11.014
Wei, N., Wei, Y., Li, B., and Pang, L. (2017). Baicalein promotes neuronal and behavioral recovery after intracerebral hemorrhage via suppressing apoptosis, oxidative stress and Neuroinflammation. Neurochem. Res. 42, 1345–1353. doi: 10.1007/s11064-017-2179-y
Weidong, W., and Zhengtang, C. (2007). The Bcl-2/Bax ratio and cell fate. Chin. J. Cancer Biother. 4, 393–396. doi: 10.3872/j.issn.1007-385X.2007.04.020
Weis, S. N., Toniazzo, A. P., Ander, B. P., Zhan, X., Careaga, M., Ashwood, P., et al. (2014). Autophagy in the brain of neonates following hypoxia-ischemia shows sex-and region-specific effects. Neuroscience 256, 201–209. doi: 10.1016/j.neuroscience.2013.10.046
Westphal, D., Kluck, R. M., and Dewson, G. (2014). Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 21, 196–205. doi: 10.1038/cdd.2013.139
White, E. (2016). Autophagy and p 53. Cold Spring Harb. Perspect. Med. 6:a026120. doi: 10.1101/cshperspect.a026120
Wu, Z., Zou, X., Zhu, W., Mao, Y., Chen, L., and Zhao, F. (2016). Minocycline is effective in intracerebral hemorrhage by inhibition of apoptosis and autophagy. J. Neurol. Sci. 371, 88–95. doi: 10.1016/j.jns.2016.10.025
Xie, Y., Zhou, Y., Wang, J., Du, L., Ren, Y., and Liu, F. (2023). Ferroptosis, autophagy, tumor and immunity. Heliyon 9:e19799. doi: 10.1016/j.heliyon.2023.e19799
Xing, S., Zhang, J., Dang, C., Liu, G., Zhang, Y., Li, J., et al. (2014). Cerebrolysin reduces amyloid-β deposits, apoptosis and autophagy in the thalamus and improves functional recovery after cortical infarction. J. Neurol. Sci. 337, 104–111. doi: 10.1016/j.jns.2013.11.028
Xing, S., Zhang, Y., Li, J., Zhang, J., Li, Y., Dang, C., et al. (2012). Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy 8, 63–76. doi: 10.4161/auto.8.1.18217
Xiong, J., Zhou, H., Lu, D., Wang, Z., Liu, H., Sun, Y., et al. (2020). Levetiracetam reduces early inflammatory response after experimental intracerebral hemorrhage by regulating the Janus kinase 2 (Jak 2)-signal transducer and activator of transcription 3 (Stat 3) signaling pathway. Med. Sci. Monit. 26:e922741. doi: 10.12659/MSM.922741
Xiu'e, W., and Liangqun, R. (2010). An experimental study on the expression levels of Bax and Bcl-2 in the brain of experimental rats with cerebral hemorrhage. Chin. J. Pract. Nerv. Dis. 13, 1–3. doi: 10.3969/j.issn.1673-5110.2010.24.001
Xu, D., Huang, P., Yu, Z., Xing, D. H., Ouyang, S., and Xing, G. (2014). Efficacy and safety of Panax notoginseng Saponin therapy for acute intracerebral hemorrhage, meta-analysis, and mini review of potential mechanisms of action. Front. Neurol. 5:274. doi: 10.3389/fneur.2014.00274
Xuefei, S., Jin, T., and Anding, X. (2011). The effect of magnesium sulfate on the expression levels of Bcl-2 and Bax in the hippocampus of rats with diffuse axonal injury. J. Int. Neurol. Neurosurg. 38, 426–431.
Yan, H., Huang, W., Rao, J., and Yuan, J. (2021). mi R-21 regulates ischemic neuronal injury via the p 53/Bcl-2/Bax signaling pathway. Aging 13, 22242–22255. doi: 10.18632/aging.203530
Yang, Z., Lin, P., Chen, B., Zhang, X., Xiao, W., Wu, S., et al. (2021). Autophagy alleviates hypoxia-induced blood-brain barrier injury via regulation of Cldn5 (claudin 5). Autophagy 17, 3048–3067. doi: 10.1080/15548627.2020.1851897
Yang, Z., Liu, B., Zhong, L., Shen, H., Lin, C., Lin, L., et al. (2015). Toll-like receptor-4-mediated autophagy contributes to microglial activation and inflammatory injury in mouse models of intracerebral haemorrhage. Neuropathol. Appl. Neurobiol. 41, e95–e106. doi: 10.1111/nan.12177
Yanling, Z., Kangning, C., Shuqin, S., and Ying, W. (2004). The correlation between apoptosis and expression level of Bcl-2 after cerebral hemorrhage in rats. J. Third Milit. Med. Univ. 18, 1635–1638. doi: 10.3321/j.issn:1000-5404.2004.18.008
Yao, J. Y., and Zou, W. (2022). Progress of researches on mechanisms of acupuncture therapy for cerebral hemorrhage. Zhenci Yanjiu. 47:7. doi: 10.13702/j.1000-0607.20210215
Yee, K. S., Wilkinson, S., James, J., Ryan, K. M., and Vousden, K. H. (2009). Puma-and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 16, 1135–1145. doi: 10.1038/cdd.2009.28
Yong, J., Guoqiang, C., Bin, H., Song, W., Kai, S., and Kai, Y. (2010). Effects of caspase inhibitors on cardiomyocyte apoptosis and expression levels of Bcl-2 and Bax in rats with ischemia-reperfusion injuries. Jinagsu Med. Jo. 36, 932–934.
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028
Yousefi, S., Perozzo, R., Schmid, I., Ziemiecki, A., Schaffner, T., Scapozza, L., et al. (2006). Calpain-mediated cleavage of Atg 5 switches autophagy to apoptosis. Nat. Cell Biol. 8, 1124–1132. doi: 10.1038/ncb1482
Yu, L., Alva, A., Su, H., Dutt, P., Freundt, E., Welsh, S., et al. (2004). Regulation of an Atg7-beclin 1 program of autophagic cell death by caspase-8. Science 304, 1500–1502. doi: 10.1126/science.1096645
Yu, F., Zhang, Q., Liu, H., Liu, J., Yang, S., Luo, X., et al. (2022). Dynamic O-Glcnacylation coordinates ferritinophagy and mitophagy to activate ferroptosis. Cell Discov. 8:40. doi: 10.1038/s41421-022-00390-6
Zalckvar, E., Berissi, H., Eisenstein, M., and Kimchi, A. (2009). Phosphorylation of Beclin 1 by dap-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-xl. Autophagy 5, 720–722. doi: 10.4161/auto.5.5.8625
Zeng, J., Zheng, S., Chen, Y., Qu, Y., Xie, J., Hong, E., et al. (2021). Puerarin attenuates intracerebral hemorrhage-induced early brain injury possibly by Pi3K/Akt signal activation-mediated suppression of Nf-κB pathway. J. Cell. Mol. Med. 25, 7809–7824. doi: 10.1111/jcmm.16679
Zhang, Y., Liu, Y., Cui, Q., Fu, Z., Yu, H., Liu, A., et al. (2022). Hydroxysafflor yellow a alleviates ischemic stroke in rats via Hif-1[formula: see text], Bnip3, and Notch1-mediated inhibition of autophagy. Am. J. Chin. Med. 50, 799–815. doi: 10.1142/S0192415X22500331
Zhang, T., Su, J., Guo, B., Zhu, T., Wang, K., and Li, X. (2014). Ursolic acid alleviates early brain injury after experimental subarachnoid hemorrhage by suppressing Tlr4-mediated inflammatory pathway. Int. Immunopharmacol. 23, 585–591. doi: 10.1016/j.intimp.2014.10.009
Zhang, Y., Yang, Y., Zhang, G.-Z., Gao, M., Ge, G.-Z., Wang, Q.-Q., et al. (2016). Stereotactic administration of edaravone ameliorates collagenase-induced intracerebral hemorrhage in rat. CNS Neurosci. Ther. 22, 824–835. doi: 10.1111/cns.12584
Zhao, L., Chen, S., Sherchan, P., Ding, Y., Zhao, W., Guo, Z., et al. (2018). Recombinant Ctrp 9 administration attenuates neuroinflammation via activating adiponectin receptor 1 after intracerebral hemorrhage in mice. J. Neuroinflammation 15:215. doi: 10.1186/s12974-018-1256-8
Zhao, J., Wang, Y., Wang, D., Yan, W., Zhang, S., Li, D., et al. (2022). MiR-124-3p attenuates brain microvascular endothelial cell injury in vitro by promoting autophagy. Histol. Histopathol. 37, 159–168. doi: 10.14670/HH-18-406
Keywords: autophagy, apoptosis, cerebral hemorrhage, Bcl-2, Bax
Citation: Wang M, Chen X, Li S, Wang L, Tang H, Pu Y, Zhang D, Fang B and Bai X (2024) A crosstalk between autophagy and apoptosis in intracerebral hemorrhage. Front. Cell. Neurosci. 18:1445919. doi: 10.3389/fncel.2024.1445919
Edited by:
Elizabeth S. Fernandes, Pelé Pequeno Príncipe Research Institute, BrazilReviewed by:
Li Zhang, Nanjing University, ChinaMohammad Hasanain, University of Miami Health System, United States
Copyright © 2024 Wang, Chen, Li, Wang, Tang, Pu, Zhang, Fang and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bangjiang Fang, ZmFuZ2JqaUAxNjMuY29t; Xue Bai, YmFpeHVlQHN3bXUuZWR1LmNvbQ==
†These authors have contributed equally to this work