 Simon Maksour1
Simon Maksour1
 Rocio K. Finol-Urdaneta2
*
Rocio K. Finol-Urdaneta2
*
 Amy J. Hulme2
Amy J. Hulme2
 Mauricio e Castro Cabral-da-Silva1
Mauricio e Castro Cabral-da-Silva1
 Helena Targa Dias Anastacio1
Rachelle Balez1
Helena Targa Dias Anastacio1
Rachelle Balez1
 Tracey Berg1
Tracey Berg1
 Calista Turner1
Calista Turner1
 Sonia Sanz Muñoz1
Martin Engel1
Predrag Kalajdzic3
Sonia Sanz Muñoz1
Martin Engel1
Predrag Kalajdzic3
 Leszek Lisowski3
,4
,5
Kuldip Sidhu6
Perminder S. Sachdev6
Leszek Lisowski3
,4
,5
Kuldip Sidhu6
Perminder S. Sachdev6
 Mirella Dottori2
*
†
Mirella Dottori2
*
†
 Lezanne Ooi1
*
†
Lezanne Ooi1
*
†
- 1School of Chemistry and Molecular Bioscience and Molecular Horizons, University of Wollongong, Wollongong, NSW, Australia
- 2School of Medical and Indigenous Health Science and Molecular Horizons, University of Wollongong, Wollongong, NSW, Australia
- 3Translational Vectorology Research Unit, Children’s Medical Research Institute, Faculty of Medicine and Health, The University of Sydney, Westmead, NSW, Australia
- 4Australian Genome Therapeutics Centre, Children’s Medical Research Institute and Sydney Children’s Hospitals Network, Westmead, NSW, Australia
- 5Laboratory of Molecular Oncology and Innovative Therapies, Military Institute of Medicine – National Research Institute, Warsaw, Poland
- 6Centre for Healthy Brain Ageing, School of Clinical Medicine, University of New South Wales, Sydney, NSW, Australia
Alzheimer’s disease (AD) is a devastating neurodegenerative condition that affects memory and cognition, characterized by neuronal loss and currently lacking a cure. Mutations in PSEN1 (Presenilin 1) are among the most common causes of early-onset familial AD (fAD). While changes in neuronal excitability are believed to be early indicators of AD progression, the link between PSEN1 mutations and neuronal excitability remains to be fully elucidated. This study examined iPSC-derived neurons (iNs) from fAD patients with PSEN1 mutations S290C or A246E, alongside CRISPR-corrected isogenic cell lines, to investigate early changes in excitability. Electrophysiological profiling revealed reduced excitability in both PSEN1 mutant iNs compared to their isogenic controls. Neurons bearing S290C and A246E mutations exhibited divergent passive membrane properties compared to isogenic controls, suggesting distinct effects of PSEN1 mutations on neuronal excitability. Additionally, both PSEN1 backgrounds exhibited higher current density of voltage-gated potassium (Kv) channels relative to their isogenic iNs, while displaying comparable voltage-gated sodium (Nav) channel current density. This suggests that the Nav/Kv imbalance contributes to impaired neuronal firing in fAD iNs. Deciphering these early cellular and molecular changes in AD is crucial for understanding disease pathogenesis.
1 Introduction
Alzheimer’s disease (AD) is a devastating, progressive neurodegenerative disease that affects memory and cognition and is characterized by the loss of neurons. Thus, an important area of research involves examining how early molecular changes may influence the cause and progression of AD. These studies focus on early changes in gene expression (Guennewig et al., 2021), energy metabolism (Johnson et al., 2020), altered neurogenesis and neuronal differentiation (Arber et al., 2021; Meyer et al., 2019), and neuronal firing (Ghatak et al., 2019; Spoleti et al., 2022). Multiple lines of evidence suggest that changes in neuronal excitability are an early phenotype of neurodegeneration that may drive disease pathology in neurodegenerative diseases, including AD (as reviewed in Targa Dias Anastacio et al., 2022) and amyotrophic lateral sclerosis (as reviewed in Do-Ha et al., 2018). Neuronal excitability changes are influenced by the two major hallmarks of AD, Aβ (Busche et al., 2012; Busche et al., 2008) and tau pathology (Busche et al., 2019; Crimins et al., 2012), whilst conversely, excitability changes have also been shown to drive both the deposition of both of these AD hallmark pathologies (Cirrito et al., 2005; Pooler et al., 2013; Wu et al., 2016; Yamamoto et al., 2015). Whilst excitatory neurons demonstrate excitability changes, there are also demonstrated contributions from inhibitory neurons (Ghatak et al., 2019; Nuriel et al., 2017; Verret et al., 2012) and glial cells (Targa Dias Anastacio et al., 2022). Furthermore, correcting neuronal activity through pharmacological or genetic intervention in AD mouse models improves memory and cognition (Martinez-Losa et al., 2018; Ping et al., 2015; Roberson et al., 2007), highlighting the role of neuronal excitability regulation in disease progression.
PSEN1 is the most common causative gene for early-onset, familial AD (fAD) and is believed to contribute to neuronal vulnerability through the overproduction of amyloid-ß (Aβ) peptides, which results in the generation of Aß plaques in the brain (Ooi et al., 2020). There are over 300 known mutations in PSEN1, many with pathogenic outcomes, however the effects of each mutation on the disease phenotype remains to be fully elucidated (PSEN1 mutations database, ALZforum).1 The implications of PSEN1 mutations on neuronal vulnerability have been assessed in animal models and human cell models of disease, including patient induced pluripotent stem cell (iPSC) derived neurons. In addition to disrupted amyloid precursor protein (APP) processing and plaque formation, PSEN1 mutations induce early changes in neurons, including, increased susceptibility to Aβ (Armijo et al., 2017) and ferroptosis (Greenough et al., 2022), dysregulated neurogenesis and differentiation (Arber et al., 2021; Hurley et al., 2023; Vanova et al., 2023), decreased neurite outgrowth (Balez et al., 2016; Dowjat et al., 1999; Furukawa et al., 1998; Ghatak et al., 2019), endosomal dysfunction (Kwart et al., 2019) and alterations in neuronal excitability (Chen et al., 2021; Hurley et al., 2023; Vitale et al., 2021). These studies highlight the complex role of presenilin-1 in multiple cellular functions, and the need to understand how specific mutations affect neuronal processes.
Human iPSCs offer an avenue to generate neurons from patients bearing disease-relevant gene mutations, and interrogate the intrinsic differences in excitability properties of neurons in the absence of late-stage AD pathology and supporting cell types. Therefore, this study aimed to use iPSC-derived neurons from two AD patients bearing pathogenic mutations in PSEN1, S290C or A246E, along with their CRISPR-corrected isogenic controls to investigate early neuronal excitability changes in disease. The S290C mutation, which results in the deletion of exon 9, was originally identified in a Finnish family and displayed typical Aß plaques, neurofibrillary tangles (NFTs) and gliosis, in addition to “cotton wool” plaques and hippocampal atrophy (Crook et al., 1998; Verkkoniemi et al., 2000). The A246E mutation was originally identified in a Canadian family, with 52 affected family members spanning eight generations, with post mortem tissue highlighting atrophy of the frontal lobe and hippocampus, neuronal loss, Aß plaques and NFTs (Nee et al., 1983). At a neuronal level, the A246E mutation alters tissue specification in organoids (Vanova et al., 2023), increased susceptibility to Aß (Armijo et al., 2017), increased vulnerability to ferroptosis (Greenough et al., 2022) and altered Ca2+, glutamate and NMDA signaling (Balez et al., 2024; Targa Dias Anastacio et al., 2024). The S290C mutation also altered AMPA signaling (Targa Dias Anastacio et al., 2024). Furthermore, the deletion of exon 9 in neurons increased vulnerability to ferroptosis (Greenough et al., 2022) and impaired endocytosis (Woodruff et al., 2016). It is currently unknown whether these mutations cause differences in intrinsic firing properties in excitatory neurons. Understanding the early common and divergent cellular and molecular changes disrupted in neurons from AD patients bearing different PSEN1 mutations will provide insight into the functional impact of disease-causing mutations and shed light on the mechanisms underlying the specific vulnerability of excitatory neurons.
2 Methods
2.1 Cell culture
2.1.1 iPSC cell lines and maintenance
Use of iPSC lines for this project was approved by the UOW Human Ethics Committee (#2017–375, 2017–382, 2020–450, 2020–451, 13–299). This study used iPSCs generated from early-onset AD patients with a PSEN1 S290C (S290C) (Supplementary Figure S1) or a A246E mutation (A246E) (Muñoz et al., 2018) and their respective CRISPR-corrected isogenic controls, S290IC (Supplementary Figure S1) and A246IC (Targa Dias Anastacio et al., 2024, in revision). The iPSCs were cultured as previously reported in Abu-Bonsrah et al. (2019), Denham and Dottori (2011), and Mattei et al. (2019). Briefly, the iPSCs were maintained in mTesR1 (StemCell Technologies, #85850) on matrigel-coated tissue culture ware, kept in normoxic conditions at 37°C with 5% CO2. Cells were passaged once every 5–7 days using 0.5 mM EDTA (Life Technologies, #AM9260G) in PBS−/− (Life Technologies, #14190250). Methods on the cell line generation and characterization can be found in the Supplementary materials.
2.2 Lentiviral production
Viral particles containing an open reading frame of Neurogenin-2 (NGN2) were produced to differentiate iPSCs into mature neurons as described in Hulme et al. (2020) and Maksour et al. (2024). Briefly, HEK293T cells were transfected with the DNA of lentiviral packaging plasmids vSVG (Addgene, USA, #8454), RSV (Addgene, #12253), pMDL (Addgene, #12251), and either the tetracycline transactivator (TTA) vector, M2rtTA (Addgene, #20342), or the NGN2 overexpression vector, TetO-NGN2-eGFP-Puro plasmid (Addgene, #79823) using Polyethyleneimine (Sigma-Aldrich, USA, #408727). DNA was added in a ratio of 4:2:1:1, transfer vector:pMDL:RSV:vSVG. The cell culture media containing viral particles was collected every 24 h over 3 days. The viral supernatant was concentrated 200× by ultracentrifugation at 66,000 × g for 2 h at 4°C. The viral pellet was resuspended in PBS and stored at −80°C until needed.
2.3 Generation of NGN2-induced neurons (iNs)
This study used a protocol published in Maksour et al. (2024) to generate mature neurons via NGN2 overexpression. Briefly, iPSCs were resuspended as single cells using Accutase for 2–3 min at RT. Single cells were plated at 15,000 cells/cm2 onto 10 μg/mL poly-D-lysine (PDL) and laminin (LAM) coated culture plate in mTeSR1 media supplemented with 10 μM Y27632. Cells were allowed to attach for 6–8 h, after which 0.5 μL of viral particles of both NGN2 overexpression and the TTA per 15,000 cells. Virus was removed 16–20 h following transduction with fresh neural media [Neurobasal medium (NBM; Life Technologies, #21103–049) supplemented with 1× N-2 supplement 1× B-27 supplement, 1× Insulin-Transferrin-Selenium-A and 2 mM L-glutamine] supplemented with 1 μg/mL doxycycline (DOX; Sigma-Aldrich, # D9891), 10 μM SB431542 and 0.1 μM LDN193189 to promote a cortical fate. After 24 h of DOX induction, 0.5 μg/mL puromycin was added daily for 3 days for selection of successfully transduced cells, in addition to DOX, SB431542 and LDN193189. Following selection fresh media supplemented with 10 μg/mL BDNF was added. Following selection, BrainPhys media [Brainphys medium (StemCell Technologies, #05790) supplemented with NeuroCult SM1 (without vitamin A; StemCell Technologies, #05731) and N2 supplement-A (StemCell Technologies, #07152)] was subsequently added in at increasing concentrations (25–100% in NM) for each media change to improve maturation. Neurons were assessed by whole-cell patch clamp between 21 and 35 days post viral transduction.
2.4 Electrophysiology
Sterile plastic coverslips, cut into 10 mm slides, were coated with PDL and LAM before plating NGN2 iNs for functional characterization. Whole-cell patch clamp recordings were performed on matured neurons aged 3–5 weeks, following the protocol outlined in Hulme et al. (2020). Recordings were conducted at room temperature (20–22°C) using a MultiClamp 700B Amplifier, digitized with a Digidata 1,440, and controlled via pClamp11 software (Molecular Devices). Whole-cell membrane currents were measured at 100 kHz, with series resistance compensated at 60–80%. Fire-polished borosilicate patch pipettes with a resistance of 2–4 MΩ were employed with an intracellular buffer composed of (in mM) 140 K-gluconate, 10 NaCl, 2 MgCl2, 10 HEPES, 5 EGTA (pH 7.2, osmolality 295 ± 5 mOsm/kg). The bath solution for current clamp experiments contained (in mM) 135 NaCl, 2 CaCl2, 2 MgCl2, 5 KCl, 10 glucose, 10 HEPES (pH 7.4, osmolality 315 ± 5 mOsm/kg).
Under current-clamp conditions, the resting membrane potential (RMP) was measured as the average membrane voltage without any current injection. Total membrane capacitance was estimated by automatically integrating the transient capacitive current produced during voltage-clamp steps, using pClamp11 (Molecular Devices). To analyze the intrinsic properties and excitability of iNs, 1-s current steps ranging from −150 to 140 pA in 10-pA increments were injected, starting from the RMP. Input resistance (Rin) was calculated from the steady-state voltage responses to 1-s hyperpolarizing currents, using the slope of the voltage–current relationship. Hyperpolarizing current induced membrane potential change (ΔMP) was defined as the difference between the minimum membrane potential recorded during the −100 pA current injection and the RMP. Rheobase was determined as the minimal current required to trigger an action potential (AP). AP characteristics were analyzed using Clampfit 11 (Molecular Devices). The AP threshold was defined as the voltage at which dV/dt exceeded 10 mV/ms, and AP amplitude was measured as the difference between the threshold and peak voltage. AP half-width was calculated as the duration at 50% of the AP amplitude. The threshold potential relative to the baseline was estimated by identifying the intersection of the AP’s rising phase and the slope leading up to its initiation, using a three-point tangent slope vector to find where the slope reached or exceeded 10 mV/ms. Under voltage-clamp conditions, total voltage-dependent sodium (INav) and potassium (IKv) currents were quantified at their peak amplitudes. INav was measured at the minimum current inflection at −10 mV, and IKv at the maximum current observed at 20 mV. These current values were then normalized by dividing by the capacitance of the respective iNs and reported as conductances in pA/pF.
2.5 Statistical analysis
Experiments were performed with 3–4 independent experimental differentiations (n = 3–4, biological replicates). Statistical analyses were conducted using GraphPad Prism software, version 10 (GraphPad Software, La Jolla, USA). Data was determined to be normally distributed using a Shapiro–Wilk normality test, where data were normally distributed, data were analyzed by a multiple t-test with Holm-Sidak method, unless stated otherwise. An α of 0.05 (p-value <0.05) was considered statistically significant.
3 Results
3.1 The iPSC derived neurons from two different fAD PSEN1 backgrounds exhibit impaired excitability
We have previously demonstrated the forced overexpression of NGN2 successfully and robustly generates functional, glutamatergic excitatory induced neurons (iNs) (Maksour et al., 2024). This study generated iNs from iPSCs of fAD patients bearing the PSEN1S290C (S290C) and PSEN1A246E (A246E) and their respective CRISPR corrected counterparts, S290IC and A246IC, to assess their electrophysiological properties (Figure 1A). Previous studies from our team have shown that iNs derived from patients with S290C and A246E mutations do not exhibit tau or Aβ pathology within 35 days in culture (Targa Dias Anastacio et al., 2024). This model allows us to investigate neuronal excitability alterations in a controlled environment without the confounding effects of disease pathology or the presence of glial cells. Therefore, it provides a unique opportunity to study the intrinsic effects of these mutations on neuronal ion channel function. Neurite analysis of fAD and isogenic corrected control iNs was performed using the Incucyte live-cell imager to assess neurite properties (Supplementary Figure S2). The neurite length and branch points of S290C iNs were significantly decreased compared to the respective controls, further supporting the observation of decreased capacitance and consistent with our previous findings in AD neurons (Balez et al., 2016). Neuronal excitability was evaluated by manual patch-clamp in the whole-cell configuration with representative recordings displayed in Figure 1B with data for all parameters summarized in Supplementary Table S2.
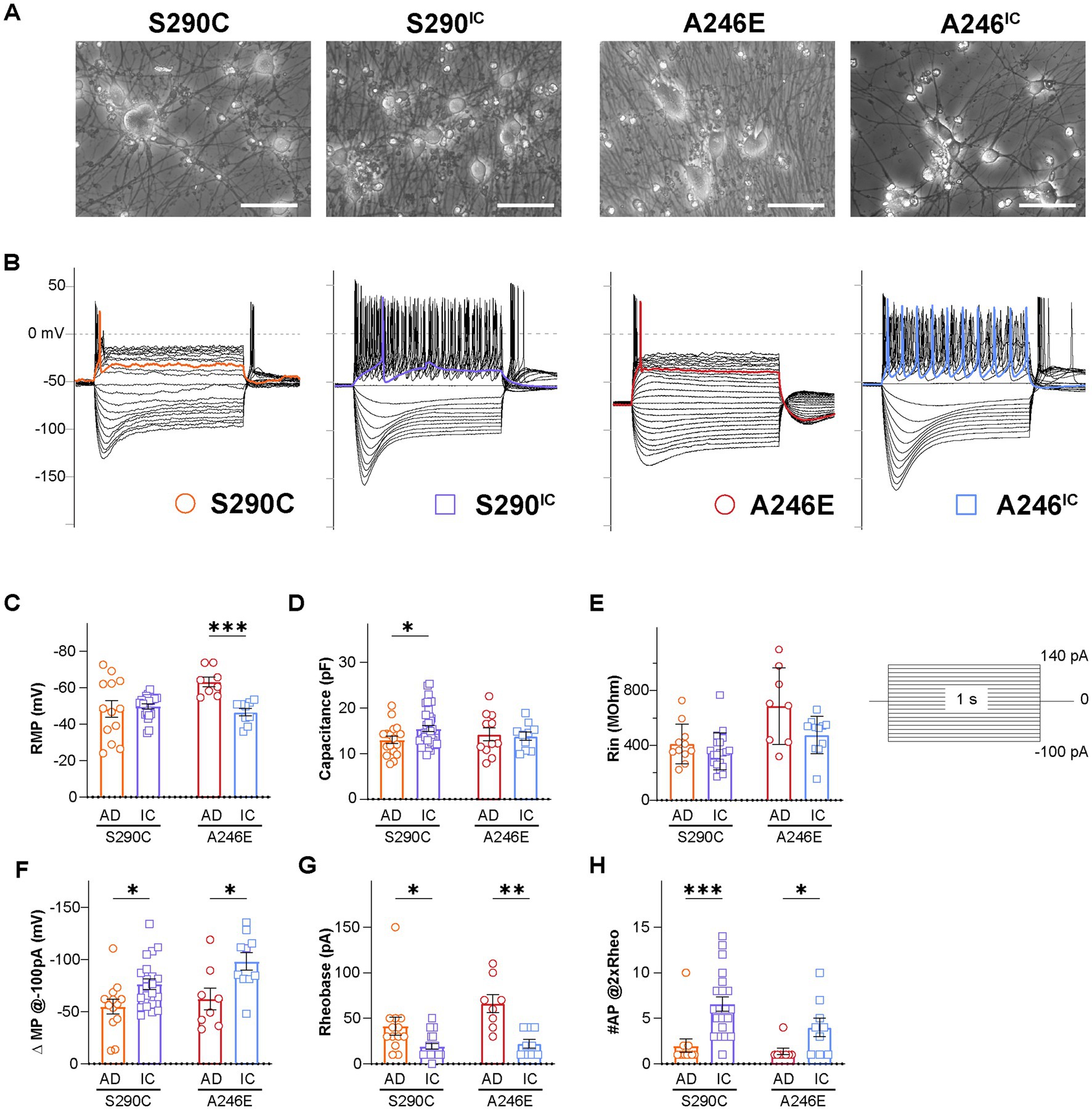
Figure 1. Neurons derived from AD patients bearing a PSEN1 mutation exhibit reduced excitability. Whole cell patch clamping was performed on day 21–35 NGN2 iNs derived from fAD patients with a PSEN1S290C (S290C) and PSEN1A246E (A246E) and their respective CRISPR-corrected isogenic controls, S290IC and A246IC (A). (B) Representative current clamp recordings of membrane potential responses of all cell lines. The stimulation protocols consisted of one-second-long current injections from −100 pA to 140 pA in 10 pA steps. The colored traces indicate firing activity at rheobase. Neurons were tested for (C) resting membrane potential (RMP), (D) capacitance, (E) input resistance (Rin), (F) hyperpolarizing current induced membrane potential change (ΔMP), (G) rheobase, and (H) the number of action potentials fired at 2 times rheobase. Data is presented as the mean ± SEM. Each data point represents an individual cell (n = 7–41), from 3 independent differentiations. Data was analyzed using multiple t-tests with Holm-Sidak for multiple comparisons where *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. AD, Alzheimer’s disease; IC, isogenic control.
The resting membrane potential (RMP) was evaluated within 2 min of switching to the current-clamp mode. RMP showed no difference between S290C and S290IC iNs, while A246E iNs had significantly lower RMP compared to their isogenic controls (−63.3 ± 2.7 mV, n = 8 vs. −46.6 ± 2.9 mV, n = 9, respectively; p < 0.001) (Figure 1C). S290C iNs had significantly lower capacitance than S290IC (13.0 ± 0.8 mV, n = 17 vs. 15.5 ± 0.6 mV, n = 41, respectively, p < 0.05) reflecting the generation of smaller iNs from this fAD line (Figure 1D), while A246E and A246IC capacitance values were comparable. The iNs were stimulated through to 1 s long step current injections ranging from −150 pA to 140 pA. A comparison of the slopes of the steady-state voltage responses hyperpolarizing currents (−150 pA to −20 pA) revealed no differences in the input resistance (Rin) between the two fAD PSEN1 mutants and their corrected controls (S290C vs. S290IC p = 0.2397; A246E vs. A246IC p = 0.1087) (Figure 1E).
Negative current injections elicited hyperpolarizing membrane responses (sags) consistent with the activation of hyperpolarization-gated cyclic nucleotide-activated ion channels (HCN). Quantification of the change in membrane potential (ΔMP) evidenced upon −100 pA current injections revealed smaller hyperpolarizing sags in fAD iNs compared to their isogenic controls (S290C vs. S290IC p = 0.016257; A246E vs. A246IC p = 0.015170) (Figure 1F).
The rheobase (the minimum current injection required to induce one action potential, AP) was higher in both fAD iNs (S290C: 41.5 ± 9.8 pA, n = 13; A246E: 66.3 ± 9.8 pA, n = 8) compared to their corrected controls S290IC (19.5 ± 3.0 pA, n = 21; p < 0.05) and A246IC (22.2 ± 4.7 pA, n = 10; p < 0.01), respectively (Figure 1G). Notably, positive current injections (up to 140 pA) elicited AP firing in all iNs, but multiple action potentials were only observed in corrected iNs (Figure 1B). Accordingly, the number of APs fired upon current injections equivalent to two times the rheobase (#AP@2xRheo) was significantly lower in both fAD lines compared to corrected iNs (Figure 1H). Thus, iNs derived from PSEN1 S290C iPSCs fired 2.0 ± 0.7 action potentials (n = 12) compared to 6.6 ± 0.8 (n = 21) APs by S290IC (p < 0.001), and PSEN1 A246E fired 1.4 ± 0.4 APs (n = 8) compared to 4.0 ± 1.0 APs (n = 9) by A246IC iNs (p < 0.05). Thus, neurons derived from fAD patients with S290C or A246E mutations in PSEN1 exhibited reduced firing capacity and excitability compared to their isogenic controls, indicating a common phenotype of reduced neuronal excitability.
3.2 iNs from different fAD PSEN1 backgrounds display distinct AP characteristics
Given the observed inter-group variability in AP waveforms, the first action potentials fired by iNs from each group were aligned at the point of initiation and averaged for illustration purposes (Figure 2A). Comparative analysis of AP shapes (at rheobase) between fAD and corrected iNs unveiled disparities in AP peak amplitude, area, half-width, rise time, rise slope, and maximal decay slope, as outlined in Supplementary Table S1. While AP peak amplitude showed no variance between S290C and S290IC neurons (Figure 2B), their AP area differed significantly, with S290C iNs exhibiting over a 5-fold smaller area than their isogenic control (2229.0 ± 423.5 mV*ms, n = 13; vs. 12036.2 ± 1390.3 mV*ms, n = 21; p < 0.001) (Figure 2C). Additionally, the AP rise slope was markedly higher in S290IC-derived neurons (59.3 ± 9.5 mV/ms, n = 21), compared to those harboring the S290C mutation (25.6 ± 4.1 mV/ms, n = 13, p < 0.05) (Figure 2E). Consequently, the AP rise time was more than 2-fold slower in S290C neurons compared to S290CIC (1.3 ± 0.2 ms, n = 13 vs. 0.6 ± 0.2 ms, n = 20, p < 0.05) (Figure 2F) with no discernible differences in AP maximal decay slope (p = 0.15) (Figure 2G).
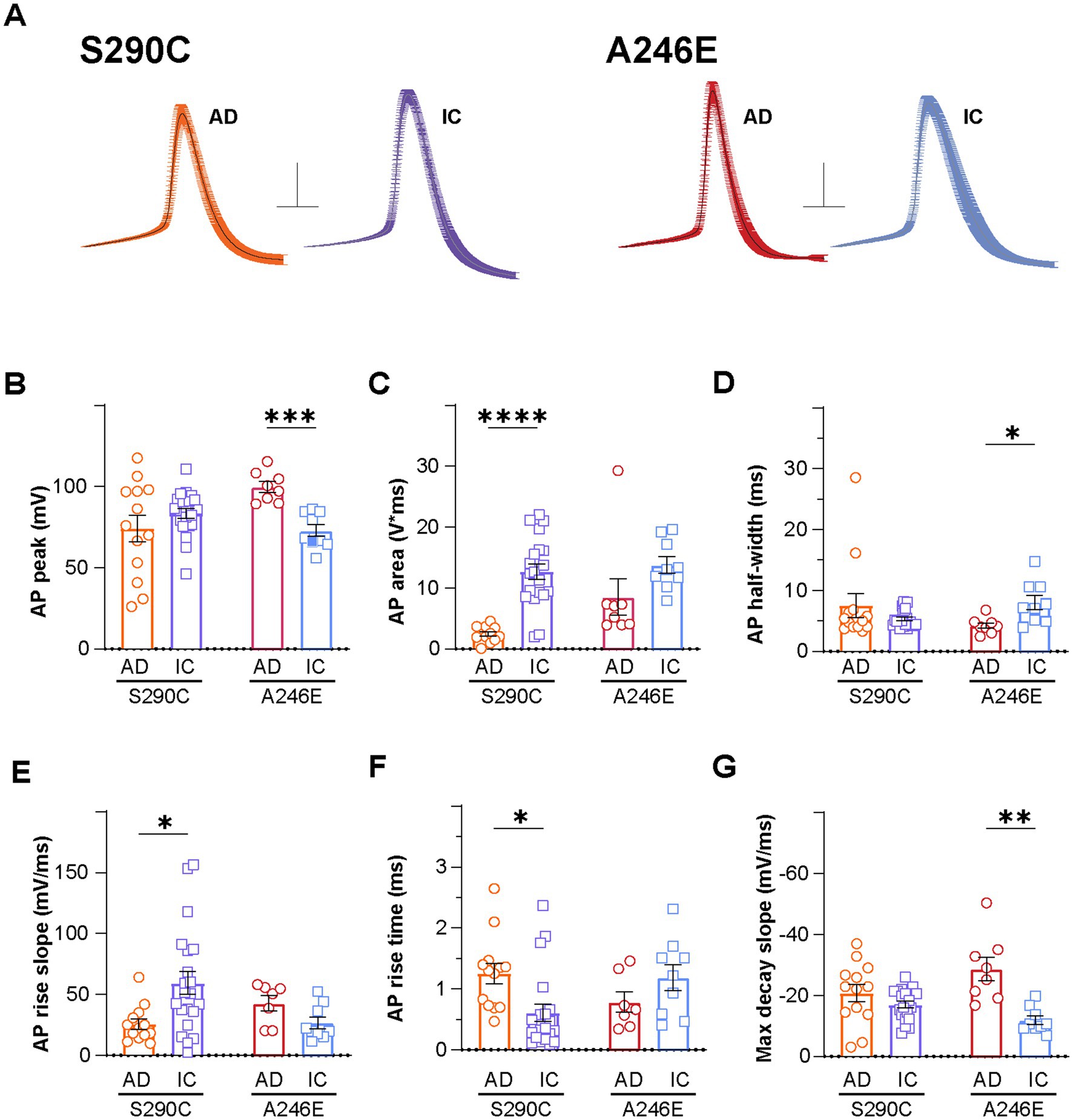
Figure 2. AD induced neurons display altered action potential properties compared to isogenic controls. Action potential properties were also compared between AD patient lines with PSEN1 mutations (S290C and A246E) and isogenic controls, including (A) action potential waveforms, where the first action potential from each neuron fired at rheobase was aligned at the point of initiation and the resulting mean ± SEM waveforms are presented (Scale: 20 mV, 5 ms), (B) peak amplitude, (C) area, (D) half-width, (E) rise slope, (F) rise time and (G) max decay slope. Data is presented as the mean ± SEM. Each data point represents an individual cell (n = 7–41), from 3 independent differentiations. Data was analyzed using multiple t-tests with Holm-Sidak for multiple comparisons where *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. AD, Alzheimer’s disease; IC, isogenic control.
In contrast, PSEN1 A246E fAD iNs exhibited higher AP peak amplitudes (99.7 ± 3.3 mV, n = 8) compared to corrected A246IC iNs (72.9 ± 3.7 mV, n = 9; p < 0.001) (Figure 2B), while showing no significant difference in AP area (p = 0.44) (Figure 2C). Despite similar AP rise parameters (Figures 1C,E,F), A246E neurons displayed narrower APs than their isogenic control (AP half-width 4.2 ± 0.5 ms, n = 8 vs. 8.0 ± 1.2 ms, n = 9, respectively; p < 0.05) (Figure 2D), accompanied by markedly larger maximal decay slopes (−28.7 ± 3.8 mV/ms, n = 8 vs. −11.9 ± 1.4 mV/ms, n = 9, p < 0.01) (Figure 2G). Thus, the action potential shape in fAD iNs appears distinctively affected by specific PSEN1 mutations.
3.3 Voltage-gated potassium currents are larger in iNs from both PSEN1 fAD patients
The shape of the action potential is intricately governed by factors, such as ion channel dynamics and the equilibrium between inward and outward currents, primarily mediated by voltage-gated sodium (Nav) and potassium (Kv) channels, respectively. Representative iN whole-cell currents elicited by 250 ms step depolarization (from −80 mV to 50 mV; Vh −80 mV, inset) recorded in voltage-clamp mode are displayed for S290C and S290IC (Figure 3A) and A246E and A246IC neurons (Figure 3B). To assess the ability of iNs to initiate and terminate action potential firing, Nav and Kv currents were analyzed. Nav channels, which drive rapid depolarization, open quickly near the threshold for action potential initiation (~ −40 to −20 mV). Sodium currents were measured at −10 mV to ensure the activation of a large fraction of available Nav channels. Kv channels, responsible for restoring the resting membrane potential and regulating action potential duration and firing frequency, activate more slowly at more depolarized potentials (~ −10 to +20 mV). Kv currents were therefore quantified at 20 mV to capture maximal outward potassium flow. Total Nav and Kv currents (at −10 mV and 20 mV, respectively) were normalized to the cell capacitance and expressed as current density (pA/pF). The disease lines and isogenic corrected controls demonstrated similar AP thresholds (Supplementary Table S2), indicating no significant differences in the voltage required to trigger an action potential. In alignment with these findings, no detectable differences in Nav channel-mediated currents were observed between these groups (Figure 3C). In contrast, Kv channel mediated currents were significantly larger in neurons derived from patients with fAD PSEN1 mutations (S290C: 161.5 ± 28.1 pA/pF, n = 17; A246E: 89.2 ± 7.1 pA/pF, n = 10), compared to their respective CRISPR-corrected isogenic controls (S290IC: 97.0 ± 8.1 pA/pF, n = 41; p < 0.01; A246IC: 50.7 ± 5.4, n = 10; p < 0.001) (Figure 3D). Consequently, the ratio between Nav and Kv current densities (INav/IKv) was significantly larger for both isogenic controls, compared to their respective fAD lines (p < 0.05) (Figure 3E). Similarly, the ratio of maximal Nav and Kv conductances (from conductance-voltage relationships) was higher in the corrected lines (Supplementary Table S2), suggesting a potential imbalance between inward and outward currents as a possible cause of reduced excitability in PSEN1 fAD iNs.
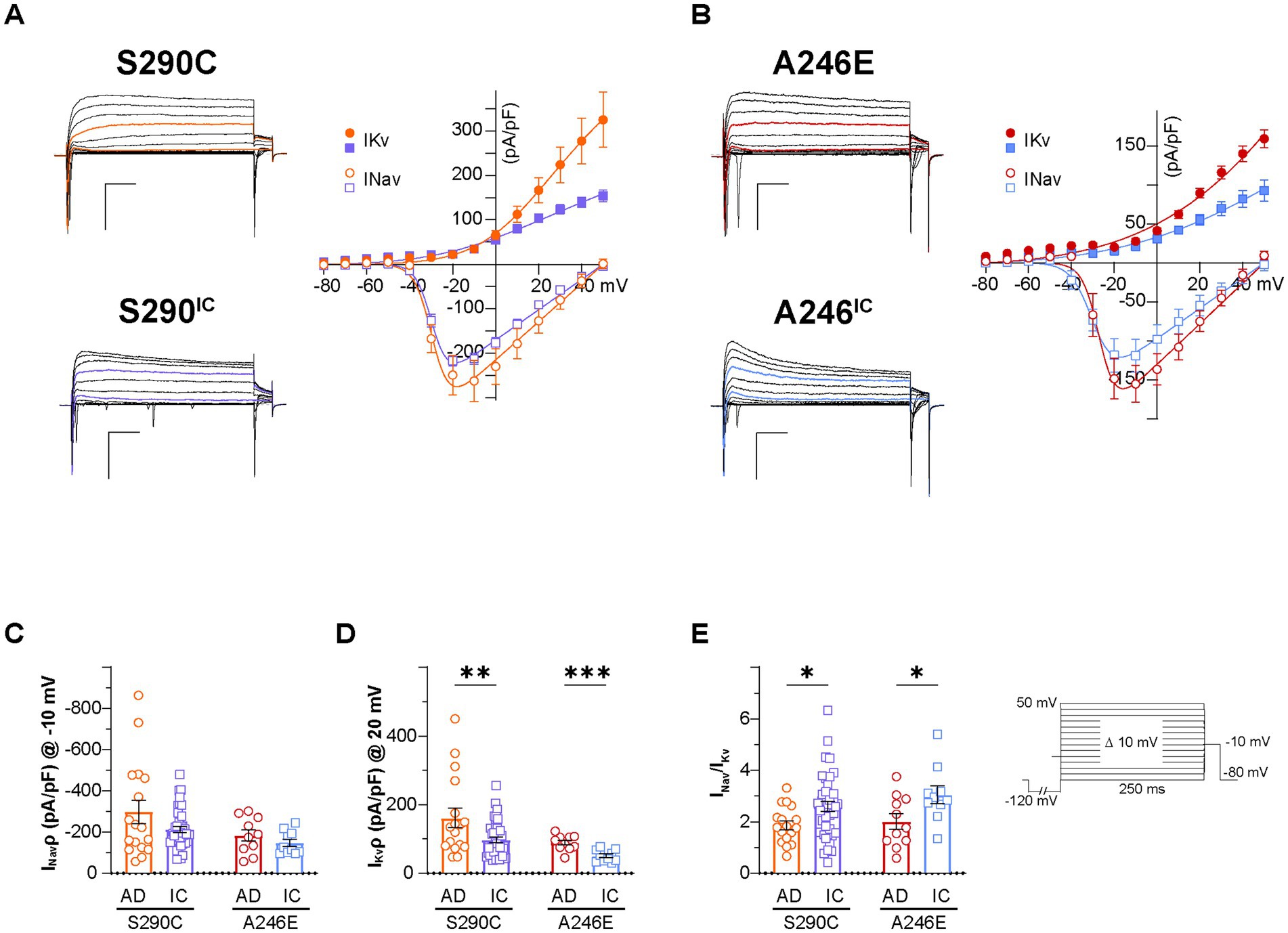
Figure 3. Voltage-dependent conductances in neurons derived from AD patients with a PSEN1 mutation and their isogenic controls. Whole cell patch-clamp recordings were performed on day 21–35 NGN2 iNs derived from S290C (PSEN1 S290C) and A246E (PSEN1 A246E) and their respective CRISPR corrected controls, S290IC and A246IC cell lines. (A) Left: representative traces of voltage-dependent currents elicited by 250 ms square pulses from −80 mV to 50 mV in 10 mV steps (Vh −80 mV, 0.1 Hz) in S290C (orange) and S290IC (purple) neurons. A 250 ms pre-pulse to −120 mV was used to minimize cumulative inactivation (inset). Right: average I–V relationships of peak inward (INav) and outward current (IKv). Data is presented ± SEM (n = 35). (B) Left: representative traces of voltage-dependent currents elicited by 250 ms square pulses from −80 mV to 50 mV in 10 mV steps (Vh −80 mV, 0.1 Hz) in A246E (red) and A246IC (blue) neurons. A 250 ms pre-pulse to −120 mV was used to minimize cumulative inactivation (inset). Right: average I–V relationships of peak inward (INav) and outward current (IKv). Data is presented ± SEM (n = 35). (C) Nav and (D) Kv current density quantification. (E) Nav to Kv current ratio. Data is presented as the mean ± SEM. Each data point represents an individual cell (n = 10–41), from 3 independent differentiations. Data was analyzed using multiple t-tests with Holm-Sidak for multiple comparisons where *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. AD, Alzheimer’s disease; IC, isogenic control.
4 Discussion
This study investigated early excitability changes in fAD using iPSC-derived neurons from patients bearing PSEN1 mutations. These neurons showed reduced firing responses suggestive of altered ionic homeostasis. Action potential properties in neurons bearing S290C or A246E mutations diverged, indicating varied disrupted pathways, however the overall impact on firing was consistent amongst the mutations. Understanding these early cellular and molecular changes may shed light on cause and progression in fAD.
There have been over 300 mutations in the PSEN1 gene identified with varying pathogenicity and effects on neuropathology and molecular pathways. This study has provided evidence of common intrinsic excitability changes that are altered in neurons with a PSEN1 S290C or A246E mutations. The S290C mutation occurs due to a missense mutation in the splice acceptor site on the boundary of intron 8 and exon 9, resulting in the skipping of exon 9, which leads to impaired APP cleavage and Aβ processing (Rovelet-Lecrux et al., 2015; Steiner et al., 1999). The deletion of exon 9 impairs cellular functions including calcium influx in hippocampal neurons (Skobeleva et al., 2022), altered mitochondrial metabolism, calcium homeostasis and inflammation in astrocytes (Oksanen et al., 2017), impaired endocytosis in neurons (Woodruff et al., 2016) and lipid metabolism (Landman et al., 2006). In this research, S290C iNs exhibited reduced capacitance, supported by reduced neurite length and branch points, and a decreased Nav/Kv current density ratio, likely underscoring the smaller AP area/rise parameters and overall impaired neuronal excitability. The A246E mutation is also pathogenic and occurs in exon 7 of PSEN1 (Sherrington et al., 1995), impeding proper APP cleavage and resulting in toxic amyloid peptide generation (Mahairaki et al., 2014; Yang et al., 2017). In a cellular context the A246E mutation results in impaired microglial differentiation (Aubert et al., 2023), premature neuronal differentiation (Yang et al., 2017), altered astrocyte metabolic function and inflammatory activation (Elsworthy et al., 2023) and increased neuronal susceptibility to Aβ (Armijo et al., 2017). In this study, iNs derived from patients with PSEN1 mutations showed decreased hyperpolarizing responses which together with an Nav/Kv imbalance would require stronger current injection (rheobase) for AP firing.
The observed reduced neuronal excitability in iNs in this study is consistent with previous findings on PSEN1 fAD mutations. For instance, cortical organoids with an L345F mutation displayed reduced extracellular network activity measured by multi-electrode array analysis compared to its isogenic control, likely due to altered notch signaling (Hurley et al., 2023). The A246E cell line used in this study, was demonstrated to have a deficiency in Notch1 in iNs, resulting in susceptibility to ferroptosis (Greenough et al., 2022). Future work will evaluate the differentiation and maturation capacity of A246E neurons. In APPSWE/PSEN1(dE9) transgenic mice, hippocampal neurons exhibited increased rheobase, decreased action potential frequency at lower current injections (30 and 50 pA), and increased frequency at 100 pA (Chen et al., 2021). Time-dependent changes in spike frequency of CA1 pyramidal neurons were observed in the APPSWE/PSEN1(dE9) transgenic mouse model, with reduced spike counts compared to wild-type at 1 month of age, no differences at 4 months and increased spikes at 10 months, suggesting excitability is altered with disease progression (Vitale et al., 2021). In an amyotrophic lateral sclerosis mouse model, it is the neurons that lose the ability to fire repetitively that become vulnerable and are lost as the disease progresses (Buskila et al., 2019). We hypothesize that PSEN1 mutations make neurons more susceptible to excitability changes, even in the absence of overt Aβ and tau pathology, which may contribute to neuronal loss in disease.
Future studies should investigate the impact of the S290C mutation on Notch signaling and neuronal differentiation, as many mutations have been linked to impaired cleavage of Notch1, resulting in impaired neurogenesis or premature differentiation, which may contribute to the altered excitability changes. Zhang et al. (2020) showed the PSEN1 S169 deletion mutation, induced AD pathology and cognitive deficits in a Notch signaling independent pathway, suggesting different PSEN1 mutations may contribute to neuronal deficits via distinct mechanisms. This phenomenon, in addition to different ion channels being dysregulated, may also explain specific impacts on action potential properties between lines. Common to both mutations was enhanced Kv conductance, likely implicated in hypoexcitability. Kv1 channels are known to regulate the repression of intrinsic excitability and synaptic transmission (Colasante et al., 2020; Thouta et al., 2021) and are upregulated in AD proteomic datasets (Askenazi et al., 2023). Nevertheless, the vast collection of potassium channels expressed in neurons and glia warrant future work identifying Kv channel expression changes in fAD PSEN1 mutant cell lines, as well as investigating the effects of introducing the same PSEN1 mutations into otherwise healthy cell lines to determine direct causal links between presenilin-1 dysfunction and Kv channel dysregulation.
In a reductionist model, this study revealed a common hypoexcitability phenotype in iNs generated from fAD patients with PSEN1 mutations in the absence of Aβ and tau disease pathology (Targa Dias Anastacio et al., 2024) and supporting cell types. Future research may employ genome editing to create cell lines with PSEN1 mutations of varying pathogenicity to establish a potential link between neuronal excitability and early onset-AD progression. In animal models, Aß appears to induce neuronal hyperexcitability, while soluble mutant tau suppresses neural activity in the rTg4510 and P301S tau transgenic mouse models (Busche et al., 2019; Marinković et al., 2019; Menkes-Caspi et al., 2015). Ghatak et al. (2019) reported hyperexcitability in co-cultures of astrocytes and iPSC-derived neurons from fAD patients. This yields an interesting observation, since astrocytes and other glial cells regulate excitability in AD (Targa Dias Anastacio et al., 2022), whereas in isolation, the excitatory neurons demonstrate higher current density of voltage-gated potassium channels. It is hypothesized that early stages of AD results in Aß-dependent hyperexcitability, preceding the onset of tau-dependent hypoactive neural circuits (Harris et al., 2020). Differences in neuronal maturity, Aß and tau generation between cell lines and co-culture may influence neural circuits in vitro. To determine the interplay between PSEN1 mutations, AD pathology and neuronal excitability, simplistic 2D models may not be sufficient and would require more advanced systems such as 3D cerebral organoids matured long-term. It is also important to consider how support cells, such as oligodendrocytes, astrocytes and microglia, influence the disruption to neuronal activity and transition between hypo- and hyperexcitability.
In summary, this study demonstrates that neurons derived from AD patients with PSEN1 mutations exhibit reduced firing activity and altered electrophysiological properties. Mechanistic understanding of the early changes disrupted in AD will provide insight into the driving forces of neurodegeneration and provide novel avenues for intervention to slow this devastating disease.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by the UOW Human Ethics Committee (#2017-375, 2017-382, 2020-450, 2020-451, 13-299). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
SM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing. RF-U: Conceptualization, Investigation, Methodology, Resources, Validation, Visualization, Writing – review & editing. AH: Data curation, Investigation, Methodology, Resources, Writing – review & editing. MC-d-S: Investigation, Writing – review & editing. HT: Investigation, Writing – review & editing. RB: Investigation, Validation, Visualization, Writing – review & editing. TB: Investigation, Writing – review & editing. CT: Investigation, Writing – review & editing. SS: Investigation, Writing – review & editing. ME: Investigation, Writing – review & editing. PK: Investigation, Writing – review & editing. LL: Investigation, Writing – review & editing. KS: Investigation, Writing – review & editing. PS: Resources, Writing – review & editing. MD: Conceptualization, Funding acquisition, Resources, Supervision, Writing – review & editing. LO: Conceptualization, Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. SM and AJH were supported by Australian Government Research Training Program scholarships. RKF-U was supported by ARC DP210102405 awarded to D.J. Adams.
Acknowledgments
The authors would like to thank the donors for their contribution to make this research possible. The authors would like to thank D.J. Adams for comments on the manuscript, providing the facilities, resources, and support for RKF-U (ARC Grant DP210102405 awarded to D.J. Adams).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2024.1406970/full#supplementary-material
Footnotes
References
Abu-Bonsrah, K. D., Viventi, S., Newgreen, D. F., and Dottori, M. (2019). “Generation of neural crest progenitors from human pluripotent stem cells” in Neural crest cells. Eds. Schwarz, Q., Wiszniak, S. (New York, NY: Humana Press), 37–47.
Arber, C., Lovejoy, C., Harris, L., Willumsen, N., Alatza, A., Casey, J. M., et al. (2021). Familial Alzheimer’s disease mutations in PSEN1 lead to premature human stem cell neurogenesis. Cell Rep. 34:108615. doi: 10.1016/j.celrep.2020.108615
Armijo, E., Gonzalez, C., Shahnawaz, M., Flores, A., Davis, B., and Soto, C. (2017). Increased susceptibility to Aβ toxicity in neuronal cultures derived from familial Alzheimer’s disease (PSEN1-A246E) induced pluripotent stem cells. Neurosci. Lett. 639, 74–81. doi: 10.1016/j.neulet.2016.12.060
Askenazi, M., Kavanagh, T., Pires, G., Ueberheide, B., Wisniewski, T., and Drummond, E. (2023). Compilation of all known protein changes in the human Alzheimer’s disease brain. bioRxiv :2023.2004.2013.536828.
Aubert, A., Mendoza-Ferri, M. G., Bramoulle, A., Stüder, F., Colombo, B. M., and Mendoza-Parra, M. A. (2023). PSEN1 M146V and PSEN1 A246E mutations associated with Alzheimer’s disease impair proper microglia differentiation. bioRxiv (preprints). 2023.2010.2008.561397
Balez, R., Steiner, N., Engel, M., Muñoz, S. S., Lum, J. S., Wu, Y., et al. (2016). Neuroprotective effects of apigenin against inflammation, neuronal excitability and apoptosis in an induced pluripotent stem cell model of Alzheimer’s disease. Sci. Rep. 6:31450. doi: 10.1038/srep31450
Balez, R., Stevens, C. H., Lenk, K., Maksour, S., Sidhu, K., Sutherland, G., et al. (2024). Increased neuronal nitric oxide synthase in Alzheimer’s disease mediates spontaneous calcium signaling and divergent glutamatergic calcium responses. Antioxid. Redox Signal. 41, 255–277. doi: 10.1089/ars.2023.0395
Busche, M. A., Chen, X., Henning, H. A., Reichwald, J., Staufenbiel, M., Sakmann, B., et al. (2012). Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 109, 8740–8745. doi: 10.1073/pnas.1206171109
Busche, M. A., Eichhoff, G., Adelsberger, H., Abramowski, D., Wiederhold, K.-H., Haass, C., et al. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science 321, 1686–1689. doi: 10.1126/science.1162844
Busche, M. A., Wegmann, S., Dujardin, S., Commins, C., Schiantarelli, J., Klickstein, N., et al. (2019). Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 22, 57–64. doi: 10.1038/s41593-018-0289-8
Buskila, Y., Kékesi, O., Bellot-Saez, A., Seah, W., Berg, T., Trpceski, M., et al. (2019). Dynamic interplay between H-current and M-current controls motoneuron hyperexcitability in amyotrophic lateral sclerosis. Cell Death Dis. 10:310. doi: 10.1038/s41419-019-1538-9
Chen, Z., Peng, L., Zhao, M., Tao, L., Zou, P., and Zhang, Y. (2021). Axon initial segment pathology in Alzheimer’s disease mouse model disturbs the action potential initiation and propagation. Research Square (Preprint).
Cirrito, J. R., Yamada, K. A., Finn, M. B., Sloviter, R. S., Bales, K. R., May, P. C., et al. (2005). Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron 48, 913–922. doi: 10.1016/j.neuron.2005.10.028
Colasante, G., Qiu, Y., Massimino, L., Di Berardino, C., Cornford, J. H., Snowball, A., et al. (2020). In vivo CRISPRa decreases seizures and rescues cognitive deficits in a rodent model of epilepsy. Brain 143, 891–905. doi: 10.1093/brain/awaa045
Crimins, J. L., Rocher, A. B., and Luebke, J. I. (2012). Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol. 124, 777–795. doi: 10.1007/s00401-012-1038-9
Crook, R., Verkkoniemi, A., Perez-Tur, J., Mehta, N., Baker, M., Houlden, H., et al. (1998). A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat. Med. 4, 452–455. doi: 10.1038/nm0498-452
Denham, M., and Dottori, M. (2011). “Neural differentiation of induced pluripotent stem cells” in Neurodegeneration: methods and protocols. eds. G. Manfredi and H. Kawamata (Totowa, NJ: Humana Press), 99–110.
Do-Ha, D., Buskila, Y., and Ooi, L. (2018). Impairments in motor neurons, interneurons and astrocytes contribute to Hyperexcitability in ALS: underlying mechanisms and paths to therapy. Mol. Neurobiol. 55, 1410–1418. doi: 10.1007/s12035-017-0392-y
Dowjat, W. K., Wisniewski, T., Efthimiopoulos, S., and Wisniewski, H. M. (1999). Inhibition of neurite outgrowth by familial Alzheimer's disease-linked presenilin-1 mutations. Neurosci. Lett. 267, 141–144. doi: 10.1016/S0304-3940(99)00351-1
Elsworthy, R. J., Finelli, M. J., Aqattan, S., Dunleavy, C., King, M., Ludlam, A., et al. (2023). Altered metabolic function induced by amyloid-beta oligomers and PSEN1-mutations in iPSC-derived astrocytes. bioRxiv (preprints). 2023.2008.2023.554346
Furukawa, K., Guo, Q., Schellenberg, G. D., and Mattson, M. P. (1998). Presenilin-1 mutation alters NGF-induced neurite outgrowth, calcium homeostasis, and transcription factor (AP-1) activation in PC12 cells. J. Neurosci. Res. 52, 618–624. doi: 10.1002/(SICI)1097-4547(19980601)
Ghatak, S., Dolatabadi, N., Trudler, D., Zhang, X., Wu, Y., Mohata, M., et al. (2019). Mechanisms of hyperexcitability in Alzheimer’s disease hiPSC-derived neurons and cerebral organoids vs isogenic controls. Elife 8:e50333. doi: 10.7554/eLife.50333
Greenough, M. A., Lane, D. J. R., Balez, R., Anastacio, H. T. D., Zeng, Z., Ganio, K., et al. (2022). Selective ferroptosis vulnerability due to familial Alzheimer’s disease presenilin mutations. Cell Death Differentiation 29, 2123–2136. doi: 10.1038/s41418-022-01003-1
Guennewig, B., Lim, J., Marshall, L., McCorkindale, A. N., Paasila, P. J., Patrick, E., et al. (2021). Defining early changes in Alzheimer's disease from RNA sequencing of brain regions differentially affected by pathology. Sci. Rep. 11:4865. doi: 10.1038/s41598-021-83872-z
Harris, S. S., Wolf, F., De Strooper, B., and Busche, M. A. (2020). Tipping the scales: peptide-dependent dysregulation of neural circuit dynamics in Alzheimer’s disease. Neuron 107, 417–435. doi: 10.1016/j.neuron.2020.06.005
Hulme, A. J., McArthur, J. R., Maksour, S., Miellet, S., Ooi, L., Adams, D. J., et al. (2020). Molecular and functional characterization of Neurogenin-2 induced human sensory neurons. Front. Cell. Neurosci. 14, –600895. doi: 10.3389/fncel.2020.600895
Hurley, E. M., Mozolewski, P., Dobrowolski, R., and Hsieh, J. (2023). Familial Alzheimer’s disease-associated PSEN1 mutations affect neurodevelopment through increased notch signaling. Stem Cell Rep. 18, 1516–1533. doi: 10.1016/j.stemcr.2023.05.018
Johnson, E. C. B., Dammer, E. B., Duong, D. M., Ping, L., Zhou, M., Yin, L., et al. (2020). Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 26, 769–780. doi: 10.1038/s41591-020-0815-6
Kwart, D., Gregg, A., Scheckel, C., Murphy, E. A., Paquet, D., Duffield, M., et al. (2019). A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP β-CTFs, not Aβ. Neuron 104, 256–270.e5. doi: 10.1016/j.neuron.2019.07.010
Landman, N., Jeong, S. Y., Shin, S. Y., Voronov, S. V., Serban, G., Kang, M. S., et al. (2006). Presenilin mutations linked to familial Alzheimer's disease cause an imbalance in phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. 103, 19524–19529. doi: 10.1073/pnas.0604954103
Mahairaki, V., Ryu, J., Peters, A., Chang, Q., Li, T., Park, T. S., et al. (2014). Induced pluripotent stem cells from familial Alzheimer's disease patients differentiate into mature neurons with amyloidogenic properties. Stem Cells Dev. 23, 2996–3010. doi: 10.1089/scd.2013.0511
Maksour, S., Ng, N., Hulme, A. J., Miellet, S., Engel, M., Muñoz, S. S., et al. (2024). REST and RCOR genes display distinct expression profiles in neurons and astrocytes using 2D and 3D human pluripotent stem cell models. Heliyon 10:e32680. doi: 10.1016/j.heliyon.2024.e32680
Marinković, P., Blumenstock, S., Goltstein, P. M., Korzhova, V., Peters, F., Knebl, A., et al. (2019). In vivo imaging reveals reduced activity of neuronal circuits in a mouse tauopathy model. Brain 142, 1051–1062. doi: 10.1093/brain/awz035
Martinez-Losa, M., Tracy, T. E., Ma, K., Verret, L., Clemente-Perez, A., Khan, A. S., et al. (2018). Nav1. 1-overexpressing interneuron transplants restore brain rhythms and cognition in a mouse model of Alzheimer’s disease. Neuron 98, 75–89.e5. doi: 10.1016/j.neuron.2018.02.029
Mattei, C., Lim, R., Drury, H., Nasr, B., Li, Z., Tadros, M. A., et al. (2019). Generation of vestibular tissue-like organoids from human pluripotent stem cells using the rotary cell culture system. Front. Cell Dev. Biol. 7:25. doi: 10.3389/fcell.2019.00025
Menkes-Caspi, N., Yamin, H. G., Kellner, V., Spires-Jones, T. L., Cohen, D., and Stern, E. A. (2015). Pathological tau disrupts ongoing network activity. Neuron 85, 959–966. doi: 10.1016/j.neuron.2015.01.025
Meyer, K., Feldman, H. M., Lu, T., Drake, D., Lim, E. T., Ling, K.-H., et al. (2019). REST and neural gene network dysregulation in iPSC models of Alzheimer’s disease. Cell Rep. 26, 1112–1127.e9. doi: 10.1016/j.celrep.2019.01.023
Muñoz, S. S., Balez, R., Berg, T., Engel, M., Bax, M., Do-Ha, D., et al. (2018). Generation and characterization of human induced pluripotent stem cell lines from a familial Alzheimer's disease PSEN1 A246E patient and a non-demented family member bearing wild-type PSEN1. Stem Cell Res. 31, 227–230. doi: 10.1016/j.scr.2018.08.006
Nee, L. E., Polinsky, R. J., Eldridge, R., Weingartner, H., Smallberg, S., and Ebert, M. (1983). A family with histologically confirmed Alzheimer's disease. Arch. Neurol. 40, 203–208. doi: 10.1001/archneur.1983.04050040033004
Nuriel, T., Angulo, S. L., Khan, U., Ashok, A., Chen, Q., Figueroa, H. Y., et al. (2017). Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat. Commun. 8:1464. doi: 10.1038/s41467-017-01444-0
Oksanen, M., Petersen, A. J., Naumenko, N., Puttonen, K., Lehtonen, Š., Olivé, M. G., et al. (2017). PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer's disease. Stem Cell Rep. 9, 1885–1897. doi: 10.1016/j.stemcr.2017.10.016
Ooi, L., Dottori, M., Cook, A. L., Engel, M., Gautam, V., Grubman, A., et al. (2020). If human brain organoids are the answer to understanding dementia, what are the questions? Neuroscientist 26, 438–454. doi: 10.1177/1073858420912404
Ping, Y., Hahm, E.-T., Waro, G., Song, Q., Vo-Ba, D.-A., Licursi, A., et al. (2015). Linking aβ42-induced hyperexcitability to neurodegeneration, learning and motor deficits, and a shorter lifespan in an Alzheimer’s model. PLoS Genet. 11:e1005025. doi: 10.1371/journal.pgen.1005025
Pooler, A. M., Phillips, E. C., Lau, D. H., Noble, W., and Hanger, D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394. doi: 10.1038/embor.2013.15
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., et al. (2007). Reducing endogenous tau ameliorates amyloid ß-induced deficits in an Alzheimer's disease mouse model. Science 316, 750–754. doi: 10.1126/science.1141736
Rovelet-Lecrux, A., Charbonnier, C., Wallon, D., Nicolas, G., Seaman, M. N. J., Pottier, C., et al. (2015). De novo deleterious genetic variations target a biological network centered on Aβ peptide in early-onset Alzheimer disease. Mol. Psychiatry 20, 1046–1056. doi: 10.1038/mp.2015.100
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 375, 754–760. doi: 10.1038/375754a0
Skobeleva, K. V., Ryazantseva, М. A., and Kaznacheyeva, Е. V. (2022). Increased calcium influx through L-type calcium channels in hippocampal neurons with exogenous expression of Presenilin-1 ΔE9 mutant. Bull. Exp. Biol. Med. 172, 785–788. doi: 10.1007/s10517-022-05478-3
Spoleti, E., Krashia, P., La Barbera, L., Nobili, A., Lupascu, C. A., Giacalone, E., et al. (2022). Early derailment of firing properties in CA1 pyramidal cells of the ventral hippocampus in an Alzheimer's disease mouse model. Exp. Neurol. 350:113969. doi: 10.1016/j.expneurol.2021.113969
Steiner, H., Romig, H., Grim, M. G., Philipp, U., Pesold, B., Citron, M., et al. (1999). The biological and pathological function of the Presenilin-1 ΔExon 9 mutation is independent of its defect to undergo proteolytic processing*. J. Biol. Chem. 274, 7615–7618. doi: 10.1074/jbc.274.12.7615
Targa Dias Anastacio, H., Matosin, N., and Ooi, L. (2022). Neuronal hyperexcitability in Alzheimer’s disease: what are the drivers behind this aberrant phenotype? Transl. Psychiatry 12:257. doi: 10.1038/s41398-022-02024-7
Targa Dias Anastacio, H., Matosin, N., and Ooi, L. (2024). Familial Alzheimer’s disease neurons bearing mutations in PSEN1 display increased calcium responses to AMPA as an early calcium dysregulation phenotype. Life 14:625. doi: 10.3390/life14050625
Thouta, S., Zhang, Y., Garcia, E., and Snutch, T. P. (2021). Kv1.1 channels mediate network excitability and feed-forward inhibition in local amygdala circuits. Sci. Rep. 11:15180. doi: 10.1038/s41598-021-94633-3
Vanova, T., Sedmik, J., Raska, J., Amruz Cerna, K., Taus, P., Pospisilova, V., et al. (2023). Cerebral organoids derived from patients with Alzheimer’s disease with PSEN1/2 mutations have defective tissue patterning and altered development. Cell Rep. 42:113310. doi: 10.1016/j.celrep.2023.113310
Verkkoniemi, A., Somer, M., Rinne, J. O., Myllykangas, L., Crook, R., Hardy, J., et al. (2000). Variant Alzheimer’s disease with spastic paraparesis. Neurology 54, 1103–1109. doi: 10.1212/WNL.54.5.1103
Verret, L., Mann, E. O., Hang, G. B., Barth, A. M., Cobos, I., Ho, K., et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. doi: 10.1016/j.cell.2012.02.046
Vitale, P., Salgueiro-Pereira, A. R., Lupascu, C. A., Willem, M., Migliore, R., Migliore, M., et al. (2021). Analysis of age-dependent alterations in excitability properties of CA1 pyramidal neurons in an APPPS1 model of Alzheimer’s disease. Front. Aging Neurosci. 13:668948. doi: 10.3389/fnagi.2021.668948
Woodruff, G., Reyna, S. M., Dunlap, M., Van Der Kant, R., Callender, J. A., Young, J. E., et al. (2016). Defective transcytosis of APP and lipoproteins in human iPSC-derived neurons with familial Alzheimer's disease mutations. Cell Rep. 17, 759–773. doi: 10.1016/j.celrep.2016.09.034
Wu, J. W., Hussaini, S. A., Bastille, I. M., Rodriguez, G. A., Mrejeru, A., Rilett, K., et al. (2016). Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 19, 1085–1092. doi: 10.1038/nn.4328
Yamamoto, K., Tanei, Z.-i., Hashimoto, T., Wakabayashi, T., Okuno, H., Naka, Y., et al. (2015). Chronic optogenetic activation augments Aβ pathology in a mouse model of Alzheimer disease. Cell Rep. 11, 859–865. doi: 10.1016/j.celrep.2015.04.017
Yang, J., Zhao, H., Ma, Y., Shi, G., Song, J., Tang, Y., et al. (2017). Early pathogenic event of Alzheimer's disease documented in iPSCs from patients with PSEN1 mutations. Oncotarget 8, 7900–7913. doi: 10.18632/oncotarget.13776
Keywords: Alzheimer’s disease, PSEN1 , neuronal excitability, iNs, iPSCs
Citation: Maksour S, Finol-Urdaneta RK, Hulme AJ, Cabral-da-Silva MC, Targa Dias Anastacio H, Balez R, Berg T, Turner C, Sanz Muñoz S, Engel M, Kalajdzic P, Lisowski L, Sidhu K, Sachdev PS, Dottori M and Ooi L (2024) Alzheimer’s disease induced neurons bearing PSEN1 mutations exhibit reduced excitability. Front. Cell. Neurosci. 18:1406970. doi: 10.3389/fncel.2024.1406970
Edited by:
Janire Urrutia, University of the Basque Country, SpainReviewed by:
Roberto Piacentini, Catholic University of the Sacred Heart, ItalyHirotaka Watanabe, Keio University, Japan
Dennis R. Tabuena, Gladstone Institutes, United States
Copyright © 2024 Maksour, Finol-Urdaneta, Hulme, Cabral-da-Silva, Targa Dias Anastacio, Balez, Berg, Turner, Sanz Muñoz, Engel, Kalajdzic, Lisowski, Sidhu, Sachdev, Dottori and Ooi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirella Dottori, bWRvdHRvcmlAdW93LmVkdS5hdQ==; Lezanne Ooi, bGV6YW5uZUB1b3cuZWR1LmF1; Rocio K. Finol-Urdaneta, cmZpbm9sdUB1b3cuZWR1LmF1
†These authors have contributed equally to this work and share senior authorship