 Amanda R. McFarlan1,2
Amanda R. McFarlan1,2 Connie Guo1,2
Connie Guo1,2 Isabella Gomez1Chaim Weinerman1Tasha A. Liang1
Isabella Gomez1Chaim Weinerman1Tasha A. Liang1 P. Jesper Sjöström1*
P. Jesper Sjöström1*- 1Centre for Research in Neuroscience, BRaIN Program, Department of Neurology and Neurosurgery, Research Institute of the McGill University Health Centre, Montreal General Hospital, Montreal, QC, Canada
- 2Integrated Program in Neuroscience, McGill University, Montreal, QC, Canada
The plasticity of inhibitory interneurons (INs) plays an important role in the organization and maintenance of cortical microcircuits. Given the many different IN types, there is an even greater diversity in synapse-type-specific plasticity learning rules at excitatory to excitatory (E→I), I→E, and I→I synapses. I→I synapses play a key disinhibitory role in cortical circuits. Because they typically target other INs, vasoactive intestinal peptide (VIP) INs are often featured in I→I→E disinhibition, which upregulates activity in nearby excitatory neurons. VIP IN dysregulation may thus lead to neuropathologies such as epilepsy. In spite of the important activity regulatory role of VIP INs, their long-term plasticity has not been described. Therefore, we characterized the phenomenology of spike-timing-dependent plasticity (STDP) at inputs and outputs of genetically defined VIP INs. Using a combination of whole-cell recording, 2-photon microscopy, and optogenetics, we explored I→I STDP at layer 2/3 (L2/3) VIP IN outputs onto L5 Martinotti cells (MCs) and basket cells (BCs). We found that VIP IN→MC synapses underwent causal long-term depression (LTD) that was presynaptically expressed. VIP IN→BC connections, however, did not undergo any detectable plasticity. Conversely, using extracellular stimulation, we explored E→I STDP at inputs to VIP INs which revealed long-term potentiation (LTP) for both causal and acausal timings. Taken together, our results demonstrate that VIP INs possess synapse-type-specific learning rules at their inputs and outputs. This suggests the possibility of harnessing VIP IN long-term plasticity to control activity-related neuropathologies such as epilepsy.
Introduction
It has long been believed that excitatory to excitatory (E→E) long-term plasticity underlies information storage in the brain (Bliss and Collingridge, 1993; Malenka and Bear, 2004; Nabavi et al., 2014) as well as circuit remapping during development (Katz and Shatz, 1996; Cline, 1998). The brain, however, is also made up of numerous inhibitory IN types that play an active role in shaping cortical circuits through plasticity (Yazaki-Sugiyama et al., 2009; Vogels et al., 2011; D’Amour and Froemke, 2015; Udakis et al., 2020). For example, several studies have shown that plasticity at excitatory to inhibitory (E→I) and I→E connections also contribute to circuit rewiring and impact E→E neurotransmission (Maffei et al., 2006; Ormond and Woodin, 2009, 2011; Vogels et al., 2011). There is, however, a paucity of literature on the long-term plasticity at I→I synapses.
Spike-timing-dependent plasticity (STDP) is a biologically plausible experimental paradigm in which the millisecond temporal ordering of pre- and postsynaptic spikes determines whether long-term potentiation (LTP) or long-term depression (LTD) is elicited (Markram et al., 2011; Feldman, 2012; Markram et al., 2012). Presynaptic spiking occurring milliseconds before postsynaptic activity is referred to as causal because here the presynaptic spiking is causally related to postsynaptic activation, whereas the opposite temporal ordering is termed acausal (McFarlan et al., 2023). For classical STDP at E→E synapses, causal spiking elicits LTP whereas acausal spiking triggers LTD (Markram et al., 1997; Bi and Poo, 1998). Classical STDP is thus in agreement with the Hebbian postulate (Hebb, 1949) that ‘cells that fire together wire together’ (Lowel and Singer, 1992; Shatz, 1992), but has the extension that synaptic weakening arises from acausal firing, i.e., when the presynaptic cell fails to excite the postsynaptic cell (Stent, 1973; Debanne et al., 1994). This acausal LTD has important functional implications, for instance to achieve synaptic competition (Song et al., 2000; Song and Abbott, 2001).
Interestingly, inhibitory synapses do not always obey the classic Hebbian STDP rule (Feldman, 2012), neither at E→I (Lu et al., 2007) nor at I→E synapses (Holmgren and Zilberter, 2001; Woodin et al., 2003). As there are many different kinds of INs (Gouwens et al., 2020), there is thus an even larger set of synapse-type-specific forms of plasticity at E→I, I→E, and I→I connections. A relatively comprehensive collection of plasticity learning rules for a given brain region — known as a plasticitome (Sjöström, 2021) — is therefore required to understand the role of plasticity in local circuits (McFarlan et al., 2023).
Several IN types receive inhibitory inputs themselves (Artinian and Lacaille, 2018; Kullander and Topolnik, 2021). These I→I synapses have important implications for network activity because disinhibition — which can be mediated by weakening I→I connections — may increase network excitation via I→I→E connectivity motifs (McFarlan et al., 2023). Vasoactive intestinal peptide (VIP) INs, which primarily target basket cells (BCs) and Martinotti cells (MCs) (Pfeffer et al., 2013; Kepecs and Fishell, 2014; Tremblay et al., 2016), have consistently been implicated in I→I→E disinhibition (Letzkus et al., 2015; Artinian and Lacaille, 2018; Kullander and Topolnik, 2021). Though several studies have demonstrated that disinhibition plays a role in plasticity and in learning (Froemke et al., 2007; Niell and Stryker, 2010; Letzkus et al., 2011; Kaneko and Stryker, 2014; Fu et al., 2015; Adler et al., 2019), few studies have explored I→I plasticity directly (Sarihi et al., 2012).
The motor cortex function is important for the execution of voluntary movement and motor learning in the healthy brain. In recent years, VIP IN-mediated suppression of SST INs in the motor cortex has been shown to have a key role in promoting motor learning (Adler et al., 2019; Ren et al., 2022). VIP IN-mediated disinhibition has additionally been implicated in diseases like epilepsy (Cunha-Reis and Caulino-Rocha, 2020). Indeed, reduced VIP IN inhibitory drive in the mouse motor cortex had a protective effect on seizure initiation and duration (Khoshkhoo et al., 2017). These are thus concrete indications that VIP INs in motor cortex constitute a promising seizure control point.
In this phenomenological study, we explored STDP of disinhibitory motor cortex VIP INs using a combination of patch-clamp electrophysiology, 2-photon imaging, extracellular stimulation, and optogenetics. We describe STDP learning rules at both inputs to and outputs from VIP IN in the mouse motor cortex.
Materials and methods
Animals and ethics statement
The animal study was approved by the Montreal General Hospital Facility Animal Care Committee and adhered to the guidelines of the Canadian Council on Animal Care. To drive expression of Channelrhodopsin-2 (ChR2) and enhanced yellow fluorescent protein (EYFP) in VIP INs, we crossed homozygous VIPTM1(cre)Zjh/J mice (JAX strain 010908) (Taniguchi et al., 2011) with homozygous B6.Cg-Gt(ROSA)26SorTM32(CAG–COP4*H134R/EYFP)Hze/J mice (also known as Ai32, JAX strain 024109) to obtain VIPCre/+;Ai32flox/+ mice, henceforth referred to as VIP-ChR2 mice. Experiments were carried out in male and female postnatal day (P)21-P40 VIP-ChR2 mice. Animals were anesthetized with isoflurane and sacrificed once the hind-limb withdrawal reflex was lost.
Acute brain slice electrophysiology
To optimize slice quality obtained from these relatively mature animals, we relied on a sucrose-based cutting solution containing (in mM) 200 sucrose, 2.5 KCl, 1.0 NH2PO4, 2.5 CaCl2, 1.3 MgCl2, 47 D-glucose and 26.2 NaHCO3. The solution was bubbled with 95% O2/5% CO2 for 10 min and cooled on ice to ∼4°C. Osmolality was adjusted to 338 mOsm with glucose, measured using Model 3300 or Osmo1 osmometers (Advanced Instruments Inc., Norwood, MA, USA).
After decapitation, the brain was removed and placed in ice-cold sucrose cutting solution. Coronal 300-μm-thick acute brain slices were prepared using a Campden Instruments 5000 mz-2 vibratome (Campden Instruments, Loughborough, UK) and ceramic blades (Lafayette Instrument, Lafayette, IN, USA). Brain slices were kept at ∼33°C in oxygenated artificial cerebrospinal fluid (ACSF), containing (in mM) 125 NaCl, 2.5 KCl, 1 MgCl2, 1.25 NaH2PO4, 2 CaCl2, 26 NaHCO3 and 25 glucose, bubbled with 95% O2/5% CO2, for ∼10 min and then allowed to cool at room temperature for at least 1 h before starting the recordings. Osmolality of the ACSF was adjusted to 338 mOsm with glucose. We carried out experiments with ACSF heated to 32–34°C with a resistive inline heater (Scientifica Ltd, Uckfield, UK), with temperature recorded and verified offline. Recordings were truncated or not used if outside this range.
An internal solution was prepared containing (in mM) 1 or 5 KCl, 115 K-Gluconate, 10 K-HEPES, 4 Mg-ATP, 0.3 Na-GTP, 10 Na2-Phosphocreatine and 0.1% biocytin. KOH was added to reach a pH of 7.2 to 7.4 and sucrose was added to reach the target osmolality of 310 mOsm. To visualize patched cells, 20 μM of Alexa 594 Hydrazide dye (Life Technologies, Eugene, OR, USA) was added to the internal solution. Patch pipettes were pulled using the P-1000 puller (Sutter Instruments, Novato, CA, USA). The pipette resistances varied between 4 and 7 MΩ.
We obtained whole-cell recordings using BVC-700A amplifiers (Dagan Corporation, Minneapolis, MN, USA) in current-clamp configuration. Amplified signals were low-pass filtered at 5 kHz and acquired at 40 kHz using PCI-6229 boards (NI, Austin, TX, USA). All data was acquired in Igor Pro 8 or 9 (WaveMetrics Inc., Lake Oswego, OR, USA) using custom software (Sjöström et al., 2001; Sjöström et al., 2003).1 We monitored input resistance, series resistance, perfusion temperature, and resting membrane potential during experiments and performed further analyses offline. We did not compensate for series resistance, nor did we account for the liquid junction potential (10 mV).
Cells were patched with a LUMPlanFL N 40 × /0.80 objective (Olympus, Olympus, Melville, NY, USA) using infrared video Dodt contrast on a custom-modified Scientifica SliceScope as previously described (Buchanan et al., 2012). A Chameleon ULTRA II (Coherent, Santa Clara, CA, USA) titanium-sapphire laser tuned to 920 or 820 nm was used to excite EYFP and Alexa 594 fluorophores, respectively. VIP INs were targeted based on EYFP expression visualized with 2-photon (2P) microscopy at 920 nm. L5 BCs and MCs were targeted based on their small round-shaped soma which were distinctly different from L5 pyramidal cells (PCs) which have a triangular-shaped soma and prominent apical dendrite. Cell identity was verified post hoc using electrophysiological and morphological properties (see below and Figure 1, Supplementary Figures 5, 7, and Supplementary Tables 1, 2). Briefly, BCs were characterized by their typical fast-spiking physiology, narrow action potential half width, and high rheobase as well as their densely branching axons and dendrites. MCs were characterized by their accommodating firing pattern and lower rheobase as well as their ascending axon and dangling dendrites (Silberberg and Markram, 2007; Buchanan et al., 2012; Sippy and Yuste, 2013; Tremblay et al., 2016).
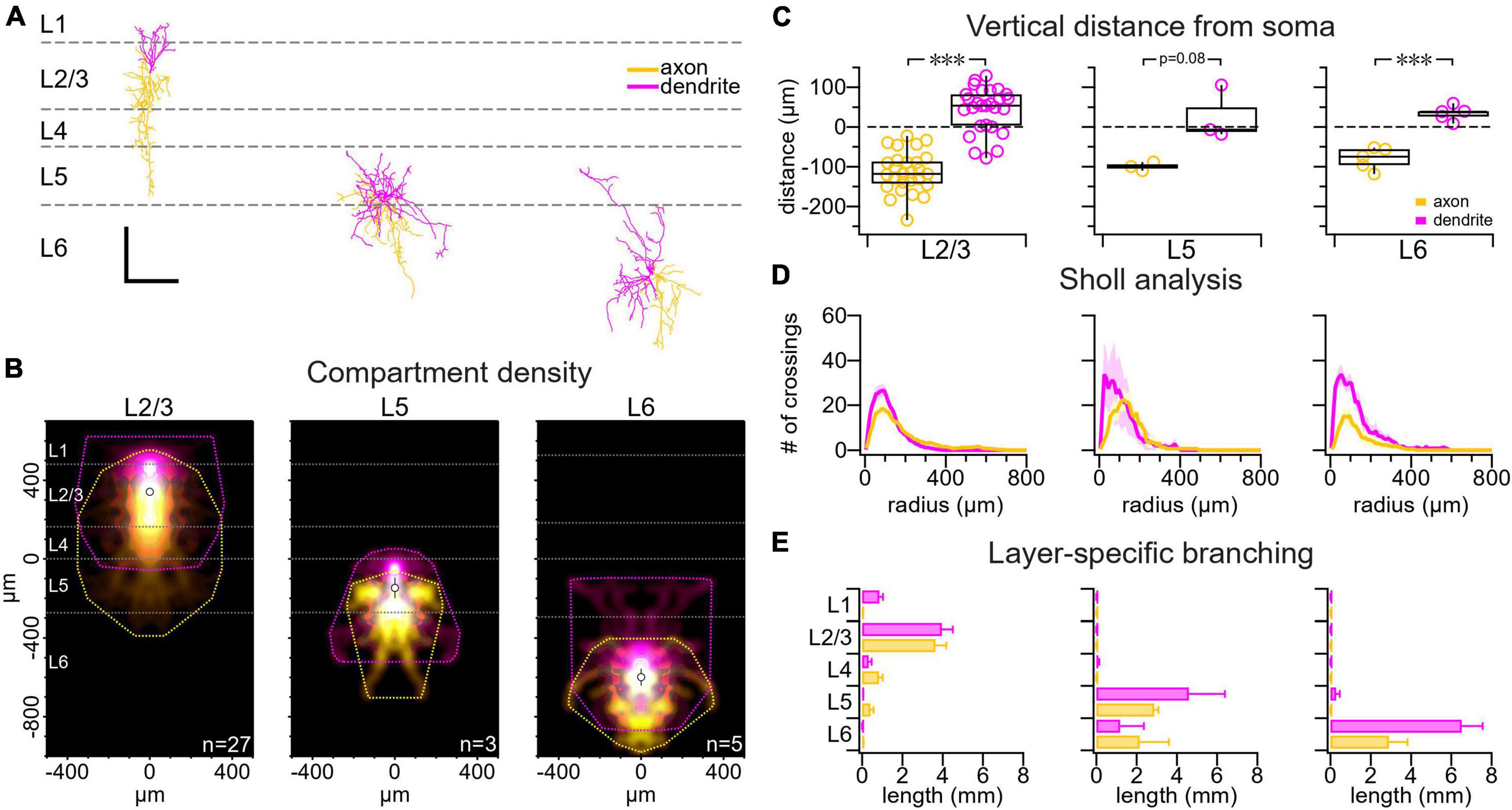
Figure 1. VIP IN morphology varied with cortical layer. (A) Sample reconstructions from a L2/3 VIP IN (left), a L5 VIP IN (middle), and a L6 VIP IN (right). Scale bar is 250 μm for both axes. Axons are labeled yellow and dendrites are labeled pink throughout the figure. (B) Compartment density heat map for L2/3 VIP INs (n = 27 cells, N = 21 animals), L5 VIP INs (n = 3 cells, N = 3 animals), and L6 VIP INs (n = 5 cells, N = 4 animals) appear to be vertically asymmetric. Heat maps are centered vertically on the boundary between L4 and L5. (C) Comparing the axonal and dendritic compartment center of mass vertically, we found that VIP IN dendrites branched upwards toward the pial surface whereas VIP IN axons chiefly projected toward deeper cortical layers (L2/3 VIP IN axon: –110 μm ± 8 μm vs. L2/3 VIP IN dendrite: 44 μm ± 10 μm, t-test p < 0.001; L5 VIP IN axon: –100 μm ± 6 μm vs. L5 VIP IN dendrite: 27 μm ± 40 μm, t-test p = 0.08; L6 VIP IN axon: –80 μm ± 10 μm vs. L6 VIP IN dendrite: 34 μm ± 8 μm, t-test p < 0.001). Dashed line: soma location. (D) Sholl analysis revealed that VIP IN dendrites and axons branch most densely at 100 μm from the soma. (E) Layer-specific branching revealed that L2/3 VIP IN axons extended into deeper cortical layers, while their dendrites were mostly localized to superficial layers. Axonal and dendritic arbors in L5 and L6 VIP INs were mostly localized to deeper cortical layers. Branching was measured as the total branch length cumulated across cells.
Long-term plasticity experiments
To explore long-term plasticity at VIP IN outputs, BCs and MCs were targeted for whole-cell recording in acute slices from P21-P40 VIP-ChR2 mice. L2/3 VIP INs were visualized using 2P microscopy at 920 nm. To activate ChR2-expressing L2/3 VIP INs, a blue laser (1-W 445-nm Blue Laser Diode Module, Item Id: 131542738201, Laserland, eBay.ca) was guided into the same light path as the 2P beam using a dichroic (FF665-Di02, Semrock Inc., Rochester, NY, USA) and controlled with a pair of 6215H 3-mm galvanometric mirrors (Cambridge Technologies, Bedford, MA, USA). The blue laser was gated by the MultiPatch software described above, thus enabling synchronization with electrophysiology acquisition. Blue laser pulses aimed onto fluorescent L2/3 VIP INs had a power of 20 mW and a duration of either 2 ms or 5 ms. L5 BCs and MCs that showed inhibitory postsynaptic potentials (IPSPs) in response to ChR2 activation were used for experiments.
To explore long-term plasticity at VIP IN inputs, L2/3 VIP INs were targeted for whole-cell recording using 2P microscopy at 920 nm. An extracellular stimulating pipette filled with ACSF was brought into the slice ∼100–200 μm from the patched cell and was used to activate VIP IN inputs. Extracellular stimulation was performed using a Biphasic Stimulation Isolator BSI-950 (Dagan Corporation, Minneapolis, MN, USA) that was manipulated via the MultiPatch software described above. Extracellular stimulation pulses were 100 μs in duration. Excitatory postsynaptic potential (EPSP) responses in patched VIP INs were inspected to ensure they were due to the activation of VIP IN inputs rather than direct stimulation of the patched VIP IN itself. A depolarization onset that emerged directly from the stimulation artifact was indicative of direct stimulation, whereas a depolarization onset that occurred 1–2 ms after the stimulation artifact was indicative of indirect stimulation. An input-output curve was used to measure the response amplitude to incremental increases in extracellular stimulation strength in the patched cell. The stimulation strength that yielded EPSPs at least 1 mV in amplitude and below the spiking threshold was used for the experiment.
For long-term plasticity experiments at VIP IN inputs and outputs, an initial pre-induction baseline consisted of two laser or extracellular stimulation pulses followed by two current pulses, both delivered at 30 Hz and offset by 700 ms, repeated 60 times over a period of 10 min. The induction protocol consisted of five laser or extracellular stimulation pulses and five current pulses delivered at 50 Hz and offset by ± 10 or +25 ms or delivered at 20 Hz and offset by ± 25 ms. The induction protocol was repeated 15 times for 2.5 min. The post-pairing baseline — which was contents-wise identical to the initial baseline — was repeated for up to 1 h. Control experiments had only presynaptic (pre only) or only postsynaptic spiking (post only) during the induction period. The time window for quantifying post-induction synaptic response amplitude started 10 min after the end of the induction and continued until the end of each individual experiment. This was compared to or normalized to the synaptic responses acquired during the pre-induction baseline period.
The paired-pulse ratio (PPR) was calculated as IPSP2/IPSP1 or EPSP2/EPSP1 for the pre-pairing and post-pairing periods. The change in PPR (ΔPPR) was calculated as PPRafter - PPRbefore. Coefficient of variation (CV) analysis was performed as previously described (Brock et al., 2020). Briefly, the mean and CV of IPSP1 or EPSP1 were calculated for the pre-pairing period, followed by normalizing mean and CV–2 to the post-pairing period. The angle (θ) was defined by the diagonal unity line and the line formed by linking the starting coordinate (1, 1) and CV analysis endpoint. θ < 0 (i.e., clockwise from diagonal) indicated a postsynaptic locus of plasticity expression, while θ > 0 indicated a presynaptic locus (Brock et al., 2020).
Identification of motor cortex layers
The motor cortex was targeted based on the location of the corpus callosum white matter tract. L1 and white matter were identified based on their lack of cell bodies. For electrophysiological experiments, we differentiated between L2/3, L5 and, L6 based on PC morphology. In L2/3, PC somata are relatively small, whereas in L5, PCs have large somata and a thick apical dendrite. L6 PCs have rounded somata and a thin apical dendrite.
Layer boundaries for immunohistochemistry and biocytin histology were informed by NeuN cell counts and in situ hybridization (ISH) data from the Allen Institute Mouse Brain Atlas (Lein et al., 2007). We selected ISH images stained for Stard8, Rorb, Bend5, and Ighm expression, which was restricted to L2/3, L4, L5, and L6, respectively. Using Fiji/ImageJ (Schindelin et al., 2012), we selected a ∼800-μm-wide linear region of interest spanning the motor cortex from pial surface to white matter and measured the intensity profile across the cortical thickness. We thus overlayed the intensity profile for each gene marker (Supplementary Figure 1). Each density profile was baseline-subtracted, peak-normalized, and box-smoothed (setting 10) in Igor Pro 9. The point of intersection between pixel intensity profiles were then used to define layer boundaries. The percentage distance normalized from pial surface to white matter was used to inform layer boundaries in individual slices.
We were concerned that fixed tissue samples might be altered, e.g., due to fixation, cover slipping, or other histology steps, which might distort the apparent cortical thickness. We therefore verified the cortical thickness in fixed tissue by comparing to the acute slice in electrophysiology experiments, which revealed a good match, thus validating the accuracy of fixed slice samples.
Immunohistochemistry
P21-P40 VIP-ChR2 mice were anesthetized with isofluorane and transcardially perfused with 0.1 M phosphate buffered saline (PBS) followed by 4% paraformaldehyde. Brains were incubated in 4% paraformaldehyde overnight, and then stored for two additional days in a 30% (w/v) sucrose solution. Next, brains were mounted in plastic cubes containing Optimal Cutting Temperature embedding medium (25608-930, VWR, Radnor, PA, USA) and then frozen using a bath of 100% EtOH and dry ice. Fixed brains were sectioned with a cryostat at 50 μm thickness through the motor cortex and sections were placed in a 0.01 M PBS solution. Sections underwent a 20-min wash in 0.01 M PBS with 1% Triton-X followed by a 90-min wash in 0.01 M PBS with 0.3% Triton-X and 10% normal donkey serum (NDS; 017-000-121 Jackson ImmunoResearch, West Grove, PA, USA). All antibody incubations were performed in 0.01 M PBS with 0.3% Triton-X and 1% NDS.
Sections were incubated overnight at 4°C in the following primary antibodies: 1:500 rabbit anti-VIP (20077, ImmunoStar, Dietzenbach, Germany), 1:100 rat anti-somatostatin (ab30788, Abcam, Boston, MA, USA), 1:1000 chicken anti-GFP (ab13970, Abcam), 1:500 mouse anti-parvalbumin (p3088, Sigma, St. Louis, MO, USA), and 1:1000 mouse anti-NeuN (ab104224, Abcam). Twenty-four hours later, tissue underwent three 15-min washes in 0.01 M PBS with 0.3% Triton-X and 1% NDS, followed by a 90-min incubation in the following Alexa Fluor secondary antibodies at 1:1000: donkey anti-rabbit 647 (711-605-152, Jackson ImmunoResearch), donkey anti-rat 594 (712-585-150, Jackson ImmunoResearch), donkey anti-chicken 488 (703-545-155, Jackson ImmunoResearch), goat anti-mouse 647 (A21240, ThermoFisher Scientific, Waltham, MA, USA), and donkey anti-mouse 568 (SAB4600075, Sigma). Next, the tissue underwent three 20-min washes in 0.01 M PBS with 0.3% Triton-X and 1% NDS. Following this procedure, coronal slices were mounted using coverslips with a 40 μl bolus of ProLong Gold Antifade Mountant (ThermoFisher Scientific).
Sections were imaged using a Fluoview FV1000 confocal laser scanning microscope and Fluoview software (Olympus Canada, Richmond Hill, ON, Canada) or a Zeiss LSM780 confocal laser scanning microscope and ZEN software (Zeiss, Oberkochen, Germany). Image analysis and quantification were performed using Fiji/ImageJ (Schindelin et al., 2012) and Igor Pro 9 (Wavemetrics). Cell counts for neurons expressing VIP, EYFP, somatostatin (SST) and parvalbumin (PV) were carried out across all six cortical layers and in both hemispheres.
Biocytin histology and morphological reconstructions
Patched VIP INs, MCs, and BCs used in long-term plasticity experiments were saved for neuronal reconstruction. Once the experiment was completed, the patch pipette was removed slowly while applying light positive pressure. Sections were then incubated in 4% paraformaldehyde overnight and were stored in 0.01 M PBS solution for up to 3 weeks before staining.
Sections underwent four 10-min washes in 0.01 M Tris-buffered saline (TBS) solution with 0.3% Triton-X followed by a 1-h wash in 0.01 M TBS with 0.3% Triton-X and 10% NDS. Sections were incubated overnight at 4°C in 0.01 M TBS with 0.3% Triton-X and 1% NDS, supplemented with 1:200 Alexa Fluor 647- or Alexa fluor 488-conjugated Streptavidin (ThermoFisher Scientific). Twenty-four hours later, tissue underwent four 10-min washes in 0.01 M TBS. Following this procedure, sections were mounted using coverslips with a 40 μl bolus of ProLong Gold Antifade Mountant (ThermoFisher Scientific). 3D image stacks were acquired using a Zeiss LSM780 confocal laser scanning microscope and ZEN software (Zeiss) and used for morphological reconstructions.
3D confocal image stacks were contrast adjusted and converted to 8 bits or 16 bits in Fiji (Schindelin et al., 2012) and then imported into Neuromantic V1.7.5 (Myatt et al., 2012) for manual tracing. Morphometry was performed in Igor Pro 9 (Wavemetrics) using the qMorph in-house custom software as previously described (Buchanan et al., 2012; Zhou et al., 2021).2
Statistics
Unless otherwise noted, results are reported as the mean ± standard error of the mean (SEM). Significance levels are denoted using asterisks (*p < 0.05, **p < 0.01, ***p < 0.001). Pairwise comparisons were carried out using a two-tailed Student’s t-test for equal means. If an equality of variances F test gave p < 0.05, we employed the unequal variances t-test. Wilcoxon-Mann-Whitney’s non-parametric test was always used in parallel to the t-test, yielding similar outcomes. Statistical tests were performed in Igor Pro 9 (Wavemetrics).
Results
The VIP-ChR2 mouse line reliably identifies VIP INs
We created a VIP-ChR2 mouse line by crossing a VIP-Cre driver line with the Ai32 ChR2/EYFP reporter line (Methods). We validated our VIP-ChR2 mice by exploring the degree of overlap between the genetic EYFP tag and VIP expression. To do so, we relied on immunohistochemistry (Methods). This revealed that ∼80% of EYFP-positive cells were also positive for VIP and that ∼88% of VIP-positive cells were also positive for EYFP (Supplementary Figure 2), demonstrating that our VIP-ChR2 mouse line was highly specific for VIP INs, in agreement with the prior literature (Taniguchi et al., 2011).
Next, we looked at the spatial distribution of VIP INs across the cortical layers. Similar to what has been shown in the barrel cortex (Prönneke et al., 2015; Almási et al., 2019) and visual cortex (Gonchar et al., 2007), we found in the motor cortex that most VIP INs were located in L2/3 (Supplementary Figure 3). We also explored whether VIP INs expressed SST or PV — molecular markers of MCs and BCs, respectively (Blackman et al., 2013) — but found that they did not (Supplementary Figure 4), in agreement with the prior literature (Prönneke et al., 2015).
L2/3, L5, and L6 VIP INs are electrophysiologically indistinguishable
We compared the electrophysiological properties of a total of 46 patched VIP INs from L2/3, L5, and L6. We found no detectable differences across layers in basic electrophysiological properties such as resting membrane potential, firing threshold, action potential height, action potential half width, rheobase, membrane time constant, and input resistance (Supplementary Table 1). We also found that VIP INs had varying spike patterns. Following the Petilla convention (Ascoli et al., 2008), we found that VIP INs exhibited three different action potential firing patterns: adapting, burst firing, and irregular firing (Supplementary Figure 5 and Supplementary Table 1).
L2/3, L5, and L6 VIP INs morphologies vary with cortical layer
We investigated the morphology of VIP INs in the mouse motor cortex. We used biocytin histology and confocal imaging to 3D reconstruct patched VIP INs in L2/3, L5 and L6. Consistent with previous findings in the barrel cortex (Prönneke et al., 2015), we found that VIP INs have dendrites that project toward the pial surface and axons that extend into deeper cortical layers (Figures 1A–C). Sholl analysis (Sholl, 1953) additionally revealed that VIP IN dendrites and axons were most densely branched around 100 μm away from the soma (Figure 1D). L2/3 VIP IN dendrites were mostly localized to superficial layers, whereas their axons extended into deeper cortical layers. L5 and L6 VIP INs dendrites and axons were mostly localized to deeper cortical layers (Figure 1E).
Long-term plasticity at VIP IN outputs
Reliable optogenetic activation of VIP INs
Because L2/3 VIP INs are mostly found in L2/3 (Supplementary Figure 3; Gonchar et al., 2007; Prönneke et al., 2015; Almási et al., 2019) and because L2/3 VIP INs are more homogeneous compared to other layers (Gouwens et al., 2020), we decided to target these INs for the subsequent experiments exploring long-term plasticity at VIP IN inputs and outputs. We first targeted L2/3 VIP INs for whole-cell recording and used blue laser light to explore whether we could reliably drive ChR2 to spike the cell. We found that blue laser light reliably evoked spiking up to 50 Hz (Supplementary Figure 6).
VIP IN→MC connections exhibited causal LTD
In acute slices from VIP-ChR2 mice, we targeted MCs for whole-cell recording and optogenetically activated presynaptic VIP INs to explore how plasticity of VIP IN→MC synapses depend on spike rate and timing (Figure 2A). We found that MC disinhibition was possible by inducing LTD at 50 Hz firing rate and causal timing difference of Δt = +10 ms (Figure 2B). In contrast, we found no plasticity at other tested timings and frequencies (pooled: 50 Hz and Δt = −10 ms, +25 ms; 20 Hz and Δt = ±25 ms) or in control experiments with no postsynaptic spiking (Figures 2C, D).
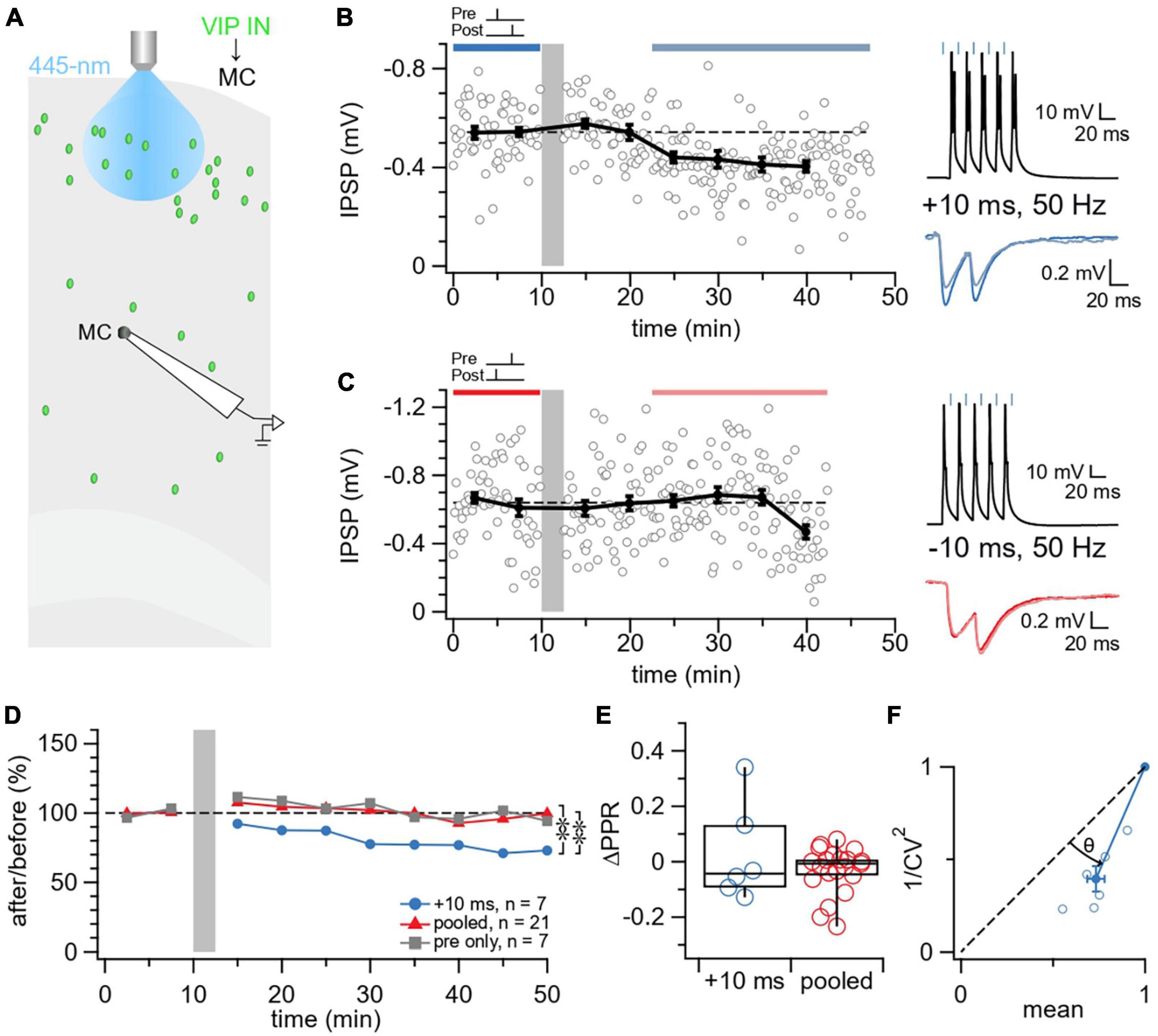
Figure 2. Causal STDP yielded LTD at VIP IN→MC synapses. (A) Schematic illustrating the patched L5 MC and laser-activated L2/3 VIP INs in the motor cortex. (B) Sample experiment from a whole-cell recording at a VIP IN→MC connection using an induction paradigm (gray bar) of 50 Hz, +10 ms revealed how LTD was elicited in the postsynaptic MC (after/before = 75%, p < 0.001). The dark blue bar (pre-pairing) and light blue bar (post-pairing) indicate the time window for plasticity quantification. Causal pre-pairing IPSPs are labeled in dark blue, causal post-pairing IPSPs are labeled in light blue. (C) Sample experiment from a whole-cell recording at a VIP IN→MC connection using an induction paradigm (gray bar) of 50 Hz, –10 ms showed that no change in IPSP response was elicited in the postsynaptic MC (after/before = 97%, p = 0.59). The red bar (pre-pairing) and pink bar (post-pairing) indicate the time window for plasticity quantification. Acausal pre-pairing IPSPs are labeled in red, acausal post-pairing IPSPs are labeled in pink. (D) Ensemble averages showed that causal LTD only occurs at VIP IN→MC synapses with an induction paradigm of 50 Hz, +10 ms (ANOVA p < 0.05; +10 ms: 78% ± 6%, n = 7 connections, N = 7 animals, vs. pooled: 98% ± 4%, n = 21 connections, N = 18 animals, t-test p < 0.01; +10 ms vs. pre only controls: 100% ± 4%, n = 7 connections, N = 7 animals, t-test p < 0.01; pooled vs. pre only controls, t-test p = 0.68). (E) VIP IN→MC LTD did not change ΔPPR compared to the pooled group (+10 ms: 0.027 ± 0.07 vs. pooled: –0.032 ± 0.02, Wilcoxon test p = 0.98), suggesting a postsynaptic locus of expression for plasticity. (F) For CV analysis, points below diagonal for VIP IN→MC LTD (Wilcoxon test, θ = 22° ± 2°, p < 0.01) suggests that IPSP suppression was due to a reduction in presynaptic release (Brock et al., 2020).
We then assessed the locus of expression of VIP IN→MC LTD. We utilized two independent methods for determining the pre- versus postsynaptic locus, synaptic response PPR (Blackman et al., 2013) and CV analysis (Brock et al., 2020). We found that VIP IN→MC LTD did not change ΔPPR compared to controls, suggesting a postsynaptic locus of expression (Figure 2E). In contrast with ΔPPR, VIP IN→MC LTD reduced 1/CV2 (Figure 2F), suggesting decreased presynaptic release (Blackman et al., 2013; Brock et al., 2020).
No STDP detected at VIP IN→BC connections
We next explored STDP at VIP IN→BC synapses. In our VIP-ChR2 mice, we targeted motor cortex L5 BCs for whole-cell recording and selectively activated L2/3 VIP INs using blue laser light (Figure 3A). Unlike VIP IN→MC synapses, we found that VIP IN→BC synapses exhibited no detectable STDP (Figures 3B–E).
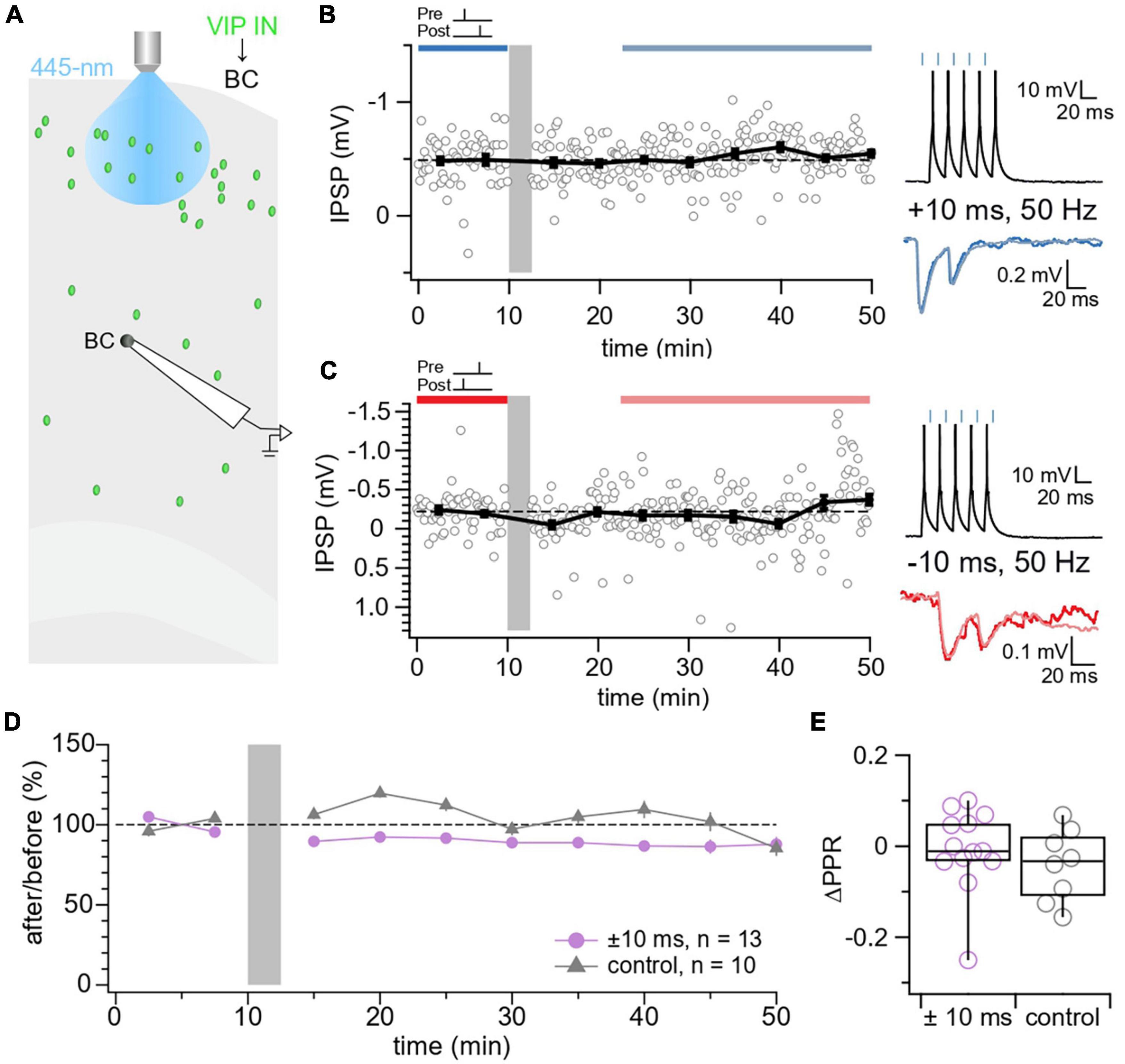
Figure 3. No STDP detected at VIP IN→BC synapses. (A) Schematic illustrating the patched L5 BC and laser-activated L2/3 VIP INs in the motor cortex. (B) Sample experiment from a whole-cell recording at a VIP IN→BC connection using an induction paradigm (gray bar) of 50 Hz, +10 ms revealed that no change in IPSP response was elicited in the postsynaptic BC (after/before = 106%, p = 0.27). The dark blue bar (pre-pairing) and light blue bar (post-pairing) indicate the time window for plasticity quantification. Causal pre-pairing IPSPs are labeled in dark blue, causal post-pairing IPSPs are labeled in light blue. (C) Sample experiment from a whole-cell recording at a VIP IN→BC connection using an induction paradigm (gray bar) of 50 Hz, –10 ms showed that no change in IPSP response was elicited in the postsynaptic BC (after/before = 93%, p = 0.75). The red bar (pre-pairing) and pink bar (post-pairing) indicate the time window for plasticity quantification. Acausal pre-pairing IPSPs are labeled in red, acausal post-pairing IPSPs are labeled in pink. (D) Ensemble averages showed that VIP IN→BC synapses did not undergo any detectable plasticity following our induction protocol ( ± 10 ms: 89% ± 6%, n = 13 connections, N = 13 animals vs. control: 102% ± 8%, n = 10 connections, N = 9 animals, t-test p = 0.22). Pre only (n = 5 connections, N = 4 animals) and post only (n = 5 connections, N = 5 animals) control conditions were indistinguishable (t-test p = 0.89) and were therefore pooled in one control group. (E) There was no change in ΔPPR at VIP IN→BC synapses ( ± 10 ms: –0.0075 ± 0.03 vs. control: –0.041 ± 0.03, t-test p = 0.35).
Post-hoc identification of MCs and BCs
MCs and BCs were identified based on their distinct electrophysiological and morphological properties (Tremblay et al., 2016). Compared to fast-spiking BCs, MCs had an adapting firing pattern with a lower rheobase, higher input resistance, larger spike half width, and longer membrane time constant (Supplementary Table 2). MCs had a single characteristically ascending axon and dangling dendrites, while BCs had highly locally branching axonal and dendritic arbors that were relatively radially symmetric (Supplementary Figure 7).
Long-term plasticity at VIP inputs
E→VIP IN connections exhibit LTP irrespective of temporal order
Next, we studied long-term plasticity at VIP IN inputs. We patched L2/3 VIP INs and used extracellular stimulation to readily recruit excitatory inputs onto VIP INs (Figure 4A). We found that E→VIP IN synapses were potentiated at 50 Hz firing rate with both causal and acausal timings of Δt = ±10 ms (Figures 4B–D).
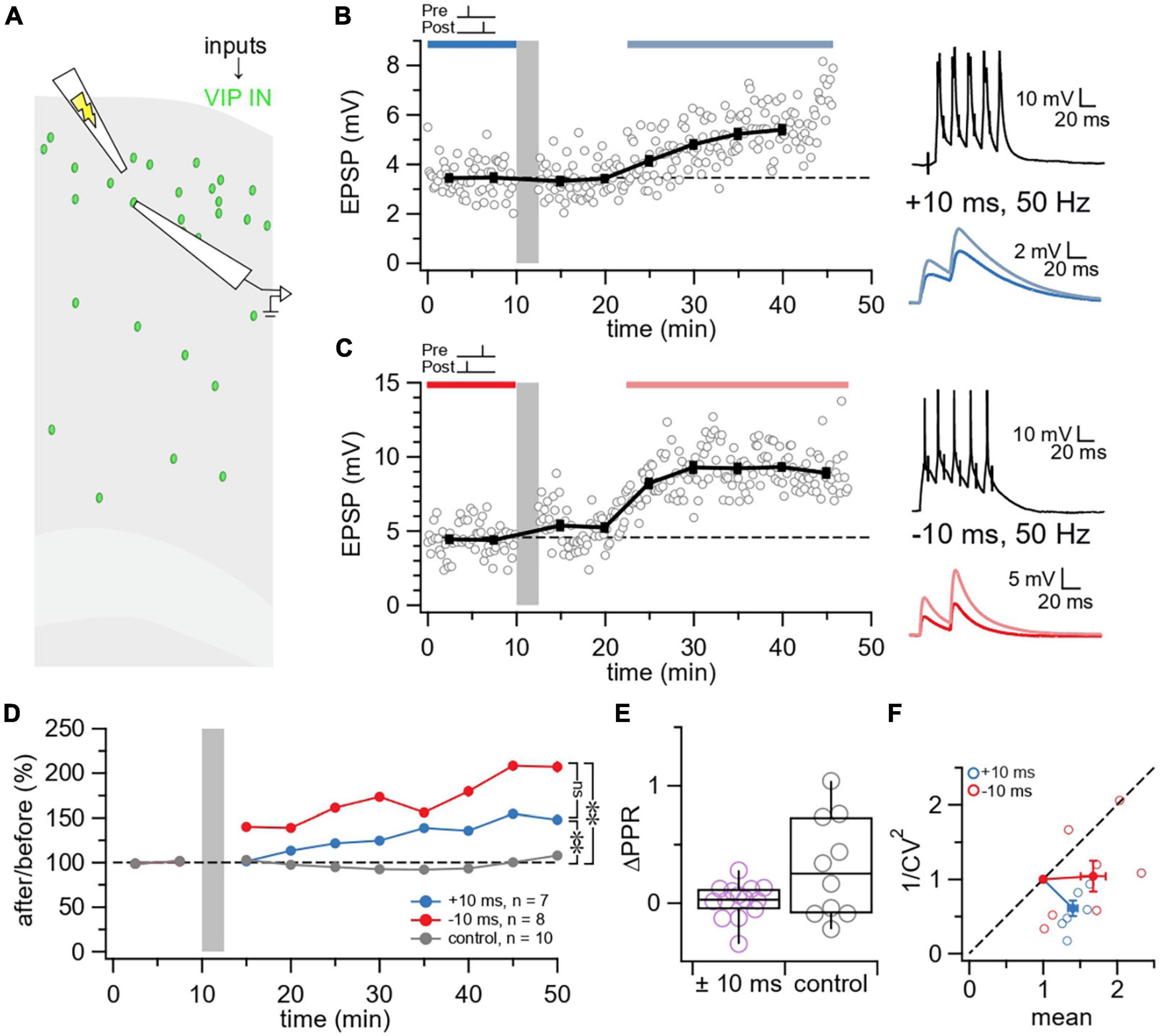
Figure 4. VIP inputs exhibit LTP irrespective of temporal order. (A) Schematic illustrating the activation of L2/3 VIP IN inputs using extracellular stimulation in the motor cortex. (B) Sample experiment from a whole-cell recording at a L2/3 VIP IN using an induction paradigm (gray bar) of 50 Hz, +10 ms revealed that LTP was elicited at this E→VIP IN synapse (after/before = 147%, p < 0.001). The dark blue bar (pre-pairing) and light blue bar (post-pairing) indicate the time window for plasticity quantification. Causal pre-pairing EPSPs are labeled in dark blue, causal post-pairing EPSPs are labeled in light blue. (C) Sample experiment from a whole-cell recording of a L2/3 VIP IN using an induction paradigm (gray bar) of 50 Hz, –10 ms revealed that LTP was elicited at this E→VIP IN synapse (after/before = 203%, p < 0.001). The red bar (pre-pairing) and pink bar (post-pairing) indicate the time window for plasticity quantification. Acausal pre-pairing EPSPs are labeled in red, acausal post-pairing EPSPs are labeled in pink. (D) Ensemble averages showed that LTP is induced at E→VIP IN synapses at 50 Hz, ± 10 ms (Brown-Forsythe ANOVA p < 0.01; +10 ms: 140% ± 6%, n = 7 connections, N = 4 animals vs. control: 99% ± 9%, n = 10 connections, N = 8 animals, Wilcoxon-Mann-Whitney test p < 0.01; –10 ms: 168% ± 17%, n = 8 connections, N = 7 animals vs. control, Wilcoxon-Mann-Whitney test p < 0.01; +10 ms vs. –10 ms, Wilcoxon-Mann-Whitney test p = 0.23). Pre only (n = 5 connections, N = 5 animals) and post only (n = 5 connections, N = 3 animals) control conditions were indistinguishable (Wilcoxon-Mann-Whitney test p = 0.15) and were therefore pooled in one control group. (E) There was no change in ΔPPR following E→VIP IN LTP ( ± 10 ms: 0.019 ± 0.04, n = 14 connections, N = 10 animals vs. control: 0.30 ± 0.1, n = 10 connections, N = 8 animals, Wilcoxon test p = 0.17). (F) For CV analysis, points below diagonal for E→VIP IN LTP (+10 ms: Wilcoxon test, θ = 266° ± 9°, p < 0.001; –10 ms: Wilcoxon test, θ = 234° ± 19°, p < 0.001) indicated that plasticity was postsynaptically expressed (Brock et al., 2020).
We then assessed whether E→VIP IN LTP was expressed presynaptically or postsynaptically. We found that E→VIP IN LTP did not change PPR (Figure 4E), in keeping with postsynaptically expressed LTP. In agreement, E→VIP IN LTP did not reduce 1/CV2 (Figure 4F), which also suggested a postsynaptic locus of expression (Blackman et al., 2013; Brock et al., 2020).
Discussion
VIP IN-mediated disinhibition has consistently been shown to boost learning and plasticity in cortical circuits (Fu et al., 2014; Fu et al., 2015; Adler et al., 2019). Yet, to our knowledge, no studies have explored the plasticity of VIP INs themselves. Here, we described the phenomenology of long-term plasticity at VIP IN inputs and outputs in the mouse motor cortex (Figure 5). We found that VIP IN→MC synapses underwent causal LTD, but we could not detect any STDP at VIP IN→BC synapses. On the input side, we found that E→VIP IN synapses potentiated for both causal and acausal timings. Taken together, our findings reveal that plasticity at VIP IN inputs and outputs is specific to synapse type.
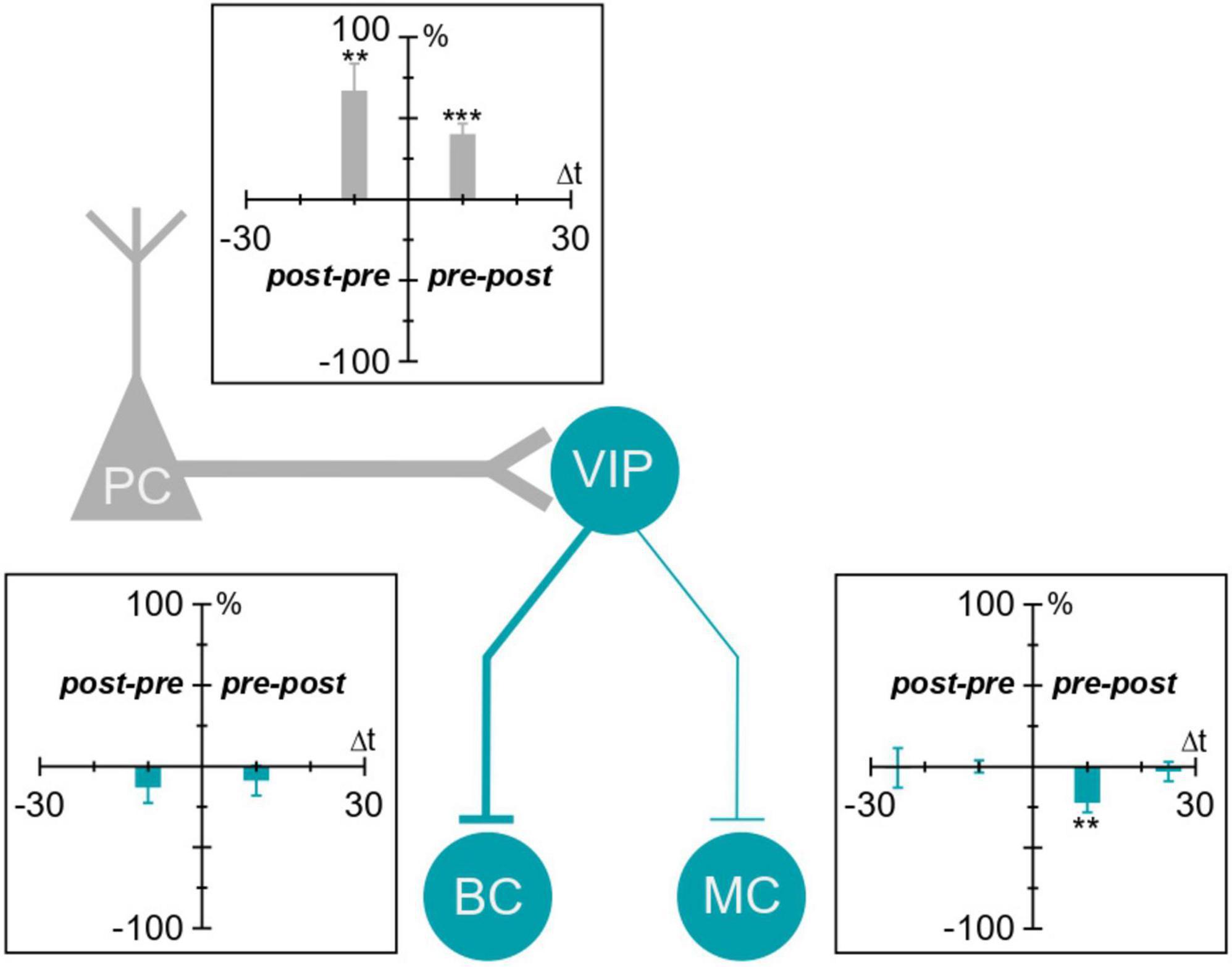
Figure 5. Plasticity of VIP IN inputs and outputs in the motor cortex. VIP IN inputs and outputs in the motor cortex undergo plasticity. We found that VIP IN inputs (labeled in gray) were potentiated with both causal and acausal timings. VIP IN outputs (labeled in blue) underwent causal LTD at VIP IN→MC synapses. VIP IN→BC synapses, on the other hand, did not exhibit any detectable plasticity. **p < 0.01, ***p < 0.001.
Synapse-type specificity
Synapse-type-specific plasticity has been reported throughout the brain (Larsen and Sjöström, 2015) and allows synapses to adapt differentially depending on factors such as target cell (Blackman et al., 2013) or functional role in the microcircuit (McFarlan et al., 2023). In our study, we found that plasticity outcomes at VIP IN outputs depend on synapse type. This synapse-type-specific plasticity has been reported at I→I synapses in L2/3 visual cortex, where presynaptic tetanic stimulation induced I→I LTP at PV IN→PV IN synapses but not at SST IN→PV IN synapses (Sarihi et al., 2012). Similarly, increased PC activity in L2/3 prefrontal cortex selectively potentiated SST IN→PC synapses but not PV IN→PC synapses (Chiu et al., 2018). Additionally, I→E synapses in the developing cortex exhibited different plasticity rules depending on the location of the postsynaptic cell. For example, auditory cortex L4 IN→L5 PC synapses exhibited causal LTP (Field et al., 2020), whereas visual cortex L4 BC→L4 PC synapses exhibited causal LTD that later switched to LTP after critical period sensory experience (Vickers et al., 2018). The switch in plasticity rules at L4 BC→L4 PC synapses suggested that these I→E synapses may be important for promoting plasticity, whereas L4 IN→L5 PC synapses may provide stability. Overall, these studies highlight the many factors that contribute to synapse-type-specific plasticity which include cell type, cell location, and synapse type.
In our study, plasticity was induced at VIP IN→MC synapses but not VIP IN→BC synapses. Considering that VIP INs weakly synapse with BCs compared to MCs (Pfeffer et al., 2013; Kepecs and Fishell, 2014; Tremblay et al., 2016; Apicella and Marchionni, 2022; Campagnola et al., 2022), it is possible that VIP INs and BCs do not have enough consistent coincident activity to induce STDP at VIP IN→BC synapses. Induction of plasticity at VIP IN→BC synapses may instead depend on pre- or postsynaptic activity alone. High frequency stimulation of PV INs, for example, resulted in E→I LTP and I→I LTP in the visual cortex (Sarihi et al., 2012; Chistiakova et al., 2019). Moreover, low frequency theta-burst stimulation at excitatory inputs onto hippocampal BCs resulted in E→I LTP, whereas high frequency theta-burst stimulation resulted in E→I LTD (Camiré and Topolnik, 2014). Other factors such as neuromodulators may be required for VIP IN→BC plasticity. It was demonstrated that the release of the neuropeptide enkephalin by hippocampal VIP INs long-term disinhibited CA2 PCs via I→E LTD at PV IN outputs. This VIP IN-mediated disinhibition allowed for enhanced information transfer between hippocampal CA3 and CA2 PCs, which was important for social memory storage (Leroy et al., 2022). Thus, this form of plasticity did not require the coincident firing of pre and postsynaptic cells, but rather a neuromodulator. Together, these results highlight the need to further explore different induction protocols to elucidate the plasticity rules that govern VIP IN→BC synapses.
VIP IN plasticity depends on spike timing
Though plasticity at a given synapse type depends on several factors including rate, timing, depolarization, and higher-order spiking statistics (Sjöström et al., 2001; Froemke and Dan, 2002; Froemke et al., 2006; Pfister and Gerstner, 2006), we focused here on how plasticity at VIP IN inputs and outputs depends on the relative timing of pre- and postsynaptic spiking. We found that LTD induction at VIP IN→MC synapses was sensitive to timing (Figure 2). At E→VIP IN synapses, however, LTP did not depend on the sign of relative timing, yet pre- or postsynaptic firing alone yielded no plasticity (Figure 4). This demonstrated that this form of LTP was still dependent on spike timing, a defining feature of STDP (Markram et al., 2012). Previous studies have additionally shown that STDP can be symmetric around the origin (Egger et al., 1999; Abbott and Nelson, 2000; Lu et al., 2007). Taken together, our findings highlight how timing is a key determinant of VIP IN STDP.
The locus of expression
The locus of expression of long-term plasticity is important because it carries with it computational implications (Costa et al., 2015; Costa et al., 2017; Mizusaki et al., 2022). Postsynaptically expressed plasticity generally alters only synaptic strength while leaving short-term dynamics unaffected, although there are exceptions to this rule (Poncer and Malinow, 2001). Presynaptically expressed long-term plasticity, however, typically alters both short-term plasticity and synaptic gain (Markram and Tsodyks, 1996; Sjöström et al., 2003; Sjöström et al., 2007). This is because during high-frequency spike trains, the readily releasable pool of vesicles is depleted, which causes short-term synaptic depression (Zucker and Regehr, 2002), although at some synapse, short-term facilitation dominates for other mechanistic reasons (Blackman et al., 2013). Either way, these short-term synaptic dynamics are generally strongly affected by pre- but not by postsynaptically expressed long-term plasticity. Short-term plasticity is functionally important as it filters information transferred by a synapse (Fortune and Rose, 2001; Fuhrmann et al., 2002). In this view, short-term facilitating connections act as high-pass filtering burst detectors (Maass and Zador, 1999; Matveev and Wang, 2000), while short-term depressing connections low-pass filter information for correlation detection and gain-control (Abbott et al., 1997; Rosenbaum et al., 2012). For instance, presynaptic LTP would be expected to increase the release probability, thereby depleting the readily-releasable pool of vesicles faster during high-frequency bursts. The ensuing increase in short-term depression thus biases the synapse toward correlation detection at the expense of burst detection (Blackman et al., 2013; Costa et al., 2017).
In our study, we analyzed PPR and CV to assess the locus of expression. For VIP IN→MC LTD, we found no change in PPR, suggesting a postsynaptic locus of expression (Figure 2E), whereas CV analysis suggested that plasticity was expressed presynaptically (Figure 2F; Brock et al., 2020). This apparent discrepancy could be explained by the paired-pulse stimulation frequency not being potent enough to sufficiently deplete the readily-releasable pool of vesicles, leading it to be inconclusive. With this interpretation, LTD at VIP IN→MC was presynaptically expressed. In this view, this form of LTD would thus be expected to alter the information filtering of VIP IN→MC synapses in addition to weakening them (Blackman et al., 2013; Costa et al., 2017).
Mechanistically, presynaptically expressed plasticity at VIP IN→MC synapses could be mediated by retrograde messengers like endocannabinoids (Castillo et al., 2012; Piette et al., 2020) or BDNF (Inagaki et al., 2008; Vickers et al., 2018). Additionally, given that the postsynaptic MC needs to be active in order to coincidentally fire with the presynaptic VIP IN, postsynaptic NMDA receptor signaling may be involved. Indeed, NMDA receptor signaling has been shown to control GABAA receptor stability at inhibitory synapses (Muir et al., 2010).
For inputs to VIP INs, on the other hand, ΔPPR and CV analysis agreed that E→VIP IN LTP had a postsynaptic locus of expression (Figure 4). The expression of postsynaptic E→E LTP is typically due to the insertion of postsynaptic AMPA receptors (Malinow and Malenka, 2002; Bredt and Nicoll, 2003; Kessels and Malinow, 2009). The NMDA receptor, which is particularly well suited for STDP given its capacity for coincidence detection, is also highly implicated in E→E LTP (Wong et al., 2021). Thus, AMPA receptor insertion and NMDA receptor signaling are strong candidates for the postsynaptic expression of E→I LTP at VIP IN inputs.
The consequences of VIP IN plasticity
To understand the consequences of long-term plasticity at VIP IN inputs and outputs, it is important to consider its impact at the circuit level (McFarlan et al., 2023). LTP of excitatory inputs to VIP INs promotes VIP IN activity, which by inhibiting BCs and MCs is expected to increase circuit activity overall. Consequently, it has been proposed that attention operates through cortical disinhibitory circuits, e.g., by neuromodulation or by top-down control (Kullander and Topolnik, 2021; Speed and Haider, 2021). It has been argued that cholinergic modulation specifically of VIP INs is required for attention (Obermayer et al., 2019), although some disagree and propose that VIP IN activation is orthogonal to attention (Myers-Joseph et al., 2023). Regardless, we show here how activity-driven VIP IN plasticity can potentially contribute to such attentional effects.
The plasticity of VIP INs may also more generally regulate learning. For instance, VIP IN-mediated suppression of SST INs has been correlated with improved motor learning in the motor cortex (Adler et al., 2019) as well as enhanced adult plasticity in the visual cortex (Fu et al., 2014; Fu et al., 2015). Thus, E→I LTP at VIP IN inputs may help to boost learning and plasticity in cortical circuits.
In addition, VIP IN→MC LTD is expected to increase inhibition specifically of PC dendrites (Wang et al., 2004). Although E→VIP IN LTP and VIP IN→MC LTD may superficially seem to oppose each other if triggered simultaneously, they would additionally redistribute inhibition across the somato-dendritic axis of pyramidal cells (Pouille and Scanziani, 2004; Blackman et al., 2013). According to influential theoretical frameworks on cortical associative learning (Larkum, 2013), such dendritic inhibition is expected to regulate neocortical information storage.
As VIP INs are able to mediate disinhibition (Artinian and Lacaille, 2018; Kullander and Topolnik, 2021), they are ideally positioned as key regulators of activity in local circuits, with implications for disease states such as epilepsy (Cunha-Reis and Caulino-Rocha, 2020). For example, it has been reported that, whereas inhibition of SST INs prolongs seizures, inhibition of VIP INs reduces seizure propensity (Khoshkhoo et al., 2017). Consequently, the protective role of VIP IN→MC LTD might be possible to harness as a therapy for epilepsy.
Caveats
One caveat in our study is the discrepancy between ΔPPR and CV analysis for determining the locus of expression of VIP IN→MC LTD. CV analysis suggested that LTD was expressed presynaptically, whereas PPR analysis suggested it was expressed postsynaptically. This discrepancy between CV and PPR analyses could occur if the 30-Hz stimulation was not sufficiently high to deplete the readily releasable pool. It is possible that depleting VIP IN→MC synapses better would reveal a change in PPR.
Another potential caveat with our experimental paradigm comes from the use of ChR2 to activate presynaptic VIP INs. Since ChR2 fluxes calcium (Nagel et al., 2003), it may directly trigger release at synaptic boutons. ChR2 may also depolarize synaptic terminals, again contributing to release. This artifact, if present, would be expected to elevate release and increase short-term depression. Consequently, this may be particularly important when using PPR as a measure for determining the locus of plasticity expression at VIP IN outputs, whereas CV analysis may be less affected. However, given the many hundreds of micrometer distance between presynaptic laser stimulation and postsynaptic cells where VIP IN synapses form (Figures 2A, 3A), this artifact seemed unlikely.
Furthermore, the lack of single-cell resolution with respect to presynaptic cell activation — we likely optogenetically stimulated more than one presynaptic L2/3 VIP IN at a time. An alternative approach for unitary synapse resolution would be paired recordings (Lalanne et al., 2016). However, given appreciable distance between L2/3 and L5, the sparsity of VIP INs in the cortex (Supplementary Figure 3), and the overall low connectivity of VIP IN outputs (Walker et al., 2016), paired recordings would be an impractical tool. Another alternative approach for unitary synapse resolution is by using 2P optogenetic activation (Chou et al., 2023), which in the future should enable the study of long-term plasticity at multiple VIP IN→MC/BC synapses in parallel.
Future directions
To further our understanding of how disinhibitory plasticity impacts the healthy brain as well as neuropathologies, future research will need to explore the plasticity at the many different I→I and I→I→E synapses, to contribute to the plasticitome of cortical INs (McFarlan et al., 2023). Because it is challenging to explore many different synapse types, this effort will likely require new high-throughput plasticity-mapping approaches (Sjöström, 2021).
We relied here on the STDP experimental paradigm, which is widely believed to be biologically plausible (Markram et al., 2011; Feldman, 2012; Markram et al., 2012). However, some have disagreed and instead argued that STDP is of limited biological relevance (Lisman and Spruston, 2005; Lisman and Spruston, 2010). On a related note, due to the experimental challenges associated with targeting these specific synapse types, we could only explore a limited plasticity induction parameter space in this study. Induction protocols that e.g., rely on local dendritic spikes (Holthoff et al., 2004; Remy and Spruston, 2007; Bittner et al., 2017) may thus reveal plasticity at VIP IN→BC synapses. We also did not explore neuromodulation, which may additionally gate plasticity (Pawlak et al., 2010), even synapse specifically for the same cell type (Edelmann et al., 2017). Our study is thus not a final verdict on VIP IN plasticity, but rather a starting point.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by the Montreal General Hospital Facility Animal Care Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
ARM: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Resources, Software, Visualization, Writing – original draft, Writing – review and editing, Project administration, Supervision, Validation. CG: Formal analysis, Funding acquisition, Investigation, Writing – review and editing, Data curation, Methodology. IG: Formal analysis, Investigation, Writing – review and editing, Data curation, Methodology. CW: Formal analysis, Investigation, Writing – review and editing, Data curation, Methodology. TAL: Formal analysis, Funding acquisition, Investigation, Writing – review and editing, Data curation, Methodology. PJS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. ARM was supported by doctoral awards from FRQS (287520) and HBHL. CG won NSERC USRA, FRQNT BPC, and RI-MUHC studentships. TAL was funded by an NSERC USRA award. PJS acknowledges funding from CFI LOF 28331, CIHR PG 156223, FRSQ CB 254033, and NSERC DG/DAS 2017-04730 as well as 2017-507818. The Montreal General Hospital Foundation kindly funded the Chameleon ULTRA II laser.
Acknowledgments
The authors thank Hovy Wong, Christina Chou, and Sjöström laboratory members for help and useful discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2024.1389094/full#supplementary-material
Footnotes
References
Abbott, L. F., and Nelson, S. B. (2000). Synaptic plasticity: Taming the beast. Nat. Neurosci. 3(Suppl.), 1178–1183. doi: 10.1038/81453
Abbott, L. F., Varela, J. A., Sen, K., and Nelson, S. B. (1997). Synaptic depression and cortical gain control. Science 275, 220–224.
Adler, A., Zhao, R., Shin, M. E., Yasuda, R., and Gan, W. B. (2019). Somatostatin-expressing interneurons enable and maintain learning-dependent sequential activation of pyramidal neurons. Neuron 102, 202–216.e7. doi: 10.1016/j.neuron.2019.01.036
Almási, Z., David, C., Witte, M., and Staiger, J. F. (2019). Distribution patterns of three molecularly defined classes of GABAergic neurons across columnar compartments in mouse barrel cortex. Front. Neuroanat. 13:45. doi: 10.3389/fnana.2019.00045
Apicella, A. J., and Marchionni, I. (2022). VIP-expressing GABAergic neurons: Disinhibitory vs. Inhibitory motif and its role in communication across neocortical areas. Front. Cell Neurosci. 16:811484. doi: 10.3389/fncel.2022.811484
Artinian, J., and Lacaille, J. C. (2018). Disinhibition in learning and memory circuits: New vistas for somatostatin interneurons and long-term synaptic plasticity. Brain Res. Bull. 141, 20–26. doi: 10.1016/j.brainresbull.2017.11.012
Ascoli, G. A., Alonso-Nanclares, L., Anderson, S. A., Barrionuevo, G., Benavides-Piccione, R., Burkhalter, A., et al. (2008). Petilla terminology: Nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat. Rev. Neurosci. 9, 557–568. doi: 10.1038/nrn2402
Bi, G. Q., and Poo, M. M. (1998). Synaptic modifications in cultured hippocampal neurons: Dependence on spike timing, synaptic strength, and postsynaptic cell type. J. Neurosci. 18, 10464–10472.
Bittner, K. C., Milstein, A. D., Grienberger, C., Romani, S., and Magee, J. C. (2017). Behavioral time scale synaptic plasticity underlies CA1 place fields. Science 357, 1033–1036.
Blackman, A. V., Abrahamsson, T., Costa, R. P., Lalanne, T., and Sjöström, P. J. (2013). Target cell-specific short-term plasticity in local circuits. Front. Synapt. Neurosci. 5:11. doi: 10.3389/fnsyn.2013.00011
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 361, 31–39.
Bredt, D. S., and Nicoll, R. A. (2003). AMPA receptor trafficking at excitatory synapses. Neuron 40, 361–379.
Brock, J. A., Thomazeau, A., Watanabe, A., Li, S. S. Y., and Sjöström, P. J. (2020). A practical guide to using CV analysis for determining the locus of synaptic plasticity. Front. Synapt. Neurosci. 12:11. doi: 10.3389/fnsyn.2020.00011
Buchanan, K. A., Blackman, A. V., Moreau, A. W., Elgar, D., Costa, R. P., Lalanne, T., et al. (2012). Target-specific expression of presynaptic NMDA receptors in neocortical microcircuits. Neuron 75, 451–466. doi: 10.1016/j.neuron.2012.06.017
Camiré, O., and Topolnik, L. (2014). Dendritic calcium nonlinearities switch the direction of synaptic plasticity in fast-spiking interneurons. J. Neurosci. 34, 3864–3877. doi: 10.1523/JNEUROSCI.2253-13.2014
Campagnola, L., Seeman, S. C., Chartrand, T., Kim, L., Hoggarth, A., Gamlin, C., et al. (2022). Local connectivity and synaptic dynamics in mouse and human neocortex. Science 375:eabj5861.
Castillo, P. E., Younts, T. J., Chavez, A. E., and Hashimotodani, Y. (2012). Endocannabinoid signaling and synaptic function. Neuron 76, 70–81.
Chistiakova, M., Ilin, V., Roshchin, M., Bannon, N., Malyshev, A., Kisvarday, Z., et al. (2019). Distinct heterosynaptic plasticity in fast spiking and non-fast-spiking inhibitory neurons in rat visual cortex. J. Neurosci. 39, 6865–6878. doi: 10.1523/JNEUROSCI.3039-18.2019
Chiu, C. Q., Martenson, J. S., Yamazaki, M., Natsume, R., Sakimura, K., Tomita, S., et al. (2018). Input-specific NMDAR-dependent potentiation of dendritic GABAergic inhibition. Neuron 97, 368-377.e3. doi: 10.1016/j.neuron.2017.12.032
Chou, C. Y. C., Wong, H. H. W., Guo, C., Boukoulou, K. E., Huang, C., Jannat, J., et al. (2023). Principles of visual cortex excitatory microcircuit organization. bioRxiv [Preprint] doi: 10.1101/2023.12.30.573666
Cline, H. T. (1998). Topographic maps: Developing roles of synaptic plasticity. Curr. Biol. 8, R836–R839.
Costa, R. P., Froemke, R. C., Sjöström, P. J., and van Rossum, M. C. W. (2015). Unified pre- and postsynaptic long-term plasticity enables reliable and flexible learning. eLife 4:e09457.
Costa, R. P., Mizusaki, B. E., Sjöström, P. J., and van Rossum, M. C. (2017). Functional consequences of pre- and postsynaptic expression of synaptic plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci 372:20160153.
Cunha-Reis, D., and Caulino-Rocha, A. (2020). VIP modulation of hippocampal synaptic plasticity: A role for VIP receptors as therapeutic targets in cognitive decline and mesial temporal lobe epilepsy. Front. Cell Neurosci. 14:153. doi: 10.3389/fncel.2020.00153
D’Amour, J. A., and Froemke, R. C. (2015). Inhibitory and excitatory spike-timing-dependent plasticity in the auditory cortex. Neuron 86, 514–528.
Debanne, D., Gahwiler, B. H., and Thompson, S. M. (1994). Asynchronous pre- and postsynaptic activity induces associative long-term depression in area CA1 of the rat hippocampus in vitro. Proc. Natl. Acad. Sci. U. S. A. 91, 1148–1152.
Edelmann, E., Cepeda-Prado, E., and Lessmann, V. (2017). Coexistence of multiple types of synaptic plasticity in individual hippocampal CA1 pyramidal neurons. Front. Synapt. Neurosci. 9:7. doi: 10.3389/fnsyn.2017.00007
Egger, V., Feldmeyer, D., and Sakmann, B. (1999). Coincidence detection and changes of synaptic efficacy in spiny stellate neurons in rat barrel cortex. Nat. Neurosci. 2, 1098–1105. doi: 10.1038/16026
Field, R. E., D’Amour, J. A., Tremblay, R., Miehl, C., Rudy, B., Gjorgjieva, J., et al. (2020). Heterosynaptic plasticity determines the set point for cortical excitatory-inhibitory balance. Neuron 106, 842–854.e4. doi: 10.1016/j.neuron.2020.03.002
Fortune, E. S., and Rose, G. J. (2001). Short-term synaptic plasticity as a temporal filter. Trends Neurosci. 24, 381–385.
Froemke, R. C., and Dan, Y. (2002). Spike-timing-dependent synaptic modification induced by natural spike trains. Nature 416, 433–438.
Froemke, R. C., Merzenich, M. M., and Schreiner, C. E. (2007). A synaptic memory trace for cortical receptive field plasticity. Nature 450, 425–429. doi: 10.1038/nature06289
Froemke, R. C., Tsay, I. A., Raad, M., Long, J. D., and Dan, Y. (2006). Contribution of individual spikes in burst-induced long-term synaptic modification. J. Neurophysiol. 95, 1620–1629. doi: 10.1152/jn.00910.2005
Fu, Y., Kaneko, M., Tang, Y., Alvarez-Buylla, A., and Stryker, M. P. (2015). A cortical disinhibitory circuit for enhancing adult plasticity. Elife 4:e05558. doi: 10.7554/eLife.05558
Fu, Y., Tucciarone, J. M., Espinosa, J. S., Sheng, N., Darcy, D. P., Nicoll, R. A., et al. (2014). A cortical circuit for gain control by behavioral state. Cell 156, 1139–1152.
Fuhrmann, G., Segev, I., Markram, H., and Tsodyks, M. (2002). Coding of temporal information by activity-dependent synapses. J. Neurophysiol. 87, 140–148.
Gonchar, Y., Wang, Q., and Burkhalter, A. (2007). Multiple distinct subtypes of GABAergic neurons in mouse visual cortex identified by triple immunostaining. Front. Neuroanat. 1:3. doi: 10.3389/neuro.05.003.2007
Gouwens, N. W., Sorensen, S. A., Baftizadeh, F., Budzillo, A., Lee, B. R., Jarsky, T., et al. (2020). Integrated morphoelectric and transcriptomic classification of cortical GABAergic cells. Cell 183, 935–953.e19.
Holmgren, C. D., and Zilberter, Y. (2001). Coincident spiking activity induces long-term changes in inhibition of neocortical pyramidal cells. J. Neurosci. 21, 8270–8277. doi: 10.1523/JNEUROSCI.21-20-08270.2001
Holthoff, K., Kovalchuk, Y., Yuste, R., and Konnerth, A. (2004). Single-shock LTD by local dendritic spikes in pyramidal neurons of mouse visual cortex. J. Physiol. 560, 27–36. doi: 10.1113/jphysiol.2004.072678
Inagaki, T., Begum, T., Reza, F., Horibe, S., Inaba, M., Yoshimura, Y., et al. (2008). Brain-derived neurotrophic factor-mediated retrograde signaling required for the induction of long-term potentiation at inhibitory synapses of visual cortical pyramidal neurons. Neurosci. Res. 61, 192–200. doi: 10.1016/j.neures.2008.02.006
Kaneko, M., and Stryker, M. P. (2014). Sensory experience during locomotion promotes recovery of function in adult visual cortex. Elife 3:e02798. doi: 10.7554/eLife.02798
Katz, L. C., and Shatz, C. J. (1996). Synaptic activity and the construction of cortical circuits. Science 274, 1133–1138.
Kepecs, A., and Fishell, G. (2014). Interneuron cell types are fit to function. Nature 505, 318–326.
Kessels, H. W., and Malinow, R. (2009). Synaptic AMPA receptor plasticity and behavior. Neuron 61, 340–350.
Khoshkhoo, S., Vogt, D., and Sohal, V. S. (2017). Dynamic, cell-type-specific roles for GABAergic interneurons in a mouse model of optogenetically inducible seizures. Neuron 93, 291–298. doi: 10.1016/j.neuron.2016.11.043
Kullander, K., and Topolnik, L. (2021). Cortical disinhibitory circuits: Cell types, connectivity and function. Trends Neurosci. 44, 643–657.
Lalanne, T., Abrahamsson, T., and Sjöström, P. J. (2016). Using multiple whole-cell recordings to study spike-timing-dependent plasticity in acute neocortical slices. Cold Spring Harb. Protoc. 2016:pdb prot091306. doi: 10.1101/pdb.prot091306
Larkum, M. (2013). A cellular mechanism for cortical associations: An organizing principle for the cerebral cortex. Trends Neurosci. 36, 141–151.
Larsen, R. S., and Sjöström, P. J. (2015). Synapse-type-specific plasticity in local circuits. Curr. Opin. Neurobiol. 35, 127–135.
Lein, E. S., Hawrylycz, M. J., Ao, N., Ayres, M., Bensinger, A., Bernard, A., et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176.
Leroy, F., de Solis, C. A., Boyle, L. M., Bock, T., Lofaro, O. M., Buss, E. W., et al. (2022). Enkephalin release from VIP interneurons in the hippocampal CA2/3a region mediates heterosynaptic plasticity and social memory. Mol. Psychiatry 27, 2879–2900. doi: 10.1038/s41380-021-01124-y
Letzkus, J. J., Wolff, S. B., and Luthi, A. (2015). Disinhibition, a circuit mechanism for associative learning and memory. Neuron 88, 264–276.
Letzkus, J. J., Wolff, S. B., Meyer, E. M., Tovote, P., Courtin, J., Herry, C., et al. (2011). A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature 480, 331–335. doi: 10.1038/nature10674
Lisman, J., and Spruston, N. (2005). Postsynaptic depolarization requirements for LTP and LTD: A critique of spike timing-dependent plasticity. Nat. Neurosci. 8, 839–841. doi: 10.1038/nn0705-839
Lisman, J., and Spruston, N. (2010). Questions About STDP as a general model of synaptic plasticity. Front. Synapt. Neurosci. 2:140. doi: 10.3389/fnsyn.2010.00140
Lowel, S., and Singer, W. (1992). Selection of intrinsic horizontal connections in the visual cortex by correlated neuronal activity. Science 255, 209–212.
Lu, J. T., Li, C. Y., Zhao, J. P., Poo, M. M., and Zhang, X. H. (2007). Spike-timing-dependent plasticity of neocortical excitatory synapses on inhibitory interneurons depends on target cell type. J. Neurosci. 27, 9711–9720.
Maass, W., and Zador, A. M. (1999). Dynamic stochastic synapses as computational units. Neural Comput. 11, 903–917.
Maffei, A., Nataraj, K., Nelson, S. B., and Turrigiano, G. G. (2006). Potentiation of cortical inhibition by visual deprivation. Nature 443, 81–84.
Malinow, R., and Malenka, R. C. (2002). AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126.
Markram, H., Gerstner, W., and Sjöström, P. J. (2011). A history of spike-timing-dependent plasticity. Front. Synapt. Neurosci. 3:4. doi: 10.3389/fnsyn.2011.00004
Markram, H., Gerstner, W., and Sjöström, P. J. (2012). Spike-timing-dependent plasticity: A comprehensive overview. Front. Synapt. Neurosci. 4:2. doi: 10.3389/fnsyn.2012.00002
Markram, H., Lübke, J., Frotscher, M., and Sakmann, B. (1997). Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275, 213–215. doi: 10.1126/science.275.5297.213
Markram, H., and Tsodyks, M. (1996). Redistribution of synaptic efficacy between neocortical pyramidal neurons. Nature 382, 807–810.
Matveev, V., and Wang, X. J. (2000). Differential short-term synaptic plasticity and transmission of complex spike trains: To depress or to facilitate? Cereb. Cortex 10, 1143–1153.
McFarlan, A. R., Chou, C. Y. C., Watanabe, A., Cherepacha, N., Haddad, M., Owens, H., et al. (2023). The plasticitome of cortical interneurons. Nat. Rev. Neurosci. 24, 80–97. doi: 10.1038/s41583-022-00663-9
Mizusaki, B. E. P., Li, S. S. Y., Costa, R. P., and Sjöström, P. J. (2022). Pre- and postsynaptically expressed spike-timing-dependent plasticity contribute differentially to neuronal learning. PLoS Comput. Biol. 18:e1009409. doi: 10.1371/journal.pcbi.1009409
Muir, J., Arancibia-Carcamo, I. L., MacAskill, A. F., Smith, K. R., Griffin, L. D., and Kittler, J. T. (2010). NMDA receptors regulate GABAA receptor lateral mobility and clustering at inhibitory synapses through serine 327 on the gamma2 subunit. Proc. Natl. Acad. Sci. U. S. A. 107, 16679–16684. doi: 10.1073/pnas.1000589107
Myatt, D. R., Hadlington, T., Ascoli, G. A., and Nasuto, S. J. (2012). Neuromantic - from semi-manual to semi-automatic reconstruction of neuron morphology. Front. Neuroinform. 6:4. doi: 10.3389/fninf.2012.00004
Myers-Joseph, D., Wilmes, K. A., Fernandez-Otero, M., Clopath, C., and Khan, A. G. (2023). Disinhibition by VIP interneurons is orthogonal to cross-modal attentional modulation in primary visual cortex. Neuron 112, 628–645.e7. doi: 10.1016/j.neuron.2023.11.006
Nabavi, S., Fox, R., Proulx, C. D., Lin, J. Y., Tsien, R. Y., and Malinow, R. (2014). Engineering a memory with LTD and LTP. Nature 511, 348–352.
Nagel, G., Szellas, T., Huhn, W., Kateriya, S., Adeishvili, N., Berthold, P., et al. (2003). Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. U. S. A. 100, 13940–13945.
Niell, C. M., and Stryker, M. P. (2010). Modulation of visual responses by behavioral state in mouse visual cortex. Neuron 65, 472–479. doi: 10.1016/j.neuron.2010.01.033
Obermayer, J., Luchicchi, A., Heistek, T. S., de Kloet, S. F., Terra, H., Bruinsma, B., et al. (2019). Prefrontal cortical ChAT-VIP interneurons provide local excitation by cholinergic synaptic transmission and control attention. Nat. Commun. 10:5280.
Ormond, J., and Woodin, M. A. (2009). Disinhibition mediates a form of hippocampal long-term potentiation in area CA1. PLoS One 4:e7224. doi: 10.1371/journal.pone.0007224
Ormond, J., and Woodin, M. A. (2011). Disinhibition-mediated LTP in the hippocampus is synapse specific. Front. Cell Neurosci. 5:17. doi: 10.3389/fncel.2011.00017
Pawlak, V., Wickens, J. R., Kirkwood, A., and Kerr, J. N. D. (2010). Timing is not everything: Neuromodulation opens the STDP Gate. Front. Synapt. Neurosci. 2:146. doi: 10.3389/fnsyn.2010.00146
Pfeffer, C. K., Xue, M., He, M., Huang, Z. J., and Scanziani, M. (2013). Inhibition of inhibition in visual cortex: The logic of connections between molecularly distinct interneurons. Nat. Neurosci. 16, 1068–1076. doi: 10.1038/nn.3446
Pfister, J. P., and Gerstner, W. (2006). Triplets of spikes in a model of spike timing-dependent plasticity. J. Neurosci. 26, 9673–9682.
Piette, C., Cui, Y., Gervasi, N., and Venance, L. (2020). Lights on endocannabinoid-mediated synaptic potentiation. Front. Mol. Neurosci. 13:132.
Poncer, J. C., and Malinow, R. (2001). Postsynaptic conversion of silent synapses during LTP affects synaptic gain and transmission dynamics. Nat. Neurosci. 4, 989–996. doi: 10.1038/nn719
Pouille, F., and Scanziani, M. (2004). Routing of spike series by dynamic circuits in the hippocampus. Nature 429, 717–723. doi: 10.1038/nature02615
Prönneke, A., Scheuer, B., Wagener, R. J., Möck, M., Witte, M., and Staiger, J. F. (2015). Characterizing VIP neurons in the barrel cortex of VIPcre/tdTomato mice reveals layer-specific differences. Cereb. Cortex 25, 4854–4868. doi: 10.1093/cercor/bhv202
Remy, S., and Spruston, N. (2007). Dendritic spikes induce single-burst long-term potentiation. Proc. Natl. Acad. Sci. U. S. A. 104, 17192–17197.
Ren, C., Peng, K., Yang, R., Liu, W., Liu, C., and Komiyama, T. (2022). Global and subtype-specific modulation of cortical inhibitory neurons regulated by acetylcholine during motor learning. Neuron 110, 2334–2350 e8. doi: 10.1016/j.neuron.2022.04.031
Rosenbaum, R., Rubin, J., and Doiron, B. (2012). Short term synaptic depression imposes a frequency dependent filter on synaptic information transfer. PLoS Comput. Biol. 8:e1002557. doi: 10.1371/journal.pcbi.1002557
Sarihi, A., Mirnajafi-Zadeh, J., Jiang, B., Sohya, K., Safari, M. S., Arami, M. K., et al. (2012). Cell type-specific, presynaptic LTP of inhibitory synapses on fast-spiking GABAergic neurons in the mouse visual cortex. J. Neurosci. 32, 13189–13199. doi: 10.1523/JNEUROSCI.1386-12.2012
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Sholl, D. A. (1953). Dendritic organization in the neurons of the visual and motor cortices of the cat. J. Anat. 87, 387–406.
Silberberg, G., and Markram, H. (2007). Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron 53, 735–746.
Sippy, T., and Yuste, R. (2013). Decorrelating action of inhibition in neocortical networks. J. Neurosci. 33, 9813–9830.
Sjöström, P. J. (2021). Grand challenge at the frontiers of synaptic neuroscience. Front. Synapt. Neurosci. 13, 748937. doi: 10.3389/fnsyn.2021.748937
Sjöström, P. J., Turrigiano, G. G., and Nelson, S. B. (2001). Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron 32, 1149–1164. doi: 10.1016/s0896-6273(01)00542-6
Sjöström, P. J., Turrigiano, G. G., and Nelson, S. B. (2003). Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39, 641–654. doi: 10.1016/s0896-6273(03)00476-8
Sjöström, P. J., Turrigiano, G. G., and Nelson, S. B. (2007). Multiple forms of long-term plasticity at unitary neocortical layer 5 synapses. Neuropharmacology 52, 176–184. doi: 10.1016/j.neuropharm.2006.07.021
Song, S., and Abbott, L. F. (2001). Cortical development and remapping through spike timing-dependent plasticity. Neuron 32, 339–350. doi: 10.1016/s0896-6273(01)00451-2
Song, S., Miller, K. D., and Abbott, L. F. (2000). Competitive Hebbian learning through spike-timing-dependent synaptic plasticity. Nat. Neurosci. 3, 919–926.
Speed, A., and Haider, B. (2021). Probing mechanisms of visual spatial attention in mice. Trends Neurosci. 44, 822–836. doi: 10.1016/j.tins.2021.07.009
Stent, G. S. (1973). A physiological mechanism for Hebb’s postulate of learning. Proc. Natl. Acad. Sci. U. S. A. 70, 997–1001.
Taniguchi, H., He, M., Wu, P., Kim, S., Paik, R., Sugino, K., et al. (2011). A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013. doi: 10.1016/j.neuron.2011.07.026
Tremblay, R., Lee, S., and Rudy, B. (2016). GABAergic interneurons in the neocortex: From cellular properties to circuits. Neuron 91, 260–292.
Udakis, M., Pedrosa, V., Chamberlain, S. E. L., Clopath, C., and Mellor, J. R. (2020). Interneuron-specific plasticity at parvalbumin and somatostatin inhibitory synapses onto CA1 pyramidal neurons shapes hippocampal output. Nat. Commun. 11:4395. doi: 10.1038/s41467-020-18074-8
Vickers, E. D., Clark, C., Osypenko, D., Fratzl, A., Kochubey, O., Bettler, B., et al. (2018). Parvalbumin-interneuron output synapses show spike-timing-dependent plasticity that contributes to auditory map remodeling. Neuron 99:e726. doi: 10.1016/j.neuron.2018.07.018
Vogels, T. P., Sprekeler, H., Zenke, F., Clopath, C., and Gerstner, W. (2011). Inhibitory plasticity balances excitation and inhibition in sensory pathways and memory networks. Science 334, 1569–1573.
Walker, F., Mock, M., Feyerabend, M., Guy, J., Wagener, R. J., Schubert, D., et al. (2016). Parvalbumin- and vasoactive intestinal polypeptide-expressing neocortical interneurons impose differential inhibition on Martinotti cells. Nat. Commun. 7:13664. doi: 10.1038/ncomms13664
Wang, Y., Toledo-Rodriguez, M., Gupta, A., Wu, C., Silberberg, G., Luo, J., et al. (2004). Anatomical, physiological and molecular properties of Martinotti cells in the somatosensory cortex of the juvenile rat. J. Physiol. 561, 65–90. doi: 10.1113/jphysiol.2004.073353
Wong, H. H., Rannio, S., Jones, V., Thomazeau, A., and Sjöström, P. J. (2021). NMDA receptors in axons: There’s no coincidence. J. Physiol. 599, 367–387. doi: 10.1113/JP280059
Woodin, M. A., Ganguly, K., and Poo, M.-M. (2003). Coincident Pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl- transporter activity. Neuron 39, 807–820. doi: 10.1016/s0896-6273(03)00507-5
Yazaki-Sugiyama, Y., Kang, S., Cateau, H., Fukai, T., and Hensch, T. K. (2009). Bidirectional plasticity in fast-spiking GABA circuits by visual experience. Nature 462, 218–221. doi: 10.1038/nature08485
Zhou, L., Nho, K., Haddad, M. G., Cherepacha, N., Tubeleviciute-Aydin, A., Tsai, A. P., et al. (2021). Rare CASP6N73T variant associated with hippocampal volume exhibits decreased proteolytic activity, synaptic transmission defect, and neurodegeneration. Sci. Rep. 11:12695.
Keywords: VIP, inhibitory interneurons, plasticity, spike-timing-dependent plasticity, motor cortex
Citation: McFarlan AR, Guo C, Gomez I, Weinerman C, Liang TA and Sjöström PJ (2024) The spike-timing-dependent plasticity of VIP interneurons in motor cortex. Front. Cell. Neurosci. 18:1389094. doi: 10.3389/fncel.2024.1389094
Received: 20 February 2024; Accepted: 09 April 2024;
Published: 19 April 2024.
Edited by:
Volkmar Lessmann, University Hospital Magdeburg, GermanyReviewed by:
Björn Kampa, RWTH Aachen University, GermanyDominique Debanne, Unité de Neurobiologie des canaux Ioniques et de la Synapse (UNIS), France
Copyright © 2024 McFarlan, Guo, Gomez, Weinerman, Liang and Sjöström. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: P. Jesper Sjöström, amVzcGVyLnNqb3N0cm9tQG1jZ2lsbC5jYQ==