 Jarnail Singh
Jarnail Singh Michelle R. Randle
Michelle R. Randle Bradley J. Walters
Bradley J. Walters Brandon C. Cox
Brandon C. Cox- 1Department of Pharmacology, Southern Illinois University School of Medicine, Springfield, IL, United States
- 2Department of Otolaryngology-Head and Neck Surgery, University of Mississippi Medical Center, Jackson, MS, United States
- 3Department of Otolaryngology, Southern Illinois University School of Medicine, Springfield, IL, United States
Introduction: Hair cells (HCs) of the cochlea are responsible for sound transduction and hearing perception in mammals. Genetic mutations in the transcription factor Pou4f3 cause non-syndromic autosomal dominant hearing loss in humans (DFNA15) which varies in the age of onset depending on the individual mutation. Mouse models with germline deletion or mutations in Pou4f3 have previously demonstrated its critical role in the maturation and survival of cochlear HCs during embryonic development. However, the role of Pou4f3 in auditory function and in the survival or maintenance of cochlear HCs after birth and during adulthood has not been studied.
Methods: Therefore, using the inducible CreER-loxP system, we deleted Pou4f3 from mouse cochlear HCs at different postnatal ages, relevant to specific stages of HC maturation and hearing function.
Results and discussion: Elevated auditory brainstem response thresholds and significant HC loss were detected in mice with Pou4f3 deletion compared to their control littermates, regardless of the age when Pou4f3 was deleted. However, HC loss occurred more rapidly when Pou4f3 was deleted from immature HCs. Additionally, HC loss caused by Pou4f3 deletion did not affect the number of cochlear supporting cells, but caused a delayed loss of spiral ganglion neurons at 4 months after the deletion. In conclusion, Pou4f3 is necessary for the survival of cochlear HCs and normal hearing at all postnatal ages regardless of their maturation state. Our data also suggest that Pou4f3 indirectly regulates the survival of spiral ganglion neurons.
1 Introduction
Mechanosensory hair cells (HCs) mediate hearing by converting sound waves into electrical signals which are relayed to the brain via spiral ganglion neurons (SGNs). The sensory epithelium of the mammalian cochlea, called the organ of Corti, contains two types of HCs: the inner HCs (IHCs) which act as primary transducers of sound and the outer HCs (OHCs) which act as amplifiers (reviewed in Fettiplace, 2017). Malformation or degeneration of cochlear HCs is one of the most common causes of congenital hearing loss derived from genetic mutations (Egilmez and Kalcioglu, 2016; Korver et al., 2017; Nicolson, 2021). Over 120 genes have been identified that cause non-syndromic hearing loss by autosomal dominant (75%−80%), autosomal recessive (20%), X-linked (2%) or mitochondrial (1%) gene mutations (Sheffield and Smith, 2019; Tollefson et al., 2023).
In humans, the autosomal dominant mutation DFNA15 lies in the transcription factor Pou4f3 which was first identified in an Israeli Jewish family (Vahava et al., 1998). Since then, various Pou4f3 mutations have been identified in humans which cause altered DNA binding, cellular localization, and/or stability of the POU4F3 protein (Weiss et al., 2003; Collin et al., 2008; Bai et al., 2020). While patients with DFNA15 are born with normal hearing, they experience progressive hearing loss between 3 and 50 years of age depending on the specific mutation (Vahava et al., 1998; Frydman et al., 2000; Kitano et al., 2017; Lin et al., 2017; Cui et al., 2020).
In loss of function experiments, both in vitro and in vivo, Pou4f3 has been shown to be important for HC development, promoting differentiation, maturation, and survival, which also impacted the innervation and survival of spiral ganglion neurons (SGNs) (Erkman et al., 1996; Xiang et al., 1997, 1998, 2003). POU4F3 protein has also been shown to act as a pioneer factor for opening closed chromatin to allow proper differentiation of HCs during embryonic development (Yu et al., 2021). Mechanistic studies have shown that Pou4f3 regulates HC survival in vivo via its target gene, growth factor independence 1 (Gfi1), which directly inhibits the expression of the stress granule protein, CAPRIN-1 (Hertzano et al., 2004; Towers et al., 2011) and indirectly induces anti-apoptotic genes such as Bcl2 and Bcl-xL (Fukada et al., 1996; Catlett-Falcone et al., 1999; Alonzi et al., 2001). Despite the usefulness of these mouse models with germline mutations to define Pou4f3's role in newly differentiating HCs during embryonic development, the delayed onset phenotypes observed in humans born with Pou4f3 mutations is quite different. Thus, using the inducible CreER-loxP system which allows temporal control of gene deletion, we aimed to investigate the role of Pou4f3 in the postnatal and adult mouse cochlea since HCs are not mature at birth and hearing onset does not occur until ~2 weeks of age in mice (Ehret, 1976).
Using mouse models for conditional deletion, we investigated the role of Pou4f3 at different stages of cochlear maturation: at birth, at 2 weeks of age, at 4 weeks of age, and at 8 weeks of age. Regardless of the postnatal age when Pou4f3 was deleted, we observed significant hearing loss and HC loss, but the rate of HC death varied across cochlear turns and was delayed when Pou4f3 was deleted from mature HCs at 8 weeks of age. However, while supporting cells remained present at normal numbers 4 months after HC loss caused by Pou4f3 deletion, we observed significant loss of SGNs. Thus, in addition to regulating HC survival throughout the lifespan of the HC, loss of Pou4f3 from HCs indirectly leads to diminished survival of auditory neurons.
2 Materials and methods
2.1 Mouse lines
Atoh1-CreER™ (Chow et al., 2006) and PrestinCreERT2(Fang et al., 2012) mice were obtained from Dr. Suzanne Baker and Dr. Jian Zuo at St. Jude Children's Research Hospital (Memphis, TN), respectively. Pou4f3loxP/loxP (stock # 10560) (Badea and Nathans, 2011) and ROSA26CAG−loxP−stop−loxP−tdTomato (ROSA26tdTomato) mice (stock # 7914; also called Ai14) (Madisen et al., 2010) were purchased from The Jackson Laboratory (Bar Harbor, ME). All genotyping was performed by Transnetyx, Inc. (Cordova, TN) and mice of both genders were used in the study. All animal work was performed in accordance with approved animal protocols from the Institutional Animal Care and Use Committee at Southern Illinois University School of Medicine.
2.2 Drug treatments
CreER recombination was induced at different postnatal ages by intraperitoneal (IP) injections of tamoxifen (Sigma-Aldrich, St. Louis, MO) dissolved in corn oil. Injections of 3 mg/40 g body weight (b.w.) were given 20–24 h apart at postnatal day (P)0 and P1 or at P12 and P13. Adult mice were injected with 9 mg/40 g b.w. tamoxifen for 2 consecutive days (20–24 h between injections) at either 4 or 8 weeks of age. Cre-negative littermates injected with tamoxifen served as controls.
2.3 Immunofluorescence
Temporal bones were post-fixed in 4% paraformaldehyde (Polysciences Inc, Warrington, PA) in 10 mM PBS for 2 h at room temperature. Samples from mice that were older than 1 week were decalcified in 120 mM EDTA (Sigma-Aldrich, St. Louis, MO) for 1–5 overnights (depending on the age of mouse at tissue collection) at room temperature (RT) using an end-over-end rotator and the EDTA solution was changed daily. Whole mount dissection and immunostaining was performed as previously described (Montgomery and Cox, 2016) with the one exception that the samples were incubated with signal enhancer (cat #I36933; Life Technologies, Waltham, MA) for 30 min at RT prior to the blocking/permeabilization step. The following primary antibodies were used: mouse anti-beta III tubulin (Tuj1, 1:500, Biolegend, cat # 801201), rabbit anti-myosin VIIa (1:200; cat. #25-6790; Proteus Biosciences, Ramona, CA), mouse IgG1 anti-Pou4f3 (1:300; cat. #sc-81980; Santa Cruz Biotechnology, Dallas, TX), and goat anti-Sox2 (1:400, cat. #sc-17320; Santa Cruz Biotechnology, Dallas, TX). Alexa-conjugated secondary antibodies (Thermo Fisher Scientific, Hampton, NH) were used at 1:1,000 and nuclei were labeled with Hoechst 33342 (1:2,000 in 10 mM PBS, cat. #H3570 Thermo Fisher Scientific, Hampton, NH). tdTomato was visualized using endogenous fluorescence. TUNEL staining was performed using the In Situ Cell Death Detection Kit, TMR Red (cat. #12156792910; Roche Applied Science, Indianapolis, IN) following the manufacturer's instructions.
2.4 Cryosections
Post-fixed temporal bones were washed three times in 10 mM PBS and decalcified in 120 mM EDTA (Sigma-Aldrich, St. Louis, MO) for 1–2 weeks at room temperature (RT) using an end-over-end rotator. EDTA solution was changed daily. The decalcified cochleae were embedded in optimal cutting temperature (OCT) media (cat #4585, Fisher Healthcare, Houston, TX) in cryo-molds placed on a slurry of dry ice in 70% ethanol. The embedded cochleae were stored at −80°C until sectioning. For analyses of SGNs, mid-modiolar cryo-sections (12 μm thickness) were obtained using an Epredia Microm HM525 NX cryostat (Thermo Fisher Scientific, Hampton, NH) as previously described (Coleman et al., 2009).
2.5 Image analysis
Samples were imaged using a Zeiss LSM800 (Oberkochen, Germany) confocal microscope and processed using Zen 2.5 lite software (Oberkochen, Germany). Myosin VIIa-positive HCs and Sox2-positive SCs were manually quantified from two representative 150 μm regions from each cochlear turn (apex, middle, and base) from cochlear whole-mounts. Tuj1-positive neuronal cell bodies from cochlear sections were quantified in a 10,000 μm2 region in the middle turn of cochleae from three alternate sections per sample. For quantification, cochlear whole mounts or sections were imaged using a 40× oil immersion objective with a numerical aperture (NA) 1.3 and a resolution of 1,024 × 1,024 pixels. The low magnification imaging of cochlear sections was done using a 10× objective with NA 0.3 and a resolution of 1,024 × 1,024 pixels. The N values represent number of mice as only one cochlea per mouse were analyzed for each immunostaining experiment type.
2.6 Auditory brainstem response recordings
Mice aged 4 weeks or older were anesthetized with avertin (250–500 mg/kg, IP) and kept on a heating pad at 37°C in a sound attenuated chamber. Auditory brainstem response (ABR) measurements were performed using an Intelligent Hearing System (IHS, Miami, FL) 4964 high frequency system using the left ear of each mouse. Therefore, N values represent the number of mice. Subdermal stainless-steel electrodes were inserted at the vertex of the skull, below the pinna of the left ear, and a ground electrode was located at the base of the tail. ABR waveforms were obtained in response to 8, 12, 16, and 22 kHz tones (5 ms tone bursts presented at a rate of 19/s and averaged over 512 presentations) given in 5 dB SPL steps decrements between 80 and 5 dB SPL. ABR thresholds were determined by the lowest sound intensity that produced a visually distinct response in wave I and II as assessed by a researcher who was blinded to the genotypes.
2.7 RNA extraction and quantitative real-time PCR
The cochlear portion of temporal bones were collected and snap frozen on dry ice and kept at −80°C until further processed for RNA isolation. Both cochleae from each mouse were pooled for RNA isolation hence N values for qPCR analysis represent the number of mice. RNA isolation and real-time quantitative polymerase chain reaction (qPCR) was performed as previously described (McGovern et al., 2018) using SYBR Green (cat #K0391, Thermo Fisher Scientific, Hampton, NH) and a CFX Connect Optics Module (Bio-Rad, Hercules, CA). The primers used for each gene are listed in Supplementary Table 1. Comparison of gene expression levels was determined using the Pfaffl method which includes the primer efficiency in the delta-delta Ct equation (Pfaffl, 2001).
2.8 Statistical analysis
All data are presented as mean ± SEM. Comparisons of ABR thresholds between genotypes and across frequencies were done using a two-way ANOVA followed by Bonferroni-corrected post-hoc testing at each frequency. HC counts from control samples across time for each dataset were tested for similarity using a one-way ANOVA which revealed no significant differences in HC numbers across controls. Thus, we used the mean number of HCs from the latest timepoint assessed for each age of Pou4f3 deletion as the control samples in Figures 1–4 and to calculate the percentage of HC loss. Percent HC loss was calculated by subtracting the HC counts from an individual sample (control or Pou4f3 cKO) from the mean HC number of the control group (N = 3, from the latest timepoint assessed for each age of Pou4f3 deletion) and dividing that number by the mean HC number of the control group. The result was then converted to a percentage. For each age of Pou4f3 deletion, a two-way ANOVA was used to assess HC loss where control samples were treated as a pseudo-pre-injection timepoint. This allowed for analysis of the main questions of interest (effect of Pou4f3 deletion, effect of time post-injection, and effect of cochlear turn) without having to test three-way interaction terms which were not the focus of these studies. Significant main effects were followed by two Tukey's post-hoc tests. One post-hoc test compared each timepoint with control as well as to other timepoints, but within each cochlear turn and the second post-hoc test compared cochlear turns within each timepoint. Note that IHCs and OHCs were compared separately for the neonatal deletion of Pou4f3 in Figure 1. The number of SCs were compared across genotype and across cochlear turn using a two-way ANOVA followed by Sidak-corrected post-hoc comparisons. The percentage of SGN loss was calculated in a similar manner as percent HC loss using the mean SGN number from control samples as the denominator. The percentage of SGN loss was compared across genotypes using a two-way ANOVA followed by Sidak's post-hoc test. qPCR data are presented in the graphs as fold change from control samples and each gene was compared to its own control using a Student's t-test. Statistical analyses were conducted using GraphPad Prism 7.0 software (La Jolla, CA).
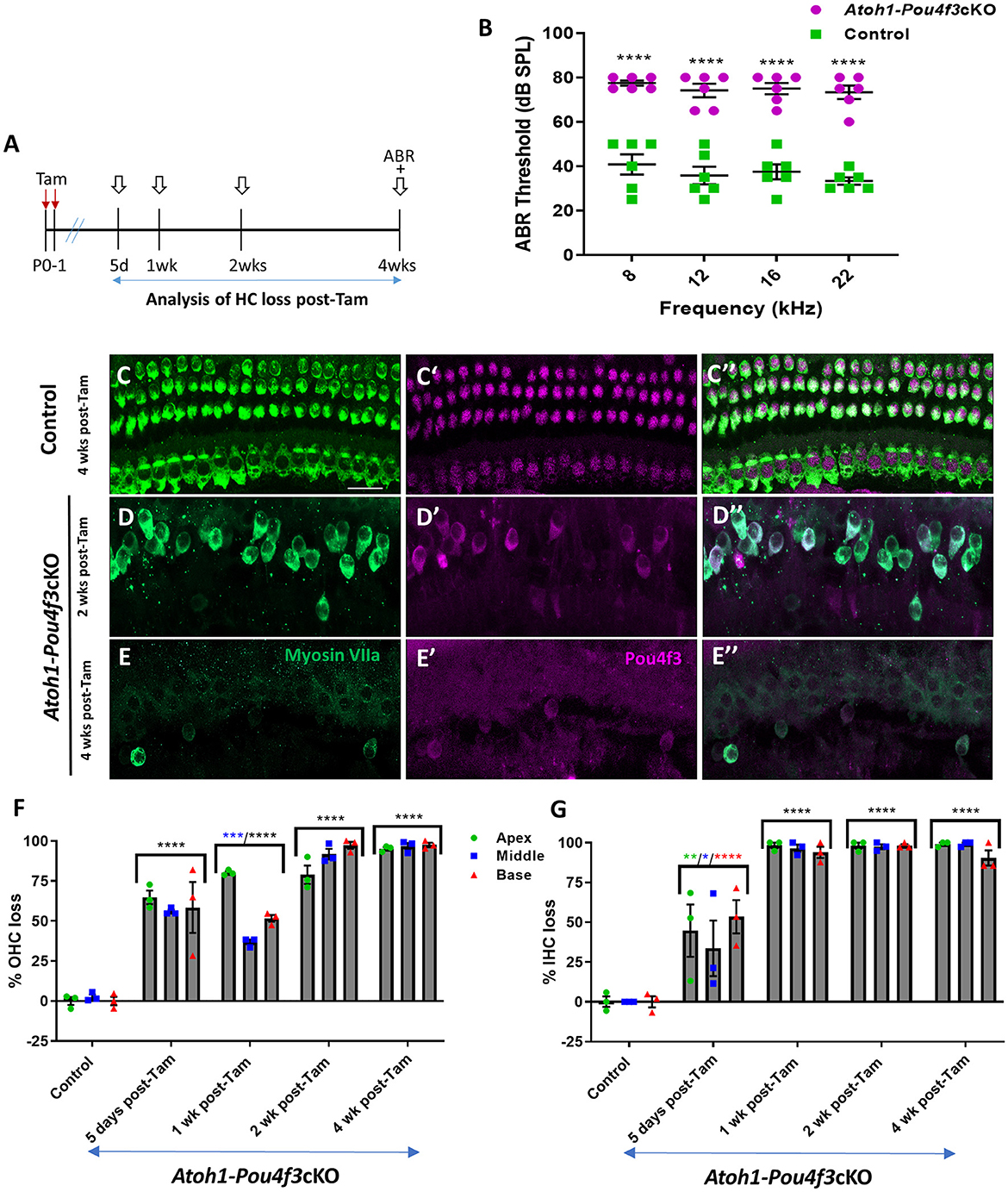
Figure 1. (A) Schematic for the experimental design using Atoh1-Pou4f3cKO mice to delete Pou4f3 from both IHCs and OHCs at P0–P1. Open arrows indicate post-tamoxifen (post-Tam) timepoints when HC loss was assessed. (B) At 4 weeks after Pou4f3 deletion, there was a significant elevation in ABR thresholds in Atoh1-Pou4f3cKO mice compared to their control littermates at all frequencies tested [N = 6; significant main effect of genotype, F(1,40) = 302.8, p < 0.0001]. Asterisks indicate comparisons between genotypes at each frequency based on a Bonferroni-corrected post-hoc test. (C–E″) Representative confocal images from control (C–C″) and Atoh1-Pou4f3cKO (D–E″) cochleae. HCs in the control cochleae remained intact (myosin VIIa, green) and had nuclear expression of POU4F3 (magenta). However, many HCs were missing in Atoh1-Pou4f3cKO cochleae at 2 and 4 weeks after Pou4f3 deletion and most of the remaining HCs exhibited POU4F3 immunoreactivity in their cytoplasm. Quantification of OHC (F) and IHC (G) loss in control and Atoh1-Pou4f3cKO cochleae (N = 3) between 5 days and 4 weeks post-Tam. For OHCs there was a significant main effect of time [F(4,10) = 142.2, p < 0.0001] and interaction between time and cochlear turn [F(8,20) = 6.697, p = 0.0003]. For IHCs there was a significant main effect of time [F(4,10) = 56.95, p < 0.0001]. In (F) and (G), differences from control within each cochlear turn are indicated by the asterisks based on a Tukey's-corrected post-hoc test. Green asterisks are p values for the apical turn, blue asterisks are p values for the middle turn, and red asterisks are p values for the basal turn. Black asterisks were used when the p value was the same for two or three turns. Comparisons across time post-Tam and across cochlear turns within the same genotype are presented in Supplementary Tables 2A,B. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. Scale bar = 20 μm. Pou4f3 deletion from immature HCs at birth causes elevated ABR thresholds and progressive HC loss.
3 Results
3.1 Pou4f3 is required for the survival of immature cochlear HCs in newborn mice
To delete Pou4f3 from HCs at birth, we used Atoh1-CreERTM mice which show CreER activity in the majority of both IHCs and OHCs after tamoxifen induction at P0 and P1 (Chow et al., 2006; Weber et al., 2008; Cox et al., 2014). We validated this expression pattern using Rosa26tdTomatoreporter mice, and found that 98%−100% of OHCs expressed tdTomato (100% ± 0% in apex, 98.5% ± 1.5% in middle, and 99.3 ± 0.7% in basal turn, N = 4, Supplementary Figure 1) and 92–100% of IHCs expressed tdTomato (100% ± 0% in apex, and 93.0% ± 6.2% in middle, and 93.7% ± 5.4% in basal turn, N = 4, Supplementary Figure 1).
HCs in the mouse cochleae are immature at birth and undergo various morphological and electrophysiological changes during the first postnatal weeks (Anniko, 1983; Eatock and Hurley, 2003). To determine the role of Pou4f3 in immature HCs, we generated Atoh1-CreERTM:Pou4f3loxP/loxP mice (referred to hereafter as Atoh1-Pou4f3cKO) and injected them with tamoxifen at P0 and P1, followed by assessment of auditory function using auditory brainstem response (ABR) measurements at 4 weeks of age (Figure 1A). Controls were CreER-negative littermates which contained the Pou4f3loxP/loxP allele and also received tamoxifen. Compared to controls, Atoh1-Pou4f3cKO mice had significant ABR threshold elevations (~35–40 dB SPL) at all four frequencies tested (Figure 1B). Analysis of the cochleae showed rapid and progressive loss of both IHCs and OHCs in Atoh1-Pou4f3cKO mice (Figures 1D–G) compared to controls (Figures 1C–C″,F–G) which had the normal pattern of four rows of HCs with POU4F3 present in all HC nuclei (Figures 1C–C″). In Atoh1-Pou4f3cKO mice, OHC loss was observed as early as 5 days post-tamoxifen with more than 50% loss in all cochlear turns (64.8 ± 4.2% in apex; 55.9% ± 1.2% in middle, and 58.4% ± 15.9% in base, N = 3), which progressed to >95% OHC loss by 4 weeks post-tamoxifen (95.2% ± 1.0% in apex, 96.7% ± 2.1% in middle, and 97.5% ± 1.3% in base, N = 3; Figure 1F, Supplementary Table 2A). IHC loss was also observed at 5 days post-tamoxifen in Atoh1-Pou4f3cKO mice, but IHCs died at a faster rate than OHCs with ~94%−98% loss by 1 week post-tamoxifen (98.2 ± 1.8% in apex; 96.6 ± 2.3% in middle; 93.9% ± 3.5% in base, N = 3; Figure 1G, Supplementary Table 2B).
3.2 Pou4f3 is required for the survival of OHCs during hearing onset in juvenile mice
HC maturation occurs over the first two postnatal weeks leading to the onset of hearing in mice at ~P12–14 (Ehret, 1976). Since Atoh1 expression is down-regulated within the first postnatal week, we instead used PrestinCreERT2mice which specifically target OHCs of the cochlea (Fang et al., 2012; Cox et al., 2014). We validated the expression pattern of PrestinCreERT2 using Rosa26tdTomato reporter mice and found that >90% of OHCs expressed tdTomato in all three turns of the cochlea after tamoxifen injections at P12 and P13, 4 weeks of age, or 8 weeks of age (Supplementary Figure 2).
To determine whether mature OHCs still require Pou4f3 for survival, we generated PrestinCreERT2::Pou4f3loxP/loxP mice (Prestin-Pou4f3cKO), injected them with tamoxifen at P12 and P13 (juvenile age), and performed similar assessments as outlined above (Figure 2A). Controls were CreER-negative littermates which contained the Pou4f3loxP/loxP allele and also received tamoxifen. Prestin-Pou4f3cKO mice also showed significant ABR threshold elevations (~33–43 dB SPL) at all four frequencies tested compared to CreER-negative control littermates when tested at 4 weeks after deletion of Pou4f3 (Figure 2B). Compared to controls, we observed progressive loss of OHCs in all three turns of Prestin-Pou4f3cKO cochleae between 1 and 6 weeks post-tamoxifen (Figures 2C–F). However, IHCs remained intact with all expressing nuclear POU4f3 (Figures 2D,E″). This was expected since PrestinCreERT2 targets OHCs exclusively. Comparison of OHC loss across cochlear turns in Prestin-Pou4f3cKO mice at 1 week post-tamoxifen showed greater loss in the basal turn (84.9 ± 2.9%) than the middle (47.6 ± 16.8%) and apical turns (24.2 ± 10.1%; Figure 2F, Supplementary Table 3). Yet, OHC loss progressed in all cochlear turns during the following weeks resulting in almost complete absence of OHCs in the middle and basal turns (97.5 ± 1.7% in middle, and 97.2 ± 1.5% in base), but only ~60% loss in the apical turn (61.5 ± 5.3%) at 6 weeks post-tamoxifen (Figure 2F, Supplementary Table 3).
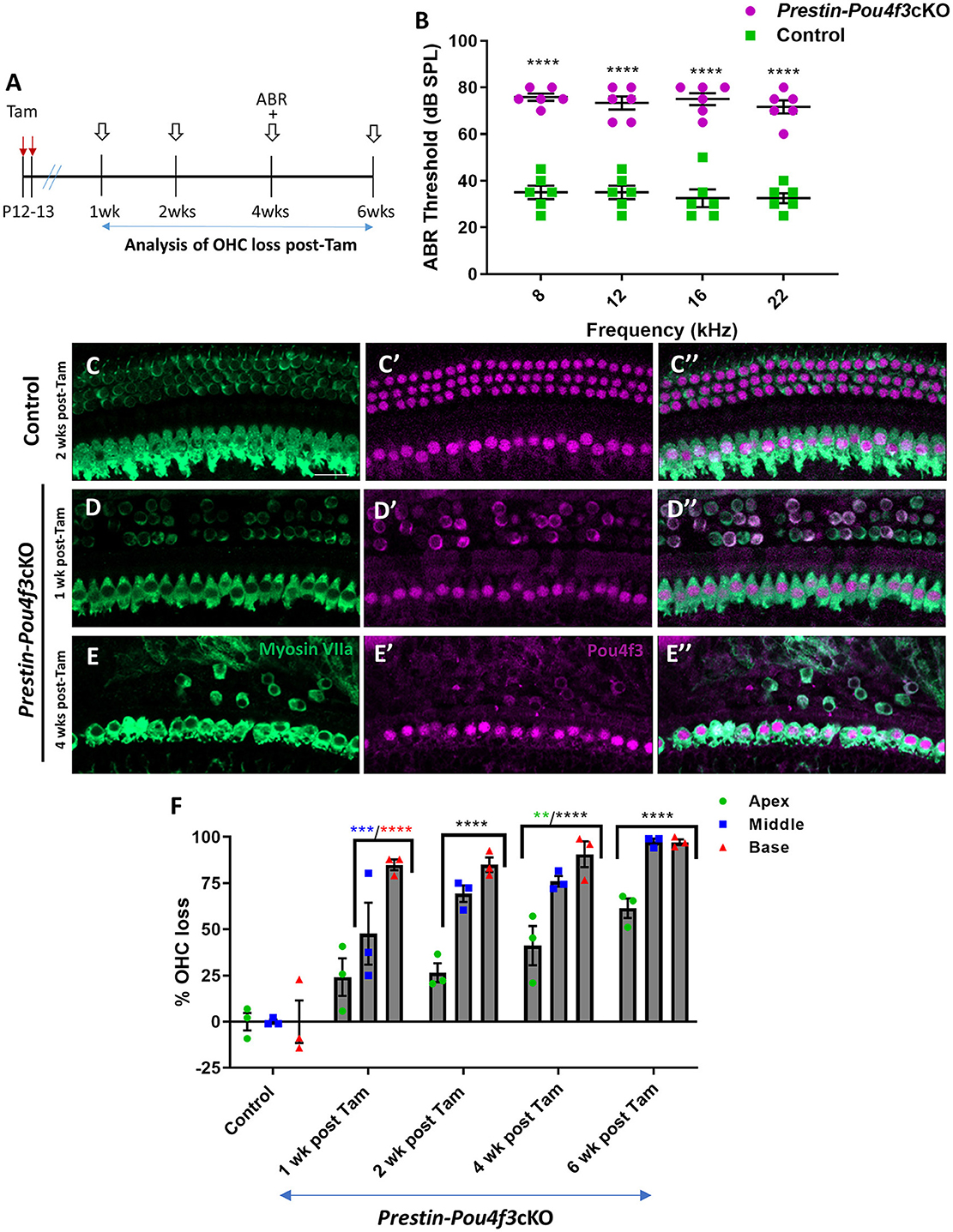
Figure 2. Pou4f3 deletion from OHCs at hearing onset causes elevated ABR thresholds and significant OHC loss. (A) Schematic for the experimental design using Prestin-Pou4f3cKO mice to delete Pou4f3 from OHCs at 2 weeks of age. Open arrows indicate post-tamoxifen (post-Tam) timepoints when OHC loss was assessed. (B) At 4 weeks (wks) after Pou4f3 deletion, there was a significant elevation in ABR thresholds in Prestin-Pou4f3cKO mice compared to their control littermates at all frequencies tested [N = 6, significant main effect of genotype, F(1,40) = 428.1, p < 0.0001]. Asterisks indicate comparisons between genotypes at each frequency based on a Bonferroni corrected post-hoc test. (C–E″) Representative confocal images from control (C–C″) and Prestin-Pou4f3cKO (D–E″) cochleae. HCs in the control cochleae and IHCs in the Prestin-Pou4f3cKO cochleae remained intact (myosin VIIa, green) and had nuclear expression of POU4F3 (magenta). However, many OHCs were missing in Prestin-Pou4f3cKO cochleae at 1 and 4 weeks after Pou4f3 deletion. Most of the remaining OHCs at 1 week post-Tam expressed POU4F3 in their cytoplasm. (F) Quantification of OHC loss in control and Prestin-Pou4f3cKO cochleae (N = 3) between 1 and 6 weeks post-Tam. There was a significant main effect of cochlear turn [F(2,20) = 43.81, p < 0.0001]; time [F(4,10) = 49.21, p < 0.0001]; and an interaction between time and cochlear turn [F(8,20) = 3.832, p = 0.0070]. Differences from control within each cochlear turn are indicated by the asterisks based on a Tukey's-corrected post-hoc test. Green asterisks are p values for the apical turn, blue asterisks are p values for the middle turn, and red asterisks are p values for the basal turn. Black asterisks were used when the p value was the same for two or three turns. Data are presented as mean ± SEM. **p < 0.01, ***p < 0.001, and ****p < 0.0001. Comparisons across time post-Tam and across cochlear turns within the same genotype are presented in Supplementary Table 3. Scale bar =20 μm.
3.3 Pou4f3 is required for the survival of OHCs in adult mice
Next, we investigated the role of Pou4f3 in OHCs when mice have mature hearing (at 4 weeks of age), or after they were sexually mature at (8 weeks of age) (reviewed in Ohlemiller et al., 2016). For both experiments, we again used Prestin-Pou4f3cKO mice to delete Pou4f3 from OHCs and performed ABR measurements and histological analyses of the cochlea at multiple timepoints (Figures 3A, 4A). Controls were CreER-negative littermates which contained the Pou4f3loxP/loxP allele and also received tamoxifen. After Pou4f3 deletion at 4 weeks of age, we observed significant elevation of ABR thresholds in Prestin-Pou4f3cKO mice at all frequencies tested at 2 weeks post-tamoxifen compared to their control littermates (Figure 3B). One week after Pou4f3 deletion, OHC numbers in the apical and middle turns of Prestin-Pou4f3cKO cochleae were not different from controls, but there was significant and robust OHC loss (85.6 ± 2.9%) in the basal turn (Figures 3C–F). Two weeks after Pou4f3 deletion, OHC loss was significantly higher in all cochlear turns compared to controls with the highest loss observed in the basal turn (85.8 ± 7.7%) followed by the middle turn (55.2 ± 13.4%), but there was only a mild loss (15.1 ± 3.4%) in the apex. OHC loss in apical and middle turns continued to progress over time with the middle turn reaching >80% loss (85.8 ± 7.7%) at 4 weeks post-tamoxifen and the apical turn reaching similar loss level (82.1 ± 5.4%) at 6 weeks post-tamoxifen (Figure 3F, Supplementary Table 4).
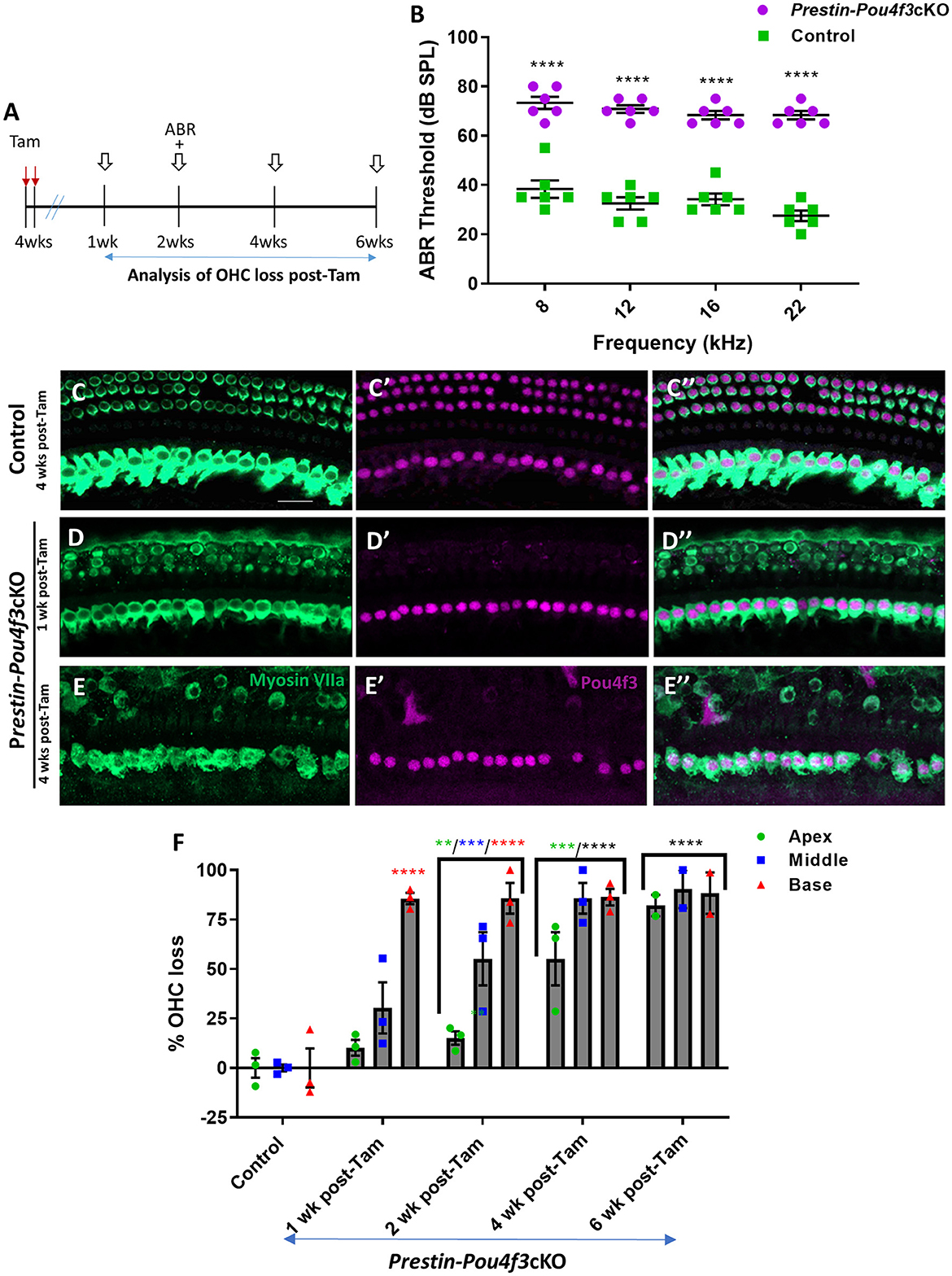
Figure 3. Pou4f3 deletion from OHCs once hearing is mature causes elevated ABR thresholds and progressive OHC loss. (A) Schematic for the experimental design using Prestin-Pou4f3cKO mice to delete Pou4f3 from OHCs at 4 weeks (wks) of age. Open arrows indicate post-tamoxifen (post-Tam) timepoints when OHC loss was assessed. (B) At 2 weeks after Pou4f3 deletion, there was a significant elevation in ABR thresholds in Prestin-Pou4f3cKO mice compared to their control littermates at all frequencies tested [N = 6; significant main effect of genotype, F(1,10) = 355.2, p < 0.0001 and frequency F(3,30) = 4.539, p = 0.0097]. Asterisks indicate comparisons between genotypes at each frequency based on a Bonferroni-corrected post-hoc test. (C–E″) Representative confocal images from control (C–C″) and Prestin-Pou4f3cKO (D–E″) cochleae. HCs in the control cochleae and IHCs in the Prestin-Pou4f3cKO cochleae remained intact (myosin VIIa, green) and had nuclear expression of POU4F3 (magenta). Analysis at 1 and 4 weeks after Pou4f3 deletion showed progressive loss of OHCs in Prestin-Pou4f3cKO cochleae over time. (F) Quantification of OHC loss in control and Prestin-Pou4f3cKO cochleae (N = 3) between 1 and 6 weeks post-Tam. There was a significant main effect of time [F(4,9) =38.51, p < 0.0001]; cochlear turn [F(2,18) = 25.62, p < 0.0001]; and an interaction between time and cochlear turn [F(8,18) = 5.625, p = 0.0011]. Differences from control within each cochlear turn are indicated by the asterisks based on a Tukey's-corrected post-hoc test. Green asterisks are p values for the apical turn, blue asterisks are p values for the middle turn, and red asterisks are p values for the basal turn. Black asterisks were used when the p value was the same for two or three turns. Data are presented as mean ± SEM. **p < 0.01, ***p < 0.001 and ****p < 0.0001. Comparisons across time post-Tam and across cochlear turns within the same genotype are presented in Supplementary Table 4. Scale bar =20 μm.
Finally, we deleted Pou4f3 from OHCs at 8 weeks of age using Prestin-Pou4f3cKO mice to assess the role of Pou4f3 in sexually mature mice and to test whether Pou4f3 is required for OHC survival throughout the lifespan (Figure 4A). Similar to Pou4f3 deletion at all other ages, we observed significant elevation in ABR thresholds in Prestin-Pou4f3cKO mice compared to control mice at 2 weeks post-tamoxifen at all frequencies tested (Figure 4B). However, OHC loss was delayed compared to Pou4f3 deletion at other ages (Figures 4D–F). There was no significant difference in the number of OHCs in Prestin-Pou4f3cKO cochleae compared to controls at 1 week post-tamoxifen. However, at 2 weeks post-tamoxifen a majority of the OHCs were missing in the basal turn (84.5 ± 9.8%; Figure 4F). By 4 weeks post-tamoxifen, OHC loss progressed to the middle (81.0 ± 17.4%) and apical (58.1 ± 15.8%) turns as well (Figure 4F, Supplementary Table 5).
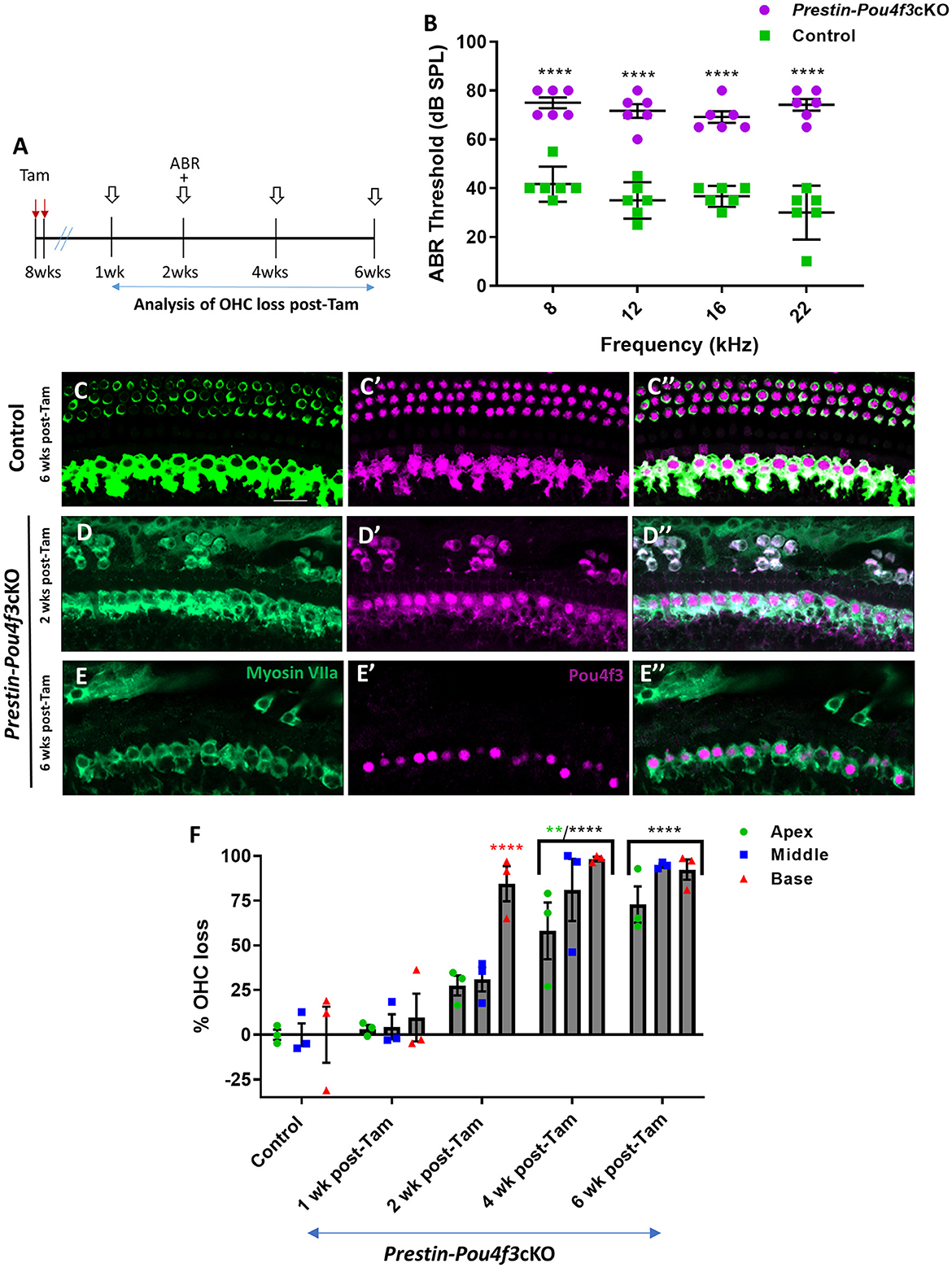
Figure 4. Pou4f3 deletion from OHCs in sexually mature mice leads to elevated ABR thresholds and progressive OHC loss. (A) Schematic for the experimental design using Prestin-Pou4f3cKO mice to delete Pou4f3 from OHCs at 8 weeks (wks) of age. Open arrows indicate post-tamoxifen (post-Tam) timepoints when OHC loss was assessed. (B) At 2 weeks after Pou4f3 deletion, there was a significant elevation in ABR thresholds in Prestin-Pou4f3cKO mice compared to their control littermates at all frequencies tested [N = 6; significant main effect of genotype, F(1,40) = 350.4, p < 0.0001]. Asterisks indicate comparisons between genotypes at each frequency based on a Bonferroni-corrected post-hoc test. (C–E″) Representative confocal images from control (C–C″) and Prestin-Pou4f3cKO (D–E″) cochleae. HCs in the control cochleae and IHCs in the Prestin-Pou4f3cKO cochleae remained intact (myosin VIIa, green) and had nuclear expression of POU4F3 (magenta). Analysis at 2 and 6 weeks after Pou4f3 deletion showed progressive loss of OHCs in Prestin Cre-Pou4f3cKO cochleae over time. The remaining OHCs at 2 weeks post-Tam expressed POU4F3 in their cytoplasm. (F) Quantification of OHC loss in control and Prestin-Pou4f3cKO cochleae (N = 3) between 1 and 6 weeks post-Tam. There was a significant main effect of time [F(4,10) = 29.01, p < 0.0001]; cochlear turn [F(2,20) = 13.88, p = 0.0002]; and an interaction between time and cochlear turn [F(8,20) = 3.68, p = 0.0085]. Differences from control within each cochlear turn are indicated by the asterisks based on a Tukey's-corrected post-hoc test. Green asterisks are p values for the apical turn and red asterisks are p values for the basal turn. Black asterisks were used when the p value was the same for two or three turns. Data are presented as mean ± SEM. **p < 0.01 and ****p < 0.0001. Comparisons across time post-Tam and across cochlear turns within the same genotype are presented in Supplementary Table 5. Scale bar = 20 μm.
3.4 Pou4f3 deletion from HCs causes cell death by apoptosis
Next, we sought to investigate the mechanism of cochlear HC death caused by Pou4f3 deletion. Using cochleae from Atoh1-Pou4f3cKO mice injected with tamoxifen at P0 and P1 and Prestin-Pou4f3cKO mice injected with tamoxifen at 4 weeks of age, we performed the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay to detect cells undergoing apoptosis. In Atoh1-Pou4f3cKO samples where CreER targeted both OHCs and IHCs, we observed TUNEL-positive HCs of both types at 5 days post-tamoxifen (Figures 5B′,B″). Similarly, 1 week after Pou4f3 deletion, we observed many TUNEL-positive OHCs in Prestin-Pou4f3cKO cochleae (Figures 5D′,D″). No TUNEL-positive HCs were observed in any of the control samples (Figures 5A–A″,C–C″).
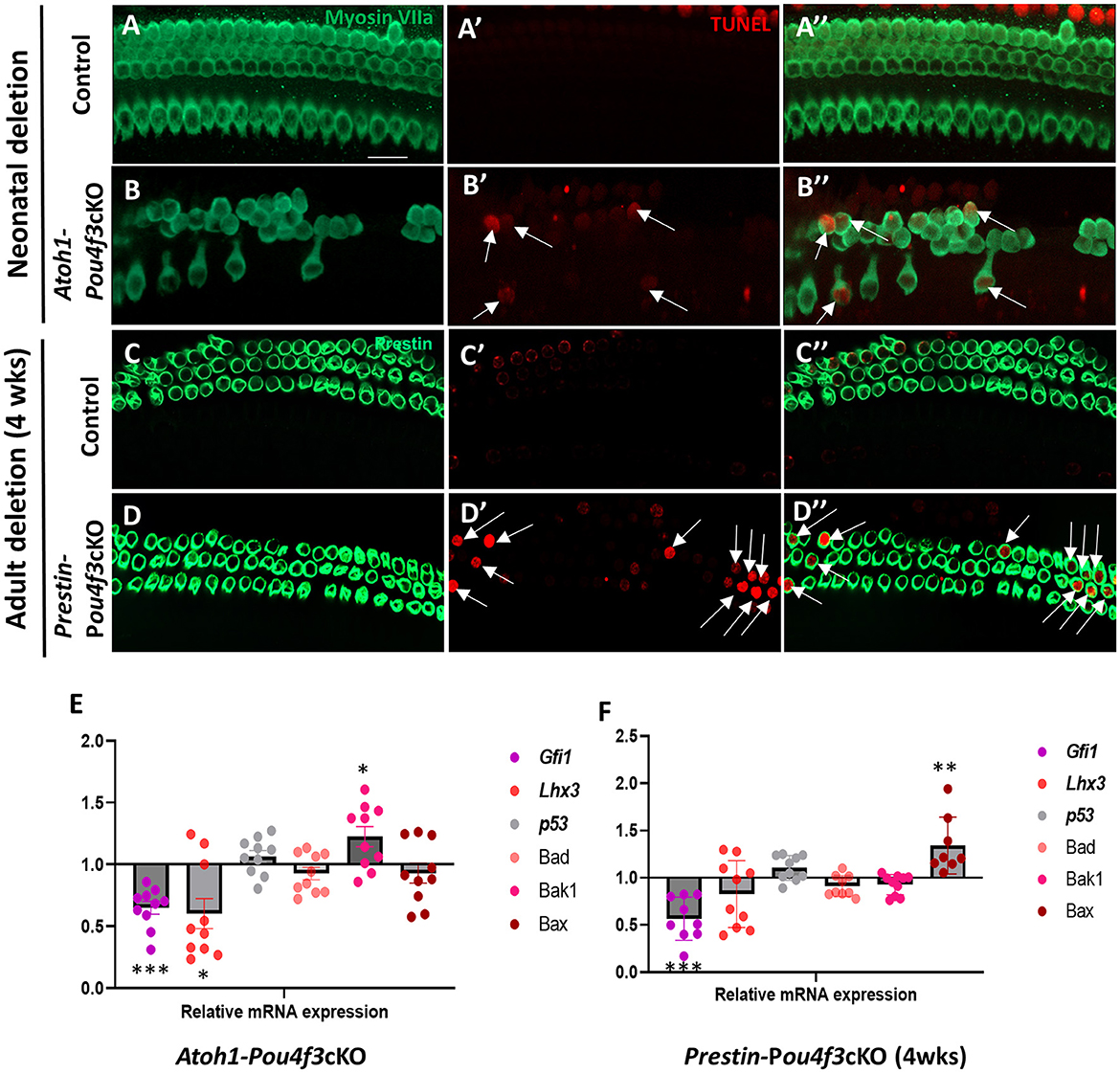
Figure 5. Pou4f3 deletion at both neonatal and adult ages causes HC death by apoptosis. (A–D″) Representative confocal images from control (A–A″, C–C″) and Atoh1-Pou4f3cKO (B–B″) or Prestin-Pou4f3cKO (D–D″) cochleae 5 days after deletion of Pou4f3 at P0/P1 or one week after deletion of Pou4f3 at 4 weeks (wks) of age respectively. IHCs and OHCs were stained using myosin VIIa [green, (A–B″)] or the OHC-specific marker prestin [green, (C–D″)]. TUNEL staining (red) was used to identify apoptotic cells. No TUNEL staining was observed in control cochleae. However, many IHCs and OHCs from Atoh1-Pou4f3cKO cochleae and OHCs from Prestin-Pou4f3cKO cochleae were TUNEL-positive (arrows). (E, F) Quantitative real-time PCR analysis of the mRNA transcripts for the Pou4f3 target genes, Gfi1 and Lhx3, as well as the pro-apoptotic genes, p53, Bad, Bak1 and Bax. Samples were analyzed 5 days after deletion of Pou4f3 at P0/P1 using Atoh1-Pou4f3cKO mice or 1 week after deletion of Pou4f3 at 4 weeks of age using Prestin-Pou4f3cKO mice. Data are expressed as fold change (mean ± SEM) from control. Each gene was compared to its own control using a Student's t-test. N = 8–10. *p < 0.05, **p < 0.01, ***p < 0.001. Scale bar = 20 μm.
To further investigate potential mediators of cell death in response to the deletion of Pou4f3, we performed real-time qPCR of pro-apoptotic genes and two known POU4F3 target genes using the same mouse models and collection timepoints as used for the TUNEL assay (Atoh1-Pou4f3cKO mice were injected with tamoxifen at P0 and P1 and samples collected 5 days post-tamoxifen; Prestin-Pou4f3cKO mice were injected with tamoxifen at 4 weeks of age and samples collected at 1-week post-tamoxifen). After deletion of Pou4f3 we observed the downregulation of two downstream targets of Pou4f3, Gfi1 (p = 0.0003 for Atoh1-Pou4f3cKO, and p = 0.0009 for Prestin-Pou4f3cKO at 4 weeks, N = 9–10) and Lhx3 (p = 0.0427 for Atoh1-Pou4f3cKO, N = 10; Figures 5E,F) (Hertzano et al., 2004, 2007). Five days after neonatal Pou4f3 deletion using Atoh1-Pou4f3cKO mice, we also observed a significant upregulation of the pro-apoptotic gene, Bak1 (p = 0.0398; Figure 5E, N = 9–10). One week after Pou4f3 deletion at 4 weeks of age using Prestin-Pou4f3cKO mice, we observed upregulation of the pro-apoptotic gene Bax (p = 0.0059, N = 8). Together these data suggest that HCs undergo apoptotic cell death after deletion of Pou4f3.
3.5 HC loss caused by Pou4f3 deletion does not affect the survival of supporting cells, but causes SGN loss
Previous studies have shown that HC loss can produce secondary effects on the organ of Corti which includes loss of supporting cells and/or delayed SGN loss (Leake et al., 1997; Izumikawa et al., 2008; Kujawa and Liberman, 2009; Barclay et al., 2011; Taylor et al., 2012; Yu et al., 2014). In addition, previous studies showed a loss of SGNs when Pou4f3 was deleted from the germline (Xiang et al., 2003; Pauley et al., 2008). Here, we investigated whether HC loss caused by Pou4f3 deletion impacts these two cell types. Using Atoh1-Pou4f3cKO mice injected with tamoxifen at P0 and P1 and Prestin-Pou4f3cKO mice injected with tamoxifen at 8 weeks of age, we collected samples at 16 weeks post-tamoxifen and performed immunostaining to detect SOX2, a supporting cell-specific marker in the mature cochlea (Figures 6A–D) (Hume et al., 2007). Quantification of SOX2-positive supporting cells revealed no significant differences from control samples for both ages of Pou4f3 deletion (Figures 6E,F).
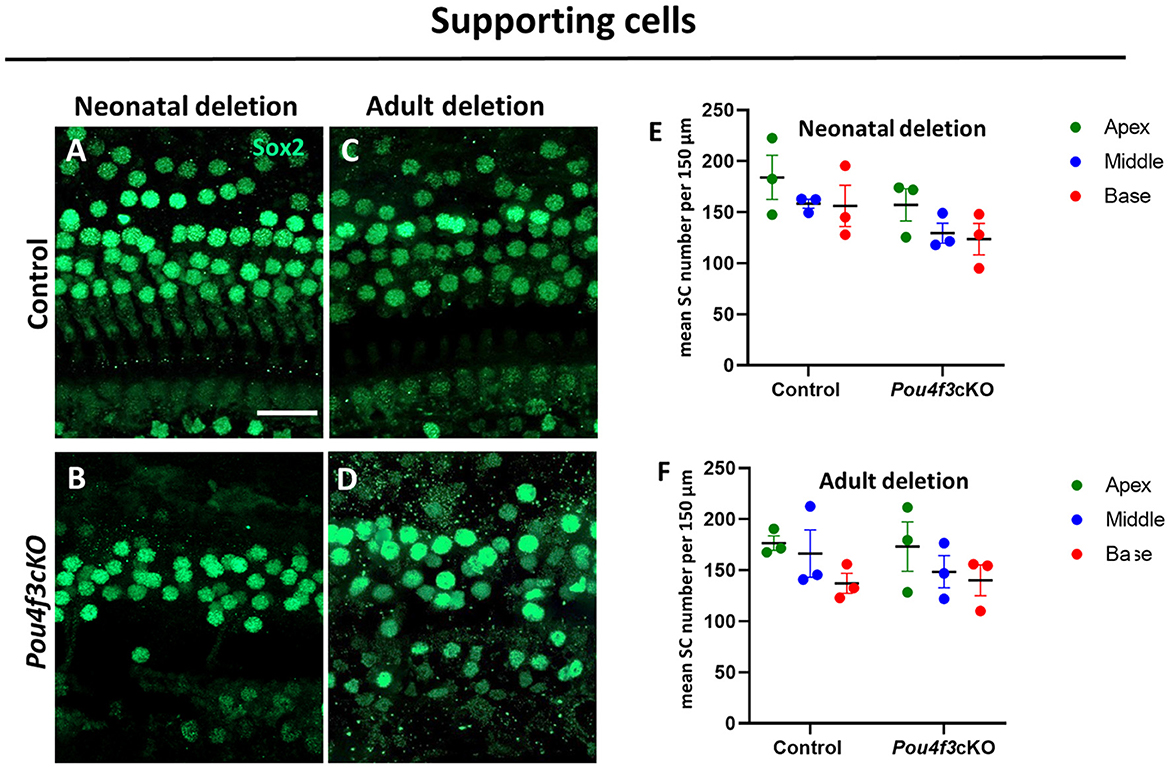
Figure 6. HC loss caused by Pou4f3 deletion does not affect the survival of SCs. (A–D) Representative confocal images of the middle turn from control (A, C) and cochleae with Pou4f3 deletion at P0 using Atoh1-Pou4f3cKO mice (B) or at 8 weeks using Prestin-Pou4f3cKO mice (D). All samples were analyzed at 4 months post-tamoxifen. (E, F) Quantification of SOX2-positive SC nuclei in each cochlear turn for control and after Pou4f3 deletion at P0/P1 (E) or 8 weeks of age (F). SCs numbers were compared to control within the same cochlear turn using a two-way ANOVA followed by Sidak's post-hoc test. There was a significant main effect of genotype [F(1,6) = 12.93, p = 0.0114] when Pou4f3 was deleted at P0/P1, but there were no main effects when Pou4f3 was deleted at 8 weeks of age and no significant differences in the post-hoc tests for either age of Pou4f3 deletion. N = 3. Scale bar = 20 μm.
To investigate the impact of Pou4f3 deletion from HCs on the SGNs, we performed mid-modiolar sectioning of the temporal bones from the same two mouse models at 16 weeks (or 4 months) post-tamoxifen and stained them with the neuron specific anti-Tuj1 antibody (Lee et al., 1990; Barclay et al., 2011; Sun et al., 2018). While the CreER-negative control samples showed normal densities of SGN cell bodies (Figures 7A,C,E), cochleae from mice where Pou4f3 was deleted from both IHCs and OHCs at P0/P1, had 85.3 ± 1.1% loss of SGN cell bodies (Figures 7B,D,K). However, when the deletion was OHC-specific and occurred at 8 weeks of age, there was a 38.9 ± 10.7% loss of SGNs (Figures 7F,K). Additionally, we observed reduction in the neuronal fibers in Pou4f3cKO cochleae (Figures 7H,J) compared to control cochleae (Figures 7G,I) at both ages of Pou4f3 deletion.
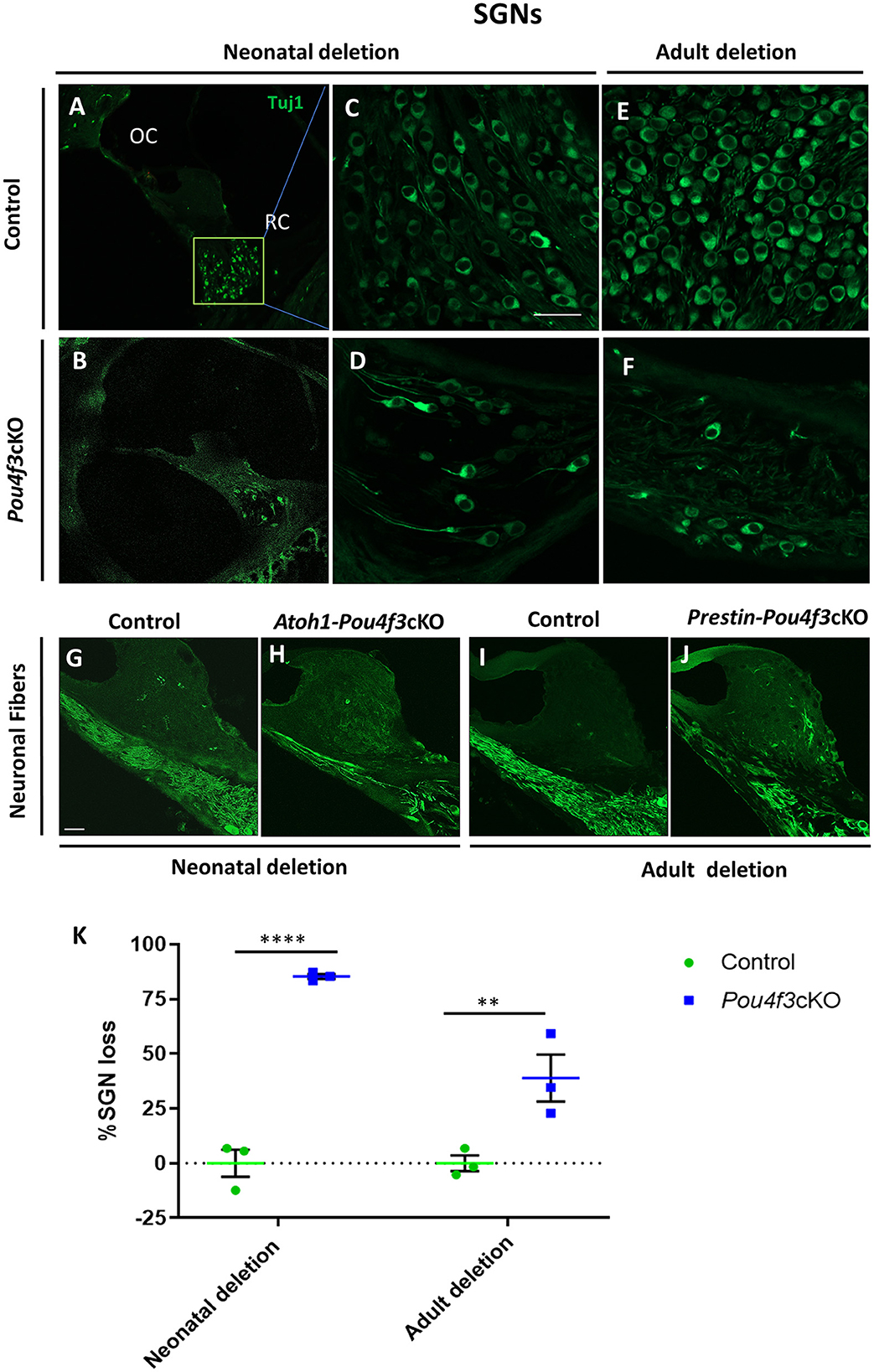
Figure 7. Pou4f3 deletion affects SGN survival in the long term. Representative confocal images of mid-modiolar cryosections from control (A, C, E) and cochleae with Pou4f3 deletion at P0/P1 using Atoh1-Pou4f3cKO mice (B, D) or at 8 weeks using Prestin-Pou4f3cKO mice (F). All samples were analyzed at 4 months post-tamoxifen. SGN cell bodies were identified using Tuj1 immunostaining (green) in the middle turn of each cochleae. Representative images of neuronal fibers projecting to the organ of Corti from control (G, I), Atoh1 Cre-Pou4f3cKO (H) and Prestin Cre-Pou4f3cKO (J) cochleae after deletion of Pou4f3 at neonatal or adult ages. (K) Quantification of Tuj1-positive SGNs after deletion of Pou4f3 at neonatal and adult ages. N = 3. There was a significant main effect of genotype [F(1,4) = 78.34, p = 0.0009]; age of Pou4f3 deletion [F(1,4) = 15.84, p = 0.0164]; and an interaction between genotype and age of Pou4f3 deletion [F(1,4) = 15.84, p = 0.0164]. Differences in the percentage of SGN loss between control samples and the respective age of deletion are indicated by the asterisks based on a Sidak's post-hoc test. OC, organ of Corti; RC, Rosenthal's canal. Scale bar = 20 μm.
4 Discussion
The present study demonstrates that the transcription factor Pou4f3 is essential for HC survival during postnatal maturation and in adulthood, which is consistent with the data from studies using germline deletion of Pou4f3 (Erkman et al., 1996; Xiang et al., 1997, 1998, 2003). After Pou4f3 deletion, HC death occurred via apoptosis as evidenced by TUNEL staining and upregulation of the pro-apoptotic genes, Bak1 and Bax. HC loss after Pou4f3 deletion did not seem to affect the number of surviving supporting cells, but small changes may not have been detected due to our sample size. This result is similar to other studies where the selective loss of cochlear HCs did not influence the survival of the adjacent supporting cells (Oesterle et al., 2008; Tong et al., 2015). However, the numbers of SGNs was reduced at 4 months after Pou4f3 deletion, with a larger negative impact when Pou4f3 was deleted from neonatal HCs.
OHCs are generally thought to be more susceptible to damage than IHCs (Stebbins et al., 1979; Rydmarker and Nilsson, 1987; Oesterle et al., 2008). While the mechanism is not fully understood, differences in mitochondrial function and intracellular calcium homeostasis have been suggested (Sha et al., 2001; Wang et al., 2019). In contrast, our data showed that after Pou4f3 deletion from HCs at birth, IHCs died at a faster rate than OHCs, with almost complete loss of IHCs within 1 week. A previous study showed that IHCs express a higher level of Pou4f3 compared to their OHC counterparts (Liu et al., 2014). Therefore, the enhanced rate of IHC loss may suggest an increased dependence on Pou4f3 to promote their survival. We unfortunately were not able to investigate the role of Pou4f3 in IHCs at older ages due to a lack of available CreER lines.
Our data also showed that immature OHCs died at a much faster rate after Pou4f3 deletion compared to mature OHCs. Specifically, when Pou4f3 was deleted at P0, >50% of the OHCs were missing within 5 days. However, when the deletion was initiated at 8 weeks of age, we observed no significant OHC loss until at least 2 weeks after Pou4f3 deletion. These findings suggest that once OHCs have matured they have a lesser dependence on Pou4f3 for their survival or perhaps the amount of POU4F3 protein present degrades at a slower rate in adult OHCs thereby leading to a delay in the amount of time it takes for POU4F3 to become sufficiently depleted. The degradation rate of POU4F3 protein may also differ as some studies have suggested changes in HC metabolism with age (Guo et al., 2022).
Many damaging insult studies using noise, aminoglycosides and cisplatin have shown that OHC loss generally follows a basal to apical gradient with OHC loss occurring faster and/or being more pronounced in the basal turn (Leake et al., 1997; Hertzano et al., 2004, 2007; Taylor et al., 2012; Kurabi et al., 2017). Consistent with these previous findings, Pou4f3 deletion from OHCs produced a similar base to apex gradient when the deletion occurred at juvenile or adult ages. Interestingly, there was a significant number of OHCs (20%−40%) remaining in the apex 6 weeks after Pou4f3 deletion was induced at the juvenile or adult ages, but all HCs were gone 4 weeks after Pou4f3 deletion was induced at birth. While we did not examine later timepoints, we suspect that these apical OHCs eventually die rather than being resistant to the effects of Pou4f3 depletion, however, future studies will be required to confirm this. Previous studies have shown that more HCs remain in the apical turn when Pou4f3 was mutated (using the ddl mutant) vs. a complete germline deletion of Pou4f3 (Xiang et al., 2003; Pauley et al., 2008). Thus, apical HCs may be sensitive to the age and level of Pou4f3 deletion.
The majority of SGNs (95%) are type I fibers which make synaptic connections with IHCs for sound transduction (Spoendlin, 1972; Perkins and Morest, 1975; Nienhuys and Clark, 1978). In contrast, OHCs are innervated by the type II SGN fibers (~5% of total SGNs), as well as a small percentage of type I fibers (Berglund and Ryugo, 1987; Raphael and Altschuler, 2003; Koundakjian et al., 2007; Coate et al., 2015; Elliott et al., 2021). Several studies have shown a critical period for SGN survival at neonatal ages where loss of HCs or loss of neurotrophic support caused SGN loss soon afterwards (Ernfors et al., 1995; Leake et al., 1997; Fritzsch et al., 1998; Rubel and Fritzsch, 2002; Barclay et al., 2011; Elliott et al., 2022). This period of sensitivity may occur because SGN innervation is still developing and being refined until ~ 2 weeks of age (Sobkowicz et al., 1986; Huang et al., 2012). When HCs were specifically ablated using diphtheria toxin (DT) in Pou4f3DTR mice, there was significant SGN loss when DT was injected at a neonatal age (P2), but not when DT was injected at P21 (Tong et al., 2015). In our model, we observed a similar amount of SGN loss (~85%) at 4 months after Pou4f3 deletion when both IHCs and OHCs were missing consistent with previous findings after germline Pou4f3 deletion (Xiang et al., 2003). However, when OHCs died after Pou4f3 was deleted at 8 weeks of age, there was a ~30%−50% loss of SGN cell bodies 4 months later. This unexpected SGN loss contrasts with the Tong et al. (2015) study. Since Pou4f3 deletion at 8 weeks of age used PrestinCreERT2, IHCs were not affected and remained intact. Thus, our results suggest that Pou4f3 may be transcribing a protein in OHCs that maintains SGN survival or inhibiting production of a protein that induces SGN cell death. In support, Pou4f3 has been shown to positively regulate expression of the growth factors, BDNF and NT-3 (Clough et al., 2004). It is also possible that the mechanism of cell death can impact SGN survival. It is unlikely that the insertion of loxP sites into the Pou4f3 locus impaired its function since our control samples were CreER-negative littermates which contained the Pou4f3loxP/loxP allele and had similar ABR thresholds and SGN densities as wild-type mice on a similar background (Huang et al., 2013; Tong et al., 2015).
Patients with DFNA15, who have one normal copy of Pou4f3, are born with normal hearing, and experience progressive hearing loss with different ages of onset, most commonly occurring after adolescence (Vahava et al., 1998; Frydman et al., 2000; Weiss et al., 2003). Thus, hearing loss could be caused by insufficient levels of POU4F3 or interference of the mutant protein with the normal POU4F3, impacting protein function, stability, cellular localization, or DNA binding (Collin et al., 2008). A recent study highlighted that expression of POU4F3 encoded by missense variants was reduced compared to wild-type POU4F3 levels. Although these mutant proteins localized to nucleus, they were vulnerable to degradation. However, a frame shift variant of Pou4f3 led to the formation of a truncated protein which primarily localized to the cytoplasm (Lee et al., 2023). In a familial case of DFNA15 where the mutation caused Pou4f3 complete deletion, haploinsufficiency is thought to be most likely the underlying cause of hearing impairment (Freitas et al., 2014). Genetic screening for Pou4f3 variants in a Japanese population demonstrated that individuals with mutations that produce a truncated POU4F3 showed earlier onset and slower progression of hearing loss than patients carrying different non-truncating mutations in Pou4f3 (Kitano et al., 2017). However other studies have shown that the age of onset varied within family members who have the same genetic mutation in the Pou4f3 gene (Pauw et al., 2008; Cui et al., 2020). Thus, while our work shows the importance of Pou4f3 in regulating the survival and maintenance of mouse cochlear HCs at postnatal and adult ages, there is still much to learn in understanding the mechanism of DFNA15-induced hearing loss.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
All animal work was conducted according to Institutional Animal Care and Use Committee approved protocol at Southern Illinois University School of Medicine. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
JS: Writing – original draft, Writing – review & editing, Formal analysis, Investigation, Methodology, Project administration, Data curation, Visualization. MR: Formal analysis, Writing – review & editing, Data curation, Methodology. BW: Conceptualization, Formal analysis, Funding acquisition, Writing – review & editing. BC: Conceptualization, Data curation, Formal analysis, Funding acquisition, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the NIDCD (R01 DC014441 to BC) and from the NIA (R01 AG073151 to BC and BW). The Southern Illinois University School of Medicine Research Imaging Facility was supported by a grant from the Office of Naval Research (N00014-15-1-2866).
Acknowledgments
We thank Dr. Suzy Baker at St. Jude Children' Research Hospital for providing the Atoh1-CreERTM mouse line and Dr. Jian Zuo at St Jude Children Hospital for providing the PrestinCreERT2 mouse line. We thank Drs. Thomas Brozoski and Don Caspary for sharing their ABR equipment and Kurt Wisner for technical assistance in performing ABR experiments.
Conflict of interest
BC is a consultant for Turner Scientific, LLC, and previously had sponsored research contracts with Decibel Therapeutics, Inc. and Otonomy, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2024.1369282/full#supplementary-material
References
Alonzi, T., Middleton, G., Wyatt, S., Buchman, V., Betz, A. K., Müller, U., et al. A. M. (2001). Role of STAT3 and PI 3-kinase/Akt in mediating the survival actions of cytokines on sensory neurons. Mol. Cell. Neurosci. 18, 270–282. doi: 10.1006/mcne.2001.1018
Anniko, M. (1983). Postnatal maturation of cochlear sensory hairs in the mouse. Anat. Embryol. 166, 355–368. doi: 10.1007/BF00305923
Badea, T. C., and Nathans, J. (2011). Morphologies of mouse retinal ganglion cells expressing transcription factors Brn3a, Brn3b, and Brn3c: analysis of wild type and mutant cells using genetically-directed sparse labeling. Vision Res. 51, 269–279. doi: 10.1016/j.visres.2010.08.039
Bai, X., Zhang, F., Xiao, Y., Jin, Y., Zheng, Q., Wang, H., et al. (2020). Identification of two novel mutations in POU4F3 gene associated with autosomal dominant hearing loss in Chinese families. J. Cell. Mol. Med. 24, 6978–6987. doi: 10.1111/jcmm.15359
Barclay, M., Ryan, A. F., and Housley, G. D. (2011). Type I vs type II spiral ganglion neurons exhibit differential survival and neuritogenesis during cochlear development. Neural Dev. 6:33. doi: 10.1186/1749-8104-6-33
Berglund, A. M., and Ryugo, D. K. (1987). Hair cell innervation by spiral ganglion neurons in the mouse. J. Comp. Neurol. 255, 560–570. doi: 10.1002/cne.902550408
Catlett-Falcone, R., Landowski, T. H., Oshiro, M. M., Turkson, J., Levitzki, A., Savino, R., et al. (1999). Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 10, 105–115. doi: 10.1016/S1074-7613(00)80011-4
Chow, L. M. L., Tian, Y., Weber, T., Corbett, M., Zuo, J., Baker, S. J., et al. (2006). Inducible Cre recombinase activity in mouse cerebellar granule cell precursors and inner ear hair cells. Dev. Dyn. 235, 2991–2998. doi: 10.1002/dvdy.20948
Clough, R. L., Sud, R., Davis-Silberman, N., Hertzano, R., Avraham, K. B., Holley, M., et al. (2004). Brn-3c (POU4F3) regulates BDNF and NT-3 promoter activity. Biochem. Biophys. Res. Commun. 324, 372–381. doi: 10.1016/j.bbrc.2004.09.074
Coate, T. M., Spita, N. A., Zhang, K. D., Isgrig, K. T., and Kelley, M. W. (2015). Neuropilin-2/Semaphorin-3F-mediated repulsion promotes inner hair cell innervation by spiral ganglion neurons. Elife 4. doi: 10.7554/eLife.07830.017
Coleman, B., Rickard, N. A., de Silva, M. G., and Shepherd, R. K. (2009). A protocol for cryoembedding the adult guinea pig cochlea for fluorescence immunohistology. J. Neurosci. Methods 176, 144–151. doi: 10.1016/j.jneumeth.2008.09.007
Collin, R. W. J., Chellappa, R., Pauw, R.-J., Vriend, G., Oostrik, J., van Drunen, W., et al. (2008). Missense mutations in POU4F3 cause autosomal dominant hearing impairment DFNA15 and affect subcellular localization and DNA binding. Hum. Mutat. 29, 545–554. doi: 10.1002/humu.20693
Cox, B. C., Chai, R., Lenoir, A., Liu, Z., Zhang, L., Nguyen, D.-H., et al. (2014). Spontaneous hair cell regeneration in the neonatal mouse cochlea in vivo. Development 141, 816–829. doi: 10.1242/dev.103036
Cui, T.-Y., Gao, X., Huang, S.-S., Sun, Y.-Y., Zhang, S.-Q., Jiang, X.-X., et al. (2020). Four novel variants in POU4F3 cause autosomal dominant nonsyndromic hearing loss. Neural Plast. 2020, 1–12. doi: 10.1155/2020/6137083
Eatock, R. A., and Hurley, K. M. (2003). Functional development of hair cells. Curr. Top. Dev. Biol. 57, 389–448. doi: 10.1016/S0070-2153(03)57013-2
Egilmez, O. K., and Kalcioglu, M. T. (2016). Genetics of nonsyndromic congenital hearing loss. Scientifica 2016:7576064. doi: 10.1155/2016/7576064
Ehret, G. (1976). Development of absolute auditory thresholds in the house mouse (Mus musculus). J. Am. Audiol. Soc. 1, 179–184.
Elliott, K. L., Fritzsch, B., Yamoah, E. N., and Zine, A. (2022). Age-related hearing loss: sensory and neural etiology and their interdependence. Front. Aging Neurosci. 14:814528. doi: 10.3389/fnagi.2022.814528
Elliott, K. L., Kersigo, J., Lee, J. H., Jahan, I., Pavlinkova, G., Fritzsch, B., et al. (2021). Developmental changes in peripherin-eGFP expression in spiral ganglion neurons. Front. Cell. Neurosci. 15:678113. doi: 10.3389/fncel.2021.678113
Erkman, L., McEvilly, R. J., Luo, L., Ryan, A. K., Hooshmand, F., O'Connell, S. M., et al. (1996). Role of transcription factors a Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature 381, 603–606. doi: 10.1038/381603a0
Ernfors, P., Van De Water, T., Loring, J., and Jaenisch, R. (1995). Complementary roles of BDNF and NT-3 in vestibular and auditory development. Neuron 14, 1153–1164. doi: 10.1016/0896-6273(95)90263-5
Fang, J., Zhang, W., Yamashita, T., Gao, J., Zhu, M., Zuo, J., et al. (2012). Outer hair cell-specific prestin-CreER T2 knockin mouse lines. Genesis 50, 124–131. doi: 10.1002/dvg.20810
Fettiplace, R. (2017). Hair cell transduction, tuning, and synaptic transmission in the mammalian cochlea. Compr. Physiol. Compr Physiol. 7, 1197–1227. doi: 10.1002/cphy.c160049
Freitas, É. L., Oiticica, J., Silva, A. G., Bittar, R. S. M., Rosenberg, C., Mingroni-Netto, R. C., et al. (2014). Deletion of the entire POU4F3 gene in a familial case of autosomal dominant non-syndromic hearing loss. Eur. J. Med. Genet. 57, 125–128. doi: 10.1016/j.ejmg.2014.02.006
Fritzsch, B., Barbacid, M., and Silos-santiago, I. (1998). The combined effects of trkB and trkC mutations on the innervation of the inner ear. Int. J. Dev. Neurosci. 16, 493–505. doi: 10.1016/S0736-5748(98)00043-4
Frydman, M., Vreugde, S., Nageris, B. I., Weiss, S., Vahava, O., Avraham, K. B., et al. (2000). Clinical characterization of genetic hearing loss caused by a mutation in the POU4F3 transcription factor. Arch. Otolaryngol.–Head Neck Surg. 126:633. doi: 10.1001/archotol.126.5.633
Fukada, T., Hibi, M., Yamanaka, Y., Takahashi-Tezuka, M., Fujitani, Y., Yamaguchi, T., et al. (1996). Two Signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity 5, 449–460. doi: 10.1016/S1074-7613(00)80501-4
Guo, D., Zhang, A., Zou, T., Ding, R., Chen, K., Pan, Y., et al. (2022). The influence of metabolic syndrome on age-related hearing loss from the perspective of mitochondrial dysfunction. Front. Aging Neurosci. 14:930105. doi: 10.3389/fnagi.2022.930105
Hertzano, R., Dror, A. A., Montcouquiol, M., Ahmed, Z. M., Ellsworth, B., Camper, S., et al. (2007). Lhx3, a LIM domain transcription factor, is regulated by Pou4f3 in the auditory but not in the vestibular system. Eur. J. Neurosci. 25, 999–1005. doi: 10.1111/j.1460-9568.2007.05332.x
Hertzano, R., Montcouquiol, M., Rashi-Elkeles, S., Elkon, R., Yücel, R., Frankel, W. N., et al. (2004). Transcription profiling of inner ears from Pou4f3ddl/ddl identifies Gfi1 as a target of the Pou4f3 deafness gene. Hum. Mol. Genet. 13, 2143–2153. doi: 10.1093/hmg/ddh218
Huang, L. C., Barclay, M., Lee, K., Peter, S., Housley, G. D., Thorne, P. R., et al. (2012). Synaptic profiles during neurite extension, refinement and retraction in the developing cochlea. Neural Dev. 7, 1–17. doi: 10.1186/1749-8104-7-38
Huang, M., Kantardzhieva, A., Scheffer, D., Liberman, M. C., and Chen, Z. Y. (2013). Hair cell overexpression of Islet1 reduces age-related and noise-induced hearing loss. J. Neurosci. 33, 15086–15094. doi: 10.1523/JNEUROSCI.1489-13.2013
Hume, C. R., Bratt, D. L., and Oesterle, E. C. (2007). Expression of LHX3 and SOX2 during mouse inner ear development. Gene Expr. Patterns 7, 798–807. doi: 10.1016/j.modgep.2007.05.002
Izumikawa, M., Batts, S. A., Miyazawa, T., Swiderski, D. L., and Raphael, Y. (2008). Response of the flat cochlear epithelium to forced expression of Atoh1. Hear. Res. 240, 52–56. doi: 10.1016/j.heares.2008.02.007
Kitano, T., Miyagawa, M., Nishio, S., Moteki, H., Oda, K., Ohyama, K., et al. (2017). POU4F3 mutation screening in Japanese hearing loss patients: massively parallel DNA sequencing-based analysis identified novel variants associated with autosomal dominant hearing loss. PLoS ONE 12:e0177636. doi: 10.1371/journal.pone.0177636
Korver, A. M. H., Smith, R. J. H., Van Camp, G., Schleiss, M. R., Bitner-Glindzicz, M. A. K., Lustig, L. R., et al. (2017). Congenital hearing loss. Nat. Rev. Dis. Primers 3:16094. doi: 10.1038/nrdp.2016.94
Koundakjian, E. J., Appler, J. L., and Goodrich, L. V. (2007). Auditory neurons make stereotyped wiring decisions before maturation of their targets. J. Neurosci. 27, 14078–14088. doi: 10.1523/JNEUROSCI.3765-07.2007
Kujawa, S. G., and Liberman, M. C. (2009). Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. 29, 14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009
Kurabi, A., Keithley, E. M., Housley, G. D., Ryan, A. F., and Wong, A. C.-Y. (2017). Cellular mechanisms of noise-induced hearing loss. Hear. Res. 349, 129–137. doi: 10.1016/j.heares.2016.11.013
Leake, P. A., Kuntz, A. L., Moore, C. M., and Chambers, P. L. (1997). Cochlear pathology induced by aminoglycoside ototoxicity during postnatal maturation in cats. Hear. Res. 113, 117–132. doi: 10.1016/S0378-5955(97)00133-0
Lee, M. K., Tuttle, J. B., Rebhun, L. I., Cleveland, D. W., and Frankfurter, A. (1990). The expression and posttranslational modification of a neuron-specific β-tubulin isotype during chick embryogenesis. Cell Motil. 17, 118–132. doi: 10.1002/cm.970170207
Lee, S. Y., Kim, M. Y., Han, J. H., Park, S. S., Yun, Y., Jee, S. C., et al. (2023). Ramifications of POU4F3 variants associated with autosomal dominant hearing loss in various molecular aspects. Sci. Rep. 13, 1–14. doi: 10.1038/s41598-023-38272-w
Lin, Y.-H., Lin, Y.-H., Lu, Y.-C., Liu, T.-C., Chen, C.-Y., Hsu, C.-J., et al. (2017). A novel missense variant in the nuclear localization signal of POU4F3 causes autosomal dominant non-syndromic hearing loss. Sci. Rep. 7:7551. doi: 10.1038/s41598-017-08236-y
Liu, Z., Fang, J., Dearman, J., Zhang, L., and Zuo, J. (2014). In vivo generation of immature inner hair cells in neonatal mouse cochleae by ectopic Atoh1 expression. PLoS ONE 9:e89377. doi: 10.1371/journal.pone.0089377
Madisen, L., Zwingman, T. A., Sunkin, S. M., Oh, S. W., Zariwala, H. A., Gu, H., et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140. doi: 10.1038/nn.2467
McGovern, M. M., Zhou, L., Randle, M. R., and Cox, B. C. (2018). Spontaneous hair cell regeneration is prevented by increased notch signaling in supporting cells. Front. Cell. Neurosci. 12:120. doi: 10.3389/fncel.2018.00120
Montgomery, S. C., and Cox, B. C. (2016). Whole mount dissection and immunofluorescence of the adult mouse cochlea. J. Vis. Exp. 107:53561. doi: 10.3791/53561-v
Nicolson, T. (2021). Navigating hereditary hearing loss: pathology of the inner ear. Front. Cell. Neurosci. 15:660812. doi: 10.3389/fncel.2021.660812
Nienhuys, T. G. W., and Clark, G. M. (1978). Frequency discrimination following the selective destruction of cochlear inner and outer hair cells. Science 199, 1356–1357. doi: 10.1126/science.628846
Oesterle, E. C., Campbell, S., Taylor, R. R., Forge, A., and Hume, C. R. (2008). Sox2 and Jagged1 expression in normal and drug-damaged adult mouse inner ear. J. Assoc. Res. Otolaryngol. 9, 65–89. doi: 10.1007/s10162-007-0106-7
Ohlemiller, K. K., Jones, S. M., and Johnson, K. R. (2016). Application of mouse models to research in hearing and balance. J. Assoc. Res. Otolaryngol. 17, 493–523. doi: 10.1007/s10162-016-0589-1
Pauley, S., Kopecky, B., Beisel, K., Soukup, G., and Fritzsch, B. (2008). Stem cells and molecular strategies to restore hearing. Panminerva Med. 50, 41–53.
Pauw, R. J., van Drunen, F. J. W., Collin, R. W. J., Huygen, P. L. M., Kremer, H., Cremers, C. W. R. J., et al. (2008). Audiometric characteristics of a dutch family linked to DFNA15 with a novel mutation (p.L289F) in POU4F3. Arch. Otolaryngol.–Head Neck Surg. 134:294. doi: 10.1001/archotol.134.3.294
Perkins, R. E., and Morest, D. K. (1975). A study of cochlear innervation patterns in cats and rats with the Golgi method and Nomarski optics. J. Comp. Neurol. 163, 129–158. doi: 10.1002/cne.901630202
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, 45e−445. doi: 10.1093/nar/29.9.e45
Raphael, Y., and Altschuler, R. A. (2003). Structure and innervation of the cochlea. Brain Res. Bull. 60, 397–422. doi: 10.1016/S0361-9230(03)00047-9
Rubel, E. W., and Fritzsch, B. (2002). Auditory system development: primary auditory neurons and their targets. Annu. Rev. Neurosci. 25, 51–101. doi: 10.1146/annurev.neuro.25.112701.142849
Rydmarker, S., and Nilsson, P. (1987). II. Effects on the inner and outer hair cells. Acta Otolaryngol. 104, 25–43. doi: 10.3109/00016488709124799
Sha, S.-H., Taylor, R., Forge, A., and Schacht, J. (2001). Differential vulnerability of basal and apical hair cells is based on intrinsic susceptibility to free radicals. Hear. Res. 155, 1–8. doi: 10.1016/S0378-5955(01)00224-6
Sheffield, A. M., and Smith, R. J. H. (2019). The epidemiology of deafness. Cold Spring Harb. Perspect. Med. 9:a033258. doi: 10.1101/cshperspect.a033258
Sobkowicz, H. M., Rose, J. E., Scott, G. L., and Levenick, C. V. (1986). Distribution of synaptic ribbons in the developing organ of corti. J. Neurocytol. 15, 693–714. doi: 10.1007/BF01625188
Spoendlin, H. (1972). Innervation densities of the cochlea. Acta Otolaryngol. 73, 235–248. doi: 10.3109/00016487209138937
Stebbins, W. C., Hawkins, J. E., Johnsson, L.-G., and Moody, D. B. (1979). Hearing thresholds with outer and inner hair cell loss. Am. J. Otolaryngol. 1, 15–27. doi: 10.1016/S0196-0709(79)80004-6
Sun, S., Babola, T., Pregernig, G., So, K. S., Nguyen, M., Su, S.-S. M., et al. (2018). Hair cell mechanotransduction regulates spontaneous activity and spiral ganglion subtype specification in the auditory system. Cell 174, 1247–1263.e15. doi: 10.1016/j.cell.2018.07.008
Taylor, R. R., Jagger, D. J., and Forge, A. (2012). Defining the cellular environment in the organ of corti following extensive hair cell loss: a basis for future sensory cell replacement in the cochlea. PLoS ONE 7:e30577. doi: 10.1371/journal.pone.0030577
Tollefson, M. R., Gogal, R. A., Weaver, A. M., Schaefer, A. M., Marini, R. J., Azaiez, H., et al. (2023). Assessing variants of uncertain significance implicated in hearing loss using a comprehensive deafness proteome. Hum. Genet. 142, 819–834. doi: 10.1007/s00439-023-02559-9
Tong, L., Strong, M. K., Kaur, T., Juiz, J. M., Oesterle, E. C., Hume, C., et al. (2015). Selective deletion of cochlear hair cells causes rapid age-dependent changes in spiral ganglion and cochlear nucleus neurons. J. Neurosci. 35, 7878–7891. doi: 10.1523/JNEUROSCI.2179-14.2015
Towers, E. R., Kelly, J. J., Sud, R., Gale, J. E., and Dawson, S. J. (2011). Caprin-1 is a target of the deafness gene Pou4f3 and is recruited to stress granules in cochlear hair cells in response to ototoxic damage. J. Cell Sci. 124, 1145–1155. doi: 10.1242/jcs.076141
Vahava, O., Morell, R., Lynch, E. D., Weiss, S., Kagan, M. E., Ahituv, N., et al. (1998). Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science 279, 1950–1954. doi: 10.1126/science.279.5358.1950
Wang, X., Zhu, Y., Long, H., Pan, S., Xiong, H., Fang, Q., et al. (2019). Mitochondrial calcium transporters mediate sensitivity to noise-induced losses of hair cells and cochlear synapses. Front. Mol. Neurosci. 11:469. doi: 10.3389/fnmol.2018.00469
Weber, T., Corbett, M. K., Chow, L. M. L., Valentine, M. B., Baker, S. J., Zuo, J., et al. (2008). Rapid cell-cycle reentry and cell death after acute inactivation of the retinoblastoma gene product in postnatal cochlear hair cells. Proc. Nat. Acad. Sci. 105, 781–785. doi: 10.1073/pnas.0708061105
Weiss, S., Gottfried, I., Mayrose, I., Khare, S. L., Xiang, M., Dawson, S. J., et al. (2003). The DFNA15 deafness mutation affects POU4F3 protein stability, localization, and transcriptional activity. Mol. Cell. Biol. 23, 7957–7964. doi: 10.1128/MCB.23.22.7957-7964.2003
Xiang, M., Gan, L., Li, D., Chen, Z. Y., Zhou, L., O'Malley, B. W., et al. (1997). Essential role of POU-domain factor Brn-3c in auditory and vestibular hair cell development. Proc. Natl. Acad. Sci. USA. 94, 9445–9450. doi: 10.1073/pnas.94.17.9445
Xiang, M., Gao, W.-Q., Hasson, T., and Shin, J. J. (1998). Requirement for Brn-3c in maturation and survival, but not in fate determination of inner ear hair cells. Development 125, 3935–3946. doi: 10.1242/dev.125.20.3935
Xiang, M., Maklad, A., Pirvola, U., and Fritzsch, B. (2003). Brn3c null mutant mice show long-term, incomplete retention of some afferent inner ear innervation. BMC Neurosci. 4, 1–16. doi: 10.1186/1471-2202-4-2
Yu, H. V., Tao, L., Llamas, J., Wang, X., Nguyen, J. D., Trecek, T., et al. (2021). POU4F3 pioneer activity enables ATOH1 to drive diverse mechanoreceptor differentiation through a feed-forward epigenetic mechanism. Proc. Nat. Acad. Sci. 118:e2105137118. doi: 10.1073/pnas.2105137118
Keywords: Pou4f3, DFNA15, hearing loss, hair cell maturation, hair cell survival
Citation: Singh J, Randle MR, Walters BJ and Cox BC (2024) The transcription factor Pou4f3 is essential for the survival of postnatal and adult mouse cochlear hair cells and normal hearing. Front. Cell. Neurosci. 18:1369282. doi: 10.3389/fncel.2024.1369282
Received: 11 January 2024; Accepted: 26 February 2024;
Published: 19 March 2024.
Edited by:
Hinrich Staecker, University of Kansas Medical Center, United StatesReviewed by:
Bernd Fritzsch, University of Nebraska Medical Center, United StatesArtur A. Indzhykulian, Massachusetts Eye & Ear Infirmary and Harvard Medical School, United States
Copyright © 2024 Singh, Randle, Walters and Cox. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brandon C. Cox, YmNveEBzaXVtZWQuZWR1