
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Cell. Neurosci. , 04 April 2024
Sec. Cellular Neuropathology
Volume 18 - 2024 | https://doi.org/10.3389/fncel.2024.1367838
This article is part of the Research Topic Pathogenic Potassium Channel Variants in Neurological Disorders: From Functional Analysis to Personalized Pharmacological Approaches View all 8 articles
 Ilaria Mosca1†
Ilaria Mosca1† Elena Freri2†
Elena Freri2† Paolo Ambrosino3Giorgio Belperio3
Paolo Ambrosino3Giorgio Belperio3 Tiziana Granata2
Tiziana Granata2 Laura Canafoglia4
Laura Canafoglia4 Francesca Ragona2Roberta Solazzi2
Francesca Ragona2Roberta Solazzi2 Ilaria Filareto2Barbara Castellotti5Giuliana Messina5Cinzia Gellera5
Ilaria Filareto2Barbara Castellotti5Giuliana Messina5Cinzia Gellera5 Jacopo C. DiFrancesco6*
Jacopo C. DiFrancesco6* Maria Virginia Soldovieri1*
Maria Virginia Soldovieri1* Maurizio Taglialatela7
Maurizio Taglialatela7Variants in KCNT1 are associated with a wide spectrum of epileptic phenotypes, including epilepsy of infancy with migrating focal seizures (EIMFS), non-EIMFS developmental and epileptic encephalopathies, autosomal dominant or sporadic sleep-related hypermotor epilepsy, and focal epilepsy. Here, we describe a girl affected by drug-resistant focal seizures, developmental delay and behavior disorders, caused by a novel, de novo heterozygous missense KCNT1 variant (c.2809A > G, p.S937G). Functional characterization in transiently transfected Chinese Hamster Ovary (CHO) cells revealed a strong gain-of-function effect determined by the KCNT1 p.S937G variant compared to wild-type, consisting in an increased maximal current density and a hyperpolarizing shift in current activation threshold. Exposure to the antidepressant drug fluoxetine inhibited currents expressed by both wild-type and mutant KCNT1 channels. Treatment of the proband with fluoxetine led to a prolonged electroclinical amelioration, with disappearance of seizures and better EEG background organization, together with an improvement in behavior and mood. Altogether, these results suggest that, based on the proband’s genetic and functional characteristics, the antidepressant drug fluoxetine may be repurposed for the treatment of focal epilepsy caused by gain-of-function variants in KCNT1. Further studies are needed to verify whether this approach could be also applied to other phenotypes of the KCNT1-related epilepsies spectrum.
Variants in KCNT1, encoding for KNa1.1 subunits forming Na+-activated K+ channels, are associated with a wide spectrum of epileptic phenotypes, mainly including epilepsy of infancy with migrating focal seizures (EIMFS), developmental and epileptic encephalopathy (DEE) other than EIMFS, and autosomal dominant or sporadic sleep-related hypermotor epilepsy (Gertler et al., 2018; Bonardi et al., 2021). Moreover, KCNT1 variants have been rarely reported in patients with focal epilepsy (Møller et al., 2015; Gertler et al., 2018).
KCNT1, similarly to highly-homologous KCNT2 (Cioclu et al., 2023) and classical voltage-gated Kv subunits, show a topological arrangement with six transmembrane segments (S1-S6) and both N- and C-termini located intracellularly. In particular, the C-terminus encompasses two regulators of K+ conductance (RCK1 and RCK2) domains located below the pore axis which form a large gating ring conferring channel opening sensitivity to intracellular Na+ ([Na]i); multiple binding sites for other cations and lipids have also been identified within this region (Zhang et al., 2023). Functional KCNT1 channels assemble as homo- or heterotetramers and contribute to single action potential repolarization, single spike afterpotentials, or to the slow afterhyperpolarization (sAHP) which follows a burst of action potentials (Bonardi et al., 2021).
Irrespective of the clinical phenotype, the large majority of epilepsy-causing variants in KCNT1 prompt gain-of-function (GoF) effects (Bonardi et al., 2021), thus suggesting that pharmacological strategies able to reduce channel function could be considered as precision-therapy approaches. Among them, KCNT1 blockers such as the antiarrhythmic drug quinidine have been tested in few patients, although contrasting results regarding quinidine anticonvulsant efficacy have been reported; moreover, drug’s clinical use is further limited by concerns regarding its cardiac toxicity (Dilena et al., 2018; Fitzgerald et al., 2019; Liu et al., 2023).
In the present study, we report a patient with drug-resistant focal epilepsy carrying a novel, de novo missense KCNT1 variant prompting GoF effects on channel function in vitro. We show that the anti-depressant fluoxetine counteracted mutation-induced functional defects in vitro, and that treatment with this drug resulted in a significant and long-lasting clinical improvement in the proband.
Written informed consent was obtained for the patient and family members. Genomic DNA was extracted from peripheral blood lymphocytes as previously reported (Campostrini et al., 2018). The patient was investigated using a multigenic NGS panel (Agilent Sure Design, Santa Clara, CA) containing 300 genes (list available upon request) correlated with developmental epileptic encephalopathy (DEE). The average coverage at 20× for this panel was greater than 99%. The resulting sequences were aligned to the GRCh37/hg19 reference genome (MiSeq software). Data analysis was obtained using MiSeq Reporter vs. 2.4.60, Variant Studio vs. 2.2 (Illumina) and CLC Genomics Workbench vs. 7.0 (Qiagen). Variants with MAF > 1% were considered benign. Other variants were classified according to ACMG criteria (Richards et al., 2015). Parental segregation was performed by direct sequencing with Applied Biosystems (Life technologies) ABI 1330 XL automated sequencer.
The p.S937G variant herein investigated was engineered in a plasmid containing the cDNA for a myc-DDK tagged human isoform 2 (Q5JUK3–2) of KCNT1 (RC214820; Origene, Rockville, MD, United States) by Quick-change mutagenesis, as previously described (Rizzo et al., 2016; Dilena et al., 2018). Mutant vector was verified by Sanger sequencing. Wild-type (WT) and mutant cDNAs were expressed in Chinese hamster ovary (CHO) cells by transient transfection using Lipofectamine 2000 (Invitrogen, Milan, Italy), as described (Rizzo et al., 2016; Dilena et al., 2018). A plasmid encoding for the Enhanced Green Fluorescent Protein (EGFP; Clontech, Palo Alto, CA) was used as a transfection marker. Total cDNA in the transfection mixture was kept constant at 4 μg.
Electrophysiological experiments were performed as previously described (Rizzo et al., 2016; Dilena et al., 2018). Briefly, macroscopic currents from transiently transfected CHO cells were recorded at room temperature (20°C-22°C) 24 h after transfection, with an Axopatch 200B amplifier (Molecular Devices, Union City, CA) using the whole-cell configuration of the patch-clamp technique. The pipette (intracellular) solution contained (in mM): 130 KCl, 10 NaCl, 10 HEPES, 5 EGTA, 5 Mg-ATP, pH 7.3–7.4 with HCl. Extracellular solution composition was (in mM): 138 NaCl, 5.4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.4 with NaOH. Data acquisition and analysis were performed as described (Rizzo et al., 2016; Dilena et al., 2018). To generate conductance-voltage curves, cells were held at −80 mV, then depolarized for 600 ms from −90 mV to +60 mV in 10-mV increments. Current densities (expressed in pA/pF) were calculated as peak K+ currents at all tested membrane potentials divided by cell capacitance (C). At each potential, peak currents (in pA) were subtracted from those measured in the previous pulse and normalized to the maximal current to obtain conductance values which were then fitted to a Boltzmann equation to obtain “V½” and “k” values. Drugs (Sigma-Aldrich, Milan, Italy) were dissolved in chloroform (quinidine; final vehicle concentration ≤ 0.01%) or DMSO (fluoxetine; final vehicle concentration ≤ 0.01%) and perfused using a fast solution exchange system as previously reported (Ambrosino et al., 2014). In these experiments, currents were activated by 1 s voltage ramps from −90 mV to +60 mV every 10 s; drug-induced current blockade was expressed as the percentage of +60 mV current inhibition produced by a 2 min drug application.
Each data point is the mean ± SEM of at least 7 determinations, each performed in different cells from at least 3 separate experiments. Statistically significant differences were evaluated with the Student’s t-test (indicated with t-test) or with the ANOVA followed by the Student–Newman–Keuls test (indicated with ANOVA-SNK), with the threshold set at p < 0.05.
The proband is a girl, now 20 years old, born after an uneventful pregnancy and delivery, without familiar history for epilepsy or other neurological diseases. Psychomotor development was normal during the first years of life.
At the age of 3 years, she started presenting focal seizures with temporal semiology (staring, feeling of terror, swallowing, abdomen tightness). EEG reported epileptic spikes on the left temporal regions, while brain MRI was unremarkable (data not shown). Treatment with topiramate led to a transient resolution of seizures, but daily episodes recurred after few months. Topiramate was than replaced with methyl bromide and diazepam, however without benefit. In the next 3 years, reduction in seizures frequency from daily to monthly was achieved with carbamazepine and clobazam. At 11 y.o., the patient experienced focal status epilepticus (SE) interrupted by intravenous midazolam. Several antiseizure medications (ASMs) including lamotrigine, nitrazepam, oxcarbazepine, acetazolamide, and clonazepam, resulted ineffective (Figure 1A). With menarche at the age of 13 y.o., seizures frequency increased during menses.
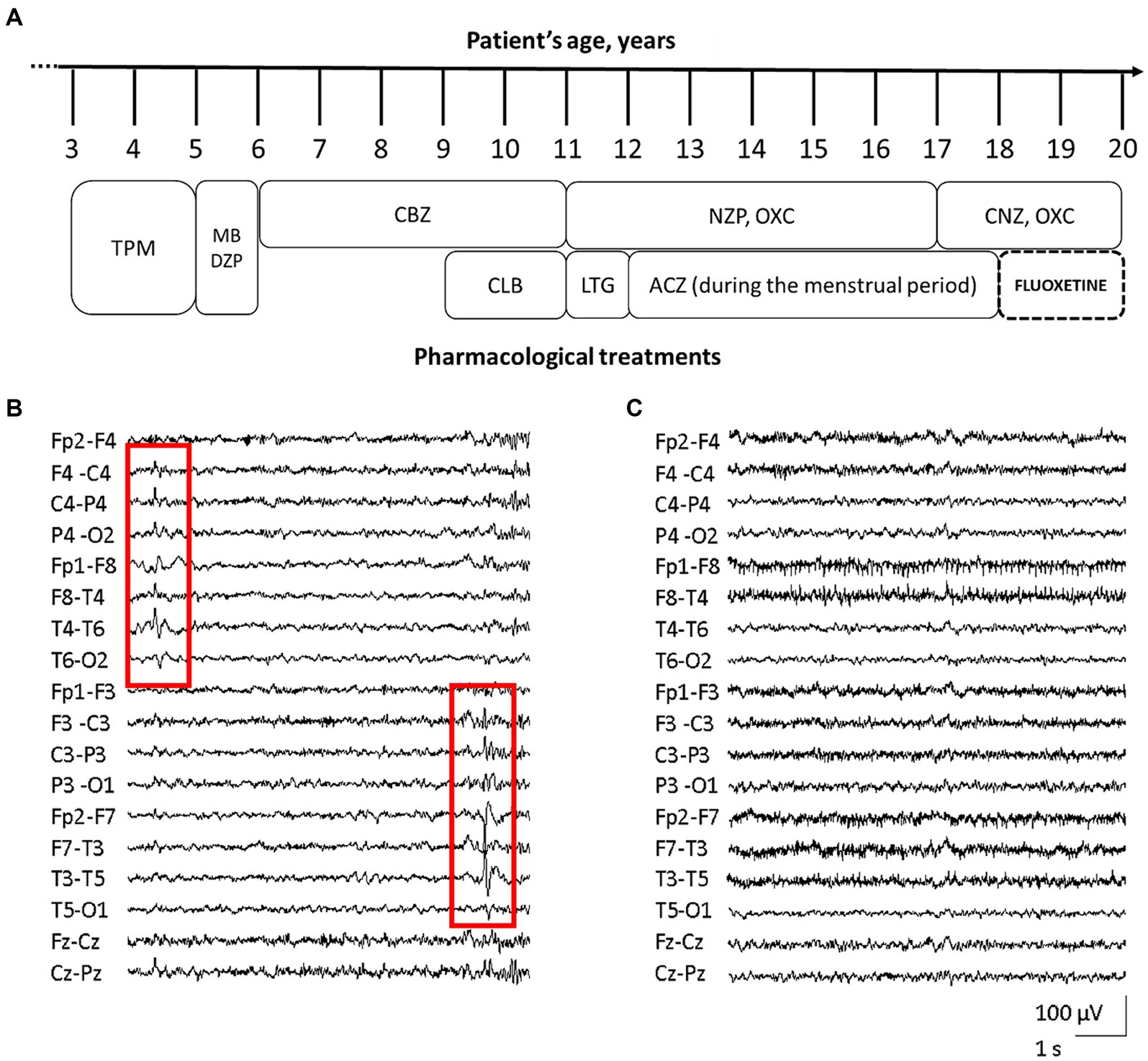
Figure 1. Patient’s pharmacological treatment, EEG traces before and after fluoxetine therapy. Schematic representation of the proband’s complex pharmacological treatment over time [legend of ASMs: acetazolamide (ACZ), carbamazepine (CBZ), clobazam (CLB), clonazepam (CNZ), diazepam (DZP), lamotrigine (LTG), methyl bromide (MB), nitrazepam (NZP), oxcarbazepine (OXC), topiramate (TPM)] (A). EEG trace assessed before fluoxetine treatment, showing diffuse, low-amplitude, theta background activity, and bitemporal epileptiform abnormalities with alternating prevalence of side (red squares, B). EEG during treatment with fluoxetine, revealing the improvement of background activity with alpha-slow rhythm of increased amplitude, together with disappearance of epileptic activity (C).
The patient came to our attention at 16 years of age. EEG showed diffuse, low-amplitude, theta background activity, and bitemporal epileptiform abnormalities with alternating prevalence of side (Figure 1B). Her neurological examination was characterized by severe intellectual disability (IQ 44), with anxiety disorder and aggressiveness. At that time the girl was experiencing daily seizures, despite treatment with oxcarbazepine and nitrazepam. Repeated brain MRI and CGH-array were unrevealing. NGS target panel for epilepsy genes detected the novel heterozygous c.2809A > G (NM_020822.2) missense variant in KCNT1, causing the p.S937G substitution (rs1554778379), resulting de novo in the proband.
The KCNT1 p.S937G variant is localized in the RCK2 domain (Figure 2A), affecting a residue highly conserved among species (Figure 2B). To investigate the functional properties of KCNT1 channels incorporating this novel variant, we performed in vitro experiments in CHO cells expressing WT or mutant KCNT1 subunits. As previously reported (Rizzo et al., 2016; Dilena et al., 2018), transfection with KCNT1 cDNA elicited outwardly-rectifying currents in response to depolarizing voltage pulses from −90 mV to +60 mV (maximal current density at +60 mV was 121.1 ± 12.9 pA/pF; n = 34; Figures 2C,D). KCNT1 currents displayed complex activation kinetics, with an instantaneous, time-independent component (IINST), followed by a slower, time-dependent one (ISTEADY-STATE – IINST). At +60 mV, the ratio between currents measured at the beginning (IINST) and at the end (ISTEADY-STATE) of the depolarizing step was 0.25 ± 0.01 (n = 46; Figure 2E). Expression of homomeric KCNT1 p.S937G channels also generated outwardly-rectifying currents. When compared to WT, current densities of mutant KCNT1 at +60 mV were significantly larger (maximal current density at +60 mV was 282.6 ± 34.0 pA/pF; n = 25; p < 0.05 versus WT, t-test; Figures 2C,D), and the IINST/ISTEADY-STATE ratio was significantly increased (IINST/ISTEADY-STATE was 0.67 ± 0.04; n = 23; p < 0.05 versus WT, t-test; Figures 2C–E). Moreover, the G/V curve was shifted in the hyperpolarizing direction (Figure 2F); indeed, Boltzmann analysis of the G/V for KCNT1 p.S937G channels revealed that the activation midpoint (V1/2) was significantly more negative when compared to WT channels (V1/2 were 13.6 ± 2.1 mV or − 33.4 ± 1.6 mV for KCNT1 or KCNT1 p.S937G channels, respectively; n = 19 or 25, respectively; p < 0.05, t-test). By contrast, no significant difference was measured in the slope (k values were 18.7 ± 1.1 or 16.7 ± 1.1 mV/efold for KCNT1 or KCNT1 p.S937G channels, respectively; p > 0.05, t-test). Qualitatively similar, although quantitatively smaller, effects were measured upon co-expression of mutant KCNT1 p.S937G with WT subunits, an experimental strategy mimicking the heterozygous condition of the proband carrying the variant on a single allele. The maximal current density at +60 mV was 192.2 ± 19.7 pA/pF (Figure 2D), the IINST/ISTEADY-STATE ratio was 0.34 ± 0.03 (Figure 2E), and the V1/2 was −9.1 ± 2.2 mV (Figure 2F; n = 22–23; p < 0.05 versus KCNT1 and KCNT1 p.S937G homomers, ANOVA-SNK).
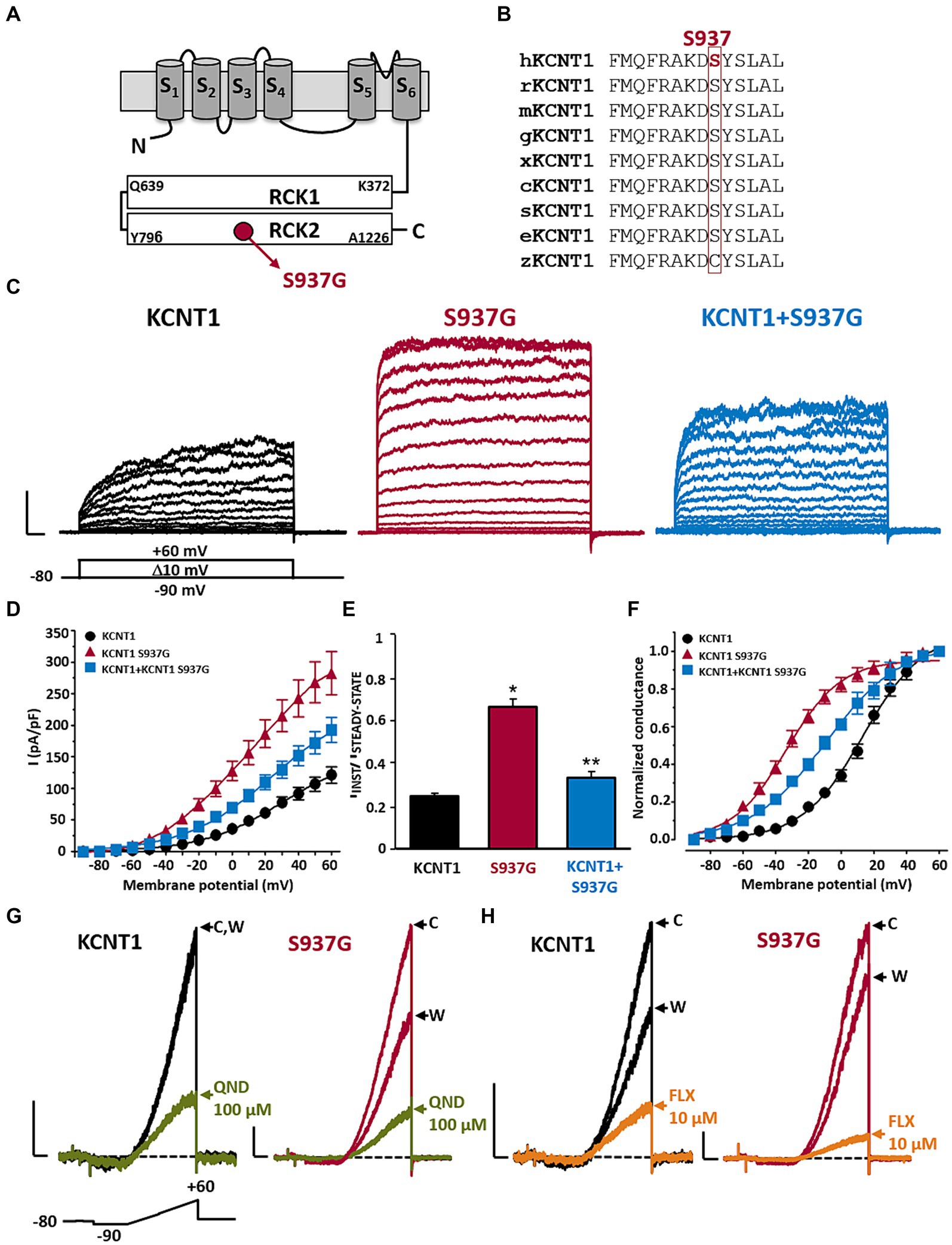
Figure 2. Functional and pharmacological characterization of KCNT1 and KCNT1 p.S937G channels. Topological representation of a KCNT1 subunit and localization of the p.S937G variant (A). Partial alignment of KCNT1 protein among species (B). Representative family traces recorded in cells expressing the indicated channels in response to the voltage protocol shown below the leftmost traces (C). Current scale: 1 nA; time scale: 50 ms. Average of IINST/ISTEADY-STATE ratios (D), current densities (E), and conductance/voltage curves (F) measured in cells expressing the indicated channels. Representative traces recorded in response to the ramp protocol shown below the leftmost traces in cells expressing the indicated channels in control solution (indicated with C), after 2-min exposure to 100 μM quinidine (QND; blue traces in panel G) or 10 μM fluoxetine (FLX; green traces in panel H), or upon drug washout (indicated with W). Current scale: 500 pA; time scale: 200 ms. * = p < 0.05 versus KCNT1; ** = p < 0.05 versus KCNT1 + S937G.
Taken together, these results suggest that the KCNT1 p.S937G variant leads to strong GoF effects by increasing the amplitude of the maximal currents and shifting their activation threshold toward hyperpolarized potentials.
To counteract the mutation-induced GoF changes, we tested in vitro the effect of different KCNT blockers, including the antiarrhythmic drug quinidine (QND). As previously demonstrated (Rizzo et al., 2016; Dilena et al., 2018), 100 μM QND inhibited WT currents at +60 mV by 70.2 ± 4.1% (n = 7; Figure 2G); KCNT1 current blockade by QND was fully reversible after ~3 min of drug washout (Figure 2G). A slight increase in current inhibition was measured in KCNT1 p.S937G currents (% of current inhibition at +60 mV was 81.6 ± 2.4, n = 12; p < 0.05 versus WT, t-test; Figure 2G), suggesting a possible therapeutic use of this drug. However, considering the poor compliance to quinidine treatment, in view of the scarce clinical efficacy and its potential cardiac toxicity (Dilena et al., 2018; Fitzgerald et al., 2019; Liu et al., 2023), we searched for alternative approaches. Among KCNT blockers, we focused on the well-known antidepressant drug fluoxetine (FLX), since we recently showed that this drug was effective in blocking currents expressed by homologous KCNT2 channels (both WT and carrying DEE-causing GoF pathogenic variants) (Cioclu et al., 2023).
Exposure to 10 μM FLX blocked 79.8 ± 2.0% of KCNT1 currents elicited at +60 mV (n = 14; Figure 2H); currents largely recovered after ~3 min of drug washout (Figure 2H). FLX was slightly more effective in blocking KCNT1 p.S937G currents (% of current inhibition at +60 mV was 86.7 ± 1.7%; n = 9; p < 0.05 versus WT, t-test; Figure 2H). Similar results were observed on KCNT1 + KCNT1 p.S937G heteromeric channels, showing a current reduction of 87.4 ± 1.7% (n = 14; p < 0.05 versus WT, p > 0.05 versus KCNT1 p.S937G, ANOVA-SNK; Figure 2H).
Considering the presence of an active anxiety disorder, a clear indication to FLX therapy, seizures not responding to several ASMs, and according to the described in vitro data showing the ability of FLX to block currents expressed by p.S937G KCNT1 channels, at the age of 18 years the patient started treatment with this drug. With an initial dose of 10 mg per day, the posology was gradually increased until 60 mg/day (1.2 mg/kg), with a plasma level of 412.6 ng/mL, resulting well tolerated.
During FLX treatment, the patient reported a clear and prolonged clinical benefit with disappearance of seizures, except for sporadic episodes during a SARS-COV-2 infection. EEG recording revealed alpha-slow background activity of greater amplitude, together with disappearance of epileptic abnormalities (Figure 1C).
After 2 years of follow-up, the patient is still under treatment with fluoxetine. Due to the absence of further seizures, acetazolamide was deemed no longer necessary, while the other ASMs were not reduced as requested by the patient and her parents. Along with amelioration of the epileptic features, the patient also reported a significant improvement in behavior and mood, and the achievement of important school milestones with high school graduation.
In the present study we report a patient with drug-resistant focal epilepsy, developmental delay and behavior disorders carrying a novel KCNT1 de novo heterozygous missense variant (p.S937G). This variant affects a highly conserved amino acid residue located in the regulator of K+ conductance-2 (RCK2) domain (Zhang et al., 2023), a critical region for channel gating. Indeed, similarly to the largest majority of previously-described KCNT1 pathogenic variants (Rizzo et al., 2016; Dilena et al., 2018; Gertler et al., 2018), mutant channels displayed significant GoF effects in terms of increased current density and leftward shift in the voltage-dependence of activation, both in homomeric and heteromeric channels when expressed together with wild-type subunits. In addition to KCNT1, GoF variants in other potassium channel genes, including KCNQ2 and KCNQ3 (Miceli et al., 2015; Sands et al., 2019), KCNT2 (Cioclu et al., 2023), among several others (Niday and Tzingounis, 2018), are associated with hyperexcitability-linked neurological diseases; how GoF variants in these channels affect neuronal and networks function is largely unknown. Among potential mechanisms, it has been hypothesized that KCNT1 GoF variants lead to network hyperexcitability by: I- selectively accelerating action potential repolarization in excitatory neurons, thereby limiting sodium channel inactivation and causing a higher spiking frequency; II- selectively reducing membrane excitability or inducing spike frequency adaptation in inhibitory neurons, resulting in disinhibition; or III- promoting developmental alterations in synaptic connectivity.
In the cortex and hippocampus, single-cell RNA sequencing data suggest that KCNT1 is robustly expressed in parvalbumin-positive (PV+) and somatostatin-positive (SOM+) interneuron subsets, almost absent in vasoactive intestinal peptide-positive (VIP+) interneurons, while it shows intermediate expression in excitatory pyramidal neurons (Cembrowski et al., 2016; Erö et al., 2018). Notably, recent results from animal models in which KCNT1 GoF variants have been introduced suggest that preferential dampening of intrinsic excitability in specific subsets of cortical (Shore et al., 2020) and hippocampal (Gertler et al., 2022) inhibitory neurons provides a major contribution to network hyperexcitability and hypersynchronization, leading to changes in synaptic connectivity, disrupted excitation/inhibition balance, and seizures (Shore et al., 2020).
In the case here reported, despite treatment with different ASMs, focal seizures persisted at high frequency; thus, we searched for pharmacological approaches to counteract the increase of KCNT1 currents caused by the novel p.S937G variant. Thus, we first tested and showed the ability of the KCNT blocker quinidine to reduce in vitro GoF effects caused by the variant. However, due to the unsatisfactory clinical response and to well-known cardiac concerns associated to quinidine treatment (Dilena et al., 2018; Cioclu et al., 2023; Zhang et al., 2023), and considering that patients with the best quinidine response have variants localized distal to the RCK2 domain (Fitzgerald et al., 2019), we searched for an alternative compound with potentially better efficacy and safety profiles.
Fluoxetine, a well-tolerated anti-depressant drug, has been suggested as a repurposed precision therapy for severe epilepsies caused by GoF variants in other neuronal Kv channels (Ambrosino et al., 2023; Cioclu et al., 2023). Thus, we tested in vitro the effect of fluoxetine on KCNT1 p.S937G channels, demonstrating that the drug’s ability to counteract the GoF features observed in channels incorporating the p.S937G variant. Thus, considering the behavioral disturbances (anxiety disorders, aggressiveness) manifested by the proband, and considering the scarce efficacy of several other ASMs, she started treatment with fluoxetine, resulting in a prolonged clinical and electroencephalographic improvement, with a complete seizure disappearance and a marked improvement in mood and behavior.
The clinical response to fluoxetine treatment observed in the herein reported transitional age patient cannot be accounted for by the disease natural history, but seems to be correlated to the drug inclusion in the therapeutic regimen. At steady-state, fluoxetine plasma level resulted within the range observed during antidepressant treatment (120–500 ng/mL), achieving a drug concentration range of 0.6–2 μM, similar to those prompting a significant block of KCNT1 currents in vitro. Being highly lipophilic, fluoxetine can reach elevated concentrations in the brain (Bolo, 2000) (brain/plasma ratio is about 10), suggesting that the block of KCNT1 current observed with this drug may have contributed to the clinical amelioration observed in the proband. However, it should be highlighted that fluoxetine cannot be considered a KCNT1-specific blocker, since this drug also blocks other potassium channels, such as KCNT2 (Cioclu et al., 2023), Kv3.1 (Choi et al., 2001) and Kv1.3 (Choi et al., 1999), with similar potency; thus, a direct causal relationship between drug-dependent blockade of KCNT1 currents and amelioration of the underlying phenotype is difficult to establish. Moreover, the occurrence of disease-related compensatory mechanisms (Pan et al., 2018), possibly including the up-regulation of additional ion channel targets contributing to treatment efficacy (Hill et al., 2022), should also be taken into account. Nonetheless, given that fluoxetine is widely-prescribed for the treatment of major depressive disorder and obsessive-compulsive disorder in pediatric patients, we hope that our results might be of help not only for adults, but also for pediatric patients with KCNT1-related disorders unresponsive to conventional AED treatment.
We herein report a gene-based approach with fluoxetine in a patient with drug-resistant focal epilepsy due to a novel KCNT1 GoF variant. In this case, fluoxetine treatment, following several “conventional” ASMs that resulted ineffective, led to a complete control of seizures. Repurposing drugs selected on the basis of probands’ genetic and functional characteristics for the treatment of severe forms of epilepsy represents one of the main strategies for a precision medicine therapy (Sisodiya, 2021). Further studies are needed to verify whether this approach could be considered also for other phenotypes of the KCNT1-related epilepsy spectrum, including EIMFS.
The data presented in the study are deposited in the Zenodo repository, accession number 10.5281/zenodo.10623783.
The studies involving humans were approved by Fondazione IRCCS Istituto Neurologico “Carlo Besta”, Milan. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. The study was conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
IM: Investigation, Writing – review & editing. EF: Investigation, Writing – review & editing. PA: Investigation, Supervision, Writing – review & editing. GB: Investigation, Writing – review & editing. TG: Conceptualization, Supervision, Writing – review & editing. LC: Data curation, Supervision, Writing – review & editing. FR: Investigation, Writing – review & editing. RS: Investigation, Writing – review & editing. IF: Investigation, Writing – review & editing. BC: Investigation, Methodology, Writing – review & editing. GM: Investigation, Writing – review & editing. CG: Supervision, Writing – review & editing. JCD: Conceptualization, Writing – original draft, Writing – review & editing. MVS: Conceptualization, Data curation, Investigation, Methodology, Supervision, Writing – original draft, Writing – review & editing. MT: Conceptualization, Investigation, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The present work was supported by the Italian Ministry for University and Research (A multiscale integrated approach to the study of the nervous system in health and disease – MNESYS; Next Generation EU, National Recovery and Resilience Plan, Mission 4 Component 2 Investment 1.3 - Project code PE0000006) to MT, and by the following Ricerca Finalizzata Projects from the Italian Ministry of Health: GR-2016-02363337 to JCD and MVS, RF-2019-12370491 to MT and BC, and PNRR-MR1-2022-12376528 to MT and MVS.
We thank the patient and her family for participating in the study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ambrosino, P., Ragona, F., Mosca, I., Vannicola, C., Canafoglia, L., Solazzi, R., et al. (2023). A novel KCNC1 gain-of-function variant causing developmental and epileptic encephalopathy: “precision medicine” approach with fluoxetine. Epilepsia 64, e148–e155. doi: 10.1111/epi.17656
Ambrosino, P., Soldovieri, M. V., De Maria, M., Russo, C., and Taglialatela, M. (2014). Functional and biochemical interaction between PPARα receptors and TRPV1 channels: potential role in PPARα agonists-mediated analgesia. Pharmacol. Res. 87, 113–122. doi: 10.1016/j.phrs.2014.06.015
Bolo, N. (2000). Brain pharmacokinetics and tissue distribution in vivo of fluvoxamine and fluoxetine by fluorine magnetic resonance spectroscopy. Neuropsychopharmacology 23, 428–438. doi: 10.1016/S0893-133X(00)00116-0
Bonardi, C. M., Heyne, H. O., Fiannacca, M., Fitzgerald, M. P., Gardella, E., Gunning, B., et al. (2021). KCNT1 -related epilepsies and epileptic encephalopathies: phenotypic and mutational spectrum. Brain 144, 3635–3650. doi: 10.1093/brain/awab219
Campostrini, G., DiFrancesco, J. C., Castellotti, B., Milanesi, R., Gnecchi-Ruscone, T., Bonzanni, M., et al. (2018). A loss-of-function HCN4 mutation associated with familial benign myoclonic epilepsy in infancy causes increased neuronal excitability. Front. Mol. Neurosci. 11:269. doi: 10.3389/fnmol.2018.00269
Cembrowski, M. S., Wang, L., Sugino, K., Shields, B. C., and Spruston, N. (2016). Hipposeq: a comprehensive RNA-seq database of gene expression in hippocampal principal neurons. eLife 5:e14997. doi: 10.7554/eLife.14997
Choi, B. H., Choi, J. S., Yoon, S. H., Rhie, D. J., Min, D. S., Jo, Y. H., et al. (2001). Effects of norfluoxetine, the major metabolite of fluoxetine, on the cloned neuronal potassium channel Kv3.1. Neuropharmacology 41, 443–453. doi: 10.1016/S0028-3908(01)00088-0
Choi, J. S., Hahn, S. J., Rhie, D. J., Yoon, S. H., Jo, Y. H., and Kim, M. S. (1999). Mechanism of fluoxetine block of cloned voltage-activated potassium channel Kv1.3. J. Pharmacol. Exp. Ther. 291, 1–6.
Cioclu, M. C., Mosca, I., Ambrosino, P., Puzo, D., Bayat, A., Wortmann, S. B., et al. (2023). KCNT2 - related disorders: phenotypes, functional, and pharmacological properties. Ann. Neurol. 94, 332–349. doi: 10.1002/ana.26662
Dilena, R., DiFrancesco, J. C., Soldovieri, M. V., Giacobbe, A., Ambrosino, P., Mosca, I., et al. (2018). Early treatment with quinidine in 2 patients with epilepsy of infancy with migrating focal seizures (EIMFS) due to gain-of-function KCNT1 mutations: functional studies, clinical responses, and critical issues for personalized therapy. Neurotherapeutics 15, 1112–1126. doi: 10.1007/s13311-018-0657-9
Erö, C., Gewaltig, M. O., Keller, D., and Markram, H. (2018). A cell atlas for the mouse brain. Front. Neuroinform. 12:84. doi: 10.3389/fninf.2018.00084
Fitzgerald, M. P., Fiannacca, M., Smith, D. M., Gertler, T. S., Gunning, B., Syrbe, S., et al. (2019). Treatment responsiveness in KCNT1-related epilepsy. Neurotherapeutics 16, 848–857. doi: 10.1007/s13311-019-00739-y
Gertler, T., Bearden, D., Bhattacharjee, A., and Carvill, G. (2018). “KCNT1-related epilepsy” in GeneReviews®. eds. M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. Bean, and K. W. Gripp, et al. (Seattle (WA): University of Washington, Seattle)
Gertler, T. S., Cherian, S., DeKeyser, J. M., Kearney, J. A., and George, A. L. (2022). KNa1.1 gain-of-function preferentially dampens excitability of murine parvalbumin-positive interneurons. Neurobiol. Dis. 168:105713. doi: 10.1016/j.nbd.2022.105713
Hill, S. F., Ziobro, J. M., Jafar-Nejad, P., Rigo, F., and Meisler, M. H. (2022). Genetic interaction between Scn8a and potassium channel genes Kcna1 and Kcnq2. Epilepsia 63, e125–e131. doi: 10.1111/epi.17374
Liu, R., Sun, L., Wang, Y., Wang, Q., and Wu, J. (2023). New use for an old drug: quinidine in KCNT1-related epilepsy therapy. Neurol. Sci. 44, 1201–1206. doi: 10.1007/s10072-022-06521-x
Miceli, F., Soldovieri, M. V., Ambrosino, P., De Maria, M., Migliore, M., Migliore, R., et al. (2015). Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of K v 7.2 and K v 7.3 Potassium Channel subunits. J. Neurosci. 35, 3782–3793. doi: 10.1523/JNEUROSCI.4423-14.2015
Møller, R. S., Heron, S. E., Larsen, L. H. G., Lim, C. X., Ricos, M. G., Bayly, M. A., et al. (2015). Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia 56, e114–e120. doi: 10.1111/epi.13071
Niday, Z., and Tzingounis, A. V. (2018). Potassium Channel gain of function in epilepsy: an unresolved paradox. Neuroscientist 24, 368–380. doi: 10.1177/1073858418763752
Pan, G., Chen, Z., Zheng, H., Zhang, Y., Xu, H., Bu, G., et al. (2018). Compensatory mechanisms modulate the neuronal excitability in a Kainic acid-induced epilepsy mouse model. Front. Neural Circuits 12:48. doi: 10.3389/fncir.2018.00048
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rizzo, F., Ambrosino, P., Guacci, A., Chetta, M., Marchese, G., Rocco, T., et al. (2016). Characterization of two de novo KCNT1 mutations in children with malignant migrating partial seizures in infancy. Mol. Cell. Neurosci. 72, 54–63. doi: 10.1016/j.mcn.2016.01.004
Sands, T. T., Miceli, F., Lesca, G., Beck, A. E., Sadleir, L. G., Arrington, D. K., et al. (2019). Autism and developmental disability caused by KCNQ3 gain-of-function variants. Ann. Neurol. 86, 181–192. doi: 10.1002/ana.25522
Shore, A. N., Colombo, S., Tobin, W. F., Petri, S., Cullen, E. R., Dominguez, S., et al. (2020). Reduced GABAergic neuron excitability, altered synaptic connectivity, and seizures in a KCNT1 gain-of-function mouse model of childhood epilepsy. Cell Rep. 33:108303. doi: 10.1016/j.celrep.2020.108303
Sisodiya, S. M. (2021). Precision medicine and therapies of the future. Epilepsia 62, S90–S105. doi: 10.1111/epi.16539
Keywords: KCNT1 , fluoxetine, next generation sequencing, drug repurposing, epilepsy
Citation: Mosca I, Freri E, Ambrosino P, Belperio G, Granata T, Canafoglia L, Ragona F, Solazzi R, Filareto I, Castellotti B, Messina G, Gellera C, DiFrancesco JC, Soldovieri MV and Taglialatela M (2024) Case report: Marked electroclinical improvement by fluoxetine treatment in a patient with KCNT1-related drug-resistant focal epilepsy. Front. Cell. Neurosci. 18:1367838. doi: 10.3389/fncel.2024.1367838
Edited by:
Luigi Catacuzzeno, University of Perugia, ItalyReviewed by:
Giulia Curia, University of Modena and Reggio Emilia, ItalyCopyright © 2024 Mosca, Freri, Ambrosino, Belperio, Granata, Canafoglia, Ragona, Solazzi, Filareto, Castellotti, Messina, Gellera, DiFrancesco, Soldovieri and Taglialatela. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacopo C. DiFrancesco, amFjb3BvLmRpZnJhbmNlc2NvQHVuaW1pYi5pdA==; Maria Virginia Soldovieri, bWFyaWF2aXJnaW5pYS5zb2xkb3ZpZXJpQHVuaW1vbC5pdA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.