
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 25 January 2024
Sec. Cellular Neurophysiology
Volume 18 - 2024 | https://doi.org/10.3389/fncel.2024.1354569
This article is part of the Research TopicCelebrating the Year of Ramon y Cajal: Cellular Biology of the RetinaView all 14 articles
 Jose A. Fernández-Albarral1†
Jose A. Fernández-Albarral1† Ana I. Ramírez1,2†
Ana I. Ramírez1,2† Rosa de Hoz1,2
Rosa de Hoz1,2 José A. Matamoros1,2
José A. Matamoros1,2 Elena Salobrar-García1,2
Elena Salobrar-García1,2 Lorena Elvira-Hurtado1
Lorena Elvira-Hurtado1 Inés López-Cuenca1,2
Inés López-Cuenca1,2 Lidia Sánchez-Puebla1,3
Lidia Sánchez-Puebla1,3 Juan J. Salazar1,2‡
Juan J. Salazar1,2‡ José M. Ramírez1,3*‡
José M. Ramírez1,3*‡Glaucoma is a neurodegenerative disease of the retina characterized by the irreversible loss of retinal ganglion cells (RGCs) leading to visual loss. Degeneration of RGCs and loss of their axons, as well as damage and remodeling of the lamina cribrosa are the main events in the pathogenesis of glaucoma. Different molecular pathways are involved in RGC death, which are triggered and exacerbated as a consequence of a number of risk factors such as elevated intraocular pressure (IOP), age, ocular biomechanics, or low ocular perfusion pressure. Increased IOP is one of the most important risk factors associated with this pathology and the only one for which treatment is currently available, nevertheless, on many cases the progression of the disease continues, despite IOP control. Thus, the IOP elevation is not the only trigger of glaucomatous damage, showing the evidence that other factors can induce RGCs death in this pathology, would be involved in the advance of glaucomatous neurodegeneration. The underlying mechanisms driving the neurodegenerative process in glaucoma include ischemia/hypoxia, mitochondrial dysfunction, oxidative stress and neuroinflammation. In glaucoma, like as other neurodegenerative disorders, the immune system is involved and immunoregulation is conducted mainly by glial cells, microglia, astrocytes, and Müller cells. The increase in IOP produces the activation of glial cells in the retinal tissue. Chronic activation of glial cells in glaucoma may provoke a proinflammatory state at the retinal level inducing blood retinal barrier disruption and RGCs death. The modulation of the immune response in glaucoma as well as the activation of glial cells constitute an interesting new approach in the treatment of glaucoma.
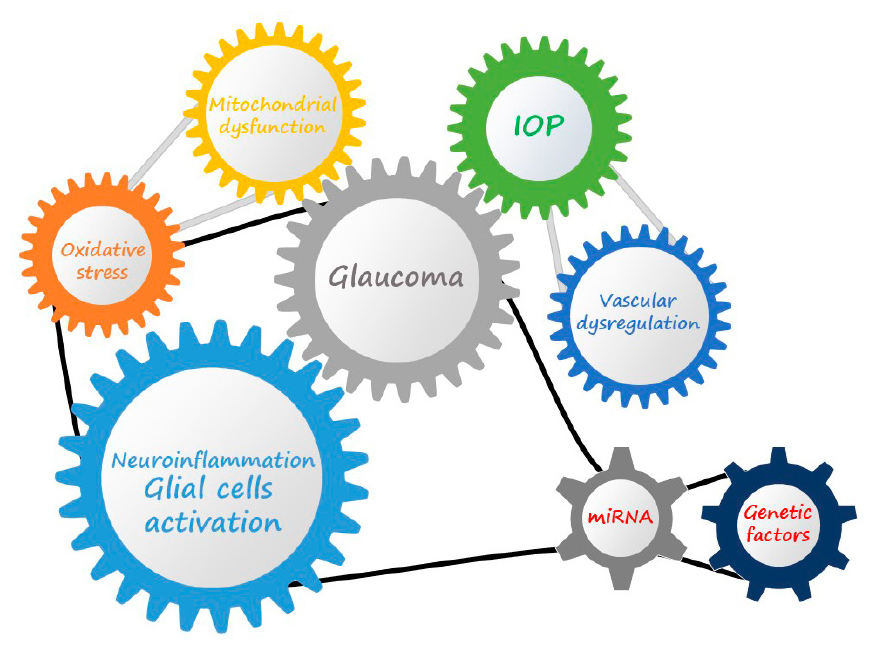
Graphical Abstract. Glaucoma is a multifactorial disease characterized by the interaction of different molecular mechanisms that can induced the onset and contribute to the progression of the pathology, and in which the dysregulation of gene transcription and genetic predisposition can also play a key role.
Glaucoma is a complex, chronic and multifactorial neurodegenerative pathology and is considered the leading cause of irreversible blindness (Weinreb et al., 2016; WHO, 2019).
In 2019, more than 60 million people were affected by glaucoma and are expected to reach nearly 95.4 million in 2030 (WHO, 2019). It is characterized by continued damage and progressive loss of RGCs and the retinal nerve fiber layer, as well as alterations of the optic disc and optic nerve head, eventually leading to progressive and irreversible constriction and loss of the visual field (Quigley and Broman, 2006; Weinreb et al., 2016).
Degeneration of RGCs and loss of their axons, as well as damage and remodeling of the lamina cribrosa constitute the main events in the pathogenesis of glaucoma (Giangiacomo and Coleman, 2009).
Different molecular pathways are involved in RGCs death, which are triggered and exacerbated as a consequence of a number of risk factors such as elevated IOP (Heijl et al., 2002; Nouri-Mahdavi et al., 2004), age (Leske et al., 2004; Lee et al., 2014), ocular biomechanics or low ocular perfusion pressure (Gordon et al., 2007; Quaranta et al., 2021).
Primary open-angle glaucoma is the most common form of glaucoma and is characterized by an alteration in the aqueous humor outflow pathways, which causes an increase in aqueous fluid outflow resistance leading to an increase in IOP. This ocular hypertension (OHT) leads to damage of the RGC axons in the optic nerve (ON) head and subsequently to concentric and progressive loss of RGCs (Mélik Parsadaniantz et al., 2020). Although most cases of glaucoma show elevated IOP values, there are patients with glaucomatous optic neuropathy whose pressure values are within the normal range (normotensive glaucoma), or who, despite elevated IOP values, do not present damage to the ON head (Jonas et al., 2014).
Current treatments are focused on OHT control, however, about 15–25% of these patients continue to exhibit disease progression despite correct IOP control (Tezel, 2013; Soto and Howell, 2014). Furthermore, in the case of normotensive glaucoma, elevated IOP cannot explain the neurodegenerative process (Wax et al., 2008).
The 70% of glaucoma cases are caused by elevated IOP, but it is clear that different molecular mechanisms are involved, such as oxidative stress, glutamate toxicity, neurotrophin deprivation, mitochondrial dysfunction, ischemia/hypoxia processes, protein misfolding, autoimmunity, dysfunction of autophagy processes, vascular dysregulation or neuroinflammation, which may also be responsible for RGC degeneration and death in cases of non-elevated IOP (Chang and Goldberg, 2012; Ramirez et al., 2017; Toft-Kehler et al., 2017; Duarte, 2021; Gutteridge, 2021).
Elevated IOP is one of the main risk factors in glaucoma and is the target of most current treatments. This increase in IOP is produced as a consequence of an increase in the outflow resistance of the aqueous humor as it is prevented from draining into the normal evacuation channels (Moreno et al., 2004).
Increased IOP leads to chronic stress on the ON head by mechanical compression of the ON head at the level of the lamina cribrosa, where the axons of the RGCs are more vulnerable to pressure-related changes. This is because the lamina cribrosa is structurally weaker than the sclera and more prone to distortion and posterior displacement (Quigley, 2005a,b; Burgoyne, 2011). In addition, at the level of the ON head, the axons of the RGCs make a 90° turn, which makes them particularly vulnerable to mechanical stress and alterations in axonal transport (Dias et al., 2022).
Analysis of the movements of the lamina cribrosa in glaucoma shows that the greatest mechanical damage is exerted on axons located in the peripheral part of the ON, which correlates with clinical findings showing that vision loss begins in the peripheral zone of the visual field (Burgoyne, 2011).
Although these physical changes in the lamina cribrosa may not be the cause of axonal loss in the early stages of glaucoma, stress and strain due to elevated IOP may accumulate, contributing to axonal damage and loss (Burgoyne et al., 2004; Burgoyne, 2011). The loss of elasticity of the lamina cribrosa with aging causes it to lose the ability to recover its normal configuration after normalization of IOP, increasing the prevalence of glaucoma with age (Albon et al., 2000).
The effect of this translaminar pressure gradient on axons leads to ischemia/hypoxia damage, blockade of axonal transport and deprivation of neurotrophic factors, ultimately resulting in apoptosis of RGCs (Nickells, 2007; Crish et al., 2010; Hirt et al., 2018). This pressure gradient alters retrograde and anterograde axonal transport, avoiding the cell bodies of RGCs to receive the contribution of neurotrophic factors such as brain derived neurotrophic factor (BDNF) or nerve growth factor (NGF) (Quigley, 1999; Almasieh et al., 2012; Križaj et al., 2014). As a consequence of this trophic deficit, abnormalities in the dendritic arborization of RGCs appear early after the increase in IOP (Weber et al., 2008).
In glaucoma, alterations in the ON head due to increased IOP also include reduced blood flow, oxidative stress, reactive gliosis, as well as remodeling of the extracellular matrix (Howell et al., 2007; Chidlow et al., 2011; Calkins et al., 2017; Tamm et al., 2017). Axonal remodeling, inflammation, glial activation and ultimately RGC death are partly dependent on intracellular Ca2+ (Crish and Calkins, 2011). Mechanical change-sensitive channels present in both the soma and dendrites of RGCs, such as P2X purinergic receptors, are activated by the tensional forces induced by increased IOP causing Ca2+ influx (Xia et al., 2012; Križaj et al., 2014). Intracellular Ca2+ triggers opening of Panexin-1 channels that allow massive ATP release. This ATP release by RGCs is associated with glial cell activation and increased expression of proinflammatory cytokines such as TNF-α, IL-1β, or IL-6 (Burnstock, 1999; Silverman et al., 2009).
Mitochondrial dysfunction can also occur as a consequence of both acute and chronic IOP elevation. Retinal hypoxia resulting from this increased pressure leads to increased production of reactive oxygen species (ROS), promoting intracellular lipid, protein, and DNA oxidation (Kamel et al., 2017).
Energy in the form of ATP (Adenosine triphosphate) is required for neurotransmitter synthesis, synaptic transmission, restoring ionic gradients, Ca2+ buffering and bidirectional transport along axons (Ito and di Polo, 2017). Due to the absence of saltatory conduction in the axons of the unmyelinated part of the ON, a greater energy supply is required in this area, and there are a large number of mitochondria to alleviate this energy deficit (Bristow et al., 2002; Wang et al., 2003; Barron et al., 2004). This high energy consumption is necessary for the generation of action potentials and restoration of the resting membrane potential (Harris and Attwell, 2012). RGCs are able to maintain this level of consumption thanks to the abundant number of mitochondria present in their soma, axons and dendrites. The highest concentration is located at the level of the lamina cribrosa in order to protect this most vulnerable area of the retina and to ensure an adequate ATP supply (Minckler et al., 1976; Fan Gaskin et al., 2021).
Retinal ganglion cells have a low tolerance to mitochondrial damage. A decrease in the number of functional mitochondria or an accumulation of dysfunctional mitochondria may be the origin of an energy deficit in RGCs, also interfering with the maintenance of synaptic integrity (Li et al., 2004; Ito and di Polo, 2017; Toft-Kehler et al., 2017). Mitochondrial dysfunction leads to the release of multiple mitochondrial damage-associated molecular patterns (DAMPs). These molecules released by the dysfunctional mitochondria may result in an innate immune response contributing to the progression of the neuroinflammatory process in glaucoma (Duarte, 2021).
Mitochondrial dysfunction, in addition to being associated with alterations in mitochondrial DNA and deficient mitophagy, also occurs as a consequence of increased oxidative stress (Wilkins et al., 2014, 2017). Under normal conditions low levels of ROS production are counteracted by antioxidants such as glutathione synthetase, superoxide dismutase or catalase, but excess ROS can lead to lipid peroxidation and apoptosis by increasing mitochondrial membrane permeability and inhibition of the mitochondrial respiratory chain (Tezel and Yang, 2004; Li and Osborne, 2008; Inman et al., 2013; Iomdina et al., 2015).
Oxidative stress is a consequence of an imbalance between the production of ROS and antioxidant agents. Although ROS participate in signaling pathways of redox processes, they can also be responsible for cell damage, necrosis, or apoptosis, as a consequence of the oxidation process of macromolecules such as lipids, proteins, nuclear DNA, and mitochondrial DNA (Jarrett et al., 2008; Fan Gaskin et al., 2021). Dysregulation of ROS production and impaired ATP production by mitochondria lead to increased oxidative stress in RGCs, being an important immunostimulatory and cell death mechanism in glaucoma (Tezel, 2006; West and Shadel, 2017; Tang et al., 2019).
The accumulation of neurotoxic levels of glutamate is another effect of ROS excess. Glutamine synthetase (GS), responsible for transforming retinal glutamate to its non-toxic form, and glutamate transporter proteins are also altered by increased ROS in models of ocular hypertension (Moreno et al., 2004; Tezel et al., 2005). In addition, glutamate excess levels can also induce oxidative stress, being implicated in numerous neurodegenerative conditions (Cassano et al., 2016). Both glutamate accumulation and decreased glutamate transporters lead to apoptosis of RGCs (Hurley et al., 2022). Increased IOP causes elevation of glutamate levels increasing glutamate-induced oxidative stress in RGCs (Nucci et al., 2005). Glutamate analogs such as N-methyl-D-aspartate (NMDA) have been used in murine models to induce an increase in calcium and ROS levels and ultimately the death of RGCs (Seitz and Tamm, 2013).
Both altered intrinsic mitochondrial activity and exposure of RGCs and retinal neuroglia to extrinsic factors such as altered blood flow, hypoxia, nutrient deficits, or calcium dysregulation are associated with ROS production in glaucoma (Satilmis et al., 2003; Malone and Hernandez, 2007; McElnea et al., 2011; Yanagi et al., 2011; Vohra et al., 2019). In addition, mechanical damage to the ON head as a consequence of elevated IOP is also responsible for neuronal oxidative stress and lipid peroxidation, factors that increase the risk of neurodegeneration (Ko et al., 2005; Ferreira et al., 2010). In the anterior segment, exposure of the trabecular meshwork to high levels of ROS leads to increased normal aqueous humor outflow resistance, contributing to increased exposure of the retina and optic nerve head to elevated IOP values (Saccà et al., 2005; Duarte, 2021).
In the retina, Müller cells perform essential functions for the maintenance of homeostasis, many of which are related to their metabolism, so that energy restriction due to oxidative stress and mitochondrial dysfunction may contribute to the pathogenesis of glaucoma and the loss of RGCs (Kawasaki et al., 2000; Skytt et al., 2012; Toft-Kehler et al., 2017).
Blood flow autoregulation in the retina involves vascular constriction or dilatation thereby increasing or decreasing resistance, thus maintaining a constant flow of nutrients in response to changes in perfusion pressure (Harris et al., 1998).
The optic nerve head is the site at which RGC axons are most vulnerable to potential ischemic events. Blood flow at this level is delicately regulated to ensure an adequate supply of oxygen and nutrients to RGC axons (Morgan, 2004). An insufficient blood supply to the optic nerve, either due to increased IOP or other vascular risk factors that may reduce ocular blood flow, will induce ischemia and hypoxia processes in the RGCs and their axons, contributing to the development of glaucomatous pathology (Flammer et al., 2002).
There are several indications postulating that reduced blood flow is one of the causes of glaucomatous pathology. Reduced ocular blood flow and its effects, such as hemorrhages at the level of the optic disc, considered hallmarks of glaucomatous processes especially in those poorly controlled, are often more pronounced in patients with normotensive glaucoma (Siegner and Netland, 1996; Healey et al., 1998; Drange et al., 2001). In addition to optic nerve hemorrhages, increased risk of venous thrombosis and retinal vasoconstriction are common in patients with glaucoma (Fan Gaskin et al., 2021).
Factors released by vascular endothelium or neural tissue contribute to the regulation of vascular tone in the retina. Nitric oxide (NO), a vasodilator factor, contributes to the autoregulation of retinal blood flow by protecting the nerve fiber layer against pathological stressors involved in glaucoma and ischemia (Toda and Nakanishi-Toda, 2007). On the other hand, the action of vasoconstrictors such as endothelin 1 (ET-1) or angiotensin II also participate in the changes in vascular tone. Alterations in NO activity and increased ET-1 expression, which could promote the weakening of the blood retinal barrier (BRB), have been observed in patients with glaucoma (Noske et al., 1997; Tezel et al., 1997; Tunny et al., 1998; Nicolela et al., 2003; Polak et al., 2007). Alterations in vascular autoregulation make the optic nerve head more vulnerable to decreased perfusion pressure, IOP increases and local metabolic demands (Moore et al., 2008).
Systemically, there is an association between cardiovascular pathologies and the development of glaucoma. Both systemic hypertension and hypotension (Schmidl et al., 2011; Zhao et al., 2014; Levine et al., 2017; de Moraes et al., 2018; Skrzypecki et al., 2019), diabetes (Gerber et al., 2015; Song et al., 2016), previous hemodynamic seizures or increased blood viscosity are considered specific risk factors for glaucoma (Fan Gaskin et al., 2021; Figure 1).
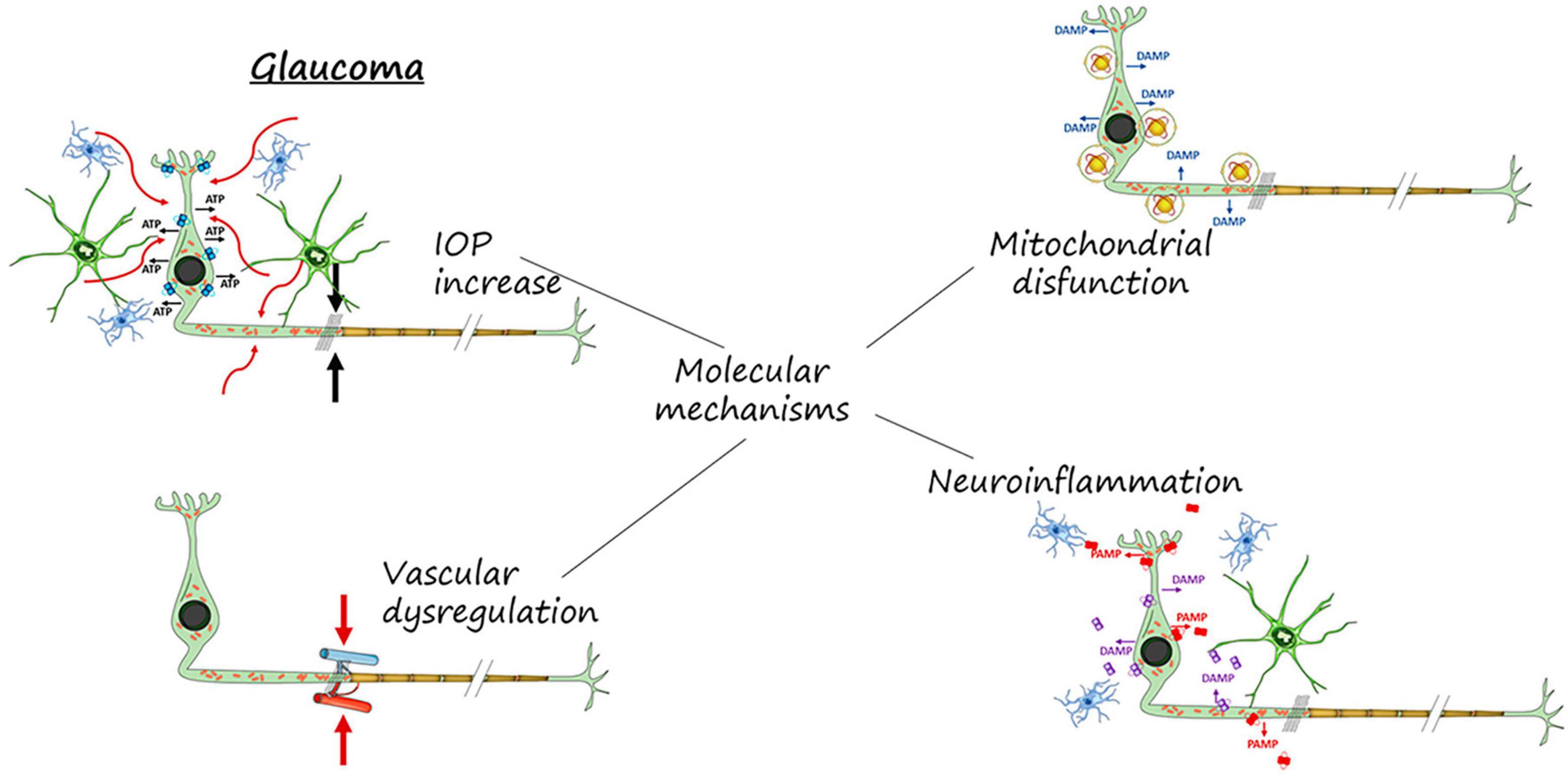
Figure 1. Molecular mechanisms involved in RGCs neurodegeneration and death. IOP increase, mitochondrial dysfunction, vascular dysregulation and neuroinflammation.
MicroRNAs (miRNAs) are small non-coding ribonucleic acid (RNA) molecules that play a key role in post-transcriptional gene regulation and are involved in a variety of biological processes, such as cell growth, differentiation, development, and apoptosis (O’Brien et al., 2018; Saliminejad et al., 2019). Dysregulated miRNAs can modulate key pathways and processes that are involved in glaucoma, such as apoptosis, autophagy, neurogenesis, aging, extracellular matrix remodeling, oxidative stress, inflammation, and angiogenesis (Cannell et al., 2008). miRNAs are involved in the maintenance of aqueous humor (AH) balance, trabecular meshwork (TM) changes, and RGC apoptosis, and studies have shown altered expression patterns of miRNAs in both glaucoma patients and animal models (Cannell et al., 2008; Greene et al., 2022).
Retinal ganglion cells apoptosis and neuroinflammation are molecular pathways regulated by differentially expressed miRNAs in glaucoma. Altered levels of different miRNAs in AH samples from glaucoma patients may regulate target genes linked to apoptosis and inflammation, such as Bcl-2, caspase-3, IL-6, IL-8, TNF, and nuclear factor κB (NFκB) (Li et al., 2010; Su et al., 2017; Wang et al., 2018; Hindle et al., 2019; Ou-Yang et al., 2020; Youngblood et al., 2020; Tabak et al., 2021).
Extracellular matrix remodeling and autophagy are also miRNA-regulated processes differentially expressed in glaucoma. These processes are involved in the degradation and renewal of the extracellular matrix and cellular components of the TM, affecting outflow resistance and IOP (Gonzalez et al., 2014; Reinehr et al., 2022). Profiles expression of several miRNAs such as miR-518d, miR-143, and miR-660 negatively modulate pathways involved in cell proliferation, cell survival, extracellular matrix (ECM) remodeling, and adherents junction function (Jayaram et al., 2017).
Mitochondrial dysfunction and oxidative stress, lead to the accumulation of reactive oxygen species and the impairment of mitochondrial function, causing cellular damage and death. Several miRNAs such as miR-7, miR-24, miR-27a, miR-29b, and miR-4295, target various genes and pathways exerting antioxidant effects in TM cells, contributing to the prevention of oxidative damage in glaucoma (Tabak et al., 2021).
The retina is an important source of miRNAs. It expresses several miRNAs that control crucial processes such as RGC survival, axonal transport, synaptic transmission and neuroinflammation (Zuzic et al., 2019). In glaucoma, both proapoptotic (miR-16, −497, −29b and let-7a) and antiapoptotic (miR-27a) miRNAs show altered levels. Apoptotic miRNAs induce neurodegeneration through Toll-like receptor signaling, or by modulating the apoptosis regulator Bcl-2. The up-regulation of miR-27a and down-regulation of pro-apoptotic miRNAs suggests a shift toward a protective anti-apoptotic phenotype of their expression profiles (Jayaram et al., 2015). The up-regulation of miRNA-96 is related to RGCs activity and activation of the caspase pathway. miRNA-96 affects survival and increases apoptosis of RGCs through interaction with caspase-2 (Kang et al., 2015). On the other hand, down-regulation of miRNA-100 reduces RGC apoptosis and promotes nerve growth through the phosphorylation pathway, controlling the intrinsic apoptotic pathway responsible for impaired mitochondrial function (Chrysostomou et al., 2013; Kong et al., 2014).
Expression profiles of miRNAs are also implicated in the microglial reactivity and RGCs survival in response to ocular hypertension. miR-93 agomir reduces cytokine expression, microglial activation, inflammation, and RGCs loss, by targeting STAT3 to exert its effect on RGC death (Wang et al., 2021).
Glaucoma exhibits significant heritability, with early onset forms inherited as autosomal dominant or Mendelian recessive traits and adult-onset disease inherited as complex traits. Genes involved in early onset glaucoma have been identified by large-family linkage analysis, whereas genes involved in adult-onset disease have been identified by genome-wide association studies (GWAS), usually requiring a large number of glaucoma cases and controls (Wiggs, 2015; Wang et al., 2022).
Linkage studies of familial and congenital glaucoma have shown the existence of genes with a significant effect on the disease, such as MYOC (myocilin) and OPTN (optineurin) for familial primary open-angle glaucoma (POAG) and CYP1B1 for congenital glaucoma (Stoilov et al., 1997; Stone et al., 1997). Variations in the MYOC gene sequence lead to the most common form, characterized by elevated IOP, whereas rare mutations in OPTN and copy number variations in TBK-1 (TANK-binding protein 1) can cause familial NTG (normal tension glaucoma) (Stuart et al., 2023).
Myocilin is expressed in various eye tissues such as the iris, retina, trabecular meshwork, and cornea (Huang et al., 2000). MYOC mutations are mainly associated with juvenile-onset POAG (Fingert et al., 2002; Fingert, 2011). The phenotype of MYOC-associated mutations is generally severe, IOP is usually very high and surgical treatment is often required (Fingert et al., 1999). Accumulation of MYOC in the trabecular meshwork caused by mutations leading to overproduction or reduced degradation is associated with increased IOP and, ultimately, with the development of glaucoma (Doucette et al., 2015).
Optineurin is expressed in several ocular tissues including the cornea, retinal ganglion cells, and the trabecular meshwork (Li et al., 1998). Among all OPTN mutations, E50K is the one most clearly shown to cause glaucoma. The E50K mutation in POAG patients is associated with an earlier age of onset, more advanced optic nerve cupping and a more frequent need for surgical intervention (Aung et al., 2005).
TBK1 plays an essential role in the regulation of inflammatory responses to foreign agents (Fingert, 2011). A duplication of the TBK1 gene has been identified in approximately 1% of normal tension POAG cases (Ritch et al., 2014; Awadalla et al., 2015; Liu and Allingham, 2017). TBK1 and OPTN regulate autophagy of damaged mitochondria (Matsumoto et al., 2015; Richter et al., 2016). Duplication of the TBK-1 associated with NTG patients could be related to autophagy dysfunction and RGCs loss (Tucker et al., 2014; Fingert et al., 2017).
Recently GWAS have implicated ABCA1 [coding for ATP-binding cassette, subfamily A (ABC1) member 1] in POAG (Wiggs, 2015). ABCA1 is expressed in glaucoma-significant ocular tissues such as the iris, ciliary body, retina, optic nerve head, optic nerve, and trabecular meshwork (Chen et al., 2014; Gharahkhani et al., 2014). It has been suggested that ABCA1 may regulate neuroinflammation and neurodegeneration in the mouse brain, which may play a role in retinal pathogenesis and POAG (Chen et al., 2014; Gharahkhani et al., 2014). ABCA1 has been implicated in retinal inflammation and retinal ganglion cell apoptosis, and ABCA1 has also been shown to play a role in IOP regulation through aqueous humor dynamics (Li et al., 2018; Hu et al., 2020).
Neuroinflammation is the inflammatory response that occurs in neural tissue in response to neuronal damage. This inflammatory process is characterized by the activation of resident glial cells, the release of proinflammatory cytokines and chemokines, and the recruitment and infiltration of peripheral cells into the CNS as a consequence of the breakdown of the blood-brain barrier (Heneka et al., 2015; Kelly et al., 2020). Neuroinflammation is a defensive mechanism that will initially protect the CNS by eliminating or inhibiting different pathogens (Wyss-Coray and Mucke, 2002).
This inflammatory response may initially exert a neuroprotective effect by promoting the repair of damaged tissue and the elimination of cellular debris, while sustained inflammation may exert a negative effect by inhibiting the regeneration of neuronal tissue (Kempuraj et al., 2016; Russo and McGavern, 2016). This sustained inflammatory process can be stimulated by both endogenous factors (genetic factors, protein aggregates) and environmental factors (infection, trauma, drugs). The activation of glial cells in chronic inflammatory processes may contribute to the progression of neurodegenerative processes (Glass et al., 2010; Kempuraj et al., 2016; Stephenson et al., 2018), with most studies in this field concluding that both astrocytes and microglia are causally involved in the pathogenesis and progression of neurodegenerative disorders (Sarlus and Heneka, 2017; Hickman et al., 2018; Yamanaka and Komine, 2018). Glial cells are essential for the maintenance of homeostasis in the CNS, both in physiological and pathological conditions, and may therefore be directly involved in the development and progression of neurodegenerative pathologies (Parpura et al., 2012). Interaction between glial cells and neurons means that dysfunction of either can lead to the development of harmful processes that can disrupt neuron-glia communication or compromise neuronal viability (Patel et al., 2019; Bernaus et al., 2020). Glial cell activation-mediated neuroinflammation or gliosis is one of the first events in response to damage in both the brain and retina (Soto and Howell, 2014; Ramirez et al., 2017).
In response to changes in the tissue environment, stress, infections, or neurodegenerative pathologies that may compromise neuronal survival or even with ageing, glial cells have the capacity to adopt different reactive phenotypes. Glial activation occurs in order to restore tissue homeostasis, repair and neuroprotection of damaged tissue. In the CNS, astrocytes and microglia are responsible for the innate immune response and their reaction is classically characterized by two opposite activation phenotypes acting as neuroprotective or neurotoxic agents (Kwon and Koh, 2020).
Microglia have been classically divided into two activation phenotypes, M1 (classical activation phenotype) and M2 (alternative activation phenotype) (Luo and Chen, 2012). The M1 phenotype is characterized by the expression of pro-inflammatory cytokines, expression of antigen presentation markers and phagocytic capacity (Mosser and Edwards, 2008), while M2, although also showing phagocytic capacity, is characterized by the expression of anti-inflammatory cytokines and extracellular matrix remodeling for tissue repair (Edwards et al., 2006). The alternative M2 activation phenotype has been subdivided into three categories M2a, M2b, and M2c based on different inductors and functions (Fan et al., 2022). These three stages are biochemically similar but show different mechanisms of activation; the STAT6 (signal transducer and activator of transcription-6) activation pathway and IL-4 induction are associated with the M2a phenotype, and the STAT3 pathway and IL-10 induction are associated with M2c (Matsukawa et al., 2003; Maier et al., 2012). On the other hand, the activation pathway of the M2b phenotype, induced by the binding of lipopolysaccharide (LPS) or IL-1β immune complexes to receptors (CD16, CD32, or CD64) on activated microglia, is not yet well defined (Moehle and West, 2015; Yang et al., 2017). Recently, the existence of an M3 activation phenotype acquired by proliferative microglia in response to colony-stimulating factor-1 (CSF-1) and IL-34 has also been suggested (Walker and Lue, 2015).
Similar to microglia, two activation phenotypes are also identified in astrocytes; A1 (neurotoxic) and A2 (neuroprotective), which are also related to the production of proinflammatory or immunomodulatory mediators according to their polarization state (Stephenson et al., 2018). On the other hand, A2 astrocytes, are considered neuroprotective and increase the expression of neurotrophic factors such as BDNF, glial cell line-derived neurotrophic factor (GDNF) and NGF, which promote synaptic growth and repair, as well as neuronal survival (Lam et al., 2019).
Astrocyte polarization toward an A1 or A2 phenotype is mainly controlled by the NFκB, toll-like receptors (TLRs), mitogen-activated protein kinase (MAPK) and the Janus kinase/STAT3 (JAK/STAT3) signaling pathway (Ding et al., 2021). The NFκB and MAPK pathways are associated with the neuroinflammatory response of astrocytes and regulate the production of inflammatory cytokines, chemokines, extracellular matrix proteins, cell adhesion molecules, ROS, and inducible nitric oxide synthase (iNOS), which are involved in leukocyte infiltration, neuronal death, and demyelination (Colombo and Farina, 2016; Perez-Nievas and Serrano-Pozo, 2018). Activation of TLR ligands on astrocytes also leads to the inflammatory response in the CNS (Azam et al., 2019). The JAK/STAT3 signaling pathway is associated with the activation of A2 astrocytes phenotype, regulating functions such as reduction of leukocyte infiltration, myelin sparing, expression of anti-inflammatory cytokines and promoting neuronal survival (Colombo and Farina, 2016; Liddelow and Barres, 2017).
However, both astrocytes and microglia are now considered to have a wide spectrum of activation phenotypes, and the loss of their neuroprotective functions toward potentially neurotoxic roles is directly related to the stage and severity of the neurodegenerative process (Bachiller et al., 2018).
Microglial cells are able to protect neural tissue against harmful stimuli such as pathogen-associated molecular patterns (PAMPs) and DAMPs, which are recognized by microglia-expressed receptors such as TLRs, oligomerization domain nuclear receptors or viral receptors (Glass et al., 2010; Stephenson et al., 2018). In response to these stimuli, microglia express proinflammatory cytokines such as TNF-α, IL-1β, IL-16 and chemokines such as monocyte chemoattractant protein-1 (MCP-1) or IL-18, which are responsible for cell recruitment and removal of pathogenic substances (Glass et al., 2010; Hickman et al., 2018).
Proinflammatory mediators released by neuroinflammatory M1 microglia, such as C1q, IL-1α, IL-1β, or TNF-α, have the ability to induce activation of the A1 astrocyte phenotype, which will be responsible for a secondary inflammatory response (Saijo et al., 2009; Liddelow and Barres, 2017). Astrocytes increase the expression of membrane receptors for IL-17 and tropomyosin receptor kinase B (TrkB) for neurotrophins during neuroinflammation. Binding of IL-17 to its receptor induces the recruitment of NFκB activator 1 (Act1) as well as the production of proinflammatory cytokines (Qian et al., 2007). In turn, transforming growth factor-β (TGF-β) expression by astrocytes inhibits NFκB signaling, helping to modulate and reduce the neuroinflammatory response (Cekanaviciute et al., 2014).
Molecular and morphological changes during astrogliosis assessed by glial fibrillary acidic protein (GFAP) expression are directly related to the severity of astroglial reactivity, being GFAP overexpression a characteristic sign of neuroinflammation in neurodegenerative processes in the CNS (Sofroniew, 2009).
As part of innate immunity mediated by glial cells and in response to PAMP and DAMP patterns, activation of pattern recognition receptors (PRRs), including TLRs, also induces the phagocytic capacity of glial cells (Underhill and Ozinsky, 2002). Microglial cells are the main cells responsible for phagocytosis in the CNS (Wolf et al., 2017), and during inflammatory processes they increase the expression of complement receptors such as CR1, CR3 and CR4 and TLRs, associated with an increase in their phagocytic capacity (Alexander et al., 2008). The phagocytic capacity of microglia may be impaired during ageing or in some pathological situations, making it necessary the action of auxiliary cells with phagocytic capacity (Abiega et al., 2016; Gabandé-Rodríguez et al., 2020).
In this context, astrocytes, although they are cells with lower phagocytic capacity and less efficient than microglia, are able to trigger a compensatory mechanism in response to phagocytic dysfunction in microglia (Konishi et al., 2020, 2022). Astrocytes have TLRs (TLR2, TLR3, TLR4, TLR5, and TLR9), increasing their expression in the presence of pro-inflammatory stimuli, acquiring the ability to engulf cellular debris and damaged or dead neurons (Bsibsi et al., 2002; Bowman et al., 2003; Morizawa et al., 2017; Ponath et al., 2017).
Like astrocytes, Müller cells express TLRs (TLR2, TLR3, TLR4, TLR5, TLR7, and TLR9) under normal conditions, and their expression increases under pathological conditions (Kumar and Shamsuddin, 2012). Expression of TLRs gives Müller cells phagocytic capacity, and they are critical for the maintenance of homeostasis, removal of neuronal debris and retinal reorganization and regeneration (Bejarano-Escobar et al., 2017; Sakami et al., 2019).
Although neuroinflammation is initially a neuroprotective mechanism, its sustained effect over time can induce a neurotoxic effect, which is directly related to neurodegenerative processes (Hickman et al., 2018).
Morphologically, microglia have the ability to adopt different morphotypes depending on their state of activation, from quiescent (branched microglia) to hyperbranched, activated, amoeboid or rod microglia (de Hoz et al., 2013; Holloway et al., 2019; Li et al., 2019).
Microglial cells are distributed in a regular mosaic and are characterized by small, rounded somas, from which long, thin, highly branched processes emerge, which are continuously extending and contracting, facilitating their immunosurveillance function (Nimmerjahn et al., 2005). Branched microglia are able to detect and respond to biochemical and bioelectrical changes in the microenvironment, acquiring the hyperbranched phenotype, characterized by a greater complexity of the processes, being more abundant, longer, and thicker, emerging from thicker and more irregularly shaped somas (Karperien et al., 2013; Torres-Platas et al., 2014).
After exposure to harmful agents, hyperbranched microglia can adopt an activated phenotype which presents similar somas to hyperbranched microglia, but showing much fewer, thicker, and retracted processes (Torres-Platas et al., 2014; Sasaki, 2017). In this state of activation, microglia are able to proliferate, induced by the presence of factors released by reactive astrocytes such as M-CSF (macrophage colony-stimulating factor) and GM-CSF (granulocyte-macrophage colony-stimulating factor), or by different cytokines; IL-1, IL-3, IL-4, IL-5, IL-10, and TNF-α (Schilling et al., 2001), as well as migration to the site of damage, in order to interact with damaged neurons and remove cellular debris or dead cells (Karlstetter et al., 2010).
After sustained exposure to these agents, activated microglia change to an amoeboid state, with a more regular morphology characterized by thicker, rounded somas and with few or even absent processes (Karperien et al., 2013; Torres-Platas et al., 2014). Amoeboid microglia, with phagocytic capacity, migrate to the site of damage to phagocytose cellular debris or dead neurons (Davis et al., 2017).
Another morphological type of activation is the rod microglia characterized by an elongated soma with very few processes, with a length similar to that of branched microglia, located close to neurons, aligned with nerve fibers (de Hoz et al., 2013; Giordano et al., 2021). This microglial morphotype is associated with neurodegeneration and neuronal circuit rearrangement (Neumann et al., 2009; Perry and O’Connor, 2010). Rod microglia run parallel to damaged neurons and disrupted axons in order to limit neuronal damage (Ziebell et al., 2012). Once recruited to the site of injury, rod microglia have the ability to degrade the extracellular matrix, to promote the retraction of dystrophic axons and to disrupt synapses, in a phenomenon known as “synaptic stripping,” decreasing synaptic activity and metabolic requirement of the damaged neurons (Perry and O’Connor, 2010; Cao et al., 2012; Bachstetter et al., 2017).
At final stages before cell death by apoptosis, microglia adopt a dystrophic state with partially or completely fragmented cytoplasm with retraction, alteration, or even complete lack of cell processes (Streit et al., 2004). In this dystrophic stage, microglia have lower neuroprotective capacity and release higher amounts of pro-inflammatory cytokines. This microglia produces chronic inflammatory mediators associated with neuroinflammation and parainflammation. Dystrophic microglial proliferation is associated with several neurodegenerative diseases such as Alzheimer’s or Huntington’s disease (Simmons et al., 2007; Streit et al., 2009; Hoare and Narita, 2013; Shahidehpour et al., 2021).
Similar to CNS, different morphotypes of microglia in the retina appear as a consequence of exposure to different cytokines, chemokines, or DAMPs (Karlstetter et al., 2015).
Quiescent astrocytes during astrogliosis, similar to microglia, are able to acquire a reactive phenotype in response to neuronal damage, undergoing different morphological changes (Sofroniew, 2009). The main changes associated with astrogliosis are cellular hypertrophy, characterized by an increase in the thickness of the soma and its cell processes, retraction, and reorientation of its primary processes, together with an increase in the number of primary and secondary processes (Peng et al., 2014). Cellular hypertrophy is accompanied by increased expression of intermediate filament constituent proteins, including vimentin, nestin and mainly GFAP, which is the main component of intermediate filaments in adult astrocytes (Pekny and Pekna, 2004).
Astrocyte proliferation occurs after sustained exposure to damage, whether in the presence of a chronic neurodegenerative process, trauma, ischemic and infectious processes (Sofroniew, 2014). Proliferation is associated with the expression of cytokines such as IFN-γ, TNF-α, M-CSF, IL-1, IL-2, and IL-6 or chemokines such as CXCL10 and CCL5 (Bajetto et al., 2002; Mohri et al., 2006; Zhang D. et al., 2009).
Astrocyte proliferation and glial scar formation play an essential role in protecting and regenerating tissue by isolating it from the site of damage, where cytotoxic molecules are released (Liu et al., 2014). However, at later stages of tissue remodeling, they can also prevent normal axonal repair and regeneration processes (Menet et al., 2003; Vay et al., 2021). Factors released by astrocytes such as TGF-β and NGF are involved in the formation of these glial scars (Escartin and Bonvento, 2008).
Müller cells in the retina, similar to astrocytes, have the capacity to react in the presence of pathological conditions, injury, or inflammation, acquiring a reactive phenotype (Wang and Wong, 2014). The main characteristics of Müller cell gliosis are cell hypertrophy, with thickening of both somas and processes, proliferation, as well as increased expression of cytoskeleton constituent proteins such as vimentin or GFAP, and decreased expression of GS (Bringmann et al., 2009).
During gliosis, Müller cells will also contribute to the formation of glial scars, which can inhibit the remodeling and normal regeneration of damaged retinal tissue (Bringmann et al., 2009; Bringmann and Wiedemann, 2012). Increased GFAP expression, as well as the expression of extracellular matrix molecules and cell adhesion molecules, can inhibit normal neurite and axon growth, and thus neuronal regeneration (Lu et al., 2011; Bringmann and Wiedemann, 2012). Growth factors released by glial cells during reactive gliosis, such as basic fibroblast growth factor (bFGF), NGF, epidermal growth factor (EGF) and platelet-derived growth factor (PDGF), as well as glutamate or cytokines such as IL-1 or IL-2, are associated with Müller cell proliferation (Lewis et al., 1999; Kruchkova et al., 2001; Bringmann et al., 2006).
In the CNS, quiescent microglia, in response to proinflammatory cytokines from pathogen debris and damaged cells, show an activated phenotype expressing proinflammatory factors such as IL-6, TNF-α, IL-1β, NO and proteases, which have negative effects on neurodegenerative processes (Liddelow and Barres, 2017). The classical M1 activation phenotype, induced by the presence of IFN-γ, released by T cells, LPS or DAMP, is characterized by a microglia with the capacity to produce inflammatory cytokines (IL-23, IL-18, IL-12, IL-1β, IL-6, TNF-α, CCL2, and CXCL10), ROS, NO and matrix metalloproteinases (MMP9 and MMP3), as well as acting as an antigen-presenting cell preventing the entry of pathogens (Cherry et al., 2014; Liu et al., 2019).
On the other hand, anti-inflammatory factors such as IL-4, IL-10, IL-13, and TGF-β lead to the neuroprotective M2a phenotype of microglia promoting the release of factors such as arginase-1, CD206 (macrophage mannose receptor), insulin-like growth factor 1 (IGF-1), Ym1 and FIZZ1, associated with neuroprotection and tissue repair (Glass et al., 2010; Sica and Mantovani, 2012; Tang and Le, 2016; Liddelow and Barres, 2017). M2b phenotype activation induced by immune complexes and TLRs agonists increases the expression of IL-1β, CD86, suppressor of cytokine signaling 3 (SOCS3), IL-6, IL-10, involved in phagocytosis and removal of damaged tissue debris (Franco and Fernández-Suárez, 2015; Lan et al., 2017). Once the immune response is weakened, the expression of TGF-β, IL-10 and glucocorticoids induces the appearance of the M2c phenotype promoting tissue regeneration (Mantovani et al., 2004).
The expression of different surface channels and receptors on microglia and their interaction with pro- or anti-inflammatory molecules will influence the density, spatial distribution, activation state and morphotype, as well as the pathogenesis of the neurodegenerative process (Ma et al., 2019). For example, IL-4 expression by microglia suppresses the release of proinflammatory cytokines such as IL-6, TNF-α, or NO, contributing to the transition of microglia toward the neuroprotective phenotype (Park et al., 2005; Zhao et al., 2006). In the retina, branched microglia show high expression of P2RY12, which is associated with immunosurveillance functions, while amoeboid microglia express high levels of CD68, a classic marker of phagocytosis (Walker et al., 2014; Dubbelaar et al., 2018; Waller et al., 2019).
Similar to microglia, pro-inflammatory reactive astrocytes increase the expression of genes related to the complement cascade, inducing the production of factors such as TNF-α, IL-1β, and NO, which can exert a neurotoxic effect (Liddelow and Barres, 2017). The production of inflammatory cytokines such as TNF-α, IL-1β, IL-6, IL-12, IL-15, IL-17, and IL-23 contribute to exacerbate the inflammatory response in the CNS (Jensen et al., 2013). Chemokines such as CCL2, CCL5, CXCL1, CXCL9, CXCL10, and CXCL12, produced by astrocytes, promote the recruitment of macrophages, monocytes, T and B lymphocytes and neutrophils (Jensen et al., 2013). In addition, neurotoxic astrocytes release NO, ROS, vascular endothelial growth factor (VEGF), and vascular cell adhesion molecules (VCAM-1), which contribute to maintaining the inflammatory microenvironment, BBB rupture and leukocyte extravasation into the CNS parenchyma (Gimenez et al., 2004; Swanson et al., 2004; Argaw et al., 2012).
Reactive astrocytes also increase the expression of neurotrophic factors and neuroprotective thrombospondins (Liddelow and Barres, 2017). Astrocytes with a neuroprotective role produce ciliary neurotrophic factor (CNTF) and BDNF increasing neuronal survival (Lee et al., 1998; Ikeda et al., 2001). Expression of glutamate transporters such as glutamate aspartate transporter (GLAST) and glutamate transporter-1 (GLT-1), retinoic acid and glutathione protect neurons against oxidative stress, ON excitotoxicity, increase anti-inflammatory character and protect the BBB (Rothstein et al., 1996; Chen et al., 2001; Mizee et al., 2014). In addition, astrocytes produce molecules with anti-inflammatory effects on microglia and monocytes (Min et al., 2006; Kostianovsky et al., 2008). The presence of anti-inflammatory cytokines such as IL-10, IL-4, or IL-13 induces the appearance of neuroprotective reactive astrocytes, which in turn have the capacity to release IL-4, IL-10, and TGF-β (Oksanen et al., 2019).
Müller cells can play both a neurotoxic and a neuroprotective role during gliosis. Müller cells are capable of producing neuroprotective factors such as pigment epithelium-derived factor (PEDF) or VEGF, or cytokines such as IL-6, which contribute to restarting the cell cycle after damage, axonal regeneration, and neuronal survival (Yoshida et al., 2001; Harada et al., 2002; Unterlauft et al., 2012; Foxton et al., 2013; Galan et al., 2014). On the other hand, Müller cells are a source of proinflammatory cytokines such as IL-1β, TNF-α, and IL-6 (Coughlin et al., 2017). Complement protein C1q and NO expression in Müller cells is associated with inflammation and RGCs loss (Neufeld, 1999; Stasi et al., 2006). Activation of the NFκB signaling pathway and increased NGF expression induce TNF-α expression by Müller cells, contributing to neurotoxic effect (Lebrun-Julien et al., 2009). Expression of both NO and TNF-α exerts a neuronal death-inducing effect (Bringmann et al., 2006).
After retinal damage, increased expression of inflammatory factors such as MCP-1 by Müller glia promotes the recruitment of monocytes/macrophages and microglial cells to the site of damage, which release proinflammatory cytokines and oxygen free radicals, leading to neuronal apoptosis and retinal degeneration (Nakazawa et al., 2006a,2007; Hollborn et al., 2008).
Glial cells are also involved in the adaptive immune response, mediated by T and B lymphocytes. APCs express MHC-II on their surface and are responsible for presenting antigens to activated T-lymphocytes, which are able to recognize antigens, although not directly. Dendritic cells, B lymphocytes and monocytes/macrophages are considered professional APCs that constitutively have high expression of MHC-II molecules on their surface. APCs degrade uptaken antigens, via endolysosomal pathway, into smaller fragments that are presented bound to MHC-II (Campana et al., 2015). In addition, they also have co-stimulatory molecules on their surface such as CD80, CD86, and CD40, which are necessary for T-cell activation (Blum et al., 2013). The presence of these co-stimulatory molecules in APCs can be induced by the expression of pro-inflammatory molecules such as IFN-γ (Rostami et al., 2020).
Glial cells, under physiological conditions, have very low or even no MHC-II levels on their surface; only microglial cells have low MHC-II expression, while it is absent in astrocytes and Müller cells (Aloisi et al., 2000a). During gliosis and in response to severe inflammatory processes, increased oxidative stress or in the presence of pro-inflammatory molecules such as IFN-γ or TNF-α, glial cells increase the expression of MHC-II, acting as non-professional APCs (Aloisi et al., 2000b; Bringmann et al., 2009).
In addition, glial cells also show increased expression of co-stimulatory molecules, which are essential for the triggering of the adaptive immune response (Nelson et al., 2002; Rostami et al., 2020). Increased MHC-II expression on reactive glial cells is directly related to inflammatory processes in the CNS (Schetters et al., 2018; Rostami et al., 2020).
Neuroinflammation in glaucoma has become an important factor due to the role of both the immune system and glial cells in the early stages of the disease (Tezel, 2013; Rizzo et al., 2017). Different studies show that glaucoma has important similarities with other neurodegenerative pathologies associated with inflammation such as amyotrophic lateral sclerosis, Alzheimer’s disease, Parkinson’s disease, or frontotemporal dementia (Ramirez et al., 2017; Fernández-Albarral et al., 2019b). The A1 phenotype of astrocytes increases the expression of genes related to the complement cascade and neurotoxins, leading to a neurotoxic effect. In addition, A1 astrocytes induce loss of synaptogenesis and neuronal and oligodendrocyte death (Liddelow and Barres, 2017).
Different genes involved in inflammatory processes increase their expression in both the retina and the glaucomatous optic nerve head (Ahmed et al., 2004; Yang et al., 2007). The first pathways involved in increasing gene expression are TLR-mediated signaling pathways, for example, heat-shock proteins (HSPs) induce an increase in major histocompatibility complex class II (MHC-II) expression and cytokine production (Luo et al., 2010). Second, activation of the nuclear factor kappa B (NFκB)-mediated pathway causes an increase in the expression of cytokines belonging to the IL-1 family that in turn promote the release of a cascade of inflammatory cytokines such as TNF-α and IL-6 (Rolle et al., 2021). In the optic nerve of patients with glaucoma, elevated levels of both TNF-α and Fas ligand (FasL), a protein with proapoptotic capacity, are observed, and both are linked to the pathogenesis of glaucoma (Yan et al., 2000; Tezel et al., 2001; Sawada et al., 2010; Gregory et al., 2011).
The onset of the inflammatory process in glaucoma is triggered by impaired communication between RGCs and glial cells, inducing the release of proinflammatory mediators, such as ROS, NO, TNF-α, and IL-1β (Nakazawa et al., 2006b; Luo et al., 2010; Chua et al., 2012; Tezel et al., 2012; Madeira et al., 2015).
The marked increase in TNF-α expression as a result of proinflammatory imbalance in glaucoma is associated with RGCs loss (Yan et al., 2000; Tezel et al., 2001). This increase is also responsible for the alteration of mitochondrial function, reducing ATP production and increasing the production of ROS, contributing to the activation of the NFκB signaling pathway, and to the production of proinflammatory signals. This neuroinflammatory stimulation in turn promotes mitochondrial dysfunction of RGCs, constituting a cycle of neuronal damage (Wilkins et al., 2017).
Both macroglia (astrocytes and Müller cells) and microglia, as well as infiltrating monocytes, are involved in the neuroinflammatory process and in the immune-mediated neurodegenerative process of RGCs that occurs in glaucoma, producing proinflammatory cytokines and complement components (Howell et al., 2012; Nickells et al., 2012; Tezel, 2013; Soto and Howell, 2014). The vision loss process in glaucoma is also related to functional and distributional changes of retinal glial cells during reactive gliosis processes (Russo et al., 2016).
Glial cells have the ability to recognize DAMPs and PAMPs which are expressed on the surface of damaged cells or can be released into the extracellular space once the processes of apoptosis and necrosis have been triggered. Among the main DAMPs associated with RGC neurodegeneration are HSPs, uric acid and high-mobility group box (HMGB) proteins (Tezel and Wax, 2000; Rifkin et al., 2005; Glass et al., 2010; Yu et al., 2010; Zhu et al., 2011).
Astrocytes and Müller cells in response to glaucoma-associated stress signals undergo an important transformation process during reactive gliosis (Wang et al., 2002; Bringmann et al., 2006; Seitz et al., 2013; Lahne et al., 2020; Fernández-Albarral et al., 2022) showing both morphological changes and a wide spectrum of molecular and functional modifications (Johnson et al., 2007; Nikolskaya et al., 2009; Qu and Jakobs, 2013; Tehrani et al., 2016; Quillen et al., 2020; Fernández-Albarral et al., 2021).
Reactive astrocytes in glaucoma show an increase in GFAP expression, an increase in the size of their cytoskeleton and a larger extension of their cellular processes, as well as migrate to the site of damage (Tezel et al., 2001; Inman and Horner, 2007; Hernandez et al., 2008; Dai et al., 2012; Fernández-Albarral et al., 2022). Hypertrophic astrocytes maintain the integrity of neuronal tissue and form a barrier that isolates healthy neurons and protects them from neuroinflammatory signals, although at the same time they activate a survival mechanism that may limit the neurotrophic, bioenergetic and antioxidant supply to RGCs. This alteration of its functions together with astrocyte-mediated remodeling of the extracellular matrix and profibrotic processes amplify the biomechanical and vascular stress associated with glaucoma occurring in the optic nerve head (Roberts et al., 2010; Burgoyne, 2011; Dai et al., 2012; Figure 2).
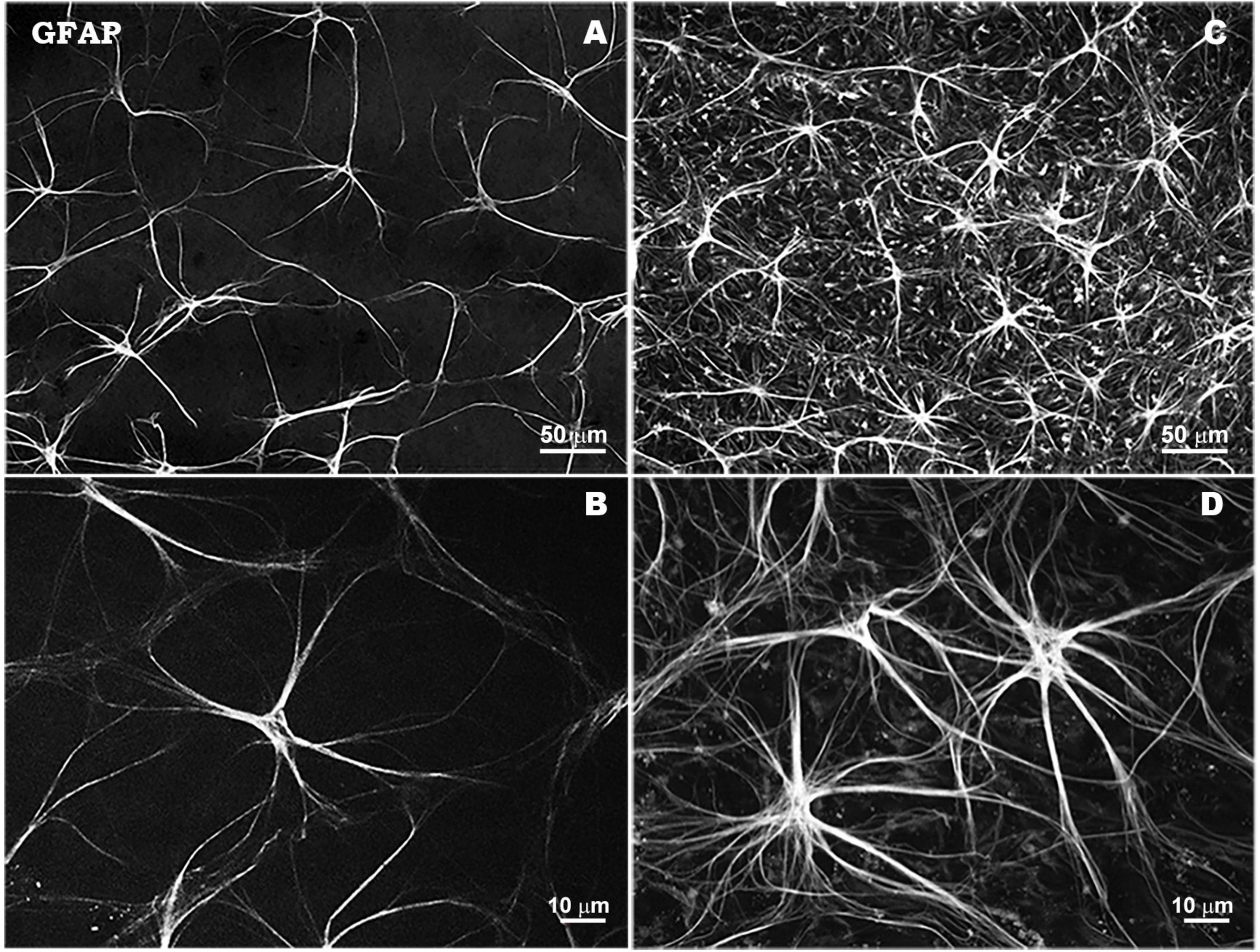
Figure 2. Morphological characteristics of astrocytes in response to elevated IOP in a laser-induced ocular hypertension experimental model in mouse. Astrocytes in hypertensive retina show an overexpression of GFAP immunostaining, a thickening of their soma size and an increase in the number and thickness of their processes. Retinal whole-mount. GFAP immunostaining. Naïve retina (A,B). Hypertensive retina (C,D). Images high magnification (B,D). Modified from Fernández-Albarral et al. (2022).
Mechanical stress induced by increased IOP in glaucoma results in increased EGF receptor expression following astrocyte activation, as well as increased levels of TNF-α, matrix metalloproteinases (MMPs), endothelins, and nitric oxide synthase 2 (NOS-2), ultimately leading to a neurodegenerative response (Liddelow et al., 2017; Lin et al., 2019). In the course of this astrogliosis in glaucoma, there is also increased expression of the phagocytosis-related Mac-2 gene and ET-1 by astrocytes, demonstrating both their role in phagocytosis of damaged or dead RGC debris and their involvement in glaucoma-associated vascular dysregulation (Nickells et al., 2012; Chong and Martin, 2015).
Müller cells are one of the first glial cells to respond to glaucoma-associated stress signals such as increased IOP (Wang et al., 2002). In response to increased IOP, Müller cells increase the expression of both GFAP and GS (Zhang S. et al., 2009). Activation of Müller glia in glaucoma occurs with the objective to maintain neuronal tissue integrity and homeostasis by increasing the expression of receptors associated with neuronal growth and survival, increasing levels of extracellular glutamate uptake, thereby protecting RGCs from glutamate excitotoxicity, and releasing NGF (Newman and Reichenbach, 1996; Bringmann et al., 2009).
On the other hand, their chronic activation induces the release of TNF-α, IL-1, and NO that contribute to RGC degeneration and death. Moreover, this chronic activation of Müller cells impairs the survival of RGCs through activation of cell death receptors and alteration of retinal potassium and water homeostasis (Bringmann et al., 2005, 2006; Iandiev et al., 2008; Xu et al., 2009; Garcia et al., 2014). Furthermore, in this context Müller cells release ROS and prostaglandin E2 that also contribute to the apoptosis of RGCs (Gao et al., 2013; García-Bermúdez et al., 2021).
Mature microglial cells take part in inflammatory processes being activated by DAMPs released by neural tissue cells and also by astrocytes and even by microglia themselves (Rifkin et al., 2005; Yu et al., 2010; Zhu et al., 2011). Among the different DAMPs associated with inflammation, HSPs are released by RGCs in response to increased IOP and Tenascin C (TN-C) increases its expression in astrocytes and is responsible for TLR activation (Luo et al., 2010; Howell et al., 2012). Another mechanism induced by damaged neurons that leads to glial cell activation is the release of HMGB proteins, which will bind to CD11b receptors present on glial cells, stimulating the release of inflammatory cytokines and promoting the acquisition of reactive phenotypes (Nakazawa et al., 2006b).
In glaucoma, reactive microglial cells initially phagocytize the debris of damaged or dead RGCs, contributing to the maintenance of a toxin-free microenvironment, and also release neurotrophic factors such as BDNF or CNTF, providing neuroprotection and promoting tissue neuroregeneration (Silverman and Wong, 2018; Alqawlaq et al., 2019). During the neuroinflammatory process in glaucoma, chronic activation of microglial cells leads to the release of cytokines and chemokines such as complement factors, TNF-α, IL-6, IL-1β, iNOS, NO, among others, which contribute to amplify the inflammatory response, induce morphological changes of different microglial phenotypes and contribute to apoptosis of RGCs (Ebneter et al., 2010; Bosco et al., 2012; Nickells et al., 2012; Wang et al., 2016; Fernández-Albarral et al., 2019a; Ramírez et al., 2020a; Fernandez-Albarral et al., 2022). In response to elevated IOP, the microglia exhibit different signs of activation, such as proliferation and migration to areas of damage, retraction, reorientation, and hyper-ramification of their processes, and the appearance of amoeboid- and rod-like microglia with macrophagic capacity, related to RGC degeneration (Ramírez et al., 2020a; Fernandez-Albarral et al., 2022; Figure 3).
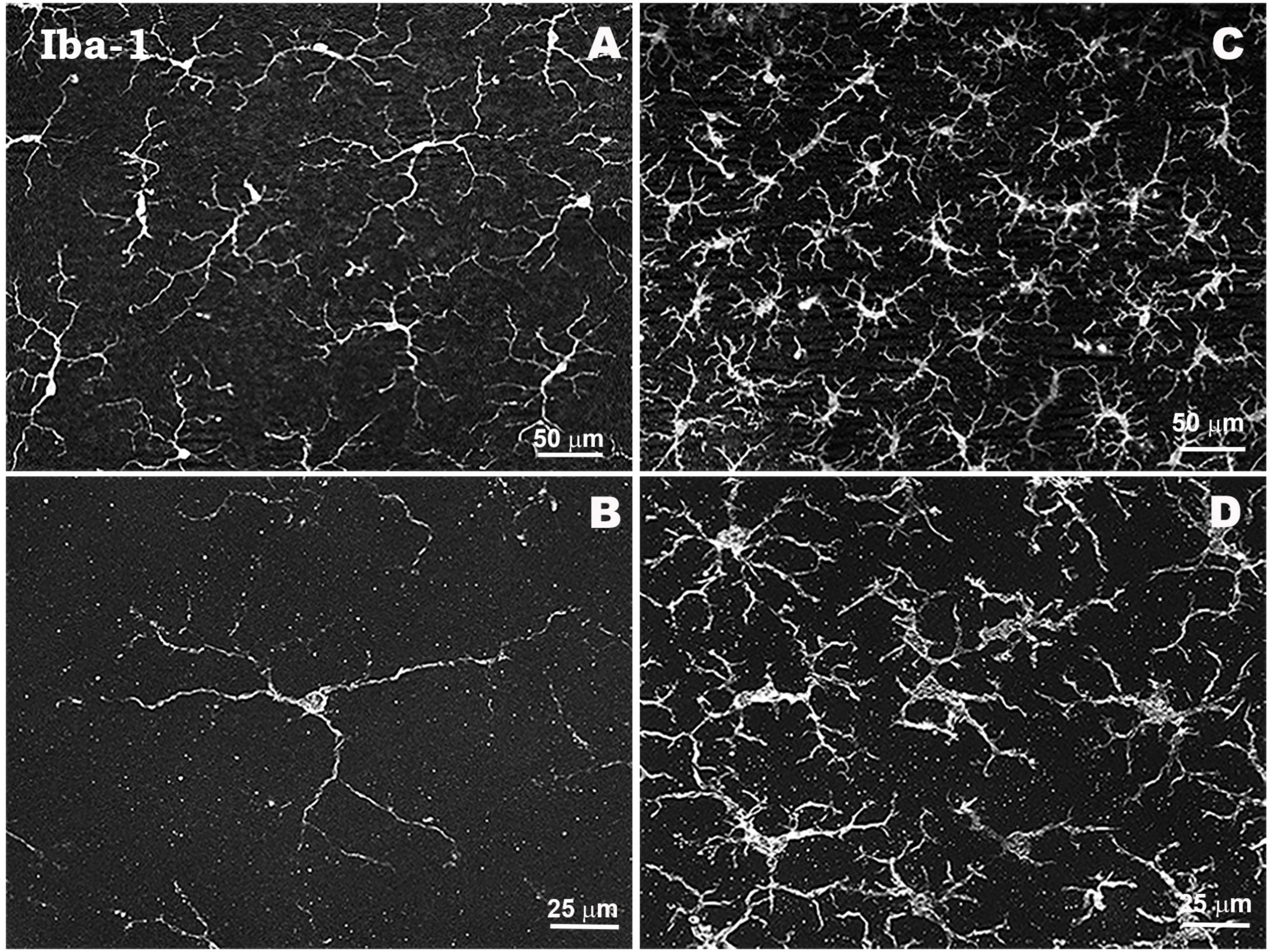
Figure 3. Morphological characteristics of microglial cells in response to elevated IOP in a laser-induced ocular hypertension experimental model in mouse. Microglia in hypertensive retina show proliferation phenomena, thickening of their soma and retraction and thickening of their processes. Retinal whole-mount. Iba-1 immunostaining. Naïve retina (A,B). Hypertensive retina (C,D). Images high magnification (B,D). Modified from Ramírez et al. (2020a).
Apoptosis is considered the main death pathway of RGCs in glaucoma and can be triggered either by signals from the cell soma, from their axons or from signals received from the extracellular environment (Quigley, 1999). Microglial and macroglial cells are involved in the activation and maintenance of apoptotic processes in glaucoma (Tezel, 2013). Increased IOP induces an inflammatory response of the glial population in the retina with the primary objective to isolate and resolve neural tissue damage by providing growth factors and metabolites to damaged RGCs, however, the chronification of this glial inflammatory response can affect glial cell function, neuronal support and extracellular glutamate buffering capacity (Martin et al., 2002), making RGCs and their axons more vulnerable and ultimately more prone to death by apoptosis (Neufeld and Liu, 2003; Weinreb and Tee Khaw, 2004; Quigley, 2005a,b; Tezel, 2011, 2013).
In addition to innate immunity, stress signals and tissue damage associated with glaucoma together with alterations resulting from mitochondrial dysfunction may also induce an adaptive immune response. The inflammatory microenvironment in glaucoma enriched by cytokines, chemokines and adhesion molecules released by glial cells is a supportive environment for glia-lymphocyte interactions and antigen presentation for an enlarged immune response (Tezel, 2022). Increased expression of MHC molecules and TLRs by glial cells allows them to participate in both innate and adaptive immunity (Yang et al., 2001; Luo et al., 2010). In glaucomatous patients, chronically activated microglial cells show increased MHC-II expression and GFAP-positive astrocytes show strong labeling with HLA-DR (human MHC-II molecule type) (Yang et al., 2001; Ramirez et al., 2017).
The increased expression of MHC-II by glial cells in both human and experimental animal models of glaucoma demonstrate the ability of these cells to act as antigen presenting cells (APCs) and stimulate an adaptive immune response (Yang et al., 2001; Ebneter et al., 2010; Howell et al., 2011; Gramlich et al., 2013; Chidlow et al., 2016). This along with the expression of proinflammatory cytokines leads to increased antigenic stimulation of T-lymphocytes. TNF-α expression by activated T cells is associated with other neurodegenerative CNS disorders and may be one of the causes of retinal changes in glaucomatous patients (Yang et al., 2011). In addition, alterations in vascular barriers, associated with glaucoma and ageing, facilitate the migration of T lymphocytes through the BRB and their contact with resident glia and autoantibodies generated by local antigens, promoting RGC damage through the toxicity of released proinflammatory cytokines (Wax et al., 2008; Gramlich et al., 2015; Chen et al., 2018).
The complement system, as part of innate immunity, plays an important role in immunosurveillance and is involved in the CNS inflammatory response to stress and damage (Tezel, 2013; Williams et al., 2016).
Associated with innate and adaptive immune responses, complement activation in glaucoma may contribute to neuroinflammation and RGCs degeneration. The complement protein C1q acts as a pattern recognition molecule tagging synapses and damaged neurons, thus initiating the classical complement activation pathway (Tezel, 2022). This complement activation appears to be important in the synaptic degeneration of RGCs in glaucoma, in a similar process to that occurring in the elimination of unwanted synapses during neuronal development (Stevens et al., 2007; Williams et al., 2016). Complement activation leads to cleavage of complement factor C3 and activation of the membrane attack complex (MAC). MAC is involved in the processes of cell lysis and necrotic death, and C3 is also associated with the processes of apoptosis (Fishelson et al., 2001; Rus et al., 2005; Harvey and Durant, 2014).
High concentrations of C5, an essential protein for MAC genesis, as well as MAC are associated with RGC apoptosis and axonal degeneration in the optic nerve head and pharmacological inhibition of the complement cascade reduces both apoptosis and MAC levels (Jha et al., 2011; Howell et al., 2013; Soto and Howell, 2014). High concentrations of C3, present in both RGCs and in the nerve fiber layer, are also associated with RGC and axon degeneration, and its inhibition under chronic elevated IOP leads to neuroprotection, suggesting a role in the development and progression of glaucoma (Kuehn et al., 2006; Bosco et al., 2018).
Increased expression of factors associated with complement activation such as C1q, C3, C5, and MAC are present in both glaucomatous patients and experimental animal models of glaucoma, noting that this increase occurs very early in response to IOP elevation (Stasi et al., 2006; Stevens et al., 2007; Howell et al., 2011; Johnson et al., 2011).
Ageing is a natural and irreversible progressive phenomenon characterized by both anatomical and functional decline. Although it is a process that can occur in different ways in each individual, several common features of the ageing process have been defined, including genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, mitochondrial dysfunction, cellular ageing, stem cell depletion and altered intercellular communication, among others (López-Otín et al., 2013).
Among all, genomic instability and DNA damage constitute one of the main biological processes associated with ageing, leading to mitochondrial dysfunction, accumulation of ROS and cellular decline (Schumacher et al., 2021). In the CNS during ageing and due to its high energy requirement, a greater accumulation of ROS will occur, leading to an increase in oxidative damage, altering mitochondrial function, neurogenesis, and the normal cell cycle (Spalding et al., 2013; Grimm and Eckert, 2017). These processes leading to neuronal decline during senescence will also contribute to the pathophysiology of neurodegenerative processes (Wang et al., 2019). Among the main markers associated with ageing are elevated oxidative stress, mentioned above, and inflammation (Lee et al., 1999; Finkel and Holbrook, 2000).
Altered cells during ageing acquire a senescence-associated secretory phenotype (SASP) characterized by the secretion of proinflammatory cytokines such as IL-6, IL-1β, TNF-α, and INF-γ, acute phase proteins, chemokines, growth factors and extracellular matrix proteases (Fulop et al., 2018). This SASP phenotype contributes to a sterile, low-grade inflammatory environment by attracting and activating immune system cells (Prata et al., 2018).
The term “inflamm-aging” is used to describe this age-related immune phenotype of inflammation, characterized by a chronic, low-grade pro-inflammatory state, which is related to both activation of the innate immune system and continued exposure to antigens and stress (Franceschi et al., 2000, 2018).
Increased cell ageing and exposure to endogenous infectious agents and harmful signals throughout life imbalance the innate and adaptive immune responses, shifting toward greater involvement of innate immune cells, to the detriment of adaptive immunity based on antigen response (Frasca and Blomberg, 2015).
With ageing, there is an alteration of the adaptive immune response affecting T and B lymphocytes, decreasing the ability to respond to antigens (Gibson et al., 2009; Grubeck-Loebenstein et al., 2009). In addition, during senescence there is an increase in circulating innate immune cells (monocytes/macrophages, neutrophils), as well as an increase in proinflammatory cytokines, indicating an increased innate immune response, which is associated with an increased inflammatory response (Roubenoff et al., 1998; Leng et al., 2005; Tan et al., 2007). Increased serum expression of inflammatory mediators such as C-reactive protein (CRP), IL-6, IL-10, or TNF-α, indicate an activation of the peripheral immune system during ageing and are associated with neurophysiological alterations (Varadhan et al., 2014; Tegeler et al., 2016).
This inflammatory status in the CNS is evidenced by the presence of oxidative stress, increased production and release of pro-inflammatory mediators and the development and progression of neurodegenerative pathologies (Barrientos et al., 2015; von Bernhardi et al., 2015).
In the CNS, as in the peripheral nervous system, there is an age-associated increase in inflammation-related genes such as NFκB, TLR-4, IL-1 receptor and GFAP. In addition, there is an increase in the expression of IL-6, TNF-α, and increased oxidative damage coupled with a reduction in antioxidant capacity (Guest et al., 2014; Primiani et al., 2014). Accumulation of free radicals and increased pro-inflammatory cytokines such as IL-1β and IL-6, combined with decreased expression of anti-inflammatory cytokines such as IL-10 and IL-4, contribute to the maintenance of the senescent inflammatory state (Ye and Johnson, 2001; Nolan et al., 2005; Sierra et al., 2007; Kuzumaki et al., 2010). Along with the above changes, chronic inflammation also compromises the integrity of the BBB, increasing permeability and making the CNS more vulnerable as a result of the entry of cytokines such as IL-1β, IL-6, and TNF-α, and immune cells from the systemic circulation, which are normally strictly regulated. Although these cytokines and immune cells are crucial for neurodevelopment and synaptic plasticity, their excess may contribute to the maintenance of inflammation and chronic activation of CNS immune cells (Steinmetz and Turrigiano, 2010; Niraula et al., 2017; Prieto and Cotman, 2017; Smith et al., 2017).
While low levels of inflammation are associated with healthy neuronal functionality and longevity, high levels of inflammation during ageing are linked to altered CNS homeostasis, physiological imbalance between proinflammatory and anti-inflammatory responses of microglia and astrocytes, as well as increased glial activation (Arai et al., 2015; Walker et al., 2017; Bachiller et al., 2018; Gamage et al., 2020).
Activation of microglial cells is one of the main features associated with CNS senescence, with changes in morphology, functionality, and molecular expression levels.
During senescence and in the absence of pathology, microglia undergo fragmentation of the cytoplasm, loss of branching, and shortening of cell processes (Streit et al., 2004), leading to a decrease in the microglial arbor area, showing a dendritic, less symmetrical, more elongated, and less circular appearance of the cells (Tremblay et al., 2012; Davies et al., 2017; Soreq et al., 2017; Zöller et al., 2018). In addition, they show reduced motility of their cytoplasmic processes, affecting the immunosurveillance processes (Damani et al., 2011; Lee et al., 2021).
Microglia, during this process, show a less uniform, uneven distribution, with less mobility, showing no alterations in cell number and density (Davies et al., 2017).
Another characteristic of microglia in ageing is the acquisition of a dystrophic phenotype characterized by a total or partial fragmentation of the cytoplasm as well as the cytoplasmic processes. The most common form of dystrophic microglia is those with an unbranched morphology, with an absence of branched cytoplasmic processes and sometimes with the spheroid formation. Abnormally tortuous and short processes may also be present (Streit et al., 2004). In addition, there is also an increase in rod microglia, a phenotype associated with altered axoplasmic outflow during ageing and neurodegeneration (Bachstetter et al., 2017; Davies et al., 2017).
With ageing, altered inflammatory signaling and phagocytic dysfunction lead to changes in the maintenance of homeostasis by microglia, affecting their capacity for immunosurveillance and cellular debris removal (Koellhoffer et al., 2017; Yuan et al., 2020). In addition, they acquire a state of greater sensitization showing an exaggerated inflammatory response, increasing the expression of pro-inflammatory cytokines and inflammation-related factors (Norden and Godbout, 2013; Niraula et al., 2017).
The secretory activity of microglia plays an essential role in the maintenance of synaptic function, the proliferation and maturation of oligodendrocytes and the recruitment of other immune cells. SASP-induced disruption of the microglial secretome results in the alteration of the homeostatic balance necessary for proper neuronal function, leading to an increase in molecules such as IL-6, IL-1β, TNF-α, an increase in inflammasome-related proteins and ROS, as well as increased expression of Nox2 by microglia (Ritzel et al., 2015; Garner et al., 2018; Mejias et al., 2018; Geng et al., 2020). In addition, microglia also undergo a reduction of inflammation suppression-related genes in senescence, such as SIRT1 (gene encoding the protein sirtuin 1), leading to an increase in the expression of pro-inflammatory cytokines such as IL-1β, also contributing to neuronal dysfunction (Cho et al., 2015).
With ageing, activated microglia also show increased expression of antigen presentation-related genes such as MHC-II and CD68, along with suppression of genes such as IL-10 or CD200. They also show increased expression of TLR receptors (1, 2, 4, and 7) and co-receptor CD14 involved in antigen recognition in innate immunity (Frank et al., 2006; Letiembre et al., 2007). In senescence, there is a clear increase in classical markers of microglial activation such as CD206, CD36, CD11b, CD14 as well as MHC-II and PRR. Both the expression of CX3CL1 and its receptor CX3CR1, essential for microglial migration, and CD200 levels that maintain microglia in their quiescent state, are decreased in senescent microglia (Frank et al., 2006; Wynne et al., 2010; Soreq et al., 2017; Zöller et al., 2018). Microglia during senescence also show significant changes in the expression of inflammatory-related genes such as NFκB, C3 or complement factor B (CFB), as well as SASP factors such as IL-1α, IL-1β, IL-6, IL-8, IL-12, TNF-α, C3, CFB, CXCL1, TGF-β, and NO, showing the involvement of senescent microglia in complement activation and immune dysregulation in the retina (Ma et al., 2013; Guillonneau et al., 2017; Ramirez et al., 2017; Sreekumar et al., 2020).
An additional feature of microglial activation in ageing is an increased reactivity to immune system stimuli (Barrientos et al., 2011). Microglia, in response to LPS stimulation, show hyperactivation with high expression of both pro-inflammatory cytokines; IL-1β, and anti-inflammatory; IL-10, extending the reduction in fractalkine receptor expression and decreasing IL-4 receptor expression (Henry et al., 2009; Xue et al., 2021). IL-10 released by microglia binds to IL-10 receptors on astrocytes, which regulate the signaling of the inflammatory phenotype of microglia through the action of TGF-β. Decreased IL-10 receptors on astrocytes as a consequence of senescence lead to dysregulation of microglial activation, showing an exacerbated response (Norden et al., 2014, 2016).
Senescent microglia show dysregulation of phagocytic capacity and may phagocytose live neurons or participate in excessive elimination of synapses via C1q, disrupting synaptic transmission (Hickman et al., 2013; Zhao et al., 2015; Liu et al., 2016). It also shows insufficient capacity to remove apoptotic bodies, protein aggregates and myelin, contributing to the accumulation of potentially toxic agents (Safaiyan et al., 2016). In addition, microglial plasticity to adopt an anti-inflammatory or pro-inflammatory phenotype is also altered as a consequence of age (Harry, 2013).
Several factors contribute to the acquisition by retinal microglia of a pro-inflammatory phenotype during ageing, including altered metabolism and reduced immunomodulatory signals from retinal neurons, such as the CX3CL1-CX3CR1 and CD200-CD200R signaling pathways, resulting in increased microglial activation (Chen et al., 2019).
As with microglial cells, astrocyte activation is also one of the hallmarks of physiological ageing (Nichols et al., 1993; Rodríguez et al., 2014; Robillard et al., 2016). The main features associated with senescent astrocytes include permanent cell cycle arrest, morphological alterations, increased GFAP and vimentin expression, chromatin alterations and the formation of senescence-associated heterochromatic foci (SAHF), increased expression of high-mobility group B (HMGB) proteins, reduced expression of neurotrophic growth factors and increased SASP factors, along with lysosomal and mitochondrial dysfunction (Bitto et al., 2010; Boccardi et al., 2015; Boisvert et al., 2018; Turnquist et al., 2019).
Senescence-associated heterochromatic foci formation and nuclear DNA alteration in senescent astrocytes results in attenuated expression of genes related to cell proliferation, expression of DNA damage response (DDR) markers, as well as increased nuclear size (Souza et al., 2015; Seoane et al., 2017; Hernandez-Segura et al., 2018; Bang et al., 2019; Gorgoulis et al., 2019). Increased HMGB protein expression induces DDR production and cellular inflammation (Enokido et al., 2008).
During senescence, there is a strong parallel between the characteristics of reactive astrocytes and SASP of senescent cells, experiencing both cellular hypertrophy and expression of pro-inflammatory molecules. Thus, during ageing, astrocytes acquire a reactive phenotype associated with low-grade neuroinflammation (Bhat et al., 2012; Clarke et al., 2018). SASP-related gene expression in astrocytes during ageing is considered a consequence of DDR production and is mediated by MAPK and NFκB pathways, as well as HMGB expression (Enokido et al., 2008; Chien et al., 2011; Salminen et al., 2011; Mombach et al., 2015; Seoane et al., 2017). SASP mediators released by senescent astrocytes such as IFN-γ, CXCL10, IL-6, and TGF-β have the ability to induce inflammation and promote microglial activation, contributing to the maintenance of the proinflammatory status characteristic of senescence (Blasko et al., 2004; Bhat et al., 2012; Taylor et al., 2018; Han et al., 2020).
With ageing, there is also an alteration in the signaling of glutamate, the main neurotransmitter in the CNS. Both the levels of glutamate transporters GLAST and GLT-1 released by astrocytes, as well as the expression of GS are altered as a consequence of the ageing process, contributing to increased glutamate excitotoxicity (Bellaver et al., 2017; Boisvert et al., 2018).
Similar to some neurodegenerative diseases, astrocyte activation during senescence is mainly observed in specific areas where there is greater synaptic loss and age-related cognitive decline, such as the hippocampus and frontal cortex frontal (Rodríguez et al., 2016; Matias et al., 2019).
During the normal ageing process, the expression of IL-1α, TNF, and C1q, pro-inflammatory cytokines released by senescent microglia, leads to the activation of astrocytes (Sierra et al., 2007; Hickman et al., 2013; Stephan et al., 2013). Most of the genes expressed by these astrocytes are characteristic of the neurotoxic A1 phenotype, classically induced by neuroinflammatory processes (Zamanian et al., 2012; Liddelow et al., 2017; Clarke et al., 2018). Neurotoxic A1 astrocytes appear with ageing and are associated with age-related cognitive decline (Clarke et al., 2018).
Glial fibrillary acidic protein and vimentin levels, two astrocyte-specific genes directly linked to reactive astrogliosis, also increase during ageing (Nichols et al., 1993; Porchet et al., 2003; Iram et al., 2016; Lye et al., 2019). Increased GFAP and vimentin have been linked to altered cell cycle and reduced cell proliferation and neurogenesis processes in senescence (Larsson et al., 2004). In addition, other signs of astrocyte activation during senescence include cellular hypertrophy with increased cytoplasmic organelles and glial filaments, reduction in the number of astrocytes, expression of proinflammatory cytokines such as IL-1, IL-6, TNF-α, and IL-1β and ROS generation (Nichols et al., 1993; Ramírez et al., 2001; Porchet et al., 2003; Mansour et al., 2008; Rodríguez et al., 2014; Weber et al., 2015; Soreq et al., 2017).
Reactive astrocytes during ageing also show increased expression of genes related to complement system activation such as C3 and C4b, as well as expression of genes related to antigen presentation such as MHC-II and markers of inflammation such as NFκB (Lee et al., 2000; Nolan et al., 2005; Campuzano et al., 2009; Carrero et al., 2012; Iram et al., 2016; Boisvert et al., 2018; Clarke et al., 2018).
Retina is also exposed to changes related to cellular ageing such as oxidative stress, cellular hypoxia, DNA alterations, epigenetic alterations, mitochondrial dysfunction, nutrient supply disruption, proteostasis dysfunction and chronic inflammation. The fact that it is constituted mostly of highly specialized post-mitotic cells such as pigment epithelial cells, photoreceptors and RGCs, cells that have a high metabolic activity and oxygen consumption, increases its susceptibility to the effects of senescence (McHugh and Gil, 2018).
Continuous exposure of the retina to visible light contributes to increased oxidative stress. With ageing, the functionality of the pigment epithelium is altered due to the accumulation of waste products such as lipofuscin, which is a potent generator of ROS, contributing to increased oxidative damage. ROS accumulation disrupts proper mitochondrial functionality, leading to reduced ATP synthesis and cell apoptosis. ROS-induced mitochondrial dysfunction alters the mitochondrial respiratory chain and leads to a secondary increase in ROS (Wassell et al., 1999; Davies et al., 2001).
Associated with ageing is also a loss of axons from RGCs, quantified in post-mortem human retinal sections at approximately 3000–7000 axons per year (Harwerth et al., 2008; Calkins, 2013). Although there is variability between different studies, axon loss corresponds to 0.3–0.6% of the total per year between the second and eighth decade of life (Budenz et al., 2007; Harwerth et al., 2008). In the mouse retina, there is a decrease in axon packing density and an increase in axonal diameter accompanied by degenerative changes associated with mitochondrial dysfunction (Zhu et al., 2018). A similar loss of RGCs also occurs in the macula during ageing. Histological samples of human retinas show a decrease in the number of RGCs by 25–43% in the interval between the second and eighth decade of life (Curcio and Drucker, 1993; Blanks et al., 1996).
Retinal glial cells, essential for the maintenance of RGC homeostasis, also undergo age-associated alterations. In senescence, microglia and macroglia acquire a state of chronic activation that results in an autoimmune response, neuroinflammation and altered BRB, ultimately leading to neurodegeneration (Chan-Ling et al., 2007; Xu et al., 2009; Chen et al., 2010).
Age is one of the main risk factors associated with the development and progression of glaucoma (Weinreb and Tee Khaw, 2004; Quigley, 2011). The mechanisms involved in physiological cellular senescence make RGCs and their axons more vulnerable to glaucoma-associated changes leading to more rapid cell loss. In aging, pathological mechanisms triggered by increased IOP will affect neuronal tissue which is less resistant to glaucomatous damage (Burgoyne, 2011).
With aging, the lamina cribrosa becomes stiffer as a consequence of collagen deposition and alterations in elastin synthesis associated with stiffening and thickening of the astrocyte basement membranes and extracellular matrix, increasing the susceptibility of the optic nerve head to glaucomatous damage (Albon et al., 2000; Kotecha et al., 2006). These morphological changes could result in reduced blood flow and nutrient supply to RGC axons as they pass through the lamina cribosa (Burgoyne and Downs, 2008; Burgoyne, 2011). In addition, it has been postulated that laminar damage is associated with oxygen deprivation and disruption of the retrograde axonal flow of neurotrophic factors leading to secondary death of RGCs (Quigley et al., 2000; Martin et al., 2006).
Mitochondrial dysfunction is one of the main features of cellular senescence and the cytotoxic effect of ROS accumulation may induce further degeneration of RGCs in glaucoma patients. Increased IOP induces the alteration of the functionality of antioxidant enzymes such as superoxide dismutase, catalase, or glutathione peroxidase, exacerbating the effects of mitochondrial damage (Moreno et al., 2004; Tezel, 2006; Zanon-Moreno et al., 2008).
Chronic activation of glial cells as part of innate immunity occurs as a result of stress factors associated with both increased IOP and aging (Xu et al., 2009). Release of TNF-α, IL-6, IL-18, IL-1β, induces apoptosis of RGCs and degeneration of their axons (Sappington and Calkins, 2008; Munemasa et al., 2010; Tezel et al., 2012). Specifically, levels of TNF-α and its receptor appear higher in the optic nerve head of aged glaucoma patients, contributing to increased axonal neurodegeneration due to increased IOP in conjunction with increased age-related susceptibility (Tezel and Wax, 2000; Yuan and Neufeld, 2000).
With aging, the immunosurveillance capacity of microglia is altered, presenting a lower number of extensions and less mobility of these, together with a lower rate of movement to the site of the lesion and a greater inflammatory response (Damani et al., 2011). Microglial activation in aging, characterized by less intense morphological changes in response to elevated IOP, is associated with this senescent altered ability of these cells to respond to damage and a reduced neuroprotective capacity (Orre et al., 2014; Ramírez et al., 2020b; Figure 4).
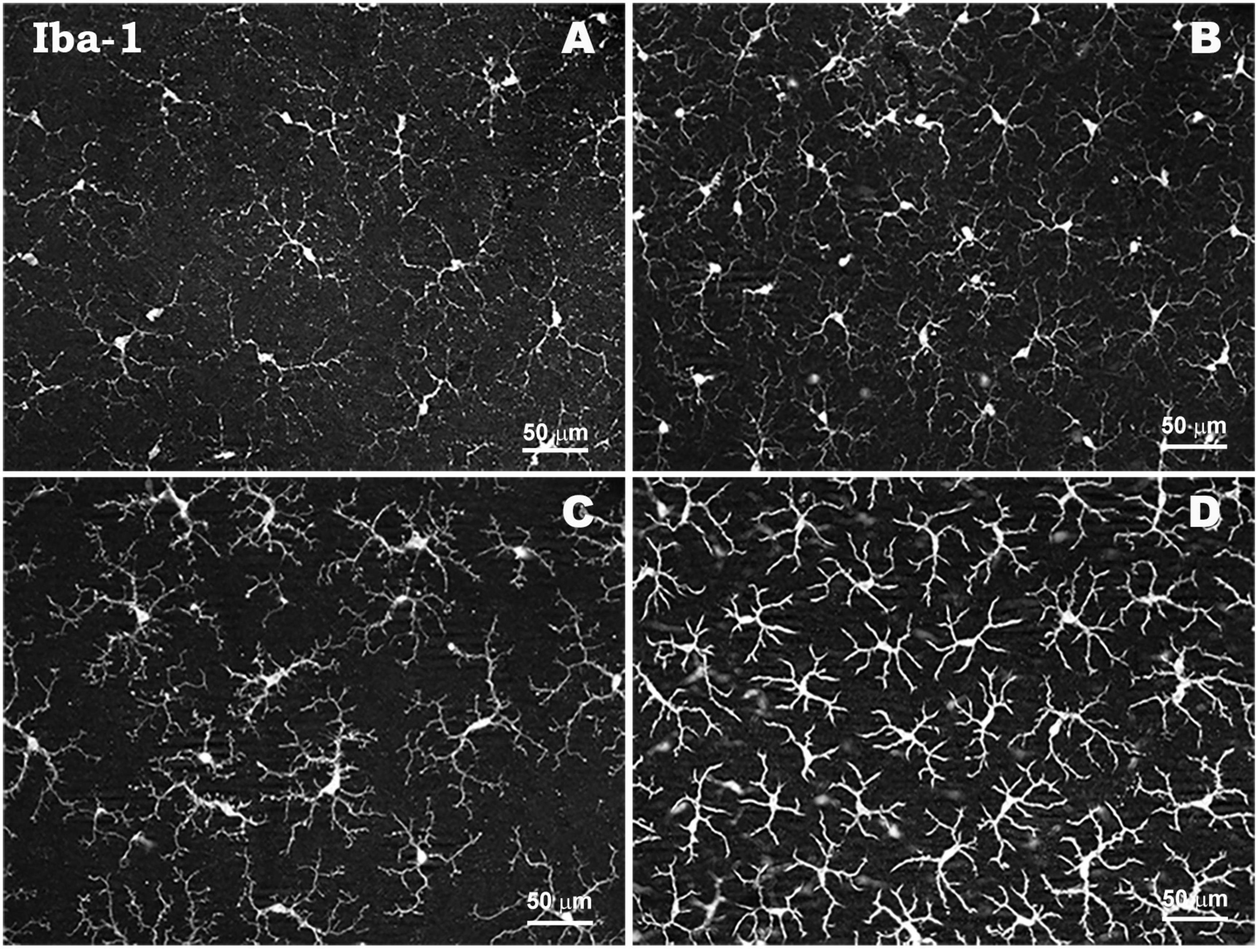
Figure 4. Morphological characteristics of microglial cells in young naïve retina (A), in young retina in response to elevated IOP in a laser-induced ocular hypertension experimental model in mouse (B), in aged naïve retina (C), and in aged retina in response to elevated IOP in a laser-induced ocular hypertension experimental model in mouse (D). Microglia in aged retinas show a more robust aspect with a higher thickening of their soma and retraction of their processes both in normal conditions and in response to IOP increase compared to young retinas. Retinal whole-mount. Iba-1 immunostaining. Modified from Ramírez et al. (2020b).
Evidence suggests that aged animals are more susceptible to RGCs loss in models of acute IOP elevation-induced ischemia, optic nerve crush, or laser-induced ocular hypertension (Ai et al., 2007; Tan et al., 2015; Ramírez et al., 2020b). In addition, in both animal models and humans, aging leads to altered mechanisms regulating the physiological turnover of degraded proteins, triggering increased intracellular accumulation that ultimately leads to apoptosis of RGCs (Wax et al., 2008; Campello et al., 2013).
In glaucoma, increased IOP and vascular dysregulation are considered two of its main risk factors. Mechanical damage caused by elevated IOP leads to altered axoplasmic flow in RGCs, while reduced blood flow and perfusion pressure at the posterior pole also contribute to neuronal damage. The main treatments for glaucoma are focused on reducing IOP; however, despite controlling IOP, in many cases glaucomatous pathology progresses, demonstrating the existence of other mechanisms involved in neurodegeneration, so that elevated IOP is not the only factor that triggers damage. Mechanisms leading to the neurodegenerative process in glaucoma include ischemia/hypoxia, mitochondrial dysfunction, oxidative stress, and neuroinflammation, among others. Increased oxidative stress along with mitochondrial dysfunction and parainflammatory state that occur during aging constitute another main risk factor related to glaucoma progression.
As in other neurodegenerative pathologies, there is a clear involvement of the immune system in neuroinflammatory processes that occur in glaucoma. The regulation of the immune response in the retina is conducted by glial cells: microglia, astrocytes, and Müller cells. Reactive glial cells, in response to damage caused by OHT, show morphological changes and release cytokines and chemokines that initially contribute to trigger the inflammatory process. Retinal glial population react in response to IOP with the primary objective to isolate and resolve neural tissue damage by providing growth factors and metabolites to damaged RGCs. Thus, a controlled response of glial cells helps to restore the functionality of retinal tissue by exerting a neuroprotective effect; however, a chronic activation of glial cells in response to elevated IOP would contribute to exacerbate the neuroinflammatory response, affecting glial cell function, neuronal support and extracellular glutamate buffering capacity, exerting a neurotoxic effect, being involved in the immune-mediated neurodegenerative process of RGCs and apoptotic death. Glaucoma is a pathology associated with aging. During this process, microglia and astrocytes undergo a state of chronic activation that can lead to neuroinflammation, blood-retinal barrier (BRB) alteration, and neurodegeneration. Consequently, with aging, there is an increased susceptibility to the inflammatory process, resulting in greater neurodegeneration associated with elevated intraocular pressure (IOP) that occurs in glaucoma.
Therefore, immune system modulation and retinal glial cell activation control is an important target in the context of neuroprotection in glaucoma.
JF-A: Writing – original draft. AR: Writing – original draft. RH: Writing – original draft. JM: Writing – review and editing. ES-G: Writing – review and editing. LE-H: Writing – review and editing. IL-C: Writing – review and editing. LS-P: Writing – review and editing. JS: Writing – original draft. JR: Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Santander-Complutense University of Madrid Research Projects (PR75/18-21560). JF-A was currently supported by a Predoctoral Fellowship (FPU17/01023) from the Spanish Ministry of Science, Innovation, and Universities; LS-P was currently supported by a Predoctoral Fellowship (CT82/20-CT83/20) and JM was currently supported by a Predoctoral Fellowship (CT58/21-CT59/21) from the Complutense University of Madrid.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abiega, O., Beccari, S., Diaz-Aparicio, I., Nadjar, A., Layé, S., Leyrolle, Q., et al. (2016). Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol. 14:e1002466. doi: 10.1371/JOURNAL.PBIO.1002466
Ahmed, F., Brown, K., Stephan, D., Morrison, J., Johnson, E., and Tomarev, S. (2004). Microarray analysis of changes in mRNA levels in the rat retina after experimental elevation of intraocular pressure. Invest. Ophthalmol. Vis. Sci. 45, 1247–1258.
Ai, L., Yuan, M., and Neufeld, A. (2007). Age-related changes in neuronal susceptibility to damage: comparison of the retinal ganglion cells of young and old mice before and after optic nerve crush. Ann. N Y Acad. Sci. 1097, 64–66. doi: 10.1196/annals.1379.027
Albon, J., Purslow, P., Karwatowski, W., and Easty, D. (2000). Age related compliance of the lamina cribrosa in human eyes. Br. J. Ophthalmol. 84, 318–323.
Alexander, J., Anderson, A., Barnum, S., Stevens, B., and Tenner, A. (2008). The complement cascade: Yin-Yang in neuroinflammation - Neuro-protection and -degeneration. J. Neurochem. 107, 1169–1187. doi: 10.1111/j.1471-4159.2008.05668.x
Almasieh, M., Wilson, A., Morquette, B., Cueva Vargas, J., and di Polo, A. (2012). The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 31, 152–181.
Aloisi, F., Ria, F., and Adorini, L. (2000a). Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunol. Today 21, 141–147. doi: 10.1016/s0167-5699(99)01512-1
Aloisi, F., Serafini, B., and Adorini, L. (2000b). Glia-T cell dialogue. J. Neuroimmunol. 107, 111–117. doi: 10.1016/s0165-5728(00)00231-9
Alqawlaq, S., Flanagan, J., and Sivak, J. (2019). All roads lead to glaucoma: induced retinal injury cascades contribute to a common neurodegenerative outcome. Exp. Eye Res. 183, 88–97. doi: 10.1016/j.exer.2018.11.005
Arai, Y., Martin-Ruiz, C., Takayama, M., Abe, Y., Takebayashi, T., Koyasu, S., et al. (2015). Inflammation, but not telomere length, predicts successful ageing at extreme old age: a longitudinal study of semi-supercentenarians. EBioMedicine 2, 1549–1558.
Argaw, A., Asp, L., Zhang, J., Navrazhina, K., Pham, T., Mariani, J., et al. (2012). Astrocyte-derived VEGF-a drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Invest. 122, 2454–2468. doi: 10.1172/JCI60842
Aung, T., Rezaie, T., Okada, K., Viswanathan, A., Child, A., Brice, G., et al. (2005). Clinical features and course of patients with glaucoma with the E50K mutation in the optineurin gene. Invest. Ophthalmol. Vis. Sci. 46, 2816–2822. doi: 10.1167/iovs.04-1133
Awadalla, M., Fingert, J., Roos, B., Chen, S., Holmes, R., Graham, S., et al. (2015). Copy number variations of TBK1 in Australian patients with primary open-angle glaucoma. Am. J. Ophthalmol. 159, 124–130.e1. doi: 10.1016/j.ajo.2014.09.044
Azam, S., Jakaria, M., Kim, I., Kim, J., Ezazul Haque, M., and Choi, D. (2019). Regulation of Toll-Like Receptor (TLR) signaling pathway by polyphenols in the treatment of age-linked neurodegenerative diseases: focus on TLR4 signaling. Front. Immunol. 10:1000. doi: 10.3389/FIMMU.2019.01000
Bachiller, S., Jiménez-Ferrer, I., Paulus, A., Yang, Y., Swanberg, M., Deierborg, T., et al. (2018). Microglia in neurological diseases: a road map to brain-disease dependent-inflammatory response. Front. Cell Neurosci. 12:488. doi: 10.3389/FNCEL.2018.00488
Bachstetter, A., Ighodaro, E., Hassoun, Y., Aldeiri, D., Neltner, J., Patel, E., et al. (2017). Rod-shaped microglia morphology is associated with aging in 2 human autopsy series. Neurobiol. Aging 52, 98–105. doi: 10.1016/j.neurobiolaging.2016.12.028
Bajetto, A., Bonavia, R., Barbero, S., and Schettini, G. (2002). Characterization of chemokines and their receptors in the central nervous system: physiopathological implications. J. Neurochem. 82, 1311–1329. doi: 10.1046/j.1471-4159.2002.01091.x
Bang, M., Kim, D., Gonzales, E., Kwon, K., and Shin, C. (2019). Etoposide induces mitochondrial dysfunction and cellular senescence in primary cultured rat astrocytes. Biomol. Ther. 27, 530–539. doi: 10.4062/biomolther.2019.151
Barrientos, R., Frank, M., Crysdale, N., Chapman, T., Ahrendsen, J., Day, H., et al. (2011). Little exercise, big effects: reversing aging and infection-induced memory deficits, and underlying processes. J. Neurosci. 31, 11578–11586. doi: 10.1523/JNEUROSCI.2266-11.2011
Barrientos, R., Kitt, M., Watkins, L., and Maier, S. (2015). Neuroinflammation in the normal aging hippocampus. Neuroscience 309, 84–99.
Barron, M., Griffiths, P., Turnbull, D., Bates, D., and Nichols, P. (2004). The distributions of mitochondria and sodium channels reflect the specific energy requirements and conduction properties of the human optic nerve head. Br. J. Ophthalmol. 88, 286–290. doi: 10.1136/bjo.2003.027664
Bejarano-Escobar, R., Sánchez-Calderón, H., Otero-Arenas, J., Martín-Partido, G., and Francisco-Morcillo, J. (2017). Müller glia and phagocytosis of cell debris in retinal tissue. J. Anat. 231, 471–483.
Bellaver, B., Souza, D., Souza, D., and Quincozes-Santos, A. (2017). Hippocampal astrocyte cultures from adult and aged rats reproduce changes in glial functionality observed in the aging brain. Mol. Neurobiol. 54, 2969–2985. doi: 10.1007/s12035-016-9880-8
Bernaus, A., Blanco, S., and Sevilla, A. (2020). Glia crosstalk in neuroinflammatory diseases. Front. Cell Neurosci. 14:209. doi: 10.3389/FNCEL.2020.00209/BIBTEX
Bhat, R., Crowe, E., Bitto, A., Moh, M., Katsetos, C., Garcia, F., et al. (2012). Astrocyte senescence as a component of Alzheimer’s disease. PLoS One 7:e45069. doi: 10.1371/JOURNAL.PONE.0045069
Bitto, A., Sell, C., Crowe, E., Lorenzini, A., Malaguti, M., Hrelia, S., et al. (2010). Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 316, 2961–2968.
Blanks, J., Torigoe, Y., Hinton, D., and Blanks, R. (1996). Retinal pathology in Alzheimer’s disease. I. ganglion cell loss in foveal/parafoveal retina. Neurobiol. Aging 17, 377–384. doi: 10.1016/0197-4580(96)00010-3
Blasko, I., Stampfer-Kountchev, M., Robatscher, P., Veerhuis, R., Eikelenboom, P., and Grubeck-Loebenstein, B. (2004). How chronic inflammation can affect the brain and support the development of Alzheimer’s disease in old age: the role of microglia and astrocytes. Aging Cell 3, 169–176. doi: 10.1111/j.1474-9728.2004.00101.x
Blum, J., Wearsch, P., and Cresswell, P. (2013). Pathways of antigen processing. Annu. Rev. Immunol. 31, 443–473.
Boccardi, V., Pelini, L., Ercolani, S., Ruggiero, C., and Mecocci, P. (2015). From cellular senescence to Alzheimer’s disease: the role of telomere shortening. Ageing Res. Rev. 22, 1–8.
Boisvert, M., Erikson, G., Shokhirev, M., and Allen, N. (2018). The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 22, 269–285.
Bosco, A., Anderson, S., Breen, K., Romero, C., Steele, M., Chiodo, V., et al. (2018). Complement C3-targeted gene therapy restricts onset and progression of neurodegeneration in chronic Mouse Glaucoma. Mol. Therapy 26, 2379–2396. doi: 10.1016/j.ymthe.2018.08.017
Bosco, A., Crish, S., Steele, M., Romero, C., Inman, D., Horner, P., et al. (2012). Early reduction of microglia activation by irradiation in a model of chronic glaucoma. PLoS One 7:e43602. doi: 10.1371/journal.pone.0043602
Bowman, C., Rasley, A., Tranguch, S., and Marriott, I. (2003). Cultured astrocytes express toll-like receptors for bacterial products. Glia 43, 281–291.
Bringmann, A., and Wiedemann, P. (2012). Müller Glial cells in retinal disease. Ophthalmologica 227, 1–19.
Bringmann, A., Iandiev, I., Pannicke, T., Wurm, A., Hollborn, M., Wiedemann, P., et al. (2009). Cellular signaling and factors involved in Müller cell gliosis: neuroprotective and detrimental effects. Prog. Retin. Eye Res. 28, 423–451. doi: 10.1016/j.preteyeres.2009.07.001
Bringmann, A., Pannicke, T., Grosche, J., Francke, M., Wiedemann, P., Skatchkov, S., et al. (2006). Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 25, 397–424.
Bringmann, A., Uckermann, O., Pannicke, T., Iandiev, I., Reichenbach, A., and Wiedemann, P. (2005). Neuronal versus glial cell swelling in the ischaemic retina. Acta Ophthalmol. Scand. 83, 528–538. doi: 10.1111/j.1600-0420.2005.00565.x
Bristow, E., Griffiths, P., Andrews, R., Johnson, M., and Turnbull, D. (2002). The distribution of mitochondrial activity in relation to optic nerve structure. Arch. Ophthalmol. 120, 791–796.
Bsibsi, M., Ravid, R., Gveric, D., and van Noort, J. (2002). Broad expression of toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 61, 1013–1021.
Budenz, D., Anderson, D., Varma, R., Schuman, J., Cantor, L., Savell, J., et al. (2007). Determinants of normal retinal nerve fiber layer thickness measured by stratus OCT. Ophthalmology 114, 1046–1052.
Burgoyne, C. (2011). A biomechanical paradigm for axonal insult within the optic nerve head in aging and glaucoma. Exp. Eye Res. 93, 120–132.
Burgoyne, C., and Downs, J. (2008). Premise and prediction – how optic nerve head biomechanics underlies the susceptibility and clinical behavior of the aged optic nerve head. J. Glaucoma 17:318. doi: 10.1097/IJG.0b013e31815a343b
Burgoyne, C., Downs, J., Bellezza, A., and Hart, R. (2004). Three-dimensional reconstruction of normal and early glaucoma monkey optic nerve head connective tissues. Invest. Ophthalmol. Vis. Sci. 45, 4388–4399.
Burnstock, G. (1999). Release of vasoactive substances from endothelial cells by shear stress and purinergic mechanosensory transduction. J. Anat. 194, 335–342. doi: 10.1046/j.1469-7580.1999.19430335.x
Calkins, D. (2013). Age-related changes in the visual pathways: blame it on the axon. Invest. Ophthalmol. Vis. Sci. 54, ORSF37–ORSF41. doi: 10.1167/iovs.13-12784
Calkins, D., Pekny, M., Cooper, M., Benowitz, L., Calkins, D., Benowitz, L., et al. (2017). The challenge of regenerative therapies for the optic nerve in glaucoma. Exp. Eye Res. 157, 28–33.
Campana, S., de Pasquale, C., Carrega, P., Ferlazzo, G., and Bonaccorsi, I. (2015). Cross-dressing: an alternative mechanism for antigen presentation. Immunol. Lett. 168, 349–354.
Campello, L., Esteve-Rudd, J., Cuenca, N., and Martín-Nieto, J. (2013). The ubiquitin–proteasome system in retinal health and disease. Mol. Neurobiol. 47, 790–810.
Campuzano, O., Castillo-Ruiz, M., Acarin, L., Castellano, B., and Gonzalez, B. (2009). Increased levels of proinflammatory cytokines in the aged rat brain attenuate injury-induced cytokine response after excitotoxic damage. J. Neurosci. Res. 87, 2484–2497. doi: 10.1002/jnr.22074
Cannell, I., Kong, Y., and Bushell, M. (2008). How do microRNAs regulate gene expression? Biochem. Soc. Trans. 36, 1224–1231.
Cao, T., Thomas, T., Ziebell, J., Pauly, J., and Lifshitz, J. (2012). Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience 225, 65–75. doi: 10.1016/j.neuroscience.2012.08.058
Carrero, I., Gonzalo, M., Martin, B., Sanz-Anquela, J., Arévalo-Serrano, J., and Gonzalo-Ruiz, A. (2012). Oligomers of beta-amyloid protein (Aβ1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Exp. Neurol. 236, 215–227.
Cassano, T., Pace, L., Bedse, G., Michele Lavecchia, A., de Marco, F., Gaetani, S., et al. (2016). Glutamate and mitochondria: two prominent players in the oxidative stress-induced neurodegeneration. Curr. Alzheimer. Res. 13, 185–197. doi: 10.2174/1567205013666151218132725
Cekanaviciute, E., Fathali, N., Doyle, K., Williams, A., Han, J., and Buckwalter, M. (2014). Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia 62, 1227–1240. doi: 10.1002/glia.22675
Chang, E., and Goldberg, J. (2012). Glaucoma 2.0: neuroprotection, neuroregeneration, neuroenhancement. Ophthalmology 119, 979–986. doi: 10.1016/j.ophtha.2011.11.003
Chan-Ling, T., Hughes, S., Baxter, L., Rosinova, E., McGregor, I., Morcos, Y., et al. (2007). Inflammation and breakdown of the blood-retinal barrier during “physiological aging” in the rat retina: a model for CNS aging. Microcirculation 14, 63–76. doi: 10.1080/10739680601073451
Chen, H., Cho, K., Vu, T., Shen, C., Kaur, M., Chen, G., et al. (2018). Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat. Commun. 9:3209.
Chen, M., Luo, C., Zhao, J., Devarajan, G., and Xu, H. (2019). Immune regulation in the aging retina. Prog. Retin. Eye Res. 69, 159–172.
Chen, M., Muckersie, E., Forrester, J. V., and Xu, H. (2010). Immune activation in retinal aging: a gene expression study. Invest. Ophthalmol. Vis. Sci. 51, 5888–5896. doi: 10.1167/iovs.09-5103
Chen, Y., Lin, Y., Vithana, E., Jia, L., Zuo, X., Wong, T., et al. (2014). Common variants near ABCA1 and in PMM2 are associated with primary open-angle glaucoma. Nat. Genet. 46, 1115–1119. doi: 10.1038/ng.3078
Chen, Y., Vartiainen, N., Ying, W., Chan, P., Koistinaho, J., and Swanson, R. (2001). Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanism. J. Neurochem. 77, 1601–1610. doi: 10.1046/j.1471-4159.2001.00374.x
Cherry, J., Olschowka, J., and O’Banion, M. (2014). Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflammation 11:98.
Chidlow, G., Ebneter, A., Wood, J., and Casson, R. (2011). The optic nerve head is the site of axonal transport disruption, axonal cytoskeleton damage and putative axonal regeneration failure in a rat model of glaucoma. Acta Neuropathol. 121, 737–751. doi: 10.1007/s00401-011-0807-1
Chidlow, G., Ebneter, A., Wood, J., and Casson, R. (2016). Evidence supporting an association between expression of major histocompatibility complex II by microglia and optic nerve degeneration during experimental glaucoma. J. Glaucoma 25, 681–691. doi: 10.1097/IJG.0000000000000447
Chien, Y., Scuoppo, C., Wang, X., Fang, X., Balgley, B., Bolden, J., et al. (2011). Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 25, 2125–2136.
Cho, S., Chen, J., Sayed, F., Ward, M., Gao, F., Nguyen, T., et al. (2015). SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1β. J. Neurosci. 35, 807–818.
Chong, R., and Martin, K. (2015). Glial cell interactions and glaucoma. Curr. Opin. Ophthalmol. 26, 73–77.
Chrysostomou, V., Rezania, F., Trounce, I., and Crowston, J. (2013). Oxidative stress and mitochondrial dysfunction in glaucoma. Curr. Opin. Pharmacol. 13, 12–15.
Chua, J., Vania, M., Cheung, C., Ang, M., Chee, S., Yang, H., et al. (2012). Expression profile of inflammatory cytokines in aqueous from glaucomatous eyes. Mol. Vis. 18, 431–438.
Clarke, L., Liddelow, S., Chakraborty, C., Münch, A., Heiman, M., and Barres, B. (2018). Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. U S A. 115, E1896–E1905.
Colombo, E., and Farina, C. (2016). Astrocytes: key regulators of neuroinflammation. Trends Immunol. 37, 608–620.
Coughlin, B., Feenstra, D., and Mohr, S. (2017). Müller cells and diabetic retinopathy. Vision Res. 139, 93–100.
Crish, S., and Calkins, D. (2011). Neurodegeneration in glaucoma: progression and calcium-dependent intracellular mechanisms. Neuroscience 176, 1–11. doi: 10.1016/j.neuroscience.2010.12.036
Crish, S., Sappington, R., Inman, D., Horner, P., and Calkins, D. (2010). Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc. Natl. Acad. Sci. 107, 5196–5201. doi: 10.1073/pnas.0913141107
Curcio, C., and Drucker, D. (1993). Retinal ganghon cells in Alzheimer’s disease and Agng. Ann. Neurol. 33, 248–257.
Dai, C., Khaw, P., Yin, Z., Li, D., Raisman, G., and Li, Y. (2012). Structural basis of glaucoma: the fortified astrocytes of the optic nerve head are the target of raised intraocular pressure. Glia 60, 13–28. doi: 10.1002/glia.21242
Damani, M., Zhao, L., Fontainhas, A., Amaral, J., Fariss, R., and Wong, W. (2011). Age-related alterations in the dynamic behavior of microglia. Aging Cell 10, 263–276.
Davies, D., Ma, J., Jegathees, T., and Goldsbury, C. (2017). Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathol. 27, 795–808. doi: 10.1111/bpa.12456
Davies, S., Elliott, M., Floor, E., Truscott, T., Zareba, M., Sarna, T., et al. (2001). Photocytotoxicity of lipofuscin in human retinal pigment epithelial cells. Free Radic. Biol. Med. 31, 256–265. doi: 10.1016/s0891-5849(01)00582-2
Davis, B., Salinas-Navarro, M., Cordeiro, M., Moons, L., and Groef, L. (2017). Characterizing microglia activation: a spatial statistics approach to maximize information extraction. Sci. Rep. 7:1576. doi: 10.1038/s41598-017-01747-8
de Hoz, R., Gallego, B., Ramírez, A., Rojas, B., Salazar, J., Valiente-Soriano, F., et al. (2013). Rod-like microglia are restricted to eyes with laser-induced ocular hypertension but absent from the microglial changes in the contralateral untreated eye. PLoS One 8:e83733. doi: 10.1371/JOURNAL.PONE.0083733
de Moraes, C., Cioffi, G., Weinreb, R., and Liebmann, J. (2018). New recommendations for the treatment of systemic hypertension and their potential implications for Glaucoma management. J. Glaucoma 27, 567–571. doi: 10.1097/IJG.0000000000000981
Dias, M., Luo, X., Ribas, V., Petrs-Silva, H., and Koch, J. (2022). The role of axonal transport in Glaucoma. Int. J. Mol. Sci. 23:3935.
Ding, Z., Song, L., Wang, Q., Kumar, G., Yan, Y., and Ma, C. (2021). Astrocytes: a double-edged sword in neurodegenerative diseases. Neural Regen. Res. 16, 1702–1710.
Doucette, L., Rasnitsyn, A., Seifi, M., and Walter, M. (2015). The interactions of genes, age, and environment in glaucoma pathogenesis. Surv. Ophthalmol. 60, 310–326.
Drange, S., Anderson, D., and Schulzer, M. (2001). Risk factors for progression of visual field abnormalities in normal-tension glaucoma. Am. J. Ophthalmol. 131, 699–708.
Duarte, J. (2021). Neuroinflammatory mechanisms of mitochondrial dysfunction and neurodegeneration in Glaucoma. J. Ophthalmol. 2021:4581909.
Dubbelaar, M., Kracht, L., Eggen, B., and Boddeke, E. (2018). The kaleidoscope of microglial phenotypes. Front. Immunol. 9:1753. doi: 10.3389/FIMMU.2018.01753
Ebneter, A., Casson, R., Wood, J., and Chidlow, G. (2010). Microglial activation in the visual pathway in experimental glaucoma: spatiotemporal characterization and correlation with axonal injury. Invest. Ophthalmol. Vis. Sci. 51, 6448–6460. doi: 10.1167/iovs.10-5284
Edwards, J., Zhang, X., Frauwirth, K., and Mosser, D. (2006). Biochemical and functional characterization of three activated macrophage populations. J. Leukoc Biol. 80, 1298–1307.
Enokido, Y., Yoshitake, A., Ito, H., and Okazawa, H. (2008). Age-dependent change of HMGB1 and DNA double-strand break accumulation in mouse brain. Biochem. Biophys. Res. Commun. 376, 128–133. doi: 10.1016/j.bbrc.2008.08.108
Escartin, C., and Bonvento, G. (2008). Targeted activation of astrocytes: a potential neuroprotective strategy. Mol. Neurobiol. 38, 231–241.
Fan Gaskin, J., Shah, M., and Chan, E. (2021). Oxidative stress and the role of NADPH oxidase in Glaucoma. Antioxidants 10:238.
Fan, W., Huang, W., Chen, J., Li, N., Mao, L., and Hou, S. (2022). Retinal microglia: functions and diseases. Immunology. 166, 268–286.
Fernández-Albarral, J., de Hoz, R., Matamoros, J., Chen, L., López-Cuenca, I., Salobrar-García, E., et al. (2022). Retinal changes in astrocytes and Müller Glia in a mouse model of laser-induced glaucoma: a time-course study. Biomedicines 10: 939. doi: 10.3390/biomedicines10050939
Fernandez-Albarral, J., Ramírez, A., de Hoz, R., and Salazar, J. (2022). Retinal microglial activation in glaucoma: evolution over time in a unilateral ocular hypertension model. Neural Regen. Res. 17:797. doi: 10.4103/1673-5374.322454
Fernández-Albarral, J., Ramírez, A., de Hoz, R., López-Villarín, N., Salobrar-García, E., López-Cuenca, I., et al. (2019a). Neuroprotective and anti-inflammatory effects of a hydrophilic saffron extract in a model of glaucoma. Int. J. Mol. Sci. 20:E4110. doi: 10.3390/ijms20174110
Fernández-Albarral, J., Salazar, J., de Hoz, R., Marco, E., Martín-Sánchez, B., Flores-Salguero, E., et al. (2021). Retinal molecular changes are associated with neuroinflammation and loss of RGCs in an experimental model of Glaucoma. Int. J. Mol. Sci. 22:2066. doi: 10.3390/ijms22042066
Fernández-Albarral, J., Salobrar-García, E., Martínez-Páramo, R., Ramírez, A., de Hoz, R., Ramírez, J., et al. (2019b). Retinal glial changes in Alzheimer’s disease – a review. J. Optom. 12, 198–207.
Ferreira, S., Fabián Lerner, S., Brunzini, R., Reides, C., Evelson, P., and Llesuy, S. (2010). Time course changes of oxidative stress markers in a rat experimental glaucoma model. Invest. Ophthalmol. Vis. Sci. 51, 4635–4640. doi: 10.1167/iovs.09-5044
Fingert, J., Héon, E., Liebmann, J., Yamamoto, T., Craig, J., Rait, J., et al. (1999). Analysis of Myocilin mutations in 1703 Glaucoma patients from five different populations. Hum. Mol. Genet. 8, 899–905. doi: 10.1093/hmg/8.5.899
Fingert, J., Miller, K., Hedberg-Buenz, A., Roos, B., Lewis, C., Mullins, R., et al. (2017). Transgenic TBK1 mice have features of normal tension glaucoma. Hum. Mol. Genet. 26, 124–132. doi: 10.1093/hmg/ddw372
Fingert, J., Stone, E., Sheffield, V., and Alward, W. (2002). Myocilin Glaucoma. Surv. Ophthalmol. 47, 547–561.
Finkel, T., and Holbrook, N. (2000). Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247.
Fishelson, Z., Attali, G., and Mevorach, D. (2001). Complement and apoptosis. Mol. Immunol. 38, 207–219.
Flammer, J., Orgül, S., Costa, V., Orzalesi, N., Krieglstein, G., Serra, L., et al. (2002). The impact of ocular blood flow in glaucoma. Prog. Retin. Eye Res. 21, 359–393.
Foxton, R., Finkelstein, A., Vijay, S., Dahlmann-Noor, A., Khaw, P., Morgan, J., et al. (2013). VEGF-a is necessary and sufficient for retinal neuroprotection in models of experimental Glaucoma. Am. J. Pathol. 182, 1379–1390. doi: 10.1016/j.ajpath.2012.12.032
Franceschi, C., Bonafè, M., Valensin, S., Olivieri, F., de Luca, M., Ottaviani, E., et al. (2000). Inflamm-aging. an evolutionary perspective on immunosenescence. Ann. N Y Acad. Sci. 908, 244–254.
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C., and Santoro, A. (2018). Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14, 576–590. doi: 10.1038/s41574-018-0059-4
Franco, R., and Fernández-Suárez, D. (2015). Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 131, 65–86.
Frank, M., Barrientos, R., Biedenkapp, J., Rudy, J., Watkins, L., and Maier, S. (2006). mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol. Aging 27, 717–722. doi: 10.1016/j.neurobiolaging.2005.03.013
Frasca, D., and Blomberg, B. (2015). Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 17, 7–19.
Fulop, T., Witkowski, J., Olivieri, F., and Larbi, A. (2018). The integration of inflammaging in age-related diseases. Semin. Immunol. 40, 17–35.
Gabandé-Rodríguez, E., Keane, L., and Capasso, M. (2020). Microglial phagocytosis in aging and Alzheimer’s disease. J. Neurosci. Res. 98, 284–298.
Galan, A., Dergham, P., Escoll, P., De-la-Hera, A., D’Onofrio, P., Magharious, M., et al. (2014). Neuronal injury external to the retina rapidly activates retinal glia, followed by elevation of markers for cell cycle re-entry and death in retinal ganglion cells. PLoS One 9:e101349. doi: 10.1371/JOURNAL.PONE.0101349
Gamage, R., Wagnon, I., Rossetti, I., Childs, R., Niedermayer, G., Chesworth, R., et al. (2020). Cholinergic modulation of glial function during aging and chronic neuroinflammation. Front. Cell Neurosci. 14:577912. doi: 10.3389/FNCEL.2020.577912
Gao, F., Ji, M., Wu, J., and Wang, Z. (2013). Roles of retinal Müller cells in health and glaucoma. Sheng Li Xue Bao 65, 654–663.
Garcia, T., Pannicke, T., Vogler, S., Berk, B., Grosche, A., Wiedemann, P., et al. (2014). Nerve growth factor inhibits osmotic swelling of rat retinal glial (Müller) and bipolar cells by inducing glial cytokine release. J. Neurochem. 131, 303–313. doi: 10.1111/jnc.12822
García-Bermúdez, M., Freude, K., Mouhammad, Z., van Wijngaarden, P., Martin, K., and Kolko, M. (2021). Glial cells in glaucoma: friends, foes, and potential therapeutic targets. Front. Neurol. 12:169. doi: 10.3389/FNEUR.2021.624983/BIBTEX
Garner, K. M., Amin, R., Johnson, R. W., Scarlett, E. J., and Burton, M. D. (2018). Microglia priming by interleukin-6 signaling is enhanced in aged mice. J. Neuroimmunol. 324, 90–99. doi: 10.1016/j.jneuroim.2018.09.002
Geng, L., Fan, L., Liu, F., Smith, C., and Li, J. (2020). Nox2 dependent redox-regulation of microglial response to amyloid-β stimulation and microgliosis in aging. Sci. Rep. 10:5294.
Gerber, A., Harris, A., Siesky, B., Lee, E., Schaab, T., Huck, A., et al. (2015). Vascular dysfunction in diabetes and glaucoma: a complex relationship reviewed. J. Glaucoma 24, 474–479. doi: 10.1097/IJG.0000000000000137
Gharahkhani, P., Burdon, K., Fogarty, R., Sharma, S., Hewitt, A., Martin, S., et al. (2014). Common variants near ABCA1, AFAP1 and GMDS confer risk of primary open-angle glaucoma. Nat. Genet. 46, 1120–1125. doi: 10.1038/ng.3079
Giangiacomo, A., and Coleman, A. (2009). “The epidemiology of Glaucoma,” in Glaucoma, eds F. Grehn and R. Stamper (Berlin: Springer).
Gibson, K., Wu, Y., Barnett, Y., Duggan, O., Vaughan, R., Kondeatis, E., et al. (2009). B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell 8, 18–25. doi: 10.1111/j.1474-9726.2008.00443.x
Gimenez, M., Sim, J., and Russell, J. (2004). TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J. Neuroimmunol. 151, 116–125. doi: 10.1016/j.jneuroim.2004.02.012
Giordano, K., Denman, C., Dubisch, P., Akhter, M., and Lifshitz, J. (2021). An update on the rod microglia variant in experimental and clinical brain injury and disease. Brain Commun. 3:fcaa227. doi: 10.1093/braincomms/fcaa227
Glass, C., Saijo, K., Winner, B., Marchetto, M., and Gage, F. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934.
Gonzalez, P., Li, G., Qiu, J., Wu, J., and Luna, C. (2014). Role of microRNAs in the trabecular meshwork. J. Ocul. Pharmacol. Ther. 30, 128–137.
Gordon, M., Torri, V., Miglior, S., Beiser, J., Floriani, I., Miller, J., et al. (2007). Validated prediction model for the development of primary open-angle glaucoma in individuals with ocular hypertension. Ophthalmology 114, 10–19.e2.
Gorgoulis, V., Adams, P., Alimonti, A., Bennett, D., Bischof, O., Bishop, C., et al. (2019). Cellular senescence: defining a path forward. Cell 179, 813–827. doi: 10.1016/j.cell.2019.10.005
Gramlich, O. W., Beck, S., von Thun und Hohenstein-Blaul, N., Boehm, N., Ziegler, A., Vetter, J. M., et al. (2013). Enhanced insight into the autoimmune component of Glaucoma: IgG autoantibody accumulation and pro-inflammatory conditions in human glaucomatous retina. PLoS One 8:e57557. doi: 10.1371/journal.pone.0057557
Gramlich, O., Ding, Q., Zhu, W., Cook, A., Anderson, M., and Kuehn, M. (2015). Adoptive transfer of immune cells from glaucomatous mice provokes retinal ganglion cell loss in recipients. Acta Neuropathol. Commun. 3:56. doi: 10.1186/s40478-015-0234-y
Greene, K., Stamer, W., and Liu, Y. (2022). The role of microRNAs in glaucoma. Exp. Eye Res. 215:108909.
Gregory, M., Hackett, C., Abernathy, E., Lee, K., Saff, R., Hohlbaum, A., et al. (2011). Opposing roles for membrane bound and soluble Fas ligand in glaucoma-associated retinal ganglion cell death. PLoS One 6:e17659. doi: 10.1371/journal.pone.0017659
Grimm, A., and Eckert, A. (2017). Brain aging and neurodegeneration: from a mitochondrial point of view. J. Neurochem. 143, 418–431.
Grubeck-Loebenstein, B., della Bella, S., Iorio, A., Michel, J., Pawelec, G., and Solana, R. (2009). Immunosenescence and vaccine failure in the elderly. Aging Clin. Exp. Res. 21, 201–209.
Guest, J., Grant, R., Mori, T., and Croft, K. (2014). Changes in oxidative damage, inflammation and [NAD(H)] with age in cerebrospinal fluid. PLoS One 9:e85335. doi: 10.1371/JOURNAL.PONE.0085335
Guillonneau, X., Eandi, C., Paques, M., Sahel, J., Sapieha, P., and Sennlaub, F. (2017). On phagocytes and macular degeneration. Prog. Retin. Eye Res. 61, 98–128.
Gutteridge, I. (2021). Normal tension glaucoma: diagnostic features and comparisons with primary open angle glaucoma. Clin. Exp. Optom. 83, 161–172.
Han, X., Zhang, T., Liu, H., Mi, Y., and Gou, X. (2020). Astrocyte senescence and Alzheimer’s disease: a review. Front. Aging Neurosci. 12:148. doi: 10.3389/FNAGI.2020.00148/BIBTEX
Harada, T., Harada, C., Kohsaka, S., Wada, E., Yoshida, K., Ohno, S., et al. (2002). Microglia-Mü ller Glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J. Neurosci. 22, 9228–9236. doi: 10.1523/JNEUROSCI.22-21-09228.2002
Harris, A., Ciulla, T., Chung, H., and Martin, B. (1998). Regulation of retinal and optic nerve blood flow. Arch. Ophthalmol. 116, 1491–1495.
Harvey, H., and Durant, S. (2014). The role of glial cells and the complement system in retinal diseases and Alzheimer’s disease: common neural degeneration mechanisms. Exp. Brain Res. 232, 3363–3377. doi: 10.1007/s00221-014-4078-7
Harwerth, R., Wheat, J., and Rangaswamy, N. V. (2008). Age-related losses of retinal ganglion cells and axons. Invest. Ophthalmol. Vis. Sci. 49, 4437–4443.
Healey, P., Mitchell, P., Smith, W., and Wang, J. J. (1998). Optic disc hemorrhages in a population with and without signs of glaucoma. Ophthalmology 105, 216–223. doi: 10.1016/s0161-6420(98)92704-x
Heijl, A., Leske, M., Bengtsson, B., Hyman, L., Bengtsson, B., and Hussein, M. (2002). Reduction of intraocular pressure and Glaucoma progression: results from the early manifest Glaucoma trial. Arch. Ophthalmol. 120, 1268–1279.
Heneka, M., Carson, M., Khoury, J., Landreth, G., Brosseron, F., Feinstein, D., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405.
Henry, C., Huang, Y., Wynne, A., and Godbout, J. (2009). Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav. Immun. 23, 309–317. doi: 10.1016/j.bbi.2008.09.002
Hernandez, M., Miao, H., and Lukas, T. (2008). Astrocytes in glaucomatous optic neuropathy. Prog. Brain Res. 173, 353–373.
Hernandez-Segura, A., Nehme, J., and Demaria, M. (2018). Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453.
Hickman, S., Izzy, S., Sen, P., Morsett, L., and Khoury, J. (2018). Microglia in neurodegeneration. Nat. Neurosci. 21, 1359–1369.
Hickman, S., Kingery, N., Ohsumi, T., Borowsky, M., Wang, L., Means, T., et al. (2013). The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905. doi: 10.1038/nn.3554
Hindle, A., Thoonen, R., Jasien, J. V., Grange, R., Amin, K., Wise, J., et al. (2019). Identification of candidate miRNA biomarkers for Glaucoma. Invest. Ophthalmol. Vis. Sci. 60, 134–146. doi: 10.1167/iovs.18-24878
Hirt, J., Porter, K., Dixon, A., McKinnon, S., and Liton, P. (2018). Contribution of autophagy to ocular hypertension and neurodegeneration in the DBA/2J spontaneous glaucoma mouse model. Cell Death Discov. 4:75. doi: 10.1038/s41420-018-0077-y
Hoare, M., and Narita, M. (2013). Transmitting senescence to the cell neighbourhood. Nature Cell Biol. 15, 887–889. doi: 10.1038/ncb2811
Hollborn, M., Francke, M., Iandiev, I., Bühner, E., Foja, C., Kohen, L., et al. (2008). Early activation of inflammation- and immune response-related genes after experimental detachment of the porcine retina. Invest. Ophthalmol. Vis. Sci. 49, 1262–1273. doi: 10.1167/iovs.07-0879
Holloway, O., Canty, A., King, A., and Ziebell, J. (2019). Rod microglia and their role in neurological diseases. Semin. Cell Dev. Biol. 94, 96–103.
Howell, G., Libby, R., Jakobs, T., Smith, R., Phalan, F., Barter, J., et al. (2007). Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J. Cell Biol. 179, 1523–1537. doi: 10.1083/jcb.200706181
Howell, G., Macalinao, D., Sousa, G., Walden, M., Soto, I., Kneeland, S., et al. (2011). Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J. Clin. Invest. 121, 1429–1444. doi: 10.1172/JCI44646
Howell, G., Soto, I., Ryan, M., Graham, L., Smith, R., and John, S. (2013). Deficiency of complement component 5 ameliorates glaucoma in DBA/2J mice. J. Neuroinflammation 10:76. doi: 10.1186/1742-2094-10-76
Howell, G., Soto, I., Zhu, X., Ryan, M., Macalinao, D., Sousa, G., et al. (2012). Radiation treatment inhibits monocyte entry into the optic nerve head and prevents neuronal damage in a mouse model of glaucoma. J. Clin. Invest. 122, 1246–1261. doi: 10.1172/JCI61135
Hu, C., Niu, L., Li, L., Song, M., Zhang, Y., Lei, Y., et al. (2020). ABCA1 regulates IOP by modulating Cav1/eNOS/NO signaling pathway. Invest. Ophthalmol. Vis. Sci. 61:33. doi: 10.1167/iovs.61.5.33
Huang, W., Jaroszewski, J., Ortego, J., Escribano, J., and Coca-Prados, M. (2000). Expression of the TIGR gene in the iris, ciliary body, and trabecular meshwork of the human eye. Ophthalmic Genet. 21, 155–169.
Hurley, D., Normile, C., Irnaten, M., and O’Brien, C. (2022). The intertwined roles of oxidative stress and endoplasmic reticulum stress in Glaucoma. Antioxidants 11:886. doi: 10.3390/antiox11050886
Iandiev, I., Wurm, A., Hollborn, M., Wiedemann, P., Grimm, C., Remé, C., et al. (2008). Müller cell response to blue light injury of the rat retina. Invest. Ophthalmol. Vis. Sci. 49, 3559–3567.
Ikeda, O., Murakami, M., Ino, H., Yamazaki, M., Nemoto, T., Koda, M., et al. (2001). Acute up-regulation of brain-derived neurotrophic factor expression resulting from experimentally induced injury in the rat spinal cord. Acta Neuropathol. 102, 239–245. doi: 10.1007/s004010000357
Inman, D., and Horner, P. (2007). Reactive nonproliferative gliosis predominates in a chronic mouse model of glaucoma. Glia 55, 942–953. doi: 10.1002/glia.20516
Inman, D., Lambert, W., Calkins, D., and Horner, P. (2013). α-Lipoic acid antioxidant treatment limits glaucoma-related retinal ganglion cell death and dysfunction. PLoS One 8:e65389. doi: 10.1371/JOURNAL.PONE.0065389
Iomdina, E. N., Khoroshilova-Maslova, I. P., Robustova, O. V., Averina, O. A., Kovaleva, N. A., Aliev, G., et al. (2015). Mitochondria-targeted antioxidant SkQ1 reverses glaucomatous lesions in rabbits. Front. Biosci. 20, 892–901. doi: 10.2741/4343
Iram, T., Trudler, D., Kain, D., Kanner, S., Galron, R., Vassar, R., et al. (2016). Astrocytes from old Alzheimer’s disease mice are impaired in Aβ uptake and in neuroprotection. Neurobiol. Dis. 96, 84–94.
Ito, Y., and di Polo, A. (2017). Mitochondrial dynamics, transport, and quality control: a bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion 36, 186–192. doi: 10.1016/j.mito.2017.08.014
Jarrett, S., Lin, H., Godley, B., and Boulton, M. (2008). Mitochondrial DNA damage and its potential role in retinal degeneration. Prog. Retin Eye Res. 27, 596–607.
Jayaram, H., Cepurna, W., Johnson, E., and Morrison, J. (2015). MicroRNA expression in the Glaucomatous retina. Invest. Ophthalmol. Vis. Sci. 56, 7971–7982.
Jayaram, H., Phillips, J., Lozano, D., Choe, T., Cepurna, W., Johnson, E., et al. (2017). Comparison of MicroRNA expression in aqueous humor of normal and primary open-angle glaucoma patients using PCR arrays: a pilot study. Invest. Ophthalmol. Vis. Sci. 58, 2884–2890.
Jensen, C., Massie, A., and de Keyser, J. (2013). Immune players in the CNS: the astrocyte. J. Neuroimmune Pharmacol. 8, 824–839.
Jha, P., Banda, H., Tytarenko, R., Bora, P., and Bora, N. (2011). Complement mediated apoptosis leads to the loss of retinal ganglion cells in animal model of glaucoma. Mol. Immunol. 48, 2151–2158. doi: 10.1016/j.molimm.2011.07.012
Johnson, E., Doser, T., Cepurna, W., Dyck, J., Jia, L., Guo, Y., et al. (2011). Cell proliferation and interleukin-6-type cytokine signaling are implicated by gene expression responses in early optic nerve head injury in rat glaucoma. Invest. Ophthalmol. Vis. Sci. 52, 504–518. doi: 10.1167/iovs.10-5317
Johnson, E., Jia, L., Cepurna, W., Doser, T., and Morrison, J. (2007). Global changes in optic nerve head gene expression after exposure to elevated intraocular pressure in a rat Glaucoma model. Invest. Ophthalmol. Vis. Sci. 48, 3161–3177. doi: 10.1167/iovs.06-1282
Jonas, J., Wang, N., Wang, Y., You, Q., Yang, D., and Xu, L. (2014). Ocular hypertension: general characteristics and estimated cerebrospinal fluid pressure. the Beijing eye study 2011. PLoS One 9:e100533. doi: 10.1371/JOURNAL.PONE.0100533
Kamel, K., Farrell, M., and O’Brien, C. (2017). Mitochondrial dysfunction in ocular disease: focus on glaucoma. Mitochondrion 35, 44–53.
Kang, Y., Jia, P., Zhao, H., Hu, C., and Yang, X. (2015). MicroRNA-26a overexpression protects RGC-5 cells against H2O2-induced apoptosis. Biochem. Biophys. Res. Commun. 460, 164–169. doi: 10.1016/j.bbrc.2015.02.164
Karlstetter, M., Ebert, S., and Langmann, T. (2010). Microglia in the healthy and degenerating retina: insights from novel mouse models. Immunobiology 215, 685–691. doi: 10.1016/j.imbio.2010.05.010
Karlstetter, M., Scholz, R., Rutar, M., Wong, W., Provis, J., and Langmann, T. (2015). Retinal microglia: just bystander or target for therapy? Prog. Retin. Eye Res. 45, 30–57.
Karperien, A., Ahammer, H., and Jelinek, H. (2013). Quantitating the subtleties of microglial morphology with fractal analysis. Front. Cell Neurosci. 7:3. doi: 10.3389/FNCEL.2013.00003
Kawasaki, A., Otori, Y., and Barnstable, C. (2000). Muller cell protection of rat retinal ganglion cells from glutamate and nitric oxide neurotoxicity. Invest. Ophthalmol. Vis. Sci. 41, 3444–3450.
Kelly, R., Joers, V., Tansey, M., McKernan, D., and Dowd, E. (2020). Microglial phenotypes and their relationship to the cannabinoid system: therapeutic implications for Parkinson’s disease. Molecules 25:453. doi: 10.3390/molecules25030453
Kempuraj, D., Thangavel, R., Natteru, P., Selvakumar, G., Saeed, D., Zahoor, H., et al. (2016). Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 1:1003.
Ko, M., Peng, P., Ma, M., Ritch, R., and Chen, C. (2005). Dynamic changes in reactive oxygen species and antioxidant levels in retinas in experimental glaucoma. Free Radic. Biol. Med. 39, 365–373. doi: 10.1016/j.freeradbiomed.2005.03.025
Koellhoffer, E., McCullough, L., and Ritzel, R. (2017). Old maids: aging and its impact on microglia function. Int. J. Mol. Sci. 18:769. doi: 10.3390/ijms18040769
Kong, N., Lu, X., and Li, B. (2014). Downregulation of microRNA-100 protects apoptosis and promotes neuronal growth in retinal ganglion cells. BMC Mol. Biol. 15:25. doi: 10.1186/S12867-014-0025-1
Konishi, H., Koizumi, S., and Kiyama, H. (2022). Phagocytic astrocytes: emerging from the shadows of microglia. Glia 70, 1009–1026. doi: 10.1002/glia.24145
Konishi, H., Okamoto, T., Hara, Y., Komine, O., Tamada, H., Maeda, M., et al. (2020). Astrocytic phagocytosis is a compensatory mechanism for microglial dysfunction. EMBO J. 39:e104464.
Kostianovsky, A., Maier, L., Anderson, R., Bruce, J., and Anderson, D. (2008). Astrocytic regulation of human monocytic/microglial activation. J. Immunol. 181, 5425–5432. doi: 10.4049/jimmunol.181.8.5425
Kotecha, A., Izadi, S., and Jeffery, G. (2006). Age-related changes in the thickness of the human lamina cribrosa. Br. J. Ophthalmol. 90, 1531–1534.
Križaj, D., Ryskamp, D., Tian, N., Tezel, G., Mitchell, C., Slepak, V., et al. (2014). From mechanosensitivity to inflammatory responses: new players in the pathology of Glaucoma. Curr. Eye Res. 39, 105–119. doi: 10.3109/02713683.2013.836541
Kruchkova, Y., Ben-Dror, I., Herschkovitz, A., David, M., Yayon, A., and Vardimon, L. (2001). Basic fibroblast growth factor: a potential inhibitor of glutamine synthetase expression in injured neural tissue. J. Neurochem. 77, 1641–1649. doi: 10.1046/j.1471-4159.2001.00390.x
Kuehn, M., Kim, C., Ostojic, J., Bellin, M., Alward, W., Stone, E., et al. (2006). Retinal synthesis and deposition of complement components induced by ocular hypertension. Exp. Eye Res. 83, 620–628. doi: 10.1016/j.exer.2006.03.002
Kumar, A., and Shamsuddin, N. (2012). Retinal Muller Glia initiate innate response to infectious stimuli via toll-like receptor signaling. PLoS One 7:e29830. doi: 10.1371/JOURNAL.PONE.0029830
Kuzumaki, N., Ikegami, D., Imai, S., Narita, M., Tamura, R., Yajima, M., et al. (2010). Enhanced IL-1beta production in response to the activation of hippocampal glial cells impairs neurogenesis in aged mice. Synapse 64, 721–728. doi: 10.1002/syn.20800
Kwon, H., and Koh, S. (2020). Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl. Neurodegener. 9:42.
Lahne, M., Nagashima, M., Hyde, D., and Hitchcock, P. (2020). Reprogramming Müller Glia to regenerate retinal neurons. Annu. Rev. Vis. Sci. 6, 171–193. doi: 10.1146/annurev-vision-121219-081808
Lam, K., Wu, Q., Hu, W., Yao, P., Wang, H., Dong, T., et al. (2019). Asarones from Acori Tatarinowii Rhizoma stimulate expression and secretion of neurotrophic factors in cultured astrocytes. Neurosci. Lett. 707:134308. doi: 10.1016/j.neulet.2019.134308
Lan, X., Han, X., Li, Q., Yang, Q., and Wang, J. (2017). Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 13, 420–433.
Larsson, Å, Wilhelmsson, U., Pekna, M., and Pekny, M. (2004). Increased cell proliferation and neurogenesis in the hippocampal dentate Gyrus of old GFAP-/- Vim-/- Mice. Neurochem. Res. 29, 2069–2073. doi: 10.1007/s11064-004-6880-2
Lebrun-Julien, F., Duplan, L., Pernet, V., Osswald, I., Sapieha, P., Bourgeois, P., et al. (2009). Excitotoxic death of retinal neurons in vivo occurs via a non-cell-autonomous mechanism. J. Neurosci. 29, 5536–5545. doi: 10.1523/JNEUROSCI.0831-09.2009
Lee, C., Klopp, R., Weindruch, R., and Prolla, T. (1999). Gene expression profile of aging and its retardation by caloric restriction. Science 285, 1390–1393.
Lee, C., Weindruch, R., and Prolla, T. (2000). Gene-expression profile of the ageing brain in mice. Nat. Genet. 25, 294–297.
Lee, J., Caprioli, J., Nouri-Mahdavi, K., Afifi, A., Morales, E., Ramanathan, M., et al. (2014). Baseline prognostic factors predict rapid visual field deterioration in Glaucoma. Invest. Ophthalmol. Vis. Sci. 55, 2228–2236. doi: 10.1167/iovs.13-12261
Lee, K., Lin, S., Copland, D., Dick, A., and Liu, J. (2021). Cellular senescence in the aging retina and developments of senotherapies for age-related macular degeneration. J. Neuroinflammation 18, 5293258. doi: 10.1186/s12974-021-02088-0
Lee, M., Kim, C., Shin, S., Moon, S., and Chun, M. (1998). Increased ciliary neurotrophic factor expression in reactive astrocytes following spinal cord injury in the rat. Neurosci. Lett. 255, 79–82. doi: 10.1016/s0304-3940(98)00710-1
Leng, S., Xue, Q., Huang, Y., Semba, R., Chaves, P., Bandeen-Roche, K., et al. (2005). Total and differential white blood cell counts and their associations with circulating interleukin-6 levels in community-dwelling older women. J. Gerontol. A Biol. Sci. Med. Sci. 60, 195–199. doi: 10.1093/gerona/60.2.195
Leske, M., Heijl, A., Hyman, L., Bengtsson, B., and Komaroff, E. (2004). Factors for progression and glaucoma treatment: the early manifest Glaucoma trial. Curr. Opin. Ophthalmol. 15, 102–106.
Letiembre, M., Hao, W., Liu, Y., Walter, S., Mihaljevic, I., Rivest, S., et al. (2007). Innate immune receptor expression in normal brain aging. Neuroscience 146, 248–254.
Levine, R., Yang, A., Brahma, V., and Martone, J. (2017). Management of blood pressure in patients with Glaucoma. Curr. Cardiol. Rep. 19:109.
Lewis, G., Mervin, K., Valter, K., Maslim, J., Kappel, P., Stone, J., et al. (1999). Limiting the proliferation and reactivity of retinal Müller cells during experimental retinal detachment: the value of oxygen supplementation. Am. J. Ophthalmol. 128, 165–172. doi: 10.1016/s0002-9394(99)00103-8
Li, G., and Osborne, N. (2008). Oxidative-induced apoptosis to an immortalized ganglion cell line is caspase independent but involves the activation of poly(ADP-ribose)polymerase and apoptosis-inducing factor. Brain Res. 1188, 35–43. doi: 10.1016/j.brainres.2007.10.073
Li, G., Luna, C., Qiu, J., Epstein, D., and Gonzalez, P. (2010). Modulation of inflammatory markers by miR-146a during replicative senescence in trabecular meshwork cells. Invest. Ophthalmol. Vis. Sci. 51, 2976–2985. doi: 10.1167/iovs.09-4874
Li, L., Xu, L., Chen, W., Li, X., Xia, Q., Zheng, L., et al. (2018). Reduced Annexin A1 secretion by ABCA1 causes retinal inflammation and ganglion cell apoptosis in a murine Glaucoma model. Front. Cell Neurosci. 12:347. doi: 10.3389/fncel.2018.00347
Li, Y., Kang, J., and Horwitz, M. (1998). Interaction of an adenovirus E3 14.7-kilodalton protein with a novel tumor necrosis factor alpha-inducible cellular protein containing leucine zipper domains. Mol. Cell Biol. 18, 1601–1610. doi: 10.1128/MCB.18.3.1601
Li, Z., Okamoto, K., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Liddelow, S., and Barres, B. (2017). Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967.
Liddelow, S., Guttenplan, K., Clarke, L., Bennett, F., Bohlen, C., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487.
Lin, F., Cheng, Y., Yu, M., Ho, J., and der Kuo, Y. (2019). The fungus-derived retinoprotectant theissenolactone C improves glaucoma-like injury mediated by MMP-9 inhibition. Phytomedicine 56, 207–214. doi: 10.1016/j.phymed.2018.11.002
Liu, C., Wang, X., Liu, C., and Zhang, H. (2019). Pharmacological targeting of microglial activation: new therapeutic approach. Front. Cell Neurosci. 13:514. doi: 10.3389/FNCEL.2019.00514
Liu, J., Copland, D., Theodoropoulou, S., Chiu, H., Barba, M., Mak, K., et al. (2016). Impairing autophagy in retinal pigment epithelium leads to inflammasome activation and enhanced macrophage-mediated angiogenesis. Sci. Rep. 6:20639. doi: 10.1038/srep20639
Liu, Y., and Allingham, R. (2017). Major review: molecular genetics of primary open-angle glaucoma. Exp. Eye Res. 160, 62–84.
Liu, Z., Li, Y., Cui, Y., Roberts, C., Lu, M., Wilhelmsson, U., et al. (2014). Beneficial effects of gfap/vimentin reactive astrocytes for axonal remodeling and motor behavioral recovery in mice after stroke. Glia 62, 2022–2033. doi: 10.1002/glia.22723
López-Otín, C., Blasco, M., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217.
Lu, Y., Iandiev, I., Hollborn, M., Korber, N., Ulbricht, E., Hirrlinger, P., et al. (2011). Reactive glial cells: increased stiffness correlates with increased intermediate filament expression. FASEB J. 25, 624–631. doi: 10.1096/fj.10-163790
Luo, C., Yang, X., Kain, A., Powell, D., Kuehn, M., and Tezel, G. (2010). Glaucomatous tissue stress and the regulation of immune response through glial toll-like receptor signaling. Invest. Ophthalmol. Vis. Sci. 51, 5697–5707. doi: 10.1167/iovs.10-5407
Luo, X., and Chen, S. D. (2012). The changing phenotype of microglia from homeostasis to disease. Transl. Neurodegener. 1:9.
Lye, J., Latorre, E., Lee, B., Bandinelli, S., Holley, J., Gutowski, N., et al. (2019). Astrocyte senescence may drive alterations in GFAPα, CDKN2A p14 ARF, and TAU3 transcript expression and contribute to cognitive decline. Geroscience 41, 561–573.
Ma, W., Cojocaru, R., Gotoh, N., Gieser, L., Villasmil, R., Cogliati, T., et al. (2013). Gene expression changes in aging retinal microglia: relationship to microglial support functions and regulation of activation. Neurobiol. Aging 34, 2310–2321. doi: 10.1016/j.neurobiolaging.2013.03.022
Ma, W., Silverman, S., Zhao, L., Villasmil, R., Campos, M., Amaral, J., et al. (2019). Absence of TGFβ signaling in retinal microglia induces retinal degeneration and exacerbates choroidal neovascularization. Elife 8:e42049.
Madeira, M., Boia, R., Santos, P., Ambrósio, A., and Santiago, A. (2015). Contribution of microglia-mediated neuroinflammation to retinal degenerative diseases. Mediators Inflamm 2015:673090.
Maier, E., Duschl, A., and Horejs-Hoeck, J. (2012). STAT6-dependent and -independent mechanisms in Th2 polarization. Eur. J. Immunol. 42, 2827–2833.
Malone, P., and Hernandez, M. (2007). 4-Hydroxynonenal, a product of oxidative stress, leads to an antioxidant response in optic nerve head astrocytes. Exp. Eye Res. 84, 444–454. doi: 10.1016/j.exer.2006.10.020
Mansour, H., Chamberlain, C., Weible, M., Hughes, S., Chu, Y., and Chan-Ling, T. (2008). Aging-related changes in astrocytes in the rat retina: imbalance between cell proliferation and cell death reduces astrocyte availability. Aging Cell 7, 526–540. doi: 10.1111/j.1474-9726.2008.00402.x
Mantovani, A., Sica, A., Sozzani, S., Allavena, P., Vecchi, A., and Locati, M. (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686. doi: 10.1016/j.it.2004.09.015
Martin, K., Levkovitch-Verbin, H., Valenta, D., Baumrind, L., Pease, M., and Quigley, H. (2002). Retinal glutamate transporter changes in experimental Glaucoma and after optic nerve transection in the rat. Invest. Ophthalmol. Vis. Sci. 43, 2236–2243.
Martin, K., Quigley, H., Valenta, D., Kielczewski, J., and Pease, M. (2006). Optic nerve dynein motor protein distribution changes with intraocular pressure elevation in a rat model of glaucoma. Exp. Eye Res. 83, 255–262. doi: 10.1016/j.exer.2005.11.025
Matias, I., Morgado, J., and Gomes, F. (2019). Astrocyte heterogeneity: impact to brain aging and disease. Front. Aging Neurosci. 11:59. doi: 10.3389/FNAGI.2019.00059
Matsukawa, A., Takeda, K., Kudo, S., Maeda, T., Kagayama, M., and Akira, S. (2003). Aberrant inflammation and lethality to septic peritonitis in mice lacking STAT3 in macrophages and neutrophils. J. Immunol. 171, 6198–6205. doi: 10.4049/jimmunol.171.11.6198
Matsumoto, G., Shimogori, T., Hattori, N., and Nukina, N. (2015). TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 24, 4429–4442. doi: 10.1093/hmg/ddv179
McElnea, E., Quill, B., Docherty, N., Irnaten, M., Siah, W., Clark, A., et al. (2011). Oxidative stress, mitochondrial dysfunction and calcium overload in human lamina cribrosa cells from glaucoma donors. Mol. Vis. 17:1182.
McHugh, D., and Gil, J. (2018). Senescence and aging: causes, consequences, and therapeutic avenues. J. Cell Biol. 217, 65–77.
Mejias, N., Martinez, C., Stephens, M., and de Rivero Vaccari, J. (2018). Contribution of the inflammasome to inflammaging. J. Inflamm. 15:1808.
Mélik Parsadaniantz, S., Réaux-le Goazigo, A., Sapienza, A., Habas, C., and Baudouin, C. (2020). Glaucoma: a degenerative optic neuropathy related to neuroinflammation? Cells 9:535.
Menet, V., Prieto, M., Privat, A., and Giménez y Ribotta, M. (2003). Axonal plasticity and functional recovery after spinal cord injury in mice deficient in both glial fibrillary acidic protein and vimentin genes. Proc. Natl. Acad. Sci. U S A. 100, 8999–9004. doi: 10.1073/pnas.1533187100
Min, K., Yang, M., Kim, S., Jou, I., and Joe, E. (2006). Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J. Neurosci. 26, 1880–1887. doi: 10.1523/JNEUROSCI.3696-05.2006
Minckler, D., McLean, I., and Tso, M. (1976). Distribution of axonal and glial elements in the rhesus optic nerve head studied by electron microscopy. Am. J. Ophthalmol. 82, 179–187. doi: 10.1016/0002-9394(76)90416-5
Mizee, M. R., Nijland, P. G., van der Pol, S. M. A., Drexhage, J. A. R., van het Hof, B., Mebius, R., et al. (2014). Astrocyte-derived retinoic acid: a novel regulator of blood-brain barrier function in multiple sclerosis. Acta Neuropathol. 128, 691–703. doi: 10.1007/s00401-014-1335-6
Moehle, M., and West, A. (2015). M1 and M2 immune activation in Parkinson’s disease: Foe and ally? Neuroscience 302, 59–73. doi: 10.1016/j.neuroscience.2014.11.018
Mohri, I., Taniike, M., Taniguchi, H., Kanekiyo, T., Aritake, K., Inui, T., et al. (2006). Prostaglandin D2-mediated microglia/astrocyte interaction enhances astrogliosis and demyelination in twitcher. J. Neurosci. 26, 4383–4393. doi: 10.1523/JNEUROSCI.4531-05.2006
Mombach, J., Vendrusculo, B., and Bugs, C. (2015). A model for p38MAPK-induced astrocyte senescence. PLoS One 10:e0125217. doi: 10.1371/JOURNAL.PONE.0125217
Moore, D., Harris, A., WuDunn, D., Kheradiya, N., and Siesky, B. (2008). Dysfunctional regulation of ocular blood flow: a risk factor for glaucoma? Clin. Ophthalmol. 2, 849–861.
Moreno, M., Campanelli, J., Sande, P., Sáenz, D., Keller Sarmiento, M., and Rosenstein, R. (2004). Retinal oxidative stress induced by high intraocular pressure. Free Radic. Biol. Med. 37, 803–812.
Morizawa, Y., Hirayama, Y., Ohno, N., Shibata, S., Shigetomi, E., Sui, Y., et al. (2017). Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 8:1598.
Mosser, D., and Edwards, J. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969.
Munemasa, Y., Kitaoka, Y., Kuribayashi, J., and Ueno, S. (2010). Modulation of mitochondria in the axon and soma of retinal ganglion cells in a rat glaucoma model. J. Neurochem. 115, 1508–1519. doi: 10.1111/j.1471-4159.2010.07057.x
Nakazawa, T., Hisatomi, T., Nakazawa, C., Noda, K., Maruyama, K., She, H., et al. (2007). Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc. Natl. Acad. Sci. 104, 2425–2430. doi: 10.1073/pnas.0608167104
Nakazawa, T., Matsubara, A., Noda, K., Hisatomi, T., She, H., Skondra, D., et al. (2006a). Characterization of cytokine responses to retinal detachment in rats. Mol. Vis. 12, 867–878.
Nakazawa, T., Nakazawa, C., Matsubara, A., Noda, K., Hisatomi, T., She, H., et al. (2006b). Tumor necrosis factor-α mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J. Neurosci. 26, 12633–12641. doi: 10.1523/JNEUROSCI.2801-06.2006
Nelson, P., Soma, L., and Lavi, E. (2002). Microglia in diseases of the central nervous system. Ann. Med. 34, 491–500.
Neufeld, A. (1999). Nitric oxide: a potential mediator of retinal ganglion cell damage in glaucoma. Surv. Ophthalmol. 43, S129–S135.
Neufeld, A., and Liu, B. (2003). Glaucomatous optic neuropathy: when glia misbehave. Neuroscientist 9, 485–495. doi: 10.1177/1073858403253460
Neumann, H., Kotter, M., and Franklin, R. (2009). Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132, 288–295. doi: 10.1093/brain/awn109
Newman, E., and Reichenbach, A. (1996). The Müller cell: a functional element of the retina. Trends Neurosci. 19, 307–312.
Nichols, N., Day, J., Laping, N., Johnson, S., and Finch, C. (1993). GFAP mRNA increases with age in rat and human brain. Neurobiol. Aging 14, 421–429.
Nickells, R. (2007). From ocular hypertension to ganglion cell death: a theoretical sequence of events leading to glaucoma. Can. J. Ophthalmol. 42, 278–287.
Nickells, R., Howell, G., Soto, I., and John, S. (2012). Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu. Rev. Neurosci. 35, 153–179. doi: 10.1146/annurev.neuro.051508.135728
Nicolela, M., Ferrier, S., Morrison, C., Archibald, M., LeVatte, T., Wallace, K., et al. (2003). Effects of cold-induced vasospasm in Glaucoma: the role of Endothelin-1. Invest. Ophthalmol. Vis. Sci. 44, 2565–2572. doi: 10.1167/iovs.02-0913
Nikolskaya, T., Nikolsky, Y., Serebryiskaya, T., Zvereva, S., Sviridov, E., Dezso, Z., et al. (2009). Network analysis of human glaucomatous optic nerve head astrocytes. BMC Med. Genomics 2:24. doi: 10.1186/1755-8794-2-24
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318.
Niraula, A., Sheridan, J., and Godbout, J. (2017). Microglia priming with aging and stress. Neuropsychopharmacology 42, 318–333.
Nolan, Y., Maher, F., Martin, D., Clarke, R., Brady, M., Bolton, A., et al. (2005). Role of interleukin-4 in regulation of age-related inflammatory changes in the hippocampus. J Biol. Chem. 280, 9354–9362.
Norden, D., and Godbout, J. (2013). Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 39, 19–34.
Norden, D., Fenn, A., Dugan, A., and Godbout, J. (2014). TGFβ produced by IL-10 redirected astrocytes attenuates microglial activation. Glia 62, 881–895.
Norden, D., Trojanowski, P., Walker, F., and Godbout, J. (2016). Insensitivity of astrocytes to interleukin 10 signaling following peripheral immune challenge results in prolonged microglial activation in the aged brain. Neurobiol. Aging 44, 22–41. doi: 10.1016/j.neurobiolaging.2016.04.014
Noske, W., Hensen, J., Wiederholt, M., Noske, W., Hensen, J., and Wiederholt, M. (1997). Endothelin-like immunoreactivity in aqueous humor of patients with primary open-angle glaucoma and cataract. Graefe’s arch. Clin. Exp. Ophthalmol. 235, 551–552. doi: 10.1007/BF00947082
Nouri-Mahdavi, K., Hoffman, D., Coleman, A., Liu, G., Li, G., Gaasterland, D., et al. (2004). Predictive factors for glaucomatous visual field progression in the advanced Glaucoma intervention study. Ophthalmology 111, 1627–1635.
Nucci, C., Tartaglione, R., Rombolà, L., Morrone, L., Fazzi, E., and Bagetta, G. (2005). Neurochemical evidence to implicate elevated glutamate in the mechanisms of high Intraocular Pressure (IOP)-induced retinal ganglion cell death in rat. Neurotoxicology 26, 935–941. doi: 10.1016/j.neuro.2005.06.002
O’Brien, J., Hayder, H., Zayed, Y., and Peng, C. (2018). Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 9:388354. doi: 10.3389/FENDO.2018.00402/BIBTEX
Oksanen, M., Lehtonen, S., Jaronen, M., Goldsteins, G., Hämäläinen, R., and Koistinaho, J. (2019). Astrocyte alterations in neurodegenerative pathologies and their modeling in human induced pluripotent stem cell platforms. Cell Mol. Life Sci. 76, 2739–2760. doi: 10.1007/s00018-019-03111-7
Orre, M., Kamphuis, W., Osborn, L., Melief, J., Kooijman, L., Huitinga, I., et al. (2014). Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol. Aging 35, 1–14. doi: 10.1016/j.neurobiolaging.2013.07.008
Ou-Yang, Y., Liu, Z., Xu, C., Wu, J., Peng, J., and Peng, Q. (2020). miR-223 induces retinal ganglion cells apoptosis and inflammation via decreasing HSP-70 in vitro and in vivo. J. Chem. Neuroanat. 104:101747. doi: 10.1016/j.jchemneu.2020.101747
Park, K., Lee, D., Joe, E., Kim, S., and Jin, B. (2005). Neuroprotective role of microglia expressing interleukin-4. J. Neurosci. Res. 81, 397–402.
Parpura, V., Heneka, M., Montana, V., Oliet, S., Schousboe, A., Haydon, P., et al. (2012). Glial cells in (patho)physiology. J. Neurochem. 121, 4–27.
Patel, D., Tewari, B., Chaunsali, L., and Sontheimer, H. (2019). Neuron–glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 20, 282–297.
Pekny, M., and Pekna, M. (2004). Astrocyte intermediate filaments in CNS pathologies and regeneration. J. Pathol. 204, 428–437.
Peng, L., Parpura, V., and Verkhratsky, A. (2014). Editorial (thematic issue: neuroglia as a central element of neurological diseases: an underappreciated target for therapeutic intervention). Curr. Neuropharmacol. 12, 303–307. doi: 10.2174/1570159X12999140829152550
Perez-Nievas, B., and Serrano-Pozo, A. (2018). Deciphering the astrocyte reaction in Alzheimer’s disease. Front. Aging Neurosci. 10:114. doi: 10.3389/FNAGI.2018.00114
Perry, V., and O’Connor, V. (2010). The role of microglia in synaptic stripping and synaptic degeneration: a revised perspective. ASN Neuro 2:e00047. doi: 10.1042/AN20100024
Polak, K., Luksch, A., Berisha, F., Fuchsjaeger-Mayrl, G., Dallinger, S., and Schmetterer, L. (2007). Altered nitric oxide system in patients with open-angle glaucoma. Arch. Ophthalmol. 125, 494–498.
Ponath, G., Ramanan, S., Mubarak, M., Housley, W., Lee, S., Sahinkaya, F., et al. (2017). Myelin phagocytosis by astrocytes after myelin damage promotes lesion pathology. Brain 140, 399–413.
Porchet, R., Probst, A., Bouras, C., Dráberová, E., Dráber, P., and Riederer, B. M. (2003). Analysis of glial acidic fibrillary protein in the human entorhinal cortex during aging and in Alzheimer’s disease. Proteomics 3, 1476–1485.
Prata, L., Ovsyannikova, I., Tchkonia, T., and Kirkland, J. (2018). Senescent cell clearance by the immune system: emerging therapeutic opportunities. Semin. Immunol. 40:101275.
Prieto, G., and Cotman, C. (2017). Cytokines and cytokine networks target neurons to modulate long-term potentiation. Cytokine Growth Factor Rev. 34, 27–33.
Primiani, C., Ryan, V., Rao, J., Cam, M., Ahn, K., Modi, H., et al. (2014). Coordinated gene expression of neuroinflammatory and cell signaling markers in dorsolateral prefrontal cortex during human brain development and aging. PLoS One 9:e110972. doi: 10.1371/JOURNAL.PONE.0110972
Qian, Y., Liu, C., Hartupee, J., Altuntas, C., Gulen, M., Jane-Wit, D., et al. (2007). The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat. Immunol. 8, 247–256. doi: 10.1038/ni1439
Qu, J., and Jakobs, T. (2013). The time course of gene expression during reactive gliosis in the optic nerve. PLoS One 8:e67094. doi: 10.1371/journal.pone.0067094
Quaranta, L., Bruttini, C., Micheletti, E., Konstas, A., Michelessi, M., Oddone, F., et al. (2021). Glaucoma and neuroinflammation: an overview. Surv. Ophthalmol. 66, 693–713.
Quigley, H. (2005a). Glaucoma: macrocosm to microcosm the friedenwald lecture. Invest. Ophthalmol. Vis. Sci. 46, 2663–2670. doi: 10.1167/iovs.04-1070
Quigley, H., and Broman, A. (2006). The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 90, 262–267.
Quigley, H., McKinnon, S., Zack, D., Pease, M., Kerrigan-Baumrind, L., Kerrigan, D., et al. (2000). Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest. Ophthalmol. Vis. Sci. 41, 3460–3466.
Quillen, S., Schaub, J., Quigley, H., Pease, M., Korneva, A., and Kimball, E. (2020). Astrocyte responses to experimental glaucoma in mouse optic nerve head. PLoS One 15:e0238104. doi: 10.1371/journal.pone.0238104
Ramírez, A., de Hoz, R., Fernández-Albarral, J., Salobrar-García, E., Rojas, B., Valiente-Soriano, F., et al. (2020a). Time course of bilateral microglial activation in a mouse model of laser-induced glaucoma. Sci. Rep. 10:4890. doi: 10.1038/s41598-020-61848-9
Ramirez, A., de Hoz, R., Salobrar-Garcia, E., Salazar, J., Rojas, B., Ajoy, D., et al. (2017). The role of microglia in retinal neurodegeneration: Alzheimer’s disease, Parkinson, and glaucoma. Front. Aging Neurosci. 9:214. doi: 10.3389/fnagi.2017.00214
Ramírez, A., Fernández-Albarral, J., de Hoz, R., López-Cuenca, I., Salobrar-García, E., Rojas, P., et al. (2020b). Microglial changes in the early aging stage in a healthy retina and an experimental glaucoma model. Prog. Brain Res. 256, 125–149. doi: 10.1016/bs.pbr.2020.05.024
Ramírez, J., Ramírez, A., Salazar, J., de Hoz, R., and Triviño, A. (2001). Changes of astrocytes in retinal ageing and age-related macular degeneration. Exp. Eye Res. 73, 601–615.
Reinehr, S., Mueller-Buehl, A., Tsai, T., and Joachim, S. (2022). Specific biomarkers in the aqueous humour of Glaucoma patients. Klin Monbl Augenheilkd 239, 169–176.
Richter, B., Sliter, D., Herhaus, L., Stolz, A., Wang, C., Beli, P., et al. (2016). Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. 113, 4039–4044. doi: 10.1073/pnas.1523926113
Rifkin, I., Leadbetter, E., Busconi, L., Viglianti, G., and Marshak-Rothstein, A. (2005). Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol. Rev. 204, 27–42.
Ritch, R., Darbro, B., Menon, G., Khanna, C., Solivan-Timpe, F., Roos, B., et al. (2014). TBK1 gene duplication and normal-tension Glaucoma. JAMA Ophthalmol. 132, 544–548.
Ritzel, R., Patel, A., Pan, S., Crapser, J., Hammond, M., Jellison, E., et al. (2015). Age- and location-related changes in microglial function. Neurobiol. Aging 36, 2153–2163. doi: 10.1016/j.neurobiolaging.2015.02.016
Rizzo, M., Greco, A., de Virgilio, A., Gallo, A., Taverniti, L., Fusconi, M., et al. (2017). Glaucoma: recent advances in the involvement of autoimmunity. Immunol. Res. 65, 207–217.
Roberts, M., Sigal, I., Liang, Y., Burgoyne, C., and Crawford Downs, J. (2010). Changes in the biomechanical response of the optic nerve head in early experimental glaucoma. Invest Ophthalmol. Vis. Sci. 51, 5675–5684.
Robillard, K., Lee, K., Chiu, K., and MacLean, A. (2016). Glial cell morphological and density changes through the lifespan of rhesus macaques. Brain Behav. Immun. 55, 60–69. doi: 10.1016/j.bbi.2016.01.006
Rodríguez, J., Butt, A., Gardenal, E., Parpura, V., and Verkhratsky, A. (2016). Complex and differential glial responses in Alzheimer’s disease and ageing. Curr. Alzheimer Res. 13, 343–358.
Rodríguez, J., Yeh, C., Terzieva, S., Olabarria, M., Kulijewicz-Nawrot, M., and Verkhratsky, A. (2014). Complex and region-specific changes in astroglial markers in the aging brain. Neurobiol. Aging 35, 15–23. doi: 10.1016/j.neurobiolaging.2013.07.002
Rolle, T., Ponzetto, A., and Malinverni, L. (2021). The role of neuroinflammation in Glaucoma: an update on molecular mechanisms and new therapeutic options. Front. Neurol. 11:612422. doi: 10.3389/fneur.2020.612422
Rostami, J., Fotaki, G., Sirois, J., Mzezewa, R., Bergström, J., Essand, M., et al. (2020). Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson’s disease brain. J. Neuroinflammation 17, 119. doi: 10.1186/s12974-020-01776-7
Rothstein, J., Dykes-Hoberg, M., Pardo, C., Bristol, L., Jin, L., Kuncl, R., et al. (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16, 675–686. doi: 10.1016/s0896-6273(00)80086-0
Roubenoff, R., Harris, T., Abad, L., Wilson, P., Dallal, G., and Dinarello, C. (1998). Monocyte cytokine production in an elderly population: effect of age and inflammation. J. Gerontol. A Biol. Sci. Med. Sci. 53, M20–M26. doi: 10.1093/gerona/53a.1.m20
Rus, H., Cudrici, C., and Niculescu, F. (2005). The role of the complement system in innate immunity. Immunol. Res. 33, 103–112.
Russo, M. V., and McGavern, D. (2016). Inflammatory neuroprotection following traumatic brain injury. Science 353, 783–785.
Russo, R., Varano, G., Adornetto, A., Nucci, C., Corasaniti, M., Bagetta, G., et al. (2016). Retinal ganglion cell death in glaucoma: exploring the role of neuroinflammation. Eur. J. Pharmacol. 787, 134–142.
Saccà, S., Pascotto, A., Camicione, P., Capris, P., and Izzotti, A. (2005). Oxidative DNA damage in the human trabecular meshwork: clinical correlation in patients with primary open-angle glaucoma. Arch. Ophthalmol. 123, 458–463. doi: 10.1001/archopht.123.4.458
Safaiyan, S., Kannaiyan, N., Snaidero, N., Brioschi, S., Biber, K., Yona, S., et al. (2016). Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 19, 995–998. doi: 10.1038/nn.4325
Saijo, K., Winner, B., Carson, C., Collier, J., Boyer, L., Rosenfeld, M., et al. (2009). A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137, 47–59. doi: 10.1016/j.cell.2009.01.038
Sakami, S., Imanishi, Y., and Palczewski, K. (2019). Müller glia phagocytose dead photoreceptor cells in a mouse model of retinal degenerative disease. FASEB J. 33, 3680–3692.
Saliminejad, K., Khorram Khorshid, H., Soleymani Fard, S., and Ghaffari, S. (2019). An overview of microRNAs: biology, functions, therapeutics, and analysis methods. J. Cell Physiol. 234, 5451–5465.
Salminen, A., Ojala, J., Kaarniranta, K., Haapasalo, A., Hiltunen, M., and Soininen, H. (2011). Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 34, 3–11.
Sappington, R., and Calkins, D. (2008). Contribution of TRPV1 to microglia-derived IL-6 and NFκB translocation with elevated hydrostatic pressure. Invest. Ophthalmol. Vis. Sci. 49, 3004–3017.
Sarlus, H., and Heneka, M. (2017). Microglia in Alzheimer’s disease. J. Clin. Invest. 127, 3240–3249.
Satilmis, M., Orgül, S., Doubler, B., and Flammer, J. (2003). Rate of progression of glaucoma correlates with retrobulbar circulation and intraocular pressure. Am. J. Ophthalmol. 135, 664–669. doi: 10.1016/s0002-9394(02)02156-6
Sawada, H., Fukuchi, T., Tanaka, T., and Abe, H. (2010). Tumor necrosis factor-α concentrations in the aqueous humor of patients with Glaucoma. Invest. Ophthalmol. Vis. Sci. 51, 903–906. doi: 10.1167/iovs.09-4247
Schetters, S., Gomez-Nicola, D., Garcia-Vallejo, J., and van Kooyk, Y. (2018). Neuroinflammation: microglia and T cells get ready to tango. Front. Immunol. 8:1905. doi: 10.3389/FIMMU.2017.01905
Schilling, T., Nitsch, R., Heinemann, U., Haas, D., and Eder, C. (2001). Astrocyte-released cytokines induce ramification and outward K+ channel expression in microglia via distinct signalling pathways. Eur. J. Neurosci. 14, 463–473. doi: 10.1046/j.0953-816x.2001.01661.x
Schmidl, D., Garhofer, G., and Schmetterer, L. (2011). The complex interaction between ocular perfusion pressure and ocular blood flow - relevance for glaucoma. Exp. Eye Res. 93, 141–155. doi: 10.1016/j.exer.2010.09.002
Schumacher, B., Pothof, J., Vijg, J., and Hoeijmakers, J. (2021). The central role of DNA damage in the ageing process. Nature 592, 695–703.
Seitz, R., and Tamm, E. (2013). N-methyl-D-aspartate (NMDA)-mediated excitotoxic damage: a mouse model of acute retinal ganglion cell damage. Methods Mol. Biol. 935, 99–109. doi: 10.1007/978-1-62703-080-9_7
Seitz, R., Ohlmann, A., and Tamm, E. (2013). The role of Müller glia and microglia in glaucoma. Cell Tissue Res. 353, 339–345.
Seoane, M., Costoya, J., and Arce, V. (2017). Uncoupling Oncogene-Induced Senescence (OIS) and DNA Damage Response (DDR) triggered by DNA hyper-replication: lessons from primary mouse embryo astrocytes (MEA). Sci. Rep. 7:12991. doi: 10.1038/s41598-017-13408-x
Shahidehpour, R. K., Higdon, R. E., Crawford, N. G., Neltner, J. H., lghodaro, E. T., Patel, E., et al. (2021). Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol. Aging 99, 19–27.
Sica, A., and Mantovani, A. (2012). Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795. doi: 10.1172/JCI59643
Siegner, S., and Netland, P. (1996). Optic disc hemorrhages and progression of glaucoma. Ophthalmology 103, 1014–1024.
Sierra, A., Gottfried-Blackmore, A., Mcewen, B., and Bulloch, K. (2007). Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55, 412–424.
Silverman, S., and Wong, W. (2018). Microglia in the retina: roles in development. maturity, and disease. Annu. Rev. Vis. Sci. 4, 45–77.
Silverman, W., de Rivero Vaccari, J., Locovei, S., Qiu, F., Carlsson, S., Scemes, E., et al. (2009). The Pannexin 1 channel activates the inflammasome in neurons and astrocytes. J. Biol. Chem. 284, 18143–18151.
Simmons, D., Casale, M., Alcon, B., Pham, N., Narayan, N., and Lynch, G. (2007). Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia 55, 1074–1084. doi: 10.1002/glia.20526
Skrzypecki, J., Ufnal, M., Szaflik, J., and Filipiak, K. (2019). Blood pressure and glaucoma: at the crossroads between cardiology and ophthalmology. Cardiol. J. 26, 8–12. doi: 10.5603/CJ.2019.0008
Skytt, D., Klawonn, A., Stridh, M., Pajecka, K., Patruss, Y., Quintana-Cabrera, R., et al. (2012). siRNA knock down of glutamate dehydrogenase in astrocytes affects glutamate metabolism leading to extensive accumulation of the neuroactive amino acids glutamate and aspartate. Neurochem. Int. 61, 490–497. doi: 10.1016/j.neuint.2012.04.014
Smith, C., Wheeler, M., Marjoram, L., Bagnat, M., Deppmann, C., and Kucenas, S. (2017). TNFa/TNFR2 signaling is required for glial ensheathment at the dorsal root entry zone. PLoS Genet. 13:e1006712. doi: 10.1371/JOURNAL.PGEN.1006712
Sofroniew, M. V. (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647.
Song, B., Aiello, L., and Pasquale, L. (2016). Presence and risk factors for Glaucoma in patients with diabetes. Curr. Diab. Rep. 16:124.
Soreq, L., Rose, J., Soreq, E., Hardy, J., Trabzuni, D., Cookson, M., et al. (2017). Major shifts in glial regional identity are a transcriptional hallmark of human brain aging. Cell Rep. 18, 557–570. doi: 10.1016/j.celrep.2016.12.011
Soto, I., and Howell, G. (2014). The complex role of neuroinflammation in glaucoma. Cold Spring Harb. Perspect. Med. 4, a017269–a017269.
Souza, D., Bellaver, B., Raupp, G., Souza, D., and Quincozes-Santos, A. (2015). Astrocytes from adult Wistar rats aged in vitro show changes in glial functions. Neurochem. Int. 90, 93–97.
Spalding, K., Bergmann, O., Alkass, K., Bernard, S., Salehpour, M., Huttner, H., et al. (2013). Dynamics of hippocampal neurogenesis in adult humans. Cell 153, 1219–1227.
Sreekumar, P., Hinton, D., Kannan, R., and Martin, P. (2020). The emerging role of senescence in ocular disease. Oxid. Med. Cell Longev. 2020:2583601.
Stasi, K., Nagel, D., Yang, X., Wang, R., Ren, L., Podos, S., et al. (2006). Complement component 1Q (C1Q) upregulation in retina of murine, primate, and human glaucomatous eyes. Invest. Ophthalmol. Vis. Sci. 47, 1024–1029.
Steinmetz, C., and Turrigiano, G. (2010). Tumor necrosis factor-α signaling maintains the ability of cortical synapses to express synaptic scaling. J. Neurosci. 30, 14685–14690. doi: 10.1523/JNEUROSCI.2210-10.2010
Stephan, A., Madison, D. V., Mateos, J., Fraser, D., Lovelett, E., Coutellier, L., et al. (2013). A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 33, 13460–13474. doi: 10.1523/JNEUROSCI.1333-13.2013
Stephenson, J., Nutma, E., van der Valk, P., and Amor, S. (2018). Inflammation in CNS neurodegenerative diseases. Immunology 154, 204–219.
Stevens, B., Allen, N., Vazquez, L., Howell, G., Christopherson, K., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178.
Stoilov, I., Akarsu, A., and Sarfarazi, M. (1997). Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum. Mol. Genet. 6, 641–647. doi: 10.1093/hmg/6.4.641
Stone, E., Fingert, J., Alward, W., Nguyen, T., Polansky, J., Sunden, S., et al. (1997). Identification of a gene that causes primary open angle glaucoma. Science 275, 668–670.
Streit, W., Braak, H., Xue, Q., and Bechmann, I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 118, 475–485. doi: 10.1007/s00401-009-0556-6
Streit, W., Sammons, N., Kuhns, A., and Sparks, D. (2004). Dystrophic microglia in the aging human brain. Glia 45, 208–212.
Stuart, K. V., Pasquale, L., Kang, J., Foster, P., and Khawaja, A. (2023). Towards modifying the genetic predisposition for glaucoma: an overview of the contribution and interaction of genetic and environmental factors. Mol. Aspects Med. 93:101203. doi: 10.1016/j.mam.2023.101203
Su, W., Li, Z., Jia, Y., Zhu, Y., Cai, W., Wan, P., et al. (2017). microRNA-21a-5p/PDCD4 axis regulates mesenchymal stem cell-induced neuroprotection in acute glaucoma. J. Mol. Cell Biol. 9, 289–301.
Swanson, R., Ying, W., and Kauppinen, T. (2004). Astrocyte influences on ischemic neuronal death. Curr. Mol. Med. 4, 193–205.
Tabak, S., Schreiber-Avissar, S., and Beit-Yannai, E. (2021). Crosstalk between MicroRNA and oxidative stress in primary open-angle Glaucoma. Int. J. Mol. Sci. 22:2421.
Tamm, E., Ethier, C., and Lasker IIoA and Glaucomatous Neurodegeneration Participants. (2017). Biological aspects of axonal damage in glaucoma: a brief review. Exp. Eye Res. 157, 5–12.
Tan, C., Hu, T., Peng, M., Liu, S., Tong, J., Ouyang, W., et al. (2015). Age of rats seriously affects the degree of retinal damage induced by acute high intraocular pressure. Curr. Eye Res. 40, 300–306. doi: 10.3109/02713683.2014.922194
Tan, Z., Beiser, A., Vasan, R., Roubenoff, R., Dinarello, C., Harris, T., et al. (2007). Inflammatory markers and the risk of Alzheimer disease: the framingham study. Neurology 68, 1902–1908.
Tang, B., Li, S., Cao, W., and Sun, X. (2019). The association of oxidative stress status with open-angle glaucoma and exfoliation Glaucoma: a systematic review and meta-analysis. J. Ophthalmol. 2019:1803619. doi: 10.1155/2019/1803619
Tang, Y., and Le, W. (2016). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53, 1181–1194.
Taylor, J., Moore, Z., Minter, M., and Crack, P. (2018). Type-I interferon pathway in neuroinflammation and neurodegeneration: focus on Alzheimer’s disease. J. Neural Transm. 125, 797–807. doi: 10.1007/s00702-017-1745-4
Tegeler, C., O’Sullivan, J., Bucholtz, N., Goldeck, D., Pawelec, G., Steinhagen-Thiessen, E., et al. (2016). The inflammatory markers CRP, IL-6, and IL-10 are associated with cognitive function–data from the Berlin aging study II. Neurobiol Aging 38, 112–117. doi: 10.1016/j.neurobiolaging.2015.10.039
Tehrani, S., Davis, L., Cepurna, W., Choe, T., Lozano, D., Monfared, A., et al. (2016). Astrocyte structural and molecular response to elevated intraocular pressure occurs rapidly and precedes axonal tubulin rearrangement within the optic nerve head in a rat model. PLoS One 11:e0167364. doi: 10.1371/JOURNAL.PONE.0167364
Tezel, G. (2006). Oxidative stress in glaucomatous neurodegeneration: mechanisms and consequences. Prog. Retin. Eye Res. 25, 490–513.
Tezel, G. (2011). The immune response in glaucoma: a perspective on the roles of oxidative stress. Exp. Eye Res. 93, 178–186. doi: 10.1016/j.exer.2010.07.009
Tezel, G. (2013). Immune regulation toward immunomodulation for neuroprotection in glaucoma. Curr. Opin. Pharmacol. 13, 23–31.
Tezel, G. (2022). Molecular regulation of neuroinflammation in glaucoma: current knowledge and the ongoing search for new treatment targets. Prog. Retin. Eye Res. 87:100998. doi: 10.1016/j.preteyeres.2021.100998
Tezel, G., and Wax, M. (2000). Increased production of tumor necrosis factor-α by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J. Neurosci. 20, 8693–8700. doi: 10.1523/JNEUROSCI.20-23-08693.2000
Tezel, G., and Yang, X. (2004). Caspase-independent component of retinal ganglion cell death. In Vitro. Invest. Ophthalmol. Vis. Sci. 45, 4049–4059.
Tezel, G., Hernandez, M., and Wax, M. (2001). In vitro evaluation of reactive astrocyte migration, a component of tissue remodeling in glaucomatous optic nerve head. Glia 34, 178–189. doi: 10.1002/glia.1052
Tezel, G., Kass, M., Kolker, A., Becker, B., and Wax, M. (1997). Plasma and aqueous humor endothelin levels in primary open-angle glaucoma. J. Glaucoma 6, 83–89.
Tezel, G., Yang, X., and Cai, J. (2005). Proteomic identification of oxidatively modified retinal proteins in a chronic pressure-induced rat model of glaucoma. Invest. Ophthalmol. Vis. Sci. 46, 3177–3187. doi: 10.1167/iovs.05-0208
Tezel, G., Yang, X., Luo, C., Cai, J., and Powell, D. (2012). An astrocyte-specific proteomic approach to inflammatory responses in experimental rat glaucoma. Invest. Ophthalmol. Vis. Sci. 53, 4220–4233. doi: 10.1167/iovs.11-9101
Toda, N., and Nakanishi-Toda, M. (2007). Nitric oxide: ocular blood flow, glaucoma, and diabetic retinopathy. Prog. Retin. Eye Res. 26, 205–238.
Toft-Kehler, A., Skytt, D., Svare, A., Lefevere, E., van Hove, I., Moons, L., et al. (2017). Mitochondrial function in Müller cells - does it matter? Mitochondrion 36, 43–51.
Torres-Platas, S., Comeau, S., Rachalski, A., Bo, G., Cruceanu, C., Turecki, G., et al. (2014). Morphometric characterization of microglial phenotypes in human cerebral cortex. J. Neuroinflammation 11:12. doi: 10.1186/1742-2094-11-12
Tremblay, M. È, Zettel, M. L., Ison, J. R., Allen, P. D., and Majewska, A. K. (2012). Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 60, 541–558. doi: 10.1002/glia.22287
Tucker, B., Solivan-Timpe, F., Roos, B., Anfinson, K., Robin, A., Wiley, L., et al. (2014). Duplication of TBK1 stimulates autophagy in iPSC-derived retinal cells from a patient with normal tension glaucoma. J. Stem Cell Res. Ther. 3:161. doi: 10.4172/2157-7633.1000161
Tunny, T., Richardson, K., and Clark, C. V. (1998). Association study of the 5’ flanking regions of endothelial-nitric oxide synthase and endothelin-1 genes in familial primary open-angle glaucoma. Clin. Exp. Pharmacol. Physiol. 25, 26–29. doi: 10.1111/j.1440-1681.1998.tb02138.x
Turnquist, C., Beck, J., Horikawa, I., Obiorah, I., von Muhlinen, N., Vojtesek, B., et al. (2019). Radiation-induced astrocyte senescence is rescued by Δ133p53. Neuro Oncol. 21, 474–485. doi: 10.1093/neuonc/noz001
Underhill, D., and Ozinsky, A. (2002). Toll-like receptors: key mediators of microbe detection. Curr. Opin. Immunol. 14, 103–110.
Unterlauft, J., Eichler, W., Kuhne, K., Mei Yang, X., Yafai, Y., Wiedemann, P., et al. (2012). Pigment epithelium-derived factor released by mü ller glial cells exerts neuroprotective effects on retinal ganglion cells. Neurochem. Res. 37, 1524–1533. doi: 10.1007/s11064-012-0747-8
Varadhan, R., Yao, W., Matteini, A., Beamer, B., Xue, Q., Yang, H., et al. (2014). Simple biologically informed inflammatory index of two serum cytokines predicts 10 year all-cause mortality in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 69, 165–173. doi: 10.1093/gerona/glt023
Vay, S., Olschewski, D., Petereit, H., Lange, F., Nazarzadeh, N., Gross, E., et al. (2021). Osteopontin regulates proliferation, migration, and survival of astrocytes depending on their activation phenotype. J. Neurosci. Res. 99, 2822–2843. doi: 10.1002/jnr.24954
Vohra, R., Dalgaard, L., Vibæk, J., Langbøl, M., Bergersen, L., Olsen, N., et al. (2019). Potential metabolic markers in glaucoma and their regulation in response to hypoxia. Acta Ophthalmol. 97, 567–576. doi: 10.1111/aos.14021
von Bernhardi, R., Eugenín-von Bernhardi, L., and Eugenín, J. (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 7:124. doi: 10.3389/FNAGI.2015.00124
Walker, D., and Lue, L. (2015). Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res. Ther. 7:56. doi: 10.1186/s13195-015-0139-9
Walker, F., Beynon, S., Jones, K., Zhao, Z., Kongsui, R., Cairns, M., et al. (2014). Dynamic structural remodelling of microglia in health and disease: a review of the models, the signals and the mechanisms. Brain Behav. Immun. 37, 1–14. doi: 10.1016/j.bbi.2013.12.010
Walker, J., Low, K., Fletcher, M., Cohen, N., Gratton, G., and Fabiani, M. (2017). Hippocampal structure predicts cortical indices of reactivation of related items. Neuropsychologia 95, 182–192. doi: 10.1016/j.neuropsychologia.2016.12.005
Waller, R., Baxter, L., Fillingham, D., Coelho, S., Pozo, J., Mozumder, M., et al. (2019). Iba-1-/CD68+ microglia are a prominent feature of age-associated deep subcortical white matter lesions. PLoS One 14:e0210888. doi: 10.1371/JOURNAL.PONE.0210888
Wang, J., Chen, S., da Zhang, X., and Jonas, J. (2016). Retinal microglia in glaucoma. J. Glaucoma 25, 459–465.
Wang, L., Cioffi, G., Cull, G., Dong, J., and Fortune, B. (2002). Immunohistologic evidence for retinal glial cell changes in human Glaucoma. Invest. Ophthalmol. Vis. Sci. 43, 1088–1094.
Wang, L., Dong, J., Cull, G., Fortune, B., and Cioffi, G. (2003). Varicosities of intraretinal ganglion cell axons in human and nonhuman primates. Invest. Ophthalmol. Vis. Sci. 44, 2–9. doi: 10.1167/iovs.02-0333
Wang, M., and Wong, W. (2014). Microglia-Müller cell interactions in the retina. Adv. Exp. Med. Biol. 801, 333–338.
Wang, Y., Chen, S., Wang, J., Liu, Y., Chen, Y., Wen, T., et al. (2021). MicroRNA-93/STAT3 signalling pathway mediates retinal microglial activation and protects retinal ganglion cells in an acute ocular hypertension model. Cell Death Dis. 12:41. doi: 10.1038/s41419-020-03337-5
Wang, Y., Xu, E., Musich, P., and Lin, F. (2019). Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci. Ther. 25, 816–824.
Wang, Y., Zhou, H., Liu, X., Han, Y., Pan, S., and Wang, Y. (2018). MIR-181a inhibits human trabecular meshwork cell apoptosis induced by H2O2 through the suppression of NF-κB and JNK pathways. Adv. Clin. Exp. Med. 27, 577–582.
Wang, Z., Wiggs, J., Aung, T., Khawaja, A., and Khor, C. (2022). The genetic basis for adult onset glaucoma: recent advances and future directions. Prog. Retin. Eye Res. 90:101066. doi: 10.1016/j.preteyeres.2022.101066
Wassell, J., Davies, S., Bardsley, W., and Boulton, M. (1999). The photoreactivity of the retinal age pigment lipofuscin. J. Biol. Chem. 274, 23828–23832.
Wax, M. B., Tezel, G., Yang, J., Peng, G., Patil, R. V., Agarwal, N., et al. (2008). Induced autoimmunity to heat shock proteins elicits glaucomatous loss of retinal ganglion cell neurons via activated T-cell-derived fas-ligand. J. Neurosci. 28, 12085–12096. doi: 10.1523/JNEUROSCI.3200-08.2008
Weber, A., Harman, C., and Viswanathan, S. (2008). Effects of optic nerve injury, glaucoma, and neuroprotection on the survival, structure, and function of ganglion cells in the mammalian retina. J. Physiol. 586, 4393–4400.
Weber, M., Wu, T., Hanson, J., Alam, N., Solanoy, H., Ngu, H., et al. (2015). Cognitive deficits, changes in synaptic function, and brain pathology in a mouse model of normal aging(1,2,3). ENeuro 2:ENEURO.0047-15.2015 doi: 10.1523/ENEURO.0047-15.2015
Weinreb, R., Leung, C., Crowston, J., Medeiros, F., Friedman, D., Wiggs, J., et al. (2016). Primary open-angle glaucoma. Nat. Rev. Dis. Primers 2:16067.
West, A., and Shadel, G. (2017). Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 17, 363–375.
Wilkins, H., Carl, S., Greenlief, A., Festoff, B., and Swerdlow, R. (2014). Bioenergetic dysfunction and inflammation in Alzheimer’s disease: a possible connection. Front. Aging Neurosci. 6:311. doi: 10.3389/FNAGI.2014.00311
Wilkins, H., Weidling, I., Ji, Y., and Swerdlow, R. (2017). Mitochondria-derived damage-associated molecular patterns in neurodegeneration. Front. Immunol. 8:508. doi: 10.3389/FIMMU.2017.00508
Williams, J., Stampoulis, D., Gunter, C., Greenwood, J., Adamson, P., and Moss, S. (2016). Regulation of C3 activation by the alternative complement pathway in the mouse retina. PLoS One 11:e0161898. doi: 10.1038/s41467-018-05681-9
Wolf, S., Boddeke, H., and Kettenmann, H. (2017). Microglia in physiology and disease. Annu. Rev. 79, 619–643.
Wynne, A., Henry, C., Huang, Y., Cleland, A., and Godbout, J. (2010). Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav. Immun. 24, 1190–1201. doi: 10.1016/j.bbi.2010.05.011
Wyss-Coray, T., and Mucke, L. (2002). Inflammation in neurodegenerative disease–a double-edged sword. Neuron 35, 419–432.
Xia, J., Lim, J., Lu, W., Beckel, J., Macarak, E., Laties, A., et al. (2012). Neurons respond directly to mechanical deformation with pannexin-mediated ATP release and autostimulation of P2X7 receptors. J. Physiol. 590, 2285–2304. doi: 10.1113/jphysiol.2012.227983
Xu, H., Chen, M., and Forrester, J. V. (2009). Para-inflammation in the aging retina. Prog. Retin. Eye Res. 28, 348–368.
Xue, Y., Nie, D., Wang, L., Qiu, H., Ma, L., Dong, M., et al. (2021). Microglial polarization: novel therapeutic strategy against ischemic stroke. Aging Dis. 12, 466–479.
Yamanaka, K., and Komine, O. (2018). The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 126, 31–38. doi: 10.1016/j.neures.2017.09.011
Yan, X., Tezel, G., Wax, M., and Edward, D. (2000). Matrix metalloproteinases and tumor necrosis factor α in glaucomatous optic nerve head. Arch. Ophthalmol. 118, 666–673. doi: 10.1001/archopht.118.5.666
Yanagi, M., Kawasaki, R., Wang, J., Wong, T., Crowston, J., and Kiuchi, Y. (2011). Vascular risk factors in glaucoma: a review. Clin. Exp. Ophthalmol. 39, 252–258.
Yang, J., Yang, P., Tezel, G., Patil, R. V., Hernandez, M. R., and Wax, M. B. (2001). Induction of HLA-DR expression in human lamina cribrosa astrocytes by cytokines and simulated ischemia. Invest. Ophthalmol. Vis. Sci. 42, 365–371.
Yang, X., Luo, C., Cai, J., Powell, D., Yu, D., Kuehn, M., et al. (2011). Neurodegenerative and inflammatory pathway components linked to TNF-α/TNFR1 signaling in the glaucomatous human retina. Invest. Ophthalmol. Vis. Sci. 52, 8442–8454. doi: 10.1167/iovs.11-8152
Yang, X., Xu, S., Qian, Y., and Xiao, Q. (2017). Resveratrol regulates microglia M1/M2 polarization via PGC-1α in conditions of neuroinflammatory injury. Brain Behav. Immun. 64, 162–172.
Yang, Z., Quigley, H., Pease, M., Yang, Y., Qian, J., Valenta, D., et al. (2007). Changes in gene expression in experimental Glaucoma and optic nerve transection: the equilibrium between protective and detrimental mechanisms. Invest. Ophthalmol. Vis. Sci. 48, 5539–5548. doi: 10.1167/iovs.07-0542
Ye, S., and Johnson, R. (2001). An age-related decline in interleukin-10 may contribute to the increased expression of interleukin-6 in brain of aged mice. Neuroimmunomodulation 9, 183–192. doi: 10.1159/000049025
Yoshida, S., Sotozono, C., Ikeda, T., and Kinoshita, S. (2001). Interleukin-6 (IL-6) production by cytokine-stimulated human Müller cells. Curr. Eye Res. 22, 341–347. doi: 10.1076/ceyr.22.5.341.5498
Youngblood, H., Cai, J., Drewry, M., Helwa, I., Hu, E., Liu, S., et al. (2020). Expression of mRNAs, miRNAs, and lncRNAs in human trabecular meshwork cells upon mechanical stretch. Invest. Ophthalmol. Vis. Sci. 61:2. doi: 10.1167/iovs.61.5.2
Yu, L., Wang, L., and Chen, S. (2010). Endogenous toll-like receptor ligands and their biological significance. J. Cell Mol. Med. 14, 2592–2603.
Yuan, C., Aierken, A., Xie, Z., Li, N., Zhao, J., and Qing, H. (2020). The age-related microglial transformation in Alzheimer’s disease pathogenesis. Neurobiol. Aging 92, 82–91.
Yuan, L., and Neufeld, A. (2000). Tumor necrosis factor-α: a potentially neurodestructive cytokine produced by glia in the human glaucomatous optic nerve head. Glia 32, 42–50.
Zamanian, J., Xu, L., Foo, L., Nouri, N., Zhou, L., Giffard, R., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410.
Zanon-Moreno, V., Marco-Ventura, P., Lleo-Perez, A., Pons-Vazquez, S., Garcia-Medina, J., Vinuesa-Silva, I., et al. (2008). Oxidative stress in primary open-angle glaucoma. J. Glaucoma 17, 263–268.
Zhang, D., Hu, X., Qian, L., Wilson, B., Lee, C., Flood, P., et al. (2009). Prostaglandin E2 released from activated microglia enhances astrocyte proliferation in vitro. Toxicol. Appl. Pharmacol. 238, 64–70. doi: 10.1016/j.taap.2009.04.015
Zhang, S., Wang, H., Lu, Q., Qing, G., Wang, N., Wang, Y., et al. (2009). Detection of early neuron degeneration and accompanying glial responses in the visual pathway in a rat model of acute intraocular hypertension. Brain Res. 1303, 131–143. doi: 10.1016/j.brainres.2009.09.029
Zhao, D., Cho, J., Kim, M., and Guallar, E. (2014). The association of blood pressure and primary open-angle glaucoma: a meta-analysis. Am. J. Ophthalmol. 158, 615–627.e9.
Zhao, L., Zabel, M., Wang, X., Ma, W., Shah, P., Fariss, R., et al. (2015). Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol. Med. 7, 1179–1197. doi: 10.15252/emmm.201505298
Zhao, W., Xie, W., Xiao, Q., Beers, D., and Appel, S. (2006). Protective effects of an anti-inflammatory cytokine, interleukin-4, on motoneuron toxicity induced by activated microglia. J. Neurochem. 99, 1176–1187. doi: 10.1111/j.1471-4159.2006.04172.x
Zhu, H., Wang, L., Ruan, Y., Zhou, L., Zhang, D., Min, Z., et al. (2011). An efficient delivery of DAMPs on the cell surface by the unconventional secretion pathway. Biochem. Biophys. Res. Commun. 404, 790–795. doi: 10.1016/j.bbrc.2010.12.061
Zhu, Y., Pappas, A., Wang, R., Seifert, P., Sun, D., and Jakobs, T. (2018). Ultrastructural morphology of the optic nerve head in aged and Glaucomatous mice. Invest. Ophthalmol. Vis. Sci. 59, 3984–3996.
Ziebell, J., Taylor, S., Cao, T., Harrison, J., and Lifshitz, J. (2012). Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J. Neuroinflammation 9:247. doi: 10.1186/1742-2094-9-247
Zöller, T., Attaai, A., Potru, P., Ruß, T., and Spittau, B. (2018). Aged mouse cortical microglia display an activation profile suggesting immunotolerogenic functions. Int. J. Mol. Sci. 19:706. doi: 10.3390/ijms19030706
Keywords: glaucoma, neuroinflammation, glial activation, microglia, astrocytes, Müller cells, ageing
Citation: Fernández-Albarral JA, Ramírez AI, de Hoz R, Matamoros JA, Salobrar-García E, Elvira-Hurtado L, López-Cuenca I, Sánchez-Puebla L, Salazar JJ and Ramírez JM (2024) Glaucoma: from pathogenic mechanisms to retinal glial cell response to damage. Front. Cell. Neurosci. 18:1354569. doi: 10.3389/fncel.2024.1354569
Received: 12 December 2023; Accepted: 10 January 2024;
Published: 25 January 2024.
Edited by:
Nicolás Cuenca, University of Alicante, SpainReviewed by:
Jorge M. Barcia, Escuela de Doctorado Universidad Católica de Valencia San Vicente Mártir, SpainCopyright © 2024 Fernández-Albarral, Ramírez, de Hoz, Matamoros, Salobrar-García, Elvira-Hurtado, Sánchez-Puebla, Salazar and Ramírez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José M. Ramírez, cmFtaXJlenNAbWVkLnVjbS5lcw==
†These authors share first authorship
‡These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.