 Sophia C. Gjervan1
Sophia C. Gjervan1- 1Department of Medical Genetics, Centre for Molecular Medicine and Therapeutics, Djavad Mowafaghian Centre for Brain Health, British Columbia Children's Hospital Research Institute, University of British Columbia, Vancouver, BC, Canada
- 2Center for Molecular Medicine and Genetics, Wayne State University School of Medicine, Detroit, MI, United States
- 3Department of Pediatrics, The University of British Columbia and BC Children's Hospital, Vancouver, BC, Canada
- 4Division of Biochemical Genetics, The University of British Columbia and BC Children's Hospital, Vancouver, BC, Canada
Claudin-11 plays a critical role in multiple physiological processes, including myelination, auditory function, and spermatogenesis. Recently, stop-loss mutations in CLDN11 have been identified as a novel cause of hypomyelinating leukodystrophy (HLD22). Understanding the multifaceted roles of claudin-11 and the potential pathogenic mechanisms in HLD22 is crucial for devising targeted therapeutic strategies. This review outlines the biological roles of claudin-11 and the implications of claudin-11 loss in the context of the Cldn11 null mouse model. Additionally, HLD22 and proposed pathogenic mechanisms, such as endoplasmic reticulum stress, will be discussed.
1 Introduction
The myelin sheath, a membrane critical for normal nerve conduction in the central nervous system (CNS), insulates axons and facilitates efficient transduction via depolarization at the nodes of Ranvier (Harry and Toews, 1998). Several diseases associated with myelin abnormalities often have a genetic basis, such as leukodystrophies, which manifest as hypomyelination, dysmyelination, or progressive demyelination (Naidu, 1999; Knaap and Bugiani, 2017). The protein products of genes implicated in leukodystrophies serve functions in myelin structure (PLP1; Tsuji, 2007), lipid metabolism (ABCD1; Powers et al., 2005), ion transport (MLC1; Knaap et al., 2012), translational regulation (EIF2B; Moon and Parker, 2018), and lysosomal function [GALC (Shin et al., 2016) and VPS11 (Skoff et al., 2021)]. These causes highlight the complexity of myelin disorders and the variability of their clinical presentations (Gow and Lazzarini, 1996; Garbern et al., 1999).
Despite the etiological heterogeneity in myelin disorders, dysfunction or degeneration of oligodendroglia is a unifying mechanistic feature. Oligodendrocytes, responsible for myelination in the CNS, produce essential proteins for myelin production, stability and maintenance (Stecca et al., 2000) . One such protein, claudin-11, constitutes approximately 7% of total myelin protein, making it the third most abundant myelin protein after proteolipid protein 1 (PLP1) and myelin basic protein (MBP) (Bronstein et al., 1997; Harry and Toews, 1998). Recent findings identify de novo mutations in CLDN11, the gene encoding claudin-11, as a novel cause of hypomyelinating leukodystrophy (HLD), specifically leading to HLD22 (Riedhammer et al., 2020). The mechanisms by which such de novo mutations in claudin-11 lead to disease remain unclear. Understanding these processes is vital for developing therapies.
This review examines the biological roles of claudin-11, the importance of myelin and claudin-11 in health and disease, and the clinical features and postulated mechanisms underlying HLD22.
2 Molecular biology of claudin-11
The claudin family comprises 30 proteins, at least 26 of which are found in humans (Mineta et al., 2011; Engelund et al., 2012). Functionally, claudins form tight junctions (TJs) between polarized cells, regulating ion transport (Colegio et al., 2002; Koziel et al., 2020). Structurally, they are tetraspan, transmembrane proteins with sizes ranging from 20–34kDa and feature two extracellular loops critical for oligomerization and TJ function (Goncalves et al., 2013). Based on sequence homology, claudins are classified into classic and non-classic types. Non-classic claudins, to which claudin-11 belongs, share less sequence similarity to classic claudins (Krause et al., 2008; Lal-Nag and Morin, 2009). While claudins typically exhibit some overlap in their patterns of expression, claudin-11 is uniquely localized to the TJs of CNS myelin, the inner ear, and testis (Gow et al., 1999; Morita et al., 1999), with rare exceptions (Wolburg et al., 2001). Claudin proteins engage in both cis (within the same membrane) and trans (across opposing membranes) interactions, either with the same claudin (homophilic), or different claudins (heterophilic) (Krause et al., 2008). Claudin-11 is known to form homophilic interactions (Figure 1).
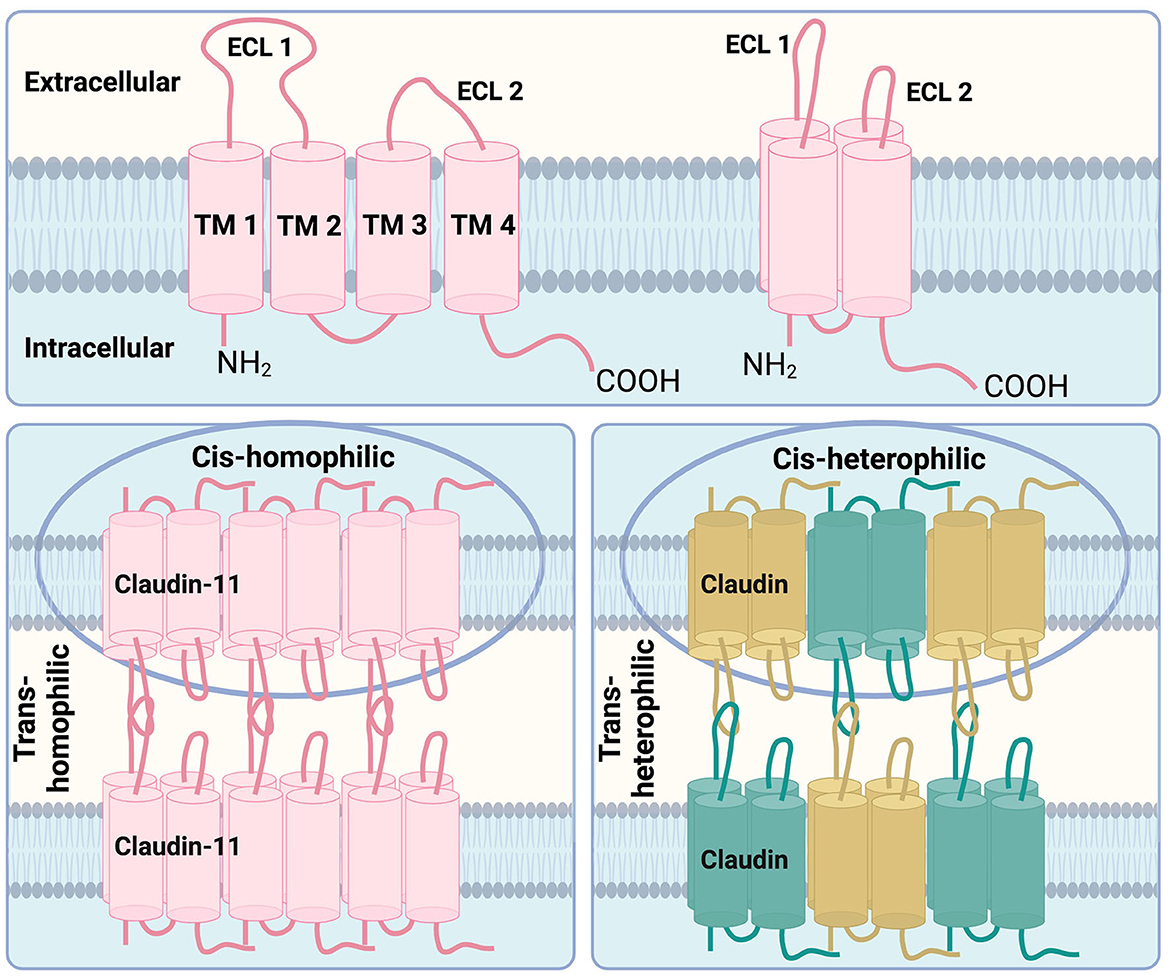
Figure 1. Structural features of claudin-11 tight junctions. Top: The key structural features of claudin family members are the four transmembrane domains (TM1, TM2, TM3, and TM4) with two extracellular loops (ECL1, ECL2) which are arranged in a 3D manner spanning across the cell membrane. Bottom left: Claudin-11, depicted in pink, is known to form homophilic interactions between cells (trans-homophilic) as well as homophilic interactions between claudin-11 proteins within the same membrane (cis-homophilic). Bottom right: Other claudin family members are known to form heterophilic interations between different claudin family proteins, shown in green and gold, within the same cell membrane (cis-heterophilic) as well as on opposing cell membranes (trans-heterophilic). Adapted from Krause et al. (2008).
2.1 Claudin-11 in the central nervous system
Initially identified as oligodendrocyte specific protein (OSP), claudin-11 expression in the CNS is specific to oligodendrocytes (Bronstein et al., 1996, 1997) (Figure 2). During early development, claudin-11 is expressed in mesenchymal cells adjacent to meningeal cartilage-rich areas, while postnatally, claudin-11 expression in the CNS is restricted to early oligodendrocyte progenitors and their lineage (Bronstein et al., 2000). Topologically, claudin-11 resembles other oligodendrocyte-expressed myelin proteins such as PLP1 (Gow et al., 1997; Krause et al., 2008; Devaux et al., 2010). Unlike PLP1, however, claudin-11 is localized exclusively within the radial component between the layers of the myelin sheath (Gow et al., 1999). This specific localization is critical for regulating ion diffusion and myelin permeability, which is essential for normal electrophysiological function (Devaux and Gow, 2008; Gow and Devaux, 2008; Rasband et al., 2012). Claudin-11 expression within the CNS is restricted to the oligodendrocyte lineage, however, outside of the CNS several other cell types are found to express this unique protein.
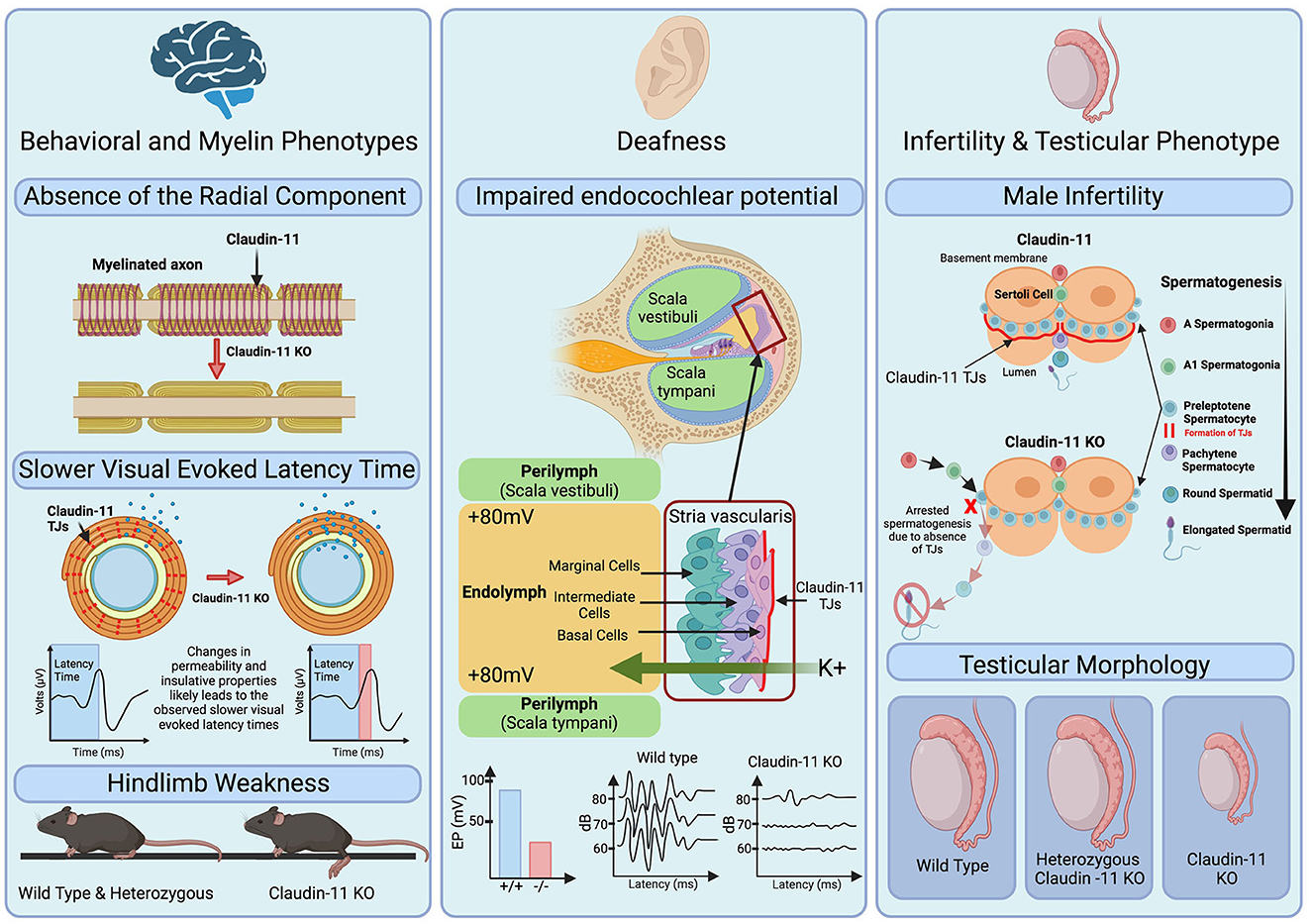
Figure 2. Role of claudin-11 tight junctions in myelin function, hearing, and male fertility. Left: Claudin-11 knock out (KO) mice demonstrate an observable absence of the radial component of the myelin sheath, depicted as the spiral. This may lead to changes in permeability and insulative properties of the myelin sheath as shown by the changes in ions (blue) present within and outside the sheath. The complete absence of claudin-11 and resulting delayed visual evoked latency time is highlighted by red in the visual evoked latency graphs. Additionally, KO mice exhibit hind limb weakness as shown in the bottom of the pannel. Middle: Deafness is a phenotype observed in claudin-11 KO mice. Graphs depict decreased endocochlear potential (EP) generated by KO mice and highlight the increased threshold of hearing for claudin-11 KO mice. Right [adapted from Smith and Braun (2012)]: Male infertility is observed in claudin-11 KO mice. During spermatogenesis, claudin-11 TJs form between sertoli cells which contributes to the blood testis barrier. In the absence of claudin-11, spermatogenesis is arrested resulting in male infertility and is accompanied by hypogonadism shown in the bottom of the pannel.
2.2 Claudin-11 in the inner ear
Outside of the CNS, claudin-11 is expressed in the basal cells of the stria vascularis of the inner-ear (Gow et al., 2004; Kitajiri et al., 2004a,b) and the Sertoli cells of the testes (Gow et al., 1999; Stanton, 2016) (Figure 2). The human inner ear contains thousands of mechanoreceptors essential for hearing (Liu et al., 2017). A potassium-rich endolymph compartment within the cochlea generates the endocochlear potential (EP), an electric potential crucial for transducing acoustic stimuli into electrical signals (Gow et al., 2004; Kitajiri et al., 2004b; Liu et al., 2017; Zhang et al., 2020). By regulating ion movement across the stria vascularis, claudin-11 serves a dual role in the cochlea as both conductor and isolator, effectively functioning as a “conventional electrochemical battery” for ion transport (Liu et al., 2017). Studies in knockout mouse models have revealed the indispensable role of claudin-11 in hearing. Despite having no clear gross morphological malformations within the inner ear, claudin-11 knockout mice are deaf as confirmed through auditory brainstem analysis (Gow et al., 2004; Kitajiri et al., 2004b). Furthermore, freeze fracture electron microscopy showed the absence of TJs in the stria vascularis, highlighting the critical role of claudin-11 TJs in hearing (Gow et al., 2004; Kitajiri et al., 2004b).
2.3 Claudin-11 in the testes
In addition to the CNS and cochlea, claudin-11 is also expressed in the testes (Gow et al., 1999) (Figure 2). Several claudin family members, including claudin-1, -3, -5, -11, -12, and -13, are present in the blood-testis barrier (Stanton, 2016). Interestingly, claudin-11 is the sole essential claudin for spermatogenesis, as its absence in knockout mice leads to infertility, whereas loss of claudin-1, -3, -12, or -13 does not affect fertility (Gow et al., 1999; Stanton, 2016). The role of claudin-5 in spermatogenesis remains unresolved due to the postnatal lethality of its knockout models (Stanton, 2016).
2.4 Claudin-11 in cancer
Outside normal physiology, claudin-11 expression has been implicated in several cancer types. Interestingly, both claudin-11 silencing and overexpression have been linked to invasiveness of cancer cells. For example, in gastric cancers, hypermethylation of CLDN11 and downregulation of its expression were associated with increased cancer cell motility and invasiveness (Agarwal et al., 2009). Similar observations were reported in nasopharyngeal carcinoma cells (Li et al., 2018). Conversely, studies investigating epithelial-mesenchymal transition (EMT) mechanism implicated in cancer metastasis report increased expression of CLDN11 correlating with less favorable patient outcomes (Li et al., 2019). These findings suggest that reduced claudin-11 expression in certain cancers, such as the gastric and nasopharyngeal types, may promote cell migration through loosening of cellular junctions (Agarwal et al., 2009; Li et al., 2018). In contrast, claudin-11 upregulation in EMT-linked cancers may promote maintenance of cell clusters, thus facilitating more efficient cancer cell dissemination (Li et al., 2019).
3 The role of claudin-11 in myelination
3.1 Expression of claudin-11 in the central nervous system
Claudin-11 is expressed in both oligodendrocyte progenitor cells (OPCs) and oligodendrocytes (OLG) (Bronstein et al., 2000). OPCs are bipolar, migratory, and proliferative cells expressing platelet-derived growth factor receptor A (PDGFRa) and neural/glial antigen 2 (NG2), which differentiate into pre-myelinating O4- and O1-positive oligodendrocytes that ultimately differentiate into non-proliferative, myelin-producing OLGs (Pouwels et al., 2014; Kuhn et al., 2019). This myelination process entails a specialized, energy-intensive membrane expansion, increasing the membrane surface area by over six thousand-fold in order to ensheath axons (Pouwels et al., 2014).
3.2 Claudin-11 in myelin
The specialized membrane of the myelin sheath contains many key transmembrane proteins such as PLP1 and claudin-11 which are critical to its integrity and function. Claudin-11 is unique in that it is the sole claudin family member expressed in CNS myelin and therefore its loss cannot be compensated for by other claudin proteins (Gow et al., 1999; Furuse, 2009). Studies using claudin-11 null mice support this notion. Indeed, while claudin-11 null mice, which lack the radial component, do not show gross changes in ultrastructural features of myelin such as thickness or compaction, they do exhibit slower conduction velocities across myelinated axons as well as hind limb weakness (Gow et al., 1999; Devaux and Gow, 2008; Gow and Devaux, 2008). These findings demonstrate that claudin-11 is critical for the maintenance of the electrochemical gradients and electrical resistance properties within the myelin sheath (Gow et al., 1999; Devaux and Gow, 2008; Gow and Devaux, 2008).
3.3 Oligodendrocyte migration
Outside its role in the radial component of myelin, claudin-11 has been linked to OLG proliferation and migration. Using a yeast two-hybrid system, claudin-11 was found to interact and form a complex with OSP/claudin-11-associated protein (OAP-1) and beta-1 integrin (Tiwari-Woodruff et al., 2001). Overexpression of claudin-11 and OAP-1 increased proliferation in a conditionally immortalized mouse oligodendrocyte (CIMO) cell line (Bronstein et al., 1998; Tiwari-Woodruff et al., 2001). Antibodies targeting claudin-11, OAP-1, and beta-1 integrin led to a marked reduction in CIMO cell migration (Tiwari-Woodruff et al., 2001). These findings suggest that the claudin-11/OAP-1/beta-1 integrin complex may be involved in the regulation of migratory and proliferative behavior of OLGs (Tiwari-Woodruff et al., 2001). Despite these in vitro observations, overt impairment or abnormal proliferation and migration of OLGs have not been reported in claudin-11 null mice (Devaux et al., 2010).
4 CLDN11-related hypomyelinating leukodystrophy
4.1 Definition of hypomyelinating leukodsytrophy
White matter diseases are classified into subcategories based on the predominant nature of the myelin pathology observed and include demyelinating, myelinolytic, and dysmyelinating conditions (Naidu, 1999; Knaap and Bugiani, 2017). Myelinolytic diseases, exemplified by Canavan disease, are characterized by unique vacuolar myelinopathy (Naidu, 1999). Demyelinating conditions, such as multiple sclerosis, involve destruction of biochemically normal myelin (Naidu, 1999). Dysmyelinating diseases, notably leukodystrophies, often have genetic causes affecting myelin formation or function (Naidu, 1999). The term “leukodystrophy,” derived from “leuko” meaning white and “dystrophy” meaning wasting, is a fitting descriptor highlighting the degeneration and atrophy of CNS white matter observed in these conditions (Vanderver et al., 2015). The first documented leukodystrophy was described in 1885 and 1910 by Pelizeaus and Merzbacher, identifying a familial progressive white matter disorder known as PMD (Koeppen and Robitaille, 2002). HLDs, such as PMD and HLD22, are progressive disorders (Naidu, 1999; Knaap and Bugiani, 2017). Clinically, they manifest as developmental delay, ataxia, spasticity, hypotonia, and varying degrees of intellectual disability (Pouwels et al., 2014; Knaap and Bugiani, 2017). A defining feature of HLDs is the abnormal development or deposition of myelin (Pouwels et al., 2014). This is typically accompanied by variety of pathological alterations, such as neuronal loss, dysmyelination, demyelination, and axonal damage, that manifest to varying degrees depending on the underlying genetic defect (Sima et al., 2009; Gruenenfelder et al., 2020). HLDs are typically diagnosed through magnetic resonance imaging (MRI) manifesting as lower T2 hypointensities without much reduction in T1 hyperintensities (the latter is typically seen in other non-HLD leukodystrophies) (Pouwels et al., 2014).
4.2 Hypomyelinating leukodystrophy 22
CLDN11 was identified as a novel cause of HLD in 2020 through trio exome sequencing in three unrelated pediatric patients (2 male, 1 female) (Riedhammer et al., 2020). These patients carried de novo, heterozygous stop-loss variants: two had a c.622T>C, p.(*208Glnext*39) variant, while the third patient had the c.622T>G, p.(*208Gluext*39) variant (Riedhammer et al., 2020). Although additional disease-associated variants are listed in databases such as ClinVar and gnomAD, with associated phenotypes like marfanoid habitus and intellectual disability (National Center for Biotechnology Information, 2022), only the stop-loss variants are categorized as pathogenic to date.
The de novo stop-loss variants are predicted to result in a 39-amino acid extension at the cytoplasmic C-terminal end of claudin-11 (Riedhammer et al., 2020). This extension is predicted to form an alpha helix that does not integrate into the cytoplasmic membrane (Riedhammer et al., 2020). Imaging findings of the patients showed that T1-weighted images had signal intensity near normal, whereas T2-weighted images showed reduced myelination of white matter structures (Riedhammer et al., 2020). All three individuals presented with delayed developmental milestones, contractures, nystagmus, drooling, hypermetropia, severe disarticulation, and require mobility support when walking and climbing stairs, features that are shared with PMD and other HLDs (Riedhammer et al., 2020). Interestingly, hypermetropia is not a common clinical feature of HLD, suggesting its potential as a diagnostic marker for HLD22 (Riedhammer et al., 2020). HLD22 affects paediatric patients and is both early onset and progressive (Riedhammer et al., 2020). The underlying mechanisms for this disease are currently not well understood, and effective treatments are currently lacking.
Since the description of the first three patients with CLDN11-related HLD22, at least three more individuals have been identified with a clinical phenotype compatible with HLD22 and with CLDN11 variants leading to an extended protein (unpublished). Currently, it is not clear whether CLDN11 variants causing other types of changes in the protein are associated with a similar phenotype in humans.
4.3 Cellular mechanisms of claudin-11 dysfunction in hypomyelinating leukodystrophy 22
4.3.1 Mouse models of claudin-11 deficiency
Mouse models, particularly the Cldn11 null and heterozygous knockouts, have provided key insights into the role of claudin-11 in myelination. Behavioral phenotyping indicates reduced anxiety in the Cldn11 null mice from an early age as assessed using open field and the marble burying tests (Maheras et al., 2018). These anxiety manifestations precede age-dependent, site-specific alterations in inhibitory and excitatory neurotransmitters, and have instead been suggested to reflect a temporal disconnection in signal transmission via myelinated axons that represent a potential mechanism of neuropsychiatric disease (Maheras et al., 2018). Motor assessments revealed hindlimb weakness in the Cldn11 null animals, although heterozygous mice were phenotypically similar to wild type controls (Gow et al., 1999). The absence of neurodegeneration markers such as N-acetylaspartate or spinal pathology suggests the behavioral changes may arise from delayed signal transduction by myelinated axons rather than axonal degeneration or neuronal loss (Maheras et al., 2018). This notion is corroborated by visual evoked potential assessments, auditory brain stem evoked potentials, and compound action potentials in the optic nerve and spinal cord showing slowed latency times in the Cldn11 null mouse model (Gow et al., 1999, 2004; Gow and Devaux, 2008). These studies showcase the utility of mouse models in understanding claudin-11 biology, and suggest that similar models may aid in gaining insights into the pathogenic mechanisms underlying HLD22 as well as evaluating potential therapeutics for the disease.
Myelin specific analysis of the Cldn11 null mouse model revealed the absence of radial tight junctions typically present in myelin, though other ultrastructural features are preserved (Gow et al., 1999). To date, no deficits in oligodendrocyte proliferation and migration have been reported in these mice (Devaux et al., 2010). Nonetheless, a comprehensive analysis of the cellular composition of the CNS of Cldn11 null mice is lacking. A detailed analysis would be required to definitively conclude that different cellular populations, including oligodendrocytes, are not affected in these mice.
In addition to myelin-related alterations, Cldn11 null mice present with two other distinct phenotypes: deafness and sterility. The absence of claudin-11 compromises endocochlear potential and mice present with deafness (Gow et al., 2004; Kitajiri et al., 2004b). The infertility is restricted to males; Cldn11 null females are able to produce litter sizes comparable to controls (Gow et al., 1999). Male infertility in Cldn11 null mice is associated with significantly smaller testis (Gow et al., 1999) and is likely due to arrested spermatogenesis (Plymate, 1994; Wu et al., 2012). Notably, this is another instance where the heterozygous mice are indistinguishable from controls, demonstrating that one functional copy of Cldn11 is sufficient for normal physiological function (Gow et al., 1999). These findings raise questions about the pathogenic mechanism(s) of HLD22, as they collectively indicate that haploinsufficiency is unlikely to cause disease.
4.3.2 Postulated mechanisms of mutant claudin-11-mediated HLD22
The endoplasmic reticulum (ER) is crucial for protein folding and post-translational modifications (Helenius et al., 1992). Given the massive scale of protein production required for myelination in oligodendrocytes (Anitei and Pfeiffer, 2006) increased ER stress has been implicated in several myelinopathies (Southwood et al., 2002; Voorn et al., 2005; Mhaille et al., 2008; Pennuto et al., 2008) [Reviewed in Lin and Popko (2009)]. The unfolded protein response (UPR) is activated when there is an accumulation of unfolded proteins within the ER (Read and Schroder, 2021). The UPR consists of three signalling pathways: the IRE1 pathway is triggered to enhance protein folding and degradation (Adams et al., 2019); the PERK pathway phosphorylates eukaryotic translation initiation factor 2α (EIF2α), reducing protein translation (Harding et al., 1999; Adams et al., 2019); and the ATF6 pathway, similar to PERK, boosts protein folding and degradation (Yoshida et al., 2001; Adams et al., 2019). Unresolved protein misfolding, despite sustained activation of these UPR pathways, leads to apoptosis of oligodendrocytes (Gow et al., 1998).
Increased ER stress and UPR activation have been implicated in PMD as a result of mutations or altered expression of PLP1, a tetraspan, secretory pathway myelin protein (Southwood et al., 2002). As claudin-11 is also a secretory pathway, tetraspan myelin protein, a similar pathogenic mechanism may underlie HLD22. Specifically, mutations in claudin-11 may lead to protein misfolding and accumulation in the ER, activation of the UPR, and ultimately oligodendrocyte death and hypomyelination. It is also possible that other gain-of-toxic function mechanisms involving aberrant claudin-11 localization or interactions may contribute to HLD22. Given that one functional claudin-11 copy suffices for normal physiology as shown in animal models, a claudin-11 loss-of-function scenario appears unlikely as the cause of HLD22, though dominant negative effects cannot be ruled out.
5 Conclusion
Claudin-11 plays a critical role in maintaining several aspects of normal physiology, including forming the radial component of the myelin sheath and facilitating nerve conduction, maintenance of the endocochlear potential required for normal hearing, and maintaining the integrity of the blood-testis barrier necessary for normal spermatogenesis. Abnormal claudin-11 function has been implicated in various pathologies ranging from cancer to behavioral and myelin abnormalities, and most notably the hypomyelinating disorder HLD22. Mouse and in vitro models serve as important tools to elucidate the disease characteristics and pathogenic mechanisms associated with claudin-11. A comprehensive understanding of the biological function of claudin-11 and the pathogenic mechanisms underlying HLD22 is essential for the development of effective treatments for this disease.
Author contributions
SG: Conceptualization, Data curation, Project administration, Visualization, Writing—original draft, Writing—review & editing. OO: Writing—review & editing. AG: Writing—review & editing. SS-I: Writing—review & editing. MP: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing—review & editing, Visualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. SG was supported by a BC Graduate Scholarship. AG was supported by grant from Wayne State University (Boost Award). MP was supported by the BC Children's Hospital Research Institute Investigator Grant Award (IGAP), a Scholar Award from the Michael Smith Health Research BC, and a grant from the Rare Diseases: Models and Mechanism Network (210327-001-001).
Acknowledgments
We thank members of the Pouladi lab for helpful discussions. The figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adams, C. J., Kopp, M. C., Larburu, N., Nowak, P. R., and Ali, M. M. U. (2019). Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front. Molec. Biosci. 6, 11. doi: 10.3389/fmolb.2019.00011
Agarwal, R., Mori, Y., Cheng, Y., Jin, Z., Olaru, A. V., Hamilton, J. P., et al. (2009). Silencing of claudin-11 is associated with increased invasiveness of gastric cancer cells. PLoS ONE 4, e8002. doi: 10.1371/journal.pone.0008002
Anitei, M., and Pfeiffer, S. E. (2006). Myelin biogenesis: sorting out protein trafficking. Curr. Biol. 16, R418–R421. doi: 10.1016/j.cub.2006.05.010
Bronstein, J., Chen, K., Tiwari–Woodruff, S., and Kornblum, H. (2000). Developmental expression of OSP/claudin–11. J. Neurosci. Res. 60, 284–290. doi: 10.1002/(SICI)1097-4547(20000501)60:3<284::AID-JNR2>3.0.CO;2-T
Bronstein, J., Micevych, P., and Chen, K. (1997). Oligodendrocyte–specific protein (OSP) is a major component of CNS myelin. J. Neuroscience Research 50, 713–720. doi: 10.1002/(SICI)1097-4547(19971201)50:5<713::AID-JNR8>3.0.CO;2-K
Bronstein, J. M., Hales, T. G., Tyndale, R. F., and Charles, A. C. (1998). A conditionally immortalized glial cell line that expresses mature myelin proteins and functional GABAA receptors. J. Neurochem. 70, 483–491. doi: 10.1046/j.1471-4159.1998.70020483.x
Bronstein, J. M., Popper, P., Micevych, P. E., and Farber, D. B. (1996). Isolation and characterization of a novel oligodendrocyte-specific protein. Neurology 47, 772–778. doi: 10.1212/WNL.47.3.772
Colegio, O. R., Itallie, C. M. V., McCrea, H. J., Rahner, C., and Anderson, J. M. (2002). Claudins create charge-selective channels in the paracellular pathway between epithelial cells. Am. J. Physiol. Cell Physiol. 283, C142–C147. doi: 10.1152/ajpcell.00038.2002
Devaux, J., Fykkolodziej, B., and Gow, A. (2010). Chapter 10 claudin proteins and neuronal function. Curr. Topics Membr. 65, 229–253. doi: 10.1016/S1063-5823(10)65010-7
Devaux, J., and Gow, A. (2008). Tight junctions potentiate the insulative properties of small CNS myelinated axons. J. Cell Biol. 183, 909–921. doi: 10.1083/jcb.200808034
Engelund, M. B., Yu, A. S. L., Li, J., Madsen, S. S., Frgeman, N. J., and Tipsmark, C. K. (2012). Functional characterization and localization of a gill-specific claudin isoform in Atlantic salmon. Am. J. Physiol. Regul. Integr. Compar. Physiol. 302, R300–R311. doi: 10.1152/ajpregu.00286.2011
Furuse, M. (2009). Knockout animals and natural mutations as experimental and diagnostic tool for studying tight junction functions in vivo. Biochim. Biophys. Acta 1788, 813–819. doi: 10.1016/j.bbamem.2008.07.017
Garbern, J., Cambi, F., Shy, M., and Kamholz, J. (1999). The molecular pathogenesis of pelizaeus-merzbacher disease. Arch. Neurol. 56, 1210–1214. doi: 10.1001/archneur.56.10.1210
Goncalves, A., Ambrsio, A. F., and Fernandes, R. (2013). Regulation of claudins in blood-tissue barriers under physiological and pathological states. Tissue Barr. 1, e24782. doi: 10.4161/tisb.24782
Gow, A., Davies, C., Southwood, C. M., Frolenkov, G., Chrustowski, M., Ng, L., et al. (2004). Deafness in claudin 11-null mice reveals the critical contribution of basal cell tight junctions to stria vascularis function. J. Neurosci. 24, 7051–7062. doi: 10.1523/JNEUROSCI.1640-04.2004
Gow, A., and Devaux, J. (2008). A model of tight junction function in central nervous system myelinated axons. Neuron Glia Biol. 4, 307–317. doi: 10.1017/S1740925X09990391
Gow, A., Gragerov, A., Gard, A., Colman, D. R., and Lazzarini, R. A. (1997). Conservation of topology, but not conformation, of the proteolipid proteins of the myelin sheath. J. Neurosci. 17, 181–189. doi: 10.1523/JNEUROSCI.17-01-00181.1997
Gow, A., and Lazzarini, R. A. (1996). A cellular mechanism governing the severity of Pelizaeus–Merzbacher disease. Nat. Genet. 13, 422–428. doi: 10.1038/ng0896-422
Gow, A., Southwood, C. M., and Lazzarini, R. A. (1998). Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of pelizaeus-merzbacher disease. J. Cell Biol. 140, 925–934. doi: 10.1083/jcb.140.4.925
Gow, A., Southwood, C. M., Li, J. S., Pariali, M., Riordan, G. P., Brodie, S. E., et al. (1999). CNS myelin and sertoli cell tight junction strands are absent in Osp/Claudin-11 null mice. Cell 99, 649–659. doi: 10.1016/S0092-8674(00)81553-6
Gruenenfelder, F. I., McLaughlin, M., Griffiths, I. R., Garbern, J., Thomson, G., Kuzman, P., et al. (2020). Neural stem cells restore myelin in a demyelinating model of Pelizaeus-Merzbacher disease. Brain 143, 1383–1399. doi: 10.1093/brain/awaa080
Harding, H. P, Zhang, Y., and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum resident kinase. Nature 397, 271–274. doi: 10.1038/16729
Harry, G. J., and Toews, A. D. (1998). “Handbook of developmental neurotoxicology,” in Part I: Cellular and Molecular Morphogenesis of the Nervous System 87–115. doi: 10.1016/B978-012648860-9/50007-8
Helenius, A., Marquardt, T., and Braakman, I. (1992). The endoplasmic reticulum as a protein-folding compartment. Trends Cell Biol. 2, 227–231. doi: 10.1016/0962-8924(92)90309-B
Kitajiri, S. I., Furuse, M., Morita, K., Saishin-Kiuchi, Y., Kido, H., Ito, J., et al. (2004a). Expression patterns of claudins, tight junction adhesion molecules, in the inner ear. Hear. Res. 187, 25–34. doi: 10.1016/S0378-5955(03)00338-1
Kitajiri, S. I., Miyamoto, T., Mineharu, A., Sonoda, N., Furuse, K., Hata, M., et al. (2004b). Compartmentalization established by claudin-11-based tight junctions in stria vascularis is required for hearing through generation of endocochlear potential. J. Cell Sci. 117, 5087–5096. doi: 10.1242/jcs.01393
Koeppen, A. H., and Robitaille, Y. (2002). Pelizaeus-merzbacher disease. J. Neuropathol. Exper. Neurol. 61, 747–759. doi: 10.1093/jnen/61.9.747
Koziel, M. J., Kowalska, K., and Piastowska-Ciesielska, A. W. (2020). Claudins: new players in human fertility and reproductive system cancers. Cancers 12, 711. doi: 10.3390/cancers12030711
Krause, G., Winkler, L., Mueller, S. L., Haseloff, R. F., Piontek, J., and Blasig, I. E. (2008). Structure and function of claudins. Biochim. Biophys Acta 1778, 631–645. doi: 10.1016/j.bbamem.2007.10.018
Kuhn, S., Gritti, L., Crooks, D., and Dombrowski, Y. (2019). Oligodendrocytes in development, myelin generation and beyond. Cells 8, 1424. doi: 10.3390/cells8111424
Lal-Nag, M., and Morin, P. J. (2009). The claudins. Genome Biol. 10, 235. doi: 10.1186/gb-2009-10-8-235
Li, C.-F., Chen, J.-Y., Ho, Y.-H., Hsu, W.-H., Wu, L.-C., Lan, H.-Y., et al. (2019). Snail-induced claudin-11 prompts collective migration for tumour progression. Nat. Cell Biol. 21, 251–262. doi: 10.1038/s41556-018-0268-z
Li, H.-P., Peng, C.-C., Wu, C.-C., Chen, C.-H., Shih, M.-J., Huang, M.-Y., et al. (2018). Inactivation of the tight junction gene CLDN11 by aberrant hypermethylation modulates tubulins polymerization and promotes cell migration in nasopharyngeal carcinoma. J. Exper. Clin. Cancer Res. 37, 102. doi: 10.1186/s13046-018-0754-y
Lin, W., and Popko, B. (2009). Endoplasmic reticulum stress in disorders of myelinating cells. Nat. Neurosci. 12, 379–385. doi: 10.1038/nn.2273
Liu, W., Schrott-Fischer, A., Glueckert, R., Benav, H., and Rask-Andersen, H. (2017). The human “cochlear battery” claudin-11 barrier and ion transport proteins in the lateral wall of the cochlea. Front. Molec. Neurosci. 10, 239. doi: 10.3389/fnmol.2017.00239
Maheras, K. J., Peppi, M., Ghoddoussi, F., Galloway, M. P., Perrine, S. A., and Gow, A. (2018). Absence of claudin 11 in CNS myelin perturbs behavior and neurotransmitter levels in mice. Sci. Rep. 8, 3798. doi: 10.1038/s41598-018-22047-9
Mhaille, A. N., McQuaid, S., Windebank, A., Cunnea, P., McMahon, J., Samali, A., et al. (2008). Increased expression of endoplasmic reticulum stress-related signaling pathway molecules in multiple sclerosis lesions. J. Neuropathol. Exper. Neurol. 67, 200–211. doi: 10.1097/NEN.0b013e318165b239
Mineta, K., Yamamoto, Y., Yamazaki, Y., Tanaka, H., Tada, Y., Saito, K., et al. (2011). Predicted expansion of the claudin multigene family. FEBS Lett. 585, 606–612. doi: 10.1016/j.febslet.2011.01.028
Moon, S. L., and Parker, R. (2018). EIF2B2 mutations in vanishing white matter disease hypersuppress translation and delay recovery during the integrated stress response. RNA 24, 841–852. doi: 10.1261/rna.066563.118
Morita, K., Sasaki, H., Fujimoto, K., Furuse, M., and Tsukita, S. (1999). Claudin-11/OSP-based tight junctions of myelin sheaths in brain and sertoli cells in testis. J. Cell Biol. 145, 579–588. doi: 10.1083/jcb.145.3.579
Naidu, S. (1999). Clinical delineation of leukodystrophies. J. Molec. Neurosci. 12, 185–192. doi: 10.1385/JMN:12:3:185
National Center for Biotechnology Information. (2022). ClinVar; [VCV000689665.1]. Available online at: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000689665.1 (accessed September 1, 2023).
Pennuto, M., Tinelli, E., Malaguti, M., Carro, U. D., D'Antonio, M., Ron, D., et al. (2008). Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in charcot-marie-tooth 1B mice. Neuron 57, 393–405. doi: 10.1016/j.neuron.2007.12.021
Plymate, S. (1994). Hypogonadism. Endocrinol. Metab. Clin. North Am. 23, 749–772. doi: 10.1016/S0889-8529(18)30065-3
Pouwels, P. J. W., Vanderver, A., Bernard, G., Wolf, N. I., Dreha Kulczewksi, S. F., Deoni, S. C. L., et al. (2014). Hypomyelinating leukodystrophies: translational research progress and prospects. Ann. Neurol. 76, 5–19. doi: 10.1002/ana.24194
Powers, J. M., Pei, Z., Heinzer, A. K., Deering, R., Moser, A. B., Moser, H. W., et al. (2005). Adreno-leukodystrophy: oxidative stress of mice and men. J. Neuropathol. Exper. Neurol. 64, 1067–1079. doi: 10.1097/01.jnen.0000190064.28559.a4
Rasband, M. N., Macklin, W. B., and Benjamins, J. A. (2012). “Basic neurochemistry (Eighth Edition),” in Part I: Cellular Neurochemistry and Neural Membranes 180–199. doi: 10.1016/B978-0-12-374947-5.00010-9
Read, A., and Schröder, M. (2021). The unfolded protein response: an overview. Biology 10, 384. doi: 10.3390/biology10050384
Riedhammer, K. M., Stockler, S., Ploski, R., Wenzel, M., Adis-Dutschmann, B., Ahting, U., et al. (2020). De novo stop-loss variants in CLDN11 cause hypomyelinating leukodystrophy. Brain 144, 411–419. doi: 10.1093/brain/awaa410
Shin, D., Feltri, M. L., and Wrabetz, L. (2016). Altered trafficking and processing of GALC mutants correlates with globoid cell leukodystrophy severity. J. Neurosci. 36, 1858–1870. doi: 10.1523/JNEUROSCI.3095-15.2016
Sima, A. A., Pierson, C. R., Woltjer, R. L., Hobson, G. M., Golden, J. A., Kupsky, W. J., et al. (2009). Neuronal loss in pelizaeus merzbacher disease differs in various mutations of the proteolipid protein 1. Acta Neuropathol. 118, 531–539. doi: 10.1007/s00401-009-0562-8
Skoff, R. P., Bessert, D., Banerjee, S., Luo, X., and Thummel, R. (2021). Characterization of the expression of vacuolar protein sorting 11 (Vps11) in mammalian oligodendrocytes. ASN Neuro 13, 17590914211009851. doi: 10.1177/17590914211009851
Smith, B. E., and Braun, R. E. (2012). Germ cell migration across sertoli cell tight junctions. Science 338, 798–802. doi: 10.1126/science.1219969
Southwood, C. M., Garbern, J., Jiang, W., and Gow, A. (2002). The unfolded protein response modulates disease severity in pelizaeus-merzbacher disease. Neuron 36, 585–596. doi: 10.1016/S0896-6273(02)01045-0
Stanton, P. G. (2016). Regulation of the blood-testis barrier. Semin. Cell Dev. Biol. 59, 166–173. doi: 10.1016/j.semcdb.2016.06.018
Stecca, B., Southwood, C. M., Gragerov, A., Kelley, K. A., Friedrich, V. L., and Gow, A. (2000). The evolution of lipophilin genes from invertebrates to tetrapods: DM-20 cannot replace proteolipid protein in CNS myelin. J. Neurosci. 20, 4002–4010. doi: 10.1523/JNEUROSCI.20-11-04002.2000
Tiwari-Woodruff, S. K., Buznikov, A. G., Vu, T. Q., Micevych, P. E., Chen, K., Kornblum, H. I., et al. (2001). Osp/Claudin-11 forms a complex with a novel member of the tetraspanin super family and integrin and regulates proliferation and migration of oligodendrocytes. J. Cell Biol. 153, 295–306. doi: 10.1083/jcb.153.2.295
Tsuji, S. (2007). “Neurobiology of disease,”? in Part A: Central nervous system, (Annual Neurology) 43–49. doi: 10.1016/B978-012088592-3/50006-2
Van De Knaap, M. S., Boor, I., and Estévez, R. (2012). Megalencephalic leukoencephalopathy with subcortical cysts: chronic white matter oedema due to a defect in brain ion and water homoeostasis. Lancet Neurol. 11, 973–985. doi: 10.1016/S1474-4422(12)70192-8
van der Knaap, M. S., and Bugiani, M. (2017). Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. 134, 351–382. doi: 10.1007/s00401-017-1739-1
Van Der Voorn, J. P., Van Kollenburg, B., Bertrand, G., Van Haren, K., Scheper, G. C., Powers, J. M., et al. (2005). The unfolded protein response in vanishing white matter disease. J. Neuropathol. Exper. Neurol. 64, 770–775. doi: 10.1097/01.jnen.0000178446.41595.3a
Vanderver, A., Prust, M., Tonduti, D., Mochel, F., Hussey, H. M., Helman, G., et al. (2015). Case definition and classification of leukodystrophies and leukoencephalopathies. Molec. Genet. Metab. 114, 494–500. doi: 10.1016/j.ymgme.2015.01.006
Wolburg, H., Wolburg-Buchholz, K., Liebner, S., and Engelhardt, B. (2001). Claudin-1, claudin-2 and claudin-11 are present in tight junctions of choroid plexus epithelium of the mouse. Neurosci. Lett. 307, 77–80. doi: 10.1016/S0304-3940(01)01927-9
Wu, X., Peppi, M., Vengalil, M. J., Maheras, K. J., Southwood, C. M., Bradley, M., et al. (2012). Transgene-mediated rescue of spermatogenesis in Cldn11-null mice. Biol. Reprod. 86, 139. doi: 10.1095/biolreprod.111.096230
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. doi: 10.1016/S0092-8674(01)00611-0
Keywords: claudin-11, tight junctions, myelin, leukodystrophy, HLD22, hearing, fertility
Citation: Gjervan SC, Ozgoren OK, Gow A, Stockler-Ipsiroglu S and Pouladi MA (2024) Claudin-11 in health and disease: implications for myelin disorders, hearing, and fertility. Front. Cell. Neurosci. 17:1344090. doi: 10.3389/fncel.2023.1344090
Received: 25 November 2023; Accepted: 31 December 2023;
Published: 17 January 2024.
Edited by:
Shirin Hosseini, Braunschweig University of Technology, GermanyReviewed by:
Janos Groh, Technical University of Munich, GermanyCopyright © 2024 Gjervan, Ozgoren, Gow, Stockler-Ipsiroglu and Pouladi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mahmoud A. Pouladi, mahmoud.pouladi@ubc.ca