
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Cell. Neurosci., 26 June 2023
Sec. Cellular Neurophysiology
Volume 17 - 2023 | https://doi.org/10.3389/fncel.2023.1212202
This article is part of the Research TopicInhibitory Interneuron Classification, Function and Dysfunction in Neural NetworkView all 12 articles
 Juliette Vivien1†
Juliette Vivien1† Anass El Azraoui1,2†
Anass El Azraoui1,2† Cloé Lheraux1
Cloé Lheraux1 Frederic Lanore3‡
Frederic Lanore3‡ Bruno Aouizerate1,3,4
Bruno Aouizerate1,3,4 Cyril Herry1‡
Cyril Herry1‡ Yann Humeau2‡
Yann Humeau2‡ Thomas C. M. Bienvenu1,3*‡
Thomas C. M. Bienvenu1,3*‡Imbalance between excitation and inhibition in the cerebral cortex is one of the main theories in neuropsychiatric disorder pathophysiology. Cortical inhibition is finely regulated by a variety of highly specialized GABAergic interneuron types, which are thought to organize neural network activities. Among interneurons, axo-axonic cells are unique in making synapses with the axon initial segment of pyramidal neurons. Alterations of axo-axonic cells have been proposed to be implicated in disorders including epilepsy, schizophrenia and autism spectrum disorder. However, evidence for the alteration of axo-axonic cells in disease has only been examined in narrative reviews. By performing a systematic review of studies investigating axo-axonic cells and axo-axonic communication in epilepsy, schizophrenia and autism spectrum disorder, we outline convergent findings and discrepancies in the literature. Overall, the implication of axo-axonic cells in neuropsychiatric disorders might have been overstated. Additional work is needed to assess initial, mostly indirect findings, and to unravel how defects in axo-axonic cells translates to cortical dysregulation and, in turn, to pathological states.
Imbalance between synaptic excitation and inhibition in cortical regions (including neocortex and hippocampus) is a key feature of many brain disorders (Marin, 2012; Sohal and Rubenstein, 2019). Activities of cortical neuron assemblies are coordinated by brain rhythms of various frequencies which are known to be critical for cognitive, emotional and motor function (Buzsaki and Draguhn, 2004). Throughout the brain, inhibition provided by GABAergic interneurons orchestrate cortical oscillations (Roux and Buzsaki, 2015). Lack of pyramidal neuron inhibition results in global hyperexcitability and highly synchronous discharges, which are hallmarks of epilepsy (EPI). Beyond hyperexcitability, dysregulation of oscillatory activities may result in more subtle alterations in cortical functions. Basal and sensory-evoked oscillatory rhythms have been found to be altered in schizophrenia (SCZ) and autism spectrum disorders (ASD; Simon and Wallace, 2016; Hunt et al., 2017). As inhibition is critical for cortical oscillations (Roux and Buzsaki, 2015), these findings collectively point toward a central implication of interneuron functional alterations in neuropsychiatric disorders (Marin, 2012; Sohal and Rubenstein, 2019).
In cortex, inhibition is provided by distinct types of GABAergic cells, with extensive diversity in structure reflected in specialized functions. Indeed, by receiving different synaptic inputs and by targeting distinct functional domains along somato-dendritic arborizations of pyramidal cells, distinct GABAergic interneuron types are thought to play highly specific roles in organizing cortical network activities (e.g., Kepecs and Fishell, 2014). Perisomatic inhibition is mainly provided by interneurons which typically emit short-duration action potentials at high frequencies and express the calcium binding protein parvalbumin (PV). PV+ interneurons are powerful inhibitors of principal neurons and regulate fast (>30 Hz) brain rhythms, such as gamma oscillations (30–80 Hz; Cardin et al., 2009; Sohal et al., 2009). This, added to their expression of disease-related genes, has led to the hypothesis that PV+ interneuron dysfunction may be implicated in neuropsychiatric disorders (reviewed in Marin, 2012).
The PV+ interneuron class has been further divided into two main types: basket cells, which axon make synapses with cell bodies and proximal dendrites of pyramidal neurons, and axo-axonic cells (AACs), which axonal boutons exclusively innervate the axon initial segment (AIS) of pyramidal neurons (Somogyi, 1977). Axo-axonic terminals are organized in vertical clusters (“cartridges”), hence the alternative name of “Chandelier cells” for AACs. This typical morphology is the most distinctive characteristic of AACs at the cellular level. In addition, PV is a classical (albeit non-selective) marker of AACs (but PV- AACs are found in cortex, e.g., Taniguchi et al., 2013). In addition, Human AACs may express calbindin (CB), which has been found to be associated (del Rio and DeFelipe, 1997), or mutually exclusive with PV (Wittner et al., 2002). At the electron microscopic level, AACs make symmetrical synapses with the axon initial segment (identified by cisternal organelles, dense undercoating, bundled microtubules). Accordingly, axo-axonic terminals contain the molecular machinery for GABA release, including glutamic acid decarboxylase (GAD), membrane GABA transporters (GAT-1) and vesicular GABA transporters (VGAT; Jung et al., 2022). At the postsynaptic level, AISs are organized by scaffold proteins with Ankyrin-G playing a decisive role (Gallo et al., 2020). AISs synapses are enriched in α2 subunit-containing GABAA-Rs (Nusser et al., 1996), although a quantitative freeze-fracture replica study found equal levels of α1 and α2 subunits (Kerti-Szigeti and Nusser, 2016). These morphological and molecular features are classically used to study AACs, including in the studies reviewed in this article.
Because action potentials are generated at the AIS, AACs have long been speculated to exert powerful control on pyramidal neurons activities, and to play a key role in regulating network excitability and neuronal assemblies. Being sole inhibitors at the AIS and showing disease-related changes, AACs might represent a cortical Achille's heel. Indeed, the potential role of AACs in network physiology has been mirrored by studies of cortical AACs in disease, pointing toward abnormalities at AACs' cell bodies and synapses in postmortem samples of patients suffering from EPI (DeFelipe et al., 1993), SCZ (Woo et al., 1998) or, more recently, ASD (Ariza et al., 2018). AACs and their implication in disease have attracted a lot of interest, as previously summarized (Wang et al., 2016; Gallo et al., 2020; Juarez and Martinez Cerdeno, 2022). However, no previous work has reviewed systematically the studies linking AACs to brain disorders. The aim of this article is to provide a systematic review of articles documenting AACs' implication in neuropsychiatric disorders (EPI, SCZ, and ASD), for scientists and clinicians studying those disorders, and to help design future studies on robust experimental grounds.
The systematic review followed PRISMA guidelines. Two independent electronic databases (Pubmed and Web of Science) were searched until July 2021 for articles dealing with AACs in EPI, SCZ and ASD with the following search terms: ≪(chandelier cell OR axo axonic) AND epilep*≫, ≪(chandelier cell OR axo axonic) AND schizophrenia≫, ≪(chandelier cell OR axo axonic) AND autism≫, respectively. Duplicates were removed, and titles and abstracts were screened for inclusion. Full texts were obtained and examined if articles potentially corresponded to eligibility criteria. Two investigators (AEA and JV) screened the literature. In case of a disagreement, individual articles were studied by all the authors, until consensus was reached. Full articles matching inclusion criteria were studied and summarized by the authors. Additional articles found by cross-referencing were also examined and included when indicated.
Eligibility was defined by the following criteria, which were defined prior to the selection of articles:
Articles investigating AACs' modifications in EPI, SCZ, ASD in brain tissue from patients (including case reports), or in animal models of those disorders (in vivo and/or in vitro) were included for analysis.
Articles written in languages other than English, review articles, and studies giving no specific results on AACs were excluded from the final analysis. Articles lacking an unambiguous (e.g., morphological) identification of AACs or their synaptic compartments at AISs were thus excluded. Likewise, studies lacking a characterization of potential AACs' alterations were excluded. All exclusions were documented and reasons for exclusions are reported.
A total of 200 articles were screened after duplicates removal (EPI 109, SCZ 77, ASD 14). Thirty-two articles were included in the final analysis (EPI 16, SCZ 13, ASD 3). The screening steps are illustrated in the corresponding flowcharts (Figures 1–3; see also Supplementary material). The results concerning AACs are summarized in Tables 1–3.
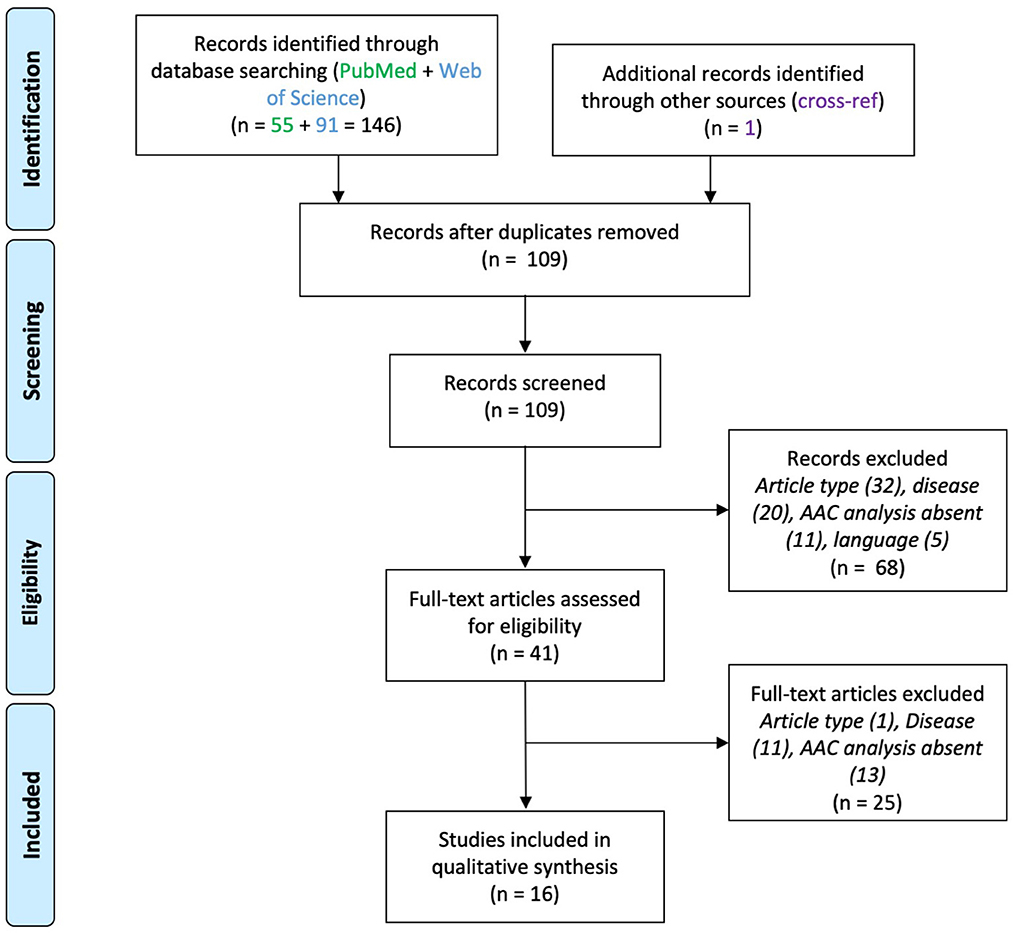
Figure 1. PRISMA flowchart for studies on axo-axonic cells in epilepsy.
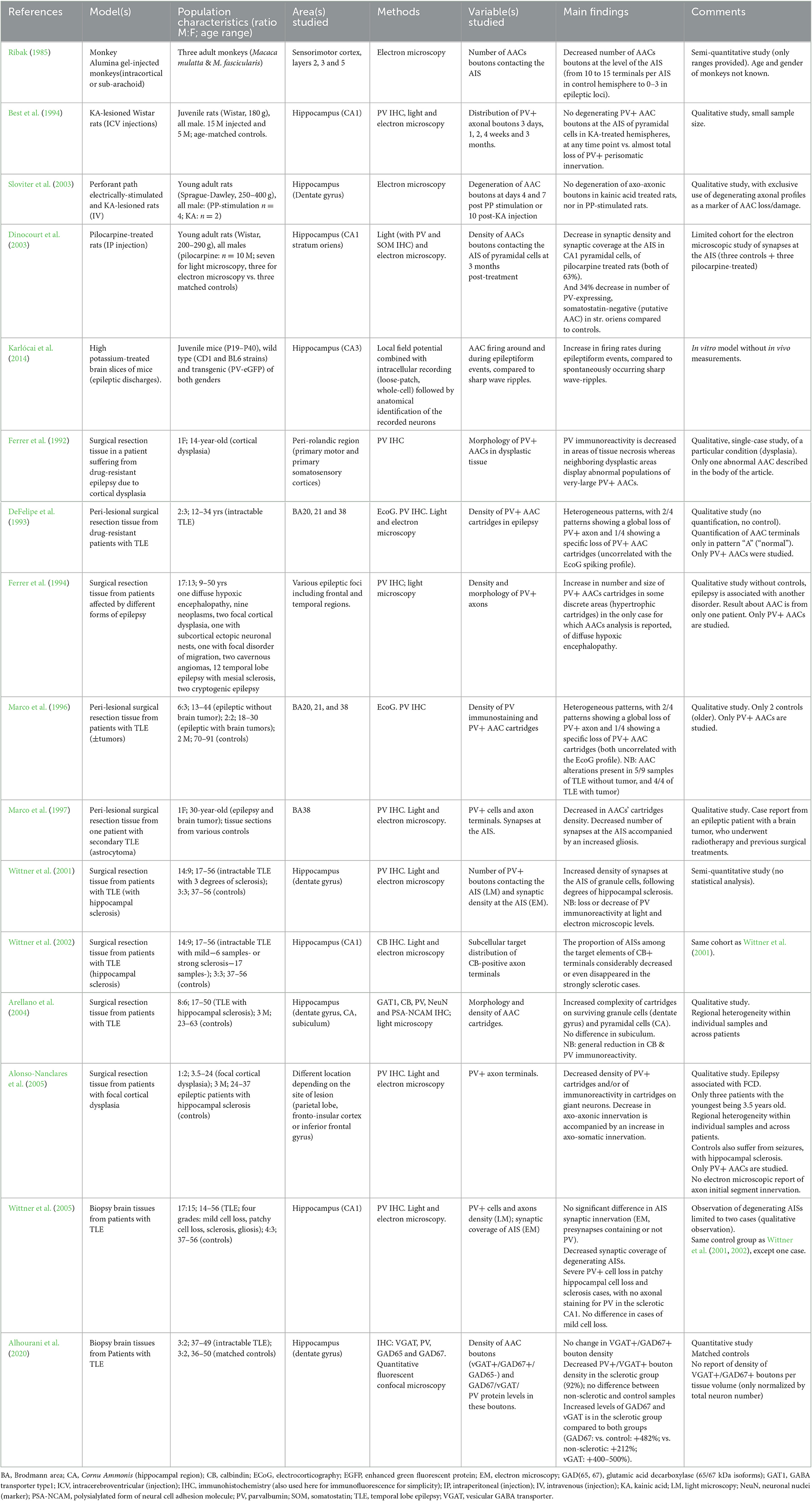
Table 1. Summary of studies on axo-axonic cells in epilepsy.
Sixteen papers were included (Figure 1). These studies used either human brain samples from patients affected by medically intractable epilepsy (n = 11) or from animal models of epilepsy (n = 5). Neocortical and hippocampal alterations in AACs were reported, which are divided below for clarity. Results are summarized in Table 1.
In the neocortex, a loss of PV+ AACs' cartridges has been reported in the anterior temporal lobe of epileptic patients, with some samples displaying pathological cytoarchitectures (DeFelipe et al., 1993; Marco et al., 1996). The decrease in PV+ AACs' cartridges has also been observed in dysplastic samples from patients suffering from EPI secondary to focal cortical dysplasia (Alonso-Nanclares et al., 2005). In a case report of a young patient with cortical dysplasia, a decrease in PV immunoreactivity was described (Ferrer et al., 1992), suggesting that the decreased density of cartridges observed in the previous studies may correspond to a decrease in PV levels. This finding was associated with abnormal morphologies of the remaining PV+ cells, including the observation of an enlarged AAC (Ferrer et al., 1992). Ferrer and colleagues also reported hypertrophic PV+ cartridges in a patient suffering from diffuse hypoxic encephalopathy associated with focal seizures (Ferrer et al., 1994). A case report of a patient suffering from seizures secondary to a temporal lobe astrocytoma also described a decrease in PV immunoreactivity and numbers of AAC boutons in the peri-tumoral tissue (Marco et al., 1997), in line with other findings. Altogether, these studies reported a decrease in PV+ AACs' cartridges and an abnormal morphology of the remaining AACs. However, these results were only qualitative with no quantification of this apparent loss, which may also correspond to a decrease in PV expression. Despite the lack of quantification, these findings are consistent with previous experimental work showing degeneration of axonal boutons contacting the AIS at epileptic foci in the somatosensory cortex in a monkey model of focal epilepsy following intracerebral administration of alumina gel (Ribak, 1985).
Studies on the human hippocampus of patients with EPI or animal models of temporal lobe EPI reported different alterations depending on the subregion studied and on the level of hippocampal sclerosis.
Wittner et al. (2005) found no change in synaptic coverage of pyramidal cells' AIS in human non-sclerotic Cornu Ammonis (CA) fields, in contrast to their somatic innervation. In sclerotic CA, these authors observed a decreased synaptic coverage of degenerating AISs, and a lack of AAC terminal staining for PV, suggesting a degeneration of AACs' cartridges. In the healthy (del Rio and DeFelipe, 1997) and mildly sclerotic CA1 region (Wittner et al., 2002), the AIS of pyramidal cells are innervated by CB-labeled boutons, which disappear in strongly sclerotic tissue, where pyramidal cells cannot be seen (Wittner et al., 2002). Similarly, in sclerotic hippocampal samples, a global loss of PV+ terminals has been described, with some PV+ cartridges remaining, of hypertrophic appearance (Arellano et al., 2004). These results collectively point toward a deficit in AAC innervation of pyramidal cells paralleling principal neuron degeneration in hippocampal sclerosis.
In the non-sclerotic Cornu Ammonis (CA), a study in rats reported that PV+ AAC boutons contacting pyramidal cells' AIS were intact and therefore resisted to kainic acid-induced seizures, in contrast to alterations in axo-somatic basket profiles (Best et al., 1994). On the contrary, in a rat model of chronic epilepsy through injection of pilocarpine, a decrease in the synaptic coverage of AIS has been observed (Dinocourt et al., 2003).
Finally, in a single physiological study using a model of interictal epileptiform discharge in CA3 slices of mice, AACs undergo increase in their firing with epileptiform discharges, suggesting an active role of AACs in generating epileptiform activity (Karlócai et al., 2014).
In the sclerotic dentate gyrus (DG) of patients with temporal lobe EPI, the morphological “complexity” of PV+ and CB+ AACs' cartridges has been found to be increased, with more boutons and axonal rows per cartridge (Arellano et al., 2004). This increased complexity of AACs' cartridges could explain the findings of increased synaptic coverage of granule cells' AIS in epileptic patients (Wittner et al., 2001). Interestingly, the increase in AACs' cartridge complexity appeared to parallel the severity of sclerosis in the human DG (Wittner et al., 2001). The most recent work on human brain tissue found that the density of AACs' PV+ boutons is preserved in epileptic DG without sclerosis, and decreased in sclerosis (Alhourani et al., 2020). However, the density of VGAT+/GAD67+ boutons (i.e., putative axo-axonic terminals) was similar to controls in both cases (Alhourani et al., 2020), suggesting that AACs' terminals might be preserved, and PV expression decreased. In addition, GAD67 and VGAT expression levels were increased in AACs' terminals in sclerotic tissue, suggestive of a compensatory mechanism. Finally, axo-axonic boutons were found intact in DG in EPI modeled with either perforant-path electric stimulation or kainic acid administration to induce seizures in rats (Sloviter et al., 2003).
Overall, these findings indicate that the DG and the CA region follow opposite directions in hippocampal sclerosis, with reduced AIS synaptic coverage in CA, and preserved innervation by more complex cartridges of surviving DG granule cells. On the other hand, AACs appear to be preserved in the non-sclerotic hippocampus. A general finding in human studies is the reduced level of PV expression in histologically abnormal tissue.
Thirteen papers were included (Figure 2), which reported molecular and functional alterations in AACs associated with SCZ (Table 2). Eight of them were post-mortem brain tissue analysis using immuno-histochemistry. The remaining five used animal models of SCZ.
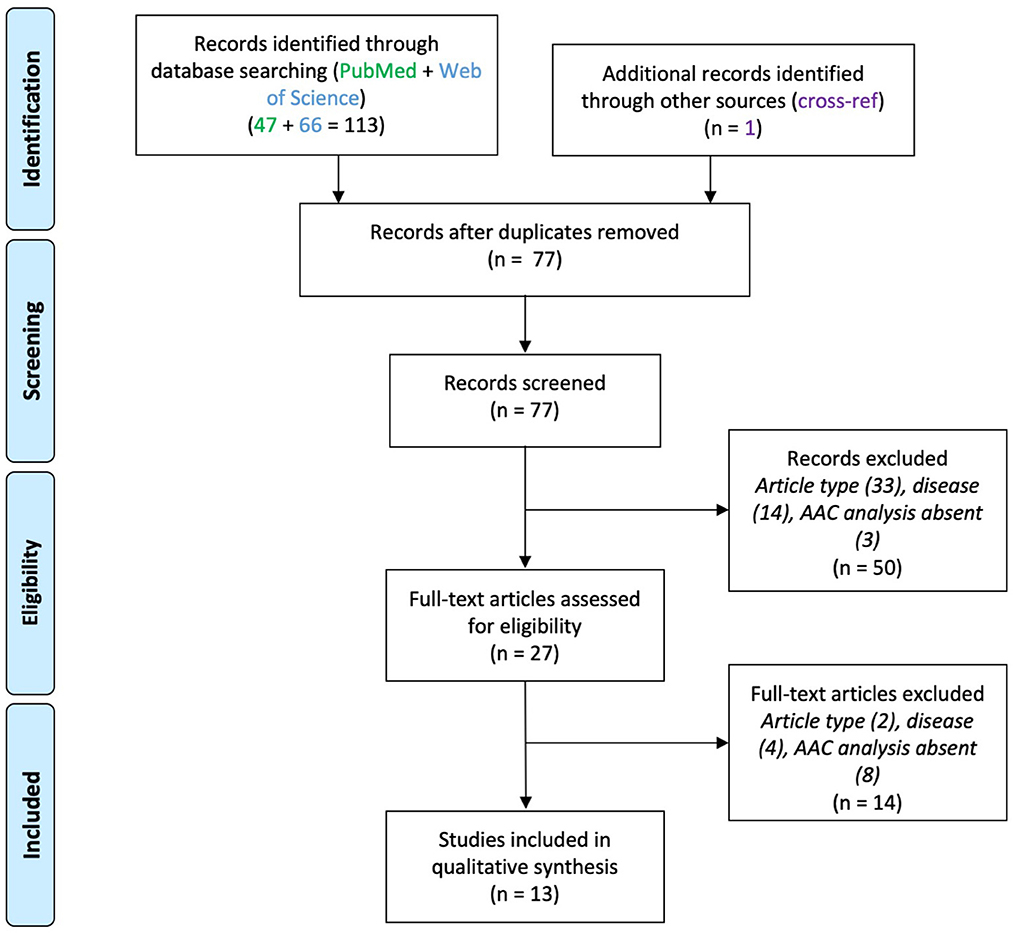
Figure 2. PRISMA flowchart for studies on axo-axonic cells in schizophrenia.
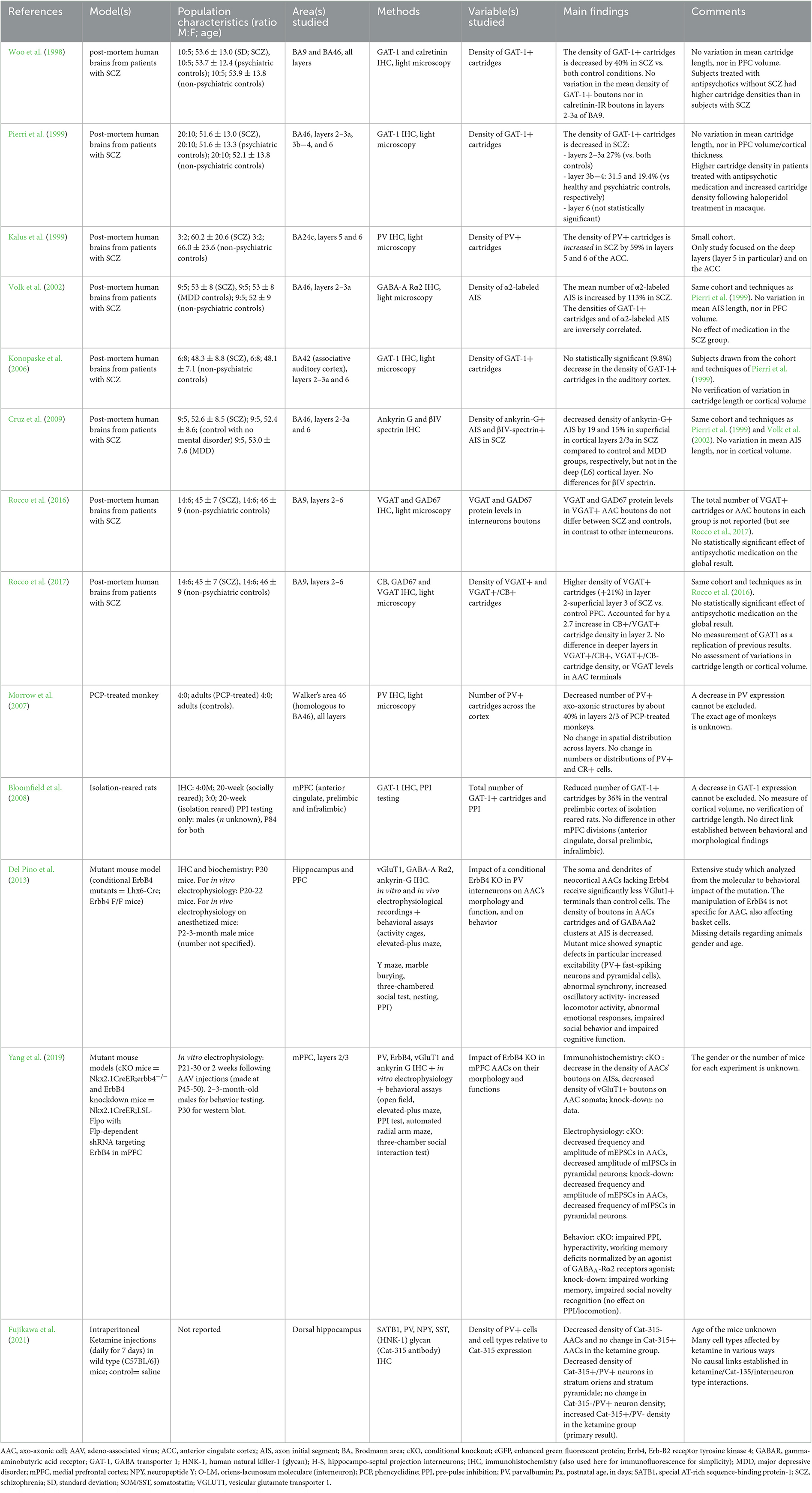
Table 2. Summary of studies on axo-axonic cells in schizophrenia.
Except for one study (Konopaske et al., 2006), all post-mortem studies were performed on the prefrontal cortex (PFC), which activity is altered in SCZ (Minzenberg et al., 2009). All reported pre- and/or post-synaptic alterations of AACs to pyramidal cell connections, albeit with conflicting conclusions.
The number of axo-axonic boutons and cartridges has been reported to be altered to various degrees in SCZ. The first article of this kind reported a 40% decrease in the density of GAT-1+ AACs' cartridges in SCZ compared to controls (Woo et al., 1998). This decrease later appeared to concern mainly layers 2–3 of the PFC in patients with SCZ, compared to controls (Pierri et al., 1999). It was then interpreted as reflecting either a decrease in cartridge density or reduced levels of GAT-1. Interestingly, this alteration displayed some degree of specificity for the PFC, since using the same approach, no significant decrease in GAT-1+ AACs' cartridges was found in the auditory cortex (Konopaske et al., 2006), and an increased density of PV+ AACs' cartridges was reported in layer 5/6 of the anterior cingulate cortex of patients with SCZ (Kalus et al., 1999). In contrast to bouton density, the expression levels of presynaptic proteins including GAD67 or VGAT were found to be preserved in SCZ subjects (Rocco et al., 2016).
In fact, the group that reported reduced PFC axo-axonic innervation in SCZ (Woo et al., 1998; Pierri et al., 1999) recently found that the number of axo-axonic boutons was unaltered in SCZ, using VGAT (instead of GAT-1) as a presynaptic marker (Rocco et al., 2017). Moreover, the density of AACs' CB+ cartridges is increased in layer 2 of PFC (Rocco et al., 2017), with no change in CB-negative cartridges density or VGAT expression levels in SCZ. Therefore, the suspected defect in AACs' cartridges (Woo et al., 1998; Pierri et al., 1999) likely reflected a decreased expression of GAT-1. Importantly, these findings did not appear to be a consequence of gender status, nicotine use, benzodiazepines and/or sodium valproate, antidepressants, or antipsychotics when accounted for in statistical analyses (Rocco et al., 2016, 2017).
The density of AISs expressing the GABAA receptor α2 subunit (GABAA-Rα2) is increased in PFC from patients with SCZ, and negatively correlated with the numbers of GAT-1+ AACs' cartridges (Volk et al., 2002). This result was interpreted as a compensatory mechanism, with increased GABAA-R α2 expression balancing a reduced number of GABAergic terminals- and in turn a likely reduced release of GABA. However, as Lewis and collaborators later showed, the number of GABAergic terminals was preserved (Rocco et al., 2017). Finally, a decrease in the density of Ankyrin-G+ AISs in the absence of β-spectrin+ (another AIS scaffold) has been reported in the PFC of patients with SCZ (Cruz et al., 2009). This result suggests a decreased expression of Ankyrin-G at AISs in SCZ, and potential defects in function since Ankyrin-G is a key scaffold protein for axo-axonic synapses at the AIS (Tai et al., 2019). Overall, evidence is limited regarding the involvement of AACs' postsynaptic changes in SCZ.
Studies on animal models of SCZ have provided evidence for structural alterations in AACs and for links between structural findings and functional or behavioral deficits.
Regarding structural studies, a monkey model of SCZ generated with repeated treatments with phencyclidine displayed a 40% decrease in the number of prefrontal PV+ axo-axonic structures (Morrow et al., 2007). In the isolation-reared rat model of SCZ, no global differences in the total number of GAT-1+ cartridges were found in the rat PFC, but a local decrease by 36% was identified in the ventral prelimbic region relative to control (Bloomfield et al., 2008). Lastly, in a ketamine model of SCZ in mice, interneurons were studied with regards to the presence of perineuronal nets detected with the marker Cat-315; (Fujikawa et al., 2021). The number of Cat-315–/PV+ AACs (identified with PV expression in the absence of neuropeptide Y or SATB1 expression) were fewer in ketamine-treated mice compared to controls, while Cat-315+/PV+ AACs were not affected (Fujikawa et al., 2021).
More direct evidence of AAC implication in SCZ has been provided by the conditional KO of ERBB4 (a SCZ risk-gene) in medial ganglionic eminence-derived interneurons (comprising PV+ cells) in mice (Del Pino et al., 2013). Among synaptic defects, Del Pino et al. found a decreased density of PV+ axo-axonic boutons and a decreased density of GABAA-R α2 clusters at the AIS contrary to human post-mortem results of Volk et al. (2002). AACs also received fewer excitatory synapses, as measured anatomically and physiologically with reduced miniature excitatory postsynaptic currents frequencies in vitro. These defects were accompanied by global increase in excitability, both of PV+ interneurons and of pyramidal cells in vitro. In vivo, this resulted in more frequent population spike occurrence in the DG, increased hippocampal oscillation power and decreased synchrony between brain regions (Del Pino et al., 2013). The latter oscillatory pattern is reminiscent of abnormalities found in SCZ (Hunt et al., 2017). Furthermore, mutant mice showed behavioral impairments consisting of increased locomotor activity, reduced anxiety, impaired working memory and reduced social interactions (Del Pino et al., 2013). However, the behavioral analysis lacked specificity for AACs. With the implementation of a novel mouse line to target selectively AACs by temporal tagging of Nkx2.1-expressing progenitors (Nkx2.1CreER; He et al., 2016), these results were confirmed by performing KO of ERBB4 selectively in AACs (Yang et al., 2019). The density of AACs was preserved, while the density of their synaptic boutons contacting the AIS was decreased. AACs also received less glutamatergic innervation (Yang et al., 2019), in agreement with previous findings on MGE-derived interneurons (Del Pino et al., 2013). Consistently, the frequency of miniature inhibitory postsynaptic currents was reduced in pyramidal neurons. Behaviorally, mice displayed increased locomotor activity, reduced paired-pulse inhibition, impaired working memory and altered social behavior (Yang et al., 2019), in line with the previous study (Del Pino et al., 2013). Behavioral deficits were rescued by systemic or intra-PFC infusion of the partial GABA-A Rα2 agonist L-838417 (Yang et al., 2019). These results were recapitulated in the Nkx2.1CreER conditional knock-down of ERBB4 in AACs restricted to the PFC, suggesting the results were not the consequence of developmental compensatory mechanisms, and that prefrontal AACs might be implicated in schizophrenia (Yang et al., 2019).
Overall, these results are consistent in showing that pyramidal neurons innervation by AACs is reduced in animal models of SCZ, and that such alterations might directly underlie pathological phenotypes via reduced functional inhibition at the AIS and perturbed brain rhythms.
Three articles were found which reported on AACs in ASD (Figure 3), studying post-mortem tissue from PFC (Table 3).
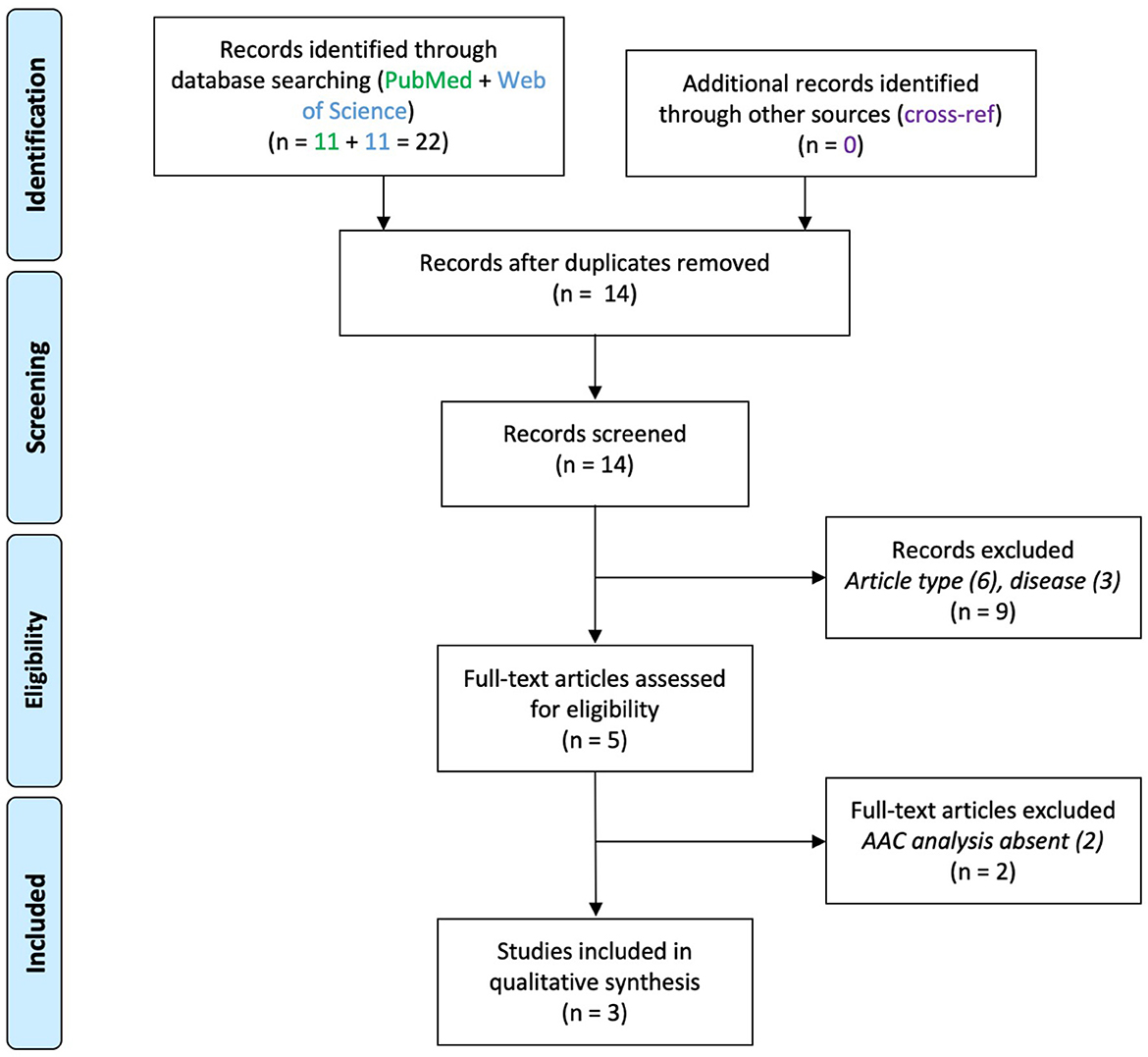
Figure 3. PRISMA flowchart for studies on axo-axonic cells in autism spectrum disorder.
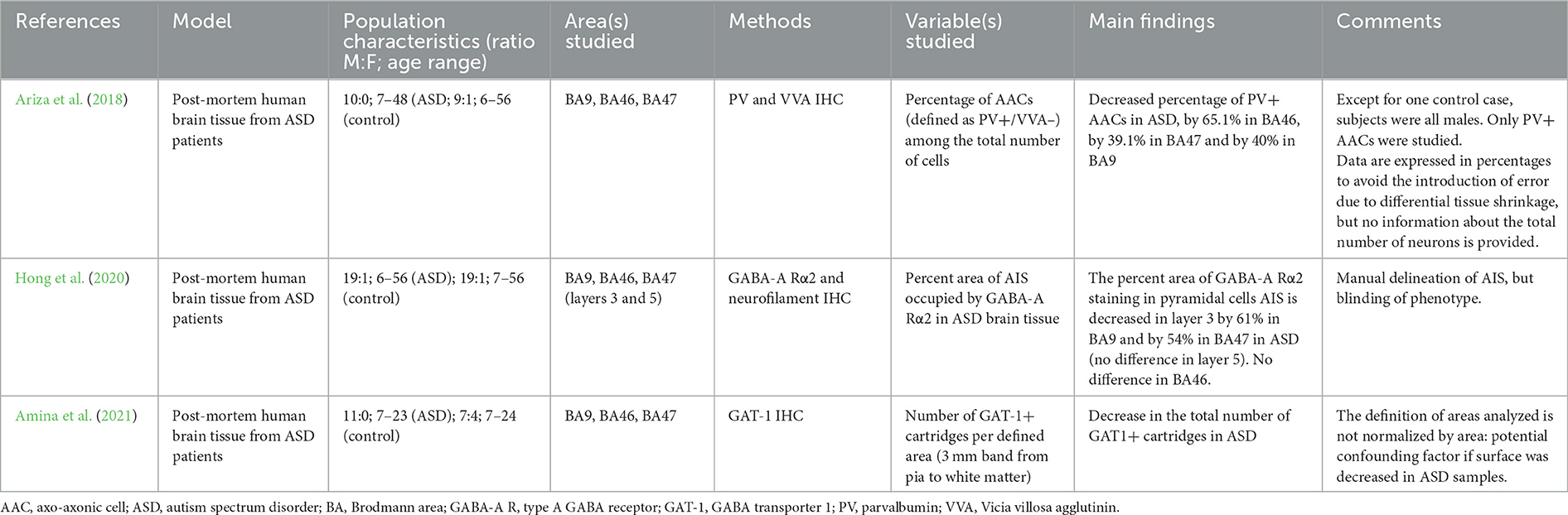
Table 3. Summary of studies on axo-axonic cells in autism spectrum disorders.
To distinguish between PV+ AACs and PV+ basket cells, Ariza and colleagues used double immunohistochemical labeling of PV and Vicia Villosa lectin and the PV+/VVA– phenotype as a proxy for AACs (Ariza et al., 2018). They found a 40–65% decrease in the absolute number of PV+ AACs while the number of PV+ basket cells was not significantly reduced (Ariza et al., 2018). The absolute number of GAT-1+ cartridges was found to be decreased by 40–60% across prefrontal areas by the same group (Amina et al., 2021). This result may support the hypothesis that prefrontal AACs' density is reduced in ASD as opposed to PV or GAT-1 immunoreactivity.
Post-synaptic alterations were also reported for AACs, with a reduction in the AIS surface covered by GABAA-Rα2 subunits in upper cortical layers of samples from patients with ASD (Hong et al., 2020). This result suggests that presynaptic loss of AACs is not compensated for by an increase in postsynaptic expression of GABAA-Rs in ASD.
The aim of this work was to provide a systematic review of AACs' alterations in neuropsychiatric disorders. Namely, articles on AACs in EPI, SCZ and ASD were studied. Review of the 32 articles relevant to this topic point to alterations in AAC to AIS synapses in brain disorders (summarized in Figure 4). Previous articles have reviewed AACs' function, and have debated their implication in disease, including recent publications (Wang et al., 2016; Gallo et al., 2020; Juarez and Martinez Cerdeno, 2022; Jung et al., 2022). However, to the best of our knowledge, none of the previous reviews has followed a systematic procedure, precluding a thorough analysis of AAC dysfunction in disease. Our review article thus provides a comprehensive summary of AACs in three main neuropsychiatric disorders, in a systematic format, for the first time. Below, we briefly summarize and articulate the main findings, before discussing their limitations and suggesting some perspectives.
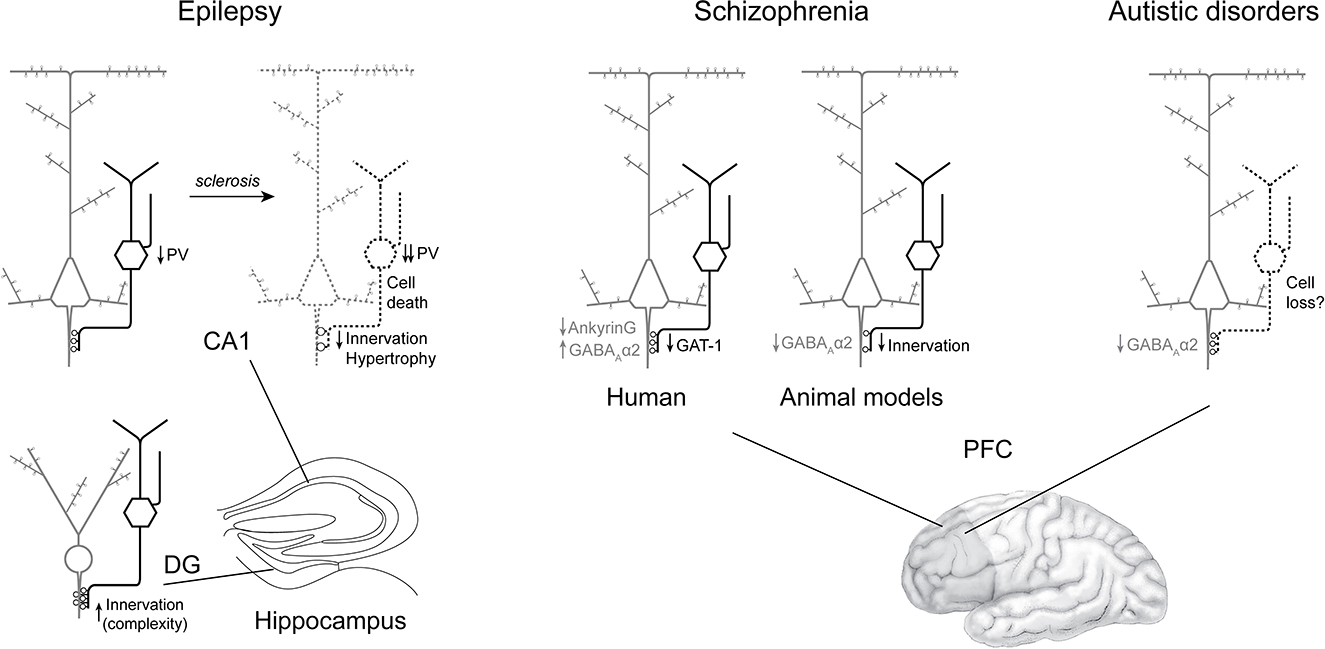
Figure 4. Summary of axo-axonic cells alterations consistently found in epilepsy, schizophrenia and autism spectrum disorder. In epilepsy (left) without sclerosis, AACs are spared in both CA1 (with reduced PV expression) and dentate gyrus (with findings of increased axo-axonic cartridge complexity). In the sclerotic CA1, PV expression, axo-axonic cell density and axo-axonic innervation appear altered, paralleling pyramidal cell loss. In brain samples from patients with schizophrenia (middle), the density of AACs seems preserved, while in their synaptic boutons, GAT-1 is downregulated. At the postsynaptic level, there are more GABAA-Rα2 clusters. In contrast, in animal models, axo-axonic innervation is reduced along with GABAA-Rα2 cluster density. In ASD (right), preliminary evidence point toward a reduced density of GABAA-Rα2 clusters, and a possible AAC loss.
Overall the systematic review of AACs' implication in EPI, SCZ, and ASD revealed some consistency, but also many contradictory findings, especially in human tissue.
In EPI, there appears to be region-specific alterations. In hippocampus, defects vary depending on the region, and with the amount of sclerosis severity. A consistent finding is reported in the sclerotic CA, with PV+ and CB+ AACs' cartridges displaying decreased density and PV expression levels, while no clear phenotype emerged from studies in non-sclerotic tissue. On the contrary, in the epileptic DG, AACs seem to be preserved, and display increased arborization complexity paralleling sclerosis. In contrast, an earlier study reported a qualitative decrease of GAT-1 immunoreactivity in the DG, including at the border between the granule cell layer and the subgranular hilus, where AISs are located (Sayin et al., 2003). Surgical samples have been reported to be heterogeneous within and across subjects of individual studies (e.g., Arellano et al., 2004), reflecting biological variability, rather than differences in techniques applied for fixation or tissue processing. Thus, it is likely that the small sample sizes and heterogeneity in the subjects analyzed may partly account for the general lack of consistency in the studies reviewed here. The loss of AACs in the sclerotic tissue might contribute to pathological activities by loss of inhibition, or by GABAergic excitation in this condition (e.g., Hristova et al., 2021, but see Krook-Magnuson et al., 2013), but could also merely reflect a global cell loss, proportional to pyramidal cell death. Intriguingly, alterations in AACs did not mirror epileptiform activities, when the latter were recorded in patients before surgical removal of the samples subsequently analyzed (DeFelipe et al., 1993; Marco et al., 1997). This suggests that AACs might not play an active role in epileptogenesis.
In SCZ, initial studies pointed to the loss of AACs' cartridges in human tissue. However, complementary analyses revealed that AACs' terminals were preserved, indicating that initial findings reflected the inability to detect molecular markers in axo-axonic boutons because of low expression levels (Rocco et al., 2017). In contrast, the density of CB-expressing cartridges may be increased (Rocco et al., 2017) in superficial layers of the PFC in SCZ, the meaning of which is uncertain. Expression of postsynaptic GABAA-Rα2, on the other hand, may be increased, the causes and consequences of which also remain to be established. Paradoxically, in animal models, consistent reductions of AIS innervation by AACs were found, along with decreased GABAA-Rα2 expression postsynaptically and decreased glutamatergic innervation presynaptically (Del Pino et al., 2013; Wang et al., 2016). Notably, one of the hypothesis on the onset of schizophrenia is a decrease of NMDA transmission, which implicate PV+ interneurons (e.g., Gawande et al., 2023), and perhaps AACs. In the animal model of ERBB4 KO in AACs, cognitive alterations were improved by a GABAA-Rα2 agonist (Yang et al., 2019). Interestingly, in an open label trial, the GABAA-Rα2-3 selective agonist MK-0777 improved cognitive function in patients with SCZ (Lewis et al., 2008). However, a subsequent randomized controlled trial failed to demonstrate any benefit (Buchanan et al., 2011). There are many potential explanations to these discrepancies between human and animal studies. For instance, the animal models employed by these authors relied on monogenetic alterations that may not reflect the full and complex spectrum of polygenic alterations found in schizophrenia (Trubetskoy et al., 2022). To date, it remains unclear how AACs are altered in SCZ, and what could be the direct contribution of these alterations to clinical symptoms.
Research on AACs' implication in ASD is still recent, and only three articles were found in the inclusion period, reporting a reduced number of AAC somata and cartridges (Ariza et al., 2018; Amina et al., 2021) and a decrease in postsynaptic GABAA-Rα2 at the AIS in the PFC of subjects with ASD (Hong et al., 2020). The indirect detection of AACs and cartridges using molecular markers as proxies calls for caution, as exemplified by the case of SCZ (and see below). The decreased density of GABAA-Rα2 at the AIS (Hong et al., 2020) is also in contrast with findings in SCZ (Volk et al., 2002), with unknown functional consequences. Despite the paucity of the literature, these initial results suggest a potential implication of the axo-axonic synapse in ASD pathophysiology.
It is intriguing that EPI, SCZ and ASD implicate overlapping brain regions, which contain relatively high densities of AACs (Inda et al., 2009). Those disorders are linked to neurodevelopmental abnormalities, while AACs have a peculiar, late developmental pattern compared with other interneurons (Taniguchi et al., 2013). Moreover, AACs undergo structural and functional maturation until young adulthood (Rinetti-Vargas et al., 2017; Steinecke et al., 2017), a period at risk of SCZ or EPI occurrence. Comorbid EPI and psychotic disorders or ASD, are highly prevalent in clinical populations (Clancy et al., 2014; Tye et al., 2018). As a result, one may speculate that common pathophysiological principles may be at stake and implicate AACs. To test this hypothesis, it would be interesting to investigate alterations of AACs in brain samples from subjects with comorbid disorders. Moreover, animal studies suggest that AACs might be implicated in other neurodevelopmental disorders. Prenatal exposure to cocaine results in decreased density of AAC cartridges in the PFC (Morrow et al., 2003). In a mouse model of Down syndrome, AACs displayed aberrant innervation of AIS with increased number of cartridges, cartridge length, bouton size and amount of GABAergic inhibition in electrophysiological recording (Liu et al., 2023). Human counterparts of these findings are currently lacking. In summary, AACs may bear common and direct pathogenic factors across EPI, SCZ and ASD, but the focus of the review on this topic may also artificially inflate this hypothesis.
The first main limitation is the result of indirect strategies to study AACs in human brain samples. Most of the histological methods applied to human tissue have so far relied on molecular markers, be it to infer the density of AACs to pyramidal cells synapses, or to evaluate the density of AACs. Clear limitations of this strategy are highlighted in the following examples. First, in SCZ, the reduction of GAT-1+ cartridges later appeared to reflect a reduction in protein levels rather than the loss of AACs (Rocco et al., 2017). Second, PV+ cartridges have been widely used as a proxy for axo-axonic synapses in EPI studies. However, the same studies consistently reported a global reduction of PV immunoreactivity in epileptic tissue (Wittner et al., 2001; Arellano et al., 2004), which might have contributed to the observation of reduced AAC cartridges density. Moreover, a significant number of AACs may be devoid of PV in the healthy brain (Taniguchi et al., 2013; Tasic et al., 2018). The case of increased CB+ axo-axonic cartridges density in SCZ (Rocco et al., 2017) or in EPI (Arellano et al., 2004) is intriguing in this context. Third, in an ASD study, the absence of perineuronal nets around PV+ cells was used to identify AACs (Ariza et al., 2018), while perineuronal nets have been extensively found around hippocampal AACs (Fujikawa et al., 2021). Alterations in molecular content of AACs may indeed be the consequence of compensatory or even confounding factors, including pharmacological treatments. In addition, most Human studies were published by one or two research groups in each disorder, calling for replication studies.
The second main limitation in knowledge is the lack of experimental evidence to causally link structure and function. Indeed, most of the articles reviewed here were reports of histopathological studies in Human, which are, by essence, correlative and cannot provide mechanisms. Animal models are instrumental to understand pathogenesis and suggest causal links between cellular alterations and symptoms. However, only very few studies were found on this topic, and data is still lacking at all levels: cells (synaptic transmission), network (assemblies, oscillations), and behavior. We have highlighted three important functional studies (Konopaske et al., 2006; Del Pino et al., 2013; Yang et al., 2019), also showing discrepancies in histological findings with human data. Animal models will thus need to be cross validated with Human data in order to improve translation. One interesting pathway is the reduced density of GABAA-Rα2 at the AIS, as suggested by animal studies in SCZ (Del Pino et al., 2013, but see Volk et al., 2002) and clinical samples in ASD (Hong et al., 2020). In addition, reduced levels of GABAA-Rα2 at the AIS (possibly from AACs) induce greater seizure susceptibility in mice (Hines et al., 2018). GABAA-Rs containing the α2 subunit show fast activation and slow deactivation kinetics (Lavoie et al., 1997). Therefore, the loss of this postsynaptic partner of AACs might induce a strong hyperexcitability in principal neurons. Until recently, AACs studies have been limited by the lack of specific genetic markers. Using single-cell RNA sequencing, such markers have been identified, including PTHLH, Uncb5 and Vipr2 (Paul et al., 2017; Tasic et al., 2018). As a result of these discoveries, genetic targeting of cortical AACs is now accessible in animal models using Nkx2.1CreER (He et al., 2016) or Vipr2-Cre mouse lines (Tasic et al., 2018) for the neocortex, or using Unc5b-2A-CreER mice for the hippocampus (Dudok et al., 2021). It is beyond the scope of this work to extensively review this, but recent work have started to unravel AACs functions in physiological conditions using these transgenic animals (e.g., Lu et al., 2017; Dudok et al., 2021; Schneider-Mizell et al., 2021; Jung et al., 2023; Seignette et al., 2023). These showed that AACs only weakly inhibited principal neurons' firing, suggesting that alterations of AACs in disease might not result in gross excitation/inhibition imbalance. In fact, AACs seem to ensure excitation homeostasis in cortical networks (Pan-Vazquez et al., 2020), a loss of which may have dramatic consequences on cortical function (e.g., overexcitation, increased local synchrony and reduced interregional coordination).
Direct examination of AACs is warranted, and may rely on complementary approaches. Detailed reconstructions of AACs at the cellular and ultrastructural levels following Golgi staining or semi-automated electron microcopy (e.g., Schneider-Mizell et al., 2021) would provide invaluable information. The study by Wittner et al. (2005) is noteworthy as being the only one in this review that systematically examined AIS innervation at the electron microscopic level regardless of PV content. Recently identified molecular markers of AACs will also prove useful in identifying AACs in human tissue (e.g., Alhourani et al., 2020). Moreover, targeting AACs with novel transgenic mouse lines represent an opportunity to directly evaluate whether a genetic mutation implicates AACs in generating a pathological phenotype (as in Yang et al., 2019). Targeting AACs for recording and opto- or chemo-genetic manipulations could also be implemented to study their implications in disease models constructed with non-genetic interventions (e.g., pharmacological, lesional). These experiments are expected to provide key evidence to causally link axo-axonic cells to brain disorders.
Although observations made in brain tissue from human subjects and in animal models indicate that axo-axonic cells may be altered in epilepsy, schizophrenia, and autism spectrum disorders, the literature is limited and mainly inconsistent, precluding clear conclusions to be drawn.
By joining clinical research and refined preclinical models, we hope that the coming years will bring significant clarification and progress in the field of AACs in disease.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
JV and AE: literature search and screening. JV, AE, and TB: article selection. JV, AE, CL, and TB: literature synthesis. JV, AE, CL, FL, BA, CH, YH, and TB: manuscript writing and revisions. TB: supervision of the project. All authors contributed to the article and approved the submitted version.
This work was funded by Fondation Bettencourt Schueller with a CCA Inserm-Bettencourt grant to TB (R20005GS). AE and CL benefited from the help of the Bordeaux Neurocampus Graduate Program, managed by the French National Research Agency reference ANR-17-EURE-0028.
We would like to dedicate this article to our mentor, Dr. Marco Capogna, who tragically passed away during the preparation of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2023.1212202/full#supplementary-material
Alhourani, A., Fish, K. N., Wozny, T. A., Sudhakar, V., Hamilton, R. L., and Richardson, R. M. (2020). GABA bouton subpopulations in the human dentate gyrus are differentially altered in mesial temporal lobe epilepsy. J. Neurophysiol. 123, 392–406. doi: 10.1152/jn.00523.2018
Alonso-Nanclares, L., Garbelli, R., Sola, R. G., Pastor, J., Tassi, L., Spreafico, R., et al. (2005). Microanatomy of the dysplastic neocortex from epileptic patients. Brain 128, 158–173. doi: 10.1093/brain/awh331
Amina, S., Falcone, C., Hong, T., Wolf-Ochoa, M. W., Vakilzadeh, G., Allen, E., et al. (2021). Chandelier cartridge density is reduced in the prefrontal cortex in autism. Cereb. Cortex 31, 2944–2951. doi: 10.1093/cercor/bhaa402
Arellano, J. I., Munoz, A., Ballesteros-Yanez, I., Sola, R. G., and DeFelipe, J. (2004). Histopathology and reorganization of chandelier cells in the human epileptic sclerotic hippocampus. Brain 127, 45–64. doi: 10.1093/brain/awh004
Ariza, J., Rogers, H., Hashemi, E., Noctor, S. C., and Martinez-Cerdeno, V. (2018). The number of chandelier and basket cells are differentially decreased in prefrontal cortex in autism. Cereb. Cortex 28, 411–420. doi: 10.1093/cercor/bhw349
Best, N., Mitchell, J., and Wheal, H. V. (1994). Ultrastructure of parvalbumin-immunoreactive neurons in the CA1 area of the rat hippocampus following a kainic acid injection. Acta Neuropathol. 87, 187–195. doi: 10.1007/BF00296189
Bloomfield, C., French, S. J., Jones, D. N., Reavill, C., Southam, E., Cilia, J., et al. (2008). Chandelier cartridges in the prefrontal cortex are reduced in isolation reared rats. Synapse 62, 628–631. doi: 10.1002/syn.20521
Buchanan, R. W., Keefe, R. S., Lieberman, J. A., Barch, D. M., Csernansky, J. G., Goff, D. C., et al. (2011). A randomized clinical trial of MK-0777 for the treatment of cognitive impairments in people with schizophrenia. Biol. Psychiatry 69, 442–449. doi: 10.1016/j.biopsych.2010.09.052
Buzsaki, G., and Draguhn, A. (2004). Neuronal oscillations in cortical networks. Science 304, 1926–1929. doi: 10.1126/science.1099745
Cardin, J. A., Carlen, M., Meletis, K., Knoblich, U., Zhang, F., Deisseroth, K., et al. (2009). Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 459, 663–667. doi: 10.1038/nature08002
Clancy, M. J., Clarke, M. C., Connor, D. J., Cannon, M., and Cotter, D. R. (2014). The prevalence of psychosis in epilepsy; a systematic review and meta-analysis. BMC Psychiatry 14, 75. doi: 10.1186/1471-244X-14-75
Cruz, D. A., Weaver, C. L., Lovallo, E. M., Melchitzky, D. S., and Lewis, D. A. (2009). Selective alterations in postsynaptic markers of chandelier cell inputs to cortical pyramidal neurons in subjects with schizophrenia. Neuropsychopharmacology 34, 2112–2124. doi: 10.1038/npp.2009.36
DeFelipe, J., Garcia Sola, R., Marco, P., del Rio, M. R., Pulido, P., and Ramon y Cajal, S. (1993). Selective changes in the microorganization of the human epileptogenic neocortex revealed by parvalbumin immunoreactivity. Cereb. Cortex 3, 39–48. doi: 10.1093/cercor/3.1.39
Del Pino, I., Garcia-Frigola, C., Dehorter, N., Brotons-Mas, J. R., Alvarez-Salvado, E., Martinez de Lagran, M., et al. (2013). Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron 79, 1152–1168. doi: 10.1016/j.neuron.2013.07.010
del Rio, M. R., and DeFelipe, J. (1997). Colocalization of parvalbumin and calbindin D-28k in neurons including chandelier cells of the human temporal neocortex. J. Chem. Neuroanat. 12, 165–173. doi: 10.1016/S0891-0618(96)00191-3
Dinocourt, C., Petanjek, Z., Freund, T. F., Ben-Ari, Y., and Esclapez, M. (2003). Loss of interneurons innervating pyramidal cell dendrites and axon initial segments in the CA1 region of the hippocampus following pilocarpine-induced seizures. J. Comp. Neurol. 459, 407–425. doi: 10.1002/cne.10622
Dudok, B., Szoboszlay, M., Paul, A., Klein, P. M., Liao, Z., Hwaun, E., et al. (2021). Recruitment and inhibitory action of hippocampal axo-axonic cells during behavior. Neuron 109, 3838–3850 e3838. doi: 10.1016/j.neuron.2021.09.033
Ferrer, I., Oliver, B., Russi, A., Casas, R., and Rivera, R. (1994). Parvalbumin and calbindin-D28k immunocytochemistry in human neocortical epileptic foci. J. Neurol. Sci. 123, 18–25. doi: 10.1016/0022-510X(94)90198-8
Ferrer, I., Pineda, M., Tallada, M., Oliver, B., Russi, A., Oller, L., et al. (1992). Abnormal local-circuit neurons in epilepsia partialis continua associated with focal cortical dysplasia. Acta Neuropathol. 83, 647–652. doi: 10.1007/BF00299415
Fujikawa, R., Yamada, J., and Jinno, S. (2021). Subclass imbalance of parvalbumin-expressing GABAergic neurons in the hippocampus of a mouse ketamine model for schizophrenia, with reference to perineuronal nets. Schizophr. Res. 229, 80–93. doi: 10.1016/j.schres.2020.11.016
Gallo, N. B., Paul, A., and Van Aelst, L. (2020). Shedding light on chandelier cell development, connectivity, and contribution to neural disorders. Trends Neurosci. 43, 565–580. doi: 10.1016/j.tins.2020.05.003
Gawande, D. Y., Narasimhan, K. K, S., Shelkar, G. P., Pavuluri, R., Stessman, H. A. F., and Dravid, S. M. (2023). GluN2D subunit in parvalbumin interneurons regulates prefrontal cortex feedforward inhibitory circuit and molecular networks relevant to schizophrenia. Biol. Psychiatry. doi: 10.1016/j.biopsych.2023.03.020. [Epub ahead of print].
He, M., Tucciarone, J., Lee, S., Nigro, M. J., Kim, Y., Levine, J. M., et al. (2016). Strategies and tools for combinatorial targeting of GABAergic neurons in mouse cerebral cortex. Neuron 92, 555. doi: 10.1016/j.neuron.2016.08.021
Hines, R. M., Maric, H. M., Hines, D. J., Modgil, A., Panzanelli, P., Nakamura, Y., et al. (2018). Developmental seizures and mortality result from reducing GABA(A) receptor alpha2-subunit interaction with collybistin. Nat. Commun. 9, 3130. doi: 10.1038/s41467-018-05481-1
Hong, T., Falcone, C., Dufour, B., Amina, S., Castro, R. P., Regalado, J., et al. (2020). GABA(A)Ralpha2 is decreased in the axon initial segment of pyramidal cells in specific areas of the prefrontal cortex in autism. Neuroscience 437, 76–86. doi: 10.1016/j.neuroscience.2020.04.025
Hristova, K., Martinez-Gonzalez, C., Watson, T. C., Codadu, N. K., Hashemi, K., Kind, P. C., et al. (2021). Medial septal GABAergic neurons reduce seizure duration upon optogenetic closed-loop stimulation. Brain 144, 1576–1589. doi: 10.1093/brain/awab042
Hunt, M. J., Kopell, N. J., Traub, R. D., and Whittington, M. A. (2017). Aberrant network activity in schizophrenia. Trends Neurosci. 40, 371–382. doi: 10.1016/j.tins.2017.04.003
Inda, M. C., DeFelipe, J., and Munoz, A. (2009). Morphology and distribution of chandelier cell axon terminals in the mouse cerebral cortex and claustroamygdaloid complex. Cereb. Cortex 19, 41–54. doi: 10.1093/cercor/bhn057
Juarez, P., and Martinez Cerdeno, V. (2022). Parvalbumin and parvalbumin chandelier interneurons in autism and other psychiatric disorders. Front. Psychiatry 13, 913550. doi: 10.3389/fpsyt.2022.913550
Jung, K., Chang, M., Steinecke, A., Berke, B., Choi, Y., Oisi, Y., et al (2023). An adaptive behavioral control motif mediated by cortical axo-axonic inhibition. Biorxiv. doi: 10.1101/2023.03.10.531767. Available online at: https://www.biorxiv.org/about/FAQ
Jung, K., Choi, Y., and Kwon, H. B. (2022). Cortical control of chandelier cells in neural codes. Front. Cell. Neurosci. 16, 992409. doi: 10.3389/fncel.2022.992409
Kalus, P., Senitz, D., Lauer, M., and Beckmann, H. (1999). Inhibitory cartridge synapses in the anterior cingulate cortex of schizophrenics. J. Neural Transm. 106, 763–771. doi: 10.1007/s007020050197
Karlócai, M. R., Kohus, Z., Kali, S., Ulbert, I., Szabo, G., Mate, Z., et al. (2014). Physiological sharp wave-ripples and interictal events in vitro: what's the difference? Brain 137, 463–485. doi: 10.1093/brain/awt348
Kepecs, A., and Fishell, G. (2014). Interneuron cell types are fit to function. Nature 505, 318–326. doi: 10.1038/nature12983
Kerti-Szigeti, K., and Nusser, Z. (2016). Similar GABAA receptor subunit composition in somatic and axon initial segment synapses of hippocampal pyramidal cells. Elife 5. doi: 10.7554/eLife.18426.020
Konopaske, G. T., Sweet, R. A., Wu, Q., Sampson, A., and Lewis, D. A. (2006). Regional specificity of chandelier neuron axon terminal alterations in schizophrenia. Neuroscience 138, 189–196. doi: 10.1016/j.neuroscience.2005.10.070
Krook-Magnuson, E., Armstrong, C., Oijala, M., and Soltesz, I. (2013). On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat. Commun. 4, 1376. doi: 10.1038/ncomms2376
Lavoie, A. M., Tingey, J. J., Harrison, N. L., Pritchett, D. B., and Twyman, R. E. (1997). Activation and deactivation rates of recombinant GABA(A) receptor channels are dependent on alpha-subunit isoform. Biophys. J. 73, 2518–2526. doi: 10.1016/S0006-3495(97)78280-8
Lewis, D. A., Cho, R. Y., Carter, C. S., Eklund, K., Forster, S., Kelly, M. A., et al. (2008). Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. Am. J. Psychiatry 165, 1585–1593. doi: 10.1176/appi.ajp.2008.08030395
Liu, H., Caballero-Floran, R. N., Hergenreder, T., Yang, T., Hull, J. M., Pan, G., et al. (2023). DSCAM gene triplication causes excessive GABAergic synapses in the neocortex in Down syndrome mouse models. PLoS Biol. 21, e3002078. doi: 10.1371/journal.pbio.3002078
Lu, J., Tucciarone, J., Padilla-Coreano, N., He, M., Gordon, J. A., and Huang, Z. J. (2017). Selective inhibitory control of pyramidal neuron ensembles and cortical subnetworks by chandelier cells. Nat. Neurosci. 20, 1377–1383. doi: 10.1038/nn.4624
Marco, P., Sola, R. G., Pulido, P., Alijarde, M. T., Sanchez, A., Ramon y Cajal, S., et al. (1996). Inhibitory neurons in the human epileptogenic temporal neocortex. An immunocytochemical study. Brain 119 (Pt 4), 1327–1347. doi: 10.1093/brain/119.4.1327
Marco, P., Sola, R. G., Ramon y Cajal, S., and DeFelipe, J. (1997). Loss of inhibitory synapses on the soma and axon initial segment of pyramidal cells in human epileptic peritumoural neocortex: implications for epilepsy. Brain Res. Bull. 44, 47–66. doi: 10.1016/S0361-9230(97)00090-7
Marin, O. (2012). Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 13, 107–120. doi: 10.1038/nrn3155
Minzenberg, M. J., Laird, A. R., Thelen, S., Carter, C. S., and Glahn, D. C. (2009). Meta-analysis of 41 functional neuroimaging studies of executive function in schizophrenia. Arch. Gen. Psychiatry 66, 811–822. doi: 10.1001/archgenpsychiatry.2009.91
Morrow, B. A., Elsworth, J. D., and Roth, R. H. (2003). Axo-axonic structures in the medial prefrontal cortex of the rat: reduction by prenatal exposure to cocaine. J. Neurosci. 23, 5227–5234. doi: 10.1523/JNEUROSCI.23-12-05227.2003
Morrow, B. A., Elsworth, J. D., and Roth, R. H. (2007). Repeated phencyclidine in monkeys results in loss of parvalbumin-containing axo-axonic projections in the prefrontal cortex. Psychopharmacology 192, 283–290. doi: 10.1007/s00213-007-0708-0
Nusser, Z., Sieghart, W., Benke, D., Fritschy, J. M., and Somogyi, P. (1996). Differential synaptic localization of two major gamma-aminobutyric acid type A receptor alpha subunits on hippocampal pyramidal cells. Proc. Natl. Acad. Sci. U. S. A. 93, 11939–11944. doi: 10.1073/pnas.93.21.11939
Pan-Vazquez, A., Wefelmeyer, W., Gonzalez Sabater, V., Neves, G., and Burrone, J. (2020). Activity-dependent plasticity of axo-axonic synapses at the axon initial segment. Neuron 10,6 265–276 e266. doi: 10.1016/j.neuron.2020.01.037
Paul, A., Crow, M., Raudales, R., He, M., Gillis, J., and Huang, Z. J. (2017). Transcriptional architecture of synaptic communication delineates GABAergic neuron identity. Cell 171, 522–539 e520. doi: 10.1016/j.cell.2017.08.032
Pierri, J. N., Chaudry, A. S., Woo, T. U., and Lewis, D. A. (1999). Alterations in chandelier neuron axon terminals in the prefrontal cortex of schizophrenic subjects. Am. J. Psychiatry 156, 1709–1719. doi: 10.1176/ajp.156.11.1709
Ribak, C. E. (1985). Axon terminals of GABAergic chandelier cells are lost at epileptic foci. Brain Res. 326, 251–260. doi: 10.1016/0006-8993(85)90034-4
Rinetti-Vargas, G., Phamluong, K., Ron, D., and Bender, K. J. (2017). Periadolescent maturation of GABAergic hyperpolarization at the axon initial segment. Cell Rep. 20, 21–29. doi: 10.1016/j.celrep.2017.06.030
Rocco, B. R., DeDionisio, A. M., Lewis, D. A., and Fish, K. N. (2017). Alterations in a unique class of cortical chandelier cell axon cartridges in schizophrenia. Biol. Psychiatry 82, 40–48. doi: 10.1016/j.biopsych.2016.09.018
Rocco, B. R., Lewis, D. A., and Fish, K. N. (2016). Markedly lower glutamic acid decarboxylase 67 protein levels in a subset of boutons in schizophrenia. Biol. Psychiatry 79, 1006–1015. doi: 10.1016/j.biopsych.2015.07.022
Roux, L., and Buzsaki, G. (2015). Tasks for inhibitory interneurons in intact brain circuits. Neuropharmacology 88, 10–23. doi: 10.1016/j.neuropharm.2014.09.011
Sayin, U., Osting, S., Hagen, J., Rutecki, P., and Sutula, T. (2003). Spontaneous seizures and loss of axo-axonic and axo-somatic inhibition induced by repeated brief seizures in kindled rats. J. Neurosci. 23, 2759–2768. doi: 10.1523/JNEUROSCI.23-07-02759.2003
Schneider-Mizell, C. M., Bodor, A. L., Collman, F., Brittain, D., Bleckert, A., Dorkenwald, S., et al. (2021). Structure and function of axo-axonic inhibition. Elife 10. doi: 10.7554/eLife.73783.sa2
Seignette, K., Jamann, N., Papale, P., Terra, H., Porneso, R. P. O., de Kraker, L., et al (2023). Visuomotor experience induces functional and structural plasticity of chandelier cells. Biorxiv. doi: 10.1101/2023.04.21.537780. Available online at: https://www.biorxiv.org/about/FAQ
Simon, D. M., and Wallace, M. T. (2016). Dysfunction of sensory oscillations in autism spectrum disorder. Neurosci. Biobehav. Rev. 68, 848–861. doi: 10.1016/j.neubiorev.2016.07.016
Sloviter, R. S., Zappone, C. A., Harvey, B. D., Bumanglag, A. V., Bender, R. A., and Frotscher, M. (2003). “Dormant basket cell” hypothesis revisited: relative vulnerabilities of dentate gyrus mossy cells and inhibitory interneurons after hippocampal status epilepticus in the rat. J. Comp. Neurol. 459, 44–76. doi: 10.1002/cne.10630
Sohal, V. S., and Rubenstein, J.L. R. (2019). Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 24, 1248–1257. doi: 10.1038/s41380-019-0426-0
Sohal, V. S., Zhang, F., Yizhar, O., and Deisseroth, K. (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702. doi: 10.1038/nature07991
Somogyi, P. (1977). A specific ‘axo-axonal' interneuron in the visual cortex of the rat. Brain Res. 136, 345–350. doi: 10.1016/0006-8993(77)90808-3
Steinecke, A., Hozhabri, E., Tapanes, S., Ishino, Y., Zeng, H., Kamasawa, N., et al. (2017). Neocortical chandelier cells developmentally shape axonal arbors through reorganization but establish subcellular synapse specificity without refinement. eNeuro 4. doi: 10.1523/ENEURO.0057-17.2017
Tai, Y., Gallo, N. B., Wang, M., Yu, J. R., and Van Aelst, L. (2019). Axo-axonic innervation of neocortical pyramidal neurons by GABAergic chandelier cells requires ankyrinG-associated L1CAM. Neuron 102, 358–372 e359. doi: 10.1016/j.neuron.2019.02.009
Taniguchi, H., Lu, J., and Huang, Z. J. (2013). The spatial and temporal origin of chandelier cells in mouse neocortex. Science 339, 70–74. doi: 10.1126/science.1227622
Tasic, B., Yao, Z., Graybuck, L. T., Smith, K. A., Nguyen, T. N., Bertagnolli, D., et al. (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78. doi: 10.1038/s41586-018-0654-5
Trubetskoy, V., Pardinas, A. F., Qi, T., Panagiotaropoulou, G., Awasthi, S., Bigdeli, T. B., et al. (2022). Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508. doi: 10.1038/s41586-022-04434-5
Tye, C., Runicles, A. K., Whitehouse, A. J. O., and Alvares, G. A. (2018). Characterizing the interplay between autism spectrum disorder and comorbid medical conditions: an integrative review. Front. Psychiatry 9, 751. doi: 10.3389/fpsyt.2018.00751
Volk, D. W., Pierri, J. N., Fritschy, J. M., Auh, S., Sampson, A. R., and Lewis, D. A. (2002). Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb. Cortex 12, 1063–1070. doi: 10.1093/cercor/12.10.1063
Wang, Y., Zhang, P., and Wyskiel, D. R. (2016). Chandelier cells in functional and dysfunctional neural circuits. Front. Neural Circ. 10, 33. doi: 10.3389/fncir.2016.00033
Wittner, L., Eross, L., Czirjak, S., Halasz, P., Freund, T. F., and Magloczky, Z. (2005). Surviving CA1 pyramidal cells receive intact perisomatic inhibitory input in the human epileptic hippocampus. Brain 128, 138–152. doi: 10.1093/brain/awh339
Wittner, L., Eross, L., Szabo, Z., Toth, S., Czirjak, S., Halasz, P., et al. (2002). Synaptic reorganization of calbindin-positive neurons in the human hippocampal CA1 region in temporal lobe epilepsy. Neuroscience 115, 961–978. doi: 10.1016/S0306-4522(02)00264-6
Wittner, L., Magloczky, Z., Borhegyi, Z., Halasz, P., Toth, S., Eross, L., et al. (2001). Preservation of perisomatic inhibitory input of granule cells in the epileptic human dentate gyrus. Neuroscience 108, 587–600. doi: 10.1016/S0306-4522(01)00446-8
Woo, T. U., Whitehead, R. E., Melchitzky, D. S., and Lewis, D. A. (1998). A subclass of prefrontal gamma-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc. Natl. Acad. Sci. U. S. A. 95, 5341–5346. doi: 10.1073/pnas.95.9.5341
Keywords: interneuron, chandelier cell, epilepsy, schizophrenia, autism
Citation: Vivien J, El Azraoui A, Lheraux C, Lanore F, Aouizerate B, Herry C, Humeau Y and Bienvenu TCM (2023) Axo-axonic cells in neuropsychiatric disorders: a systematic review. Front. Cell. Neurosci. 17:1212202. doi: 10.3389/fncel.2023.1212202
Received: 25 April 2023; Accepted: 09 June 2023;
Published: 26 June 2023.
Edited by:
Dominique Debanne, INSERM U1072 Neurobiologie des canaux Ioniques et de la Synapse, FranceReviewed by:
Rocco Pizzarelli, European Brain Research Institute, ItalyCopyright © 2023 Vivien, El Azraoui, Lheraux, Lanore, Aouizerate, Herry, Humeau and Bienvenu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas C. M. Bienvenu, dGhvbWFzLmJpZW52ZW51QHUtYm9yZGVhdXguZnI=
‡ORCID: Frederic Lanore orcid.org/0000-0003-4316-6855
Cyril Herry orcid.org/0000-0003-2973-0843
Yann Humeau orcid.org/0000-0003-0471-3414
Thomas C. M. Bienvenu orcid.org/0000-0002-7416-0504
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.