 Shuang Zou1†
Shuang Zou1† Yu-Long Lan
Yu-Long Lan Yiwei Gong
Yiwei Gong Zhong Chen
Zhong Chen Cenglin Xu
Cenglin Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 13 February 2023
Sec. Non-Neuronal Cells
Volume 17 - 2023 | https://doi.org/10.3389/fncel.2023.1143956
The ATP1A3 gene, which encodes the Na+/K+-ATPase α3 catalytic subunit, plays a crucial role in both physiological and pathological conditions in the brain, and mutations in this gene have been associated with a wide variety of neurological diseases by impacting the whole infant development stages. Cumulative clinical evidence suggests that some severe epileptic syndromes have been linked to mutations in ATP1A3, among which inactivating mutation of ATP1A3 has been intriguingly found to be a candidate pathogenesis for complex partial and generalized seizures, proposing ATP1A3 regulators as putative targets for the rational design of antiepileptic therapies. In this review, we introduced the physiological function of ATP1A3 and summarized the findings about ATP1A3 in epileptic conditions from both clinical and laboratory aspects at first. Then, some possible mechanisms of how ATP1A3 mutations result in epilepsy are provided. We think this review timely introduces the potential contribution of ATP1A3 mutations in both the genesis and progression of epilepsy. Taken that both the detailed mechanisms and therapeutic significance of ATP1A3 for epilepsy are not yet fully illustrated, we think that both in-depth mechanisms investigations and systematic intervention experiments targeting ATP1A3 are needed, and by doing so, perhaps a new light can be shed on treating ATP1A3-associated epilepsy.
The electrogenic activity of sodium (Na+)/potassium (K+)-ATPase, an ionic pump driven by ATP hydrolysis, is essential for normal neuronal physiology, homeostasis, and signaling by maintaining Na+ and K+ gradients across the plasma membrane. The basic structure of Na+/K+-ATPase has been previously elucidated; it contains one large catalytic subunit (α) and two small auxiliary subunits (β, FXYD), which are composed of a Na+/K+-ATPase complex (Holm et al., 2016). The enriched α-subunits in vertebrate brains are encoded by paralogous genes (ATP1A1 to 3) that share a common gene promoter (McGrail et al., 1991). Among these genes, ATP1A3 encodes the Na+/K+-ATPase α3 isozyme, which is mainly expressed in the brain and highly expressed in neurons, and serves as the catalytic component of the pump because it contains binding sites for Na+, K+, ATP, and cardiac glycosides (pump inhibitors). The ATP1A3 isoform is particularly crucial for restoring electrochemical gradients in neuronal activities (Laird et al., 2015; Holm et al., 2016) since ATP1A3 is expressed exclusively in neurons, while others are not (McGrail et al., 1991).
Despite physiological conditions, substantial evidence has suggested that pathogenic variants in ATP1A3 are potential causes of various neurological disorders (Smedemark-Margulies et al., 2016; Salles et al., 2021; Vezyroglou et al., 2022). Interestingly, the disease onsets seem to extensively span among all ages, even though those disorders are associated with genetic mutations in ATP1A3, suggesting that dysfunction of vulnerable ATP1A3 in various forms could occur in childhood as the brain develops, but its impact may also emerge in the later adulthood. Single-cell exploration has pointed toward a critical role for ATP1A3 in human brain development and a cell type basis for ATP1A3-associated neurological disorders (Smith et al., 2021). Multiple studies have already shown the causal significance of ATP1A3 polymorphisms in developing various neurological diseases with a common inheritance mode (Heinzen et al., 2014; Rosewich et al., 2017; Carecchio et al., 2018; Capuano et al., 2020). Here, we mainly focus on the specific functions of ATP1A3 in epilepsy which is a common and intractable chronic neurological disease. Delighted by related findings from both clinical and laboratory aspects, in this review, the alterations of ATP1A3 in epilepsy were initially introduced, followed by some related mechanisms. Then, we raised some prospections in translational therapeutic views for both controlling seizures and interfering with epileptogenesis caused by ATP1A3 mutations.
Currently, various studies have demonstrated that 0.3% of the entire population is affected by genetic generalized epilepsies (GGEs), nearly accounting for 30% of all the patients who suffer from epilepsies (Reid et al., 2009). Genetic generalized epilepsies are found to be polygenic and primarily include juvenile myoclonic epilepsy (JME) or epilepsy with generalized tonic-clonic seizures (EGTCS) alone (Weber and Lerche, 2008). All of these share a high incidence of pharmacoresistance, and the management of intractable pharmacoresistant epilepsy emerges as a great burden for those patients with epilepsy (PWE) (Xu et al., 2022). Thus, revealing the exact genetic mutations and related pathogenesis factors of genetic epilepsy would largely benefit those patients for both prognosis and management purposes.
It must be admitted that most previous efforts have focused on the mutations in genes encoding ion channels or synaptic receptors which directly cause abnormal hyperexcitability in the brain (Hanada, 2020). The existence of ATP1A3 mutations in epilepsy seems to be the “elephant in the room”. Although somehow limited direct data have been presented, various clinical evidence could still support the essential roles of ATP1A3 in the incidence of epilepsy. Until now, more than 243 mutations (missense, 214; fusion, 1; nonsense, 8; splice, 14; FS del, 4; FS ins, 2) in ATP1A3 have been reported since the initial reports appeared in 2003 and 2004 (De Fusco et al., 2003; De Carvalho Aguiar et al., 2004) (cBioPortal data, http://www.cbioportal.org, last accessed January 2023). Among these, various mutations could be of great potential in inducing early-onset epilepsy (Ishii et al., 2013; Paciorkowski et al., 2015; Marzin et al., 2018; Schirinzi et al., 2018; Younes et al., 2018; Ishihara et al., 2019) (Table 1). Some researchers have proposed a phenotypic continuum for the autosomal dominant diseases caused by ATP1A3 mutations, encompassing alternating hemiplegia of children (AHC), rapid-onset dystonia-parkinsonism (RDP), relapsing encephalopathy with cerebellar ataxia (RECA), cerebellar ataxia, areflexia, pes cavus, optic atrophy and sensorineural hearing loss (CAPOS), and developmental and epileptic encephalopathies (DEE) (Rosewich et al., 2014; Dard et al., 2015; Paciorkowski et al., 2015).
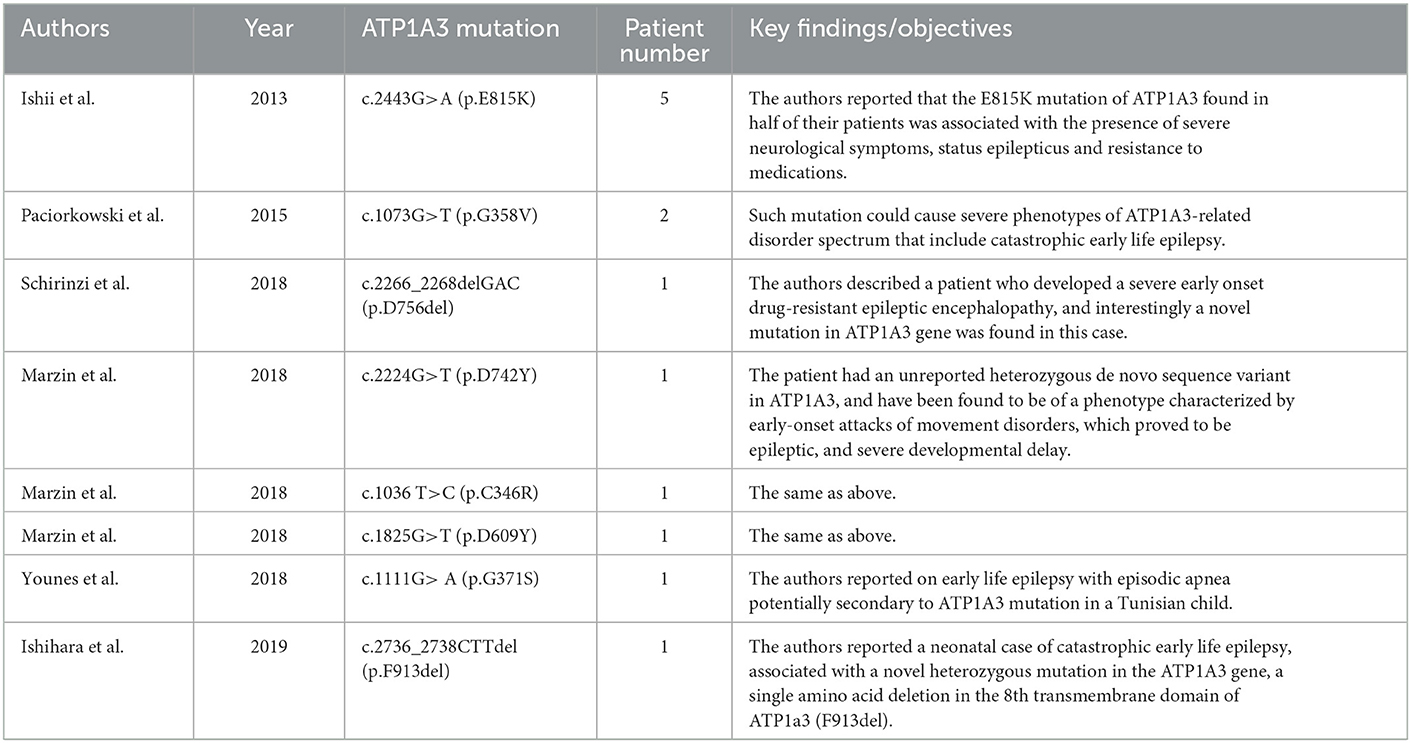
Table 1. Summary of clinical findings involving ATP1A3-related epilepsy.
Multiple autosomal dominant neurological diseases with limited overlap have been linked to ATP1A3 mutations in constitutional heterozygotes (Dard et al., 2015). Previous research has linked ATP1A3 mutations to the most severe form of infantile epileptic encephalopathy, characterized by seizures beginning at infancy, episodic apnea, poor survival, and profound developmental retardation (Sasaki et al., 2014; Paciorkowski et al., 2015). Patients with RDP are less likely to have epileptic seizures than those with AHC in which more than half of individuals would experience seizures (Bøttger et al., 2012). Patients with clinically diagnosed AHC have been found to suffer untreatable newborn seizures, status epilepticus, and early-life epilepsy (Saito et al., 2010; Ishii et al., 2013; Rosewich et al., 2014). To ascertain whether these frequent variations play significant roles in the susceptibility to generalized genetic epilepsies, Qu et al. (2015) screened 484 patients with GGE and 284 healthy controls for eight tagSNPs in ATP1A2 and ATP1A3. According to the authors' hypothesis, frequent variations of ATP1A3 increase one's risk of developing GGEs. Evidences have been found linking a common variation of ATP1A3 (ATP1A3 rs8107107) to a higher risk of developing epilepsy, and in particular, epilepsy with generalized tonic-clonic seizures. These results proved that the ATP1A3 gene is involved in the etiology of generalized genetic epilepsies. The following studies further demonstrated that severe epilepsies have been reported in people with ATP1A3 mutations, and intellectual impairment may concomitantly occur with FHM and AHC (Paciorkowski et al., 2015; Liu et al., 2018).
In addition to those PWEs whose epilepsy was directly caused by the mutation in the ATP1A3 isoform, patients with other ATP1A3-associated neurological diseases are also reported to be at high risk for concomitant seizures. An early study published in 2015 estimated that more than 50–80% of all alternating hemiplegia of childhood (AHC) cases in the literature had a history of seizures (Paciorkowski et al., 2015), while the incidence rate is lower (26.1%) in a later study (Li et al., 2018). This difference may be attributed to the different sample sizes; however, the incidence rate of epilepsy was significantly higher than in normal populations (nearly 1%). Despite the heterogeneity of ATP1A3-related seizure types, specific patterns have emerged. Seizures begin in childhood and range from the focal or generalized tonic, tonic-clonic to myoclonic attacks and are triggered by secondary external factors, such as stress, excitement, temperature extremes, water exposure, physical effort, and changes in lighting (Younes et al., 2018).
Thus, generally speaking, as a mutational susceptible gene, ATP1A3 alteration could easily lead to developmental epilepsy, normally characterized by more frequent seizures, more serious symptoms as well as poor treatment efficacy. While the decreased activity of ATP1A3 may participate, current existing evidence is still not sufficient to totally explain the observed clinical phenomenon. When ATP1A3 is mutated, it causes neurological abnormalities that range widely in symptomatology and severity. This might be the reason why ATP1A3-related epilepsy usually shows a worse prognosis. Interestingly, D923N, related to hypotonia or dystonia with an average onset age of 3 years, and L924P, which is associated with severe infantile epilepsy and profound disability, have already been examined (Arystarkhova et al., 2021). In this study, the authors showed that the eIF2α inhibition, ER retention, lactacystin response, and membrane redistribution on sucrose density gradients were all affected differently in humans with these two ATP1A3 mutations. As the L924P mutation has a more severe phenotype, misfolding during biosynthesis likely contributes to pathogenicity in addition to the enzyme's loss of activity (Einholm et al., 2010; Holm et al., 2016). Based on the clinical evidence, we have already recognized the potential roles of ATP1A3 in the incidence of epilepsy clinically. However, the critical roles of ATP1A3 still need further validation on more and larger population groups, especially focusing on the clear elaboration of ATP1A3 mutation-related family gene expression pattern.
Although decreased activity may play a role, a molecular foundation for the epilepsy-promoting effects of ATP1A3 mutations is yet unknown. Most mutations had no clear relationship between the mutation site and phenotype (Arystarkhova et al., 2019). The protein misfolding and pump deactivation are thought to have unique biological implications (Arystarkhova et al., 2019). The authors showed that whereas a mutation that affects Na+/K+-ATPase function would cause moderate illness, one that triggers the unfolded protein response might cause severe disease and, perhaps, the death of neurons even if the ion pump is not disabled.
Dominant ATP1A3 mutations may cause a broad spectrum of neurological diseases. Predictions regarding the factors in the epilepsy disease severity of ATP1A3 mutations can be made, and at least four ways things may go wrong (Arystarkhova et al., 2019). To start, a mutant protein with similar folding properties to the wild-type allele should be transported to the membrane (Figure 1). That is the worst-case scenario if a mutation has a gain-of-toxic function, such as ion leakage, but not if it is inactive. Second, a mutation that prevents the C-terminal third of the protein from folding (the obligatory β binding sites location) may have less of an effect: the α from the mutant allele will not compete with the normal allele if it is unable to interact with β. Similar to what has been described for several Drosophila Na+/K+-ATPase mutations, this might result in more β being accessible from the good allele to the developing α chains, leading to an improved outcome beyond that of haploinsufficiency (Ashmore et al., 2009). The unfolded mutant α will be extruded from the ER into the cytoplasm with the help of chaperones like BiP, where it will be ubiquitinated and degraded by the proteasome endoplasmic reticulum-associated degradation (ERAD), a mechanism that is always active to cope with the normal background of translation and folding mistakes. Third, even if faulty, misfolded proteins may try to traffic to the Golgi and beyond by engaging in calnexin- or calreticulin-mediated refolding cycles. The α subunit lacks the N-glycans necessary to associate with these chaperones. The unfolded protein response (UPR) involves the adaptive expansion of the ER and upregulation of proteins that promote folding in response to an increasing load of misfolded protein (Karagöz et al., 2019). This may modify the outcome of mutations that result in a gain-of-toxic function. Misfolded protein aggregates will trigger the UPR defensive reaction autophagy. Finally, the UPR will cause apoptosis, leading to the death of neuronal cells, if ER enlargement and autophagy are inadequate (Figure 1).
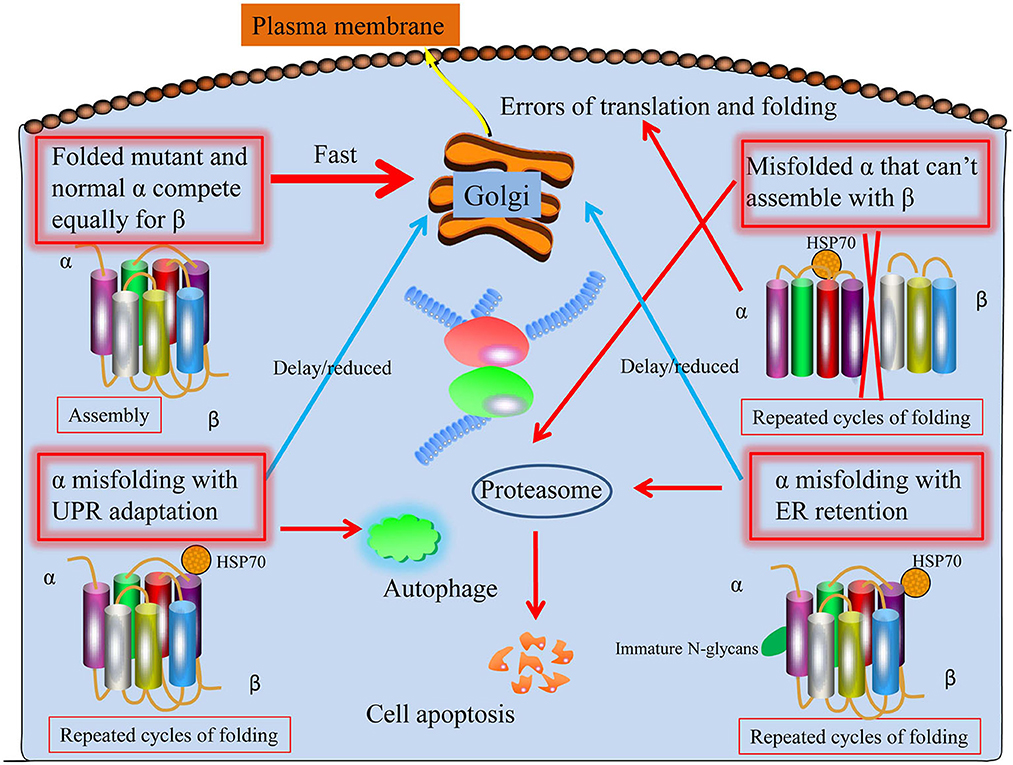
Figure 1. Proposed four potential ways ATP1A3 mutations may proceed through the biosynthetic process. First, the protein folds and biosynthesizes normally; second, it undergoes irreversible misfolding and is subsequently degraded by proteasomes (ERAD); third, the presence of a chaperone assists biosynthesis; and fourth, the chaperone assistance is ineffective. As with HSP70, the shown chaperone is meant to stand in for various chaperone classes. Mutant and normal ATP1A3 alleles compete for the β subunit, making it difficult to anticipate the effects of ATP1A3 mutations. It is believed that every particular mutation that might worsen epilepsy disease severity may lie among these four extremes.
It has now been established that protein-modifying genetic variations in ATP1A3 are uncommon in the general population and that when they do exist, they carry a very high risk of causing severe neurological disorders (Heinzen et al., 2014). Consequently, researchers have begun looking into the possibility that mutations in ATP1A3 cause other disorders. If they exist, the potential involvement of ATP1A3 mutations in a broader spectrum of phenotypes may likely become evident as NGS becomes more frequently employed in routine clinical practice. More research is necessary to fully understand the vast spectrum of illness severity reported with ATP1A3 mutations.
Anti-epileptic drugs (AEDs) are routinely used to control seizures in PWEs. However, one-third of the PWEs cannot achieve desirable outcomes, which is diagnosed as pharmacoresistant epilepsy (Kwan and Brodie, 2000). Pharmacoresistant epilepsy usually shares more severe pathological conditions including hyperexcitable circuitry and abnormally activated molecular pathways (Xu et al., 2019, 2021), resulting in the intractability of successful management. With increased clinical findings, it is somehow convincible that epilepsy with ATP1A3 mutation shares a high incidence of being pharmacoresistant. Gasser et al. (2020) analyzed the frequency of their usage and the effectiveness of these treatments in individuals with ATP1A3-related seizures. Although the therapeutic responses to AEDs are seldom recorded in individuals with ATP1A3-related seizures. As a result, very few patients were included in their study based on their systematic reports and patient data. However, among the total 21 patients, only 12 (57%) reported a reduction in both the frequency and severity of seizures. In 43% of the patients, no substantial response to seizure medication was observed, which is far higher than normal PWEs (30% of which would become pharmacoresistant). According to Schirinzi et al., in a patient with severe early-onset drug-resistant epileptic encephalopathy, another new mutation in the ATP1A3 gene was also discovered (Schirinzi et al., 2018), supporting the hypothesis that ATP1A3 mutation would induce poor responses to AEDs. The answers to why ATP1A3 mutations resulted in poor responses to current AEDs may be attributed to that when the ATP1A3 mutations are placed in their proper context, a possible chain of events may be envisioned: some people are predisposed to status epilepticus (SE) because of pathogenic ATP1A3 gene variants; SE may increase the risk of recurrent seizures; and damage the mesial temporal lobe in a way that reduces GABA-mediated inhibition or misdirects regeneration, leading to a novel recurrent excitatory circuit (Sills, 2007). Also, our very recent research has already confirmed that Na+/K+-ATPase emerges as a potential anti-seizure target (Zhao et al., 2022). It is plausible to speculate that dysfunction of the ATP1A3 gene, which is a crucial part of Na+/K+-ATPase, would lead to a more seizure-susceptible and hyper-excitable brain that would directly lead to pharmacoresistance (Xu et al., 2019). However, the precise mechanisms regarding the roles of ATP1A3 in the drug sensitivity of epilepsy still need more evidence.
Intriguingly, epileptic seizures of an ATP1A3 mutated PWE were controlled when the ketogenic diet (KD) was started (Schirinzi et al., 2018). These findings further proposed the use of KD, which has become more and more popular as a treatment for drug-resistant epilepsy and other neurological diseases, as an innovative treatment for ATP1A3-related diseases. Although definitive confirmatory trials of KD are still required. The anti-epileptic effect of KD has been reported to be related to several factors, including direct anti-seizure action, the restoration of a new neurotransmitter balance, alterations in mitochondrial and cellular metabolism, and an antioxidant influence (Rho, 2017). However, the unique activity of ketones on ATP-sensitive potassium channels (KATP channels) may be responsible for the success of KD in the above situation. KATP channels (activity altered by ATP1A3 malfunction) are thought to be responsible for the beneficial effects of ketosis on epilepsy (ref), which makes sense that dysfunction of the Na+/K+-ATPase caused by ATP1A3 mutation, which induces hyperexcitability in neurons, would also occasionally lead to inhibition of KATP channels (Tanner et al., 2011). Additionally, KD therapy may reverse this condition to reduce neuronal excitability. Their patient's clinical path and the success of KD imply the possibility of using this approach to treat ATP1A3-related epilepsy or other disorders. However, more systematic clinical trials with enough sample sizes are urgently needed.
However, most current papers on pharmaco-resistant epilepsy associated with ATP1A3 mutations did not provide any tips on how to manage seizures. The current treatment experiences are often ambiguous and inadequate for properly assessing the efficacy of AEDs. Consequently, further research is required to first identify the specific types of AEDs associated with ATP1A3 mutations and to seek a more prominent combination of multiple treatment strategies in the clinical arena.
Besides clinical investigations, animal studies provide great platforms for both intervention convenience and in-depth mechanism insights for addressing the role of ATP1A3 as well as Na+-K+-ATPase in epilepsy from the bench side.
Currently, increasing numbers of laboratory studies have unveiled the critical roles of Na+-K+-ATPase in epilepsy (Ygberg et al., 2021). In rodent models of epilepsy, the activity of Na+-K+-ATPase has been reported to be altered, and pharmacological inhibition of Na+-K+-ATPase would cause epileptic seizures in rodents (Holm and Lykke-Hartmann, 2016). Mechanically, Na+-K+-ATPase inhibition could lead to intracellular Na+ accumulation, which will further result in the increased NMDA receptor-mediated excitation and inhibition of inward transportation of GABA, as well as the accumulation of intercellular free Ca2+ through the reversion of intercellular Na+-Ca2+ exchanger (Sun et al., 2022). In addition, Na+-K+-ATPase dysfunction could also be related to a decrease in intracellular K+ homeostasis, resulting in impaired glutamate clearance and a depolarized change in resting membrane potential (Shao et al., 2021). Furthermore, as we recently reported, activation of astrocytic Na+-K+-ATPase would achieve great anti-seizure outcomes in neocortical epileptic mice (Zhao et al., 2022). With such substantial reported data, Na+-K+-ATPase is certainly a crucial contributor to brain excitability in epileptic brains (Sun et al., 2022). Importantly, the presence of epileptic symptoms in various neurological disorders caused by Na+-K+-ATPase subunits mutations suggested the causal relationship between Na+-K+-ATPase activity and epilepsy (Vezyroglou et al., 2022).
With regard to ATP1A3, clinical literature has demonstrated that, as a mutational susceptibility gene, ATP1A3 alteration could easily lead to developmental epilepsy, which normally exhibits more frequent seizures, more serious symptoms, and poor AEDs responses. Besides clinicians, laboratory researchers also devote their efforts to investigating the role and mechanisms of ATP1A3 in epileptic animals. The beginning of related studies can be traced back to the 2000s, and various early explorations have indicated the important roles of ATP1A3 in epileptogenesis and epilepsy, and have tried to elucidate the precise mechanisms (Li and Stys, 2001; Chu et al., 2009; Clapcote et al., 2009), although most of which were theoretical speculation. To determine whether significant reductions in Na+/K+-ATPase in the paramicrogyral cortex can contribute to epileptogenesis, Chu et al. (2009) studied the expression of the α3 isoform of Na+/K+-ATPase in the freezing lesion (FL) microgyrus model of developing epileptogenesis. Both spatial and temporal altered protein expressions including ATP1A3 in the cortex have been linked to hyperexcitability, which is a key factor in the pathophysiology of seizure activities. In addition, the functional importance of the ATP1A3 in regulating epileptiform activity and seizure behavior was shown in a mouse mutagenesis screen, where the ATP1A3 mutation was found to be associated with seizures (Clapcote et al., 2009). Although previous research has linked ATP1A3 mutations to the most severe form of infantile epileptic encephalopathy, however, the precise affected brain area of ATP1A3-related epilepsy currently remain unknown. To address this issue, when comparing ATP1A3−/− and wild-type (ATP1A3+/+) littermates, Ikeda et al. (2017) discovered a considerably increased c-Fos-expressing cells number in several brain regions, with distinct distribution in the cerebellum. Other distribution areas that were strongly positive for ATP1A3 also include Purkinje cell progenitors in the simple lobule and hemisphere and medial cerebellar nuclei. Brains of ATP1A3−/− mice have been compared to those of ATP1A3+/+ mice, and it has been discovered that the ATP1A3−/− brains had greater levels of monoamine neurotransmitters, particularly dopamine and noradrenaline. These findings point to an essential function for ATP1A3 in the developing and newborn brains. Furthermore, they have shown that ATP1A3−/− consistently has elevated levels of noradrenaline (NA), dopamine (DA), 5-hydroxytryptamine (5-HT), and their metabolites, suggesting that the propulsion afforded by the α3 subunit is essential for monoamine signaling. Drugs that control monoamine signaling may be effective in treating ATP1A3 mutation-related neurological diseases, where it has been hypothesized that high monoamine levels are a consequence of seizure activity rather than a cause (Ikeda et al., 2017).
The expression of ATP1A3 is restricted in neurons. These neurons are especially abundant in the cerebellum and basal ganglia, two brain regions responsible for regulating motor activity, memory, and spatial learning. The ATP1A3 mutation in neurons, as shown by Kinoshita et al. (2016), reduces the Na+/K+-ATPase activity and increases intracellular Na+, which raises cellular excitability and, thus, affects neuronal activities (Figure 2). Indeed, previous studies have revealed that depolarization, enhanced action potential firing, and further glutamate release may follow from a decrease in the α3 subunit in excitatory neurons (Li and Stys, 2001). Neurotransmitter release may also be affected by a lack of α3 at presynaptic terminals. The impact of ATP1A3 mutation on epilepsy prevalence may partly be mediated via these mechanisms. Treatment options may include flunarizine and topiramate. Flunarizine is a Ca+ and Na+ channel blocker and was used to treat migraine (Kinoshita et al., 2016). Similarly, the mechanism and efficacy of the action of topiramate is unknown and may include inhibition of 2-amino-3-(3-hydroxyl-5-methyl-4-isoxazolyl) propionic acid (AMPA) receptors as well the carbonic anhydrase, thereby lowering extracellular pH (Hoei-Hansen et al., 2014; Yang et al., 2014). Both of them could be of great potential to be treatment options.
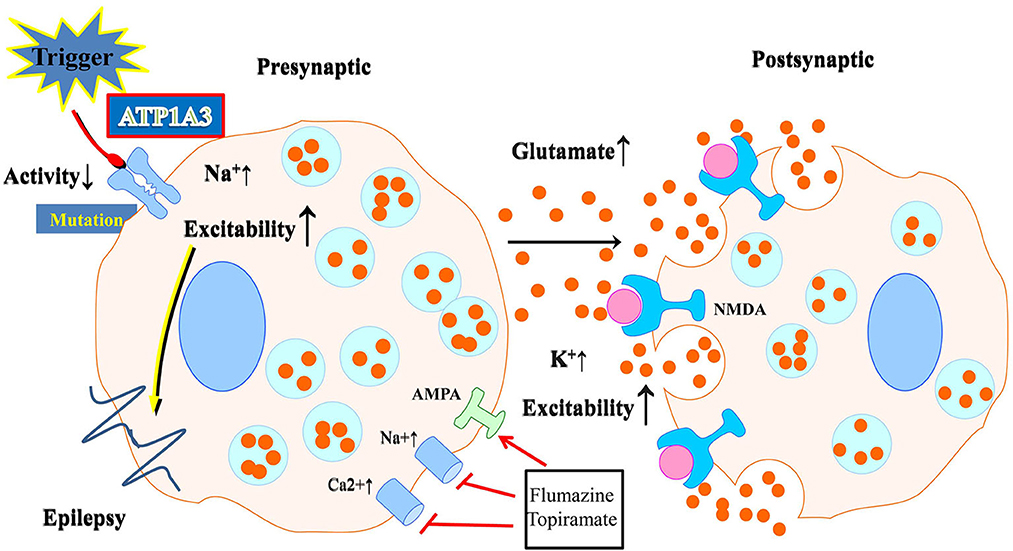
Figure 2. Schematic illustration of the effect of neuronal ATP1A3 mutations on glutamatergic system activity. Abnormalities in Na+/K+-ATPase may result from mutations. Neurons with an ATP1A3 mutation have increased cellular excitability due to decreased Na+/K+-ATPase activity and increased intracellular Na+. Flunarizine and topiramate could be used as a treatment in some relevant disease cases.
Although substantial hints lead researchers to make such theoretical speculations that ATP1A3 mutation impacts the normal ion exchange and homeostasis within and outside the neuronal membrane, leading to hyperexcitation and following the occurrence of epilepsy, more evidence obtained from multifaceted experiments, including in vitro/in vivo electrophysiology and genetical interferences, are still needed. On the contrary, deficits in glutamatergic control may contribute to various CNS disorders brought on by age-related and mutagenic changes in Na+/K+-ATPase function. Changes in brain metabolism have been associated with many poorly controlled neurodegenerative disorders including epilepsy. Research into Na+/K+-ATPase, including how it is regulated by the environment, epigenome, and genome, is expected to be useful in developing novel pharmacological and non-pharmaceutical therapies because of its key role in improving synaptic functioning and energy control (Kinoshita et al., 2016).
Given that disrupting ATP1A3 functions would directly induce neuronal hyperexcitability and result in seizures, it provides an alternative way to reproduce epileptic animals through genetic modification of ATP1A3. Previously, Clapcote et al. described an epileptic mouse model in which a mutation in ATP1A3 caused seizures (Clapcote et al., 2009). Through in vivo mutagenesis with N-nitroso-N-ethylurea (ENU), they were able to generate a mutant termed Myshkin (Myk), which displays convulsions and neuronal hyperexcitability in heterozygotes (Myk−/+) and neonatal death in homozygotes (Myk/Myk). Complex partial and secondary generalized seizures in these mice are caused by a single mutation in the ATP1A3 gene (I810N in exon 18), which is unique to the Myshkin allele and accounts for all of the diseases. They found that ATP1A3 plays a crucial role in regulating epileptiform activity and seizure behavior. Seizures (spontaneous and those triggered by vestibular stress), medial temporal sclerosis, sleep disturbances, and various cognitive, motor, social, and behavioral impairments are presented in Myk−/+ mice (Clapcote et al., 2009; Kirshenbaum et al., 2016). Consistent with the involvement of ATP1A3 in the fast extrusion of Na+ following high neuronal activity, the hippocampal CA3-CA1 pathway becomes hyperexcitable in hippocampal slices from Myk−/+ mice when subjected to high-frequency synaptic activity, such as that induced by stress (Clapcote et al., 2009; Azarias et al., 2013). In addition, when exposed to vestibular stress, Myk−/+ mice backcrossed 20 generations to C57BL/6NCr (N20) exhibited a considerably reduced drop in adult brain Na+/K+-ATPase activity and did not demonstrate seizure activity in electrocorticography recordings, in contrast to N12 C57BL/6NCr animals (Kirshenbaum et al., 2011). Two crosses (N2) to the seizure-prone FVB/NCr strain (Goelz et al., 1998) resulted in significant levels of focal reactive astrogliosis and microglial activation in the hippocampus. In contrast, these changes were not seen in N12 C57BL/6NCr Myk−/+ mice, providing further evidence for the notion that genetic background can influence phenotype (Clapcote et al., 2009). Generally speaking, these mutation-related phenotypes could provide more optimized animal models for exploring the roles of ATP1A3 in epilepsy, and furthermore, could provide a useful platform for screening effective treatments of certain epileptic phenotypes.
Undoubtedly, Na+/K+-ATPase function is closely related to the emergence of epileptiform activity, due to its crucial role in maintaining normal ion homeostasis. As this review introduces, mutations in ATP1A3, which is a crucial gene encoding the key α subunit, somehow contribute to epileptogenesis and concomitant seizure activities. In this review, we focused on the role of ATP1A3 alterations in epilepsy, followed by potential mechanisms, by summarizing both clinical and laboratory findings. Our current review highlights that inactivating mutation of ATP1A3 is a plausible mechanism for human epilepsy due to the high degree of similarity between the various Na+/K+-ATPase isoforms of mice and humans. Based on current research, more efforts should be directed toward launching more large-scale clinical research to further explore ATP1A3 alteration significance in the incidence of epilepsy, disease severity, or other specific clinical characteristics. In addition, more laboratory efforts were also needed to in-depth clarify the precise mechanisms, mainly the close association between ATP1A3 dysfunction and the genesis of epileptic excitatory circuits. It could be proposed that inhibiting ATP1A3 mutations might be a potential treatment for severe epilepsy, and more efforts should be directed toward clarifying the side effects of certain strategies via impeding ATP1A3 mutations. In addition, systematic research regarding the ATP1A3 gene mutations in different populations could be of great significance and need to be further explored in the near future. Epilepsy is currently demonstrated to be one consequence of ATP1A3 alteration; however, whether epilepsy could be also a cause of ATP1A3 alteration still need more research. Finally, the most anticipated research of great significance would be seeking potential therapeutic strategies targeting ATP1A3 in near future.
SZ and Y-LL wrote the manuscript. YG, CX, and ZC contributed to the revision of the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by grants from the National Natural Science Foundation of China (Nos. 82103480 and 82173796) and the Zhejiang Provincial Natural Science Foundation (No. LQ22H090018).
We acknowledge TCGA and CGGA databases for providing their platform and contributors for uploading their meaningful datasets.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Arystarkhova, E., Haq, I. U., Luebbert, T., Mochel, F., Saunders-Pullman, R., Bressman, S. B., et al. (2019). Factors in the disease severity of ATP1A3 mutations: Impairment, misfolding, and allele competition. Neurobiol. Dis. 132, 104577. doi: 10.1016/j.nbd.2019.104577
Arystarkhova, E., Ozelius, L. J., Brashear, A., and Sweadner, K. J. (2021). Misfolding, altered membrane distributions, and the unfolded protein response contribute to pathogenicity differences in Na,K-ATPase ATP1A3 mutations. J. Biol. Chem. 296, 100019. doi: 10.1074/jbc.RA120.015271
Ashmore, L. J., Hrizo, S. L., Paul, S. M., Van Voorhies, W. A., Beitel, G. J., Palladino, M. J., et al. (2009). Novel mutations affecting the Na, K ATPase alpha model complex neurological diseases and implicate the sodium pump in increased longevity. Hum. Genet. 126, 431–447. doi: 10.1007/s00439-009-0673-2
Azarias, G., Kruusmägi, M., Connor, S., Akkuratov, E. E., Liu, X. L., Lyons, D., et al. (2013). A specific and essential role for Na,K-ATPase α3 in neurons co-expressing α1 and α3. J. Biol. Chem. 288, 2734–43. doi: 10.1074/jbc.M112.425785
Bøttger, P., Doǧanlı, C., and Lykke-Hartmann, K. (2012). Migraine- and dystonia-related disease-mutations of Na+/K+-ATPases: relevance of behavioral studies in mice to disease symptoms and neurological manifestations in humans. Neurosci. Biobehav. Rev. 36, 855–71. doi: 10.1016/j.neubiorev.2011.10.005
Capuano, A., Garone, G., Tiralongo, G., and Graziola, F. (2020). Alternating hemiplegia of childhood: understanding the genotype–phenotype relationship of ATP1A3 variations. Appl. Clin. Genet. 13, 71–81. doi: 10.2147/TACG.S210325
Carecchio, M., Zorzi, G., Ragona, F., Zibordi, F., and Nardocci, N. (2018). ATP1A3-related disorders: an update. Eur. J. Paediatr. Neurol. 22, 257–263. doi: 10.1016/j.ejpn.2017.12.009
Chu, Y., Parada, I., and Prince, D. A. (2009). Temporal and topographic alterations in expression of the alpha3 isoform of Na+, K(+)-ATPase in the rat freeze lesion model of microgyria and epileptogenesis. Neuroscience 162, 339–348. doi: 10.1016/j.neuroscience.2009.04.003
Clapcote, S. J., Duffy, S., Xie, G., Kirshenbaum, G., Bechard, A. R., Rodacker Schack, V., et al. (2009). Mutation I810N in the α3 isoform of Na+, K+-ATPase causes impairments in the sodium pump and hyperexcitability in the CNS. Proc. Natl. Acad. Sci. U.S.A. 106, 14085–14090. doi: 10.1073/pnas.0904817106
Dard, R., Mignot, C., Durr, A., Lesca, G., Sanlaville, D., Roze, E., et al. (2015). Relapsing encephalopathy with cerebellar ataxia related to an ATP1A3 mutation. Dev. Med. Child. Neurol. 57, 1183–1186. doi: 10.1111/dmcn.12927
De Carvalho Aguiar, P., Sweadner, K. J., Penniston, J. T., Zaremba, J., Liu, L., Caton, M., et al. (2004). Mutations in the Na+/K+-ATPase a3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 43, 169–175. doi: 10.1016/j.neuron.2004.06.028
De Fusco, M., Marconi, R., Silvestri, L., Atorino, L., Rampoldi, L., Morgante, L., et al. (2003). Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump a2 subunit associated with familial hemiplegic migraine type 2. Nat. Genet. 33, 192–196. doi: 10.1038/ng1081
Einholm, A. P., Toustrup-Jensen, M. S., Holm, R., Andersen, J. P., and Vilsen, B. (2010). The rapid-onset dystonia parkinsonism mutation D923N of the Na+,K+-ATPase α3 isoform disrupts Na+ interaction at the third Na+ site. J. Biol. Chem. 285, 26245–26254. doi: 10.1074/jbc.M110.123976
Gasser, M., Boonsimma, P., Netbaramee, W., Wechapinan, T., Srichomthomg, C., Ittiwut, C., et al. (2020). ATP1A3-related epilepsy: Report of seven cases and literature-based analysis of treatment response. J. Clin. Neurosci.72, 31–38. doi: 10.1016/j.jocn.2020.01.041
Goelz, M. F., Mahler, J., Harry, J., Myers, P., Clark, J., Thigpen, J. E., et al. (1998). Neuropathologic findings associated with seizures in FVB mice. Lab. Anim. Sci. 48, 34–37. doi: 10.1007/s10709-004-1439-3
Hanada, T. (2020). Ionotropic glutamate receptors in epilepsy: a review focusing on AMPA and NMDA Receptors. Biomolecules 10, 464. doi: 10.3390/biom10030464
Heinzen, E. L., Arzimanoglou, A., Brashear, A., Clapcote, S. J., Gurrieri, F., Goldstein, D. B., et al. (2014). Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol. 13, 503–514. doi: 10.1016/S1474-4422(14)70011-0
Hoei-Hansen, C. E., Dali, C. Í., Lyngbye, T. J., Duno, M., and Uldall, P. (2014). Alternating hemiplegia of childhood in Denmark: clinical manifestations and ATP1A3 mutation status. Eur. J. Paediatric Neurol. 18, 50–54. doi: 10.1016/j.ejpn.2013.08.007
Holm, R., Toustrup-Jensen, M. S., Einholm, A. P., Schack, V. R., Andersen, J. P., Vilsen, B., et al. (2016). Neurological disease mutations of α3 Na+,K+-ATPase: Structural and functional perspectives and rescue of compromised function. Biochim. Biophys. Acta 1857, 1807–1828. doi: 10.1016/j.bbabio.2016.08.009
Holm, T. H., and Lykke-Hartmann, K. (2016). Insights into the pathology of the α3 Na+/K+-ATPase ion pump in neurological disorders; lessons from animal models. Front. Physiol. 7, 209. doi: 10.3389/fphys.2016.00209
Ikeda, K., Onimaru, H., and Kawakami, K. (2017). Knockout of sodium pump α3 subunit gene (Atp1a3-/-) results in perinatal seizure and defective respiratory rhythm generation. Brain Res. 1666, 27–37. doi: 10.1016/j.brainres.2017.04.014
Ishihara, N., Inagaki, H., Miyake, M., Kawamura, Y., Yoshikawa, T., Kurahashi, H., et al. (2019). A case of early onset life-threatening epilepsy associated with a novel ATP1A3 gene variant. Brain Dev. 41, 285–291. doi: 10.1016/j.braindev.2018.10.008
Ishii, A., Saito, Y., Mitsui, J., Ishiura, H., Yoshimura, J., Arai, H., et al. (2013). Identification of ATP1A3 mutations by exome sequencing as the cause of alternating hemiplegia of childhood in Japanese patients. PLoS ONE 8, e56120. doi: 10.1371/journal.pone.0056120
Karagöz, G. E., Acosta-Alvear, D., and Walter, P. (2019). The unfolded protein response: detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulum. cold spring harb. Perspect Biol. 11, a033886. doi: 10.1101/cshperspect.a033886
Kinoshita, P. F., Leite, J. A., Orellana, A. M., Vasconcelos, A. R., Quintas, L. E., Kawamoto, E. M., et al. (2016). The influence of Na(+), K(+)-ATPase on glutamate signaling in neurodegenerative diseases and senescence. Front. Physiol. 7, 195. doi: 10.3389/fphys.2016.00195
Kirshenbaum, G. S., Clapcote, S. J., Duffy, S., Burgess, C. R., Petersen, J., Jarowek, K. J., et al. (2011). Mania-like behavior induced by genetic dysfunction of the neuronspecific Na+,K+-ATPase α3 sodium pump. Proc. Natl. Acad. Sci. U.S.A. 108, 18144–18149. doi: 10.1073/pnas.1108416108
Kirshenbaum, G. S., Idris, N. F., Dachtler, J., Roder, J. C., and Clapcote, S. J. (2016). Deficits in social behavioral tests in a mouse model of alternating hemiplegia of childhood. J. Neurogenet. 30, 42–49. doi: 10.1080/01677063.2016.1182525
Kwan, P., and Brodie, M. J. (2000). Early identification of refractory epilepsy. N. Engl. J. Med. 342, 314–319. doi: 10.1056/NEJM200002033420503
Laird, J. G., Pan, Y., Modestou, M., Yamaguchi, D. M., Song, H., Sokolov, M., et al. (2015). Identification of a VxP Targeting Signal in the Flagellar Na+/K+-ATPase. Traffic 16, 1239–1253. doi: 10.1111/tra.12332
Li, S., and Stys, P. K. (2001). Na+-K+-ATPase inhibition and depolarization induce glutamate release via reverse Na+-dependent transport in spinal cord white matter. Neuroscience. 107, 675–83. doi: 10.1016/s0306-4522(01)00385-2
Li, S. P., Zhang, Y. H., Yang, X. L., Chen, J. Y., Zeng, Q., Zhang, J., et al. (2018). Genotype-phenotype correlation in patients with alternating hemiplegia of childhood. Zhonghua Er Ke Za Zhi 56, 811–817. doi: 10.3760/cma.j.issn.0578-1310.2018.11.004
Liu, J., Tong, L., Song, S., Niu, Y., Li, J., Wu, X., et al. (2018). Novel and de novo mutations in pediatric refractory epilepsy. Mol. Brain 11, 18. doi: 10.1186/s13041-018-0392-5
Marzin, P., Mignot, C., Dorison, N., Dufour, L., Ville, D., Kaminska, A., et al. (2018). Early-onset encephalopathy with paroxysmal movement disorders and epileptic seizures without hemiplegic attacks: About three children with novel ATP1A3 mutations. Brain Dev. 40, 768–774. doi: 10.1016/j.braindev.2018.05.008
McGrail, K. M., Phillips, J. M., and Sweadner, K. J. (1991). Immunofluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: Both neurons and glia can express more than one Na,K-ATPase. J. Neurosci. 11, 381–391. doi: 10.1523/JNEUROSCI.11-02-00381.1991
Paciorkowski, A. R., McDaniel, S. S., Jansen, L. A., Tully, H., Tuttle, E., Ghoneim, D. H., et al. (2015). Novel mutations in ATP1A3 associated with catastrophic early life epilepsy, episodic prolonged apnea, and postnatal microcephaly. Epilepsia 56, 422–430. doi: 10.1111/epi.12914
Qu, J., Yang, Z. Q., Zhang, Y., Mao, C. X., Wang, Z. B., Mao, X. Y., et al. (2015). Common variants of ATP1A3 but not ATP1A2 are associated with Chinese genetic generalized epilepsies. J. Neurol. Sci. 354, 56–62. doi: 10.1016/j.jns.2015.04.045
Reid, C. A., Berkovic, S. F., and Petrou, S. (2009). Mechanisms of human inherited epilepsies. Prog. Neurobiol. 87, 41–57. doi: 10.1016/j.pneurobio.2008.09.016
Rho, J. M. (2017). How does the ketogenic diet induce anti-seizure effects? Neurosci. Lett. 637, 4–10. doi: 10.1016/j.neulet.2015.07.034
Rosewich, H., Ohlenbusch, A., Huppke, P., Schlotawa, L., Baethmann, M., Carrilho, I., et al. (2014). The expanding clinical and genetic spectrum of ATP1A3-related disorders. Neurology 82, 945–955. doi: 10.1212/WNL.0000000000000212
Rosewich, H., Sweney, M. T., DeBrosse, S., Ess, K., Ozelius, L., Andermann, E., et al. (2017). Research conference summary from the 2014 International task force on ATP1A3 -related disorders. Neurol. Genet. 3, e139. doi: 10.1212/NXG.0000000000000139
Saito, Y., Inui, T., Sakakibara, T., Sugai, K., Sakuma, H., Sasaki, M., et al. (2010). Evolution of hemiplegic attacks and epileptic seizures in alternating hemiplegia of childhood. Epilepsy Res. 90, 248–258. doi: 10.1016/j.eplepsyres.2010.05.013
Salles, P. A., Mata, I. F., Brünger, T., Lal, D., and Fernandez, H. H. (2021). ATP1A3-related disorders: an ever-expanding clinical spectrum. Front. Neurol. 12, 637890. doi: 10.3389/fneur.2021.637890
Sasaki, M., Ishii, A., Saito, Y., Morisada, N., Iijima, K., Takada, S., et al. (2014). Genotypephenotype correlations in alternating hemiplegia of childhood. Neurology 82, 482–490. doi: 10.1212/WNL.0000000000000102
Schirinzi, T., Graziola, F., Cusmai, R., Fusco, L., Nicita, F., Elia, M., et al. (2018). ATP1A3-related epileptic encephalopathy responding to ketogenic diet. Brain Dev. 40, 433–438. doi: 10.1016/j.braindev.2018.01.002
Shao, L. R., Janicot, R., and Stafstrom, C. E. (2021). Na+-K+-ATPase functions in the developing hippocampus: regional differences in CA1 and CA3 neuronal excitability and role in epileptiform network bursting. J. Neurophysiol. 125, 1–11. doi: 10.1152/jn.00453.2020
Sills, G. J. (2007). Seizures beget seizures: a lack of experimental evidence and clinical relevance fails to dampen enthusiasm. Epilepsy Curr. 7, 103–104. doi: 10.1111/j.1535-7511.2007.00189.x
Smedemark-Margulies, N., Brownstein, C. A., Vargas, S., Tembulkar, S. K., Towne, M. C., Shi, J., et al. (2016). A novel de novo mutation in ATP1A3 and childhoodonset schizophrenia. Cold Spring Harb. Mol. Case Stud. 2, a001008–a001013. doi: 10.1101/mcs.a001008
Smith, R. S., Florio, M., Akula, S. K., Neil, J. E., Wang, Y., Hill, R. S., et al. (2021). Early role for a Na+,K+-ATPase (ATP1A3) in brain development. Proc. Natl. Acad. Sci. U.S.A. 118, e2023333118. doi: 10.1073/pnas.2023333118
Sun, J., Zheng, Y., Chen, Z., and Wang, Y. (2022). The role of Na+ -K+ -ATPase in the epileptic brain. CNS Neurosci. Ther. 28, 1294–1302. doi: 10.1111/cns.13893
Tanner, G. R., Lutas, A., Marti'nez-Francois, J. R., Yellen, G., and Single, K. (2011). ATP channel opening in response to action potential firing in mouse dentate granule neurons. J. Neurosci. 31, 8689–8696. doi: 10.1523/JNEUROSCI.5951-10.2011
Vezyroglou, A., Akilapa, R., Barwick, K., Koene, S., Brownstein, C. A., Holder-Espinasse, M., et al. (2022). The Phenotypic Continuum of ATP1A3-Related Disorders. Neurology 99, e1511–e1526. doi: 10.1212/WNL.0000000000200927
Weber, Y. G., and Lerche, H. (2008). Genetic mechanisms in idiopathic epilepsies. Dev. Med. Child. Neurol. 50, 648–654. doi: 10.1111/j.1469-8749.2008.03058.x
Xu, C., Gong, Y., Wang, Y., and Chen, Z. (2022). New advances in pharmacoresistant epilepsy towards precise management-from prognosis to treatments. Pharmacol. Ther. 233, 108026. doi: 10.1016/j.pharmthera.2021.108026
Xu, C., Wang, Y., Zhang, S., Nao, J., Liu, Y., Wang, Y., et al. (2019). Subicular pyramidal neurons gate drug resistance in temporal lobe epilepsy. Ann. Neurol. 86, 626–640. doi: 10.1002/ana.25554
Xu, C., Zhang, S., Gong, Y., Nao, J., Shen, Y., Tan, B., et al. (2021). Subicular Caspase-1 Contributes to Pharmacoresistance in Temporal Lobe Epilepsy. Ann. Neurol. 90, 377–390. doi: 10.1002/ana.26173
Yang, X., Gao, H., Zhang, J., Xu, X., Liu, X., Wu, X., et al. (2014). ATP1A3 mutations and genotype-phenotype correlation of alternating hemiplegia of childhood in Chinese patients. PLoS ONE 9, e97274. doi: 10.1371/journal.pone.0097274
Ygberg, S., Akkuratov, E. E., Howard, R. J., Taylan, F., Jans, D. C., Mahato, D. R., et al. (2021). A missense mutation converts the Na+,K+-ATPase into an ion channel and causes therapy-resistant epilepsy. J. Biol. Chem. 297, 101355. doi: 10.1016/j.jbc.2021.101355
Younes, T. B., Benrhouma, H., Klaa, H., Rouissi, A., Chaabouni, M., Kraoua, I., et al. (2018). Early life epilepsy and episodic apnea revealing an ATP1A3 mutation: report of a pediatric case and literature review. Neuropediatrics 49, 339–341. doi: 10.1055/s-0038-1667024
Keywords: ATP1A3, epilepsy, Na+/K+-ATPase, animal models, mutation, mechanism, treatments ATP1A3, treatments
Citation: Zou S, Lan Y-L, Gong Y, Chen Z and Xu C (2023) The role of ATP1A3 gene in epilepsy: We need to know more. Front. Cell. Neurosci. 17:1143956. doi: 10.3389/fncel.2023.1143956
Received: 13 January 2023; Accepted: 23 January 2023;
Published: 13 February 2023.
Edited by:
Xun Wang, Northern Theater General Hospital, ChinaReviewed by:
Jinming Han, Capital Medical University, ChinaCopyright © 2023 Zou, Lan, Gong, Chen and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu-Long Lan,  bGFueXVsb25nQHpqdS5lZHUuY24=; Cenglin Xu, eHVjZW5nbGluNXp6QHpqdS5lZHUuY24=
bGFueXVsb25nQHpqdS5lZHUuY24=; Cenglin Xu, eHVjZW5nbGluNXp6QHpqdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.