
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci., 31 March 2023
Sec. Cellular Neurophysiology
Volume 17 - 2023 | https://doi.org/10.3389/fncel.2023.1120651
This article is part of the Research TopicRecent Advances in Measuring and Controlling Synaptic CommunicationView all 10 articles
 Marius Seidenthal1,2
Marius Seidenthal1,2 Barbara Jánosi1,2
Barbara Jánosi1,2 Nils Rosenkranz1,2Noah Schuh1,2
Nils Rosenkranz1,2Noah Schuh1,2 Nora Elvers1,2Miles Willoughby1,2
Nora Elvers1,2Miles Willoughby1,2 Xinda Zhao1,2
Xinda Zhao1,2 Alexander Gottschalk1,2*
Alexander Gottschalk1,2*pH-sensitive fluorescent proteins are widely used to study synaptic vesicle (SV) fusion and recycling. When targeted to the lumen of SVs, fluorescence of these proteins is quenched by the acidic pH. Following SV fusion, they are exposed to extracellular neutral pH, resulting in a fluorescence increase. SV fusion, recycling and acidification can thus be tracked by tagging integral SV proteins with pH-sensitive proteins. Neurotransmission is generally activated by electrical stimulation, which is not feasible in small, intact animals. Previous in vivo approaches depended on distinct (sensory) stimuli, thus limiting the addressable neuron types. To overcome these limitations, we established an all-optical approach to stimulate and visualize SV fusion and recycling. We combined distinct pH-sensitive fluorescent proteins (inserted into the SV protein synaptogyrin) and light-gated channelrhodopsins (ChRs) for optical stimulation, overcoming optical crosstalk and thus enabling an all-optical approach. We generated two different variants of the pH-sensitive optogenetic reporter of vesicle recycling (pOpsicle) and tested them in cholinergic neurons of intact Caenorhabditis elegans nematodes. First, we combined the red fluorescent protein pHuji with the blue-light gated ChR2(H134R), and second, the green fluorescent pHluorin combined with the novel red-shifted ChR ChrimsonSA. In both cases, fluorescence increases were observed after optical stimulation. Increase and subsequent decline of fluorescence was affected by mutations of proteins involved in SV fusion and endocytosis. These results establish pOpsicle as a non-invasive, all-optical approach to investigate different steps of the SV cycle.
Chemical synaptic transmission, the release of neurotransmitters into the synaptic cleft, depends on synaptic vesicle (SV) exocytosis (Sudhof, 2013). To efficiently sustain neurotransmitter release during phases of (high) neuronal activity, SV-associated proteins and lipids must be recycled from the plasma membrane, thus allowing regeneration of ready-to-release SVs (Gan and Watanabe, 2018; Chanaday et al., 2019). Several modes of SV recycling have been uncovered, including classical clathrin-mediated endocytosis, activity-dependent bulk endocytosis, kiss-and-run release and the recently described ultrafast endocytosis (Heuser and Reese, 1973; Kittelmann et al., 2013; Watanabe et al., 2013a,b; Morton et al., 2015; Watanabe and Boucrot, 2017; Shin et al., 2021). It is still under debate which of these processes happen under which conditions, and whether some of these represent short-cuts in the SV cycle, e.g., bypassing the endosome. Also, the exact involvement of known, and the discovery of novel, recycling factors mediating these events is the subject of ongoing research (Gan and Watanabe, 2018; Yu et al., 2018). To study SV fusion and recycling, methods such as electron microscopy (EM), measurement of membrane capacitance, and super-resolution microscopy are used. However, these methods are either limited in their temporal resolution (EM) or applicability to different neuronal cell types (membrane capacitance measurements and super-resolution microscopy) (von Gersdorff and Matthews, 1999; Yu et al., 2018; Shin et al., 2021).
Another method to indirectly visualize and quantify exocytosis and recycling of proteins is through tagging with pH-sensitive fluorescent proteins (Miesenbock et al., 1998). SVs acidification precedes and is required for refilling with neurotransmitters during the recycling process (Egashira et al., 2015; Gowrisankaran and Milosevic, 2020). For this reason, the fluorescence of pH-sensitive proteins, such as the green fluorescent pHluorin, when targeted to the acidic intraluminal side of the SV membrane, is quenched by the low-pH environment (Sankaranarayanan et al., 2000). Typically, the utilized reporters are either fused to the vesicular glutamate transporter or inserted into loops of the tetraspan membrane protein synaptophysin (Voglmaier et al., 2006; Luo et al., 2021). Upon SV fusion with the plasma membrane, the fluorescence increases due to exposure to the neutral extracellular medium and dequenching of the fluorophore (Figure 1). Following stimulation, the fluorescence decreases depending on the rate of endocytosis, sorting and reacidification of SVs. This way, positive or negative influences on the speed of SV retrieval can be quantified (Morton et al., 2015; Watanabe et al., 2018). Several variants of pH-sensitive fluorescent proteins have been created, covering almost the entire spectrum of visible light (Miesenbock et al., 1998; Shen et al., 2014; Liu et al., 2021). This opens the door for multiplexing with other fluorophores or optical actuators (Li and Tsien, 2012; Jackson and Burrone, 2016).
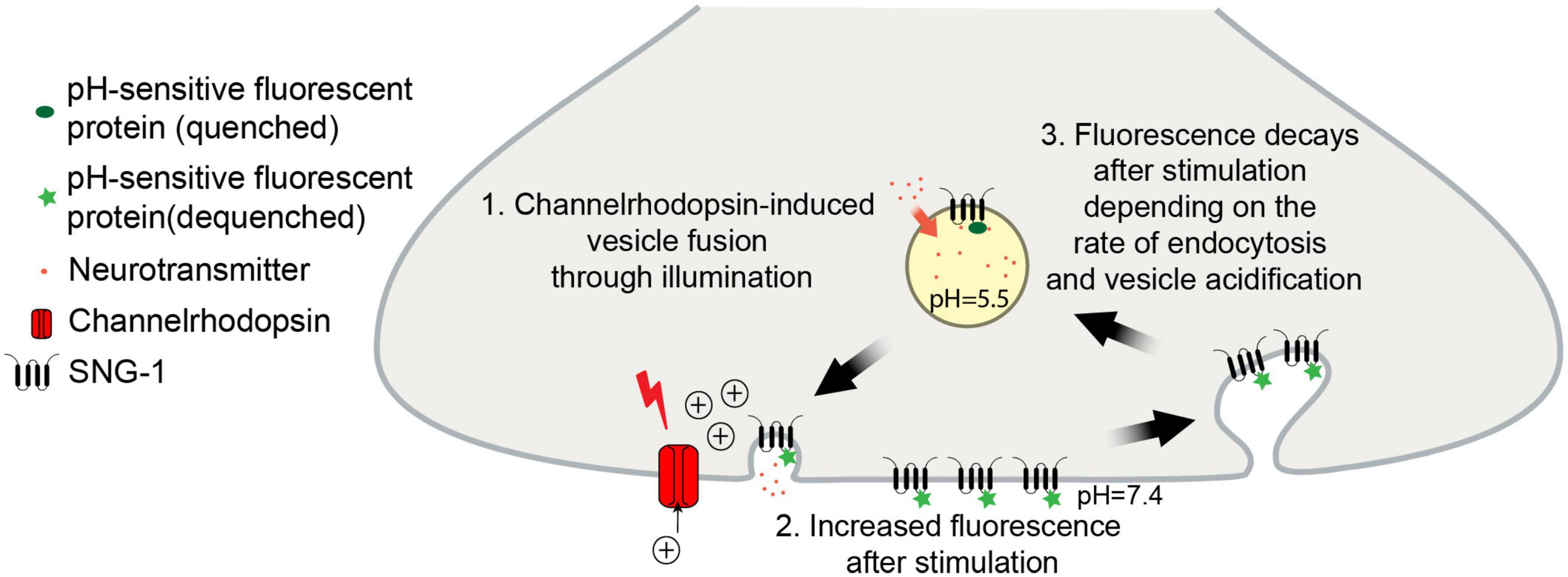
Figure 1. Schematic of the pOpsicle assay. pH-sensitive fluorescent proteins fused to SNG-1 are exposed to the neutral extracellular medium in response to optogenetic stimulation which leads to an increase in fluorescence.
Most studies utilizing pH-sensitive fluorescent proteins in mammalian organisms are performed using cultured neurons or semi-intact preparations such as hippocampal slices (Rose et al., 2013; Watanabe et al., 2018). In vivo studies are rare and usually performed in translucent non-mammalian model systems such as Danio rerio and Drosophila melanogaster larvae or Caenorhabditis elegans (Poskanzer et al., 2003; Koudelka et al., 2016; Ventimiglia and Bargmann, 2017; Seitz and Rizzoli, 2019). Electrical stimulation can be used to trigger neurotransmission, but this is comparatively invasive and labor-intensive, particularly if it involves dissection. Alternatively, exposure to stimuli such as odors can be used. However, this is difficult to control, and limited to applications in sensory neurons (Choi et al., 2021). Thus, an all-optical solution that is not limited to certain cell types would be ideal, e.g., a combination of genetically encoded non-invasive tools for in vivo stimulation of neurons, and pH-sensitive fluorescent proteins. One possibility to manipulate neurotransmission is through transgenic expression of channelrhodopsins (ChRs), which are light-gated cation channels that can be used to depolarize neurons (Nagel et al., 2003, 2005; Boyden et al., 2005; Liewald et al., 2008). Light absorption leads to isomerization of the chromophore all-trans retinal (ATR) and opening of the channel pore. A variety of ChRs that are activated by different wavelengths have been discovered or engineered (Guru et al., 2015; Schild and Glauser, 2015; Chang, 2019). This enables multiplexing with both short- or long-wavelength absorbing fluorophores (Wabnig et al., 2015; Hawk et al., 2021; Vierock et al., 2021). In this work, we characterize two different combinations of ChRs with pH-sensitive fluorescent proteins in living C. elegans nematodes. We first tested pHuji, a recently described red fluorescent protein, together with the well described blue light-gated ChR2 (Nagel et al., 2005; Shen et al., 2014). To determine whether the increase in fluorescence during stimulation is dependent on SV fusion, we analyzed reduction-of-function mutants of synaptobrevin (SNB-1), which is an essential vesicular soluble N-ethylmaleimide-sensitive-factor attachment receptor (v-SNARE) and thus involved in SV fusion (Nonet et al., 1998; Liu et al., 2018). Similarly, to examine whether the rate of the fluorescence decay after stimulation depended on the rate of SV recycling, we analyzed mutants lacking the established SV recycling factors synaptojanin/UNC-26, endophilin/UNC-57 and synaptotagmin/SNT-1, which are involved in different steps of SV recycling, such as membrane bending and clathrin-uncoating (Jorgensen et al., 1995; Harris et al., 2000; Schuske et al., 2003; Kittelmann et al., 2013; Watanabe et al., 2018; Yu et al., 2018; Mochida, 2022). SNT-1 is the primary sensor of calcium for exocytosis, but also regulates endocytosis, by recruiting adapter complex 2 (AP2) to the plasma membrane (Poskanzer et al., 2003; Yu et al., 2013; Mochida, 2022). The pHuji approach could be used to estimate the extent of SV release but was unable to efficiently report recycling kinetics. We thus swapped both the actuator and the sensor to different excitation wavelengths. We used the recently described red-light activated ChR ChrimsonSA and the well-established green fluorescent pHluorin (Miesenbock et al., 1998; Oda et al., 2018; Seidenthal et al., 2022). Using this combination, we could stimulate and visualize SV exo- and endocytosis in an all-optical, non-invasive manner in vivo. We termed this approach the pH-sensitive optogenetic reporter of vesicle recycling (pOpsicle). We tested the pOpsicle method in cholinergic motor neurons and in the glutamatergic/tyraminergic interneuron RIM. pOpsicle should be applicable to most neuronal cell types and is, to our knowledge, the only all-optical approach to study SV recycling using ChRs and pH-sensitive fluorescent proteins in living animals to date. Our approach expands the possibilities to study SV recycling at the C. elegans neuromuscular junction (NMJ) which previously could only be done by indirect measurement of postsynaptic effects using electrophysiology, Ca2+ imaging, or by (non-)time-resolved EM (Liewald et al., 2008; Kittelmann et al., 2013; Wabnig et al., 2015; Steuer Costa et al., 2017; Yu et al., 2018).
For the expression of SNG-1 fusion constructs and channelrhodopsins in C. elegans, the punc-17 promotor (cholinergic motor neurons) and a short version of the ptdc-1 promotor (RIM interneurons) were used. pcDNA3-SypHluorin 4x (S4x) was a gift from Stephen Heinemann & Yongling Zhu (Addgene plasmid #370051; RRID:Addgene_37005). pJB14 (TOPO vector::2xpHluorin) was generated using the TOPO cloning kit (Thermo Fisher Scientific Inc., USA) by amplifying two copies of pHluorin cDNA from the pcDNA3-SypHluorin 4x (S4x) plasmid with primers oBJ51 (5′-ATATCGAACCGTCTTCAGATATGGATCTAGCCACC-3′) and oBJ62 (5′-TATATTCGCCGTCTTCTCCACCGCATGTGATTCGA GCTCC-3′). pJB10 (punc-17::sng-1::unc-54-3′UTR) was generated through Gibson assembly by digesting pRM348 with BmtI and BsiWI (punc-17 and backbone), by amplifying pAG52 (sng-1) with primers oBJ58 (5′-TCAGGAGGACCCTTGGCTAGATGGAGAACGTGCGTGCTTA TG-3′) and oBJ59 (5′-ATGACTCGAGCTAATAACCATATCCTTC CGACTGAG-3′) and by amplifying pAH03 (unc-54-3′UTR) with oBJ60 (5′-ATATGGTTATTAGCTCGAGTC ATGGTCGACAAG-3′), and oBJ61 (5′-AAACGCGCGAG ACGAAAGGGCCCAAACAGTTATGTTTGGTATATTGGG-3′). pJB11 (punc-17:: sng-1::2xpHluorin::unc-54-3′UTR) was generated by digestion of pJB10 and pJB14 with BbsI and subsequent ligation to introduce two copies of pHluorin cDNA into the sequence encoding the first intraluminal loop of SNG-1. pDisplay-pHuji was a gift from Robert Campbell (Addgene plasmid #615562; RRID:Addgene_61556). pDisplay-pHuji was amplified using primers oBJ104 (5′-GCAGAAGAAAACCATGGGCTG-3′) and oBJ105 (5′-CAGCCCATGGTTTTCTTCTGC-3′) to remove the BbsI restriction site to generate pJB24. pJB25 (punc-17::sng-1::pHuji::unc-54-3′UTR) was generated via Gibson assembly by digesting pJB10 with BbsI and amplifying pJB24 with primers oBJ107 (5′-ATATCGAAAAGTCTTCAGGTGGAGGTGGAAGTATGGTGAG CAAGGGCGAG-3′) and oBJ108 (5′-TATATTCGCCGTC TTCGGTGGAGGTGGAAGTCTTGTACAGCTCGTCCATG-3′) which contain the sequence for a GGGGS linker to add in front of the coding region of pHuji. pJB26 (punc-17::ChR2(H134R)::myc) was generated by amplifying ChR2(H134R)::myc using primers oBJ113 (5′-GAACGCTAGCACCACTAGATCCATCTAGAG-3′) and oBJ114 (5′-GCATGCTAGCCACCAGACAAGTTGGTAA-3′) which was introduced into pRM348 by restriction digest with NheI and subsequent ligation. pMSE01 (punc-17::ChrimsonSA::unc-54-3′UTR) was generated by amplifying pDV07 (punc-17::ChrimsonWT::unc-54-3′UTR) with oMSE16 (5′-CGAGTGGCTGCTGGCTTGCCCCGTGAT-3′) and oMSE017 (5′-ATCACGGGGCAAGCCAGCAGCCACTCG-3′) to introduce the point mutation (S169A). pMSE23 (ptdc-1s::ChrimsonSA::unc-54-3′UTR) was generated by amplifying ptdc-1s from pXY07 (ptdc-1s::GFP) with primers oMSE105 (5′-TCCCGGCCGCCATGGCCGCGATTTCTGTATGAGCCGCCCG-3′) and oMSE106 (5′-AAAGACTTTCGATGAATTACTTGGGCG GTCCTGAAAAATG-3′), amplifying the ChrimsonSA backbone from pMSE01 with oMSE107 (5′-CATTTTTCAGGACCGCCCAAGTAATTCATCGAAAGTCTTT CTATTTTCCGCATCTCTTGTTCAAGGGATTGG-3′) and oMSE108 (5′-CGGGCGGCTCATACAGAAATCGCGGCCATGGC GGCCG-3′) and combined the fragments using Gibson assembly. pMSE24 (ptdc-1s::sng-1::pHluorin::unc-54-3′UTR) was generated by amplifying the sng-1::pHluorin backbone from pBJ11 with oMSE114 (5′-AGGGTCGACCATGACTCGAGCTAATAACCATATCCTTC-3′) and oMSE115 (5′-GTAATTCATCGAAAGTCTTTCTATTTTCCG CATCTCTTGTTCAAGGGATTGG-3′) and with oMSE108 and oMSE113 (5′-GAAGGATATGGTTATTAGCTCGAGTCATGGT CGACCCT-3′) and fusing these fragments with the ptdc-1s using Gibson assembly.
Caenorhabditis elegans strains were kept under standard conditions on nematode growth medium (NGM) plates seeded with the Escherichia coli strain OP50, obtained from the Caenorhabditis Genetics Center (CGC, University of Minnesota, USA), at 20°C (Brenner, 1974). The N2 Bristol strain was provided by the CGC and used as wild type. Transgenic animals were generated as described previously (Fire, 1986). An overview of transgenic and mutant strains used or generated in this work can be found in Table 1. For experiments, well-fed L4 larvae were picked ∼18 h before the assays. For RIM experiments, only animals showing marker fluorescence were used. Animals were supplemented with ATR (Sigma-Aldrich, USA) by adding 100 μl OP50 containing 200 μM ATR to 10 ml NGM agar dishes. Experiments were performed on at least two separate days with animals picked from different plates.
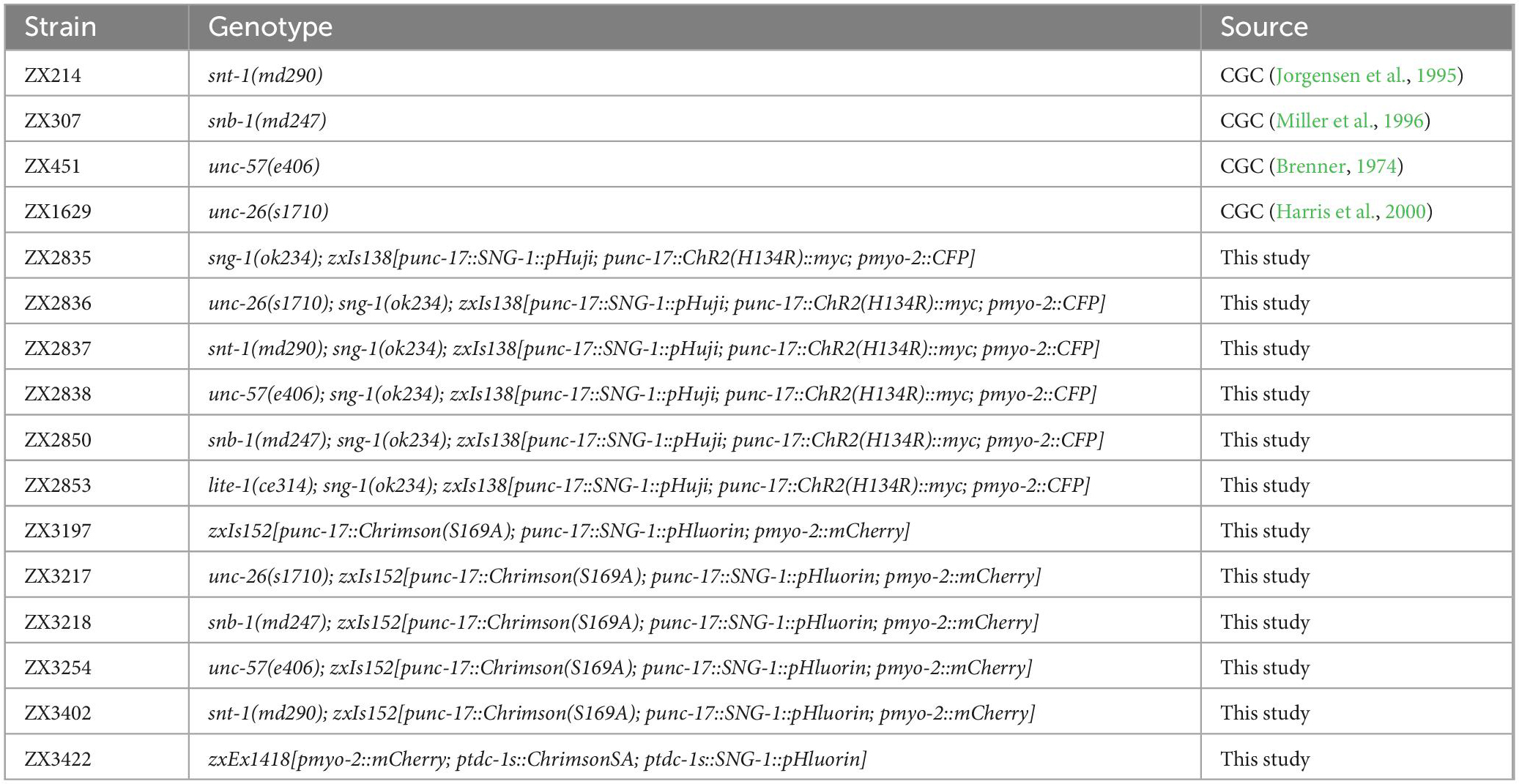
Table 1. Caenorhabditis elegans strains.
Body length assays were performed as described previously (Liewald et al., 2008; Seidenthal et al., 2022). Briefly, light from a 50 W HBO lamp was filtered with a 450–490 nm bandpass excitation filter for ChR2(H134R) and with a 568.5–591.5 nm filter for ChrimsonSA stimulation. ChR2(H134R) was stimulated at 340 μW/mm2. ChrimsonSA was stimulated and tested for crosstalk at a light intensity of 40 μW/mm2. Brightfield light was filtered with a 665–715 nm filter to avoid unwanted activation of channelrhodopsins. Videos of single animals were acquired and then analyzed using the WormRuler software (Seidenthal et al., 2022). Body length of each worm was normalized to the 5 s period before stimulation and values >120 or <80% of the initial body length were discarded as these are biomechanically impossible and result from artifacts in the background correction.
Videos of crawling animals were acquired as described previously (Vettkötter et al., 2022) and crawling speed measured using the multi-worm-tracker (MWT) setup (Swierczek et al., 2011). Animals were washed three times with M9 buffer to remove OP50 bacteria. They were then transferred to unseeded NGM plates and kept in darkness for 15 min. A light stimulus was applied using a custom-build LED ring (Alustar 3W 30°, ledxon, 623 nm) which was controlled by an Arduino Uno (Arduino, Italy) device running a custom-written Arduino script. Videos were acquired using a high-resolution camera (Falcon 4M30, DALSA) and crawling speed of single animals as well as reversal count (in bins of 10 s) were extracted using “Choreography” software (Swierczek et al., 2011) and summarized using a custom Python script.
For fluorescence imaging, animals were placed upon 7% agarose pads in M9 buffer. Animals were immobilized using a 20 mM Levamisole-hydrochloride (Sigma-Aldrich, USA) solution in M9 and visualized on an Axio Observer Z1 microscope (Zeiss, Germany) equipped with a 100× oil objective. Fluorescent proteins and channelrhodopsins were excited using a 460 and a 590 nm LED system (Lumen 100, Prior Scientific, UK) coupled via a beamsplitter. pHuji and ChR2(H134R) were excited using a double band pass filter (460–500 and 570–600 nm) combined with a 605 nm beam splitter (AHF Analysentechnik, Germany). 460 nm LED light to stimulate ChR2(H134R) was set to 340 μW/mm2 intensity. pHuji fluorescence was excited at 280 μW/mm2 and filtered using a 615–680 nm emission filter and visualized using an EMCCD camera (Evolve 512 Delta, Teledyne Photometrics, USA). pHluorin and ChrimsonSA were excited using a 450–490/555–590 nm double band pass filter combined with a GFP/mCherry beamsplitter (AHF Analysentechnik, Germany). 590 nm LED light intensity to stimulate ChrimsonSA and excite pHluorin was set to 40 μW/mm2. pHluorin fluorescence was filtered using a 502.5–537.5 nm band pass emission filter and visualized using a sCMOS camera (Kinetix 22, Teledyne Photometrics, USA). For increased ChrimsonSA stimulation at 280 μW/mm2, we used the same excitation filter as for pHuji experiments but combined with a FITC/TxRed double band pass beamsplitter and a 515–555 nm emission filter (AHF Analysentechnik, Germany). pHluorin fluorescence was again excited with 40 μW/mm2. The dorsal nerve cord (DNC) was visualized using the basal pHuji or pHluorin fluorescence. For cholinergic neurons, a region in the posterior third of the animal was imaged, where an abundance of synaptic puncta can be found. For RIM experiments, fluorescent neuronal extensions in the head region were visualized. Videos were captured using the μManager v.1.4.22 software (Edelstein et al., 2014). pHuji – pOpsicle experiments were performed with 50 ms exposure time (20 fps), pHluorin – pOpsicle experiments with 200 ms exposure (5 fps). Stimulation of channelrhodopsins was triggered using a custom written Autohotkey script to activate and deactivate LEDs. Representative images displaying entire worms were acquired using a 40× oil objective and stitched together using the ImageJ Stitching Plugin (Preibisch et al., 2009). The representative image of RIM neurons (Figure 5A) was made using the Z Project function to generate a projection of slices acquired throughout the head region.
Example images were processed, and fluorescence was quantified using ImageJ v1.53 (Schindelin et al., 2012). A region of interest (ROI) was placed on the DNC or RIM neuron using the Segmented Line tool. Pixel width of the line was adjusted according to the width of the fluorescent signal. A background ROI was set in close proximity to the imaging ROI, inside the worm (but avoiding gut autofluorescence) and fluorescence was quantified using the Multi Measure function. XY-drift was corrected using the Template Matching ImageJ plugin, if necessary. Animals that moved excessively or drifted in the focal plane were discarded. Fluorescence was normalized to the average fluorescence before stimulation (F0) to compare different animals:
A custom written python script was used for background subtraction, normalization and (if needed) filtering of animals according to whether they show a strong response during stimulation (available on GitHub3). For this, the maximum background corrected fluorescence during stimulation was calculated (as a moving average of 1 s). If this was higher than the average background corrected fluorescence before the stimulation +3 × standard deviations (of background corrected fluorescence before stimulation; minimum of two arbitrary units of fluorescence increase), the animal was counted as a strong responder (adapted from Choi et al., 2021); see Supplementary Figure 6A for example traces that fit or do not fit these parameters; animals not fitting the cut-off showed no discernable light-evoked effect on DNC fluorescence. Also, animals that showed an increase of the fluorescence after the end of the stimulus, or animals showing spontaneous events, were excluded. These measures were necessary for the calculation of fluorescence rise and decay kinetics because data from “non-responders” could not be properly fitted. Fluorescence was not corrected for bleaching since bleaching rates were difficult to estimate robustly in the presence of alternating light protocols. Attempts to correct for bleaching led to a progressive deviation toward the end of the acquisition. However, the measured background fluorescence bleached with a similar rate as the fluorescence in the DNC. Thus, subtraction of background fluorescence was sufficient to correct for bleaching in most cases. Fluorescence signal increases in the pOpsicle assay were calculated using the mean of the normalized fluorescence at the first second after stimulation (±SEM) for pHuji experiments or the mean of seconds 15–20 (±SEM) for pHluorin experiments. Regression analysis, to calculate the rate of fluorescence rise and decay, was performed using Graphpad Prism 9.4.1. The equations “Plateau followed by one-phase exponential association” (1) and “Plateau followed by one-phase exponential decay” (2) were fitted to the fluorescence increase during stimulation and decay after stimulation and the time constants τ calculated for each animal. The curve fits were constrained between minimum and maximum fluorescence values to receive reasonable τ values:
where. t, time (in seconds); t0, timepoint of experimental intervention; set to the first timepoint after the start or stop of stimulation; f0, value of f(t) at t0; set to 0 (for Equation 1) or maximum fluorescence values (for Equation 2, as a moving average of five frame intervals); Max, maximum fluorescence during or immediately after stimulation as a moving average of five frame intervals (for Equation 1); Min, minimum fluorescence after stimulation as a moving average of 5 s intervals (for Equation 2); τ, time constant (in seconds; higher τ values indicate a slower rise or decay).
As above, each fit was inspected. Individual fits that showed no increase during stimulation were discarded from analysis as they could not be fitted properly (Supplementary Figure 6A). Similarly, data sets that displayed an increase rather than a decay after stimulation were also discarded.
For the preparation of C. elegans primary cell culture, established protocols were adapted and modified (Christensen et al., 2002; Strange et al., 2007). Gravid adult worms were grown on enriched peptone plates with nystatin (NEP agar) seeded with Na22 E. coli (CGC; Zhang et al., 2011). Worms were washed off the plate using double-distilled water (ddH2O) and transferred to 15 ml centrifuge tubes. A total of 2 ml of household bleach as well as 1 ml of 5 M NaOH solution were added to 7 ml of worm suspension. The solution was vortexed for at least 5 min at maximum speed to get rid of adult worm bodies. All the following steps were performed under a sterile workbench. The solution, now containing only eggs, was centrifuged at 500 g for 1 min. Excess liquid was removed, and the pellet was resuspended in ddH2O. Washing was repeated three times. The egg pellet was resuspended in 500 μl freshly thawed chitinase (1 U/ml, Sigma-Aldrich, USA) and transferred to a 1.5 ml tube. The tube was placed into a shaker for 90 min at room temperature to digest the chitin shell of the eggs. The chitinase reaction was stopped with 800 μl L-15 full medium (Gibco, USA) containing 10% fetal calf serum (FCS) as well as Pen/Strep (50 U/ml penicillin + 50 μg/ml streptomycin; Sigma-Aldrich, USA). After centrifugation at 900 g, excess liquid was discarded, and the pellet was resuspended in 500 μl L-15 full medium. Using a 2 ml syringe with an 18-gauge needle, the solution was aspirated and released back into the tube 15–20 times to dissociate the cells. After dissociation, 1 ml L-15 full medium was added to the tube and taken up into the syringe. With the cell solution inside the syringe, the needle was replaced by a 5 μm filter (Millipore, Germany). The solution was released through the filter into a fresh tube. The original tube was refilled with 1 ml L-15 full medium, the filter was replaced by the needle and the procedure was repeated to release the solution into another tube. This was repeated four to six times, depending on the initial number of eggs (more eggs = less repetitions). Filtered cell solutions were centrifuged at 900 g and most of the supernatant was discarded. Cell pellets were resuspended in the remaining medium and pooled. A total of 500–1,000 μl L-15 full medium were added to the suspension and the solution was seeded on 1–2 peanut lectin (Sigma-Aldrich, USA) coated glass bottom petri dishes (ibidi, Germany). Petri dishes were filled with 1 ml L-15 full medium and cells were allowed to adhere for 24 h in a 20°C sterile incubator (Memmert, Germany) before exchanging the medium. Treatment with ATR was performed after two days. Medium was replaced with a 10 μM ATR solution in L15 full medium and the cells incubated for at least 12 h at 20°C. Prior to measurement, cells were washed two times with L-15 full medium without ATR. Neurons were imaged three to four days after seeding. The filter setup was identical to the one used to visualize pHluorin in intact animals, yet neurons were visualized using a 40× objective and 200 ms exposure time. To reduce bleaching during NH4Cl treatment frame rate was reduced to one frame every 5 s. The camera shutter and 460 nm LED were controlled using a custom-written Arduino script (kindly provided by Ichiro Aoki). Buffers used to either de-quench or quench pHluorin fluorescence were adapted from Dittman and Kaplan (2006) and added manually by pipetting 1 ml of the respective solution onto the petri dish and then removing the same amount of liquid. Before starting a new acquisition, cells were washed three times using the control saline buffer (pH 7.4). Fluorescence was quantified as in living animals. A ROI was set on top of neurite extensions using the ImageJ “Segmented line” tool with a background ROI set in proximity. The releasable fraction of SVs (RF) was calculated for individual cells as the ratio of the mean normalized fluorescence during optogenetic stimulation to the normalized fluorescence during treatment with NH4Cl containing buffer (mean of 85–90 s; Egashira et al., 2016):
Statistical analysis and plotting of graphs were done using Graphpad Prism 9.4.1. Unpaired t-tests were performed if two normally distributed datasets were compared and one-way ANOVA (with Dunnett’s or Tukey’s correction) or two-way ANOVA (with Šídák’s correction) for three or more datasets. Fluorescence rise and decay constants were compared using the Mann–Whitney test (for two datasets) and the Kruskal–Wallis tests (with Dunn’s correction) or by applying multiple Mann–Whitney tests (with Holm–Šídák’s correction) for three or more datasets.
To enable an all-optical method for analysis of SV exo- and endocytosis, we used proteins with presumably low optical crosstalk. First, we tested the recently described red fluorescent pHuji protein which shows a 22-fold increase of fluorescence when transferred from intravesicular to extracellular pH (Shen et al., 2014), combined with the blue light-gated ChR2(H134R; hereafter called ChR2) for stimulation of neurotransmission (Nagel et al., 2005). Overlap of the pHuji and ChR2 excitation spectra is minimal (Azimi Hashemi et al., 2014; Shen et al., 2014; Lambert, 2019; Supplementary Figure 1A). C. elegans proteins can be targeted to, e.g., cholinergic or GABAergic motor neurons using specific promoters, and for subcellular localization of pHuji we chose the ubiquitous (in neurons) integral SV membrane protein synaptogyrin-1 (SNG-1; Zhao and Nonet, 2001; Abraham et al., 2006). SNG-1 was previously used to target proteins to SVs (Liu et al., 2019; Vettkötter et al., 2022). We inserted pHuji into the second intraluminal loop of SNG-1 (Figure 1) and co-expressed it with ChR2 in cholinergic motor neurons using the promotor of unc-17, encoding the vesicular acetylcholine transporter (Alfonso et al., 1993; Miller et al., 1996; Liewald et al., 2008). SNG-1::pHuji could be observed throughout the cholinergic nervous system, i.e., nerve ring, ventral and dorsal nerve cords (VNC/DNC; Supplementary Figure 1B) and dorsal neuronal extensions (Supplementary Figure 1C). Fluorescent puncta in the DNC indicate accumulation of SNG-1::pHuji at NMJs (Sieburth et al., 2005). Next, we analyzed whether neurotransmission can be activated in these animals using ChR2, which typically leads to muscle contraction and reduction of body length through acetylcholine release (Liewald et al., 2008). As expected, blue light caused a decrease of body length of animals when grown in the presence of ATR (Supplementary Figure 1D), but not in no-ATR controls. Next, we quantified fluorescence of SNG-1::pHuji in the posterior DNC where NMJs are abundant (Sieburth et al., 2005) before, during and after ChR2 photostimulation (Supplementary Figures 1C, E). Since pHuji fluorescence was relatively dim, we could not quantify fluorescence during the stimulation period, as blue light increased autofluorescence and/or further excited pHuji, thus erroneously increasing the measured signal. Alternating light protocols also failed, likely due to the photoswitching nature of pHuji (Liu et al., 2021). However, we could compare the fluorescence before and after stimulation. Animals treated with ATR showed a significantly increased fluorescence after stimulation (19 ± 3% ΔF/F0), compared to animals without ATR (2 ± 2%, ***p < 0.001; Supplementary Figure 1E). This increase gradually declined toward the fluorescence level in animals grown without ATR, most likely indicating recycling of externalized SNG-1::pHuji (and thus of SVs). We termed the pHuji-ChR2 combination “red pOpsicle.” To determine whether the red-pOpsicle assay reports on altered SV exo- and endocytosis, we crossed the line carrying the ChR2 and SNG-1::pHuji transgene into mutants known to affect the SV cycle (see section “1. Introduction”; Supplementary Figure 2). Compared to wild type, snb-1(md247) mutants showed a significantly lower fluorescence increase after stimulation with blue light [wild type: 15 ± 2%, snb-1(md247): 2 ± 2%; Supplementary Figures 2A, B], indicating lower amounts of SVs containing SNG-1::pHuji being exocytosed. Deletion mutants of SV recycling factors, unc-26(s1710) and unc-57(e406), displayed a higher increase in pHuji fluorescence than wild type animals [Supplementary Figure 2C: wild type 17 ± 3%, unc-26(s1710) 31 ± 5%, *p < 0.05; Supplementary Figure 2D: wild type 16 ± 3%, unc-57(e406) 30 ± 4%, *p < 0.05]. This was surprising since both mutants were previously shown to be depleted of ready-to-release SVs and should therefore not be able to externalize as many SVs as wild type animals (Harris et al., 2000; Schuske et al., 2003). Nevertheless, they showed prolonged increased fluorescence during acquisition compared to wild type. snt-1(md290) knockout mutants displayed a similar signal increase and decay trend as wild type [Supplementary Figure 2E: wild type 14 ± 3%, snt-1(md290) 18 ± 3%, ns p > 0.05]. We calculated the kinetics of fluorescence decay after stimulation using a one-phase exponential decay model (Supplementary Figures 2F–H). However, no significant difference in the rate of the fluorescence decay was observed. We thus wondered if the pHuji/ChR2 combination works properly. One concern was that photophobic reactions to blue light, mediated by the photoreceptor LITE-1, may affect the measured fluorescence increase and recycling rate (Edwards et al., 2008). However, we did not observe any significant difference between lite-1(ce314) knockout mutants and wild type animals in the pOpsicle assay, independent of ATR treatment (Supplementary Figures 2I, J). We suggest that the previously reported photo-switching behavior of pHuji in blue light (Liu et al., 2021) likely explains the slight increase in fluorescence in animals without ATR after stimulation and could also influence the decay kinetics in animals with ATR (Supplementary Figures 1E, 2I). Since pHuji appeared unable to properly track SV recycling, we investigated another protein combination for the pOpsicle assay.
We tested the more commonly used green fluorescent pHluorin as a pH-sensitive fluorophore which moreover features brighter fluorescence (Miesenbock et al., 1998; Li et al., 2022). Since pHluorin has overlapping excitation spectra with ChR2, we needed to exchange ChR2 with a red light activated channelrhodopsin. The recently described ChrimsonSA (for super red-shifted and accelerated), which is a mutated variant (S169A) of Chrimson, seemed to be a suitable candidate (Oda et al., 2018). Previously, we could show that ChrimsonSA can be used to depolarize C. elegans motor neurons upon stimulation with red light (Seidenthal et al., 2022; Figure 2A). Two copies of pHluorin were inserted into the second intraluminal loop of SNG-1 as multiple pHluorin insertions have been shown to increase the signal-to-noise ratio (Zhu et al., 2009). We co-expressed this fusion construct with ChrimsonSA in cholinergic motor neurons and observed basal pHluorin fluorescence throughout the cholinergic nervous system (Figure 2B). SNG-1::pHluorin also localizes to neuronal extensions of DA and DB neurons innervating dorsal muscle cells (Figure 2C). Fluorescent puncta in the DNC indicate an accumulation of pHluorin at NMJs. We then tested for crosstalk of pHluorin imaging light with ChrimsonSA activation using contraction assays (Figures 2D, E). The intensity of blue light used to visualize pHluorin (470 nm, 40 μW/mm2) did not lead to a significant decrease in body length of animals treated with ATR. In contrast to this, 40 μW/mm2 of 580 nm light was sufficient to trigger contraction of body wall muscles. This indicates that ChrimsonSA is functioning properly to depolarize cholinergic motor neurons yet is not pre-activated by blue light. Next, we performed pOpsicle assays with pHluorin animals (“green pOpsicle”). We observed spontaneous pHluorin increases in some animals, independent of treatment with ATR or light stimulation (Supplementary Figure 3A), verifying that pHluorin imaging can report on exocytosis. During photostimulation, DNC fluorescence gradually increased by 21 ± 2% upon continuous 590 nm stimulation in ATR-treated animals (Figures 2F–I, see Supplementary Figure 3B for a representation of all experiments, and Supplementary Movie 1). The signal approached a plateau after ∼10 s, indicating that most SNG-1::pHluorin was externalized at this point. Animals without ATR showed no increase upon stimulation (0 ± 1%, ***p < 0.001; Figures 2H, I). The increase in fluorescence was especially high in synaptic puncta along the DNC, indicating locations of highly active SV release sites (Figures 2F, G). Following the end of the stimulation, like in red pOpsicle experiments, the signal gradually decreased toward levels before stimulation. We also tested a higher light intensity to stimulate ChrimsonSA (280 μW/mm2) and observed a slightly higher increase in fluorescence yet overall similar trend (Supplementary Figure 3C; 30 ± 3%, *p < 0.05 compared to 40 μW/mm2). Similarly, stronger light stimulation led to a linearly increasing contraction in behavioral experiments (Supplementary Figure 3D). For subsequent analyses we used 40 μW/mm2 since it seemed sufficient to release most SVs and stronger stimulation may trigger unphysiological effects.
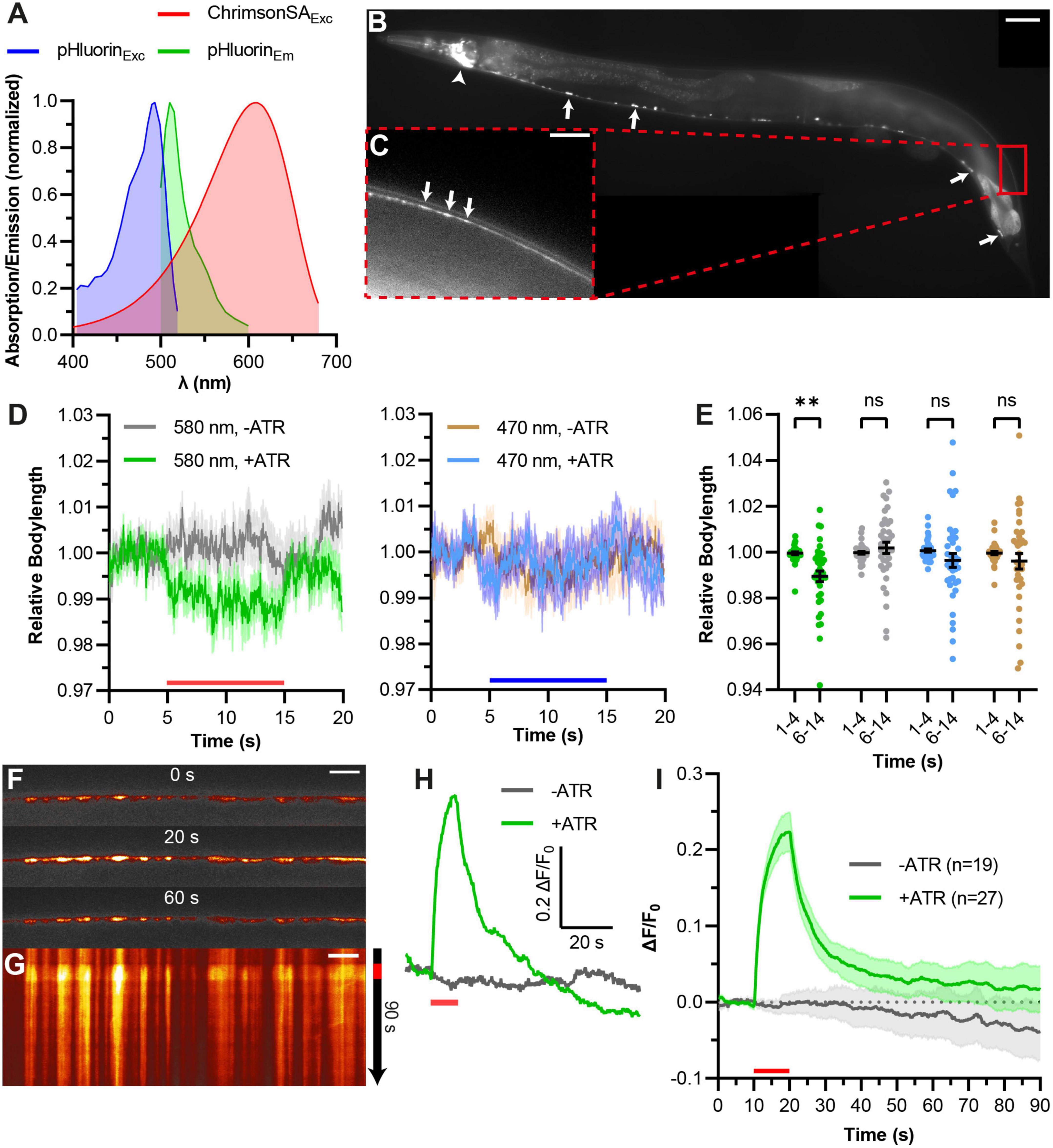
Figure 2. Combining ChrimsonSA with pHluorin (“green pOpsicle”) for stimulation and visualization of exo- and endocytosis. (A) Relative excitation and emission spectra of ChrimsonSA and super-ecliptic pHluorin, normalized to the maximum absorption/emission amplitude. (B) Representative image of C. elegans expressing SNG-1::pHluorin in cholinergic neurons. Arrows represent cell bodies of A- and B-type motor neurons (ventral nerve cord). Arrowhead represents cholinergic neurons in the head ganglia. 40× magnification. Scale bar, 50 μm. (C) Enlarged view of the DNC in panel (B). Arrows represent fluorescent puncta, representing en passant synaptic terminals. Scale bar, 10 μm. (D) Mean relative body length (±SEM) of animals expressing ChrimsonSA and pHluorin optionally treated with ATR, as indicated, normalized to the average body length before stimulation. A 10 s continuous light pulse (580 or 470 nm as indicated, 40 μW/mm2; indicated by red and blue bars) was applied after 5 s. Number of animals (n), accumulated from N = 3 biological replicates. (E) Statistical analysis of animals shown in panel (D). Each dot represents the mean normalized body length of a single animal during the indicated period. Two-way ANOVA with Šídák’s correction (nsp > 0.05, **p < 0.01). (F) Representative images of pHluorin fluorescence in the DNC of an animal treated with ATR at different time points during the pOpsicle assay, as indicated. A 10 s continuous light pulse (590 nm, 40 μW/mm2) was applied after 10 s. The ImageJ Smart Look-Up-Table was used. 100× magnification. Scale bar, 5 μm. (G) Kymograph representing the change in fluorescence of the DNC represented in panel (F) over a time course of 90 s. The red bar indicates the period of light stimulus. Scale bar, 5 μm. (H) Representative traces of normalized DNC fluorescence of individual animals with or without ATR. A 10 s continuous light pulse (590 nm, 40 μW/mm2) was applied after 10 s (red bar). (I) Mean (±SEM) change in DNC fluorescence of animals supplemented with and without ATR. A 10 s continuous light pulse (590 nm, 40 μW/mm2) was applied after 10 s (red bar). Number of animals (n), accumulated from N = 4 (+ATR), and N = 3 (–ATR) biological replicates.
To further characterize parameters of the “green pOpsicle” assay, e.g., releasable fraction of total pHluorin, we replicated it in neurons of primary cell cultures from dissociated C. elegans embryos (Supplementary Figures 4A, B). pHluorin fluorescence could be observed in neurite extensions and in cell bodies, surrounding nuclei (Supplementary Figure 4A). This fluorescence increased in some neurons when illuminated (and when treated with ATR) even though the signal size was lower than in intact animals, where this was the case for each individual recording (Figure 2I and Supplementary Figure 4B). A total of 18 of 52 neurons showed a strong response (for a definition of “strong response,” see section “Materials and methods”). Next, we calculated the fraction of releasable SVs by ChrimsonSA stimulation. To this end, we first stimulated cultured neurons with light and then applied a pH 7.4 NH4Cl buffer (Supplementary Figure 4C), which can be used to increase the pH within SVs and calculate the SV-associated portion of pHluorins (Sankaranarayanan et al., 2000; Dittman and Kaplan, 2006; Rose et al., 2013; Egashira et al., 2016). To minimize bleaching, we reduced the exposure to blue light by decreasing the frame rate of pHluorin acquisition. When exposed to NH4Cl (pH 7.4), pHluorin fluorescence in neurites rapidly increased (Supplementary Figures 4C–E). The same culture was then treated with a low pH buffer (pH 5.6) to quench the surface fraction of pHluorin (Supplementary Figures 4C, F). The fluorescence immediately decreased to levels below the basal fluorescence, demonstrating the pH-dependency of pHluorin fluorescence. The fraction of releasable SVs was determined by calculation of the ratio of maximum fluorescence during light stimulation and during NH4Cl treatment (Supplementary Figure 4G): 10 s continuous light stimulation released 20.20% (±3.72%) of SNG-1::pHluorin molecules attached to SVs, which is comparable to previous measurements in hippocampal slices that used synaptophysin::pHluorin as a reporter (Rose et al., 2013). To test a different, less vigorous stimulation regime, which may be more reminiscent to endogenous activity, we applied 2 Hz pulsed light (Supplementary Figure 4C). This led to a reduced release of SVs compared to continuous light (Supplementary Figure 4G, 10.61 ± 1.87%). Even though the fraction of releasable SVs by optogenetic stimulation is within a typical range compared to other reports, we conclude that in intact animals the signal size is higher and the assay more reliable in living animals indicating that a higher portion of SVs is exocytosed (Figure 2I and Supplementary Figure 4B).
Next, we used green pOpsicle to analyze mutants affecting SV fusion (snb-1(md247), snt-1(md290)) and/or recycling (unc-26(s1710), unc-57(e406), snt-1(md290)) (Figure 3). In snb-1(md247) mutants, the signal increase during stimulation was almost completely abolished (Figures 3A, B and Supplementary Figure 5A). Only 2 of 19 animals showed a relevant response (compared to 23/23 in wild type), demonstrating that the increase of fluorescence during stimulation reports on SV exocytosis. Synaptojanin (unc-26(s1710)) mutant animals also exhibited a reduced signal compared to wild type (Figures 3C, D and Supplementary Figure 5B). Thus, green pOpsicle faithfully revealed the expected depletion of SVs in unc-26(s1710) mutants (Harris et al., 2000), unlike pHuji that displayed a higher fluorescence after stimulation (Supplementary Figure 2C). To allow calculating the kinetics of fluorescence rise and decay, animals which showed no significant increase in fluorescence or no decay after stimulation were removed from our analyses (Supplementary Figure 6A). We observed a significantly slower rise of fluorescence in unc-26(s1710) mutant compared to wild type animals, as could be seen by the increased time constants of the signal curves obtained from single animals, when fitted to a one-phase exponential association kinetic (τRise, wild type = 2.3 s, τRise, unc–26 = 3.6 s; Figure 3E). This could indicate an exocytosis defect, or it could be due to the reduced number of SVs that are available for fusion in the unc-26(s1710) mutant (Dong et al., 2015). The kinetics of the fluorescence decay after stimulation showed a strong reduction, as expected for the SV recycling mutant (Figure 3F). Consequently, the calculated time constants of one-phase exponential decay were significantly increased in unc-26(s1710) mutants (τDecay, wild type = 15.2 s, τDecay, unc–26 = 24.2 s), closely matching previous results measured in mammalian cortical neurons lacking synaptojanin after strong stimulation (Cao et al., 2017). Wild type animals on the other hand showed decay time constants which are in the range of previous measurements in C. elegans sensory neurons or mammalian hippocampal neurons (Kwon and Chapman, 2011; Ventimiglia and Bargmann, 2017; Watanabe et al., 2018; Li et al., 2022). Endophilin (unc-57(e406)) mutants exhibited similar trends as unc-26(s1710) with a significantly reduced signal and significantly slower association and decay kinetics compared to wild type (τRise, wild type = 2.7 s, τRise, unc–57 = 3.3 s, τDecay, wild type = 15.2 s, τDecay, unc–57 = 24.7 s; Figures 3G–J and Supplementary Figure 5C). The time constants obtained for unc-57(e406) mutants closely matched those of unc-26(s1710) mutants, in line with the function of synaptojanin and endophilin in similar cellular processes (Harris et al., 2000; Dong et al., 2015; Watanabe et al., 2018). Synaptotagmin [snt-1(md290)] mutants also displayed a smaller increase in fluorescence (Figures 3K, L and Supplementary Figure 5D). The time constants of fluorescence rise were significantly increased in mutant animals (τRise,wild type = 3.1 s, τRise,snt–1 = 5.6 s; Figure 3M), demonstrating the role of SNT-1 in SV fusion (Liewald et al., 2008; Yu et al., 2013). We further observed a significantly delayed decrease of fluorescence after stimulation, indicating that SNT-1 is involved in SV recycling at C. elegans NMJs (Poskanzer et al., 2003; Mochida, 2022; τDecay, wild type = 11.1 s, τDecay, snt–1 = 31.1 s; Figure 3N). Since the increase in fluorescence was generally lower in mutant animals, we wondered whether higher time constants of decay were caused by a lower activation of synaptic transmission rather than decreased SV recycling rates. We thus compared decay time constants of individual animals with the respective signal size. However, there was no significant correlation within any of the analyzed genotypes (Supplementary Figures 6B–D). Therefore, recycling rates likely do not depend on the amount of SV fusion in this assay.
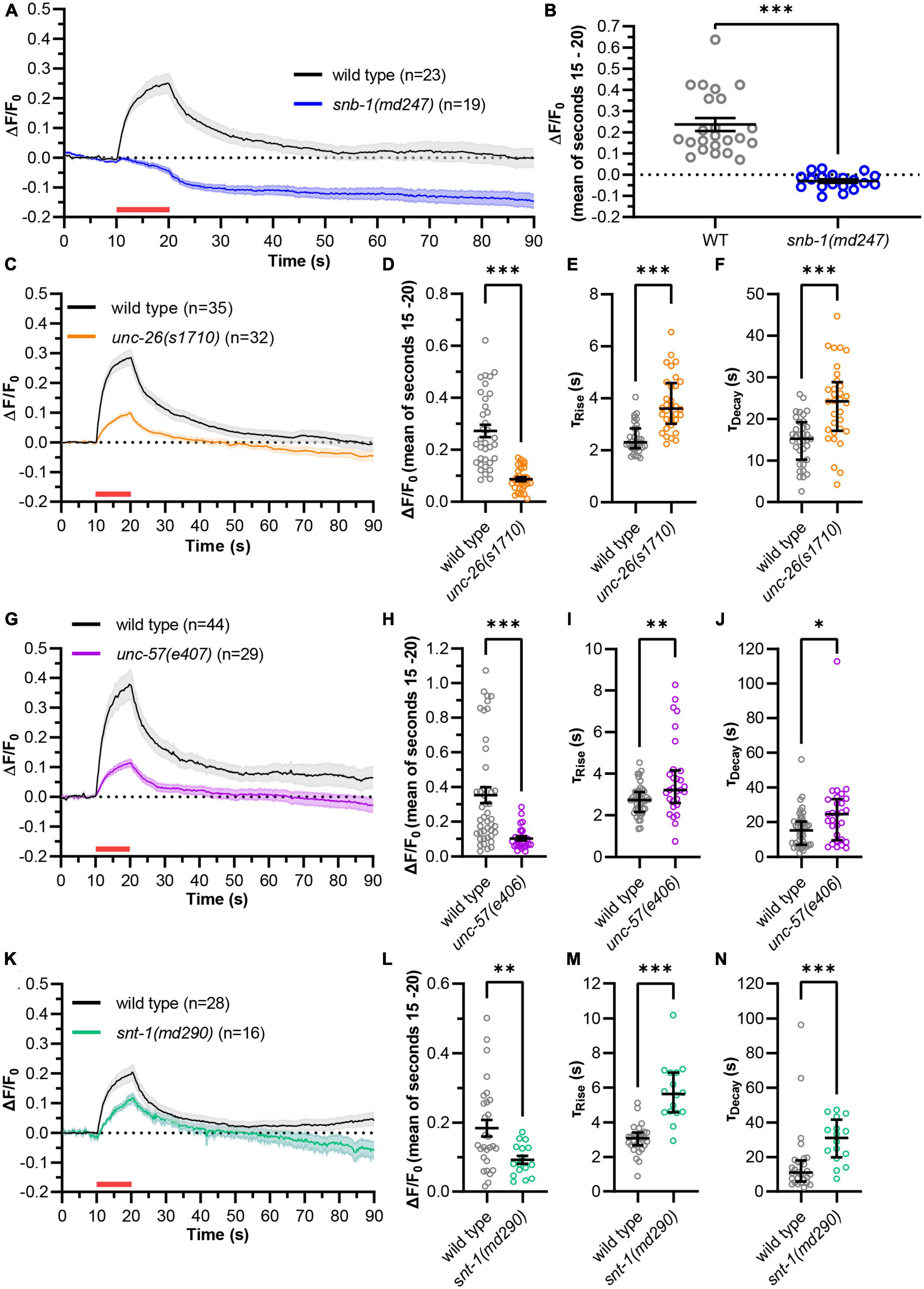
Figure 3. “Green” pOpsicle reports on mutations affecting SV fusion and endocytosis. (A,C,G,K) Mean (±SEM) change of fluorescence of SNG-1::pHluorin co-expressed with ChrimsonSA in cholinergic motor neurons. DNC of wild type and mutant animals, as indicated. A 10 s continuous light pulse (590 nm, 40 μW/mm2; indicated by a red bar) was applied after 10 s. Number of animals (n), accumulated from N = 4–6 biological replicates. (B,D,H,L) Fluorescent signal of individual wild type and mutant animals at the end of stimulation (15–20 s). Mean (±SEM). Unpaired t-test (**p < 0.01, ***p < 0.001). (E,I,M) Calculated fluorescence rise constants of single animals using a one-phase exponential fit during stimulation (10–20 s). Median with interquartile range. Mann–Whitney test (**p < 0.01, ***p < 0.001). (F,J,N) Calculated fluorescence decay constants of single animals using a one-phase exponential fit after stimulation (20–90 s). Median with interquartile range. Mann–Whitney test (*p < 0.05, ***p < 0.001). In panels (C–N), animals showing an increase <3 standard deviations during stimulation, or no decay of fluorescence following stimulation, were excluded [wild type: 4 of 111 animals, unc-26(s1710): 7 of 39, unc-57(e406): 18 of 47, snt-1(md290): 23 of 39].
Continuous photostimulation induces maximal depolarization and transmitter release, likely causing bulk endocytosis as the extreme form of ultrafast endocytosis (Kittelmann et al., 2013). To assess whether less vigorous, possibly more physiological activation also affects slower recycling, we applied 2 Hz pulsed stimulation (100 ms light pulses; Figure 4). The mutant strains again showed significantly reduced signal amplitudes compared to wild type (Figures 4A, B and Supplementary Figure 7) but no significantly increased time constants of fluorescence rise (τRise, wild type = 4.2 s, τRise, unc–26 = 6.0 s, τRise, unc–57 = 4.7 s, τRise, snt–1 = 4.7 s; Figure 4C). When comparing pulsed and continuous stimulation we observed a tendency toward decreased fluorescence amplitudes and significantly increased rise time constants, indicating that pulsed stimulation leads to a reduced activation of neurotransmission (Supplementary Figures 8A, B). For the recycling phase, pulsed stimulation again resulted in significantly larger decay time constants in unc-57(e406) and unc-26(s1710) mutants compared to wild type (τDecay,wild type = 20.9 s, τDecay, unc–26 = 41.1 s, τDecay, unc–57 = 45.1 s; Figure 4D), however, snt-1(md290) mutant animals showed no significant difference (τDecay, snt–1 = 28.6 s). Possibly, SNT-1 is dispensable for recycling at lower levels of stimulation. Decay time constants for pulsed stimulation were significantly increased in unc-57(e406) and unc-26(s1710) mutants indicating that a compensatory mechanism, which is independent of endophilin and synaptojanin, may be activated after strong stimulation (Supplementary Figure 8C).
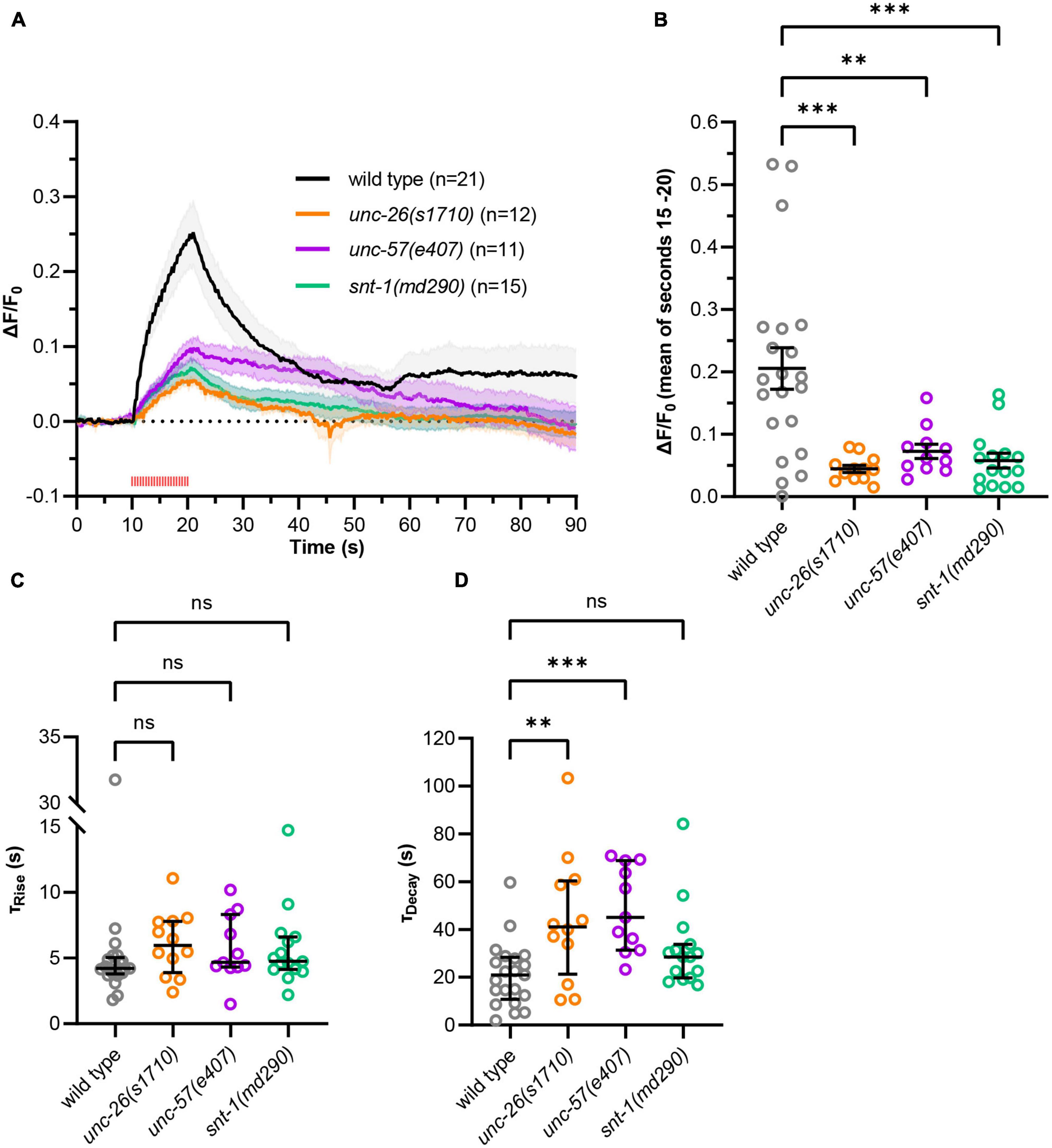
Figure 4. Using pulsed, more physiological optogenetic stimulation. (A) As in Figure 3, but using 2 Hz pulsed light stimulation (100 ms pulses, 590 nm, 40 μW/mm2, red tick marks) applied after 10 s for 10 s. Using this stimulation regime, more animals were excluded from analysis [wild type: 17 of 38 animals, unc-26(s1710): 18 of 30, unc-57(e406): 22 of 33, snt-1(md290): 39 of 54] accumulated from N = 5–6 biological replicates. (B) Fluorescent signal of individual wild type and mutant animals at the end of stimulation (15–20 s). Mean (±SEM). One-way ANOVAs with Dunnett’s correction (**p < 0.01, ***p < 0.001). (C) Calculated fluorescence rise constants of single animals using a one-phase exponential fit during stimulation (10–20 s). Median with interquartile range. Kruskal–Wallis test with Dunn’s correction (nsp > 0.05). (D) Calculated fluorescence decay constants of single animals using a one-phase exponential fit after stimulation (20–90 s). Median with interquartile range. Kruskal–Wallis test with Dunn’s correction (nsp > 0.05, **p < 0.01, ***p < 0.001).
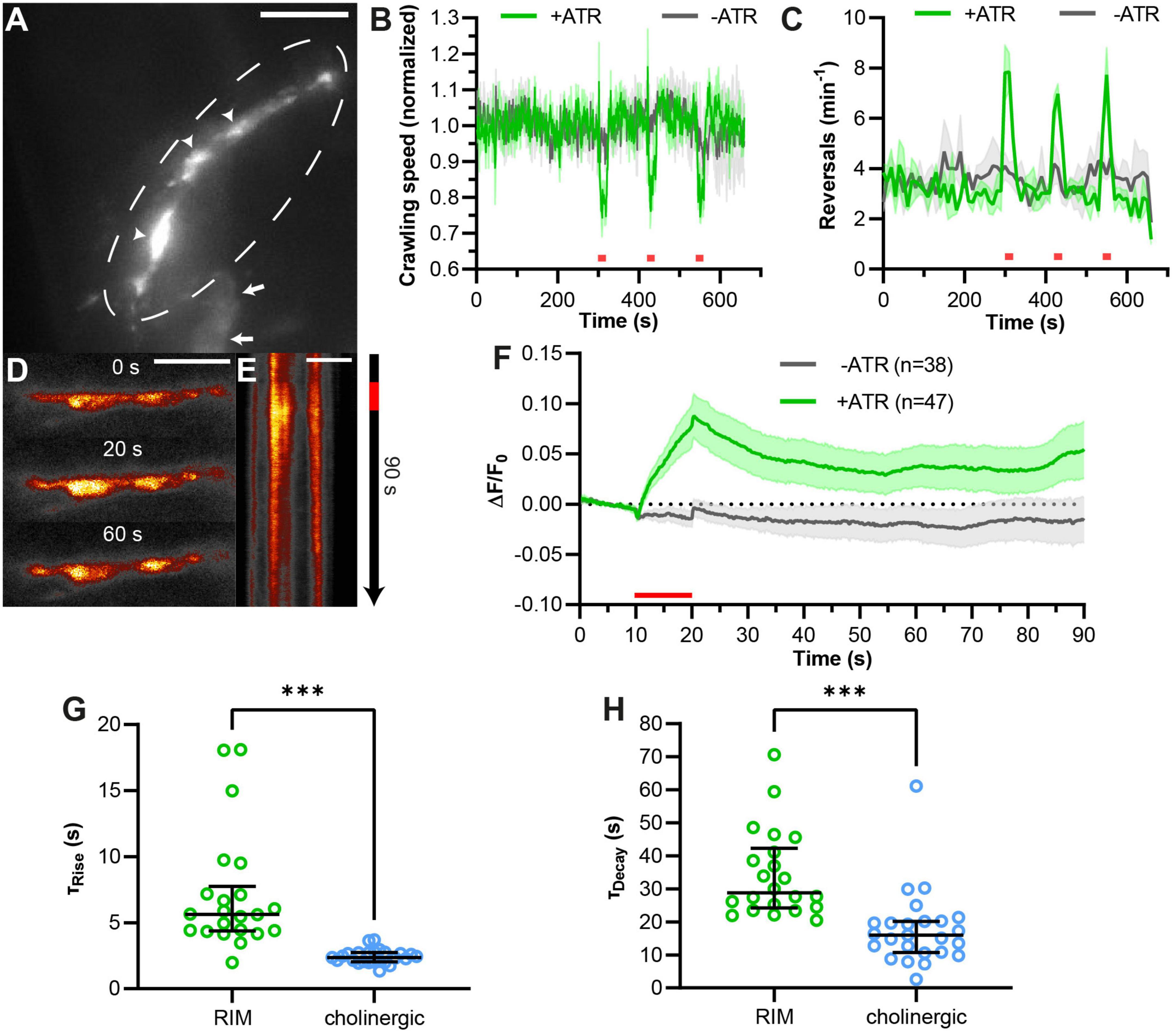
Figure 5. pOpsicle assay in the RIM interneuron pair. (A) Representative Z-projected image of C. elegans expressing SNG-1::pHluorin in RIM neurons using the promotor of tdc-1. 40× magnification. Arrows represent cell bodies of RIM neurons. Circle represents RIM axon in the nerve ring. Arrowheads represent fluorescent puncta, representing en passant synaptic terminals. 40× magnification. Scale bar, 5 μm. (B) Mean crawling speed (±SEM) of animals expressing ChrimsonSA and pHluorin in RIM neurons normalized to the average before the first light pulse. Three 20 s light pulses (623 nm, 400 μW/mm2) were applied at 300, 420, and 540 s as indicated by red bars. (C) Mean (±SEM) number of reversals per minute per animal. Light stimulation as in panel (B). In panels (B,C), N = 3 populations of animals were tested. (D) Representative images acquired at different time points during the pOpsicle assay, pHluorin fluorescence in RIM neurons, animal treated with ATR. A 10 s continuous light pulse (590 nm, 40 μW/mm2) was applied after 10 s. The ImageJ Smart Look-Up-Table was used. 100× magnification. Scale bar, 5 μm. (E) Kymograph representing the change in fluorescence in RIM neurons as shown in panel (D) over a time course of 90 s. The red bar indicates the period of light stimulus. Scale bar, 5 μm. (F) Mean (±SEM) pHluorin fluorescence in RIM neurons of animals with and without ATR. A 10 s continuous light pulse (590 nm, 40 μW/mm2) was applied after 10 s. Number of animals (n), accumulated from N = 5 (+ATR) or N = 4 (–ATR) biological replicates. (G) Comparison of fluorescence rise constants of single animals expressing pHluorin and ChrimsonSA in RIM neurons or in cholinergic neurons, using a one-phase exponential fit during stimulation (10–20 s). Median with interquartile range. Mann–Whitney test (***p < 0.001). (H) Comparison of fluorescence decay constants of single animals as in panel (G), using a one-phase exponential fit after stimulation (20–90 s). Median with interquartile range. Mann–Whitney test (***p < 0.001). (G,H) Only animals showing a strong response during stimulation and a decay of fluorescence after stimulation were taken into consideration (RIM: 22 of 47 animals, cholinergic neurons: 25 of 27 animals as depicted in Figure 2).
While the green pOpsicle assay worked well in cholinergic motor neurons, it remained to be shown that this system works in other neuronal cell types. The RIM interneuron pair integrates signals from sensory neurons to regulate forward and reversal locomotion, using gap junctions as well as glutamate and tyramine signaling (Piggott et al., 2011; Li et al., 2020; Sordillo and Bargmann, 2021). We expressed SNG-1::pHluorin and ChrimsonSA in RIM using the tdc-1 promotor. Green fluorescence could be observed in neuronal extensions surrounding the pharynx, suggesting correct localization of SNG-1::pHluorin (Figure 5A). To explore ChrimsonSA functionality in this neuron pair, we measured animal crawling speed (Figure 5B). Optogenetic depolarization of RIM neurons previously induced reversals and reduced crawling speed (Guo et al., 2009; Li et al., 2020; Sordillo and Bargmann, 2021). Consistent with this, illumination with red light slowed down crawling speed of RIM pOpsicle animals treated with ATR and the number of reversals was increased (Figures 5B, C). This indicated that ChrimsonSA depolarized RIM neurons when activated. Consequently, SNG-1::pHluorin fluorescence in synaptic puncta was significantly increased by ChrimsonSA activation (Figures 5D, E, Supplementary Figure 9, and Supplementary Movie 2), while control animals without ATR showed no change (+ATR 6 ± 2%, −ATR −1 ± 1%, ***p < 0.001; Figure 5F). The signal increase was significantly slower than in cholinergic neurons (τRise, RIM = 5.7 s, τRise, cholinergic = 2.4 s; Figure 5G), as was the decay of fluorescence following the end of stimulation (τDecay, RIM = 42.3 s, τDecay, cholinergic = 16.0 s; Figure 5H). These results indicate that different classes of neurons may have diverging kinetics of SV exo- and endocytosis in C. elegans. Thus, the pOpsicle assay can be adapted to different neuronal cell types.
Here, we present the first all-optical method to investigate SV recycling in vivo by combining pH-sensitive fluorescent proteins with ChRs. With pOpsicle, factors that influence the extent and rate of exo- and endocytosis can be investigated with minor experimental effort and equipment. We described two approaches using different pH-sensitive fluorescent proteins. pHuji could only be used to quantify the extent of exocytosis after stimulation. The low quantum yield and photoswitching behavior of this protein influenced the emitted fluorescence in a way that precluded quantification of exo- and endocytosis kinetics (Shen et al., 2014; Liu et al., 2021). Using pHluorin and ChrimsonSA, however, solved these problems, enabling calculation of fluorescence rise and decay time constants, which characterize different rates of exo- and endocytosis (Li et al., 2022). Neuronal primary culture of pHluorin expressing neurons could further open the way for investigation of exocytosis independent of SV recycling, e.g., by applying pharmacological agents such as bafilomycin A to inhibit SV acidification (Subramanian and Morozov, 2011; Li et al., 2022).
The pOpsicle system should be applicable to various neuronal cell types with minor modifications as exemplified by expression in cholinergic neurons and RIM interneurons. Establishing pOpsicle in various cell types could unveil disparities in SV exo- and endocytosis between different neuron classes, as has been shown for sensory neurons in C. elegans (Ventimiglia and Bargmann, 2017). Hitherto, electrophysiological recordings or Ca2+ imaging in body wall muscles were the method of choice to quantify neurotransmitter release in C. elegans in a time-resolved manner (Liewald et al., 2008; Wabnig et al., 2015). However, these only report postsynaptic effects which might be altered by unrelated phenomena such as neurotransmitter-receptor upregulation (Hammond-Weinberger et al., 2020). By using pOpsicle, a direct observation of the presynaptic SV cycle was achieved. This way, we could observe slowed SV fusion and endocytosis in RIM interneurons compared to cholinergic motor neurons. The faster release and recycling in cholinergic neurons may be in line with their function in mediating locomotion, and the likely high SV turnover needed, while the slower release rate could be important to the dual role of RIM in regulating reversal behavior (Li et al., 2020; Sordillo and Bargmann, 2021; Bach et al., 2023). While RIM promotes reversals through activation AVA and AVE neurons via gap junctions, and by mutual interaction with RIS, glutamate signaling inhibits reversal probability by reducing the amplitude of Ca2+ spikes within AVA and AVE and tyramine inhibits RIS (Li et al., 2020; Bach et al., 2023). A delayed release of glutamate and tyramine from RIM may thus promote a fast reaction to noxious stimuli by initiation of reversals. Observation of pHluorin dynamics in freely moving worms may solve this issue. However, we note that it was imperative for the assay to work that the animals were kept as immobile as possible.
With green pOpsicle, we could reveal differences in recycling kinetics of synaptojanin and endophilin knockout mutants between continuous and pulsed stimulation. Discrepancies in the rate of recycling may be caused by different degrees of stimulation which trigger distinct routes of SV retrieval (Watanabe and Boucrot, 2017). Activity-dependent bulk endocytosis (ADBE) after strong optogenetic stimulation was shown to occur in unc-57(e406) and unc-26(s1710) knockout mutants and may be the main pathway of retrieval after continuous stimulation (Kittelmann et al., 2013; Nicholson-Fish et al., 2016; Yu et al., 2018). Moderate, pulsed stimulation however may trigger other endocytic mechanisms such as ultrafast and clathrin-mediated endocytosis, which are dependent on endophilin and synaptojanin (Milosevic et al., 2011; Watanabe et al., 2018). snt-1(md290) mutants displayed more severe recycling defects after strong stimulation, indicating that it is dispensable for recycling at lower activity. This is in stark contrast to previous results in mammalian hippocampal neurons in which synaptotagmin promotes slow small-scale endocytosis, while inhibiting bulk retrieval during sustained neurotransmission (Chen et al., 2022). Although a role of SNT-1 in the slow clathrin-mediated endocytosis is likely also in C. elegans and simply not efficiently reported by pOpsicle, a role in inhibition of bulk endocytosis might not be conserved between nematodes and mammals. Previously, snt-1(md290) mutants have shown earlier fatigue of postsynaptic currents during strong optogenetic depolarization of cholinergic motor neurons indicating a compensatory SV recycling defect during strong sustained neurotransmission (Liewald et al., 2008; Wabnig et al., 2015).
Finally, SNG-1::pHluorin can further be used as a sensor for spontaneous neuronal activity and SV endocytosis in C. elegans, independent of optogenetic stimulation (Supplementary Figure 3A). This opens the way for multiplexing with other fluorescent reporters of neuronal activity such as genetically encoded Ca2+ or voltage indicators (Dreosti and Lagnado, 2011; Jackson and Burrone, 2016; Azimi Hashemi et al., 2019). The combination of simultaneous imaging of SV dynamics and membrane potential changes could help to unravel the complicated interplay of interneurons in the control of locomotion by differentiating between electrical and chemical transmission.
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
MS, BJ, and XZ created the plasmids and generated the strains. MS performed the pOpsicle, cell culture, and contraction assays. MW tested the ChrimsonSA in cholinergic neurons. NS and NE generated and tested the ChrimsonSA/pHluorin strains. NR generated the primary neuronal cell cultures. MS, BJ, and AG designed and coordinated the study. MS and AG wrote the manuscript. AG supervised the work. All authors read and approved the final manuscript.
This project was funded by Deutsche Forschungsgemeinschaft, Collaborative Research Centre 1080 project B02 (grant DFG CRC1080/B2 to AG).
We are indebted to Franziska Baumbach, Hans-Werner Müller, and Eva Dunkel for their expert technical assistance, and members of the Gottschalk Group for critical comments regarding the manuscript. We thank Dennis Vettkötter for providing MWT software and Ichiro Aoki for Arduino scripts and μManager settings to control LEDs and camera shutter for pulsed stimulation. We thank Johannes Vierock for providing ChrimsonSA absorption spectra. We also thank Maximilian Bach for advice on RIM experiments, and the Caenorhabditis Genetics Center (CGC), which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440), for providing strains.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2023.1120651/full#supplementary-material
Abraham, C., Hutter, H., Palfreyman, M. T., Spatkowski, G., Weimer, R. M., Windoffer, R., et al. (2006). Synaptic tetraspan vesicle membrane proteins are conserved but not needed for synaptogenesis and neuronal function in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U S A. 103, 8227–8232. doi: 10.1073/pnas.0509400103
Alfonso, A., Grundahl, K., Duerr, J. S., Han, H. P., and Rand, J. B. (1993). The Caenorhabditis elegans unc-17 gene: a putative vesicular acetylcholine transporter. Science 261, 617–619. doi: 10.1126/science.8342028
Azimi Hashemi, N., Bergs, A. C. F., Schuler, C., Scheiwe, A. R., Steuer Costa, W., Bach, M., et al. (2019). Rhodopsin-based voltage imaging tools for use in muscles and neurons of Caenorhabditis elegans. Proc. Natl. Acad. Sci. U S A. 116, 17051–17060. doi: 10.1073/pnas.1902443116
Azimi Hashemi, N., Erbguth, K., Vogt, A., Riemensperger, T., Rauch, E., Woodmansee, D., et al. (2014). Synthetic retinal analogues modify the spectral and kinetic characteristics of microbial rhodopsin optogenetic tools. Nat. Commun. 5:5810. doi: 10.1038/ncomms6810
Bach, M., Bergs, A., Mulcahy, B., Zhen, M., and Gottschalk, A. (2023). Coordinated electrical and chemical signaling between two neurons orchestrates switching of motor states. bioRxiv [preprint] doi: 10.1101/2023.01.04.522780
Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G., and Deisseroth, K. (2005). Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268. doi: 10.1038/nn1525
Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. doi: 10.1093/genetics/77.1.71
Cao, M., Wu, Y., Ashrafi, G., McCartney, A. J., Wheeler, H., Bushong, E. A., et al. (2017). Parkinson sac domain mutation in synaptojanin 1 impairs clathrin uncoating at synapses and triggers dystrophic changes in dopaminergic axons. Neuron 93, 882–896 e5. doi: 10.1016/j.neuron.2017.01.019.
Chanaday, N. L., Cousin, M. A., Milosevic, I., Watanabe, S., and Morgan, J. R. (2019). The synaptic vesicle cycle revisited: new insights into the modes and mechanisms. J. Neurosci. 39, 8209–8216. doi: 10.1523/JNEUROSCI.1158-19.2019
Chang, R. B. (2019). Optogenetic control of the peripheral nervous system. Cold Spring Harb. Perspect. Med. 9:a034397. doi: 10.1101/cshperspect.a034397
Chen, Y., Hu, S., Wu, X., Xie, Z., Wang, Y., Wang, B., et al. (2022). Synaptotagmin-1 is a bidirectional Ca(2+) sensor for neuronal endocytosis. Proc. Natl. Acad. Sci. U S A. 119:e2111051119. doi: 10.1073/pnas.2111051119
Choi, J. H., Horowitz, L. B., and Ringstad, N. (2021). Opponent vesicular transporters regulate the strength of glutamatergic neurotransmission in a C. elegans sensory circuit. Nat. Commun. 12:6334. doi: 10.1038/s41467-021-26575-3
Christensen, M., Estevez, A., Yin, X., Fox, R., Morrison, R., McDonnell, M., et al. (2002). A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron 33, 503–514. doi: 10.1016/s0896-6273(02)00591-3
Dittman, J. S., and Kaplan, J. M. (2006). Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. Proc. Natl. Acad. Sci. U S A. 103, 11399–11404.
Dong, Y., Gou, Y., Li, Y., Liu, Y., and Bai, J. (2015). Synaptojanin cooperates in vivo with endophilin through an unexpected mechanism. eLife 4:e05660. doi: 10.7554/eLife.05660
Dreosti, E., and Lagnado, L. (2011). Optical reporters of synaptic activity in neural circuits. Exp. Physiol. 96, 4–12. doi: 10.1113/expphysiol.2009.051953
Edelstein, A. D., Tsuchida, M. A., Amodaj, N., Pinkard, H., Vale, R. D., and Stuurman, N. (2014). Advanced methods of microscope control using muManager software. J. Biol. Methods 1:e10. doi: 10.14440/jbm.2014.36
Edwards, S. L., Charlie, N. K., Milfort, M. C., Brown, B. S., Gravlin, C. N., Knecht, J. E., et al. (2008). A novel molecular solution for ultraviolet light detection in Caenorhabditis elegans. PLoS Biol. 6:e198. doi: 10.1371/journal.pbio.0060198
Egashira, Y., Takase, M., and Takamori, S. (2015). Monitoring of vacuolar-type H+ ATPase-mediated proton influx into synaptic vesicles. J. Neurosci. 35, 3701–3710. doi: 10.1523/JNEUROSCI.4160-14.2015
Egashira, Y., Takase, M., Watanabe, S., Ishida, J., Fukamizu, A., Kaneko, R., et al. (2016). Unique pH dynamics in GABAergic synaptic vesicles illuminates the mechanism and kinetics of GABA loading. Proc. Natl. Acad. Sci. U S A. 113, 10702–10707. doi: 10.1073/pnas.1604527113
Fire, A. (1986). Integrative transformation of Caenorhabditis elegans. EMBO J. 5, 2673–2680. doi: 10.1002/j.1460-2075.1986.tb04550.x
Gan, Q., and Watanabe, S. (2018). Synaptic vesicle endocytosis in different model systems. Front. Cell Neurosci. 12:171. doi: 10.3389/fncel.2018.00171
Gowrisankaran, S., and Milosevic, I. (2020). Regulation of synaptic vesicle acidification at the neuronal synapse. IUBMB Life 72, 568–576. doi: 10.1002/iub.2235
Guo, Z. V., Hart, A. C., and Ramanathan, S. (2009). Optical interrogation of neural circuits in Caenorhabditis elegans. Nat. Methods 6, 891–896. doi: 10.1038/nmeth.1397
Guru, A., Post, R. J., Ho, Y. Y., and Warden, M. R. (2015). Making sense of optogenetics. Int. J. Neuropsychopharmacol. 18:yv079. doi: 10.1093/ijnp/pyv079
Hammond-Weinberger, D. R., Wang, Y., Glavis-Bloom, A., and Spitzer, N. C. (2020). Mechanism for neurotransmitter-receptor matching. Proc. Natl. Acad. Sci. U S A. 117, 4368–4374. doi: 10.1073/pnas.1916600117
Harris, T. W., Hartwieg, E., Horvitz, H. R., and Jorgensen, E. M. (2000). Mutations in synaptojanin disrupt synaptic vesicle recycling. J. Cell Biol. 150, 589–600.
Hawk, J. D., Wisdom, E. M., Sengupta, T., Kashlan, Z. D., and Colon-Ramos, D. A. (2021). A genetically encoded tool for reconstituting synthetic modulatory neurotransmission and reconnect neural circuits in vivo. Nat. Commun. 12:4795. doi: 10.1038/s41467-021-24690-9
Heuser, J. E., and Reese, T. S. (1973). Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J. Cell Biol. 57, 315–344. doi: 10.1083/jcb.57.2.315
Jackson, R. E., and Burrone, J. (2016). Visualizing presynaptic calcium dynamics and vesicle fusion with a single genetically encoded reporter at individual synapses. Front. Synaptic Neurosci. 8:21. doi: 10.3389/fnsyn.2016.00021
Jorgensen, E. M., Hartwieg, E., Schuske, K., Nonet, M. L., Jin, Y., and Horvitz, H. R. (1995). Defective recycling of synaptic vesicles in synaptotagmin mutants of Caenorhabditis elegans. Nature 378, 196–199. doi: 10.1038/378196a0
Kittelmann, M., Liewald, J. F., Hegermann, J., Schultheis, C., Brauner, M., Steuer Costa, W., et al. (2013). In vivo synaptic recovery following optogenetic hyperstimulation. Proc. Natl. Acad. Sci. U S A. 110, E3007–E3016. doi: 10.1073/pnas.1305679110
Koudelka, S., Voas, M. G., Almeida, R. G., Baraban, M., Soetaert, J., Meyer, M. P., et al. (2016). Individual neuronal subtypes exhibit diversity in CNS myelination mediated by synaptic vesicle release. Curr. Biol. 26, 1447–1455. doi: 10.1016/j.cub.2016.03.070
Kwon, S. E., and Chapman, E. R. (2011). Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron 70, 847–854. doi: 10.1016/j.neuron.2011.04.001
Lambert, T. J. (2019). FPbase: a community-editable fluorescent protein database. Nat. Methods 16, 277–278. doi: 10.1038/s41592-019-0352-8
Li, H., Datunashvili, M., Reyes, R. C., and Voglmaier, S. M. (2022). Inositol hexakisphosphate kinases differentially regulate trafficking of vesicular glutamate transporters 1 and 2. Front. Cell Neurosci. 16:926794. doi: 10.3389/fncel.2022.926794
Li, Y., and Tsien, R. W. (2012). pHTomato, a red, genetically encoded indicator that enables multiplex interrogation of synaptic activity. Nat. Neurosci. 15, 1047–1053. doi: 10.1038/nn.3126
Li, Z., Zhou, J., Wani, K., Yu, T., Ronan, E. A., Piggott, B. J., et al. (2020). A C. elegans neuron both promotes and suppresses motor behavior to fine tune motor output. bioRxiv [preprint] doi: 10.1101/2020.11.02.354472
Liewald, J. F., Brauner, M., Stephens, G. J., Bouhours, M., Schultheis, C., Zhen, M., et al. (2008). Optogenetic analysis of synaptic function. Nat. Methods 5, 895–902. doi: 10.1038/nmeth.1252
Liu, A., Huang, X., He, W., Xue, F., Yang, Y., Liu, J., et al. (2021). pHmScarlet is a pH-sensitive red fluorescent protein to monitor exocytosis docking and fusion steps. Nat. Commun. 12:1413. doi: 10.1038/s41467-021-21666-7
Liu, H., Li, L., Wang, W., Gong, J., Yang, X., and Hu, Z. (2018). Spontaneous vesicle fusion is differentially regulated at cholinergic and GABAergic synapses. Cell Rep. 22, 2334–2345. doi: 10.1016/j.celrep.2018.02.023
Liu, Q., Sinnen, B. L., Boxer, E. E., Schneider, M. W., Grybko, M. J., Buchta, W. C., et al. (2019). A photoactivatable botulinum neurotoxin for inducible control of neurotransmission. Neuron 101, 863–875.e6. doi: 10.1016/j.neuron.2019.01.002.
Luo, J. K., Melland, H., Nithianantharajah, J., and Gordon, S. L. (2021). Postsynaptic neuroligin-1 mediates presynaptic endocytosis during neuronal activity. Front. Mol. Neurosci. 14:744845. doi: 10.3389/fnmol.2021.744845
Miesenbock, G., De Angelis, D. A., and Rothman, J. E. (1998). Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394, 192–195. doi: 10.1038/28190
Miller, K. G., Alfonso, A., Nguyen, M., Crowell, J. A., Johnson, C. D., and Rand, J. B. (1996). A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc. Natl. Acad. Sci. U S A. 93, 12593–12598. doi: 10.1073/pnas.93.22.12593
Milosevic, I., Giovedi, S., Lou, X., Raimondi, A., Collesi, C., Shen, H., et al. (2011). Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron 72, 587–601. doi: 10.1016/j.neuron.2011.08.029
Mochida, S. (2022). Mechanisms of synaptic vesicle exo- and endocytosis. Biomedicines 10:1593. doi: 10.3390/biomedicines10071593
Morton, A., Marland, J. R., and Cousin, M. A. (2015). Synaptic vesicle exocytosis and increased cytosolic calcium are both necessary but not sufficient for activity-dependent bulk endocytosis. J. Neurochem. 134, 405–415. doi: 10.1111/jnc.13132
Nagel, G., Brauner, M., Liewald, J. F., Adeishvili, N., Bamberg, E., and Gottschalk, A. (2005). Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr. Biol. 15, 2279–2284. doi: 10.1016/j.cub.2005.11.032
Nagel, G., Szellas, T., Huhn, W., Kateriya, S., Adeishvili, N., Berthold, P., et al. (2003). Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. U S A. 100, 13940–13945.
Nicholson-Fish, J. C., Smillie, K. J., and Cousin, M. A. (2016). Monitoring activity-dependent bulk endocytosis with the genetically-encoded reporter VAMP4-pHluorin. J. Neurosci. Methods 266, 1–10. doi: 10.1016/j.jneumeth.2016.03.011
Nonet, M. L., Saifee, O., Zhao, H., Rand, J. B., and Wei, L. (1998). Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J. Neurosci. 18, 70–80. doi: 10.1523/JNEUROSCI.18-01-00070.1998
Oda, K., Vierock, J., Oishi, S., Rodriguez-Rozada, S., Taniguchi, R., Yamashita, K., et al. (2018). Crystal structure of the red light-activated channelrhodopsin Chrimson. Nat. Commun. 9:3949. doi: 10.1038/s41467-018-06421-9
Piggott, B. J., Liu, J., Feng, Z., Wescott, S. A., and Xu, X. Z. (2011). The neural circuits and synaptic mechanisms underlying motor initiation in C. elegans. Cell 147, 922–933. doi: 10.1016/j.cell.2011.08.053
Poskanzer, K. E., Marek, K. W., Sweeney, S. T., and Davis, G. W. (2003). Synaptotagmin I is necessary for compensatory synaptic vesicle endocytosis in vivo. Nature 426, 559–563. doi: 10.1038/nature02184
Preibisch, S., Saalfeld, S., and Tomancak, P. (2009). Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25, 1463–1465. doi: 10.1093/bioinformatics/btp184
Rose, T., Schoenenberger, P., Jezek, K., and Oertner, T. G. (2013). Developmental refinement of vesicle cycling at Schaffer collateral synapses. Neuron 77, 1109–1121. doi: 10.1016/j.neuron.2013.01.021
Sankaranarayanan, S., De Angelis, D., Rothman, J. E., and Ryan, T. A. (2000). The use of pHluorins for optical measurements of presynaptic activity. Biophys. J. 79, 2199–2208. doi: 10.1016/S0006-3495(00)76468-X
Schild, L. C., and Glauser, D. A. (2015). Dual color neural activation and behavior control with chrimson and CoChR in Caenorhabditis elegans. Genetics 200, 1029–1034. doi: 10.1534/genetics.115.177956
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Schuske, K. R., Richmond, J. E., Matthies, D. S., Davis, W. S., Runz, S., Rube, D. A., et al. (2003). Endophilin is required for synaptic vesicle endocytosis by localizing synaptojanin. Neuron 40, 749–762. doi: 10.1016/s0896-6273(03)00667-6
Seidenthal, M., Vettkötter, D., and Gottschalk, A. (2022). WormRuler: a software to track body length used to characterize a super red-shifted channelrhodopsin in Caenorhabditis elegans. MicroPubl. Biol. 2022:10.17912/micropub.biology.000607. doi: 10.17912/micropub.biology.000607
Seitz, K. J., and Rizzoli, S. O. (2019). GFP nanobodies reveal recently-exocytosed pHluorin molecules. Sci. Rep. 9:7773. doi: 10.1038/s41598-019-44262-8
Shen, Y., Rosendale, M., Campbell, R. E., and Perrais, D. (2014). pHuji, a pH-sensitive red fluorescent protein for imaging of exo- and endocytosis. J. Cell Biol. 207, 419–432. doi: 10.1083/jcb.201404107
Shin, W., Wei, L., Arpino, G., Ge, L., Guo, X., Chan, C. Y., et al. (2021). Preformed Omega-profile closure and kiss-and-run mediate endocytosis and diverse endocytic modes in neuroendocrine chromaffin cells. Neuron 109, 3119–3134.e5. doi: 10.1016/j.neuron.2021.07.019
Sieburth, D., Ch’ng, Q., Dybbs, M., Tavazoie, M., Kennedy, S., Wang, D., et al. (2005). Systematic analysis of genes required for synapse structure and function. Nature 436, 510–517. doi: 10.1038/nature03809
Sordillo, A., and Bargmann, C. I. (2021). Behavioral control by depolarized and hyperpolarized states of an integrating neuron. eLife 10, e67723. doi: 10.7554/eLife.67723
Steuer Costa, W., Yu, S. C., Liewald, J. F., and Gottschalk, A. (2017). Fast cAMP modulation of neurotransmission via neuropeptide signals and vesicle loading. Curr. Biol. 27, 495–507. doi: 10.1016/j.cub.2016.12.055
Strange, K., Christensen, M., and Morrison, R. (2007). Primary culture of Caenorhabditis elegans developing embryo cells for electrophysiological, cell biological and molecular studies. Nat. Protoc. 2, 1003–1012. doi: 10.1038/nprot.2007.143
Subramanian, J., and Morozov, A. (2011). Erk1/2 inhibit synaptic vesicle exocytosis through L-type calcium channels. J. Neurosci. 31, 4755–4764. doi: 10.1523/JNEUROSCI.6594-10.2011
Sudhof, T. C. (2013). Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690. doi: 10.1016/j.neuron.2013.10.022
Swierczek, N. A., Giles, A. C., Rankin, C. H., and Kerr, R. A. (2011). High-throughput behavioral analysis in C. elegans. Nat. Methods 8, 592–598. doi: 10.1038/nmeth.1625
Ventimiglia, D., and Bargmann, C. I. (2017). Diverse modes of synaptic signaling, regulation, and plasticity distinguish two classes of C. elegans glutamatergic neurons. eLife 6:e31234. doi: 10.7554/eLife.31234
Vettkötter, D., Schneider, M., Goulden, B. D., Dill, H., Liewald, J., Zeiler, S., et al. (2022). Rapid and reversible optogenetic silencing of synaptic transmission by clustering of synaptic vesicles. Nat. Commun. 13:7827. doi: 10.1038/s41467-022-35324-z
Vierock, J., Rodriguez-Rozada, S., Dieter, A., Pieper, F., Sims, R., Tenedini, F., et al. (2021). BiPOLES is an optogenetic tool developed for bidirectional dual-color control of neurons. Nat. Commun. 12:4527. doi: 10.1038/s41467-021-24759-5
Voglmaier, S. M., Kam, K., Yang, H., Fortin, D. L., Hua, Z., Nicoll, R. A., et al. (2006). Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron 51, 71–84. doi: 10.1016/j.neuron.2006.05.027
von Gersdorff, H., and Matthews, G. (1999). Electrophysiology of synaptic vesicle cycling. Annu. Rev. Physiol. 61, 725–752. doi: 10.1146/annurev.physiol.61.1.725
Wabnig, S., Liewald, J. F., Yu, S. C., and Gottschalk, A. (2015). High-Throughput all-optical analysis of synaptic transmission and synaptic vesicle recycling in Caenorhabditis elegans. PLoS One 10:e0135584. doi: 10.1371/journal.pone.0135584
Watanabe, S., and Boucrot, E. (2017). Fast and ultrafast endocytosis. Curr. Opin. Cell Biol. 47, 64–71. doi: 10.1016/j.ceb.2017.02.013
Watanabe, S., Liu, Q., Davis, M. W., Hollopeter, G., Thomas, N., Jorgensen, N. B., et al. (2013a). Ultrafast endocytosis at Caenorhabditis elegans neuromuscular junctions. eLife 2:e00723. doi: 10.7554/eLife.00723
Watanabe, S., Rost, B. R., Camacho-Perez, M., Davis, M. W., Sohl-Kielczynski, B., Rosenmund, C., et al. (2013b). Ultrafast endocytosis at mouse hippocampal synapses. Nature 504, 242–247. doi: 10.1038/nature12809
Watanabe, S., Mamer, L. E., Raychaudhuri, S., Luvsanjav, D., Eisen, J., Trimbuch, T., et al. (2018). Synaptojanin and endophilin mediate neck formation during ultrafast endocytosis. Neuron 98, 1184–1197.e6. doi: 10.1016/j.neuron.2018.06.005.
Yu, S. C., Janosi, B., Liewald, J. F., Wabnig, S., and Gottschalk, A. (2018). Endophilin A and B join forces with clathrin to mediate synaptic vesicle recycling in Caenorhabditis elegans. Front. Mol. Neurosci. 11:196. doi: 10.3389/fnmol.2018.00196
Yu, S. C., Klosterman, S. M., Martin, A. A., Gracheva, E. O., and Richmond, J. E. (2013). Differential roles for snapin and synaptotagmin in the synaptic vesicle cycle. PLoS One 8:e57842. doi: 10.1371/journal.pone.0057842
Zhang, S., Banerjee, D., and Kuhn, J. R. (2011). Isolation and culture of larval cells from C. elegans. PLoS One 6:e19505. doi: 10.1371/journal.pone.0019505
Zhao, H., and Nonet, M. L. (2001). A conserved mechanism of synaptogyrin localization. Mol. Biol. Cell 12, 2275–2289. doi: 10.1091/mbc.12.8.2275
Keywords: neurotransmission, exo- and endocytosis, optogenetics, Caenorhabditis elegans, synaptic plasticity
Citation: Seidenthal M, Jánosi B, Rosenkranz N, Schuh N, Elvers N, Willoughby M, Zhao X and Gottschalk A (2023) pOpsicle: An all-optical reporter system for synaptic vesicle recycling combining pH-sensitive fluorescent proteins with optogenetic manipulation of neuronal activity. Front. Cell. Neurosci. 17:1120651. doi: 10.3389/fncel.2023.1120651
Received: 10 December 2022; Accepted: 15 March 2023;
Published: 31 March 2023.
Edited by:
Jacopo Lamanna, Vita-Salute San Raffaele University, ItalyReviewed by:
David Perrais, Centre National de la Recherche Scientifique (CNRS), FranceCopyright © 2023 Seidenthal, Jánosi, Rosenkranz, Schuh, Elvers, Willoughby, Zhao and Gottschalk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander Gottschalk, YS5nb3R0c2NoYWxrQGVtLnVuaS1mcmFua2Z1cnQuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.