 Stephanie Bigsby†
Stephanie Bigsby† Joseph Neapetung
Joseph Neapetung Verónica A. Campanucci
Verónica A. Campanucci- Department of Anatomy, Physiology and Pharmacology (APP), College of Medicine, University of Saskatchewan, Saskatoon, SK, Canada
Voltage-gated sodium channels (NaV) are the main contributors to action potential generation and essential players in establishing neuronal excitability. NaV channels have been widely studied in pain pathologies, including those that develop during diabetes. Diabetic sensory neuropathy (DSN) is one of the most common complications of the disease. DSN is the result of sensory nerve damage by the hyperglycemic state, resulting in a number of debilitating symptoms that have a significant negative impact in the quality of life of diabetic patients. Among those symptoms are tingling and numbness of hands and feet, as well as exacerbated pain responses to noxious and non-noxious stimuli. DSN is also a major contributor to the development of diabetic foot, which may lead to lower limb amputations in long-term diabetic patients. Unfortunately, current treatments fail to reverse or successfully manage DSN. In the current review we provide an updated report on NaV channels including structure/function and contribution to DSN. Furthermore, we summarize current research on the therapeutic potential of targeting NaV channels in pain pathologies, including DSN.
Introduction
Diabetic neuropathies are amongst the most common chronic complications of diabetes, affecting approximately 50–60% of diabetic patients (Boulton et al., 2005; Zakin et al., 2019). Neuropathy results from diabetes-induced damage to peripheral nerves, and up to 30% of those patients develop a sensory form of neuropathy (diabetic sensory neuropathy or DSN) (Boulton et al., 2005; Tesfaye et al., 2013). DSN can present with a wide spectrum of clinical symptoms from tingling, numbness, weakness, and sensory loss, to exacerbated pain perception (Callaghan et al., 2012; Kobayashi and Zochodne, 2018). Patients suffering from DSN can display one or more types of stimulus-evoked pain, such as exacerbated responses to noxious (hyperalgesia) or innocuous (allodynia) stimuli (Hong et al., 2004; Tesfaye et al., 2010). DSN involves sensory abnormalities that most commonly manifest in a symmetrical “stocking-and-glove” distribution, being experienced in the distal extremities (e.g., hands and feet) and progresses proximally towards the torso over the course of diabetes (Callaghan et al., 2012). Consequently, DSN patients are at a higher risk of developing “diabetic foot,” a condition characterized by the development of unhealing foot ulcers, which can eventually contribute to lower limb amputations (Callaghan et al., 2012; Kobayashi and Zochodne, 2018). Ultimately, pathological damage to the peripheral nerves in DSN is debilitating, resulting in a considerable reduction in patient quality of life. Despite a strong relationship between blood glucose levels and neuropathy, the underlying mechanisms contributing to the pathology of DSN remain unclear. Unfortunately, no therapeutic strategy has succeeded in halting, reversing or preventing the development of DSN (Dyck et al., 1999; Callaghan et al., 2012; Kobayashi and Zochodne, 2018).
Some symptoms characteristic of DSN, such as numbness and hyposensitivity, are usually associated with long-standing diabetes and the progressive degeneration of peripheral nerves and loss peripheral innervation (Chantelau, 2015). Although painful symptoms are also consequence of nerve damage and inflammation, they have been linked to biochemical changes affecting membrane proteins and signaling pathways in sensory neurons. Amongst the many proteins that may contribute to the development and/or progression of DSN, voltage-gated sodium channels (NaV) have been particularly considered. NaV channels are essential for the initiation and propagation of action potentials, and thus, are responsible for electrical signaling (Bagal et al., 2015). Activation of NaV channels carry the increase in sodium conductance during action potentials, allowing the movement of sodium ions down their electrochemical gradient and into the cell following local depolarization (Catterall, 1992; De Lera Ruiz and Kraus, 2015). This large influx of sodium ions causes further depolarization, which activates more NaV channels triggering a positive feedback leading ultimately to the rising phase of the action potential (Chong and Ruben, 2008). Various NaV channels participate in pain perception, which has made them subjects of extensive investigation into the pathophysiology of pain, including their contribution to pain symptoms in DSN (Dib-Hajj et al., 2010).
NaV function is intimately linked to channel structure and tissue distribution
NaV channels are heteromeric complexes composed of a pore-forming α subunit and at least one associated β subunit (Bagal et al., 2015; O’Malley and Isom, 2015). The α subunit family members (NaV1.1 - 1.9) are highly homologous in amino acid sequence and display tissue specificity throughout the body (De Lera Ruiz and Kraus, 2015). They are encoded by the genes SCN1A-SCN5A and SCN8A-SCN11A (Hoeijmakers et al., 2015). These subunits are large single polypeptide chains of approximately 260 kDa, composed of 4 homologous domains each containing a voltage sensor, a pore region, and a selectivity filter (De Lera Ruiz and Kraus, 2015).
NaV channels are commonly characterized by their differential sensitivity to tetrodotoxin (TTX), NaV1.1–1.4, 1.6, and 1.7 are inhibited by this toxin with an IC50 in the low nanomolar concentration range, and thus are considered TTX-sensitive (TTX-S) (Baer et al., 1976; De Lera Ruiz and Kraus, 2015). TTX-S channels carry faster activating and inactivating currents (Wang et al., 2011; Yin et al., 2016). Meanwhile, NaV 1.5, 1.8, and 1.9 are inhibited by TTX with an IC50 in the micromolar range, and hence are considered as TTX-resistant (TTX-R) (Baer et al., 1976; De Lera Ruiz and Kraus, 2015). TTX-R currents show slower activation and inactivation kinetics (Wang et al., 2011; Yin et al., 2016). NaV1.1 and 1.3 are expressed in both the central (CNS) and the peripheral (PNS) nervous systems (Cummins and Rush, 2007; Bagal et al., 2015). NaV 1.1 expression in peripheral dorsal root ganglion (DRG) neurons is high in large-diameter neurons, moderate in medium-diameter neurons, and low in small diameter neurons. A small portion of NaV1.1-positive neurons co-express isolectin B4 (IB4), a marker for nociceptive neurons, suggesting this isoform may play some role in pain transmission (Wang et al., 2011). NaV1.3 expression in DRGs is highest in the embryonic period of development, and downregulates postnatally (Cummins and Rush, 2007; Bennett et al., 2019). NaV1.2, on the other hand, is highly expressed in the CNS. It is predominantly found in dendrites, unmyelinated axons, and pre-myelinated axons (Wang et al., 2011) predominantly in the embryonic period (Wang et al., 2011; Bennett et al., 2019). Other NaV subunits are almost exclusively expressed in embryonic muscle (Bennett et al., 2019), NaV 1.4 in skeletal (Cummins and Rush, 2007) and Nav 1.5 in the cardiac muscle (Rogers et al., 2006; Cummins and Rush, 2007).
The rest of the NaV isoforms (Nav 1.6–1.9) are expressed within the DRGs and play important roles in nociception (Rush et al., 2007; Chen et al., 2018). NaV1.6 expression in DRG, as well as in motor neurons, is preferentially targeted to the nodes of Ranvier in myelinated fibers and along unmyelinated C- fiber (Wittmack et al., 2005; Bennett et al., 2019). Nav1.7 is the most expressed TTX-S isoform, mostly found in small diameter Aδ and C-fibers (Black et al., 1996; Toledo-Aral et al., 1997; Berta et al., 2008; Dib-Hajj et al., 2010, 2013; Ho and O’Leary, 2011), including 85% of functionally identified nociceptors (Djouhri et al., 2003). Nav1.8 and Nav1.9, are also highly expressed in nociceptive neurons (Akopian et al., 1997; Dib-Hajj et al., 1998; Bennett et al., 2019). NaV1.8 is the major contributor to the rising phase of the action potential; carrying 80–90% of the inward sodium current during the upstroke of the action potential (Renganathan et al., 2001), and its ability to rapidly recover from inactivation allows for repetitive, high frequency firing (Waxman, 2012). NaV1.9 is almost exclusively expressed in the PNS (Cummins and Rush, 2007), particularly in C-fiber nociceptors and with moderated expression in medium diameter (Aδ-fibers) and low in large diameter (Aβ-fibers) (Rogers et al., 2006; Priest and Kaczorowski, 2007). NaV 1.9 contribute to the amplification of subthreshold inputs, but do not contribute to the upstroke or amplitude of the action potential (Bennett et al., 2019). These are considered threshold channels due to their activation at hyperpolarized potentials (near the resting membrane potential), where other NaV channels remain inactive (De Lera Ruiz and Kraus, 2015; Bennett et al., 2019).
Most NaV channels contain associated β subunits; although they were traditionally considered as “auxiliary,” growing evidence suggests they are critical for channel function. The β subunits promote channel trafficking (Schmidt and Catterall, 1986; Chen et al., 2002), and modulate channel biophysical properties (Calhoun and Isom, 2014). The 4 known β subunits (β1- β4) (Qin et al., 2003; Brackenbury and Isom, 2011) are encoded by genes SCN1B-SCN4B. Mutations in these genes, as well as changes in expression levels, have been linked to the development of many isoform-specific pathologies such as epilepsy, Huntington’s disease, cardiac arrhythmias, and various neuropathies (Meadows et al., 2002; Lucas et al., 2005; Oyama et al., 2006; Medeiros-Domingo et al., 2007; Brackenbury and Isom, 2011; Alsaloum et al., 2019). These subunits have a much smaller molecular weights than their α counterparts (30–40 kDa, Priest and Kaczorowski, 2007), and are members of the immunoglobulin superfamily of cell adhesion molecules (De Lera Ruiz and Kraus, 2015). β1 and β3 noncovalently bind to the α-subunit, while β2 and β4 covalently attach to the α-subunit via disulfide bonds (Namadurai et al., 2015). The immunoglobulin domain modulates expression and gating properties of α subunits, while the transmembrane domain influences their voltage dependence (Rogers et al., 2006; De Lera Ruiz and Kraus, 2015). β subunits are found in the CNS, PNS, heart, and skeletal muscle (excepting β2) (Isom et al., 1992; Brackenbury and Isom, 2011). β1 and β4 can modulate channel kinetics by enhancing the rate of inactivation and recovery from inactivation (Namadurai et al., 2015). β4 is the promoter of the resurgent current, which flows through channels that reopen in response to negative voltage changes due to the decay of the macroscopic sodium current, but when the inactivation gates have yet to close (Raman and Bean, 1997). In contrast, β3 is mostly expressed in heart muscle (Brackenbury and Isom, 2011) and has been linked to cardiac arrhythmias and cardiac conduction problems (O’Malley and Isom, 2015).
Contribution of NaV channels to diabetic sensory neuropathy
The induction of experimental diabetes in rodents using streptozotocin (STZ), a pancreatic β-cell-specific cytotoxin, results in hyperglycemia due to lack of insulin production (Islam and Loots, 2009). STZ-induced diabetic animals develop painful neuropathy accompanied by a typical increase in NaV1.3, 1.6, and 1.7 expression (Cummins et al., 1998, 2001; Herzog et al., 2003; Hong et al., 2004) and DRG neuron hyperexcitability (Hirade et al., 1999). The pivotal role of NaV1.7 in nociception (Black et al., 1996; Toledo-Aral et al., 1997; Berta et al., 2008; Ho and O’Leary, 2011; Dib-Hajj et al., 2013) has been well documented, particularly by the effects of mutations. While gain-of-function mutations in the NaV1.7 subunit lead to hereditary pain disorders, such as primary erythromelalgia, paroxysmal extreme pain disorder and small fiber neuralgia (De Lera Ruiz and Kraus, 2015; Tibbs et al., 2016), loss-of-function mutations lead to congenital insensitivity to pain (De Lera Ruiz and Kraus, 2015). In the DRG of diabetic rodents, Nav1.7 channel expression increased robustly and triggered evoked pain symptoms of thermal hyperalgesia and mechanical allodynia (Hong et al., 2004). Consistently, symptoms of thermal hyperalgesia and mechanical allodynia in these animals were attenuated by the miRNA-mediated knockdown of the Nav1.7 α subunit (Chattopadhyay et al., 2012). Furthermore, electrophysiological studies in DRG neurons from diabetic rats revealed that the TTX-S current showed an increased current density, a negatively shifted voltage-dependent activation, and delayed inactivation kinetics (Hong and Wiley, 2006). The latter are consistent with expression and function changes reported in NaV1.7, which is the predominant TTX-S isoform expressed in DRG neurons (Black et al., 1996; Toledo-Aral et al., 1997; Berta et al., 2008; Ho and O’Leary, 2011; Dib-Hajj et al., 2013).
Isoforms NaV1.2, 1.3, and 1.9 are upregulated in the DRG of STZ-induced diabetic rats (Hong and Wiley, 2006). The detection of NaV1.2 and 1.3 in adult diabetic rodents contrasts with physiological expression levels of these subunits, since they are normally higher in embryonic neurons (Cummins and Rush, 2007; Wang et al., 2011; Bennett et al., 2019). The latter suggest that the diabetic environment triggers a pathological resurgence in the expression of these embryonic channels. It has been reported that NaV1.3 is upregulated in the adult spinal cord of rats after peripheral nerve injury (Black et al., 2004; Cummins and Rush, 2007) and in the DRG after axotomy (Hains et al., 2003). And more importantly, the knockdown of NaV1.3 expression successfully reduced evoked tactile allodynia and hyperexcitability in the dorsal horn neurons of STZ-induced diabetic rats (Tan et al., 2015). These findings suggest that NaV1.3 could be a suitable therapeutic target in the treatment of DSN.
Contrasting with other NaVs, the expression levels of NaV1.6 and 1.8 decreased in DRG homogenates from diabetic rats (Hong et al., 2004). Although the mechanisms for this downregulation is still unclear, it has been reported that reactive oxygen species (ROS) reduced NaV1.8 peak current in DRG neurons (Schink et al., 2016). This finding is consistent with the well documented generation of ROS in diabetes from mitochondria as well as by enzymatic and non-enzymatic glucose oxidation, and constitute the most explored hypothesis for the effects of diabetes on the nervous system (Russell et al., 2002; Vincent et al., 2005; Tomlinson and Gardiner, 2008; Campanucci et al., 2010; Chandna et al., 2015; Lam et al., 2018; Momeni et al., 2021).
Much less, however, is known about the possible contribution of β subunits to DSN. Genetic analysis of the β2 subunit gene from a diabetic patient, who presented symptoms of painful neuropathy, revealed a gain-of-function mutation of an aspartic acid substituted by asparagine mutation, D109N. This point mutation lead to DRG neuron hyperexcitability (Alsaloum et al., 2019). These findings were in line with the increase in β2 subunit expression in neuropathic pain models of injured and uninjured DRG neurons (Pertin et al., 2005), highlighting this subunit as a potential player in the development of varied pain pathologies.
The therapeutic potential of NaV channels
Table 1 summarizes the current knowledge on approved therapeutic strategies for DSN targetting NaV channels, as well as mechanisms with the potential to modulate NaV function in DSN. Tricyclic antidepressants (TCAs), such as amitriptyline, imipramine, nortriptyline, and duloxetine, are Food and Drug Administration (FDA) approved drugs that are effective for the treatment of painful forms of DSN (Berger et al., 2005). A recent report (Horishita et al., 2017) demonstrate that the analgesic effects of some of these drugs in neuropathic pain pathology is mediated by the inhibition of NaV channels. Particularly, they strongly inhibited NaV1.7, and NaV1.8 in DRG, as well as upregulated NaV1.3 in the DRG of models of peripheral nerve injury (Lindia et al., 2005; Fukuoka et al., 2008). These drugs also inhibited, but in a weaker fashion, NaV channels highly expressed in the CNS, such as NaV1.6 and NaV1.2 (Horishita et al., 2017). Another therapeutic approach, not first considered as targeting NaV channels, is the pungent ingredient in “hot” chili peppers, capsaicin. Capsaicin is an agonist of the transient receptor potential vanilloid 1 (TRPV1), a non-selective cation channel (Caterina et al., 1997). TRPV1 plays a central role in pain transduction, and its inhibition alleviates thermal hyperalgesia and mechanical allodynia in animal models of peripheral nerve injury (Walker et al., 2003; Kasama et al., 2007; Sugimoto et al., 2013). Interestingly, capsaicin can also induce analgesia. In fact, the topical application of capsaicin has been used for the treatment of localized pain (Deal et al., 1991; Hersh et al., 1994; Gratton and Cusson, 1995; Winocur et al., 2000; Melis et al., 2019), and the use of capsaicin as a therapeutic approach for diabetic patients with symptoms of painful neuropathy has been recently reviewed by Dludla et al. (2022). It is well accepted that repeated exposure to capsaicin induces a calcium-mediated desensitization of TRPV1 channels (Jancsó et al., 1967; Hains et al., 2003). However, the analgesic effects of capsaicin are mediated in part by the inhibition of NaV channels through second messengers, such as cAMP (Liu et al., 2001). Although, the identity of the specific NaV isoforms modulated by capsaicin remain to be explored. Another approach targetting NaV channels in DSN is the use of the antithrombotic agent cilostazol (Cheng et al., 2022). The interest on cilostazol for DSN symptoms stems from its known protection of human endothelial cells via activation of ERK1/2 and p38 MAPKs (MAPK) (Lim et al., 2009), and neuroprotection in animal models of cerebral ischemia (Iwama et al., 2007). When tested on diabetic rats, oral administration of cilostazol successfully decreased withdrawal threshold to mechanical stimuli and attenuated neuropathic pain symptoms. More importantly, it reduced expression levels of multiple NaV channels (NaV1.1, 1.2, 1.6, and 1.7); and restored expression levels of NaV1.8, which was markedly reduced in STZ rats. The anticonvulsant gabapentin, has also been testes to treat DSN. Gabapentin induced analgesia in STZ-induced diabetic rats, it successfully reverted mechanical allodynia and thermal hyperalgesia in these animals, which correlated with reduced expression of the NaV1.7 isoform and phosphorylated ERK1/2 in DRG neurons (Zhang et al., 2013). Similar findings come from pioglitazone, a proliferator-activated receptors (PPARs) agonist usually prescribed to type 2 diabetic patients, and ranolazine, an adjuvant in chronic angina medication (Elkholy et al., 2020). Both drugs successfully reverted symptoms of mechanical allodynia and thermal hyperalgesia in type 2 diabetic rats. Furthermore, these drugs individually reduced the expression levels of NaV1.7 in DRG neurons to control levels (Elkholy et al., 2020).
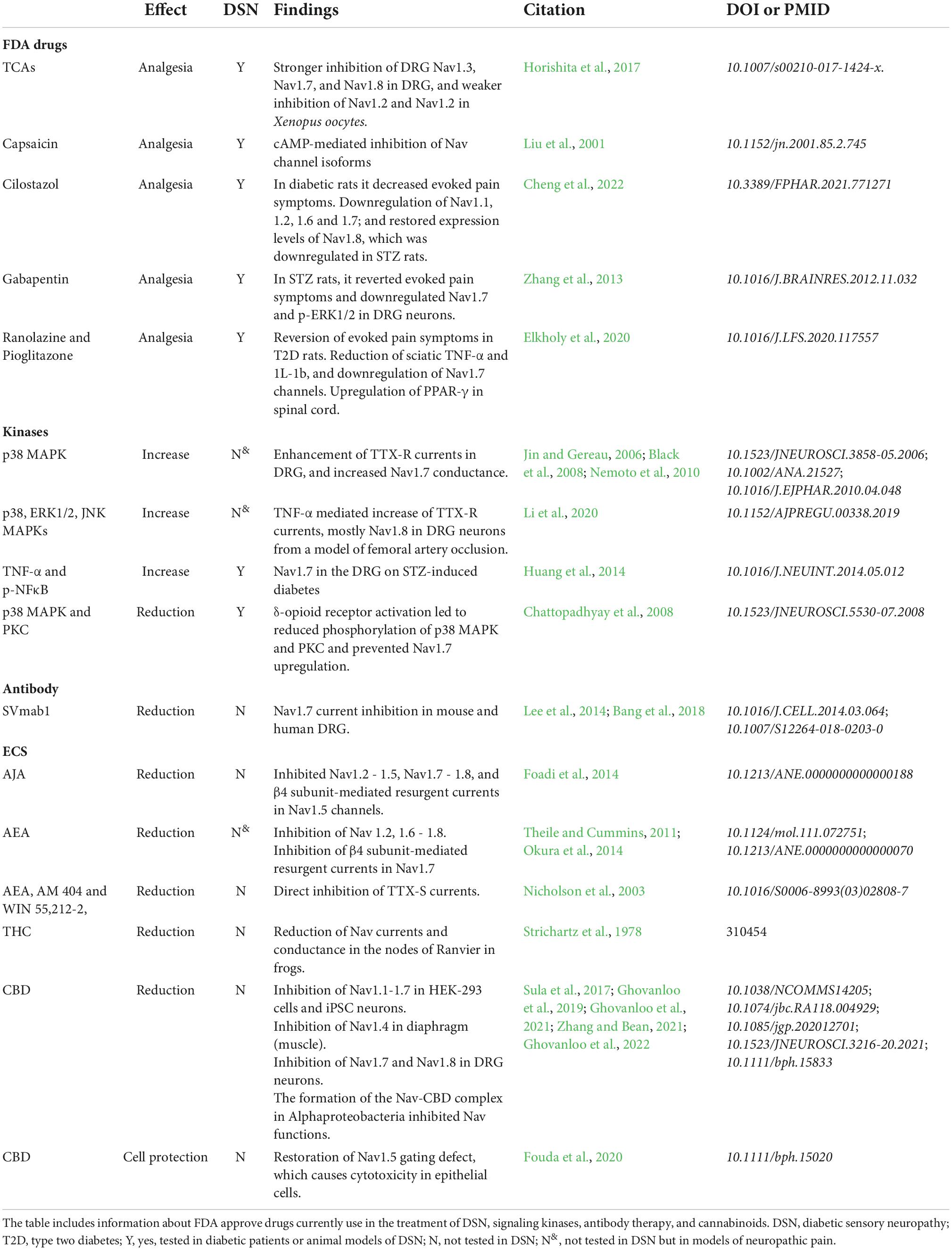
Table 1. Modulation of NaV expression and channel function as treatment for DSN.
Targeting signaling kinases such as those from the mitogen-activated protein kinase (MAP) and protein kinase (PK) families in the context of DSN, is supported by reports highlighting their role regulating NaV expression and function in other pain pathologies. For instance, in human painful neuromas, multiple NaV isoforms (NaV1.1 - 1.3, NaV1.6 - 1.9) co-localized with the activated MAPKs p38 and extracellular signal-regulated kinases 1 and 2 (ERK1/2), suggesting these signaling proteins could potentially modulate NaV channel function in painful neuropathies (Black et al., 2008). In fact, in chromaffin cells, p38 and ERK1/2 primed NaV1.7 function and increased ion conductance (Nemoto et al., 2010). Activation of the p38 MAPK pathway by tumor necrosis factor-α (TNF-α) enhanced TTX-R currents in isolated DRG neurons and induced mechanical hypersensitivity in mice, which was prevented by the pharmacological inhibition of p38 (Jin and Gereau, 2006). Similarly, in DRG neurons from a model of femoral artery occlusion, TNF-α mediated the increase of TTX-R currents, mostly through NaV1.8; and this effect was prevented by the pharmacological inhibition of p38, ERK1/2, and c-Jun N-terminal kinases (JNKs) (Li et al., 2020).
Consistently, in STZ-induced diabetic rats with symptoms of DSN, the upregulation of NaV1.7 in the DRGs was prevented by inhibiting the synthesis or by blocking the action of TNF-α and p-nucleus factor-kappa B (p-NFκB)(Huang et al., 2014). In DRG neurons exposed to hyperglycemic conditions in vitro, the enkephalin-mediated activation of δ-opioid receptors prevented NaV1.7 upregulation. The later was accompanied by reduction of phosphorylation of p38 MAPK and PKC (Chattopadhyay et al., 2008).
A provocative new approach proposes the use of monoclonal antibodies to inhibit NaV1.7 channels. This novel strategy revealed that the antibody SVmab1, selectively inhibited NaV1.7 currents in mouse and human DRG neurons (Lee et al., 2014; Bang et al., 2018). Moreover, the antibody effectively supressed inflammatory and neuropathic pain in mouse models, and unveiled a significant role of NaV1.7 in itch sensation (Lee et al., 2014), suggesting this strategy may be effective in the management of chronic itch in humans (Bang et al., 2018). Although, this antibody therapy has yet to be tested in the context of DSN, the ability of SVmab1 to reduce excitatory transmission in pain-sensitive neurons may relief diabetes-related pain symptoms.
More recently, targeting NaV channels through the endocannabinoid system (ECS) has been proposed as a therapeutic tool in pain pathologies. The ECS is composed of endocannabinoids, their receptors, and the enzymatic pathways required for endocannabinoids’ synthesis. The endocannabinoids, N arachidonoylethanolamine (AEA), and 2-arachidonoylglycerol (2-AG) bind to two well-characterized G-protein coupled receptors cannabinoid receptors 1 and 2 (CB1) and 2 (CB2) (Howlett et al., 2004). Numerous animal and clinical studies have shown the potential of synthetic and naturally occurring cannabinoids to effectively attenuate inflammatory and neuropathic pain, including DSN pain (Howlett et al., 2004; Rahn and Hohmann, 2009). Some of the beneficial effect of cannabinoids on DSN could be explained by their action on NaV channel function. The synthetic cannabinoid ajulemic acid (AJA), which has been reported to induce analgesia in inflammatory pain in humans (Burstein et al., 2004), inhibited NaV1.2 - 1.5, NaV1.7 - 1.8 channels, as well the β4 subunit-mediated resurgent currents in NaV1.5 channels (Foadi et al., 2014). Similarly, the endocannabinoid anandamide (AEA) has been reported to inhibit β4 subunit-mediated resurgent currents in NaV1.7 (Theile and Cummins, 2011). Cannabinoids are known to modulate presynaptic Ca2+ and K+ channels (Nicholson et al., 2003), but also to inhibit NaV channels either through CB1 receptor signaling or by their direct action on the channel protein. AEA, as well as the synthetic cannabinoids AM 404 and WIN 55,212-2, directly bound to TTX-S NaV channels (Nicholson et al., 2003). Moreover, AEA was reported to inhibit NaV 1.2, 1.6 - 1.8 (Okura et al., 2014) by unknown mechanisms. The phytocannabinoid Δ9-tetrahydrocannabinol (THC), the principal psychoactive constituent of cannabis, has previously been reported to decrease peak Na+ current and conductance in the nodes of Ranvier in frogs (Strichartz et al., 1978). Furthermore, cannabidiol (CBD), a phytocannabinoid lacking psychoactive effect, has been reported to block NaV channels. CBD inhibited human NaV1.1-1.7 currents expressed in human embryonic kidney 293 (HEK-293) cells and in induced pluripotent stem cell (iPSC)-derived neurons. The mechanisms of action of CBD seemed to be indirect, mediated by its interaction with membrane lipids resulting in loss of NaV channel activity (Ghovanloo et al., 2019). Consistently, in DRG neurons, CBD inhibited NaV1.7 leading to reduced neuronal excitability (Ghovanloo et al., 2022), a mechanisms with relevant potential in pain pathologies. CBD also inhibited NaV1.4 and 1.8 by similar mechanisms. In muscle, CBD stabilized the inactivated state of NaV1.4 (Ghovanloo et al., 2021). In the DRGs, CBD showed preferential binding to the slow inactivated state of NaV1.8, which directly inhibited repetitive firing of nociceptors (Zhang and Bean, 2021). Moreover, CBD was shown to block NaV currents by physically interacting with the channels. The high-resolution crystal structure of the NaV-CBD complex was studied in NaV channels from M. marinus (Sula et al., 2017), which exhibits similar function, sequence and structural homologies to mammalian NaV channels (Sula and Wallace, 2017; Sula et al., 2017), demonstrated that CBD interacts with the channel at a novel site at the location of the central hydrophobic cavity of the channel (Sait et al., 2020). Furthermore, CBD has been shown to restore gating defects in NaV 1.5 caused by reactive oxygen species in high glucose conditions, which protected against high glucose-induced oxidative stress and cytotoxicity in the Chinese hamster ovary (CHO) epithelial cell line (Fouda et al., 2020). More importantly, the beneficial effect of engaging the ECS has been documented in patients with painful DSN. A randomized, double-blinded, placebo-controlled crossover study revealed improved spontaneous and evoked pain scores between placebo and patients receiving treatment (Wallace et al., 2015), which may be mediated at least in part by the modulation of NaV channels.
Conclusion
In the current review, we focused on NaV channels and their role in DSN, not only due to their unequivocal relevance in cell excitability, but also because several recent studies point at NaVs as potential therapeutic targets in pain pathologies. Here we discussed the important role that some NaVs play in DSN and their yet largely potential for pain management in diabetes. Novel strategies using NaVs as a therapeutic tool may involve (1) FDA approved drugs with effects on NaV channel function; (2) signaling kinases that regulate NaV expression/function; (3) monoclonal antibody therapy; and (4) modulation by the ECS. Figure 1 summarizes the therapeutic potential of NaV channels for the treatment of DSN symptoms, including strategies that have yet to be tested in the context of diabetes.
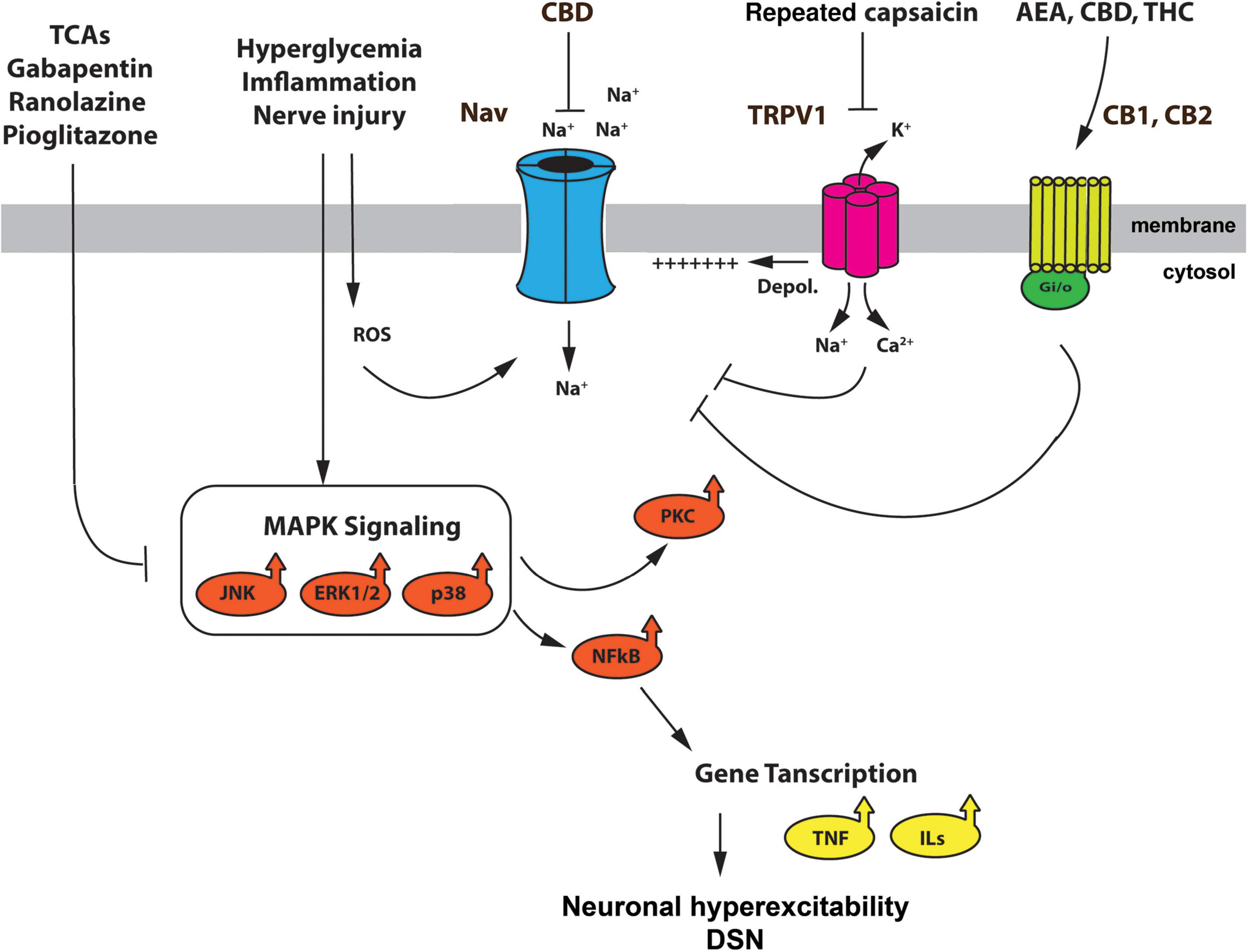
Figure 1. Schematic representation summarizing the mechanisms modulating NaV channel function in DSN, and the effect of therapeutic agents. Hyperglycemia, nerve injury, and inflammation activate the MAPK kinase pathway and generate reactive oxygen species (ROS). The p38, ERK1/2 and JNK MAPKs, as well as ROS, have been reported to mediate upregulation and enhancement of different NaV channel subunits. Furthermore, MAPK signaling leads to PKC activation and expression NFkB. The latter supports the expression of interleukins and TNF-a further enhancing an inflammatory environment in DSN. TRPV1 channels, which are upregulated by hyperglycemia, inflammatory mediators, and ROS (Lam et al., 2018; Nair et al., 2021), depolarize DRG neurons and contribute to excitability in DSN (Jin et al., 2004; Hong and Wiley, 2005; Melli and Höke, 2009) by assisting the cell in reaching threshold voltage for activation of NaV channels. The use of FDA approved drugs for the treatment of DSN such as TCA, gabapentin, ranolazine and pioglitazone, reduce the activation of the MAPK pathway, preventing the upregulation of some NaV channels, such as NaV1.7. Furthermore, tonic (repeated) application of capsaicin prevents the upregulation of NaV channels by cAMP mediated signaling pathways. Agonist of the ECS, such as AEA, THC, and CBD, can prevent NaV channel upregulation by actions through they receptors (CB1, CB2), or by direct action on the NaV channel. Although these effects of cannabinoids have yet to be tested in the context of DSN, they do, however, provide a mechanistic explanation of some of the beneficial effects of cannabinoid therapy in diabetic patients with symptoms of DSN. TCAs, tricyclic antidepressants; ROS, reactive oxygen species; ECS, endocannabinoid system; AEA, N arachidonoylethanolamine, CBD, cannabidiol; THC, Δ9-tetrahydrocannabinol; CB1, cannabinoid receptor 1; CB2, cannabinoid receptor 2.
Author contributions
SB and VC conceptualized the manuscript. SB, JN, and VC wrote the manuscript. JN and VC edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the NSERC (grant RGPIN-2021-04252) and CoMRAD (University of Saskatchewan, College of Medicine) to VC, and a NSERC-USRA scholarship to SB.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akopian, A. N., Souslova, V., Sivilotti, L., and Wood, J. N. (1997). Structure and distribution of a broadly expressed atypical sodium channel. FEBS Lett. 400, 183–187. doi: 10.1016/S0014-5793(96)01389-0
Alsaloum, M., Estacion, M., Almomani, R., Gerrits, M. M., Bönhof, G. J., Ziegler, D., et al. (2019). A gain-of-function sodium channel β 2-subunit mutation in painful diabetic neuropathy. Mol. Pain 15:174480691984980. doi: 10.1177/1744806919849802
Baer, M., Best, P. M., and Reuter, H. (1976). Voltage-dependent action of tetrodotoxin in mammalian cardiac muscle. Nature 263, 344–345. doi: 10.1038/263344a0
Bagal, S. K., Marron, B. E., Owen, R. M., Storer, R. I., and Swain, N. A. (2015). Voltage gated sodium channels as drug discovery targets. Channels 9, 360–366. doi: 10.1080/19336950.2015.1079674
Bang, S., Yoo, J., Gong, X., Liu, D., Han, Q., Luo, X., et al. (2018). Differential Inhibition of Nav1.7 and neuropathic pain by hybridoma-produced and recombinant monoclonal antibodies that target Nav1.7: Differential activities of Nav1.7-targeting monoclonal antibodies. Neurosci. Bull. 34:22. doi: 10.1007/S12264-018-0203-0
Bennett, D. L., Clark, X. A. J., Huang, J., Waxman, S. G., and Dib-Hajj, S. D. (2019). The role of voltage-gated sodium channels in pain signaling. Physiol. Rev. 99, 1079–1151. doi: 10.1152/physrev.00052.2017
Berger, A., Dukes, E., Hagiwara, M., Edelsberg, J., Stacey, B., and Oster, G. (2005). Use of tricyclic antidepressants in patients with diabetic peripheral neuropathy. J. Pain 6:S35. doi: 10.1016/j.jpain.2005.01.135
Berta, T., Poirot, O., Pertin, M., Ji, R. R., Kellenberger, S., and Decosterd, I. (2008). Transcriptional and functional profiles of voltage-gated Na(+) channels in injured and non-injured DRG neurons in the SNI model of neuropathic pain. Mol. Cell. Neurosci. 37, 196–208. doi: 10.1016/J.MCN.2007.09.007
Black, J. A., Dib-Hajj, S., McNabola, K., Jeste, S., Rizzo, M. A., Kocsis, J. D., et al. (1996). Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Brain Res. Mol. Brain Res. 43, 117–131. doi: 10.1016/S0169-328X(96)00163-5
Black, J. A., Liu, S., Tanaka, M., Cummins, T. R., and Waxman, S. G. (2004). Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108, 237–247. doi: 10.1016/j.pain.2003.12.035
Black, J. A., Nikolajsen, L., Kroner, K., Jensen, T. S., and Waxman, S. G. (2008). Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann. Neurol. 64, 644–653. doi: 10.1002/ANA.21527
Boulton, A. J. M., Vinik, A. I., Arezzo, J. C., Bril, V., Feldman, E. L., Freeman, R., et al. (2005). Diabetic neuropathies: A statement by the American Diabetes Association. Diabetes Care 28, 956–962. doi: 10.2337/DIACARE.28.4.956
Brackenbury, W. J., and Isom, L. L. (2011). Na+ channel β subunits: Overachievers of the ion channel family. Front. Pharmacol. 2:53. doi: 10.3389/fphar.2011.00053
Burstein, S. H., Karst, M., Schneider, U., and Zurier, R. B. (2004). Ajulemic acid: A novel cannabinoid produces analgesia without a “high.”. Life Sci. 75, 1513–1522. doi: 10.1016/J.LFS.2004.04.010
Calhoun, J. D., and Isom, L. L. (2014). The role of non-pore-forming b subunits in physiology and pathophysiology of voltage-gated sodium channels. Handb. Exp. Pharmacol. 221, 51–89. doi: 10.1007/978-3-642-41588-3_4
Callaghan, B. C., Cheng, H. T., Stables, C. L., Smith, A. L., and Feldman, E. L. (2012). Diabetic neuropathy: Clinical manifestations and current treatments. Lancet Neurol. 11, 521–534. doi: 10.1016/S1474-4422(12)70065-0
Campanucci, V., Krishnaswamy, A., and Cooper, E. (2010). Diabetes depresses synaptic transmission in sympathetic ganglia by inactivating nAChRs through a conserved intracellular cysteine residue. Neuron 66, 827–834. doi: 10.1016/j.neuron.2010.06.010
Caterina, M. J., Schumacher, M. A., Tominaga, M., Rosen, T. A., Levine, J. D., Julius, D., et al. (1997). The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 389, 816–824. doi: 10.1038/39807
Catterall, W. A. (1992). Cellular and molecular biology of voltage-gated sodium channels. Physiol. Rev. 72, S15–S48. doi: 10.1152/physrev.1992.72.suppl_4.s15
Chandna, A. R., Nair, M., Chang, C., Pennington, P. R., Yamamoto, Y., Mousseau, D. D., et al. (2015). RAGE mediates the inactivation of nAChRs in sympathetic neurons under high glucose conditions. Eur. J. Neurosci. 41, 341–351. doi: 10.1111/EJN.12795
Chantelau, E. A. (2015). Nociception at the diabetic foot, an uncharted territory. World J. Diabetes 6:391. doi: 10.4239/WJD.V6.I3.391
Chattopadhyay, M., Mata, M., and Fink, D. J. (2008). Continuous delta-opioid receptor activation reduces neuronal voltage-gated sodium channel (NaV1.7) levels through activation of protein kinase C in painful diabetic neuropathy. J. Neurosci. 28, 6652–6658. doi: 10.1523/JNEUROSCI.5530-07.2008
Chattopadhyay, M., Zhou, Z., Hao, S., Mata, M., and Fink, D. J. (2012). Reduction of voltage gated sodium channel protein in DRG by vector mediated miRNA reduces pain in rats with painful diabetic neuropathy. Mol. Pain 8:17. doi: 10.1186/1744-8069-8-17
Chen, C., Bharucha, V., Chen, Y., Westenbroek, R. E., Brown, A., Malhotra, J. D., et al. (2002). Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel β2-subunits. Proc. Natl. Acad. Sci. U.S.A. 99, 17072–17077. doi: 10.1073/pnas.212638099
Chen, L., Huang, J., Zhao, P., Persson, A. K., Dib-Hajj, F. B., Cheng, X., et al. (2018). Conditional knockout of NaV1.6 in adult mice ameliorates neuropathic pain. Sci. Rep. 81, 1–17. doi: 10.1038/s41598-018-22216-w
Cheng, K. I., Wang, H. C., Tseng, K. Y., Wang, Y. H., Chang, C. Y., Chen, Y. J., et al. (2022). Cilostazol ameliorates peripheral neuropathic pain in streptozotocin-induced Type I diabetic rats. Front. Pharmacol. 12:771271. doi: 10.3389/FPHAR.2021.771271
Chong, H. L., and Ruben, P. C. (2008). Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels 2, 407–412. doi: 10.4161/chan.2.6.7429
Cummins, T. R., Aglieco, F., Renganathan, M., Herzog, R. I., Dib-Hajj, S. D., and Waxman, S. G. (2001). Nav1.3 sodium channels: Rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J. Neurosci. 21, 5952–5961. doi: 10.1523/JNEUROSCI.21-16-05952.2001
Cummins, T. R., and Rush, A. M. (2007). Voltage-gated sodium channel blockers for the treatment of neuropathic pain. Expert Rev. Neurother. 7, 1597–1612. doi: 10.1586/14737175.7.11.1597
Cummins, T. R., Howe, J. R., and Waxman, S. G. (1998). Slow closed-state inactivation: A novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J. Neurosci. 18, 9607–9619. doi: 10.1523/JNEUROSCI.18-23-09607.1998
De Lera Ruiz, M., and Kraus, R. L. (2015). Voltage-gated sodium channels: Structure, function, pharmacology, and clinical indications. J. Med. Chem. 58, 7093–7118. doi: 10.1021/jm501981g
Deal, C. L., Schnitzer, T. J., Lipstein, E., Seibold, J. R., Stevens, R. M., Levy, M. D., et al. (1991). Treatment of arthritis with topical capsaicin: A double-blind trial. Clin. Ther. 13, 383–395.
Dib-Hajj, S. D., Cummins, T. R., Black, J. A., and Waxman, S. G. (2010). Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 33, 325–347. doi: 10.1146/ANNUREV-NEURO-060909-153234
Dib-Hajj, S. D., Tyrrell, L., Black, J. A., and Waxman, S. G. (1998). NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc. Natl. Acad. Sci. U.S.A. 95, 8963–8968. doi: 10.1073/PNAS.95.15.8963
Dib-Hajj, S. D., Yang, Y., Black, J. A., and Waxman, S. G. (2013). The Na(V)1.7 sodium channel: From molecule to man. Nat. Rev. Neurosci. 14, 49–62. doi: 10.1038/NRN3404
Djouhri, L., Newton, R., Levinson, S. R., Berry, C. M., Carruthers, B., and Lawson, S. N. (2003). Sensory and electrophysiological properties of guinea-pig sensory neurones expressing Nav 1.7 (PN1) Na+ channel alpha subunit protein. J. Physiol. 546, 565–576. doi: 10.1113/JPHYSIOL.2002.026559
Dludla, P. V., Nkambule, B. B., Cirilli, I., Marcheggiani, F., Mabhida, S. E., Ziqubu, K., et al. (2022). Capsaicin, its clinical significance in patients with painful diabetic neuropathy. Biomed. Pharmacother. 153:113439. doi: 10.1016/J.BIOPHA.2022.113439
Dyck, P. J., Davies, J. L., Wilson, D. M., Service, F. J., Melton, L. J., and O’Brien, P. C. (1999). Risk factors for severity of diabetic polyneuropathy: Intensive longitudinal assessment of the Rochester Diabetic Neuropathy Study cohort. Diabetes Care 22, 1479–1486. doi: 10.2337/DIACARE.22.9.1479
Elkholy, S. E., Elaidy, S. M., El-Sherbeeny, N. A., Toraih, E. A., and El-Gawly, H. W. (2020). Neuroprotective effects of ranolazine versus pioglitazone in experimental diabetic neuropathy: Targeting Nav1.7 channels and PPAR-γ. Life Sci. 250:117557. doi: 10.1016/J.LFS.2020.117557
Foadi, N., Berger, C., Pilawski, I., Stoetzer, C., Karst, M., Haeseler, G., et al. (2014). Inhibition of voltage-gated na+ channels by the synthetic cannabinoid ajulemic acid. Anesth. Analg. 118, 1238–1245. doi: 10.1213/ANE.0000000000000188
Fouda, M. A., Ghovanloo, M. R., and Ruben, P. C. (2020). Cannabidiol protects against high glucose-induced oxidative stress and cytotoxicity in cardiac voltage-gated sodium channels. Br. J. Pharmacol. 177, 2932–2946. doi: 10.1111/bph.15020
Fukuoka, T., Kobayashi, K., Yamanaka, H., Obata, K., Dai, Y., and Noguchi, K. (2008). Comparative study of the distribution of the alpha-subunits of voltage-gated sodium channels in normal and axotomized rat dorsal root ganglion neurons. J. Comp. Neurol. 510, 188–206. doi: 10.1002/CNE.21786
Ghovanloo, M. R., Choudhury, K., Bandaru, T. S., Fouda, M. A., Rayani, K., Rusinova, R., et al. (2021). Cannabidiol inhibits the skeletal muscle Nav1.4 by blocking its pore and by altering membrane elasticity. J. Gen. Physiol. 153:e202012701. doi: 10.1085/jgp.202012701
Ghovanloo, M. R., Estacion, M., Higerd-Rusli, G. P., Zhao, P., Dib-Hajj, S., and Waxman, S. G. (2022). Inhibition of sodium conductance by cannabigerol contributes to a reduction of dorsal root ganglion neuron excitability. Br. J. Pharmacol. 179, 4010–4030. doi: 10.1111/bph.15833
Ghovanloo, M. R., Shuart, N. G., Mezeyova, J., Dean, R. A., Ruben, P. C., and Goodchild, S. J. (2019). Inhibitory effects of cannabidiol on voltage-dependent sodium currents. J. Biol. Chem. 293, 16546–16558. doi: 10.1074/jbc.RA118.004929
Gratton, N., and Cusson, J. R. (1995). Topical capsaicin and joint pain: A therapeutic break-through? Union. Med. Can. 124, 2–4.
Hains, B. C., Klein, J. P., Saab, C. Y., Craner, M. J., Black, J. A., and Waxman, S. G. (2003). Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J. Neurosci. 23, 8881–8892. doi: 10.1523/JNEUROSCI.23-26-08881.2003
Hersh, E. V., Pertes, R. A., and Ochs, H. A. (1994). Topical capsaicin - Pharmacology and potential role in the treatment of temporomandibular pain. J. Clin. Dent. 5, 54–59.
Herzog, R. I., Cummins, T. R., Ghassemi, F., Dib-Hajj, S. D., and Waxman, S. G. (2003). Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J. Physiol. 551, 741–750. doi: 10.1113/JPHYSIOL.2003.047357
Hirade, M., Yasuda, H., Omatsu-Kanbe, M., Kikkawa, R., and Kitasato, H. (1999). Tetrodotoxin-resistant sodium channels of dorsal root ganglion neurons are readily activated in diabetic rats. Neuroscience 90, 933–939. doi: 10.1016/S0306-4522(98)00486-2
Ho, C., and O’Leary, M. E. (2011). Single-cell analysis of sodium channel expression in dorsal root ganglion neurons. Mol. Cell. Neurosci. 46, 159–166. doi: 10.1016/J.MCN.2010.08.017
Hoeijmakers, J. G. J., Faber, C. G., Merkies, I. S. J., and Waxman, S. G. (2015). Painful peripheral neuropathy and sodium channel mutations. Neurosci. Lett. 596, 51–59. doi: 10.1016/j.neulet.2014.12.056
Hong, S., and Wiley, J. W. (2005). Early painful diabetic neuropathy is associated with differential changes in the expression and function of vanilloid receptor 1. J. Biol. Chem. 280, 618–627. doi: 10.1074/jbc.M408500200
Hong, S., and Wiley, J. W. (2006). Altered expression and function of sodium channels in large DRG neurons and myelinated A-fibers in early diabetic neuropathy in the rat. Biochem. Biophys. Res. Commun. 339, 652–660. doi: 10.1016/j.bbrc.2005.11.057
Hong, S., Morrow, T. J., Paulson, P. E., Isom, L. L., and Wiley, J. W. (2004). Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J. Biol. Chem. 279, 29341–29350.
Horishita, T., Yanagihara, N., Ueno, S., Okura, D., Horishita, R., Minami, T., et al. (2017). Antidepressants inhibit Nav1.3, Nav1.7, and Nav1.8 neuronal voltage-gated sodium channels more potently than Nav1.2 and Nav1.6 channels expressed in Xenopus oocytes. Naunyn. Schmiedebergs. Arch. Pharmacol. 390, 1255–1270. doi: 10.1007/S00210-017-1424-X/FIGURES/10
Howlett, A. C., Breivogel, C. S., Childers, S. R., Deadwyler, S. A., Hampson, R. E., and Porrino, L. J. (2004). Cannabinoid physiology and pharmacology: 30 Years of progress. Neuropharmacology 47, 345–358. doi: 10.1016/j.neuropharm.2004.07.030
Huang, Y., Zang, Y., Zhou, L., Gui, W., Liu, X., and Zhong, Y. (2014). The role of TNF-alpha/NF-kappa B pathway on the up-regulation of voltage-gated sodium channel Nav1.7 in DRG neurons of rats with diabetic neuropathy. Neurochem. Int. 75, 112–119. doi: 10.1016/J.NEUINT.2014.05.012
Islam, M. S., and Loots, D. T. (2009). Experimental rodent models of type 2 diabetes: A review. Methods Find. Exp. Clin. Pharmacol. 31, 249–261. doi: 10.1358/MF.2009.31.4.1362513
Isom, L. L., De Jongh, K. S., Patton, D. E., Reber, B. F. X., Offord, J., Charbonneau, H., et al. (1992). Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256, 839–842. doi: 10.1126/SCIENCE.1375395
Iwama, D., Miyamoto, K., Miyahara, S., Tamura, H., Tsujikawa, A., Yamashiro, K., et al. (2007). Neuroprotective effect of cilostazol against retinal ischemic damage via inhibition of leukocyte–endothelial cell interactions. J. Thromb. Haemost. 5, 818–825. doi: 10.1111/J.1538-7836.2007.02425.X
Jancsó, N., Jancsó-Gábor, A., and Szolcsányi, J. (1967). Direct evidence for neurogenic inflammation and its prevention by denervation and by pretreatment with capsaicin. Br. J. Pharmacol. Chemother. 31, 138–151. doi: 10.1111/J.1476-5381.1967.TB01984.X
Jin, X., and Gereau, R. W. IV (2006). Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-α. J. Neurosci. 26, 246–255. doi: 10.1523/JNEUROSCI.3858-05.2006
Jin, X., Morsy, N., Winston, J., Pasricha, P. J., Garrett, K., and Akbarali, H. I. (2004). Modulation of TRPV1 by nonreceptor tyrosine kinase, c-Src kinase. Am. J. Physiol. Cell Physiol. 287, C558–C563. doi: 10.1152/ajpcell.00113.2004
Kasama, S., Kawakubo, M., Suzuki, T., Nishizawa, T., Ishida, A., and Nakayama, J. (2007). RNA interference-mediated knock-down of transient receptor potential vanilloid 1 prevents forepaw inflammatory hyperalgesia in rat. Eur. J. Neurosci. 25, 2956–2963. doi: 10.1111/J.1460-9568.2007.05584.X
Kobayashi, M., and Zochodne, D. W. (2018). Diabetic neuropathy and the sensory neuron: New aspects of pathogenesis and their treatment implications. J. Diabetes Investig. 9, 1239–1254. doi: 10.1111/JDI.12833
Lam, D., Momeni, Z., Theaker, M., Jagadeeshan, S., Yamamoto, Y., Ianowski, J. P., et al. (2018). RAGE-dependent potentiation of TRPV1 currents in sensory neurons exposed to high glucose. PLoS One 13:e0193312. doi: 10.1371/journal.pone.0193312
Lee, J. H., Park, C. K., Chen, G., Han, Q., Xie, R. G., Liu, T., et al. (2014). A monoclonal antibody that targets a NaV1.7 channel voltage sensor for pain and itch relief. Cell 157, 1393–1404. doi: 10.1016/J.CELL.2014.03.064
Li, Q., Qin, L., and Li, J. (2020). Enhancement by TNF-α of TTX-resistant Na V current in muscle sensory neurons after femoral artery occlusion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 318, R772–R780. doi: 10.1152/AJPREGU.00338.2019
Lim, J. H., Woo, J. S., and Shin, Y. W. (2009). Cilostazol Protects Endothelial Cells Against Lipopolysaccharide-Induced Apoptosis Through ERK1/2- and P38 MAPK-Dependent Pathways. Korean J. Intern. Med. 24, 113–122. doi: 10.3904/KJIM.2009.24.2.113
Lindia, J. A., Köhler, M. G., Martin, W. J., and Abbadie, C. (2005). Relationship between sodium channel NaV1.3 expression and neuropathic pain behavior in rats. Pain 117, 145–153. doi: 10.1016/J.PAIN.2005.05.027
Liu, L., Oortgiesen, M., Li, L., and Simon, S. A. (2001). Capsaicin inhibits activation of voltage-gated sodium currents in capsaicin-sensitive trigeminal ganglion neurons. J. Neurophysiol. 85, 745–758. doi: 10.1152/jn.2001.85.2.745
Lucas, P. T., Meadows, L. S., Nicholls, J., and Ragsdale, D. S. (2005). An epilepsy mutation in the beta1 subunit of the voltage-gated sodium channel results in reduced channel sensitivity to phenytoin. Epilepsy Res. 64, 77–84. doi: 10.1016/J.EPLEPSYRES.2005.03.003
Meadows, L. S., Malhotra, J., Loukas, A., Thyagarajan, V., Kazen-Gillespie, K. A., Koopman, M. C., et al. (2002). Functional and biochemical analysis of a sodium channel beta1 subunit mutation responsible for generalized epilepsy with febrile seizures plus type 1. J. Neurosci. 22, 10699–10709. doi: 10.1523/JNEUROSCI.22-24-10699.2002
Medeiros-Domingo, A., Kaku, T., Tester, D. J., Iturralde-Torres, P., Itty, A., Ye, B., et al. (2007). SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation 116, 134–142. doi: 10.1161/CIRCULATIONAHA.106.659086
Melis, M., Di Giosia, M., and Colloca, L. (2019). Ancillary factors in the treatment of orofacial pain: A topical narrative review. J. Oral Rehabil. 46, 200–207. doi: 10.1111/joor.12736
Melli, G., and Höke, A. (2009). Dorsal root ganglia sensory neuronal cultures: A tool for drug discovery for peripheral neuropathies. Exp. Opin. Drug Discov. 4, 1035–1045. doi: 10.1517/17460440903266829
Momeni, Z., Bautista, M., Neapetung, J., Urban, R., Yamamoto, Y., Krishnan, A., et al. (2021). RAGE signaling is required for AMPA receptor dysfunction in the hippocampus of hyperglycemic mice. Physiol. Behav 229:113255. doi: 10.1016/J.PHYSBEH.2020.113255
Nair, M., Jagadeeshan, S., Katselis, G., Luan, X., Momeni, Z., Henao-Romero, N., et al. (2021). Lipopolysaccharides induce a RAGE-mediated sensitization of sensory neurons and fluid hypersecretion in the upper airways. Sci. Rep. 11:8336. doi: 10.1038/s41598-021-86069-6
Namadurai, S., Yereddi, N. R., Cusdin, F. S., Huang, C. L. H., Chirgadze, D. Y., and Jackson, A. P. (2015). A new look at sodium channel β subunits. Open Biol. 5:140192. doi: 10.1098/rsob.140192
Nemoto, T., Miyazaki, S., Kanai, T., Maruta, T., Satoh, S., Yoshikawa, N., et al. (2010). Nav1.7-Ca2+ influx-induced increased phosphorylations of extracellular signal-regulated kinase (ERK) and p38 attenuate tau phosphorylation via glycogen synthase kinase-3beta: Priming of Nav1.7 gating by ERK and p38. Eur. J. Pharmacol. 640, 20–28. doi: 10.1016/J.EJPHAR.2010.04.048
Nicholson, R. A., Liao, C., Zheng, J., David, L. S., Coyne, L., Errington, A. C., et al. (2003). Sodium channel inhibition by anandamide and synthetic cannabimimetics in brain. Brain Res. 978, 194–204. doi: 10.1016/S0006-8993(03)02808-7
O’Malley, H. A., and Isom, L. L. (2015). Sodium channel β subunits: Emerging targets in channelopathies. Annu. Rev. Physiol. 77, 481–504. doi: 10.1146/annurev-physiol-021014-071846
Okura, D., Horishita, T., Ueno, S., Yanagihara, N., Sudo, Y., Uezono, Y., et al. (2014). The endocannabinoid anandamide inhibits voltage-gated sodium channels Nav1.2, Nav1.6, Nav1.7, and Nav1.8 in xenopus oocytes. Anesth. Analg. 118, 554–562. doi: 10.1213/ANE.0000000000000070
Oyama, F., Miyazaki, H., Sakamoto, N., Becquet, C., Machida, Y., Kaneko, K., et al. (2006). Sodium channel beta4 subunit: Down-regulation and possible involvement in neuritic degeneration in Huntington’s disease transgenic mice. J. Neurochem. 98, 518–529. doi: 10.1111/J.1471-4159.2006.03893.X
Pertin, M., Ji, R. R., Berta, T., Powell, A. J., Karchewski, L., Tate, S. N., et al. (2005). Upregulation of the voltage-gated sodium channel β2 subunit in neuropathic pain models: Characterization of expression in injured and non-injured primary sensory neurons. J. Neurosci. 25, 10970–10980. doi: 10.1523/JNEUROSCI.3066-05.2005
Priest, B. T., and Kaczorowski, G. J. (2007). Blocking sodium channels to treat neuropathic pain. Exp. Opin. Ther. Targets 11, 291–306. doi: 10.1517/14728222.11.3.291
Qin, N., D’Andrea, M. R., Lubin, M. L., Shafaee, N., Codd, E. E., and Correa, A. M. (2003). Molecular cloning and functional expression of the human sodium channel β1B subunit, a novel splicing variant of the β1 subunit. Eur. J. Biochem. 270, 4762–4770. doi: 10.1046/J.1432-1033.2003.03878.X
Rahn, E. J., and Hohmann, A. G. (2009). Cannabinoids as pharmacotherapies for neuropathic pain: From the bench to the bedside. Neurotherapeutics 6, 713–737. doi: 10.1016/J.NURT.2009.08.002
Raman, I. M., and Bean, B. P. (1997). Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J. Neurosci. 17, 4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997
Renganathan, M., Cummins, T. R., and Waxman, S. G. (2001). Contribution of Nav 1.8 sodium channels to action potential electrogenesis in DRG neurons. J. Neurophysiol. 86, 629–640. doi: 10.1152/jn.2001.86.2.629
Rogers, M., Tang, L., Madge, D. J., and Stevens, E. B. (2006). The role of sodium channels in neuropathic pain. Semin. Cell Dev. Biol. 17, 571–581. doi: 10.1016/j.semcdb.2006.10.009
Rush, A. M., Cummins, T. R., and Waxman, S. G. (2007). Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. 579, 1–14. doi: 10.1113/jphysiol.2006.121483
Russell, J. W., Golovoy, D., Vincent, A. M., Mahendru, P., Olzmann, J. A., Mentzer, A., et al. (2002). High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 16, 1738–1748. doi: 10.1096/fj.01-1027com
Sait, L. G., Sula, A., Ghovanloo, M. R., Hollingworth, D., Ruben, P. C., and Wallace, B. A. (2020). Cannabidiol interactions with voltage-gated sodium channels. Elife 9, 1–17. doi: 10.7554/eLife.58593
Schink, M., Leipold, E., Schirmeyer, J., Schönherr, R., Hoshi, T., and Heinemann, S. H. (2016). Reactive species modify NaV1.8 channels and affect action potentials in murine dorsal root ganglion neurons. Pflugers Arch. Eur. J. Physiol. 468, 99–110. doi: 10.1007/s00424-015-1735-z
Schmidt, J. W., and Catterall, W. A. (1986). Biosynthesis and processing of the α subunit of the voltage-sensitive sodium channel in rat brain neurons. Cell 46, 437–445. doi: 10.1016/0092-8674(86)90664-1
Strichartz, G. R., Chiu, S. Y., and Ritchie, J. M. (1978). The effect of Δ9-tetrahydrocannabinol on the activation of sodium conductance in node of Ranvier. J. Pharmacol. Exp. Ther. 207, 801–809.
Sugimoto, Y., Kojima, Y., Inayoshi, A., Inoue, K., Miura-Kusaka, H., Mori, K., et al. (2013). K-685, a TRPV1 antagonist, blocks PKC-sensitized TRPV1 activation and improves the inflammatory pain in a rat complete Freund’s adjuvant model. J. Pharmacol. Sci. 123, 256–266. doi: 10.1254/JPHS.13088FP
Sula, A., and Wallace, B. A. (2017). Interpreting the functional role of a novel interaction motif in prokaryotic sodium channels. J. Gen. Physiol. 149, 613–622. doi: 10.1085/JGP.201611740
Sula, A., Booker, J., Ng, L. C. T., Naylor, C. E., Decaen, P. G., and Wallace, B. A. (2017). The complete structure of an activated open sodium channel. Nat. Commun. 8:14205. doi: 10.1038/NCOMMS14205
Tan, A. M., Samad, O. A., Dib-Hajj, S. D., and Waxman, S. G. (2015). Virus-mediated knockdown of Nav1.3 in dorsal root ganglia of STZ-induced diabetic rats alleviates tactile allodynia. Mol. Med. 21, 544–552. doi: 10.2119/molmed.2015.00063
Tesfaye, S., Boulton, A. J. M., and Dickenson, A. H. (2013). Mechanisms and management of diabetic painful distal symmetrical polyneuropathy. Diabetes Care 36, 2456–2465. doi: 10.2337/DC12-1964
Tesfaye, S., Boulton, A. J. M., Dyck, P. J., Freeman, R., Horowitz, M., Kempler, P., et al. (2010). Diabetic neuropathies: Update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care 33, 2285–2293. doi: 10.2337/DC10-1303
Theile, J. W., and Cummins, T. R. (2011). Inhibition of Navβ4 peptide-mediated resurgent sodium currents in Nav1.7 channels by carbamazepine, riluzole, and anandamide. Mol. Pharmacol. 80, 724–734. doi: 10.1124/mol.111.072751
Tibbs, G. R., Posson, D. J., and Goldstein, P. A. (2016). Voltage-gated ion channels in the PNS: Novel therapies for neuropathic pain? Trends Pharmacol. Sci. 37, 522–542. doi: 10.1016/j.tips.2016.05.002
Toledo-Aral, J. J., Moss, B. L., He, Z. J., Koszowski, A. G., Whisenand, T., Levinson, S. R., et al. (1997). Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc. Natl. Acad. Sci. U.S.A. 94, 1527–1532. doi: 10.1073/PNAS.94.4.1527
Tomlinson, D. R., and Gardiner, N. J. (2008). Glucose neurotoxicity. Nat. Rev. Neurosci. 9, 36–45. doi: 10.1038/nrn2294
Vincent, A. M., Stevens, M. J., Backus, C., McLean, L. L., and Feldman, E. L. (2005). Cell culture modeling to test therapies against hyperglycemia-mediated oxidative stress and injury. Antioxid. Redox Signal. 7, 1494–1506. doi: 10.1089/ars.2005.7.1494
Walker, K. M., Urban, L., Medhurst, S. J., Patel, S., Panesar, M., Fox, A. J., et al. (2003). The VR1 antagonist capsazepine reverses mechanical hyperalgesia in models of inflammatory and neuropathic pain. J. Pharmacol. Exp. Ther. 304, 56–62. doi: 10.1124/JPET.102.042010
Wallace, M. S., Marcotte, T. D., Umlauf, A., Gouaux, B., and Atkinson, J. H. (2015). Efficacy of Inhaled Cannabis on Painful Diabetic Neuropathy. J. Pain 16, 616–627. doi: 10.1016/j.jpain.2015.03.008
Wang, W., Gu, J., Li, Y. Q., and Tao, Y. X. (2011). Are voltage-gated sodium channels on the dorsal root ganglion involved in the development of neuropathic pain? Mol. Pain 7:16. doi: 10.1186/1744-8069-7-16
Waxman, S. G. (2012). Sodium channels, the electrogenisome and the electrogenistat: Lessons and questions from the clinic. J. Physiol. 590, 2601–2612. doi: 10.1113/jphysiol.2012.228460
Winocur, E., Gavish, A., Halachmi, M., Eli, I., and Gazit, Z. (2000). Topical application of capsaicin for the treatment of localized pain in the temporomandibular joint area. J. Orofac. Pain 14, 31–36.
Wittmack, E. K., Rush, A. M., Hudmon, A., Waxman, S. G., and Dib-Hajj, S. D. (2005). Voltage-gated sodium channel Nav1.6 is modulated by p38 mitogen-activated protein kinase. J. Neurosci. 25, 6621–6630. doi: 10.1523/JNEUROSCI.0541-05.2005
Yin, R., Liu, D., Chhoa, M., Li, C. M., Luo, Y., Zhang, M., et al. (2016). Voltage-gated sodium channel function and expression in injured and uninjured rat dorsal root ganglia neurons. Int. J. Neurosci. 126, 182–192. doi: 10.3109/00207454.2015.1004172
Zakin, E., Abrams, R., and Simpson, D. M. (2019). Diabetic Neuropathy. Semin. Neurol. 39, 560–569. doi: 10.1055/S-0039-1688978
Zhang, H. X. B., and Bean, B. P. (2021). Cannabidiol inhibition of murine primary nociceptors: Tight binding to slow inactivated states of nav1.8 channels. J. Neurosci. 41, 6371–6387. doi: 10.1523/JNEUROSCI.3216-20.2021
Zhang, J. L., Yang, J. P., Zhang, J. R., Li, R. Q., Wang, J., Jan, J. J., et al. (2013). Gabapentin reduces allodynia and hyperalgesia in painful diabetic neuropathy rats by decreasing expression level of Nav1.7 and p-ERK1/2 in DRG neurons. Brain Res. 1493, 13–18. doi: 10.1016/J.BRAINRES.2012.11.032
Keywords: diabetic neuropathy (painful), Nav channels, modulation, therapeutics, sensory neurons
Citation: Bigsby S, Neapetung J and Campanucci VA (2022) Voltage-gated sodium channels in diabetic sensory neuropathy: Function, modulation, and therapeutic potential. Front. Cell. Neurosci. 16:994585. doi: 10.3389/fncel.2022.994585
Received: 15 July 2022; Accepted: 11 October 2022;
Published: 17 November 2022.
Edited by:
Xin Qi, Case Western Reserve University, United StatesReviewed by:
Rayaz A. Malik, Weill Cornell Medicine - Qatar, QatarMohammad-Reza Ghovanloo, Yale University, United States
Tamer M. Gamal El-Din, University of Washington, United States
Copyright © 2022 Bigsby, Neapetung and Campanucci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Verónica A. Campanucci, dmVyb25pY2EuY2FtcGFudWNjaUB1c2Fzay5jYQ==
†These authors have contributed equally to this work