
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci., 03 August 2022
Sec. Cellular Neuropathology
Volume 16 - 2022 | https://doi.org/10.3389/fncel.2022.972029
This article is part of the Research TopicAdvances in Spinal Muscular AtrophyView all 9 articles
 Lucia Chehade1,2,3
Lucia Chehade1,2,3 Marc-Olivier Deguise1,2,3,4
Marc-Olivier Deguise1,2,3,4 Yves De Repentigny1Rebecca Yaworski1Ariane Beauvais1
Yves De Repentigny1Rebecca Yaworski1Ariane Beauvais1 Sabrina Gagnon1
Sabrina Gagnon1 Niko Hensel1
Niko Hensel1 Rashmi Kothary1,2,3,5,6*
Rashmi Kothary1,2,3,5,6*Spinal muscular atrophy (SMA) is a monogenic neuromuscular disease caused by low levels of the Survival Motor Neuron (SMN) protein. Motor neuron degeneration is the central hallmark of the disease. However, the SMN protein is ubiquitously expressed and depletion of the protein in peripheral tissues results in intrinsic disease manifestations, including muscle defects, independent of neurodegeneration. The approved SMN-restoring therapies have led to remarkable clinical improvements in SMA patients. Yet, the presence of a significant number of non-responders stresses the need for complementary therapeutic strategies targeting processes which do not rely solely on restoring SMN. Dysregulated cell death pathways are candidates for SMN-independent pathomechanisms in SMA. Receptor-interacting protein kinase 1 (RIPK1) and RIPK3 have been widely recognized as critical therapeutic targets of necroptosis, an important form of programmed cell death. In addition, Caspase-1 plays a fundamental role in inflammation and cell death. In this study, we evaluate the role of necroptosis, particularly RIPK3 and Caspase-1, in the Smn2B/− mouse model of SMA. We have generated a triple mutant (TKO), the Smn2B/−; Ripk3−/−; Casp1−/− mouse. TKO mice displayed a robust increase in survival and improved motor function compared to Smn2B/− mice. While there was no protection against motor neuron loss or neuromuscular junction pathology, larger muscle fibers were observed in TKO mice compared to Smn2B/− mice. Our study shows that necroptosis modulates survival, motor behavior and muscle fiber size independent of SMN levels and independent of neurodegeneration. Thus, small-molecule inhibitors of necroptosis as a combinatorial approach together with SMN-restoring drugs could be a future strategy for the treatment of SMA.
Spinal muscular atrophy (SMA) is a devastating neuromuscular disease characterized by low levels of the ubiquitously expressed Survival Motor Neuron (SMN) protein. The SMN protein is produced by the Survival motor neuron 1 (SMN1) gene located on chromosome 5q13 (Lefebvre et al., 1995). The genetic basis of SMA involves a mutation or homozygous deletion of the SMN1 gene and retention of at least one copy of the paralogous gene SMN2 (Lefebvre et al., 1995). The latter undergoes alternative splicing resulting in insufficient SMN protein to compensate for SMN1 loss (Lorson et al., 1999; Monani et al., 1999). In most forms of the disease, patients will exhibit variable severity of progressive muscle weakness and atrophy (Sumner, 2006).
SMN depletion leads to the profound loss of motor neurons of the spinal cord (Lefebvre et al., 1997) and peripheral organ pathology including the muscle (reviewed in Shababi et al., 2014). While the underlying mechanisms are not fully understood, there is growing evidence of cell death pathway activation (Simic et al., 2000, 2007; Shafey et al., 2005; Tsai et al., 2006, 2008; Dachs et al., 2011; Mutsaers et al., 2011; Hayhurst et al., 2012; Martínez-Hernández et al., 2013; Fayzullina and Martin, 2014; Simon et al., 2017; Carrasco et al., 2019; Courtney et al., 2019; Deguise et al., 2020a; Weissman et al., 2021) and inflammation in several tissues in SMA (Deguise and Kothary, 2017; Deguise et al., 2017; Wan et al., 2018; Salucci et al., 2021). As such, investigating cell death mechanisms could shed light on the specific pathogenic mechanisms underlying motor neuron death and muscle atrophy.
There is mounting evidence that apoptosis plays a role in motor neuron death in patients and mouse models of SMA. In spinal cords, a significant number of neurons appear to be dying from apoptosis accompanied by dysregulation of apoptosis regulators, specifically p53 (Simic et al., 2000, 2007; Soler-Botija et al., 2005; Tsai et al., 2006, 2008; Simon et al., 2017). While p53 activity has been traditionally linked to apoptosis, numerous studies have identified p53 involvement in other forms of cell death including necroptosis (Wang et al., 2016). In fact, a recent study shows that p53 induction mediates neuronal death via necroptosis (Peek et al., 2022). SMN depleted muscles also display dysregulation of cell death pathways and apoptosis (Shafey et al., 2005; Dachs et al., 2011; Mutsaers et al., 2011; Martínez-Hernández et al., 2013). Importantly, cell death and defects in skeletal muscle precedes overt spinal cord pathology such as denervation and motor neuron loss (Fayzullina and Martin, 2014; Sheng et al., 2018). Beyond the neuromuscular system, a potential role of cell death is supported by digital and cutaneous necrosis in SMA infants (Carrasco et al., 2019; Weissman et al., 2021). Likewise, in the Smn2B/− mice, a mouse model of severe SMA (Bowerman et al., 2012), spleen, tail, and ear necrosis are observed (Deguise et al., 2020a). Altogether, this suggests a scenario in which cell death mechanisms contribute to overall SMA pathology.
Cell death is known to play a role in the regulation of inflammation but may also result from inflammation (Yang et al., 2015). For example, in the central nervous system (CNS), microglial cells sample the environment and recognize pattern recognition receptors and cell damage-associated molecular patterns, triggering an inflammatory response and subsequent cell death (Schroder and Tschopp, 2010; Boucher et al., 2018). There is neuroinflammation resulting from defective microglial and astroglial cells in SMA that is suggested to contribute to the activation of cell death pathways (reviewed in Abati et al., 2020). These observations are not exclusive to motor neurons and have been made outside the CNS. As such, inflammation pathways may contribute to cell death in neurons, muscle, and other affected organs in SMA.
Necroptosis is a regulated form of cell death mimicking features of both necrosis and apoptosis. Under pathological conditions, upregulation of death receptor family ligands can sensitize cells in the CNS to necroptosis (Ransohoff, 2016; Dhuriya and Sharma, 2018). Necroptosis is mediated by the activation of receptor-interacting protein kinases (RIPK1 and RIPK3) leading to the phosphorylation of mixed lineage kinase domain-like protein (MLKL) (Rodriguez et al., 2016). RIPK3 has been identified as a core component of this pathway since it is critical for the oligomerization of MLKL that culminates with robust inflammation and the release of cell damage-associated molecular patterns (Dhuriya and Sharma, 2018; Zhu et al., 2018). Depending on the cellular environment, inflammasome activation will occur which associates with Caspase-1, leading to the cleavage of IL-1β into the mature form and thus the secretion of pro-inflammatory cytokines (Dhuriya and Sharma, 2018).
In seeking to understand the pathways that could lead to neuron and muscle death in SMA, we turned to investigate necroptosis, a disease process in which cell death and inflammation have been linked. Given the strong evidence of programmed cell death and inflammation occurring in SMA, we generated a mouse model to study the pathway by knocking out Ripk3 and Casp1. RIPK3 is a crucial mediator of necroptosis and homozygous deletion of Ripk3 should target the necroptosis pathway (Zhang et al., 2011; Rodriguez et al., 2016). Caspase 1 knockout provides significant protection against IL-1β mediated inflammation (Schroder and Tschopp, 2010; Boucher et al., 2018). Therefore, we generated a triple mutant model, namely the Smn2B/−; Ripk3−/−; Casp1−/− mice (henceforth referred to as TKO mice). While we did not observe protection against motor neuron loss nor neuromuscular junction (NMJ) pathology, there was a significant increase in survival and motor function improvement in TKO mice compared to Smn2B/− mice. Interestingly, we also observed larger muscle fibers without any significant changes in neurogenic muscle atrophy markers. Our study shows that necroptosis plays a role in muscle fiber size indicating an intrinsic mechanistic relevant in skeletal muscle development. Understanding this mechanism is important given the availability of small-molecule inhibitors of necroptosis which could be combined with SMN-restoring drugs as a novel approach in the early treatment of SMA.
Smn+/− mice were crossed to Smn2B/2B mice to obtain Smn2B/+ and Smn2B/− animals maintained on the C57BL6/J background (Bowerman et al., 2012; Eshraghi et al., 2016). The Smn2B/− mice are a model of SMA and the asymptomatic heterozygous Smn2B/+ mice are used as controls in these experiments. Casp1−/−; Ripk3−/− knockout mice (Shutinoski et al., 2020) were provided by Dr. Sad's laboratory (University of Ottawa, Ottawa, Canada). The Smn2B/−; Casp1−/−; Ripk3−/− triple mutant mice (TKO) were derived after multiple generations of crossing Smn+/−; Casp1−/−; Ripk3−/− and Smn2B/2B; Casp1−/−; Ripk3−/− mice on the C57BL6/J background. Establishment of the line was confirmed by genotyping the DNA extracted from mouse ear biopsies using multiplex PCR (see Supplementary Table 1 for primers used). Both male and female mice were included in the studies. Mice were bred and housed at the University of Ottawa Animal Facility and cared for under protocol OHRI-3343 according to the Canadian Council on Animal Care.
Mice were tattooed by Animal Care Facility staff between P4 and P6 for longitudinal assessment of survival, body weight change and motor function tests. Mice were weighed every 2 days. For longitudinal assessment of survival, mice were weaned at P21 into cages containing DietGel 76A (ClearH2O), a purified dietary supplement. Additional animals were sacrificed at P9 or P19 for tissue collection and analysis.
Spinal reflexes were assessed using the righting reflex test. Motor balance and coordination was assessed using the pen test. Forelimb and hindlimb muscle strength were measured using an inverted mesh grip test. Testing was administered in the following order: (1) Righting reflex, (2) Pen test, and (3) Inverted mesh/wire grip test on the same day, every 2 days. A maximum of 30 s on the pen test and 60 s on the inverted mesh grip was considered the maximal threshold. Three measurements were taken in succession and the average was recorded for analysis using the lab's previously reported protocols (Deguise et al., 2020b).
Tissues for quantitative biochemical analysis were harvested from pre-symptomatic mice at P9 and symptomatic mice at P19 and preserved as previously described (Deguise et al., 2020b). For paraffin embedding, the tibialis anterior (TA) muscles were fixed for 24 h while livers, hearts, and spinal cords were fixed for 48 h in 1:10 diluted buffered formalin (Thermo Fisher Scientific, Waltham, MA) at 4°C and then transferred to 70% ethanol at 4°C until processing. All samples were processed, sectioned and stained at the Department of Pathology and Laboratory Medicine (University of Ottawa) as previously described (Deguise et al., 2020b). For NMJ analysis, the transverse abdominal muscle (TVA) was dissected and isolated under a dissection microscope according to protocol detailed in (Murray et al., 2014). Briefly, the TVA was incubated with TRITC conjugated alpha-bungarotoxin for 30 min at room temperature. Muscles were subsequently permeabilized with 2% (v/v) Triton X-100 (Sigma), and blocked at room temperature with 4% BSA, 1% (v/v) Triton X-100 in PBS. Tissues were then incubated overnight at 4°C in blocking reagent containing primary antibodies for neurofilament and synaptic vesicle protein 2 (Supplementary Table 2). The following day, tissues were incubated in secondary antibodies (Supplementary Table 2) for 2 h at room temperature in the same antibody diluent. Slides were mounted with fluorescent mounting medium (DAKO mounting media S302380-2). Images were acquired using a Zeiss Axio Imager M1 microscope mounted with a camera.
Quantification of spinal cord and muscle histology was performed in a blinded fashion. N number refers to individual mice and is described in each figure legend. Representative images of H&E-stained sections were acquired using the ZEISS Axio Scan Z1 slide scanner for brightfield at different magnifications. Muscle fiber size was quantified using ImageJ. About 100–200 muscle fibers in randomly selected areas of the muscle were counted to ensure proper coverage. The area of each fiber was measured to calculate a mean fiber size for each animal. Quantification of motor neuron number and size was performed on a total of 5 sections separated by 10 sections each per animal from the H&E-stained lumbar spinal cord. To ensure proper counts, motor neurons were identified by location in the ventral horn, presence of a nucleolus, and cells having cross-sectional area >350 μm2. For quantification of choline acetyltransferase (ChAT) positive motor neurons, lumbar spinal cords were dissected, postfixed in 4% paraformaldehyde overnight at 4°C, then prepared for cryosectioning as previously described (Deguise et al., 2020b). The number of ChAT-positive motor neurons per ventral horn was recorded for four different sections per animal, each separated by at least 100 μm to avoid counting the same motor neuron twice. Only ChAT-positive motor neurons (diameter ≥20 μm) were counted. NMJs were quantified as either normal or displaying neurofilament accumulation, and endplates were noted as either occupied or unoccupied.
Total protein was collected by homogenization of flash frozen whole spinal cord, liver, heart, tibialis anterior (TA), and hindlimb muscles in RIPA lysis buffer (Cell Signaling) in the presence of PMSF (Cell Signaling) and proteinase and phosphatase inhibitors (Abcam). Protein concentrations were determined using Pierce BCA Protein Assay kit (ThermoFisher). Thirty ug protein per sample was separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis. Proteins were transferred to a PVDF membrane (Immobilon-P, Millipore) and blocked for 1 h at room temperature in 5% (w/v) skim milk in TBST. Membranes were then incubated in primary antibodies and secondary antibodies (see Supplementary Table 2). Proteins were visualized using Pierce ECL Western Blotting Substrate (Thermo Scientific) and SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific). Protein quantification was by densitometric analysis using ImageJ software (Version 1.51, Free Java software provided by National Institutes of Health, USA) by taking the mean gray value of bands for the target protein, normalized to loading control (α-tubulin or GAPDH) from the same blot.
Total RNA from the TA muscle was extracted using Qiagen RNeasy Mini kit, quantified and reverse transcribed using RT2 first strand kit (Qiagen) according to the manufacturer's protocol. qPCR was performed in technical triplicates for each sample using mouse primers obtained from Bio-Rad targeting Atrogin-1 (Fbxo32), MuRF1 (Trim63) and Gapdh. Each qPCR reaction contained 300 ng of cDNA, 2x SsoFast EvaGreen (Bio-Rad), RNase/DNase-free water, and respective primers in a final volume of 20 μl. Three negative controls containing water instead of cDNA were included in every qPCR plate. Results were quantified using 2−ΔΔCt method. Results were normalized to Gapdh.
All data were expressed as mean ± standard error of the mean. Survival data was represented with Kaplan-Meier survival curves and statistically significant differences between groups were evaluated using the Mantel-Cox test. One-way analysis of variance (ANOVA) with Tukey post-hoc test was used to distinguish differences between more than two groups when multiple comparisons were necessary. Matched two-way ANOVA with Geisser–Greenhouse correction was used with complete data sets. Matched two-way ANOVA with mixed effects model was performed where values were missing (in case of death) with Sidak's or Tukey's method for multiple comparisons depending on the characteristics of the dataset. Statistical tests were performed using GraphPad Prism V.9.0 (GraphPad Software, San Diego, CA). Significance was set at P < 0.05.
All data will be made available upon contacting the corresponding author.
To investigate necroptosis in an SMA mouse model, we generated a knockout of the necroptotic pathway in the Smn2B/− mice. This was achieved via genetic crosses of mice to introduce the Smn2B allele onto the Ripk3−/−; Casp1−/− double knockout (Figure 1A). Two separate breeding schemes were initiated to obtain the Smn+/−; Ripk3−/−; Casp1−/− and Smn2B/2B; Ripk3−/−; Casp1−/− mice. These mice were paired and gave a progeny of triple mutant mice (TKO, Smn2B/−; Ripk3−/−; Casp1−/−) and littermate double knockout (DKO, Smn2B/+; Ripk3−/−; Casp1−/−) control mice with necroptotic pathway ablation (Figure 1A). The Smn2B/+ and Smn2B/− mice were included as additional controls. Genetic background has a substantial effect on the phenotype and survival of severe mouse models of SMA (Eshraghi et al., 2016). Therefore, all mice used in this study were derived and maintained on a pure C57BL6/J background to exclude any potential effect related to a mixed genetic background. Reduced tail length and ear necrosis are two morphological canonical features present in the Smn2B/− mice when compared to Smn2B/+ controls. These pathological features were preserved in the TKO mice compared to DKO mice (Figure 1B). Extra-neuronal pathology has been previously described in mouse models of SMA. In the Smn2B/− mouse model, defects in fatty acid metabolism led to non-alcoholic fatty liver disease (NAFLD), which is a macroscopic pathological feature (Deguise et al., 2021). TKO livers were unchanged compared to the Smn2B/− mice on gross morphology (Figure 1C) and histological evaluation confirmed that necroptotic pathway ablation does not impact fat accumulation in the liver (Supplementary Figures 1A–D). The Smn2B/− mice express 15% of SMN protein in the spinal cord compared to wild type (Eshraghi et al., 2016), which we confirmed by western blot analysis (Figures 1D,E). Levels of SMN in TKO mice did not change from those observed in Smn2B/− mice across all tissues assessed (Figures 1D–G). This means that any potential changes in phenotype are independent of SMN levels in TKO mice. Altogether, we conclude that genetic ablation of necroptotic cell death pathways does not alter SMN levels in TKO mice and that any phenotypic alterations may result from an SMN-independent mechanism.
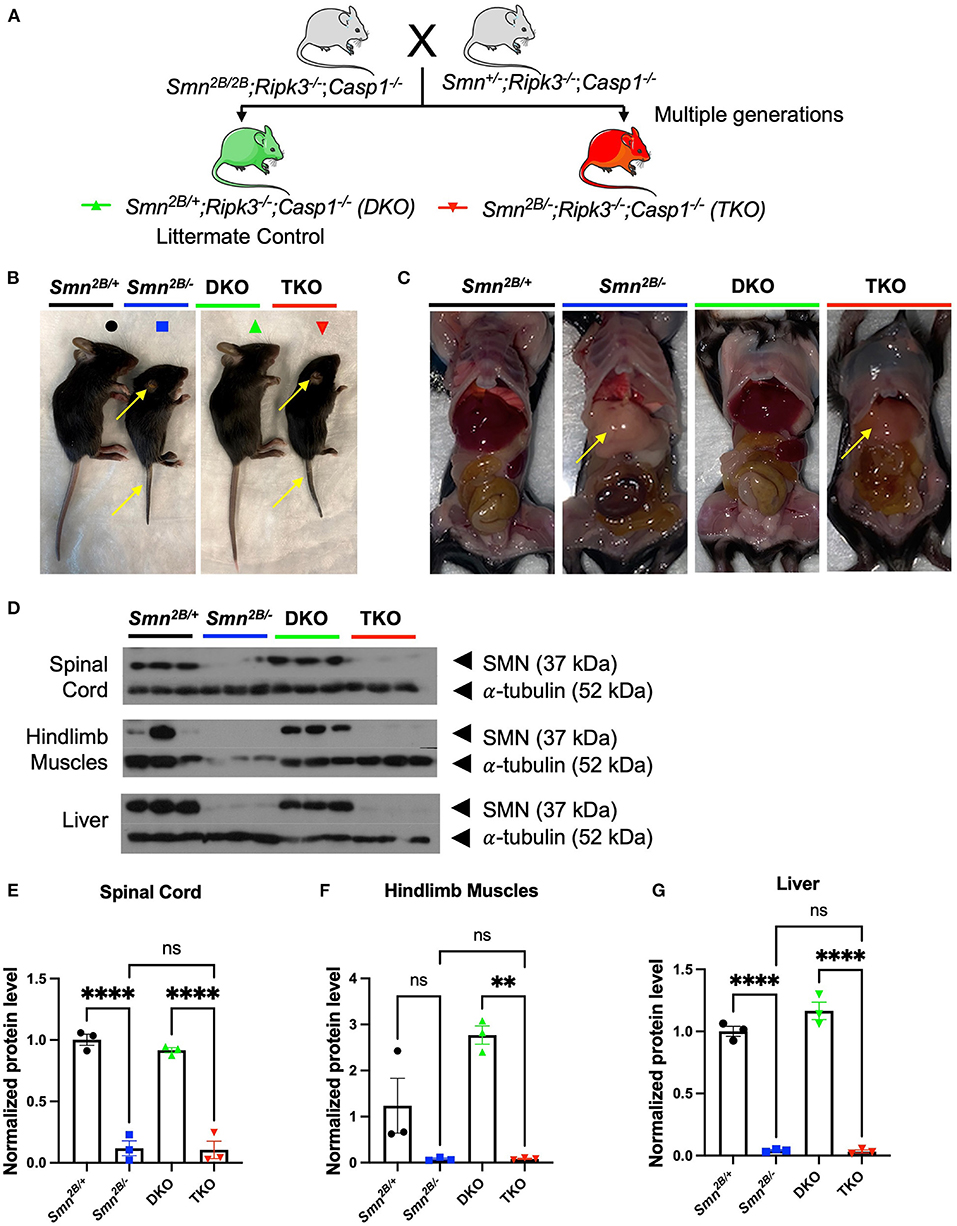
Figure 1. Generation of necroptotic cell death pathway knockout mice on the Smn2B/− SMA mouse model background. (A) Schematic representation of the breeding scheme to generate the necroptotic pathway knockout on Smn2B/+ background (Smn2B/+; Ripk3−/−; Casp1−/−). These double knockout mice (DKO, Green) do not develop an SMA-like phenotype and serve as controls. Smn2B/−; Ripk3−/−; Casp1−/− triple mutant mice (TKO, Red) harbor a necroptotic pathway knockout on the Smn2B/− mouse background. Parental strains were generated by multiple generations of crossings. Mice were compared to the Smn2B/+ (control) and Smn2B/− (mouse model of severe SMA). (B,C) Representative images of Smn2B/+ (Black), Smn2B/− (Blue), DKO (Green) and TKO (Red) at postnatal day 19 (P19) and gross phenotypic changes as denoted by the yellow arrows pointing to tail, ear necrosis, and fatty liver. (D) SMN and α-tubulin immunoblots of Smn2B/+ control, Smn2B/−, DKO, and TKO mice spinal cord, muscle, and liver extracts. (E–G) Densitometric analysis of SMN western blots normalized to α-tubulin levels. Data represents means ± SEM with η = 4 per genotype. An ordinary one-way ANOVA with Tukey post-hoc test for multiple comparison was performed with p < 0.001 for **, p < 0.0001 for ****, and ns, non-significant.
TKO mice showed a mean increased survival of 6 days in comparison to the Smn2B/− mice (median survival of 27 days compared to 21 days) (Figure 2A). In fact, by the time all the Smn2B/− mice are dead, 95% of TKO mice remained alive, with a few animals surviving up to the very end of the observational period of 45 days (Figure 2A). The increased survival was independent of sex, since we could not observe a differential effect between male and female mice in the TKO mice (Supplementary Figure 2A). To determine whether increased survival correlates with improved weight gain, we assessed body weight every second day starting at P7. Similar to the Smn2B/− mice, we noted a slowing down in weight gain in the TKO mice regardless of prolonged survival (Figure 2B).
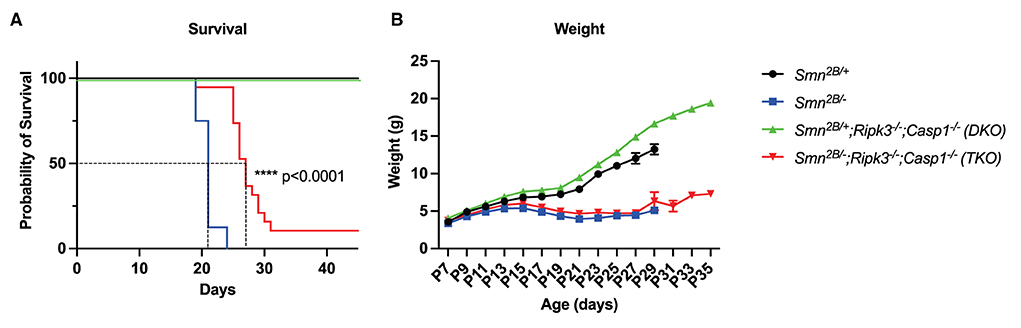
Figure 2. Impact of necroptotic pathway ablation on survival and weight gain in Smn2B/− mice. (A) Kaplan–Meier curve comparing survival across genotypes up to 50 days displays increased survival in TKO mice compared to Smn2B/− (p < 0.001). (B) Analysis of weight beginning at P7 to P35, shows a reduction in weight in TKO mice comparable to Smn2B/−. η = 11−19 per genotype. Data represents means ± SEM, statistical analysis (A) Mantel–Cox test and (B) An ordinary two-way ANOVA with Sidak's post-hoc test for multiple comparison. P < 0.0001 for ****.
Qualitative observation revealed an increased mobility in TKO mice (data not shown). We therefore employed a battery of age-specific tests tailored to follow motor function in our mice (Figure 3A). Spinal reflexes, measured by the righting reflex, were slightly improved in TKO mice compared to Smn2B/− mice at P7 and P9 (Figure 3B). In contrast, no changes were observed with the inverted mesh grip, which focuses on the distal muscle strength (Figure 3C). However, TKO mice did display significant improvements in motor function revealed by the pen test (Figure 3D). This test measures motor balance and coordination. The improved performance of TKO mice in this test is in line with the qualitative observations of a better gait. Altogether, these findings demonstrate that genetic ablation of necroptotic cell death pathways ameliorates survival and modestly improves some motor function in SMA mice.
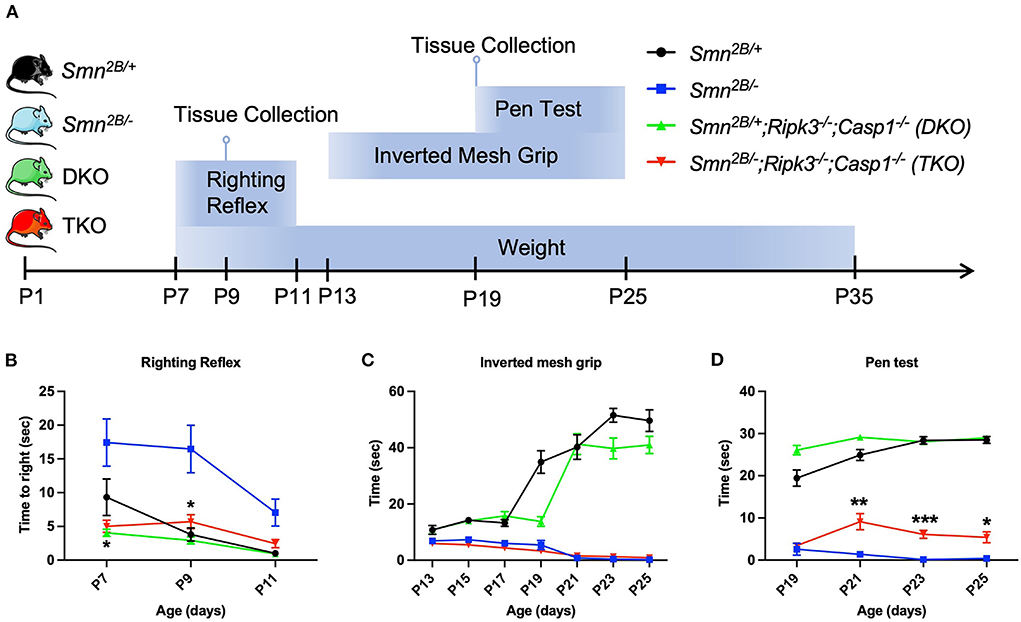
Figure 3. Impact of genetic ablation of necroptotic cell death pathways on motor function in Smn2B/− mice. (A) Schematic representation of experimental design and evaluation of motor function. Motor function and weight were evaluated every second day in each group with (B) righting reflex from P7 to P11, (C) inverted mesh grip test from P13 to P25, and (D) pen test from P19 to P25. Minor motor improvement could be identified in the TKO mice on the righting reflex and the pen test while no improvement was seen on inverted mesh grip test. η = 11−19 per genotype. Data represents means ± SEM. An ordinary two-way ANOVA with Sidak's post-hoc test for multiple comparison. P < 0.05 for *, P < 0.001 for **, and P < 0.0002 for ***.
Next, we investigated whether attenuation of motor neuron loss and NMJ pathology, classical hallmarks of SMA, underly the improved motor function in TKO mice. H&E staining of the lumbar spinal cord did not show any changes in motor neuron number in TKO mice compared to Smn2B/− animals (Figures 4A–E). This was further validated via choline acetyltransferase (ChAT) immunostaining, a specific marker of motor neurons (Figures 4G–K). No difference between DKO and TKO motor neuron count was seen in both ChAT and H&E-stained lumbar spinal cord sections. Perhaps this is due to the tendency for a decrease in DKO motor neuron count when compared to Smn2B/+ animals, and a tendency for an increase in motor neuron count between TKO and Smn2B/− mice. Since we cannot explain this observation, we measured motor neuron size as an outcome for degeneration and we see no changes in motor neuron size in TKO mice compared to Smn2B/− animals (Figure 4F). NMJ pathology has been shown to precede motor neuron death in Smn2B/− mice (Buettner et al., 2021). Given that NMJ pathology is another readout for assessing the degeneration of the neuromuscular system in the Smn2B/− mice, we quantified neurofilament accumulation and NMJ denervation (Figure 5). A comparison of Smn2B/− with TKO animals revealed no difference in neurofilament accumulation or end plate denervation (Figures 5E,F). Altogether, we conclude that necroptosis pathway ablation does not influence the expected neuromuscular phenotype in SMA.
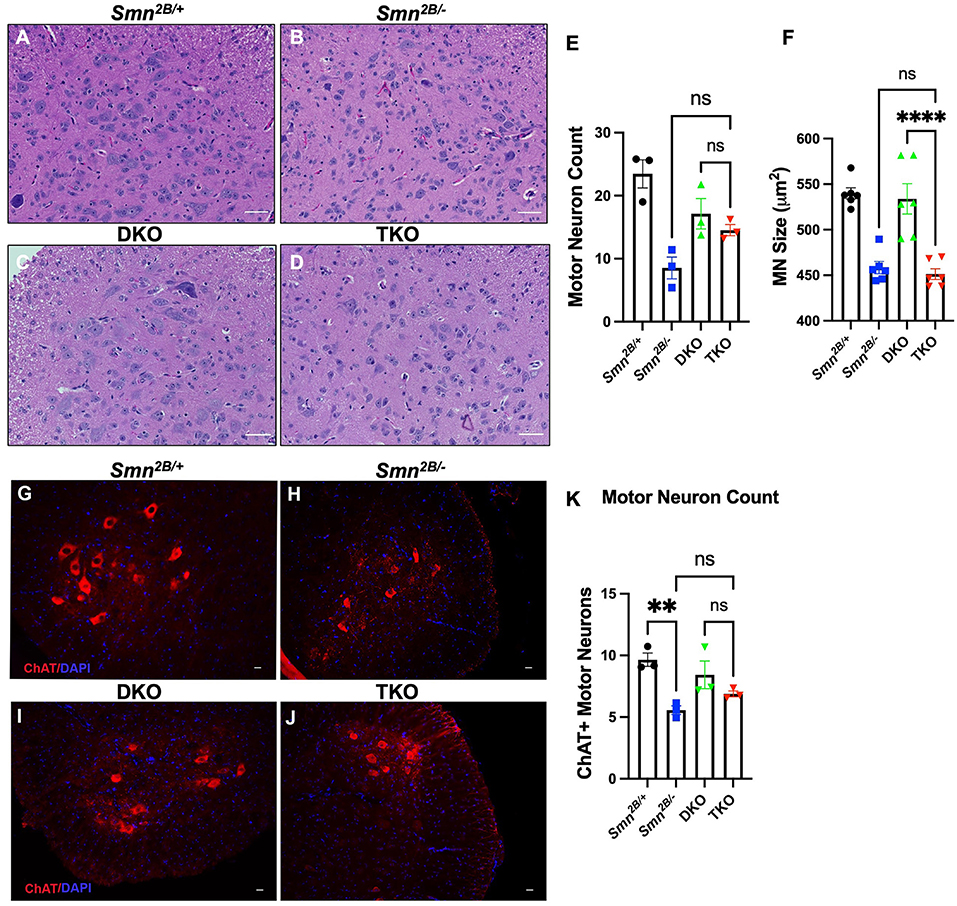
Figure 4. Genetic ablation of necroptotic cell death pathways does not rescue motor neuron degeneration in Smn2B/− mice. (A–D) Representative H&E images (Scale bars = 50 μm) of sections of lumbar spinal cord from P19 mice and quantification of (E,F) motor neuron cell body number and size, identified by location in the ventral horn, presence of nucleolus, and cells bodies >350 μm2 in cross-sectional area. (G–J) Representative immunofluorescent images of lumbar spinal cord anterior horns stained for ChAT (red) and DAPI (blue) from P19 mice (Scale bars = 20 μm). (K) Quantification of ChAT positive motor neuron cell body number. The n value for each study is as follows; (A–F) η = 6 − 9 per genotype, (G–K) η = 3 per genotype. Note that a total of 5 sections separated by 10 sections each per animal from the H&E-stained lumbar spinal cord were analyzed for (A–F) and 4 sections separated by at least 100 μm per animal for (G–K). Data represent mean ± SEM. An ordinary one-way ANOVA with Tukey post-hoc test for multiple comparison. ns, non-significant. P < 0.001 for **, and P < 0.0001 for ****.
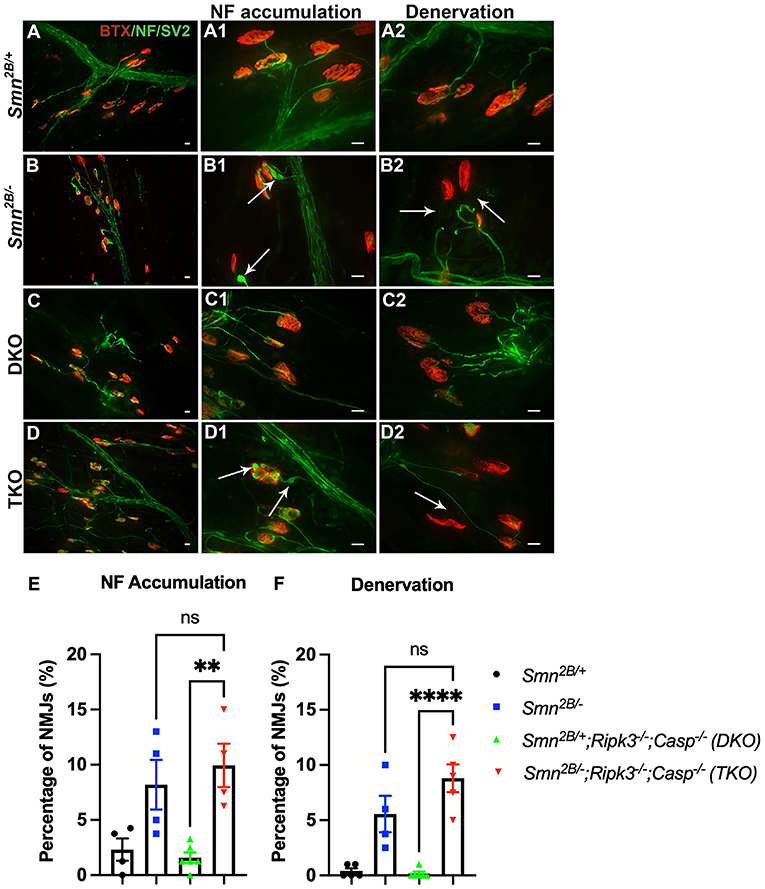
Figure 5. Genetic ablation of necroptotic cell death pathways does not improve NMJ pathology in Smn2B/− mice. (A–D) Representative immunofluorescent images of the transversus abdominis (TVA) muscle stained with bungarotoxin (BTX; red) and for neurofilament (NF; green) and synaptic vesicle protein 2 (SV2; green). Quantification for NF accumulation (E) and denervation (F) in NMJs from P19 mice. White arrows show NF accumulation in (A1–D1) and end plate denervation in (A2–D2). Scale bars = 20 μm. η = 4−6 per genotype. Data represent mean ± SEM. An ordinary one-way ANOVA with Tukey post-hoc test for multiple comparison. ns, non-significant. P < 0.001 for **, and P < 0.0001 for ****.
Given the lack of protection imparted by necroptosis at the motor neuron and NMJ level, we directly assessed muscle morphology in the TA muscle and whether muscle intrinsic mechanisms could be underlying the observed motor function improvements. Interestingly, TKO mice displayed larger muscle fibers compared to the Smn2B/− mice at symptomatic stage (Figures 6A–E). Additional analysis of fiber size distribution revealed that there is an increased number of larger fibers in the TKO (Figure 6F). This effect is also observed in DKO mice when compared to Smn2B/+ controls, indicating an SMA/SMN independent effect. These findings suggest that genetic ablation of necroptosis pathways may modulate muscle intrinsic pathways involved in the regulation of muscle fiber size. Finally, muscle atrophy pathways have been previously shown to contribute to the pathophysiology of SMA (Deguise et al., 2016). Following denervation, the expression of the proteasomal E3 ligases, Atrogin-1 and MuRF1, is generally upregulated in Smn2B/− mice compared to Smn2B/+ controls (Figures 6G,H). In TKO mice, mRNA levels of both Atrogin-1 and MuRF1 remained upregulated. Therefore, other mechanisms must be contributing to the amelioration of motor function in TKO mice.
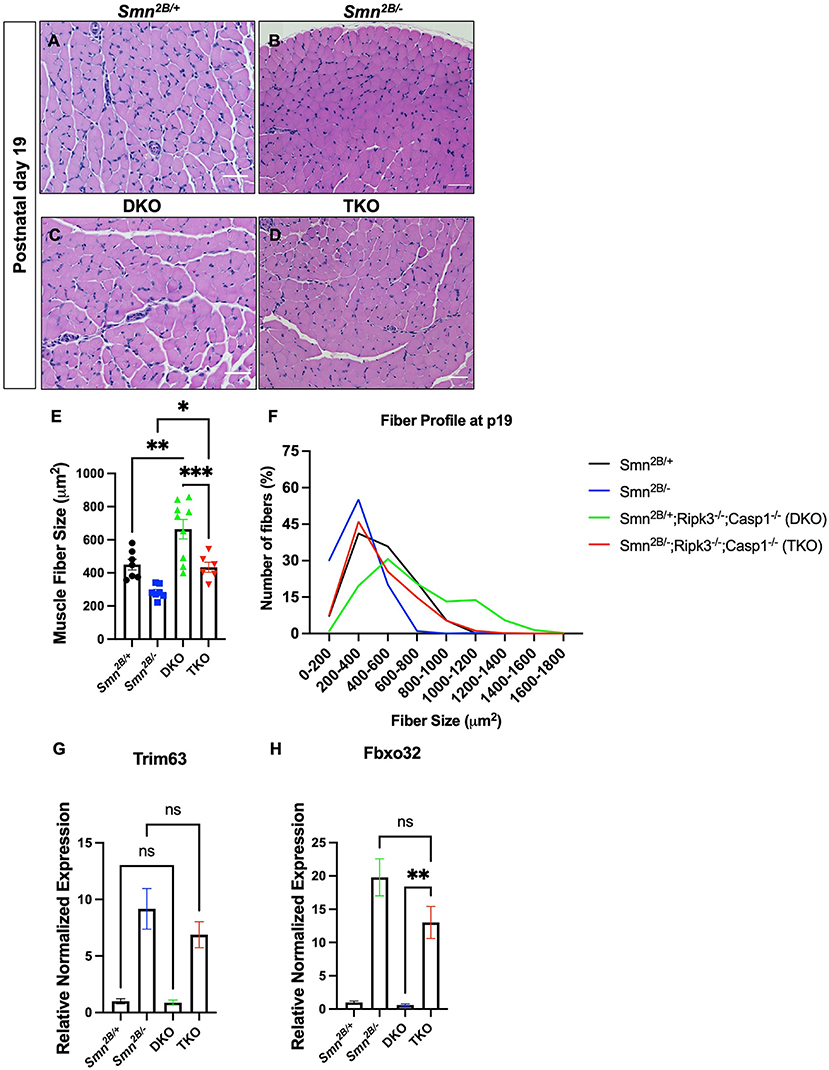
Figure 6. Genetic ablation of necroptotic cell death pathways increases muscle fiber size in both Smn2B/+ and Smn2B/− mice. (A–D) Representative images of H&E-stained tibialis anterior (TA) muscle from P19 mice and (E) quantification of muscle fiber size area size. The fiber size profile distribution (F) is shifted to the right in the DKO and TKO muscle. Primers targeting Atrogin-1 (Fbxo32), MuRF1 (Trim63), and Gapdh were used to examine whether muscle fiber size was rescued due to reduction of muscle atrophy. (G,H) Levels of Atrogin-1 and MuRF1 mRNA are not significantly changed at symptomatic stage (P19) in the hindlimb of TKO mice compared to Smn2B/−. Scale bars = 50 μm. The n value for each study is as follows; (A–E) η = 6 − 9 per genotype, (G,H) η = 4 per genotype. Data represent mean ± SEM. An ordinary one-way ANOVA with Tukey post-hoc test for multiple comparison. P < 0.05 for *, P < 0.001 for **, and P < 0.0002 for ***. ns, non-significant.
Since there is mounting evidence that p53 plays a role in motor neuron death in different SMA mouse models (Simon et al., 2017; Courtney et al., 2019) and that p53 mediates neuronal cell death by necroptosis (Peek et al., 2022), it is important to investigate the contribution of necroptosis to motor neuron and muscle pathology in SMA. Initially, we attempted to quantify the expression of key mediators of necroptosis, RIPK1/RIPK3, MLKL, and Caspase-1, in the tissues of the Smn2B/+ and Smn2B/− mice. Using, RT-qPCR analysis, we detected no change in the expression of RIPK1/RIPK3 and other mediators of necroptosis in the Smn2B/− mice (data not shown). Further, we were unable to detect basal levels of these mediators nor their phosphorylated forms in tissue homogenates through western blotting. Our inability to detect any changes may be due to transient or temporal upregulation that we were unable to pick up given that our tissues were analyzed only at pre-symptomatic and symptomatic time points. Additionally, changes may be occurring at a cell-specific level that are difficult to pick up with analysis of whole tissue homogenates. Despite the lack of direct evidence in this model, it is relevant for the field to understand the impact of targeting necroptosis in a whole-body model.
In the present study, we generated a triple mutant (TKO) mouse of the necroptotic pathway in the Smn2B/− mice, to gain a better understanding of inflammatory cell death by necroptosis in SMA. This model has an SMA phenotype comparable to the one in the Smn2B/− mice. However, TKO mice had a notable increase in mean survival by 6 days, which is independent of any changes in SMN protein levels. This was accompanied by ameliorated motor function. However, necroptotic pathway ablation did not provide protection against motor neuron loss or NMJ pathology. Accordingly, markers of neurogenic atrophy remained elevated. Yet, we have identified muscle intrinsic changes following ablation of necroptosis, namely increased muscle fiber size in TKO mice.
Necroptosis has been shown to be involved in skeletal muscle pathology of other diseases (Bencze et al., 2018; Morgan et al., 2018; Kamiya et al., 2022). A first study in dystrophin-deficient muscles, identified RIPK3 as a key player in the degeneration of skeletal muscle (Morgan et al., 2018). Muscular dystrophy patients and dystrophin-deficient mice displayed an upregulation of RIPK3 which indicates necroptosis activation (Morgan et al., 2018). Ripk3 ablation in the mdx mouse model improved motor function, which was attributed to a reduction in muscle degeneration, inflammation, and fibrosis (Morgan et al., 2018). In the context of inflammatory myopathies, suppression of necroptosis improved muscle weakness and cell death via reduction of pro-inflammatory cytokines and the NF-κβ pathway (Kamiya et al., 2022). As a result of necroptotic pathway ablation, apoptosis of muscle satellite cells was reduced (Kamiya et al., 2022). The beneficial effects of suppressing necroptosis thereby seem to be restricted to chronic muscle impairments since inhibition of necroptosis led to significant muscle regeneration defects in the context of acute toxin induced muscle injury (Zhou et al., 2020).
Here, we report an impact of necroptosis on SMA muscle which is independent of neuromuscular denervation. This indicates that necroptosis modulates muscle intrinsic defects in spinal muscular atrophy. This is supported by (i) findings of inflammation, (ii) dysregulation of muscle satellite cell homeostasis and (iii) increased apoptotic activity in SMA. While there appears to be no clear signs of muscle-intrinsic inflammatory processes, there is evidence of systemic inflammation. Reported inflammation across different tissues from SMA patients and mice (Deguise and Kothary, 2017; Deguise et al., 2017; Wan et al., 2018; Salucci et al., 2021) is suggested to aggravate atrophy of skeletal muscle and promotes cell damage. In muscle from SMA patients, smaller and disorganized myotubes are indicative of impaired growth and maturation (Martínez-Hernández et al., 2013). In vitro experiments in SMN-depleted muscle satellite cells, have identified satellite cell impairments, notably, accelerated differentiation and failure in forming myotubes (Le et al., 2011; Hayhurst et al., 2012). Moreover, apoptotic and cell death proteins are dysregulated in skeletal muscle from SMA patients and mouse models (Cifuentes-Diaz et al., 2001; Shafey et al., 2005; Dachs et al., 2011; Mutsaers et al., 2011; Martínez-Hernández et al., 2013; Armbruster et al., 2016).
Altogether, SMA patients and model mice exhibit systemic inflammation, impaired satellite cells, and skeletal muscle apoptosis, all of which can be modulated by necroptosis. In our study, muscle fiber size was improved in both DKO and TKO mice, implying an effect of necroptosis that is independent of SMA and SMN levels. This effect could be explained by reduced levels of pro-inflammatory cytokines and improved satellite cell function. Therefore, we suggest that necroptosis may have an indirect influence on muscle growth and maturation via reduction of pro-inflammatory cytokines and suppression of other forms of cell death happening in SMA muscle.
Preclinical models of severe SMA have a significantly reduced median lifespan of 10, 14, and 21 days in the Taiwanese, SMN7 and the Smn2B/− mouse models, respectively (Le et al., 2005; Gogliotti et al., 2010; Deguise et al., 2019). The remarkable increase in survival of TKO mice could relate to an amelioration of cell death in other tissues. Thus, effects of necroptosis on cell death pathways in other tissues besides muscle cannot be excluded. Abnormal apoptotic regulation is reported in spinal cord from SMA patients and mice (Simic et al., 2000, 2007; Soler-Botija et al., 2005; Tsai et al., 2006, 2008). Necrosis is also seen in cutaneous tissues in SMA infants (Araujo et al., 2009; Rudnik-Schöneborn et al., 2010; Carrasco et al., 2019; Weissman et al., 2021) and immune organs of Smn2B/− mice (Deguise et al., 2017). Further, modulation of these cell death pathways alters survival, motor neuron loss, motor function, and muscle atrophy in mice (Tsai et al., 2006, 2008; Piras et al., 2017; Simon et al., 2017). Therefore, potential influences of necroptosis ablation in other tissues on these other reported cell death mechanisms cannot be ruled out. Besides, necroptosis has been described in chronic organ injury (Zhao et al., 2015). Peripheral organ defects are present in the Smn2B/− mice, therefore the inhibition of necroptosis may protect against or attenuate damage to solid organs such as the kidneys, gut, lungs, and liver. Specifically, RIPK3 deficiency in an experimental model of NAFLD improved hepatic inflammation and hepatocyte injury without reducing the accumulation of fat in the liver (Afonso et al., 2021). As previously mentioned, the Smn2B/− mice display a NAFLD phenotype. As such, a potential decrease in the expression of pro-inflammatory mediators and inflammatory cell infiltration in the livers of TKO mice resulting from a knockout of RIPK3 and Caspase 1, could contribute to the observed improved survival. Additional analysis is required to understand the underlying molecular mechanisms of necroptosis involved in protection of defective peripheral organs. In NAFLD, RIPK3 expression correlates with caspase-1 expression in the liver (Afonso et al., 2021). Therefore, RIPK3 knockout alone may be responsible for the seen effects in the TKO mice, however further work is required to tease out the contribution of each pathway in the TKO mice.
SMA is no longer considered the leading cause of infant mortality due to major advances in therapeutics which focus solely on restoring SMN levels (Kingsmore et al., 2020). Yet, SMN restoration does not fully rescue the phenotype, as seen in humans and animal mouse models (Gavrilina et al., 2008; Hua et al., 2011, 2015; Gogliotti et al., 2012; Reilly et al., 2022). Additionally, the mechanisms by which motor neuron loss and muscle atrophy occur in SMA remain unclear. Here, SMN levels in the TKO mice were the same to those measured in the Smn2B/− mouse model. Yet, TKO mice had improved survival compared to the Smn2B/− mice, a significant observation given the short lifespan of these mice. This means that our current findings are independent of SMN levels. While further research on the mechanism of necroptotic pathways in SMA is necessary, pharmacological inhibition could potentially serve as a treatment in combination with SMN-enhancing drugs.
Cell death is an essential process in physiology, pathology, and development. Previous studies have exposed the contribution cell death by apoptosis, necrosis, and autophagy in SMA. However, no link to necroptosis has been previously made as a possible pathological mechanism contributing to CNS and peripheral pathology. Here, we report that targeting necroptosis pathways modulates survival, motor function and muscle fiber size independent of SMN levels and independent of the neurodegeneration. Commercially available small-molecule inhibitors of necroptosis such as Nec-1 could be combined with SMN-restoring drugs as a novel approach to ameliorate muscle function via modulation of muscle fiber size. Our results highlight the importance of further research to understand the mechanisms that could be modulated independently of SMN as a treatment for SMA.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by University of Ottawa Animal Facility.
LC, M-OD, and RK designed the research. LC, M-OD, YD, and RY performed experiments. AB and SG provided support for experiments. LC, M-OD, and NH analyzed the data. LC, NH, and RK wrote the manuscript with input from all authors. All authors have read and agreed to the published version of the manuscript.
RK was supported by Muscular Dystrophy Association (USA) (grant number 575466), Muscular Dystrophy Canada, and Canadian Institutes of Health Research (CIHR) (grant number PJT-156379). LC was supported by a CIHR Vanier Canada Graduate Scholarship. M-OD was supported by Fredrick Banting and Charles Best CIHR Doctoral Research Award.
We thank Dr. Sad's Laboratory at the University of Ottawa for providing us with the double knockout mice (Cas1−/−; Ripk3−/−) which were used to generate the TKO mouse model of necroptosis. We extend our gratitude to the Animal Care and Veterinary Services staff of the University of Ottawa for their assistance with experiments. We are thankful for our laboratory members and the colleagues who work in this field for their important contributions.
M-OD received honoraria and travel accommodations from Biogen for speaking engagements at the SMA Summit 2018 held in Montreal, Canada and SMA Academy 2019 held in Toronto, Canada. RK received honoraria and travel accommodations from Roche as an invited speaker at their global and national board meetings in 2019. RK and the Ottawa Hospital Research Institute have a licensing agreement with Biogen for the Smn2B/− mouse model. This COI is outside the scope of this study.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2022.972029/full#supplementary-material
Abati, E., Citterio, G., Bresolin, N., Comi, G. P., and Corti, S. (2020). Glial cells involvement in spinal muscular atrophy: could SMA be a neuroinflammatory disease? Neurobiol. Dis. 140, 1–12. doi: 10.1016/j.nbd.2020.104870
Afonso, M. B., Rodrigues, P. M., Mateus-Pinheiro, M., Simão, A. L., Gaspar, M. M., Majdi, A., et al. (2021). RIPK3 acts as a lipid metabolism regulator contributing to inflammation and carcinogenesis in non-alcoholic fatty liver disease. Gut 70, 2359–2372. doi: 10.1136/gutjnl-2020-321767
Araujo, A., Prufer De, Q. C., Araujo, M., and Swoboda, K. J. (2009). Vascular perfusion abnormalities in infants with spinal muscular atrophy. J. Pediatrics 155, 292–294. doi: 10.1016/j.jpeds.2009.01.071
Armbruster, N., Lattanzi, A., Jeavons, M., van Wittenberghe, L., Gjata, B., Marais, T., et al. (2016). Efficacy and biodistribution analysis of intracerebroventricular administration of an optimized scAAV9-SMN1 vector in a mouse model of spinal muscular atrophy. Mol. Therapy Methods Clin. Develop. 3, 1–8. doi: 10.1038/mtm.2016.60
Bencze, M., Pini, V., Mariot, V., Dumonceaux, J., Conti, F., Muntoni, F., et al. (2018). Necroptosis, a programmed form of necrosis, participates in muscle degeneration in Duchenne muscular dystrophy. Morphologie 102, 145–146. doi: 10.1016/j.morpho.2018.07.110
Boucher, D., Monteleone, M., Coll, R. C., Chen, K. W., Ross, C. M., Teo, J. L., et al. (2018). Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 215, 827–840. doi: 10.1084/jem.20172222
Bowerman, M., Murray, L. M., Beauvais, A., Pinheiro, B., and Kothary, R. (2012). A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul. Disord. 22, 263–276. doi: 10.1016/j.nmd.2011.09.007
Buettner, J. M., Sime Longang, J. K., Gerstner, F., Apel, K. S., Blanco-Redondo, B., Sowoidnich, L., et al. (2021). Central synaptopathy is the most conserved feature of motor circuit pathology across spinal muscular atrophy mouse models. iScience 24, 1–27. doi: 10.1016/j.isci.2021.103376
Carrasco, D., Magoulas, P., Scull, J. C., Jarrell, J. A., Lalani, S. R., and Wangler, M. F. (2019). Digital necrosis in an infant with severe spinal muscular atrophy. Neurol. Genet. 5, 1–2. doi: 10.1212/NXG.0000000000000361
Cifuentes-Diaz, C., Frugier, T., Tiziano, F. D., Lacène, E., Roblot, N., Joshi, V., et al. (2001). Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J. Cell Biol. 152, 1107–1114. doi: 10.1083/jcb.152.5.1107
Courtney, N. L., Mole, A. J., Thomson, A. K., and Murray, L. M. (2019). Reduced P53 levels ameliorate neuromuscular junction loss without affecting motor neuron pathology in a mouse model of spinal muscular atrophy. Cell Death Dis. 10, 1–14. doi: 10.1038/s41419-019-1727-6
Dachs, E., Hereu, M., Piedrafita, L., Casanovas, A., Calderó, J., and Esquerda, J. E. (2011). Defective neuromuscular junction organization and postnatal myogenesis in mice with severe spinal muscular atrophy. J. Neuropathol. Exp. Neurol. 70, 444–461. doi: 10.1097/NEN.0b013e31821cbd8b
Deguise, M.-O., De Repentigny, Y., Tierney, A., Beauvais, A., Michaud, J., Chehade, L., et al. (2020b). Motor transmission defects with sex differences in a new mouse model of mild spinal muscular atrophy. EBioMedicine 55, 1–14. doi: 10.1016/j.ebiom.2020.102750
Deguise, M.-O., Pileggi, C., De Repentigny, Y., Beauvais, A., Tierney, A., Chehade, L., et al. (2021). SMN depleted mice offer a robust and rapid onset model of nonalcoholic fatty liver disease. Cell. Mol. Gastroenterol. Hepatol. 12, 354–377. doi: 10.1016/j.jcmgh.2021.01.019
Deguise, M. O., Beauvais, A., Schneider, B. L., and Kothary, R. (2020a). Blood flow to the spleen is altered in a mouse model of spinal muscular atrophy. J. Neuromuscul. Dis. 7, 315–322. doi: 10.3233/JND-200493
Deguise, M. O., Boyer, J. G., McFall, E. R., Yazdani, A., De Repentigny, Y., and Kothary, R. (2016). Differential induction of muscle atrophy pathways in two mouse models of spinal muscular atrophy. Sci. Rep. 6, 1–13. doi: 10.1038/srep28846
Deguise, M. O., Chehade, L., Tierney, A., Beauvais, A., and Kothary, R. (2019). Low fat diets increase survival of a mouse model of spinal muscular atrophy. Ann. Clin. Transl. Neurol. 6, 2340–2346. doi: 10.1002/acn3.50920
Deguise, M. O., De Repentigny, Y., McFall, E., Auclair, N., Sad, S., and Kothary, R. (2017). Immune dysregulation may contribute to disease pathogenesis in spinal muscular atrophy mice. Hum. Mol. Genet. 26, 801–819. doi: 10.1093/hmg/ddw434
Deguise, M. O., and Kothary, R. (2017). New insights into SMA pathogenesis: immune dysfunction and neuroinflammation. Ann. Clin. Transl. Neurol. 4, 522–530. doi: 10.1002/acn3.423
Dhuriya, Y. K., and Sharma, D. (2018). Necroptosis: a regulated inflammatory mode of cell death. J. Neuroinflamm. 15, 1–9. doi: 10.1186/s12974-018-1235-0
Eshraghi, M., McFall, E., Gibeault, S., and Kothary, R. (2016). Effect of genetic background on the phenotype of the Smn2B/- mouse model of spinal muscular atrophy. Hum. Mol. Genet. 25, 4494–4506. doi: 10.1093/hmg/ddw278
Fayzullina, S., and Martin, L. J. (2014). Skeletal muscle DNA damage precedes spinal motor neuron DNA damage in a mouse model of spinal muscular atrophy (SMA). PLoS ONE 9, 1–16. doi: 10.1371/journal.pone.0093329
Gavrilina, T. O., McGovern, V. L., Workman, E., Crawford, T. O., Gogliotti, R. G., Didonato, C. J., et al. (2008). Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum. Mol. Genet. 17, 1063–1075. doi: 10.1093/hmg/ddm379
Gogliotti, R. G., Hammond, S. M., Lutz, C., and DiDonato, C. J. (2010). Molecular and phenotypic reassessment of an infrequently used mouse model for spinal muscular atrophy. Biochem. Biophys. Res. Commun. 391, 517–522. doi: 10.1016/j.bbrc.2009.11.090
Gogliotti, R. G., Quinlan, K. A., Barlow, C. B., Heier, C. R., Heckman, C. J., and DiDonato, C. J. (2012). Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J. Neurosci. 32, 3818–3829. doi: 10.1523/JNEUROSCI.5775-11.2012
Hayhurst, M., Wagner, A. K., Cerletti, M., Wagers, A. J., and Rubin, L. L. (2012). A cell-autonomous defect in skeletal muscle satellite cells expressing low levels of survival of motor neuron protein. Develop. Biol. 368, 323–334. doi: 10.1016/j.ydbio.2012.05.037
Hua, Y., Liu, Y. H., Sahashi, K., Rigo, F., Frank Bennett, C., and Krainer, A. R. (2015). Motor neuron cell-nonautonomous rescue of spinal muscular atrophy phenotypes in mild and severe transgenic mouse models. Genes Develop. 29, 288–297. doi: 10.1101/gad.256644.114
Hua, Y., Sahashi, K., Rigo, F., Hung, G., Horev, G., Bennett, C. F., et al. (2011). Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478, 123–126. doi: 10.1038/nature10485
Kamiya, M., Mizoguchi, F., Kawahata, K., Wang, D., Nishibori, M., Day, J., et al. (2022). Targeting necroptosis in muscle fibers ameliorates inflammatory myopathies. Nat. Commun. 13, 1–12. doi: 10.1038/s41467-021-27875-4
Kingsmore, S. F., Henderson, A., Owen, M. J., Clark, M. M., Hansen, C., Dimmock, D., et al. (2020). Measurement of genetic diseases as a cause of mortality in infants receiving whole genome sequencing. NPJ Genomic Med. 5, 1–10. doi: 10.1038/s41525-020-00155-8
Le, T. T., McGovern, V. L., Alwine, I. E., Wang, X., Massoni-Laporte, A., Rich, M. M., et al. (2011). Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet. 20, 3578–3591. doi: 10.1093/hmg/ddr275
Le, T. T., Pham, L. T., Butchbach, M. E. R., Zhang, H. L., Monani, U. R., Coovert, D. D., et al. (2005). SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 14, 845–857. doi: 10.1093/hmg/ddi078
Lefebvre, S., Bürglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165.
Lefebvre, S., Burlet, P., Liu, Q., Bertrandy, S., Clermont, O., Munnich, A., et al. (1997). Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 16, 265–269. doi: 10.1038/NG0797-265
Lorson, C. L., Hahnen, E., Androphy, E. J., and Wirth, B. (1999). A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U. S. A. 96, 6307–6311. doi: 10.1073/pnas.96.11.6307
Martínez-Hernández, R., Bernal, S., Also-Rallo, E., Alías, L., Barcelõ, M., Hereu, M., et al. (2013). Synaptic defects in type i spinal muscular atrophy in human development. J. Pathol. 229, 49–61. doi: 10.1002/path.4080
Monani, U. R., Lorson, C. L., Parsons, D. W., Prior, T. W., Androphy, E. J., Burghes, A. H. M., et al. (1999). A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183. doi: 10.1093/HMG/8.7.1177
Morgan, J. E., Prola, A., Mariot, V., Pini, V., Meng, J., Hourde, C., et al. (2018). Necroptosis mediates myofibre death in dystrophin-deficient mice. Nat. Commun. 9, 1–10. doi: 10.1038/s41467-018-06057-9
Murray, L., Gillingwater, T. H., and Kothary, R. (2014). Dissection of the transversus abdominis muscle for whole-mount neuromuscular junction analysis. J. Vis. Exp. 83, 1–6. doi: 10.3791/51162
Mutsaers, C. A., Wishart, T. M., Lamont, D. J., Riessland, M., Schreml, J., Comley, L. H., et al. (2011). Reversible molecular pathology of skeletal muscle in spinal muscular atrophy. Hum. Mol. Genet. 20, 4334–4344. doi: 10.1093/hmg/ddr360
Peek, S. L., Bosch, P. J., Bahl, E., Iverson, B. J., Parida, M., Bais, P., et al. (2022). p53-mediated neurodegeneration in the absence of the nuclear protein Akirin2. iScience 25, 1–24. doi: 10.1016/j.isci.2022.103814
Piras, A., Schiaffino, L., Boido, M., Valsecchi, V., Guglielmotto, M., de Amicis, E., et al. (2017). Inhibition of autophagy delays motoneuron degeneration and extends lifespan in a mouse model of spinal muscular atrophy. Cell Death Dis. 8, 1–16. doi: 10.1038/s41419-017-0086-4
Ransohoff, R. M. (2016). How neuroinflammation contributes to neurodegeneration. Science 353, 777–783. doi: 10.1126/science.aag2590
Reilly, A., Deguise, M.-O., Beauvais, A., Yaworski, R., Thebault, S., Tessier, D. R., et al. (2022). Central and peripheral delivered AAV9-SMN are both efficient but target different pathomechanisms in a mouse model of spinal muscular atrophy. Gene Ther. 2022, 1–11. doi: 10.1038/s41434-022-00338-1
Rodriguez, D., Weinlich, R., Brown, S., Guy, C., Fitzgerald, P., Dillon, C., et al. (2016). Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 23, 76–88. doi: 10.1038/cdd.2015.70
Rudnik-Schöneborn, S., Vogelgesang, S., Armbrust, S., Graul-Neumann, L., Fusch, C., and Zerres, K. (2010). Digital necroses and vascular thrombosis in severe spinal muscular atrophy. Muscle Nerve 42, 144–147. doi: 10.1002/mus.21654
Salucci, S., Bartoletti, A. S., Battistelli, M., Burattini, S., Bavelloni, A., Cocco, L. I., et al. (2021). How inflammation pathways contribute to cell death in neuro-muscular disorders. Biomolecules 11, 1–17. doi: 10.3390/biom11081109
Schroder, K., and Tschopp, J. (2010). The Inflammasomes. Cell 140, 821–832. doi: 10.1016/j.cell.2010.01.040
Shababi, M., Lorson, C. L., and Rudnik-Schöneborn, S. S. (2014). Spinal muscular atrophy: a motor neuron disorder or a multi-organ disease? J. Anat. 224, 15–28. doi: 10.1111/joa.12083
Shafey, D., Côté, P. D., and Kothary, R. (2005). Hypomorphic Smn knockdown C2C12 myoblasts reveal intrinsic defects in myoblast fusion and myotube morphology. Exp. Cell Res. 311, 49–61. doi: 10.1016/j.yexcr.2005.08.019
Sheng, L., Wan, B., Feng, P., Sun, J., Rigo, F., Frank Bennett, C., et al. (2018). Downregulation of survivin contributes to cell-cycle arrest during postnatal cardiac development in a severe spinal muscular atrophy mouse model. Hum. Mol. Genet. 27, 486–498. doi: 10.1093/hmg/ddx418
Shutinoski, B., Patel, R., Tomlinson, J. J., Schlossmacher, M. G., and Sad, S. (2020). Ripk3 licenced protection against microbial infection in the absence of Caspase1-11 inflammasome. Microb. Infect. 22, 40–45. doi: 10.1016/j.micinf.2019.08.002
Simic, G., Mladinov, M., Simic, D. S., Milosevic, N. J., Islam, A., Pajtak, A., et al. (2007). Abnormal motoneuron migration, differentiation, and axon outgrowth in spinal muscular atrophy. Acta Neuropathol. 115, 313–326. doi: 10.1007/s00401-007-0327-1
Simic, G., Seso-Simic, D., Lucassen, P. J., Islam, A., Krsnik, Z., Cviko, A., et al. (2000). Ultrastructural analysis and TUNEL demonstrate motor neuron apoptosis in werdnig-hoffmann disease. J. Neuropathol. Exp. Neurol. 59, 398–407. doi: 10.1093/jnen/59.5.398
Simon, C. M., Dai, Y., van Alstyne, M., Koutsioumpa, C., Pagiazitis, J. G., Chalif, J. I., et al. (2017). Converging mechanisms of p53 activation drive motor neuron degeneration in spinal muscular atrophy. Cell Rep. 21, 3767–3780. doi: 10.1016/j.celrep.2017.12.003
Soler-Botija, C., Cuscó, I., Caselles, L., López, E., Baiget, M., and Tizzano, E. F. (2005). Implication of fetal SMN2 expression in type I SMA pathogenesis: protection or pathological gain of function? J. Neuropathol. Exp. Neurol. 64, 215–223. doi: 10.1093/jnen/64.3.215
Sumner, C. J. (2006). Therapeutics development for spinal muscular atrophy. NeuroRX 3, 235–245. doi: 10.1016/j.nurx.2006.01.010
Tsai, L. K., Tsai, M. S., Ting, C. H., Wang, S. H., and Li, H. (2008). Restoring Bcl-xL levels benefits a mouse model of spinal muscular atrophy. Neurobiol. Dis. 31, 361–367. doi: 10.1016/j.nbd.2008.05.014
Tsai, M. S., Chiu, Y. T., Wang, S. H., Hsieh-Li, H. M., Lian, W. C., and Li, H. (2006). Abolishing bax-dependent apoptosis shows beneficial effects on spinal muscular atrophy model mice. Mol. Therapy 13, 1149–1155. doi: 10.1016/j.ymthe.2006.02.008
Wan, B., Feng, P., Guan, Z., Sheng, L., Liu, Z., and Hua, Y. (2018). A severe mouse model of spinal muscular atrophy develops early systemic inflammation. Hum. Mol. Genet. 27, 4061–4076. doi: 10.1093/hmg/ddy300
Wang, K., Liu, F., Liu, C. Y., An, T., Zhang, J., Zhou, L. Y., et al. (2016). The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell Death Differ. 23, 1394–1405. doi: 10.1038/cdd.2016.28
Weissman, A. S., Kennedy, K. R., Powell, M. R., and Davis, L. S. (2021). Skin necrosis in spinal muscular atrophy: case report and review of the literature. Pediatric Dermatol. 38, 632–636. doi: 10.1111/pde.14538
Yang, Y., Jiang, G., Zhang, P., and Fan, J. (2015). Programmed cell death and its role in inflammation. Mil. Med. Res. 2, 1–12. doi: 10.1186/s40779-015-0039-0
Zhang, H., Zhou, X., McQuade, T., Li, J., Ka-Ming Chan, F., and Zhang, J. (2011). Functional complementation between FADD and RIP1 in embryos and lymphocytes HHS public access. Nature 471, 373–376. doi: 10.1038/nature09878
Zhao, H., Jaffer, T., Eguchi, S., Wang, Z., Linkermann, A., and Ma, D. (2015). Role of necroptosis in the pathogenesis of solid organ injury. Cell Death Dis. 6, e1975–e1985. doi: 10.1038/cddis.2015.316
Zhou, S., Zhang, W., Cai, G., Ding, Y., Wei, C., Li, S., et al. (2020). Myofiber necroptosis promotes muscle stem cell proliferation via releasing Tenascin-C during regeneration. Cell Res. 30, 1063–1077. doi: 10.1038/s41422-020-00393-6
Keywords: neuroinflammation, necroptosis, motor neuron disease (MND), multi-system disease, mouse genetic models
Citation: Chehade L, Deguise M-O, De Repentigny Y, Yaworski R, Beauvais A, Gagnon S, Hensel N and Kothary R (2022) Suppression of the necroptotic cell death pathways improves survival in Smn2B/− mice. Front. Cell. Neurosci. 16:972029. doi: 10.3389/fncel.2022.972029
Received: 17 June 2022; Accepted: 11 July 2022;
Published: 03 August 2022.
Edited by:
Jianli Sun, Delaware State University, United StatesReviewed by:
Matthew E. R. Butchbach, Nemours Children's Health Delaware, United StatesCopyright © 2022 Chehade, Deguise, De Repentigny, Yaworski, Beauvais, Gagnon, Hensel and Kothary. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rashmi Kothary, cmtvdGhhcnlAb2hyaS5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.