 Victorio M. Pozo Devoto
Victorio M. Pozo Devoto Isaac G. Onyango1
Isaac G. Onyango1 Gorazd B. Stokin
Gorazd B. Stokin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 05 August 2022
Sec. Cellular Neuropathology
Volume 16 - 2022 | https://doi.org/10.3389/fncel.2022.959598
This article is part of the Research Topic Cellular CNS Repair Strategies, Technologies and Therapeutic Developments View all 21 articles
Axonal homeostasis is maintained by processes that include cytoskeletal regulation, cargo transport, synaptic activity, ionic balance, and energy supply. Several of these processes involve mitochondria to varying degrees. As a transportable powerplant, the mitochondria deliver ATP and Ca2+-buffering capabilities and require fusion/fission to maintain proper functioning. Taking into consideration the long distances that need to be covered by mitochondria in the axons, their transport, distribution, fusion/fission, and health are of cardinal importance. However, axonal homeostasis is disrupted in several disorders of the nervous system, or by traumatic brain injury (TBI), where the external insult is translated into physical forces that damage nervous tissue including axons. The degree of damage varies and can disconnect the axon into two segments and/or generate axonal swellings in addition to cytoskeletal changes, membrane leakage, and changes in ionic composition. Cytoskeletal changes and increased intra-axonal Ca2+ levels are the main factors that challenge mitochondrial homeostasis. On the other hand, a proper function and distribution of mitochondria can determine the recovery or regeneration of the axonal physiological state. Here, we discuss the current knowledge regarding mitochondrial transport, fusion/fission, and Ca2+ regulation under axonal physiological or pathological conditions.
Mitochondria's main role in the cell is to generate chemically usable energy in the form of ATP (Chang and Reynolds, 2006a). Among several other functions, mitochondria participate in reactive oxygen species (ROS) homeostasis, trigger apoptosis, and intervene in the buffering of the intracellular Ca2+. To ensure their health and proper distribution, mitochondria rely on the processes such as biogenesis, transport, fusion/fission, and recycling, collectively known as mitochondrial dynamics (Mishra and Chan, 2016; Seager et al., 2020).
Mitochondrial DNA (mtDNA) codes for only 13 proteins, which are essential, but not sufficient for respiratory chain function, and thus, mitochondria depend on the nuclear DNA, other organelles, and axonal translation for most of their proteins (Mishra and Chan, 2014; Shigeoka et al., 2016). In flat, spherical, or elongated shaped cells, mitochondria are localized few micrometers away from the nucleus or the endoplasmic reticulum (ER). Conversely, in neurons, the highly polarized morphology would constitute a constrain to axonal development and function if mitochondrial biogenesis and maintenance would occur only in the soma. At a mean anterograde speed of 0.5 μm/s (Misgeld and Schwarz, 2017), a fully functional mitochondrion in the soma would need almost 23 days to reach the tip of a 1-m axon, whereas in a typical epithelial columnar cell, it would need less than a minute to move from the basal to the apical membrane. Thus, processes responsible for functional maintenance, movement, distribution, and recycling of mitochondrial populations far away from the soma are particularly relevant in a neuron (Amiri and Hollenbeck, 2008). In the axon, the mitochondrial roles are key for neuronal function and include providing energy for the maintenance of the membrane potential, axonal transport, synaptic activity, neurotransmitter uptake and recycling, and buffering of the presynaptic Ca2+ (Howarth et al., 2012; Sheng and Cai, 2012).
The cardinal importance of mitochondria in the axons is evidenced by the strong association between mitochondrial homeostasis and axonal degeneration in neurodegenerative diseases. Axonal damage or degeneration manifests in Alzheimer's disease (AD) (Huang et al., 2007), Parkinson's disease (PD) (Cheng et al., 2010), amyotrophic lateral sclerosis (ALS) (Graham et al., 2004), multiple sclerosis (MS) (Ferguson et al., 1997), and Charcot–Marie–Tooth (CMT) disease (Verhoeven et al., 2006). Some of the familial forms of neurodegenerative diseases present mutations in genes that have association with mitochondrial function such as PTEN-induced putative kinase (PINK1), Parkin, DJ-1, alpha-synuclein, mitofusin (MFN) 2, and Cu/Zn superoxide dismutase (SOD1) among others (Schon and Przedborski, 2011). In general, even if these proteins have an impact on energy production, they do not participate directly in oxidative phosphorylation, but in processes linked to mitochondrial recycling, ROS, and fusion/fission. Besides the strong correlation between these diseases and mitochondrial dysfunction, it is not completely clear yet if the dysfunction originates in the axon, or if the pathology begins in the soma and then propagates to the axon. Detailed and extensive reviews are dedicated to these topics (Lingor et al., 2012; Burté et al., 2015; Paß et al., 2021; Wang et al., 2021).
Pathologies that result from direct impact from external insults to the axonal bundles include traumatic brain injury (TBI), spinal cord injury (SCI), and peripheral nerve injury (PNI). TBI is caused by a blow or a shake to the head, with possible long-term consequences such as cognitive deficits, emotional and behavioral problems, and increased risk of developing AD (Wilson et al., 2017). The physical trauma or the acceleration forces applied to the head produce internal pressure gradients that stretch and damage axons. This results in diffuse axonal injury (DAI), characterized by a multifocal damage to white matter tracts in regions such as the corpus callosum, brain stem, and the gray-white matter junctions (Blennow et al., 2016). The main histopathological hallmark of DAI is the presence of axonal dilations along the axonal tracts, known as axonal swellings (Bruggeman et al., 2021). In SCI or PNI, a trauma in the form of contusion, compression, or laceration affects white matter tracts in the spinal cord or peripheral nerves, respectively (McDonald and Sadowsky, 2002; Scheib and Höke, 2013). To a different degree, and depending on the type of injury, TBI, SCI, or PNI all produce also a secondary injury consisting of events that develop as a consequence of the primary impact such as hemorrhage, edema, excitotoxicity, and immune activation. Many experimental models devoted to understanding the molecular events that occur after axonal trauma or axotomy highlight the role of mitochondria as a player in axonal degeneration and regeneration. An increasing number of studies on axonal mitochondrial distribution, fusion/fission, transport, and function under physiological or pathological settings are revealing a strong interconnection among these processes, which not only govern mitochondrial homeostasis but, in several cases, also axonal fate.
In this work, we examine the current knowledge regarding the roles of mitochondrial transport, fusion/fission, and Ca2+ handling, when axonal homeostasis is disrupted, emphasizing the studies that focus on axonal mitochondria whenever possible. We focus particularly on the axonal pathological conditions derived from physical traumatic events and to a lesser extent on neurodegenerative disorders that present axonal pathologies.
Proper mitochondrial distribution along the axon is essential to deliver ATP and Ca2+-buffering capacity to the regions in need. Their movement is relevant to maintain a population of functional and active mitochondria, particularly in axons where extremely long distances from the soma must be reached. In general, movement toward the axonal tip replenishes energy supply that is essential for neuronal viability, whereas the movement back to the soma is required for the elimination of damaged and dysfunctional mitochondria (Chen et al., 2016). A number of studies using different types of neurons observed that mitochondria populate the axonal shaft at a density near to 1 per 10 μm (Shepherd and Harris, 1998; Smit-Rigter et al., 2016; Pozo Devoto et al., 2017). However, the presence of mitochondria is higher in some presynaptic terminals (Palay, 1956; Li et al., 2020), branching regions (Spillane et al., 2013), and the juxtaparanode region of myelinated axons (Figure 1A; Chang et al., 2006; Ohno et al., 2011; Chavan et al., 2015). Compared to other common axonal cargoes such as lysosomes, vesicles, and endosomes, mitochondrial general mobility is the lowest (Maday et al., 2014; Lewis et al., 2016; Misgeld and Schwarz, 2017). In vitro, only 10% of the mitochondria are in movement at any given moment (Obashi and Okabe, 2013), but this proportion can increase to 40% depending on the time window and the model used (Overly et al., 1996; Misgeld and Schwarz, 2017). In addition, factors such as age of the neurons and distance from the soma are among the variables that influence mitochondrial motility. Several studies show that mitochondria move more in young or developing than in mature or older axons (Chang and Reynolds, 2006b; Lewis et al., 2016) and that motility decreases with increasing distance to the soma (Plucińska et al., 2012). Furthermore, electrical activity of the axon modulates its movement, enhancing or reducing mitochondrial mobility depending on the location in the axon (e.g., nodes of Ranvier, presynapses, etc.) (Ohno et al., 2011; Obashi and Okabe, 2013; Sajic et al., 2013). Increased Ca2+ and ADP levels that result from electrical activity are at least partially responsible for the inhibition of mitochondrial movement (Mironov, 2007; Wang and Schwarz, 2009).
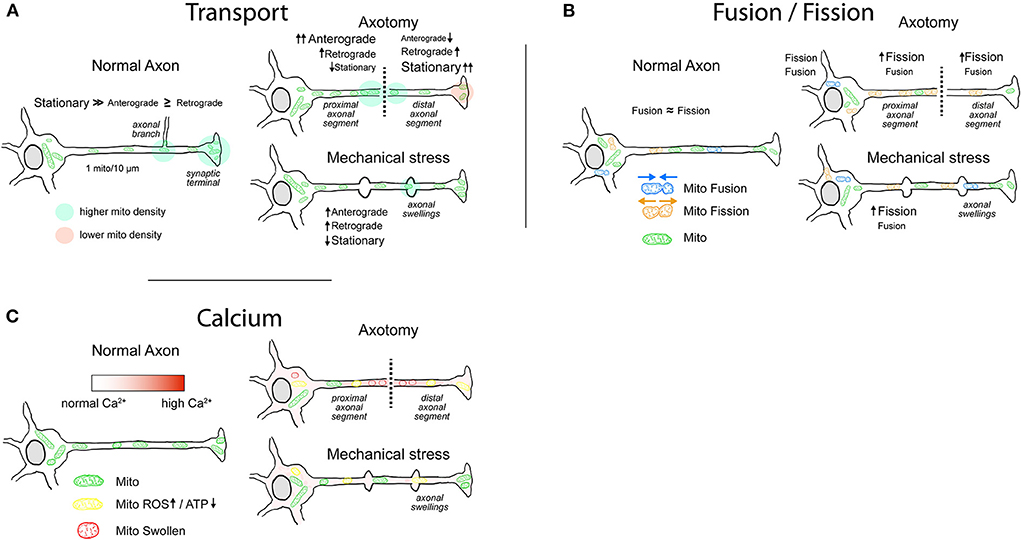
Figure 1. Mitochondrial behavior in normal and injured axons. (A) Mitochondrial transport. Most of the axonal mitochondria in physiological conditions are stationary, with increased density in axonal branches and in some presynaptic sites. After injury, the axons that have a higher chance of regeneration exhibit an increased mitochondrial motility. In axotomy, the mitochondrial anterograde movement increases in the proximal segment, generating a buildup of ATP near the lesioned region that favors regeneration. Mitochondria in the distal segment are mainly stationary with some increases in retrograde movement, leading to an accumulation in the injury site. Typically, distal segment will go through Wallerian degeneration. Axons that are damaged but maintain their integrity display axonal swellings, which can present disruption or rearrangement of cytoskeletal components. Evidence shows that the transport through axonal swellings is not always impaired and that immediately after injury mitochondrial movement is moderately increased. (B) Mitochondrial fusion/fission. In physiological conditions, the rates of fusion/fission in axons are balanced. Fission favors the movement and degradation of mitochondria, whereas fusion favors replenishment of mitochondrial proteins and enhances Ca2+-buffering capacity. Following axonal injury, the fission of mitochondria is enhanced, more notably after axotomy. It is yet not clear whether the enhanced fission in the proximal axonal segment favors regeneration. (C) Mitochondria and Ca2+. Mitochondria in the axon play a role in Ca2+ buffering, particularly in presynaptic sites. Immediately following axotomy, intra-axonal Ca2+ levels rise with a gradient that is maximum at the injury site. Moderate and sustained increase in Ca2+ levels lead to an impairment of electron transport chain with decrease in ATP production and increase in ROS generation. High intra-axonal Ca2+ levels result in mitochondrial Ca2+ overload, activation of the mPTP, and consequent loss of mitochondrial membrane potential. Mitochondria display swollen morphology. After mechanical stress, rise in intra-axonal Ca2+ levels varies in magnitude and duration, impairing mitochondrial respiration and ROS generation to varying degrees.
Long-distance movement of mitochondria in the axon is executed by ATP hydrolyzing motor proteins that walk on microtubule tracks. The unique axonal disposition of the microtubules with their plus-ends toward the axonal tips and minus-ends toward the soma (Burton and Paige, 1981) dictates the directionality of the motor protein movement with an anterograde movement (toward the microtubule plus-end) and a retrograde movement (toward the microtubule minus-end). Kinesin (heavy chains of the kinesin-1 family: KIF5A, B, and C) and dynein motor (cytoplasmic dynein) are responsible for the anterograde and the retrograde movement, respectively (Hirokawa et al., 2010). The link between these motor proteins and mitochondria is mediated by several adaptor proteins such as the mitochondrial Rho-GTPase protein (MIRO) (Guo et al., 2005), syntabulin (Cai et al., 2005), metaxins (MTX) (Zhao et al., 2021), fasciculation, and elongation protein zeta-1 (FEZ) (Ikuta et al., 2007), armadillo repeat-containing genes located in X chromosome (Armcx) (López-Doménech et al., 2012), Ran-binding protein 2 (RanBP2) (Cho et al., 2007), and actin-related protein 10 (Actr10) (Drerup et al., 2017). The interaction between MIRO and the motor heavy chains is mediated by Milton (Stowers et al., 2002), which in mammals has two family members TRAK 1 and 2 that are present in the axons and dendrites, respectively (van Spronsen et al., 2013). Interestingly, MIRO not only has adaptor properties, but it can also regulate mitochondrial movement through intracellular Ca2+ levels. MIRO presents two EF-hand motifs that when bound to Ca2+ favor the interaction with kinesin heavy chain motor domain. As a result, it abrogates interaction between the motor and microtubules leading to inhibition of mitochondrial movement (Wang and Schwarz, 2009). Additionally, syntaphilin (SNPH) through interactions with microtubules and with kinesin-1 motor participates in the immobilization (docking) of mitochondria in a MIRO-Ca2+-dependent manner (Kang et al., 2008; Chen and Sheng, 2013). Mitochondrial movement in actin filaments, which in many cases acts by opposing microtubule-based movement, is further controlled by myosin motor proteins such as MYO5, MYO6, and MYO19 (Quintero et al., 2009; Pathak et al., 2010; Li et al., 2020). The complexity of the molecular players that participate in this organelle movement and positioning can be found in extensive reviews dedicated to the topic (Saxton and Hollenbeck, 2012; Cheng and Sheng, 2021; Kruppa and Buss, 2021).
Direct insults to the axons resulting from SCI, PNI, or TBI can lead to axotomy or to axonal damage without disconnection. Axotomy is a severe process where the axon is cut in a region of the shaft leading to detachment from the soma of the distal segment and regeneration or degeneration of the proximal segment. The detached segment undergoes a process of programmed axonal death called Wallerian degeneration. A number of studies show that mitochondria increase their movement after axotomy in the proximal axon segment (Figure 1A; Misgeld et al., 2007; Mar et al., 2014; Cartoni et al., 2016). The first response is an increase in anterograde movement that starts almost immediately after injury, followed in time by a subsequent increase in retrograde transport (Misgeld et al., 2007; Mar et al., 2014). After intercostal nerve transection in mice, a robust 80% increase in anterograde transport in the proximal segment of the axotomized axons was observed, lasting for 48 h, and declining only slightly in the following weeks. Retrograde transport followed the increase in anterograde transport with a delay of 12 h following transection. In contrast, in the distal segment of the axon, the anterograde mitochondrial transport is almost immediately reduced, whereas retrograde transport lasts between 6 and 12 h after injury (Misgeld et al., 2007). Indeed, delayed Wallerian degeneration caused by JNK inhibition enhances mitochondrial movement in the distal axon segment (Shin et al., 2012). The buildup of mitochondria in the proximal segment of the axon that results from the increased anterograde transport showed to be determinant in the regeneration process (Han et al., 2016; Zhou et al., 2016). In C. elegans, axotomy in GABA motoneurons shows a significantly higher regeneration in the axons with more than 10 mitochondria per 100 μm when compared to the axons with lower mitochondrial density. Moreover, regeneration of the axons depends directly on the energy supply that results from increased density of mitochondria (Han et al., 2016; Zhou et al., 2016).
In contrast to axotomy, mechanical stress or stretching applied to the axon can injure the axon without an initial disconnection. Depending on the severity and the location, it has been reported that such injuries produce breakage of microtubules (Tang-Schomer et al., 2010), neurofilament compaction (Chen et al., 1999), and temporary increase in axonal membrane permeability (Farkas et al., 2006), among other effects (Datar et al., 2019). Moreover, following TBI, the formation of focal enlargements of the axonal shaft, called axonal swellings, occurs particularly within the white matter tracts (Ziogas and Koliatsos, 2018). Some studies report the spatial coincidence of the cytoskeletal disruption and the formation of axonal swellings (Chen et al., 1999; Tang-Schomer et al., 2010; Datar et al., 2019), leading to an accumulation of mitochondria and other cargoes in regions with axonal swellings (Maxwell, 1996; Tang-Schomer et al., 2012; Cross et al., 2019). Based on these studies, mitochondria are stuck in axonal swelling regions with less anterograde or retrograde movement across them. Supporting these observations, primary neuronal cultures subjected to fluid shear stress injury show that mitochondria concentrate at the locations of axonal swellings (Kilinc et al., 2008). In vivo, axonal swellings that appear after 24 h of a closed cortical impact (CCI) injury show degraded neurofilament or microtubule architecture, and some of them present electron-dense bodies and swollen mitochondria without clearly delineated cristae (Ziogas and Koliatsos, 2018). However, most of these observations were collected by transmission electron microscopy, which makes it impossible to obtain a complete picture of the mitochondrial distribution across the whole axon. To our knowledge, there is a lack of studies that assess in vivo mitochondrial transport or distribution in the axons after a TBI or similar injury. Alternatively, axonal swellings generated after a TBI may present cytoskeletal rearrangement that does not completely impair transport. On the one hand, mitochondria are not always present in the axonal swellings (Stone et al., 2001) whereas on the other hand, the accumulation of organelles does not necessarily mean that they cannot cross through the axonal swellings (Datar et al., 2019; Pozo Devoto et al., 2022). Furthermore, the in vitro real-time models of mechanical stress show that there is no drastic impairment on mitochondrial transport immediately after injury (Figure 1A; Gu et al., 2017; Pozo Devoto et al., 2022). Applying a controlled mechanical injury in axons derived from human neurons results in immediate formation of axonal swellings, characterized by microtubule and neurofilament rearrangements. In parallel, anterograde and retrograde proportions of moving mitochondria during and 5 min after injury are significantly increased (Pozo Devoto et al., 2022). These results are in line with mitochondrial behavior after axotomy in proximal axonal segments and suggest that as a result of axonal damage, mobilization of mitochondria favors axonal repair and regeneration.
In contrast, axonal transport of the mitochondria is generally impaired in neurodegenerative diseases. In ALS, motoneurons develop an early decrease in anterograde transport, resulting in a significant reduction of axonal mitochondrial population with the concomitant increase in somatic mitochondrial density (De Vos et al., 2007; Smith et al., 2019). Amyloid-β peptide (Aβ) accumulation observed in AD pathology also reduces mitochondrial anterograde transport and its speed in the axons, causing a reduction in the presynaptic mitochondrial density (Du et al., 2010; Calkins and Reddy, 2011; Guo et al., 2013; Zhang et al., 2018). Moreover, in neurons derived from patients with alpha-synuclein duplication (a model of PD), the proportion of moving axonal mitochondria is significantly decreased due to the oligomerization of alpha-synuclein (Prots et al., 2013). As an exception, in MS models where demyelination of the axons occurs, mitochondrial mobilization is significantly enhanced, particularly the anterograde movement that delivers mitochondria from soma to the axon (Licht-Mayer et al., 2020). Several studies show that the resulting higher mitochondrial density in axons promotes the survival of demyelinated axons (Witte et al., 2009; Zambonin et al., 2011; Ohno et al., 2014). Altogether, the evidence suggests that the impairment in mitochondrial movement observed in neurodegenerative diseases corresponds to a preexisting pathophysiological process that may be affecting their normal function or dynamics.
The movement behavior of mitochondria in the axon is determined by signaling pathways that target molecular regulators of mitochondrial transport. For example, in axonal injury, modifying different molecular regulators of mitochondrial motility promotes axonal regeneration (Kiryu-Seo et al., 2016; Zhou et al., 2016). The C. elegans mutant for ric-7, a gene that is essential for mitochondrial localization in the axons, leads to an impaired mitochondrial transport in injured axons which degenerate rapidly. Such degeneration can be suppressed by forcing mitochondria into axons through a direct link between tomm7 and kinesin-1 (Rawson et al., 2014). Overexpression of Armcx1, an adaptor that promotes mitochondrial motility, also enhances axonal regeneration and neuronal survival in vivo (Cartoni et al., 2016). Moreover, if SNPH, a protein that mediates the docking and axonal retention of mitochondria (Kang et al., 2008), is overexpressed, the mitochondrial movement is significantly suppressed. On the other hand, in axonal injury, its downregulation leads to a reduction in stationary mitochondria, contributing to the removal of damaged mitochondria and enhancing nerve regeneration (Zhou et al., 2016; Lin et al., 2017; Han et al., 2020). Physiologically, this response is mediated by AKT signaling. Indeed, after axonal damage due to ischemic injury, there is an activation of AKT, a kinase that acts on several prosurvival signaling pathways. One of the targets activated by AKT is p21-activated kinase 5 (PAK5), which in turn phosphorylates SNPH to release and promote remobilization of anchored mitochondria (Cotteret et al., 2003; Huang et al., 2021). In C. elegans neurons, axotomy or axonal breakage is followed by an increase of about 2-folds in mitochondrial density. This effect is mainly a consequence of increased mitochondrial transport from the soma mediated by MIRO and the signaling of dual-leucine zipper kinase-1 (DLK-1) pathway (Han et al., 2016). DLK-1 activation through CEBP-1 is in fact sufficient to increase axonal mitochondrial density in a MIRO-independent manner. DLK-1 injury-signaling pathway is a key determinant of axonal regeneration in C. elegans, Drosophila, and mice; however, the specific molecular connection to mitochondria remains unknown.
Fusion and fission of mitochondria are the processes involved in determining the size, the distribution, and the health of mitochondria. Fusion favors biogenesis through the exchange of new proteins and mitochondrial DNA between the merging organelles, restoring functional proteins to dysfunctional mitochondria. By splitting one mitochondria into two, the fission process reduces mitochondrial size and leads to enhanced mitochondrial axonal transport, distribution, and mitophagy (Itoh et al., 2013; Chan, 2020).
In physiological conditions, the size of axonal mitochondria is regulated by a balance between fusion and fission (Figure 1B; Amiri and Hollenbeck, 2008), an active process that in axons presents a rate of fission comparable to that of fusion (Cagalinec et al., 2013; Di Meo et al., 2021). In C. elegans neurons, this balance is regulated by aging, with a drastic increase in size occurring in early adulthood, followed by a progressive reduction in length through the rest of the lifespan (Morsci et al., 2016). Moreover, modulation of the molecular players of fusion/fission impacts directly on mitochondrial distribution in the axon (Spillane et al., 2013; Berthet et al., 2014). As an example, inhibition of an important component of the fission machinery results in a 50–60% decrease in mitochondrial content in the axon (Spillane et al., 2013). Finally, besides the specific mutations in the fusion/fission molecular machinery that lead to CMT disease or dominant optic atrophy (DOA), changes in mitochondrial size have also been observed in neurodegenerative diseases such as PD, Huntington's disease, and AD (Itoh et al., 2013) and in other neuropathological conditions (Ineichen et al., 2021).
Regulation of mitochondrial morphology is mediated by interactions between the proteins controlling fusion and fission. The fusion process is driven by MFN1 and MFN2, proteins from the dynamin-related GTPase family (Meeusen et al., 2004). MFN is located in the outer mitochondrial membrane (OMM) and expression of MFN2 in the nervous tissue is more abundant when compared to that of MFN1 (Lee et al., 2012; Zhou et al., 2019). MFN is responsible for the fusion of the OMM, whereas optic dominant atrophy 1 (OPA1) is responsible for the fusion of the inner mitochondrial membrane (IMM) (Alexander et al., 2000). Joining together two mitochondria requires the sequential activation of the outer and inner membrane fusion to yield a functional mitochondrion (Song et al., 2009). On the other hand, fission led by the activity of another dynamin-related GTPase protein (DRP1), which oligomerizes and forms a ring-shape structure around mitochondria, acting as a constricting diaphragm at the mitochondrial membrane (Pitts et al., 1999). Given that DRP1 is a cytosolic protein, its recruitment to mitochondria is mediated by different receptor-like proteins including mitochondrial fission factor (Mff), mitochondrial fission 1 (Fis1), and mitochondrial dynamic proteins MiD49s (MiD49) and MiD51 (MiD51) (Osellame et al., 2016). For an in-depth description of the molecular components involved in the mitochondrial fusion/fission, we direct the readers to the following reviews (Chan, 2020; Giacomello et al., 2020).
The relevance of mitochondrial fusion process in axonal homeostasis is manifested in diseases such as CMT and DOA, caused by mutations in MFN2 and OPA1, respectively (Burté et al., 2015). These mutations have a deleterious effect on the axons, particularly the long ones, with neuropathy in peripheral motor or sensory axons, or degeneration of retinal ganglion cells (RGC) and optic nerves. A plethora of studies shows that loss of function mutations in MFN2 affects several mitochondrial processes occurring in the axon ranging from size reduction and transport impairment to altered distribution, clustering, and functional deficit (Detmer et al., 2008; Cartoni et al., 2010; Byrne et al., 2019; Zhou et al., 2019). Furthermore, the effects can go beyond mitochondria. As an example, knockdown of MFN2 in motoneurons derived from human embryonic stem cells results not only in smaller mitochondria but also in the loss of mitochondrial potential, aggregation of phosphorylated neurofilaments, decreased transport, reduced levels of motor proteins, and formation of axonal swellings (Mou et al., 2021). The impact of these processes increases with age, and in most cases, dysfunction hastens axonal loss (Byrne et al., 2019). The observation that these mutations affect primarily peripheral motor and sensory axons points to the relevance of mitochondrial function and distribution for proper maintenance of the axon throughout all its length. It is not clear, however, whether the wide spectrum of perturbations resulting from disruption in MFN2 can be attributed completely to the decreased fusion of mitochondria. In dorsal root ganglia neurons, the fusion impairment due to the overexpression of MFN2 disease-linked mutant leads to axonal degeneration, whereas fusion impairment due to depletion of OPA1 does not (Misko et al., 2012). The role of MFN2 in mitophagy and mitochondrial transport can be an alternative mechanism underlying the deleterious effects of its mutations (Dorn, 2020).
In mice, the absence of DRP1 is embryonic lethal, whereas the conditional knockout in the nervous tissue is perinatal lethal (Ishihara et al., 2009). Nevertheless, the ablation of DRP1 in forebrain neurons of adult mice leads to minor changes in the branching of the hippocampal neurites with no reduction in spine or synaptic numbers. At the same time, mitochondria accumulate in the perikarya of hippocampal neurons, suggesting that the delivery of mitochondria to neurites is impaired (Oettinghaus et al., 2016). In fact, loss of mitochondrial mass in axons due to reduced DRP1 activity has been observed in several settings. For example, in Drosophila DRP1 mutant flies, motoneurons show reduced levels of mitochondria in the neuropil and clumps in the somas, with mitochondria largely absent from synapses (Verstreken et al., 2005; El Fissi et al., 2018). DRP1-null mice selective for dopaminergic neurons show a drastic decrease in mitochondrial mass in the axons as well as impaired transport (Berthet et al., 2014). Surprisingly, the consequences of the mitochondrial mass reduction in axons are not always deleterious and depend on the neuronal type (Shields et al., 2015). In dopaminergic neurons of the substantia nigra, the decrease of mitochondria in nerve terminals leads to a progressive degeneration of synaptic terminals and cell loss, whereas dopaminergic neurons in the ventral tegmental area survive, despite the depletion of mitochondria in their axons (Berthet et al., 2014). Furthermore, in Drosophila, the lack of mitochondria at presynaptic sites of neuromuscular junctions (NMJ) has no major effect on basal neurotransmission, showing impairments only under intense stimulation (Verstreken et al., 2005). On the other hand, the effects of Mff downregulation are less drastic than the ones observed when DRP1 is downregulated. Mff knockdown increases axonal mitochondrial size, without affecting their localization in pre-synapses (Lewis et al., 2018). The bigger mitochondria in pre-synapses result in enhanced Ca2+ buffering, less cytosolic Ca2+ and reduced evoked neurotransmitter release. A further effect of reducing Mff levels is the decreased rate at which mitochondria enter the axon, explaining at least in part the diminished axonal mitochondrial mass observed in fission impairment models (Lewis et al., 2018).
Size, transport, and distribution of mitochondria are the results of a fine balance between fusion- and fission-dedicated proteins. Depletion of mitochondria in NMJ of motoneurons caused by the MFN R364W mutation, which enhances MFN activity, can be reverted by overexpressing DRP1 (El Fissi et al., 2018), whereas simultaneous disruption of fusion and fission proteins rescues the morphology and the functionality of the neuron (Byrne et al., 2019). MFN or OPA knockdown in flies produces a reduction in motility and density of mitochondria in distal axons, together with a decrease in respiration rate, ATP synthesis, and mitochondrial membrane potential. Simultaneous knockdown of DRP1 with OPA restores morphology and density of mitochondria, but the functional parameters remain impaired. The opposite occurs with simultaneous knockdown of DRP and MFN, where the function of mitochondria is restored, recovering also fly viability and indicating that mitochondrial function is more important for survival than the morphology or the distribution (Trevisan et al., 2018).
Several studies report a decrease in the size of axonal mitochondria after axotomy (Figure 1B; Kiryu-Seo et al., 2016; Arrázola et al., 2019; Kedra et al., 2021). As an example, there is a decrease in mitochondrial size in sciatic nerve explants that progress during the 48 h after injury (Arrázola et al., 2019). A similar outcome has been observed in mice after sciatic nerve transection, showing a significant decrease in size 7 days after the injury and recovering to normal sizes 56 days after the injury (Kiryu-Seo et al., 2016). Using the same mouse model, it was shown that mitochondria located in the soma of motoneurons lack significant changes in size after axotomy (Tamada et al., 2017), suggesting that smaller mitochondria in axons are the result of locally enhanced fission process and not of smaller mitochondria delivered from the soma. In support of this finding, it was observed in a SCI model that the size of mitochondria is smaller the closer they are to the injury site (Kedra et al., 2021). However, the linkage between fragmentation of mitochondria and nerve regeneration is not completely clear. Reducing the fragmentation of mitochondria after axotomy in vitro by incubation with Mdivi (a pharmacological inhibitor of DRP1) results in a partial but significant decrease in axonal degeneration (Arrázola et al., 2019). In line with this, after a SCI in vivo, incubation with Mdivi decreases the lesion size and the number of retraction bulbs present in the axons (Kedra et al., 2021). Furthermore, in C. elegans, loss of function for OPA1 shows impairment in axonal regrowth, despite that both show mitochondrial fragmentation (Knowlton et al., 2017). Opposing effects were observed in a mouse model harboring a conditional knockout for DRP1, where after sciatic nerve injury, mitochondria are bigger, show decreased transport, and have lower membrane potential, altogether leading to a significant degeneration of the axons after 14 days (Kiryu-Seo et al., 2016).
Studies of the fusion/fission balance after TBI are scarce. In vivo, the effects of TBI related to fusion/fission have not been assessed directly in the axons, but by brain homogenates or in isolated mitochondria. In rats, a mild TBI induces the changes in the protein expression levels within the first 120 h, with upregulation of the fusion-related proteins (MFN1/2, OPA1) and downregulation of the fission proteins (DRP1, FIS1). Surprisingly, the opposite is observed if the TBI is stronger, showing downregulation of fusion and upregulation of fission components (Di Pietro et al., 2017). In line with this last result, a CCI injury in mice produces a mild but significant increase in the size of hippocampal mitochondria 24 h after injury, evolving to a decrease in size at 72 h. This correlates with an increased localization of DRP1 in the mitochondria at 72 h, which when blocked by the administration of Mdivi results in mitochondria with normal size, decreased death in dentate gyrus neurons and memory improvement (Fischer et al., 2016). Taken together with the observed mitochondrial behavior in axotomy, the evidence suggests that the balance favors the fission of mitochondria after axonal damage (Figure 1B). The increased fission may induce rapid transport (Misgeld et al., 2007) and delivery of mitochondria, as well as their homogenous distribution along the axonal shaft. Furthermore, in an in vitro TBI model, phosphoproteomic analysis performed immediately after axonal injury revealed changed regulation of fusion/fission-related proteins in the soma. Among them, mitochondrial elongation factor 1 (MIEF1 or MiD51), a receptor for DRP1, mitochondrial fission regulator 1-like protein (MTFR1L or FAM54B), and HECT, UBA, and WWE domain-containing E3 ubiquitin protein ligase 1 (HUWE1), which ubiquitinates MFN2 to negatively regulate mitochondrial fusion (Pozo Devoto et al., 2022). These fast post-translational modifications of fusion/fission molecular machinery could be interpreted as an early response of the neurons to axonal stress, which could be followed by slower de novo synthesis or degradation.
In neurodegenerative diseases, a plethora of studies reports fusion/fission imbalance, which is particularly well-described in PD and ALS (Cho et al., 2009; Zilocchi et al., 2018; Smith et al., 2019). Indeed, most of the studies observe increased fragmentation of mitochondria. For example, in PD, several disease-associated mutations in alpha-synuclein, PINK, Parkin, and LRRK2 have been shown to impact fusion/fission regulators or exert their effects directly by binding to the mitochondrial membrane (Yang et al., 2008; Glauser et al., 2011; Wang et al., 2012; Pozo Devoto et al., 2017). In ALS, models including SOD1 and TDP-43 disease-associated mutations show an expression profile that tilts the balance toward the increased expression of DRP1 or Fis1 (Ferri et al., 2010; Xu et al., 2010). However, these phenomena involve many factors that could be influencing the balance between fusion and fission including the changes in the soma and dendrites to the regulation of mitophagy.
Ca2+ is a fundamental regulator of axonal function and homeostasis, participating in the processes such as synaptic neurotransmitter release, cargo transport, cytoskeletal remodeling, and axon pathfinding (Henley and Poo, 2004; Sudhof, 2004; Rosenberg and Spitzer, 2011; Niescier et al., 2018). Axonal Ca2+ levels also regulate mitochondrial transport as well as fusion/fission (Saxton and Hollenbeck, 2012) and mitochondrial energy metabolism with Ca2+ acting as a co-activator for some tricarboxylic acid cycle enzymes (Wan et al., 1989). Importantly, due to its Ca2+-buffering role in presynaptic terminals, mitochondria naturally cope with transient increases in Ca2+ concentrations (Devine and Kittler, 2018). However, above a certain threshold, increased intra-mitochondrial Ca2+ uncouples the electron transport chain resulting in a greater production of oxidative stress by increasing free electrons, which are easily trapped by oxygen. The increased levels of ROS are responsible for oxidizing mitochondrial membrane phospholipids such as cardiolipin that are essential to maintain the selectivity and permeability of the IMM (Bayir et al., 2007). In cases where high long-lasting Ca2+ and ROS levels are present, such as in excitotoxic conditions, mitochondrial Ca2+ uptake is overloaded and leads to an activation of the mitochondrial permeability transition pore (mPTP), loss of mitochondrial membrane potential, ATP depletion, and cell loss (Bernardi et al., 2006; Barrientos et al., 2011).
Aside from excitotoxicity, the excessive increase in intracellular Ca2+ levels has been observed in injuries that produce mechanical damage to the axon as well as in specific pathologies. Several studies have shown an intra-axonal increase in Ca2+ levels after TBI in in vitro models (Figure 1C; Wolf et al., 2001; Yuen et al., 2009; Staal et al., 2010; Tang-Schomer et al., 2010; Gu et al., 2017; Pozo Devoto et al., 2022). The axonal entry of Ca2+ can occur through mechanosensitive channels (Gu et al., 2017), voltage-gated Ca2+ channels (VGCCs) (Yuen et al., 2009), and the Na+-Ca2+ exchanger (Wolf et al., 2001) and can even result from Ca2+ release from intracellular stores (Staal et al., 2010). In vivo, the Ca2+ increase observed in TBI models such as CCI or fluid percussion injury (FPI) is supported by indirect evidence from spectrin cleavage by specific Ca2+-dependent proteases (Büki et al., 2000; Johnson et al., 2016) or attenuation of axonal degeneration by Ca2+ channel inhibitors (Stirling et al., 2014; Ribas et al., 2017). In axotomy, a Ca2+ increase is also detected almost immediately after the ablation (Figure 1C; Villegas et al., 2014), and depending on the model, this increase can happen as a short peak of 100 s (Vargas et al., 2015) or it can be longer than 10 min (Kedra et al., 2021) or even hours (Stirling et al., 2014). Altogether, most of the experimental evidence points to a fundamental role of Ca2+ in the axonal collapse, which can be the result of the impact of Ca2+ on mitochondrial homeostasis.
The increase in axonal Ca2+ impacts the mitochondrial behavior directly. On the one hand, it can modify their transport and localization (Yi et al., 2004). Several experiments have shown that increased cytosolic Ca2+ in the axon leads to mitochondrial arrest through MIRO1 (Saotome et al., 2008; Macaskill et al., 2009; Wang and Schwarz, 2009). When cytosolic Ca2+ levels are high, the MIRO EF-hand domains that bind Ca2+ modify their conformation and promote the interaction with kinesin-1 motor domains leading to mitochondrial arrest (Wang and Schwarz, 2009). The cessation of mitochondrial movement due to high Ca2+ levels is particularly relevant to retain mitochondria in presynaptic terminals where they deliver ATP and exert the Ca2+-buffering function. However, under pathological conditions, the arrest of mitochondria due to the elevated Ca2+ levels can lead to axonal degeneration. In Drosophila, Ca2+ elevation triggered by CaMKII significantly reduces mitochondrial transport, resulting in axonal degeneration (Woolums et al., 2020). Surprisingly, the overexpression of a MIRO mutant unable to bind Ca2+ reduces axonal degeneration, suggesting that impaired transport contributes to degeneration. It is possible that sustained high Ca2+ levels are achieved in the distal axonal segment after axotomy. This part of the axon presents a decrease in mitochondrial transport and coincidentally also requires sustained high Ca2+ levels for axonal collapse (Wallerian degeneration) (Misgeld et al., 2007; Coleman and Höke, 2020).
On the other hand, several studies showed that increased cytosolic Ca2+ levels in neurites produce an upregulation in DRP1 activity, causing increased mitochondrial fragmentation (Bao et al., 2018). In rat hippocampal neurons, DRP1 becomes phosphorylated as a result of CaMKII activation (Godoy et al., 2014) and in primary neurons, DRP1 becomes activated by MIRO's Ca2+-binding activity (Saotome et al., 2008). Furthermore, blockage of mitochondrial calcium uniporter channel (MCU) inhibits the entry of Ca2+ into mitochondria and reduces their fragmentation, indicating that mitochondrial fission can result from Ca2+ overload (Kedra et al., 2021).
The capability of mitochondria to buffer intracellular Ca2+ in the axon is well-documented, particularly in the presynapses where it exerts a fundamental role in neurotransmitter release (Guo et al., 2005; Kang et al., 2008; Li et al., 2020; Stavsky et al., 2021). In the axons, evoked action potentials lead to an increased Ca2+ concentration in the mitochondrial matrix, a buffer effect even more notable in mature axons than in the young ones (Lewis et al., 2016).
In pathological conditions, the massive rise of Ca2+ in the axon resulting from axotomy (Vargas et al., 2015) or from intracellular store release (Villegas et al., 2014) leads to axonal degeneration. It has been shown that Ca2+ increase spreads from the injury site through the shaft and is followed by a wave of mitochondrial oxidation which results in mitochondrial swelling and fragmentation (Figure 1C; Gitler and Spira, 1998; Breckwoldt et al., 2014). Molecularly, the Ca2+ overload triggers the opening of the mPTP, a protein complex formed mainly by the voltage-dependent anion channel (VDAC) in the OMM, the adenine nucleotide translocase (ANT) in the IMM and cyclophilin D in the mitochondrial matrix. The opening of the pore consists in the interaction of these proteins in an irreversible conformation, leading to the massive release of Ca2+, disruption of the IMM, and loss of mitochondrial membrane potential, resulting in ATP depletion and ROS production (Vargas et al., 2015). The flow of solutes through the IMM that result from the mPTP formation leads to an increase in the volume of the mitochondria, which ultrastructurally can be seen as swollen mitochondria (Arrázola et al., 2015). Diverse methods to block the mitochondrial incorporation and overload of Ca2+, such as blocking the MCU, have proven not only to protect mitochondria, but also to reduce axonal degeneration (Villegas et al., 2014). Alternatively, inhibiting the formation of the mPTP genetically by cyclophilin D knockdown (Barrientos et al., 2011; Breckwoldt et al., 2014) or pharmacologically by cyclosporin A (Villegas et al., 2014) prevents the swelling of mitochondria and protects the axons from degeneration.
In TBI, mitochondria harvested from the forebrain of mice following a CCI present significantly higher Ca2+ levels 6 h post-injury, resulting in the impairment of the electron transport chain (Figure 1C; Xiong et al., 1997). The decreased oxidative phosphorylation and ATP production in injured mitochondria occur within 24 h post-injury and can last up to 14 days in experimental models of TBI (Gilmer et al., 2009). In these models, post-injury administration of cyclosporin A or NIM811 (a non-immunosuppressive cyclosporin A analog) has shown to decrease the neuronal and axonal damages, improving both motor skills and cognition when compared to non-treated animals (Büki et al., 1999; Mbye et al., 2009; Readnower et al., 2011). Also in vitro, a uniaxial strain on hippocampal axons shows decreased mitochondrial membrane potential 1 h after the injury. However, the mitochondrial potential is maintained if Ca2+ entry to mitochondria is reduced by pharmacological incubation with a Na+/H+ exchanger inhibitor (Toda et al., 2007; Dollé et al., 2014). These data suggest that the mitochondrial damage in TBI may occur in severe cases and way later after the injury (Hånell et al., 2015).
Ca2+ imbalance has a well-studied role in the pathogenesis of the neurodegenerative diseases and extensive reviews dwell on the topic; however, studies directed exclusively to the axons are more limited (Cali et al., 2012; Schrank et al., 2020; Jadiya et al., 2021). Aβ was shown to interact with several components that can be implicated in the regulation of Ca2+ levels in the axon. There is evidence that incubation of hippocampal slices with Aβ increases intracellular Ca2+ originating from the extracellular space through different types of VGCC (Ueda et al., 1997; Ramsden et al., 2002). Alternatively, intracellular Ca2+ levels have been shown to increase either through ER stores depletion by Aβ (Ferreiro et al., 2008; Sanz-Blasco et al., 2008) or through enhancing the gating of the inositol trisphosphate receptor by presenilin mutations associated with AD (Cheung et al., 2008). Furthermore, Aβ peptides can translocate to the mitochondrial matrix through TOM40 where they interact with specific intra-mitochondrial proteins such as the cyclophilin D (Hansson Petersen et al., 2008). Axons derived from primary hippocampal neurons reveal that after translocation to mitochondria, Aβ induces an opening of mPTP with consequent increase in Ca2+ and ROS levels, reducing the number of synapses (Guo et al., 2013). Similarly to Aβ behavior, PD linked mutation in alpha-synuclein modulates the activity of N-type VGCC in the presynapses leading to an increased Ca2+ levels and reduced axonal arborization in mouse caudate-putamen (Sgobio et al., 2019). PINK1 deficiency increases Ca2+ accumulation in mitochondrial matrix and ROS production by diminishing the Ca2+ efflux through the Na+/Ca2+ exchanger of the mitochondria (NCLX) (Gandhi et al., 2009).
There is an intricate relationship between the axons and mitochondria. Changes in the axons impact mitochondrial transport, fusion/fission, and health, but also the changes in mitochondrial behavior and health contribute to axonal homeostasis. With few exceptions, an axon devoid of mitochondria is destined to degenerate (Kitay et al., 2013; Berthet et al., 2014; Rawson et al., 2014). Immediately after axonal injury, several changes happen in mitochondria. In several models, mitochondrial anterograde transport is enhanced, which is followed by an increase in retrograde transport (Figure 1A; Misgeld et al., 2007; Zhou et al., 2016; Pozo Devoto et al., 2022). Mobilization of mitochondria together with proper delivery of ATP to regions in need (Han et al., 2016; Zhou et al., 2016) seems to be important factors that influence axonal regeneration (Xu et al., 2017; Wang et al., 2019). Favoring the increased movement, the fusion/fission balance tilts to the fission side, thus generating smaller mitochondria that are more prone to move (Figure 1B; Kiryu-Seo et al., 2016). However, not all of the evidence points to a positive role of mitochondrial fragmentation in axonal rescue after injury (Kedra et al., 2021). Even though fission favors the mobilization of mitochondria, the fusion is involved in stability and increased biogenesis (Chen et al., 2016). It is possible that factors such as high local ATP requirements that may need an abundant mitochondrial mass located in specific region would favor a fusion strategy. On the other hand, the need for increased distribution along the axon or the damage of a big number of mitochondria can be the factors defining the fission as a best strategy to promote survival and/or regeneration. The two main organelles involved in Ca2+ regulation in the axons are the ER and mitochondria. In contrast to ER, mitochondrial function can be irreversibly impaired by Ca2+ overload (Figure 1C). Even more, the loss of mitochondrial potential can trigger signals that result in apoptosis or a degenerative process. For this reason, the pharmacological inhibition of Ca2+ influx through the MCU or the blockage of the mPTP formation protects mitochondrial and axonal stability in several cases (Breckwoldt et al., 2014; Villegas et al., 2014). In milder conditions, a surge in intra-axonal Ca2+ levels has shown to increase ROS production, which directly results in decreased ATP production through the oxidation of the components of the electron transport chain or increased leakage of the IMM (Xiong et al., 1997; Breckwoldt et al., 2014).
Each of the different pathologies that lead to axonal degeneration presents characteristics that makes them unique. For example, an axon that is either stretched or crushed requires repair of the damaged regions, whereas an axon that is transected requires regrowth of its proximal segment and reinnervation of its target to maintain its previous function. Furthermore, the distal axonal segment that results from axotomy undergoes Wallerian degeneration. The discovery of delayed degeneration of the distal segment of cut axons in the Wallerian degeneration slow (WldS) transgenic mouse model (Lunn et al., 1989) led to the identification of nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) and sterile alpha and TIR motif-containing protein 1 (SARM1) as the two main players responsible for this delay (Gilley and Coleman, 2010; Osterloh et al., 2012; Gerdts et al., 2015). NMNAT2 is an axonal protein that catalyzes the conversion of nicotinamide mononucleotide (NMN) to nicotinamide adenine dinucleotide (NAD+). Both depletion of NAD+ and increase in NMN trigger axonal degeneration through SARM1 (Angeletti et al., 2022). Axotomy reduces the axonal availability of NMNAT2, which is a short-lived enzyme transported from the soma, leading to NAD+ depletion and NMN increase. However, the connection between this pathway and mitochondria is not completely clear (Merlini et al., 2022). In Drosophila motoneurons, it has been shown that the presence of mitochondria accelerates the axonal Wallerian degeneration but is not necessary for its execution (Kitay et al., 2013). Conversely, in the olfactory receptor neurons of Drosophila, the degeneration triggered by mitochondrial depletion from the axon cannot be rescued by NMNAT overexpression (Kitay et al., 2013). The involvement of NMNAT2 or SARM1 as executors in axonal degeneration is well supported (Feng et al., 2010; Ziogas and Koliatsos, 2018), but several studies suggest that axonal homeostasis is further maintained by the pathways independent of NMNAT2/SARM1 (Kitay et al., 2013; Liu et al., 2021).
In axonal mechanical injury without detachment, axons present microtubule disruption or rearrangement, which can affect mitochondrial mobility. It is generally accepted that an axon that regenerates is an axon that exhibits more mitochondrial movement. So, the integrity of microtubule tracks is a key for proper distribution and delivery of mitochondria to places in need of ATP and Ca2+-buffering capacity. Axonal swellings are present in many pathological settings, from TBI (Weber et al., 2019) to neurodegenerative diseases (Stokin et al., 2005; Adalbert et al., 2009; Nikić et al., 2011; Babij et al., 2013) and last for long periods or immediately precede axonal collapse (Rawson et al., 2014). It is still not clear whether the axonal swellings represent regions of cytoskeletal damage, particularly of microtubules and neurofilaments (Tang-Schomer et al., 2012). Several studies observe that axonal swellings can form and last for a really short period of time (Gu et al., 2017), or that they can present cytoskeletal rearrangement that permits the movement of cargo across them (Datar et al., 2019; Pozo Devoto et al., 2022). It has been proposed that the specific local increases of Ca2+ occurring during axonal injury contribute to the formation or maintenance of axonal swellings in time (Staal et al., 2010; Stirling et al., 2014). Mechanistically, this could have an impact not only on cytoskeletal remodeling by Ca2+-dependent signaling, but also on the movement and fission of mitochondria if high Ca2+ concentrations are maintained. An important amount of evidence points to a major role of Ca2+ as a determinant of axonal fate after injury, with effects that are mitochondria-dependent and mitochondria-independent.
An increasing knowledge on the molecular pathways that regulate and connect processes such as mitochondrial transport and fusion/fission with biogenesis and recycling of mitochondrial components keeps advancing our understanding of the mitochondrial homeostasis in a challenging structure such as the axon.
VMPD: conceptualization, literature research, manuscript writing, and figure. IGO: literature research,manuscript writing, and editing. GBS: conceptualization, supervision, reviewing, and editing. All authors contributed to the article and approved the submitted version.
This work was supported by the European Regional Development Funds No. CZ.02.1.01/0.0/0.0/16_019/0000868 ENOCH grant and the European Regional Development Fund-Project MAGNET No. CZ.02.1.01/0.0/0.0/15_003/0000492.
We would like to thank Ms. Monica Feole and Katja Klosterman for their assistance and feedback.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adalbert, R., Nogradi, A., Babetto, E., Janeckova, L., Walker, S. A., Kerschensteiner, M., et al. (2009). Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain 132, 402–416. doi: 10.1093/brain/awn312
Alexander, C., Votruba, M., Pesch, U. E., Thiselton, D. L., Mayer, S., Moore, A., et al. (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215. doi: 10.1038/79944
Amiri, M., and Hollenbeck, P. J. (2008). Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev. Neurobiol. 68, 1348–1361. doi: 10.1002/dneu.20668
Angeletti, C., Amici, A., Gilley, J., Loreto, A., Trapanotto, A. G., Antoniou, C., et al. (2022). SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated bymultiple physiologically relevant NAD metabolites. iScience 25, 103812. doi: 10.1016/j.isci.2022.103812
Arrázola, M. S., Saquel, C., Catalán, R. J., Barrientos, S. A., Hernandez, D. E., Martínez, N. W., et al. (2019). Axonal degeneration is mediated by necroptosis activation. J. Neurosci. 39, 3832–3844. doi: 10.1523/JNEUROSCI.0881-18.2019
Arrázola, M. S., Silva-Alvarez, C., and Inestrosa, N. C. (2015). How the Wnt signaling pathway protects from neurodegeneration: the mitochondrial scenario. Front. Cell. Neurosci. 9, 166–166. doi: 10.3389/fncel.2015.00166
Babij, R., Lee, M., Cortés, E., Vonsattel, J.-P. G., Faust, P. L., and Louis, E. D. (2013). Purkinje cell axonal anatomy: quantifying morphometric changes in essential tremor versus control brains. Brain 136, 3051–3061. doi: 10.1093/brain/awt238
Bao, F., Shi, H., Gao, M., Yang, L., Zhou, L., Zhao, Q., et al. (2018). Polybrene induces neural degeneration by bidirectional Ca2+ influx-dependent mitochondrial and ER-mitochondrial dynamics. Cell Death Dis. 9, 966. doi: 10.1038/s41419-018-1009-8
Barrientos, S. A., Martinez, N. W., Yoo, S., Jara, J. S., Zamorano, S., Hetz, C., et al. (2011). Axonal degeneration is mediated by the mitochondrial permeability transition pore. J. Neurosci. 31, 966–978. doi: 10.1523/JNEUROSCI.4065-10.2011
Bayir, H., Tyurin, V. A., Tyurina, Y. Y., Viner, R., Ritov, V., Amoscato, A. A., et al. (2007). Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis. Ann. Neurol. 62, 154–169. doi: 10.1002/ana.21168
Bernardi, P., Krauskopf, A., Basso, E., Petronilli, V., Blachly-Dyson, E., Blalchy-Dyson, E., et al. (2006). The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273, 2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x
Berthet, A., Margolis, E. B., Zhang, J., Hsieh, I., Zhang, J., Hnasko, T. S., et al. (2014). Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J. Neurosci. 34, 14304–14317. doi: 10.1523/JNEUROSCI.0930-14.2014
Blennow, K., Brody, D. L., Kochanek, P. M., Levin, H., McKee, A., Ribbers, G. M., et al. (2016). Traumatic brain injuries. Nat. Rev. Dis. Primers 2, 16084. doi: 10.1038/nrdp.2016.84
Breckwoldt, M. O., Pfister, F. M., Bradley, P. M., Marinković, P., Williams, P. R., Brill, M. S., et al. (2014). Multiparametric optical analysis of mitochondrial redox signals during neuronal physiology and pathology in vivo. Nat. Med. 20, 555–560. doi: 10.1038/nm.3520
Bruggeman, G. F., Haitsma, I. K., Dirven, C. M. F., and Volovici, V. (2021). Traumatic axonal injury (TAI): definitions, pathophysiology and imaging-a narrative review. Acta Neurochir. 163, 31–44. doi: 10.1007/s00701-020-04594-1
Büki, A., Okonkwo, D. O., and Povlishock, J. T. (1999). Postinjury cyclosporin A administration limits axonal damage and disconnection in traumatic brain injury. J. Neurotrauma 16, 511–521. doi: 10.1089/neu.1999.16.511
Büki, A., Okonkwo, D. O., Wang, K. K., and Povlishock, J. T. (2000). Cytochrome c release and caspase activation in traumatic axonal injury. J. Neurosci. 20, 2825–2834. doi: 10.1523/JNEUROSCI.20-08-02825.2000
Burté, F., Carelli, V., Chinnery, P. F., and Yu-Wai-Man, P. (2015). Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 11, 11–24. doi: 10.1038/nrneurol.2014.228
Burton, P. R., and Paige, J. L. (1981). Polarity of axoplasmic microtubules in the olfactory nerve of the frog. Proc. Natl. Acad. Sci. U.S.A. 78, 3269–3273. doi: 10.1073/pnas.78.5.3269
Byrne, J. J., Soh, M. S., Chandhok, G., Vijayaraghavan, T., Teoh, J. S., Crawford, S., et al. (2019). Disruption of mitochondrial dynamics affects behaviour and lifespan in Caenorhabditis elegans. Cell. Mol. Life Sci. 76, 1967–1985. doi: 10.1007/s00018-019-03024-5
Cagalinec, M., Safiulina, D., Liiv, M., Liiv, J., Choubey, V., Wareski, P., et al. (2013). Principles of the mitochondrial fusion and fission cycle in neurons. J. Cell Sci. 126, 2187–2197. doi: 10.1242/jcs.118844
Cai, Q., Gerwin, C., and Sheng, Z. H. (2005). Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J. Cell Biol. 170, 959–969. doi: 10.1083/jcb.200506042
Cali, T., Ottolini, D., and Brini, M. (2012). Mitochondrial Ca(2+) and neurodegeneration. Cell Calcium 52, 73–85. doi: 10.1016/j.ceca.2012.04.015
Calkins, M. J., and Reddy, P. H. (2011). Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer's disease neurons. Biochim. Biophys. Acta 1812, 507–513. doi: 10.1016/j.bbadis.2011.01.007
Cartoni, R., Arnaud, E., Médard, J. J., Poirot, O., Courvoisier, D. S., Chrast, R., et al. (2010). Expression of mitofusin 2(R94Q) in a transgenic mouse leads to charcot-marie-tooth neuropathy type 2A. Brain 133, 1460–1469. doi: 10.1093/brain/awq082
Cartoni, R., Norsworthy, M. W., Bei, F., Wang, C., Li, S., Zhang, Y., et al. (2016). The mammalian-specific protein armcx1 regulates mitochondrial transport during axon regeneration. Neuron 92, 1294–1307. doi: 10.1016/j.neuron.2016.10.060
Chan, D. C (2020). Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 15, 235–259. doi: 10.1146/annurev-pathmechdis-012419-032711
Chang, D. T., Honick, A. S., and Reynolds, I. J. (2006). Mitochondrial trafficking to synapses in cultured primary cortical neurons. J. Neurosci. 26, 7035–7045. doi: 10.1523/JNEUROSCI.1012-06.2006
Chang, D. T., and Reynolds, I. J. (2006a). Mitochondrial trafficking and morphology in healthy and injured neurons. Prog. Neurobiol. 80, 241–268. doi: 10.1016/j.pneurobio.2006.09.003
Chang, D. T. W., and Reynolds, I. J. (2006b). Differences in mitochondrial movement and morphology in young and mature primary cortical neurons in culture. Neuroscience 141, 727–736. doi: 10.1016/j.neuroscience.2006.01.034
Chavan, V., Willis, J., Walker, S. K., Clark, H. R., Liu, X., Fox, M. A., et al. (2015). Central presynaptic terminals are enriched in ATP but the majority lack mitochondria. PLoS ONE 10, e0125185. doi: 10.1371/journal.pone.0125185
Chen, M., Li, Y., Yang, M., Chen, X., Chen, Y., Yang, F., et al. (2016). A new method for quantifying mitochondrial axonal transport. Protein Cell 7, 804–819. doi: 10.1007/s13238-016-0268-3
Chen, X. H., Meaney, D. F., Xu, B. N., Nonaka, M., McIntosh, T. K., Wolf, J. A., et al. (1999). Evolution of neurofilament subtype accumulation in axons following diffuse brain injury in the pig. J. Neuropathol. Exp. Neurol. 58, 588–596. doi: 10.1097/00005072-199906000-00003
Chen, Y., and Sheng, Z. H. (2013). Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J. Cell Biol. 202, 351–364. doi: 10.1083/jcb.201302040
Cheng, H. C., Ulane, C. M., and Burke, R. E. (2010). Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 67, 715–725. doi: 10.1002/ana.21995
Cheng, X. T., and Sheng, Z. H. (2021). Developmental regulation of microtubule-based trafficking and anchoring of axonal mitochondria in health and diseases. Dev. Neurobiol. 81, 284–299. doi: 10.1002/dneu.22748
Cheung, K.-H., Shineman, D., Müller, M., Cárdenas, C., Mei, L., Yang, J., et al. (2008). Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58, 871–883. doi: 10.1016/j.neuron.2008.04.015
Cho, D. H., Nakamura, T., Fang, J., Cieplak, P., Godzik, A., Gu, Z., et al. (2009). S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102–105. doi: 10.1126/science.1171091
Cho, K. I., Cai, Y., Yi, H., Yeh, A., Aslanukov, A., and Ferreira, P. A. (2007). Association of the kinesin-binding domain of RanBP2 to KIF5B and KIF5C determines mitochondria localization and function. Traffic 8, 1722–1735. doi: 10.1111/j.1600-0854.2007.00647.x
Coleman, M. P., and Höke, A. (2020). Programmed axon degeneration: from mouse to mechanism to medicine. Nat. Rev. Neurosci. 21, 183–196. doi: 10.1038/s41583-020-0269-3
Cotteret, S., Jaffer, Z. M., Beeser, A., and Chernoff, J. (2003). p21-Activated kinase 5 (Pak5) localizes to mitochondria and inhibits apoptosis by phosphorylating BAD. Mol. Cell. Biol. 23, 5526–5539. doi: 10.1128/MCB.23.16.5526-5539.2003
Cross, D. J., Meabon, J. S., Cline, M. M., Richards, T. L., Stump, A. J., Cross, C. G., et al. (2019). Paclitaxel reduces brain injury from repeated head trauma in mice. J. Alzheimers Dis. 67, 859–874. doi: 10.3233/JAD-180871
Datar, A., Ameeramja, J., Bhat, A., Srivastava, R., Mishra, A., Bernal, R., et al. (2019). The roles of microtubules and membrane tension in axonal beading, retraction, and atrophy. Biophys. J. 117, 880–891. doi: 10.1016/j.bpj.2019.07.046
De Vos, K. J., Chapman, A. L., Tennant, M. E., Manser, C., Tudor, E. L., Lau, K. F., et al. (2007). Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 16, 2720–2728. doi: 10.1093/hmg/ddm226
Detmer, S. A., Vande Velde, C., Cleveland, D. W., and Chan, D. C. (2008). Hindlimb gait defects due to motor axon loss and reduced distal muscles in a transgenic mouse model of charcot-marie-tooth type 2A. Hum. Mol. Genet. 17, 367–375. doi: 10.1093/hmg/ddm314
Devine, M. J., and Kittler, J. T. (2018). Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 19, 63–80. doi: 10.1038/nrn.2017.170
Di Meo, D., Ravindran, P., Sadhanasatish, T., Dhumale, P., and Püschel, A. W. (2021). The balance of mitochondrial fission and fusion in cortical axons depends on the kinases SadA and SadB. Cell Rep. 37, 110141. doi: 10.1016/j.celrep.2021.110141
Di Pietro, V., Lazzarino, G., Amorini, A. M., Signoretti, S., Hill, L. J., Porto, E., et al. (2017). Fusion or fission: the destiny of mitochondria in traumatic brain injury of different severities. Sci. Rep. 7, 9189. doi: 10.1038/s41598-017-09587-2
Dollé, J. P., Morrison, B., Schloss, R. S., and Yarmush, M. L. (2014). Brain-on-a-chip microsystem for investigating traumatic brain injury: axon diameter and mitochondrial membrane changes play a significant role in axonal response to strain injuries. Technology 2, 106. doi: 10.1142/S2339547814500095
Dorn, G. W (2020). Mitofusin 2 dysfunction and disease in mice and men. Front. Physiol. 11, 782. doi: 10.3389/fphys.2020.00782
Drerup, C. M., Herbert, A. L., Monk, K. R., and Nechiporuk, A. V. (2017). Regulation of mitochondria-dynactin interaction and mitochondrial retrograde transport in axons. Elife 6, e22234. doi: 10.7554/eLife.22234.021
Du, H., Guo, L., Yan, S., Sosunov, A. A., McKhann, G. M., and Yan, S. S. (2010). Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc. Natl. Acad. Sci. U.S.A. 107, 18670–18675. doi: 10.1073/pnas.1006586107
El Fissi, N., Rojo, M., Aouane, A., Karatas, E., Poliacikova, G., David, C., et al. (2018). Mitofusin gain and loss of function drive pathogenesis in Drosophila models of CMT2A neuropathy. EMBO Rep. 19, e45241. doi: 10.15252/embr.201745241
Farkas, O., Lifshitz, J., and Povlishock, J. T. (2006). Mechanoporation induced by diffuse traumatic brain injury: an irreversible or reversible response to injury. J. Neurosci. 26, 3130–3140. doi: 10.1523/JNEUROSCI.5119-05.2006
Feng, Y., Yan, T., Zheng, J., Ge, X., Mu, Y., Zhang, Y., et al. (2010). Overexpression of Wld(S) or Nmnat2 in mauthner cells by single-cell electroporation delays axon degeneration in live zebrafish. J. Neurosci. Res. 88, 3319–3327. doi: 10.1002/jnr.22498
Ferguson, B., Matyszak, M. K., Esiri, M. M., and Perry, V. H. (1997). Axonal damage in acute multiple sclerosis lesions. Brain 120, 393–399. doi: 10.1093/brain/120.3.393
Ferreiro, E., Oliveira, C. R., and Pereira, C. M. F. (2008). The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 30, 331–342. doi: 10.1016/j.nbd.2008.02.003
Ferri, A., Fiorenzo, P., Nencini, M., Cozzolino, M., Pesaresi, M. G., Valle, C., et al. (2010). Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum. Mol. Genet. 19, 4529–4542. doi: 10.1093/hmg/ddq383
Fischer, T. D., Hylin, M. J., Zhao, J., Moore, A. N., Waxham, M. N., and Dash, P. K. (2016). Altered mitochondrial dynamics and TBI pathophysiology. Front. Syst. Neurosci. 10, 29. doi: 10.3389/fnsys.2016.00029
Gandhi, S., Wood-Kaczmar, A., Yao, Z., Plun-Favreau, H., Deas, E., Klupsch, K., et al. (2009). PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 33, 627–638. doi: 10.1016/j.molcel.2009.02.013
Gerdts, J., Brace, E. J., Sasaki, Y., DiAntonio, A., and Milbrandt, J. (2015). Neurobiology. SARM1 activation triggers axon degeneration locally via NAD? destruction. Science 348, 453–457. doi: 10.1126/science.1258366
Giacomello, M., Pyakurel, A., Glytsou, C., and Scorrano, L. (2020). The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 21, 204–224. doi: 10.1038/s41580-020-0210-7
Gilley, J., and Coleman, M. P. (2010). Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 8, e1000300-. doi: 10.1371/journal.pbio.1000300
Gilmer, L. K., Roberts, K. N., Joy, K., Sullivan, P. G., and Scheff, S. W. (2009). Early mitochondrial dysfunction after cortical contusion injury. J. Neurotrauma 26, 1271–1280. doi: 10.1089/neu.2008.0857
Gitler, D., and Spira, M. E. (1998). Real time imaging of calcium-induced localized proteolytic activity after axotomy and its relation to growth cone formation. Neuron 20, 1123–1135. doi: 10.1016/S0896-6273(00)80494-8
Glauser, L., Sonnay, S., Stafa, K., and Moore, D. J. (2011). Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J. Neurochem. 118, 636–645. doi: 10.1111/j.1471-4159.2011.07318.x
Godoy, J. A., Arrázola, M. S., Ordenes, D., Silva-Alvarez, C., Braidy, N., and Inestrosa, N. C. (2014). Wnt-5a ligand modulates mitochondrial fission-fusion in rat hippocampal neurons. J. Biol. Chem. 289, 36179–36193. doi: 10.1074/jbc.M114.557009
Graham, J. M., Papadakis, N., Evans, J., Widjaja, E., Romanowski, C. A., Paley, M. N., et al. (2004). Diffusion tensor imaging for the assessment of upper motor neuron integrity in ALS. Neurology 63, 2111–2119. doi: 10.1212/01.WNL.0000145766.03057.E7
Gu, Y., Jukkola, P., Wang, Q., Esparza, T., Zhao, Y., Brody, D., et al. (2017). Polarity of varicosity initiation in central neuron mechanosensation. J. Cell Biol. 216, 2179–2199. doi: 10.1083/jcb.201606065
Guo, L., Du, H., Yan, S., Wu, X., McKhann, G. M., Chen, J. X., et al. (2013). Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer's neurons. PLoS ONE 8, e54914. doi: 10.1371/journal.pone.0054914
Guo, X., Macleod, G. T., Wellington, A., Hu, F., Panchumarthi, S., Schoenfield, M., et al. (2005). The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47, 379–393. doi: 10.1016/j.neuron.2005.06.027
Hånell, A., Greer, J. E., McGinn, M. J., and Povlishock, J. T. (2015). Traumatic brain injury-induced axonal phenotypes react differently to treatment. Acta Neuropathol. 129, 317–332. doi: 10.1007/s00401-014-1376-x
Han, Q., Xie, Y., Ordaz, J. D., Huh, A. J., Huang, N., Wu, W., et al. (2020). Restoring cellular energetics promotes axonal regeneration and functional recovery after spinal cord injury. Cell Metab. 31, 623–641.e8. doi: 10.1016/j.cmet.2020.02.002
Han, S. M., Baig, H. S., and Hammarlund, M. (2016). Mitochondria localize to injured axons to support regeneration. Neuron 92, 1308–1323. doi: 10.1016/j.neuron.2016.11.025
Hansson Petersen, C. A., Alikhani, N., Behbahani, H., Wiehager, B., Pavlov, P. F., Alafuzoff, I., et al. (2008). The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. U.S.A. 105, 13145–13150. doi: 10.1073/pnas.0806192105
Henley, J., and Poo, M. M. (2004). Guiding neuronal growth cones using Ca2+ signals. Trends Cell Biol. 14, 320–330. doi: 10.1016/j.tcb.2004.04.006
Hirokawa, N., Niwa, S., and Tanaka, Y. (2010). Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68, 610–638. doi: 10.1016/j.neuron.2010.09.039
Howarth, C., Gleeson, P., and Attwell, D. (2012). Updated energy budgets for neural computation in the neocortex and cerebellum. J. Cereb. Blood Flow Metab. 32, 1222–1232. doi: 10.1038/jcbfm.2012.35
Huang, J., Friedland, R. P., and Auchus, A. P. (2007). Diffusion tensor imaging of normal-appearing white matter in mild cognitive impairment and early Alzheimer disease: preliminary evidence of axonal degeneration in the temporal lobe. Am. J. Neuroradiol. 28, 1943–1948. doi: 10.3174/ajnr.A0700
Huang, N., Li, S., Xie, Y., Han, Q., Xu, X. M., and Sheng, Z. H. (2021). Reprogramming an energetic AKT-PAK5 axis boosts axon energy supply and facilitates neuron survival and regeneration after injury and ischemia. Curr. Biol. 31, 3098–3114.e7. doi: 10.1016/j.cub.2021.04.079
Ikuta, J., Maturana, A., Fujita, T., Okajima, T., Tatematsu, K., Tanizawa, K., et al. (2007). Fasciculation and elongation protein zeta-1 (FEZ1) participates in the polarization of hippocampal neuron by controlling the mitochondrial motility. Biochem. Biophys. Res. Commun. 353, 127–132. doi: 10.1016/j.bbrc.2006.11.142
Ineichen, B. V., Zhu, K., and Carlström, K. E. (2021). Axonal mitochondria adjust in size depending on g-ratio of surrounding myelin during homeostasis and advanced remyelination. J. Neurosci. Res. 99, 793–805. doi: 10.1002/jnr.24767
Ishihara, N., Nomura, M., Jofuku, A., Kato, H., Suzuki, S. O., Masuda, K., et al. (2009). Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 11, 958–966. doi: 10.1038/ncb1907
Itoh, K., Nakamura, K., Iijima, M., and Sesaki, H. (2013). Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 23, 64–71. doi: 10.1016/j.tcb.2012.10.006
Jadiya, P., Garbincius, J. F., and Elrod, J. W. (2021). Reappraisal of metabolic dysfunction in neurodegeneration: Focus on mitochondrial function and calcium signaling. Acta Neuropathol. Commun. 9, 124. doi: 10.1186/s40478-021-01224-4
Johnson, V. E., Stewart, W., Weber, M. T., Cullen, D. K., Siman, R., and Smith, D. H. (2016). SNTF immunostaining reveals previously undetected axonal pathology in traumatic brain injury. Acta Neuropathol. 131, 115–135. doi: 10.1007/s00401-015-1506-0
Kang, J. S., Tian, J. H., Pan, P. Y., Zald, P., Li, C., Deng, C., et al. (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148. doi: 10.1016/j.cell.2007.11.024
Kedra, J., Lin, S., Pacheco, A., Gallo, G., and Smith, G. M. (2021). Axotomy induces Drp1-dependent fragmentation of axonal mitochondria. Front. Mol. Neurosci. 14, 668670. doi: 10.3389/fnmol.2021.668670
Kilinc, D., Gallo, G., and Barbee, K. A. (2008). Mechanically-induced membrane poration causes axonal beading and localized cytoskeletal damage. Exp. Neurol. 212, 422–430. doi: 10.1016/j.expneurol.2008.04.025
Kiryu-Seo, S., Tamada, H., Kato, Y., Yasuda, K., Ishihara, N., Nomura, M., et al. (2016). Mitochondrial fission is an acute and adaptive response in injured motor neurons. Sci. Rep. 6, 28331. doi: 10.1038/srep28331
Kitay, B. M., McCormack, R., Wang, Y., Tsoulfas, P., and Zhai, R. G. (2013). Mislocalization of neuronal mitochondria reveals regulation of Wallerian degeneration and NMNAT/WLD(S)-mediated axon protection independent of axonal mitochondria. Hum. Mol. Genet. 22, 1601–1614. doi: 10.1093/hmg/ddt009
Knowlton, W. M., Hubert, T., Wu, Z., Chisholm, A. D., and Jin, Y. (2017). A select subset of electron transport chain genes associated with optic atrophy link mitochondria to axon regeneration in Caenorhabditis elegans. Front. Neurosci. 11, 263. doi: 10.3389/fnins.2017.00263
Kruppa, A. J., and Buss, F. (2021). Motor proteins at the mitochondria-cytoskeleton interface. J. Cell Sci. 134, jcs226084. doi: 10.1242/jcs.226084
Lee, S., Sterky, F. H., Mourier, A., Terzioglu, M., Cullheim, S., Olson, L., et al. (2012). Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 21, 4827–4835. doi: 10.1093/hmg/dds352
Lewis, T. L., Kwon, S. K., Lee, A., Shaw, R., and Polleux, F. (2018). MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat. Commun. 9, 5008. doi: 10.1038/s41467-018-07416-2
Lewis, T. L., Turi, G. F., Kwon, S. K., Losonczy, A., and Polleux, F. (2016). Progressive decrease of mitochondrial motility during maturation of cortical axons in vitro and in vivo. Curr. Biol. 26, 2602–2608. doi: 10.1016/j.cub.2016.07.064
Li, S., Xiong, G. J., Huang, N., and Sheng, Z. H. (2020). The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat. Metab. 2, 1077–1095. doi: 10.1038/s42255-020-00289-0
Licht-Mayer, S., Campbell, G. R., Canizares, M., Mehta, A. R., Gane, A. B., McGill, K., et al. (2020). Enhanced axonal response of mitochondria to demyelination offers neuroprotection: implications for multiple sclerosis. Acta Neuropathol. 140, 143–167. doi: 10.1007/s00401-020-02179-x
Lin, M. Y., Cheng, X. T., Tammineni, P., Xie, Y., Zhou, B., Cai, Q., et al. (2017). Releasing syntaphilin removes stressed mitochondria from axons independent of mitophagy under pathophysiological conditions. Neuron 94, 595–610.e6. doi: 10.1016/j.neuron.2017.04.004
Lingor, P., Koch, J. C., Tönges, L., and Bähr, M. (2012). Axonal degeneration as a therapeutic target in the CNS. Cell Tissue Res. 349, 289–311. doi: 10.1007/s00441-012-1362-3
Liu, P., Huang, H., Fang, F., Liu, L., Li, L., Feng, X., et al. (2021). Neuronal NMNAT2 overexpression does not achieve significant neuroprotection in experimental autoimmune encephalomyelitis/optic neuritis. Front. Cell. Neurosci. 15, 754651. doi: 10.3389/fncel.2021.754651
López-Doménech, G., Serrat, R., Mirra, S., D'Aniello, S., Somorjai, I., Abad, A., et al. (2012). The Eutherian Armcx genes regulate mitochondrial trafficking in neurons and interact with Miro and Trak2. Nat. Commun. 3, 814–814. doi: 10.1038/ncomms1829
Lunn, E. R., Perry, V. H., Brown, M. C., Rosen, H., and Gordon, S. (1989). Absence of wallerian degeneration does not hinder regeneration in peripheral nerve. Eur. J. Neurosci. 1, 27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x
Macaskill, A. F., Rinholm, J. E., Twelvetrees, A. E., Arancibia-Carcamo, I. L., Muir, J., Fransson, A., et al. (2009). Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61, 541–555. doi: 10.1016/j.neuron.2009.01.030
Maday, S., Twelvetrees, A. E., Moughamian, A. J., and Holzbaur, E. L. (2014). Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84, 292–309. doi: 10.1016/j.neuron.2014.10.019
Mar, F. M., Simões, A. R., Leite, S., Morgado, M. M., Santos, T. E., Rodrigo, I. S., et al. (2014). CNS axons globally increase axonal transport after peripheral conditioning. J. Neurosci. 34, 5965–5970. doi: 10.1523/JNEUROSCI.4680-13.2014
Maxwell, W. L (1996). Histopathological changes at central nodes of Ranvier after stretch-injury. Microsc. Res. Tech. 34, 522–535. doi: 10.1002/(SICI)1097-0029(19960815)34:6<522::AID-JEMT4>3.0.CO;2-L
Mbye, L. H., Singh, I. N., Carrico, K. M., Saatman, K. E., and Hall, E. D. (2009). Comparative neuroprotective effects of cyclosporin A and NIM811, a nonimmunosuppressive cyclosporin A analog, following traumatic brain injury. J. Cereb. Blood Flow Metab. 29, 87–97. doi: 10.1038/jcbfm.2008.93
McDonald, J. W., and Sadowsky, C. (2002). Spinal-cord injury. Lancet 359, 417–425. doi: 10.1016/S0140-6736(02)07603-1
Meeusen, S., McCaffery, J. M., and Nunnari, J. (2004). Mitochondrial fusion intermediates revealed in vitro. Science 305, 1747–1752. doi: 10.1126/science.1100612
Merlini, E., Coleman, M. P., and Loreto, A. (2022). Mitochondrial dysfunction as a trigger of programmed axon death. Trends Neurosci. 45, 53–63. doi: 10.1016/j.tins.2021.10.014
Mironov, S. L (2007). ADP regulates movements of mitochondria in neurons. Biophys. J. 92, 2944–2952. doi: 10.1529/biophysj.106.092981
Misgeld, T., Kerschensteiner, M., Bareyre, F. M., Burgess, R. W., and Lichtman, J. W. (2007). Imaging axonal transport of mitochondria in vivo. Nat. Methods 4, 559–561. doi: 10.1038/nmeth1055
Misgeld, T., and Schwarz, T. L. (2017). Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron 96, 651–666. doi: 10.1016/j.neuron.2017.09.055
Mishra, P., and Chan, D. C. (2014). Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 15, 634–646. doi: 10.1038/nrm3877
Mishra, P., and Chan, D. C. (2016). Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 212, 379–387. doi: 10.1083/jcb.201511036
Misko, A. L., Sasaki, Y., Tuck, E., Milbrandt, J., and Baloh, R. H. (2012). Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 32, 4145–4155. doi: 10.1523/JNEUROSCI.6338-11.2012
Morsci, N. S., Hall, D. H., Driscoll, M., and Sheng, Z.-H. (2016). Age-Related phasic patterns of mitochondrial maintenance in adult caenorhabditis elegans neurons. J. Neurosci. 36, 1373–1385. doi: 10.1523/JNEUROSCI.2799-15.2016
Mou, Y., Dein, J., Chen, Z., Jagdale, M., and Li, X. J. (2021). MFN2 deficiency impairs mitochondrial transport and downregulates motor protein expression in human spinal motor neurons. Front. Mol. Neurosci. 14, 727552. doi: 10.3389/fnmol.2021.727552
Niescier, R. F., Hong, K., Park, D., and Min, K. T. (2018). MCU interacts with miro1 to modulate mitochondrial functions in neurons. J. Neurosci. 38, 4666–4677. doi: 10.1523/JNEUROSCI.0504-18.2018
Nikić, I., Merkler, D., Sorbara, C., Brinkoetter, M., Kreutzfeldt, M., Bareyre, F. M., et al. (2011). A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 17, 495–499. doi: 10.1038/nm.2324
Obashi, K., and Okabe, S. (2013). Regulation of mitochondrial dynamics and distribution by synapse position and neuronal activity in the axon. Eur. J. Neurosci. 38, 2350–2363. doi: 10.1111/ejn.12263
Oettinghaus, B., Schulz, J. M., Restelli, L. M., Licci, M., Savoia, C., Schmidt, A., et al. (2016). Synaptic dysfunction, memory deficits and hippocampal atrophy due to ablation of mitochondrial fission in adult forebrain neurons. Cell Death Differ. 23, 18–28. doi: 10.1038/cdd.2015.39
Ohno, N., Chiang, H., Mahad, D. J., Kidd, G. J., Liu, L., Ransohoff, R. M., et al. (2014). Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc. Natl. Acad. Sci. U.S.A. 111, 9953–9958. doi: 10.1073/pnas.1401155111
Ohno, N., Kidd, G. J., Mahad, D., Kiryu-Seo, S., Avishai, A., Komuro, H., et al. (2011). Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of Ranvier. J. Neurosci. 31, 7249–7258. doi: 10.1523/JNEUROSCI.0095-11.2011
Osellame, L. D., Singh, A. P., Stroud, D. A., Palmer, C. S., Stojanovski, D., Ramachandran, R., et al. (2016). Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell Sci. 129, 2170–2181. doi: 10.1242/jcs.185165
Osterloh, J. M., Yang, J., Rooney, T. M., Fox, A. N., Adalbert, R., Powell, E. H., et al. (2012). dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. doi: 10.1126/science.1223899
Overly, C. C., Rieff, H. I., and Hollenbeck, P. J. (1996). Organelle motility and metabolism in axons vs dendrites of cultured hippocampal neurons. J. Cell Sci. 109, 971–980. doi: 10.1242/jcs.109.5.971
Palay, S. L (1956). Synapses in the central nervous system. J. Biophys. Biochem. Cytol. 2, 193–202. doi: 10.1083/jcb.2.4.193
Paß, T., Wiesner, R. J., and Pla-Martín, D. (2021). Selective neuron vulnerability in common and rare diseases-mitochondria in the focus. Front. Mol. Biosci. 8, 676187. doi: 10.3389/fmolb.2021.676187
Pathak, D., Sepp, K. J., and Hollenbeck, P. J. (2010). Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria. J. Neurosci. 30, 8984–8992. doi: 10.1523/JNEUROSCI.1621-10.2010
Pitts, K. R., Yoon, Y., Krueger, E. W., and McNiven, M. A. (1999). The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Mol. Biol. Cell 10, 4403–4417. doi: 10.1091/mbc.10.12.4403
Plucińska, G., Paquet, D., Hruscha, A., Godinho, L., Haass, C., Schmid, B., et al. (2012). In vivo imaging of disease-related mitochondrial dynamics in a vertebrate model system. J. Neurosci. 32, 16203–16212. doi: 10.1523/JNEUROSCI.1327-12.2012
Pozo Devoto, V. M., Dimopoulos, N., Alloatti, M., Pardi, M. B., Saez, T. M., Otero, M. G., et al. (2017). αSynuclein control of mitochondrial homeostasis in human-derived neurons is disrupted by mutations associated with Parkinson's disease. Sci. Rep. 7, 5042. doi: 10.1038/s41598-017-05334-9
Pozo Devoto, V. M., Lacovich, V., Feole, M., Bhat, P., Chovan, J., Carna, M., et al. (2022). Unraveling axonal mechanisms of traumatic brain injury cold spring harbor laboratory. bioRxiv. doi: 10.1101/2022.03.30.486433
Prots, I., Veber, V., Brey, S., Campioni, S., Buder, K., Riek, R., et al. (2013). α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem. 288, 21742–21754. doi: 10.1074/jbc.M113.451815
Quintero, O. A., DiVito, M. M., Adikes, R. C., Kortan, M. B., Case, L. B., Lier, A. J., et al. (2009). Human Myo19 is a novel myosin that associates with mitochondria. Curr. Biol. 19, 2008–2013. doi: 10.1016/j.cub.2009.10.026
Ramsden, M., Henderson, Z., and Pearson, H. A. (2002). Modulation of Ca2+ channel currents in primary cultures of rat cortical neurones by amyloid beta protein (1-40) is dependent on solubility status. Brain Res. 956, 254–261. doi: 10.1016/S0006-8993(02)03547-3
Rawson, R. L., Yam, L., Weimer, R. M., Bend, E. G., Hartwieg, E., Horvitz, H. R., et al. (2014). Axons degenerate in the absence of mitochondria in C. elegans. Curr. Biol. 24, 760–765. doi: 10.1016/j.cub.2014.02.025
Readnower, R. D., Pandya, J. D., McEwen, M. L., Pauly, J. R., Springer, J. E., and Sullivan, P. G. (2011). Post-injury administration of the mitochondrial permeability transition pore inhibitor, NIM811, is neuroprotective and improves cognition after traumatic brain injury in rats. J. Neurotrauma 28, 1845–1853. doi: 10.1089/neu.2011.1755
Ribas, V. T., Koch, J. C., Michel, U., Bähr, M., and Lingor, P. (2017). Attenuation of axonal degeneration by calcium channel inhibitors improves retinal ganglion cell survival and regeneration after optic nerve crush. Mol. Neurobiol. 54, 72–86. doi: 10.1007/s12035-015-9676-2
Rosenberg, S. S., and Spitzer, N. C. (2011). Calcium signaling in neuronal development. Cold Spring Harb. Perspect. Biol. 3, a004259. doi: 10.1101/cshperspect.a004259
Sajic, M., Mastrolia, V., Lee, C. Y., Trigo, D., Sadeghian, M., Mosley, A. J., et al. (2013). Impulse conduction increases mitochondrial transport in adult mammalian peripheral nerves in vivo. PLoS Biol. 11, e1001754. doi: 10.1371/journal.pbio.1001754
Sanz-Blasco, S., Valero, R. A., Rodríguez-Crespo, I., Villalobos, C., and Núñez, L. (2008). Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE 3, e2718. doi: 10.1371/journal.pone.0002718
Saotome, M., Safiulina, D., Szabadkai, G., Das, S., Fransson, A., Aspenstrom, P., et al. (2008). Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. U.S.A. 105, 20728–20733. doi: 10.1073/pnas.0808953105
Saxton, W. M., and Hollenbeck, P. J. (2012). The axonal transport of mitochondria. J. Cell Sci. 125, 2095–2104. doi: 10.1242/jcs.053850
Scheib, J., and Höke, A. (2013). Advances in peripheral nerve regeneration. Nat. Rev. Neurol. 9, 668–676. doi: 10.1038/nrneurol.2013.227
Schon, E. A., and Przedborski, S. (2011). Mitochondria: the next (neurode)generation. Neuron 70, 1033–1053. doi: 10.1016/j.neuron.2011.06.003
Schrank, S., Barrington, N., and Stutzmann, G. E. (2020). Calcium-handling defects and neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 12, a035212. doi: 10.1101/cshperspect.a035212
Seager, R., Lee, L., Henley, J. M., and Wilkinson, K. A. (2020). Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signal 4, NS20200008. doi: 10.1042/NS20200008
Sgobio, C., Sun, L., Ding, J., Herms, J., Lovinger, D. M., and Cai, H. (2019). Unbalanced calcium channel activity underlies selective vulnerability of nigrostriatal dopaminergic terminals in parkinsonian mice. Sci. Rep. 9, 4857. doi: 10.1038/s41598-019-41091-7
Sheng, Z.-H., and Cai, Q. (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77–93. doi: 10.1038/nrn3156
Shepherd, G. M., and Harris, K. M. (1998). Three-dimensional structure and composition of CA3–>CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J. Neurosci. 18, 8300–8310. doi: 10.1523/JNEUROSCI.18-20-08300.1998
Shields, L. Y., Kim, H., Zhu, L., Haddad, D., Berthet, A., Pathak, D., et al. (2015). Dynamin-related protein 1 is required for normal mitochondrial bioenergetic and synaptic function in CA1 hippocampal neurons. Cell Death Dis. 6, e1725. doi: 10.1038/cddis.2015.94
Shigeoka, T., Jung, H., Jung, J., Turner-Bridger, B., Ohk, J., Lin, J. Q., et al. (2016). Dynamic axonal translation in developing and mature visual circuits. Cell 166, 181–192. doi: 10.1016/j.cell.2016.05.029
Shin, J. E., Miller, B. R., Babetto, E., Cho, Y., Sasaki, Y., Qayum, S., et al. (2012). SCG10 is a JNK target in the axonal degeneration pathway. Proc. Natl. Acad. Sci. U.S.A. 109, E3696–E3705. doi: 10.1073/pnas.1216204109
Smith, E. F., Shaw, P. J., and De Vos, K. J. (2019). The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 710, 132933. doi: 10.1016/j.neulet.2017.06.052
Smit-Rigter, L., Rajendran, R., Silva, C. A., Spierenburg, L., Groeneweg, F., Ruimschotel, E. M., et al. (2016). Mitochondrial dynamics in visual cortex are limited in vivo and not affected by axonal structural plasticity. Curr. Biol. 26, 2609–2616. doi: 10.1016/j.cub.2016.07.033
Song, Z., Ghochani, M., McCaffery, J. M., Frey, T. G., and Chan, D. C. (2009). Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 20, 3525–3532. doi: 10.1091/mbc.e09-03-0252
Spillane, M., Ketschek, A., Merianda, T. T., Twiss, J. L., and Gallo, G. (2013). Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Rep. 5, 1564–1575. doi: 10.1016/j.celrep.2013.11.022
Staal, J. A., Dickson, T. C., Gasperini, R., Liu, Y., Foa, L., and Vickers, J. C. (2010). Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury. J. Neurochem. 112, 1147–1155. doi: 10.1111/j.1471-4159.2009.06531.x
Stavsky, A., Stoler, O., Kostic, M., Katoshevsky, T., Assali, E. A., Savic, I., et al. (2021). Aberrant activity of mitochondrial NCLX is linked to impaired synaptic transmission and is associated with mental retardation. Commun. Biol. 4, 666. doi: 10.1038/s42003-021-02312-w
Stirling, D. P., Cummins, K., Wayne Chen, S. R., and Stys, P. (2014). Axoplasmic reticulum Ca(2+) release causes secondary degeneration of spinal axons. Ann. Neurol. 75, 220–229. doi: 10.1002/ana.24099
Stokin, G. B., Lillo, C., Falzone, T. L., Brusch, R. G., Rockenstein, E., Mount, S. L., et al. (2005). Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307, 1282–1288. doi: 10.1126/science.1105681
Stone, J. R., Singleton, R. H., and Povlishock, J. T. (2001). Intra-axonal neurofilament compaction does not evoke local axonal swelling in all traumatically injured axons. Exp. Neurol. 172, 320–331. doi: 10.1006/exnr.2001.7818
Stowers, R. S., Megeath, L. J., Górska-Andrzejak, J., Meinertzhagen, I. A., and Schwarz, T. L. (2002). Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36, 1063–1077. doi: 10.1016/S0896-6273(02)01094-2
Sudhof, T. C (2004). The synaptic vesicle cycle. Annu. Rev. Neurosci. 27, 509–547. doi: 10.1146/annurev.neuro.26.041002.131412
Tamada, H., Kiryu-Seo, S., Hosokawa, H., Ohta, K., Ishihara, N., Nomura, M., et al. (2017). Three-dimensional analysis of somatic mitochondrial dynamics in fission-deficient injured motor neurons using FIB/SEM. J. Comp. Neurol. 525, 2535–2548. doi: 10.1002/cne.24213
Tang-Schomer, M. D., Johnson, V. E., Baas, P. W., Stewart, W., and Smith, D. H. (2012). Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp. Neurol. 233, 364–372. doi: 10.1016/j.expneurol.2011.10.030
Tang-Schomer, M. D., Patel, A. R., Baas, P. W., and Smith, D. H. (2010). Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J. 24, 1401–1410. doi: 10.1096/fj.09-142844
Toda, T., Kadono, T., Hoshiai, M., Eguchi, Y., Nakazawa, S., Nakazawa, H., et al. (2007). Na+/H+ exchanger inhibitor cariporide attenuates the mitochondrial Ca2+ overload and PTP opening. Am. J. Physiol. Heart Circ. Physiol. 293, H3517–H3523. doi: 10.1152/ajpheart.00483.2006
Trevisan, T., Pendin, D., Montagna, A., Bova, S., Ghelli, A. M., and Daga, A. (2018). Manipulation of mitochondria dynamics reveals separate roles for form and function in mitochondria distribution. Cell Rep. 23, 1742–1753. doi: 10.1016/j.celrep.2018.04.017
Ueda, K., Shinohara, S., Yagami, T., Asakura, K., and Kawasaki, K. (1997). Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J. Neurochem. 68, 265–271. doi: 10.1046/j.1471-4159.1997.68010265.x
van Spronsen, M., Mikhaylova, M., Lipka, J., Schlager, M. A., Kuijpers, M., Wulf, P. S., et al. (2013). TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 77, 485–502. doi: 10.1016/j.neuron.2012.11.027
Vargas, M. E., Yamagishi, Y., Tessier-Lavigne, M., and Sagasti, A. (2015). Live imaging of calcium dynamics during axon degeneration reveals two functionally distinct phases of calcium influx. J. Neurosci. 35, 15026–15038. doi: 10.1523/JNEUROSCI.2484-15.2015
Verhoeven, K., Claeys, K. G., Züchner, S., Schröder, J. M., Weis, J., Ceuterick, C., et al. (2006). MFN2 mutation distribution and genotype/phenotype correlation in charcot-marie-tooth type 2. Brain 129, 2093–2102. doi: 10.1093/brain/awl126
Verstreken, P., Ly, C. V., Venken, K. J., Koh, T. W., Zhou, Y., and Bellen, H. J. (2005). Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47, 365–378. doi: 10.1016/j.neuron.2005.06.018
Villegas, R., Martinez, N. W., Lillo, J., Pihan, P., Hernandez, D., Twiss, J. L., et al. (2014). Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J. Neurosci. 34, 7179–7189. doi: 10.1523/JNEUROSCI.4784-13.2014
Wan, B., LaNoue, K. F., Cheung, J. Y., and Scaduto, R. C. (1989). Regulation of citric acid cycle by calcium. J. Biol. Chem. 264, 13430–13439. doi: 10.1016/S0021-9258(18)80015-1
Wang, B., Huang, M., Shang, D., Yan, X., Zhao, B., and Zhang, X. (2021). Mitochondrial behavior in axon degeneration and regeneration. Front. Aging Neurosci. 13, 650038. doi: 10.3389/fnagi.2021.650038
Wang, Q., Tian, J., Chen, H., Du, H., and Guo, L. (2019). Amyloid beta-mediated KIF5A deficiency disrupts anterograde axonal mitochondrial movement. Neurobiol. Dis. 127, 410–418. doi: 10.1016/j.nbd.2019.03.021
Wang, X., and Schwarz, T. L. (2009). The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. doi: 10.1016/j.cell.2008.11.046
Wang, X., Yan, M. H., Fujioka, H., Liu, J., Wilson-Delfosse, A., Chen, S. G., et al. (2012). LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 21, 1931–1944. doi: 10.1093/hmg/dds003
Weber, M. T., Arena, J. D., Xiao, R., Wolf, J. A., and Johnson, V. E. (2019). CLARITY reveals a more protracted temporal course of axon swelling and disconnection than previously described following traumatic brain injury. Brain Pathol. 29, 437–450. doi: 10.1111/bpa.12677
Wilson, L., Stewart, W., Dams-O'Connor, K., Diaz-Arrastia, R., Horton, L., Menon, D. K., et al. (2017). The chronic and evolving neurological consequences of traumatic brain injury. Lancet Neurol. 16, 813–825. doi: 10.1016/S1474-4422(17)30279-X
Witte, M. E., Bø, L., Rodenburg, R. J., Belien, J. A., Musters, R., Hazes, T., et al. (2009). Enhanced number and activity of mitochondria in multiple sclerosis lesions. J. Pathol. 219, 193–204. doi: 10.1002/path.2582
Wolf, J. A., Stys, P. K., Lusardi, T., Meaney, D., and Smith, D. H. (2001). Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J. Neurosci. 21, 1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001
Woolums, B. M., McCray, B. A., Sung, H., Tabuchi, M., Sullivan, J. M., Ruppell, K. T., et al. (2020). TRPV4 disrupts mitochondrial transport and causes axonal degeneration via a CaMKII-dependent elevation of intracellular Ca2. Nat. Commun. 11, 2679. doi: 10.1038/s41467-020-16411-5
Xiong, Y., Gu, Q., Peterson, P. L., Muizelaar, J. P., and Lee, C. P. (1997). Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J. Neurotrauma 14, 23–34. doi: 10.1089/neu.1997.14.23
Xu, Y., Chen, M., Hu, B., Huang, R., and Hu, B. (2017). In vivo imaging of mitochondrial transport in single-axon regeneration of zebrafish mauthner cells. Front. Cell. Neurosci. 11, 4. doi: 10.3389/fncel.2017.00004
Xu, Y. F., Gendron, T. F., Zhang, Y. J., Lin, W. L., D'Alton, S., Sheng, H., et al. (2010). Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 30, 10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010
Yang, Y., Ouyang, Y., Yang, L., Beal, M. F., McQuibban, A., Vogel, H., et al. (2008). Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl. Acad. Sci. U.S.A. 105, 7070–7075. doi: 10.1073/pnas.0711845105
Yi, M., Weaver, D., and Hajnóczky, G. (2004). Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J. Cell Biol. 167, 661–672. doi: 10.1083/jcb.200406038
Yuen, T. J., Browne, K. D., Iwata, A., and Smith, D. H. (2009). Sodium channelopathy induced by mild axonal trauma worsens outcome after a repeat injury. J. Neurosci. Res. 87, 3620–3625. doi: 10.1002/jnr.22161
Zambonin, J. L., Zhao, C., Ohno, N., Campbell, G. R., Engeham, S., Ziabreva, I., et al. (2011). Increased mitochondrial content in remyelinated axons: implications for multiple sclerosis. Brain 134, 1901–1913. doi: 10.1093/brain/awr110
Zhang, L., Trushin, S., Christensen, T. A., Tripathi, U., Hong, C., Geroux, R. E., et al. (2018). Differential effect of amyloid beta peptides on mitochondrial axonal trafficking depends on their state of aggregation and binding to the plasma membrane. Neurobiol. Dis. 114, 1–16. doi: 10.1016/j.nbd.2018.02.003
Zhao, Y., Song, E., Wang, W., Hsieh, C. H., Wang, X., Feng, W., et al. (2021). Metaxins are core components of mitochondrial transport adaptor complexes. Nat. Commun. 12, 83. doi: 10.1038/s41467-020-20346-2
Zhou, B., Yu, P., Lin, M. Y., Sun, T., Chen, Y., and Sheng, Z. H. (2016). Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J. Cell Biol. 214, 103–119. doi: 10.1083/jcb.201605101
Zhou, Y., Carmona, S., Muhammad, A. K. M. G., Bell, S., Landeros, J., Vazquez, M., et al. (2019). Restoring mitofusin balance prevents axonal degeneration in a charcot-marie-tooth type 2A model. J. Clin. Invest. 129, 1756–1771. doi: 10.1172/JCI124194
Zilocchi, M., Finzi, G., Lualdi, M., Sessa, F., Fasano, M., and Alberio, T. (2018). Mitochondrial alterations in Parkinson's disease human samples and cellular models. Neurochem. Int. 118, 61–72. doi: 10.1016/j.neuint.2018.04.013
Keywords: mitochondria, axonal degeneration, traumatic brain injury, mitochondrial dynamics, mitochondrial transport, calcium homeostasis
Citation: Pozo Devoto VM, Onyango IG and Stokin GB (2022) Mitochondrial behavior when things go wrong in the axon. Front. Cell. Neurosci. 16:959598. doi: 10.3389/fncel.2022.959598
Received: 07 June 2022; Accepted: 18 July 2022;
Published: 05 August 2022.
Edited by:
Shuxin Li, Temple University, United StatesReviewed by:
Regina Armstrong, Uniformed Services University of the Health Sciences, United StatesCopyright © 2022 Pozo Devoto, Onyango and Stokin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gorazd B. Stokin, Z2JzdG9raW5AYWx1bW5pLnVjc2QuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.