 Thomas J. Waller
Thomas J. Waller Catherine A. Collins
Catherine A. Collins- 1Department of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI, United States
- 2Department of Neurosciences, Case Western Reserve University, Cleveland, OH, United States
Axons are considered to be particularly vulnerable components of the nervous system; impairments to a neuron’s axon leads to an effective silencing of a neuron’s ability to communicate with other cells. Nervous systems have therefore evolved plasticity mechanisms for adapting to axonal damage. These include acute mechanisms that promote the degeneration and clearance of damaged axons and, in some cases, the initiation of new axonal growth and synapse formation to rebuild lost connections. Here we review how these diverse processes are influenced by the therapeutically targetable enzyme SARM1. SARM1 catalyzes the breakdown of NAD+, which, when unmitigated, can lead to rundown of this essential metabolite and axonal degeneration. SARM1’s enzymatic activity also triggers the activation of downstream signaling pathways, which manifest numerous functions for SARM1 in development, innate immunity and responses to injury. Here we will consider the multiple intersections between SARM1 and the injury signaling pathways that coordinate cellular adaptations to nervous system damage.
Introduction
Axons enable neural communication over great distances in the nervous system. Tuned to the vulnerability of long axons, decades of studies (including work by Ramon y Cajal and Augustus Waller in prior centuries) have showcased the plasticity of the nervous system through the varied responses made by individual cells adapting to nervous system damage. These responses include the ability to regrow damaged axons, to repair lost connections and/or sprout new axonal branches. Also critical for nervous system plasticity is the degeneration and clearance of the distal axon “stump” which has lost connectivity with the cell body.
Unless the refusion of axons can occur [which occurs robustly in C. elegans (Neumann et al., 2011)], the distal stump has no clear function in its original circuit. Despite this, continued existence of the severed axon may be enabled by localized protein synthesis, energy supply from axonally localized mitochondria (and other organelles), and support from local glia.
While it is logical that the stump should ultimately degenerate from the lack of new organelles or newly expressed proteins transported from the cell body, there is increasing evidence that the mechanism by which most axons degenerate is not simply a passive rundown of cellular function. Rather, axons degenerate through an active and regulated “self-destruction” pathway akin to apoptosis, but involving distinct cellular machinery. A key component of this machinery is the sterile alpha TIR motif containing protein SARM1, whose enzymatic activity drives the loss of NAD+, a central electron carrier. This enzymatic activity is essential for SARM1’s ability to drive axonal degeneration. SARM1 has also been shown to function as a regulator of intracellular signaling pathways, which can intersect in multiple ways to drive degeneration. This review focuses on these points of intersection, which help to inform a global, albeit complex view, of how this therapeutically targetable enzyme works to coordinate responses to nervous system damage in multiple cell types.
SARM1-regulated signaling mediates diverse functions, including responses to nervous system injury
First we review some of the multiple known functions of SARM1 in different cell types. SARM1 is broadly expressed in many different tissues, and within the nervous system it is expressed within glial and immune cells as well as neurons (Hu et al., 2021). Prior to the discovery of SARM1’s best-known role in axonal degeneration, studies in multiple model organisms have identified SARM1 as an upstream regulator of MAP (mitogen activated kinase) Kinase signaling (Liberati et al., 2004; Chuang and Bargmann, 2005; Kim et al., 2007; Chen et al., 2011). Its homolog in C. elegans was first discovered for its role in regulating left-right asymmetric cell fate choices between two synaptically connected olfactory neurons (Chuang and Bargmann, 2005; Inoue et al., 2013; Gerdts et al., 2016). This choice is driven by MAP Kinase signaling downstream of the Apoptosis signal-regulating kinase (ASK1), whose activation and localization at synapses requires SARM1’s homolog TIR-1 (Chuang and Bargmann, 2005; Inoue et al., 2013; Gerdts et al., 2016). Additional roles for SARM1-gated MAP Kinase signaling include regulation of neuronal cytoskeleton and neurite structure (Kim et al., 2007; McLaughlin et al., 2016; Izadifar et al., 2021), and the clearance of apoptotic cells by glia (McLaughlin et al., 2019). Both SARM1 and ASK1 regulate Toll-like receptor (TLR) signaling, a key signaling arm of innate immunity (Liu et al., 2014; Matsuzawa, 2017), and act via JNK to promote “forgetting” in C. elegans olfactory neurons (Inoue et al., 2013). Loss of SARM1 also leads to impairments in synaptic plasticity (Lin et al., 2014) and loss of parvalbumin neurons associated with autism-like behaviors (Xiang et al., 2022).
Several of SARM1’s signaling functions are engaged following axonal injury. These functions (cartooned in Figure 1) are particularly interesting to consider as they tie together multiple responses in different cell types to axonal damage. First, SARM1 functions within injured neurons (colored violet in Figure 1) to regulate several cell autonomous responses to axonal damage. These include SARM1’s most well-known function in promoting Wallerian degeneration of the distal axon stump (Osterloh et al., 2012). Another cell autonomous response is the triggering of cytokine production and release from axotomized DRG neurons following sciatic nerve injury, which is mediated by Jun N-terminal kinase signaling (an arm of MAP kinase signaling) (Wang et al., 2018). This places Sarm1 upstream of an important inflammatory response to nerve damage, relevant for pain and also for the ability of axons to regenerate (Jessen and Mirsky, 2016; Sommer et al., 2018). In contrast to the degeneration of axons severed from the cell body, this response involves changes in gene expression, hence the cell body. A third cell autonomous injury response gated by Sarm1 was recently described in axotomized C. elegans motor axons, where Sarm1 inhibits the ability of injured axons to initiate regenerative axon growth (Julian and Byrne, 2020). This response requires the ASK1 kinase and a p38-mediated arm of MAP Kinase signaling.
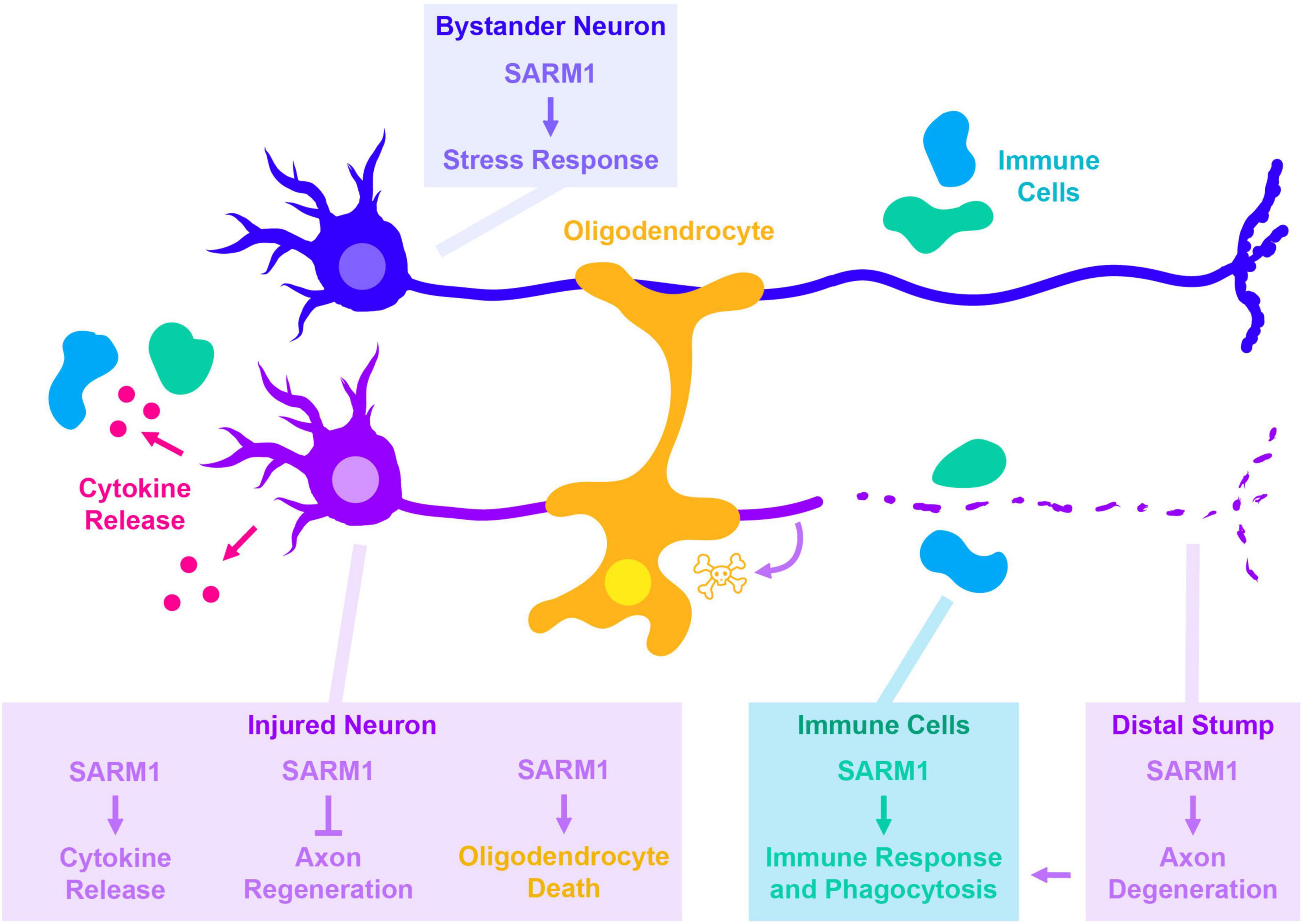
Figure 1. Cell autonomous and non-cell autonomous roles for SARM1 in responses to axonal injury. Each box indicates the cell type SARM1 is acting within, with text color indicating the cell type being impacted by SARM1’s function (same colors indicate cell autonomous, different colors indicate non-autonomous). SARM1’s cell autonomous roles in injured neurons are shown in magenta. In addition to its most well-known for its role in promoting degeneration of the distal stump, SARM1 is also required for additional responses made by injured neurons. These include the JNK- and c-Jun-dependent release of cytokines (Wang et al., 2018), the ability of neurons to regenerate in C. elegans (Julian and Byrne, 2020), and in promoting death of oligodendrocytes in a glaucoma model (Ko et al., 2020). SARM1 also functions within uninjured cells that participate in responses to injury, including uninjured “bystander” neurons (dark blue blue) (Hsu et al., 2021), and immune cells that react to the damage (light blue) (McLaughlin et al., 2019).
Events downstream of SARM1 activation also trigger responses in other cells. For example, Wallerian degeneration leads to the generation of axonal debris, which contributes to the initiation and recruitment of immune response. This immune response is hypothesized to be important for axonal regeneration in the peripheral nervous system; supporting this idea, multiple studies have noted that axonal regeneration in the peripheral nervous system is impaired when Wallerian degeneration is inhibited (Lunn et al., 1989; Bisby and Chen, 1990; Brown et al., 1994, 1991; McKerracher et al., 1994; Mukhopadhyay et al., 1994; Schäfer et al., 1996; Rotshenker, 2011). Another example of non-cell autonomous roles for SARM1 was recently shown in a model of glaucoma, where necroptosis signaling leads to the activation of SARM1 (Ko et al., 2020). In this model, knockout of SARM1 not only rescules axonal degeneration, but also the death of neighboring oligodendrocytes, which may be a consequence of the axonal degeneration.
In addition to the above roles in injured neurons, SARM1-regulated signaling also acts in other non-injured cells to regulate responses to injury (Figure 1). Hsu et al. (2021) documented a role for Drosophila SARM1 in slowing axonal transport in non-injured “bystander” axons. Signaling downstream of SARM1 also regulates the phagocytic capacity of glial cells during Drosophila development (McLaughlin et al., 2019), which may potentially be relevant for their ability to clear debris following injury.
The SARM1 enzyme
While TIR domains typically mediate protein-protein interactions with other TIR domains to scaffold intramolecular complexes, SARM1’s TIR domains have potent enzymatic activity (Gerdts et al., 2015). In its activated form, the SARM1 enzyme cleaves Nicotinamide Adenine Dinucleotide (NAD+) into Nicotinamide Mononucleotide (NAM) and cyclic (and non-cyclic) ADP-Ribose (cADPR) (Essuman et al., 2017). The rundown of NAD+, a key electron carrier for cellular metabolism, is expected to lead to metabolic catastrophe due to failure of NAD-dependent oxidative reactions and eventual ATP synthesis (Zhao et al., 2019; Figley et al., 2021). SARM1 is required and responsible for axon degeneration under a variety of stress and disease conditions (Coleman and Höke, 2020; Figley and DiAntonio, 2020; Sarkar et al., 2021), and also serves as an executioner of a form of cell death named “sarmoptosis” (reviewed in Fricker et al., 2018). SARM1-mediated neuron death can be triggered by mitochondrial toxins (Summers et al., 2014), and TLR signaling (Mukherjee et al., 2015, 2013). It has also been documented in non-neuronal cells including as a form of programmed cell death to terminate activated T-cells (Panneerselvam et al., 2013).
In addition to NAD+ loss, the products of SARM1’s enzymatic reaction may also carry out additional functions, either to facilitate degeneration or additional roles in cellular signaling. Cyclic ADPR, a robustly measurable product of SARM1 enzymatic activity, is a known regulator of the ER Ryanodine receptor and TRPM2 calcium channels (Galione and Ruas, 2005; Galione and Chuang, 2020). This function makes cADPR a prime candidate to mediate intracellular signaling, as well as calcium influx. Of note, the final stages of axonal degeneration are closely accompanied by an increase in intracellular calcium (Geffen and Hughes, 1972; Schlaepfer and Bunge, 1973; Schlaepfer, 1974; Schlaepfer and Hasler, 1979; Mishra et al., 2013; Ko et al., 2021). While some findings did not detect a role for cADPR in Wallerian degeneration of injured axons in vitro (Sasaki et al., 2020), recent work has shown a role for cADPR in triggering calcium flux and degeneration in a model of chemotherapy-induced peripheral neuropathy (Li et al., 2021). Hence cADPR may also contribute to the degenerative outcome of SARM1 activation.
Beyond the traditional breakdown of NAD+ into NAM and cADPR, recent work has shown that the SARM1 enzyme can carry out multiple base exchange reactions (Angeletti et al., 2022). This includes base exchanges between NAD (and NADP) and the pyrimidines 3-acetylpyridine, vacour, and nicotinic acid. In addition to being a point of vulnerability to neurotoxins (Loreto et al., 2021; Wu et al., 2021), the wide range of reactants and products may enable SARM1 to influence multiple signaling pathways and cellular responses.
Is SARM1’s enzymatic activity an essential property of its signaling mechanism? Structure-functions studies indicate that its TIR domains are essential (Chuang and Bargmann, 2005; Gerdts et al., 2015; Herrmann et al., 2022), and recent studies in Drosophila found that dSarm’s enzymatic activity tracks with its signaling functions (Brace et al., 2022; Herrmann et al., 2022). However, Herrmann et al. (2022) also found some differences: axonal degeneration requires additional domains of SARM1 that are dispensable for its signaling functions. Hence Sarm1 may regulate degeneration and signaling through distinct biochemical states.
Regulation of SARM1
Since unmitigated SARM1 enzymatic activity can drive cellular degeneration, SARM1’s enzymatic activity is tightly controlled in healthy cells. The catalytic domain of the enzyme is comprised of an octomeric ring formed by the oligomerization of TIR domains from multiple subunits (Bratkowski et al., 2020; Jiang et al., 2020; Sporny et al., 2020). Under normal conditions, the TIR domains are held in an inactive state through intramolecular interactions with SARM1’s ARM domain. The ARM domain contains an allosteric binding site for NAD + and NMN, which either stabilize (for NAD +) or inhibit (for NMN) the inhibitory interaction. Strikingly, the allosteric regulation provides a compelling explanation for the long-held observation that axonal degeneration and SARM1 activation are strongly inhibited by the NAD + biosynthesis enzyme NMNAT (NAM Adenylyl Transferase). NMNAT can regulate SARM1 through its enzymatic activity: it consumes SARM1’s activator NMN to produce NAD +, which inhibits SARM1.
By inhibiting SARM1 activation, NMNAT enzymes are critical “survival” factors for cells. The levels and localization of the cytoplasmic isoform, NMNAT2, are highly regulated in neurons. Multiple mechanisms are known to regulate the stability, turnover and/or localization of NMNAT enzymes, and some of these are also linked to or signaling pathways that become activated by axonal damage (Figure 2). Most notably, this includes JNK signaling, which regulates NMNAT levels (Milde et al., 2013; Walker et al., 2017), and evolutionarily conserved E3 ubiquitin ligase, Phr1 (Xiong et al., 2012; Babetto et al., 2013).
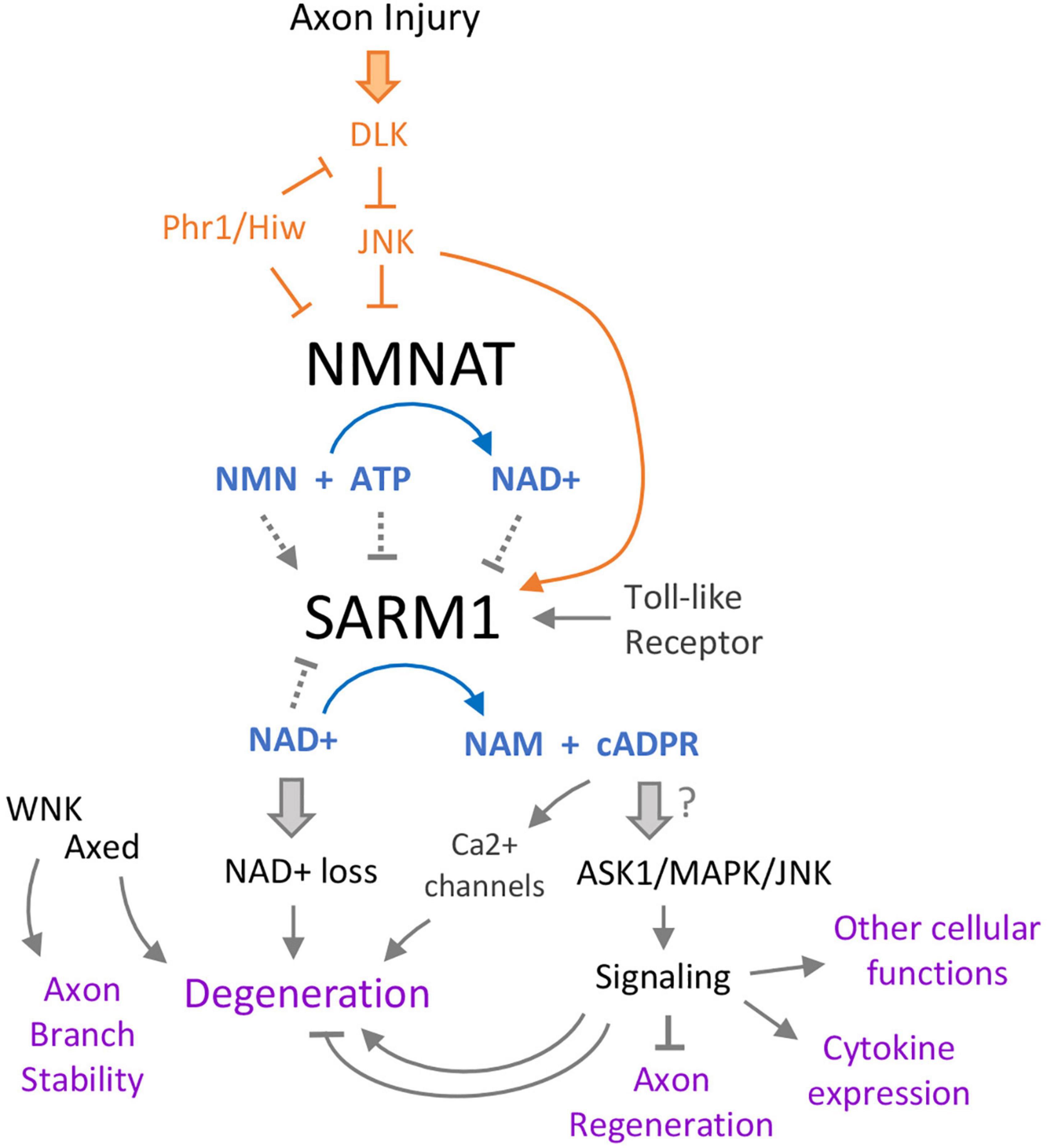
Figure 2. Intertwining relationships between SARM1 and axonal damage signaling. Multiple pathways converge on SARM1, which itself has several effects. Following axon injury, the Duel-leucine kinase (DLK) initiates a kinase cascade that both promotes SARM1 activation and turnover of the NAD+ biosynthetic enzyme NMNAT. Loss of NMNAT further promotes activation of SARM1 via a shift in the ratio of NAD+ to its precursor NMN, both of which are allosteric regulators of SARM1. SARM1’s catalytic state is therefore potently tuned by the function of the NMNAT, which is a critical protective factor in axons and neurons (Gilley and Coleman, 2010). This regulation also enables a feed-forward mechanism of SARM1 activation in injured axons and cells that lack NMNAT function, since once SARM1 is activated it can promote its own further activation by lowering NAD+ levels. Downstream of SARM1, loss of NAD+ leads to axon breakdown via an unclear mechanism, but is thought to be tied to rapid ATP loss and catastrophic metabolic rundown. One of SARM1’s products, cyclic ADP-ribose (cADPR) has increasingly gained prominence in the model of SARM1’s signaling. Acting on multiple calcium channels, cADPR promotes degeneration via Ca2+ flux in the axon. There have also been links between this metabolite and SARM1’s protein signaling activities, which act through several MAP kinases to promote axon regeneration (from the axon segment connected to the cell body), cytokine expression, neurite development, and other functions.
It is expected that similarly to NMNAT, there should be additional mechanisms that directly regulate SARM1. SARM1 is phosphorylated, which has been shown to stimulate its NAD + cleavage activity (Murata et al., 2018; Xue et al., 2021). Recent work suggests that SARM1’s activation state tracks with a phase transition; in C. elegans intestinal epithelial cells, fluorescently labeled SARM1 ortholog (TIR-1) could be observed to coalesce into visible puncta during conditions of stress that activate its downstream signaling (Loring et al., 2021). The phase transition and activation of downstream signaling can be triggered in genetic conditions that result in cholesterol scarcity, suggesting a cellular mechanism for how conditions of cellular stress can lead to SARM1 activation (Peterson et al., 2022).
SARM1 activation also occurs in additional conditions of high translational relevance, which include treatment with chemotherapy agents that induce neuropathy (Bosanac et al., 2021), and TNF-alpha triggered inflammation in an inflammatory model of glaucoma (Ko et al., 2020). Importantly, SARM1 inhibition rescues axon function and slows both axon degeneration and cell death. In the Ko et al. (2020) study, the induction of necroptosis signaling via Mixed Lineage Kinase domain-Like pseudokinase (MLKL) was shown to be responsible for SARM1 activation, and subsequent SARM1-mediated axonal degeneration. This SARM1 activation can be rescued by inhibiting MAP Kinase/JNK signaling (Ko et al., 2020), which, as discussed above, can regulate the levels NMNAT2 (Walker et al., 2017). Therefore SARM1 activation during necroptosis signaling may converge with its regulation by NAD+ /NMN. To summarize, we know thus far of multiple mechanisms that control SARM1’s enzymatic activity, but most converge on NMNAT, and MAP Kinase signaling pathways activated by axonal damage.
Axonal damage signaling
Axonal damage triggers the activation of multiple signaling pathways within the damaged neurons, which influence cellular responses ranging from neuronal death to the initiation of new axonal growth for facilitating repair and/or the ability to form alternative synaptic connections. Stress-associated signaling via the dileucine zipper kinase DLK and downstream Jun N-terminal Kinases (JNKs) are known to be activated from multiple aspects of axonal injury response (Asghari Adib et al., 2018; Jin and Zheng, 2019). These kinases have been observed appearing within damaged axons at short time periods after nerve injury (Cavalli et al., 2005; Lindwall and Kanje, 2005; Yang et al., 2015).
Multiple observations have documented an intimate web of interactions between axonal damage signaling and axonal degeneration machinery (Figure 2). First, several studies have pointed to roles for JNK signaling in the execution of distal stump degeneration. Acute application of JNK inhibitors to axomized DRG neurons in culture concomitant with injury is capable of delaying the onset of degeneration (Miller et al), suggesting a local and acute role for JNK in axotomized axons. One pro-degenerative role for JNK is promoting the turnover of NMNAT2, as discussed above (Walker et al., 2017). In addition, all 3 JNK kinase isoforms are capable of phosphorylating SARM1, and phosphorylation of SARM1 activates its enzymatic activity and subsequent neurodegeneration (Murata et al., 2018; Xue et al., 2021). While the above studies suggest upstream regulatory roles for JNK signaling in driving the activation of SARM1, other studies have suggested a converse relationship where JNK signaling activation in damaged axons may be triggered downstream of SARM1 activation (Yang et al., 2015; Wang et al., 2018).
It is also plausible that signaling gated by SARM1 acts together with its enzymatic activity to drive degeneration. One study has posited a role for JNK downstream of SARM1, since genetic disruption of all 3 JNK isoforms (or both MKK4/7) could inhibit degeneration caused by ectopic activation of SARM1’s TIR domains (Yang et al., 2015). The dual upstream and downstream relationships of JNK signaling with SARM1 (and vice versa) suggests the possibility that they cycle together in a feed-forward relationship, acting to both promote axonal degeneration and detect the presence, likelihood, or possibility of axonal degeneration following SARM1 activation. These relationships suggest intimate links between axonal damage signaling and SARM1 that we have yet to understand on a molecular level (Figley et al., 2021; Waller and Collins, 2021).
One more set of twists in the intertwined relationship of JNK signaling with SARM1 come from observations in invertebrates that JNK signaling activation following axonal damage can have protective functions in neurons. This includes the inhibition of degeneration in both axons and dendrites (Xiong and Collins, 2012; Hao et al., 2019), which contrasts with the pro-degenerative role described above for JNK in mammalian axons. The mechanism for this protective role is not yet clear. However, it appears to require the transcription factor Fos and may therefore be mediated by downstream changes in gene expression. This could possibly promote multiple changes to axonal metabolic mechanisms or cytoskeleton that influence the propensity of axons to degenerate. While it is plausible that the Fos-dependent pathway could influence the expression level or localization of NMNAT enzyme isoforms(s) (Chen et al., 2016), a recent study has described an NMNAT-independent form of JNK-mediated protective signaling (Hao et al., 2019).
Potential new players in SARM1 signaling mechanisms
Recent work has brought to light some additional players in NMNAT-SARM1 signaling. One intriguing player is Axed, short for “Axundead,” which was discovered in a forward genetic screen in Drosophila (Neukomm et al., 2017). Similar to mutants in dSarm, neurons lacking axed function fail to undergo Wallerian degeneration. Remarkably, axed mutants inhibit axonal degeneration even in conditions that are expected to result in unmitigated NADase enzymatic activity by dSarm. This includes deletion of NMNAT, which normally leads to axonal and neuronal degeneration, but is rescued by mutation of Axed. From these striking genetic data it is proposed that Axed promotes axonal degeneration downstream of SARM1’s enzymatic activity.
How Axed may do this remains mysterious. It contains a BTB domain, which could participate in scaffolding signaling interactions, but has no known enzymatic activity of its own. Axed protein is abundantly localized near presynaptic terminals in Drosophila motoneurons. There are four orthologs of Axed in mammals: BTBD1, BTBD2, BTBD3, and BTBD6 (Neukomm et al., 2017). Potential functional overlap or redundancy has made it hard to determine whether any of these proteins functionally analogously to Axed in mammals.
Another intriguing new set of players are the Lysine Deficient Protein WNK (abbreviated for “with-no-lysine K”) kinases. WNK kinases are broadly expressed, best known for roles in the regulation of ion transport and blood pressure regulation (Murthy et al., 2017). However a recent study, also resulting from a genetic screen in Drosophila, identified developmental roles for dWnk in stabilizing axonal branches, in addition to regulating branching during neurite outgrowth (Izadifar et al., 2021). Knockdown of dWnk leads to both axon branching defects and early onset degeneration. This degeneration is dependent on Axed and is rescued by dNMNAT activity (similarly to SARM1-mediated Wallerian degeneration). In axon maintenance, dWnk promotes dNMNAT activity while negatively regulating both Axed and dSarm based on genetic epistasis data. Therefore WNK appears to be an important regulator of at least two functions for signaling surrounding SARM1. Future study of the cellular mechanism of its interactions with SARM1 may provide important insights into SARM1’s functions in signaling as well as degeneration.
Conclusion
The SARM1 NADase enzyme is an attractive druggable target for intervening neuropathies and a variety of neurodegenerative diseases. SARM1’s enzymatic activity drives the degeneration of axons and, in addition, regulates signaling pathways that influence development, inflammation, and multiple responses to axonal injury. Further consideration of SARM1 as an enzymatic target requires a deeper understanding of SARM1’s roles in development and the crosstalk of SARM1’s multiple functions in the context of nervous system injury.
Author contributions
TW and CC worked in collaboration to carry out the conception and writing and revising of this review. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institutes of Health (RO1 NS069844).
Acknowledgments
We thank the reviewers, Heather Broihier, and Dan Yellajoshyula for helpful comments to improve the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Angeletti, C., Amici, A., Gilley, J., Loreto, A., Trapanotto, A. G., Antoniou, C., et al. (2022). SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated by multiple physiologically relevant NAD metabolites. iScience 25:103812. doi: 10.1016/j.isci.2022.103812
Asghari Adib, E., Smithson, L. J., and Collins, C. A. (2018). An axonal stress response pathway: Degenerative and regenerative signaling by DLK. Curr. Opin. Neurobiol. 53, 110–119. doi: 10.1016/j.conb.2018.07.002
Babetto, E., Beirowski, B., Russler, E. V., Milbrandt, J., and DiAntonio, A. (2013). The Phr1 Ubiquitin Ligase Promotes Injury-Induced Axon Self-Destruction. Cell Rep. 3, 1422–1429. doi: 10.1016/j.celrep.2013.04.013
Bisby, M. A., and Chen, S. (1990). Delayed wallerian degeneration in sciatic nerves of C57BL/Ola mice is associated with impaired regeneration of sensory axons. Brain Res. 530, 117–120.
Bosanac, T., Hughes, R. O., Engber, T., Devraj, R., Brearley, A., Danker, K., et al. (2021). Pharmacological SARM1 inhibition protects axon structure and function in paclitaxel-induced peripheral neuropathy. Brain 144, 3226–3238. doi: 10.1093/brain/awab184
Brace, E. J., Essuman, K., Mao, X., Palucki, J., Sasaki, Y., Milbrandt, J., et al. (2022). Distinct developmental and degenerative functions of SARM1 require NAD+ hydrolase activity. PLoS Genet. 18:e1010246. doi: 10.1371/journal.pgen.1010246
Bratkowski, M., Xie, T., Thayer, D. A., Lad, S., Mathur, P., Yang, Y.-S., et al. (2020). Structural and Mechanistic Regulation of the Pro-degenerative NAD Hydrolase SARM1. Cell Rep. 32:107999. doi: 10.1016/j.celrep.2020.107999
Brown, M. C., Perry, V. H., Hunt, S. P., and Lapper, S. R. (1994). Further studies on motor and sensory nerve regeneration in mice with delayed Wallerian degeneration. Eur. J. Neurosci. 6, 420–428.
Brown, M. C., Perry, V. H., Lunn, E. R., Gordon, S., and Heumann, R. (1991). Macrophage dependence of peripheral sensory nerve regeneration: Possible involvement of nerve growth factor. Neuron 6, 359–370. doi: 10.1016/0896-6273(91)90245-u
Cavalli, V., Kujala, P., Klumperman, J., and Goldstein, L. S. B. (2005). Sunday Driver links axonal transport to damage signaling. J. Cell Biol. 168, 775–787. doi: 10.1083/jcb.200410136
Chen, C.-Y., Lin, C.-W., Chang, C.-Y., Jiang, S.-T., and Hsueh, Y.-P. (2011). Sarm1, a negative regulator of innate immunity, interacts with syndecan-2 and regulates neuronal morphology. J. Cell Biol. 193, 769–784. doi: 10.1083/jcb.201008050
Chen, L., Nye, D. M., Stone, M. C., Weiner, A. T., Gheres, K. W., Xiong, X., et al. (2016). Mitochondria and Caspases Tune Nmnat-Mediated Stabilization to Promote Axon Regeneration. PLoS Genet. 12:e1006503. doi: 10.1371/journal.pgen.1006503
Chuang, C.-F., and Bargmann, C. I. (2005). A Toll-interleukin 1 repeat protein at the synapse specifies asymmetric odorant receptor expression via ASK1 MAPKKK signaling. Genes Dev. 19, 270–281. doi: 10.1101/gad.1276505
Coleman, M. P., and Höke, A. (2020). Programmed axon degeneration: From mouse to mechanism to medicine. Nat. Rev. Neurosci. 21, 183–196.
Essuman, K., Summers, D. W., Sasaki, Y., Mao, X., DiAntonio, A., and Milbrandt, J. (2017). The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 93, 1334–1343.e5. doi: 10.1016/j.neuron.2017.02.022
Figley, M. D., and DiAntonio, A. (2020). The SARM1 axon degeneration pathway: Control of the NAD+ metabolome regulates axon survival in health and disease. Curr. Opin. Neurobiol. 63, 59–66. doi: 10.1016/j.conb.2020.02.012
Figley, M. D., Gu, W., Nanson, J. D., Shi, Y., Sasaki, Y., Cunnea, K., et al. (2021). SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration. Neuron 109, 1118–1136.e11. doi: 10.1016/j.neuron.2021.02.009
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal Cell Death. Physiol. Rev. 98, 813–880.
Galione, A., and Chuang, K.-T. (2020). Pyridine Nucleotide Metabolites and Calcium Release from Intracellular Stores. Adv. Exp. Med. Biol. 1131, 371–394.
Geffen, L. B., and Hughes, C. C. (1972). Degeneration of sympathetic nerves in vitro and development of smooth muscle supersensitivity to noradrenaline. J. Physiol. 221, 71–84. doi: 10.1113/jphysiol.1972.sp009739
Gerdts, J., Brace, E. J., Sasaki, Y., DiAntonio, A., and Milbrandt, J. (2015). SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 348, 453–457. doi: 10.1126/science.1258366
Gerdts, J., Summers, D. W., Milbrandt, J., and DiAntonio, A. (2016). Axon Self-Destruction: New Links among SARM1, MAPKs, and NAD+ Metabolism. Neuron 89, 449–460. doi: 10.1016/j.neuron.2015.12.023
Gilley, J., and Coleman, M. P. (2010). Endogenous nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 8:e1000300. doi: 10.1371/journal.pbio.1000300
Hao, Y., Waller, T. J., Nye, D. M., Li, J., Zhang, Y., Hume, R. I., et al. (2019). Degeneration of injured axons and dendrites requires restraint of a protective JNK signaling pathway by the transmembrane protein Raw. J. Neurosci. 39, 8457–8470. doi: 10.1523/JNEUROSCI.0016-19.2019
Herrmann, K. A., Liu, Y., Llobet-Rosell, A., Mc Laughlin, C. N., Neukomm, L. J., Coutinho-Budd, J. C., et al. (2022). Divergent signaling requirements of dSARM in injury-induced degeneration and developmental glial phagocytosis. PLoS Genet. 18:e1010257. doi: 10.1371/journal.pgen.1010257
Hsu, J.-M., Kang, Y., Corty, M. M., Mathieson, D., Peters, O. M., and Freeman, M. R. (2021). Injury-Induced Inhibition of Bystander Neurons Requires dSarm and Signaling from Glia. Neuron 109, 473–487.e5. doi: 10.1016/j.neuron.2020.11.012
Hu, Y., Tattikota, S. G., Liu, Y., Comjean, A., Gao, Y., Forman, C., et al. (2021). DRscDB: A single-cell RNA-seq resource for data mining and data comparison across species. Comput. Struct. Biotechnol. J. 19, 2018–2026. doi: 10.1016/j.csbj.2021.04.021
Inoue, A., Sawatari, E., Hisamoto, N., Kitazono, T., Teramoto, T., Fujiwara, M., et al. (2013). Forgetting in C. elegans is accelerated by neuronal communication via the TIR-1/JNK-1 pathway. Cell Rep. 3, 808–819. doi: 10.1016/j.celrep.2013.02.019
Izadifar, A., Courchet, J., Virga, D. M., Verreet, T., Hamilton, S., Ayaz, D., et al. (2021). Axon morphogenesis and maintenance require an evolutionary conserved safeguard function of wnk kinases antagonizing sarm and axed. Neuron 109, 2864–2883.e8. doi: 10.1016/j.neuron.2021.07.006
Jessen, K. R., and Mirsky, R. (2016). The repair Schwann cell and its function in regenerating nerves. J. Physiol. 594, 3521–3531.
Jiang, Y., Liu, T., Lee, C.-H., Chang, Q., Yang, J., and Zhang, Z. (2020). The NAD+-mediated self-inhibition mechanism of pro-neurodegenerative SARM1. Nature 588, 658–663. doi: 10.1038/s41586-020-2862-z
Jin, Y., and Zheng, B. (2019). Multitasking: Dual Leucine Zipper-Bearing Kinases in Neuronal Development and Stress Management. Annu. Rev. Cell Dev. Biol. 35, 501–521. doi: 10.1146/annurev-cellbio-100617-062644
Julian, V., and Byrne, A. B. (2020). TIR-1/SARM1 Inhibits Axon Regeneration. bioRxiv [Preprint]. doi: 10.1101/2020.06.23.165852
Kim, Y., Zhou, P., Qian, L., Chuang, J.-Z., Lee, J., Li, C., et al. (2007). MyD88-5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. J. Exp. Med. 204, 2063–2074. doi: 10.1084/jem.20070868
Ko, K. W., Devault, L., Sasaki, Y., Milbrandt, J., and DiAntonio, A. (2021). Live imaging reveals the cellular events downstream of SARM1 activation. Elife 10:e71148. doi: 10.7554/eLife.71148
Ko, K. W., Milbrandt, J., and DiAntonio, A. (2020). SARM1 acts downstream of neuroinflammatory and necroptotic signaling to induce axon degeneration. J. Cell Biol. 219:e201912047. doi: 10.1083/jcb.201912047
Li, Y., Pazyra-Murphy, M. F., Avizonis, D., de Sa Tavares Russo, M., Tang, S., Bergholz, J. S., et al. (2021). Activation of Sarm1 produces cADPR to increase intra-axonal calcium and promote axon degeneration in CIPN. bioRxiv [Preprint]. doi: 10.1101/2021.04.15.440024
Liberati, N. T., Fitzgerald, K. A., Kim, D. H., Feinbaum, R., Golenbock, D. T., and Ausubel, F. M. (2004). Requirement for a conserved Toll/interleukin-1 resistance domain protein in the Caenorhabditis elegans immune response. Proc. Natl. Acad. Sci. U.S.A. 101, 6593–6598. doi: 10.1073/pnas.0308625101
Lindwall, C., and Kanje, M. (2005). Retrograde axonal transport of JNK signaling molecules influence injury induced nuclear changes in p-c-Jun and ATF3 in adult rat sensory neurons. Mol. Cell. Neurosci. 29, 269–282. doi: 10.1016/j.mcn.2005.03.002
Liu, H.-Y., Chen, C.-Y., and Hsueh, Y.-P. (2014). Innate immune responses regulate morphogenesis and degeneration: Roles of Toll-like receptors and Sarm1 in neurons. Neurosci. Bull. 30, 645–654. doi: 10.1007/s12264-014-1445-5
Lin, C.-W., Chen, C.-Y., Cheng, S.-J., Hu, H.-T., and Hsueh, Y.-P. (2014). Sarm1 deficiency impairs synaptic function and leads to behavioral deficits, which can be ameliorated by an mGluR allosteric modulator. Front. Cell. Neurosci. 8:87. doi: 10.3389/fncel.2014.00087
Loreto, A., Angeletti, C., Gu, W., Osborne, A., Nieuwenhuis, B., Gilley, J., et al. (2021). Neurotoxin-mediated potent activation of the axon degeneration regulator SARM1. Elife 10:e72823. doi: 10.7554/eLife.72823
Loring, H. S., Czech, V. L., Icso, J. D., O’Connor, L., Parelkar, S. S., Byrne, A. B., et al. (2021). A phase transition enhances the catalytic activity of SARM1, an NAD+ glycohydrolase involved in neurodegeneration. Elife 10:e66694. doi: 10.7554/eLife.66694
Lunn, E. R., Perry, V. H., Brown, M. C., Rosen, H., and Gordon, S. (1989). Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur. J. Neurosci. 1, 27–33.
Matsuzawa, A. (2017). Physiological roles of ASK family members in innate immunity and their involvement in pathogenesis of immune diseases. Adv. Biol. Regul. 66, 46–53. doi: 10.1016/j.jbior.2017.10.007
McKerracher, L., David, S., Jackson, D. L., Kottis, V., Dunn, R. J., and Braun, P. E. (1994). Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 13, 805–811. doi: 10.1016/0896-6273(94)90247-x
McLaughlin, C. N., Nechipurenko, I. V., Liu, N., and Broihier, H. T. (2016). A Toll receptor-FoxO pathway represses Pavarotti/MKLP1 to promote microtubule dynamics in motoneurons. J. Cell Biol. 214, 459–474. doi: 10.1083/jcb.201601014
McLaughlin, C. N., Perry-Richardson, J. J., Coutinho-Budd, J. C., and Broihier, H. T. (2019). Dying Neurons Utilize Innate Immune Signaling to Prime Glia for Phagocytosis during Development. Dev. Cell 48, 506–522.e6. doi: 10.1016/j.devcel.2018.12.019
Milde, S., Gilley, J., and Coleman, M. P. (2013). Subcellular localization determines the stability and axon protective capacity of axon survival factor Nmnat2. PLoS Biol. 11:e1001539. doi: 10.1371/journal.pbio.1001539
Mishra, B., Carson, R., Hume, R. I., and Collins, C. A. (2013). Sodium and potassium currents influence Wallerian degeneration of injured Drosophila axons. J. Neurosci. 33, 18728–18739. doi: 10.1523/JNEUROSCI.1007-13.2013
Mukherjee, P., Winkler, C. W., Taylor, K. G., Woods, T. A., Nair, V., Khan, B. A., et al. (2015). SARM1, Not MyD88, Mediates TLR7/TLR9-Induced Apoptosis in Neurons. J. Immunol. 195, 4913–4921. doi: 10.4049/jimmunol.1500953
Mukherjee, P., Woods, T. A., Moore, R. A., and Peterson, K. E. (2013). Activation of the innate signaling molecule MAVS by bunyavirus infection upregulates the adaptor protein SARM1, leading to neuronal death. Immunity 38, 705–716. doi: 10.1016/j.immuni.2013.02.013
Mukhopadhyay, G., Doherty, P., Walsh, F. S., Crocker, P. R., and Filbin, M. T. (1994). A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13, 757–767.
Murata, H., Khine, C. C., Nishikawa, A., Yamamoto, K.-I., Kinoshita, R., and Sakaguchi, M. (2018). c-Jun N-terminal kinase (JNK)-mediated phosphorylation of SARM1 regulates NAD+ cleavage activity to inhibit mitochondrial respiration. J. Biol. Chem. 293, 18933–18943. doi: 10.1074/jbc.RA118.004578
Murthy, M., Kurz, T., and O’Shaughnessy, K. M. (2017). WNK signalling pathways in blood pressure regulation. Cell. Mol. Life Sci. 74, 1261–1280.
Neukomm, L. J., Burdett, T. C., Seeds, A. M., Hampel, S., Coutinho-Budd, J. C., Farley, J. E., et al. (2017). Axon death pathways converge on axundead to promote functional and structural axon disassembly. Neuron 95, 78–91.e5. doi: 10.1016/j.neuron.2017.06.031
Neumann, B., Nguyen, K. C. Q., Hall, D. H., Ben-Yakar, A., and Hilliard, M. A. (2011). Axonal regeneration proceeds through specific axonal fusion in transected C. elegans neurons. Dev. Dyn. 240, 1365–1372. doi: 10.1002/dvdy.22606
Osterloh, J. M., Yang, J., Rooney, T. M., Fox, A. N., Adalbert, R., Powell, E. H., et al. (2012). DSARM/SARM1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. doi: 10.1126/science.1223899
Panneerselvam, P., Singh, L. P., Selvarajan, V., Chng, W. J., Ng, S. B., Tan, N. S., et al. (2013). T-cell death following immune activation is mediated by mitochondria-localized SARM. Cell Death Differ. 20, 478–489. doi: 10.1038/cdd.2012.144
Peterson, N. D., Icso, J. D., Salisbury, J. E., Rodríguez, T., Thompson, P. R., and Pukkila-Worley, R. (2022). Pathogen infection and cholesterol deficiency activate the C. elegans p38 immune pathway through a TIR-1/SARM1 phase transition. Elife 11:e74206. doi: 10.7554/eLife.74206
Rotshenker, S. (2011). Wallerian degeneration: The innate-immune response to traumatic nerve injury. J. Neuroinflammation 8:109.
Sarkar, A., Kumari, N., and Mukherjee, P. (2021). The curious case of SARM1: Dr. Jekyll and Mr. Hyde in cell death and immunity? FEBS J. [Epub ahead of print]. doi: 10.1111/febs.16256
Sasaki, Y., Engber, T. M., Hughes, R. O., Figley, M. D., Wu, T., Bosanac, T., et al. (2020). cADPR is a gene dosage-sensitive biomarker of SARM1 activity in healthy, compromised, and degenerating axons. Exp. Neurol. 329:113252. doi: 10.1016/j.expneurol.2020.113252
Schäfer, M., Fruttiger, M., Montag, D., Schachner, M., and Martini, R. (1996). Disruption of the gene for the myelin-associated glycoprotein improves axonal regrowth along myelin in C57BL/Wlds mice. Neuron 16, 1107–1113. doi: 10.1016/s0896-6273(00)80137-3
Schlaepfer, W. W. (1974). Calcium-induced degeneration of axoplasm in isolated segments of rat peripheral nerve. Brain Res. 69, 203–215. doi: 10.1016/0006-8993(74)90002-x
Schlaepfer, W. W., and Bunge, R. P. (1973). Effects of calcium ion concentration on the degeneration of amputated axons in tissue culture. J. Cell Biol. 59, 456–470. doi: 10.1083/jcb.59.2.456
Schlaepfer, W. W., and Hasler, M. B. (1979). Characterization of the calcium-induced disruption of neurofilaments in rat peripheral nerve. Brain Res. 168, 299–309. doi: 10.1016/0006-8993(79)90171-9
Sommer, C., Leinders, M., and Üçeyler, N. (2018). Inflammation in the pathophysiology of neuropathic pain. Pain 159, 595–602.
Sporny, M., Guez-Haddad, J., Khazma, T., Yaron, A., Dessau, M., Shkolnisky, Y., et al. (2020). Structural basis for SARM1 inhibition and activation under energetic stress. Elife 9:e62021. doi: 10.7554/eLife.62021
Summers, D. W., DiAntonio, A., and Milbrandt, J. (2014). Mitochondrial Dysfunction Induces Sarm1-Dependent Cell Death in Sensory Neurons. J. Neurosci. 34, 9338–9350. doi: 10.1523/JNEUROSCI.0877-14.2014
Walker, L. J., Summers, D. W., Sasaki, Y., Brace, E. J., Milbrandt, J., and DiAntonio, A. (2017). MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. Elife 6:e22540. doi: 10.7554/eLife.22540
Waller, T. J., and Collins, C. A. (2021). An NAD+/NMN balancing act by SARM1 and NMNAT2 controls axonal degeneration. Neuron 109, 1067–1069. doi: 10.1016/j.neuron.2021.03.021
Wang, Q., Zhang, S., Liu, T., Wang, H., Liu, K., Wang, Q., et al. (2018). Sarm1/Myd88-5 Regulates Neuronal Intrinsic Immune Response to Traumatic Axonal Injuries. Cell Rep. 23, 716–724. doi: 10.1016/j.celrep.2018.03.071
Wu, T., Zhu, J., Strickland, A., Ko, K. W., Sasaki, Y., Dingwall, C. B., et al. (2021). Neurotoxins subvert the allosteric activation mechanism of SARM1 to induce neuronal loss. Cell Rep. 37:109872. doi: 10.1016/j.celrep.2021.109872
Xiong, X., and Collins, C. A. (2012). A conditioning lesion protects axons from degeneration via the Wallenda/DLK MAP kinase signaling cascade. J. Neurosci. 32, 610–615. doi: 10.1523/JNEUROSCI.3586-11.2012
Xiang, L., Wu, Q., Sun, H., Miao, X., Lv, Z., Liu, H., et al. (2022). SARM1 deletion in parvalbumin neurons is associated with autism-like behaviors in mice. Cell Death Dis. 13:638. doi: 10.1038/s41419-022-05083-2
Xiong, X., Hao, Y., Sun, K., Li, J., Li, X., Mishra, B., et al. (2012). The Highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein. PLoS Biol. 10:e1001440. doi: 10.1371/journal.pbio.1001440
Xue, T., Sun, Q., Zhang, Y., Wu, X., Shen, H., Li, X., et al. (2021). Phosphorylation at S548 as a Functional Switch of Sterile Alpha and TIR Motif-Containing 1 in Cerebral Ischemia/Reperfusion Injury in Rats. Mol. Neurobiol. 58, 453–469. doi: 10.1007/s12035-020-02132-9
Yang, J., Wu, Z., Renier, N., Simon, D. J., Uryu, K., Park, D. S., et al. (2015). Pathological Axonal Death through a MAPK Cascade that Triggers a Local Energy Deficit. Cell 160, 161–176. doi: 10.1016/j.cell.2014.11.053
Keywords: injury signaling, Nicotinamide Adenine Dinucleotide (NAD), MAP Kinase, axonal transport, axon degeneration, SARM1, TIR domain
Citation: Waller TJ and Collins CA (2022) Multifaceted roles of SARM1 in axon degeneration and signaling. Front. Cell. Neurosci. 16:958900. doi: 10.3389/fncel.2022.958900
Received: 01 June 2022; Accepted: 09 August 2022;
Published: 25 August 2022.
Edited by:
Gerardo Morfini, University of Illinois at Chicago, United StatesReviewed by:
Julia Margaret Edgar, University of Glasgow, United KingdomValeria Cavalli, Washington University in St. Louis, United States
Copyright © 2022 Waller and Collins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine A. Collins, Y3hjMTIxNUBjYXNlLmVkdQ==