 Momoka Hikosaka
Momoka Hikosaka Takeo Kawano
Takeo Kawano Yayoi Wada
Yayoi Wada Tomoki Maeda
Tomoki Maeda Takeshi Sakurai
Takeshi Sakurai Gen Ohtsuki
Gen Ohtsuki
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 22 July 2022
Sec. Cellular Neurophysiology
Volume 16 - 2022 | https://doi.org/10.3389/fncel.2022.925493
This article is part of the Research Topic Cerebellum-Related Learning and Psychiatric Diseases View all 6 articles
Immune cells play numerous roles in the host defense against the invasion of microorganisms and pathogens, which induces the release of inflammatory mediators (e.g., cytokines and chemokines). In the CNS, microglia is the major resident immune cell. Recent efforts have revealed the diversity of the cell types and the heterogeneity of their functions. The refinement of the synapse structure was a hallmark feature of the microglia, while they are also involved in the myelination and capillary dynamics. Another promising feature is the modulation of the synaptic transmission as synaptic plasticity and the intrinsic excitability of neurons as non-synaptic plasticity. Those modulations of physiological properties of neurons are considered induced by both transient and chronic exposures to inflammatory mediators, which cause behavioral disorders seen in mental illness. It is plausible for astrocytes and pericytes other than microglia and macrophage to induce the immune-triggered plasticity of neurons. However, current understanding has yet achieved to unveil what inflammatory mediators from what immune cells or glia induce a form of plasticity modulating pre-, post-synaptic functions and intrinsic excitability of neurons. It is still unclear what ion channels and intracellular signaling of what types of neurons in which brain regions of the CNS are involved. In this review, we introduce the ubiquitous modulation of the synaptic efficacy and the intrinsic excitability across the brain by immune cells and related inflammatory cytokines with the mechanism for induction. Specifically, we compare neuro-modulation mechanisms by microglia of the intrinsic excitability of cerebellar Purkinje neurons with cerebral pyramidal neurons, stressing the inverted directionality of the plasticity. We also discuss the suppression and augmentation of the extent of plasticity by inflammatory mediators, as the meta-plasticity by immunity. Lastly, we sum up forms of immune-triggered plasticity in the different brain regions with disease relevance. Together, brain immunity influences our cognition, sense, memory, and behavior via immune-triggered plasticity.
Brain immunity is the defense system for the invasion of microorganisms and viruses. Such immune system to protect our body from diseases and infections is mainly ascribed to the functions of immune cells: microglia, macrophage, myeloid cells, granulocytes, dendritic cells, natural killer cells, T cells, and B cells (Medzhitov and Janeway, 2000; Strecker et al., 2017). Aberrance of the brain immune system is one of the promising features of psychiatric disorders as well as neurodegenerative diseases (Yolken and Torrey, 1995; Gruol and Nelson, 1997; Parker-Athill and Tan, 2010; Khandaker et al., 2015; Colonna and Butovsky, 2017; Nimgaonkar et al., 2017; Barichello et al., 2019; Pape et al., 2019; Ozaki et al., 2021; Segawa et al., 2021). The immune system of mammals, including humans, is divided into adaptive immunity and innate immunity. Adaptive immunity (i.e., acquired immunity) is acquired in response to the stimulation of foreign substances. And it is the specific antibodies and T cell receptors that recognize foreign substances through antigen presentation. Immunological memory is an important characteristic of adaptive immunity, and this acquired system enables us to respond instantly to the identical antigen that had invaded our body previously. On the other hand, innate immunity is another dominant immune system, which recognizes foreign substances against targeting molecular patterns peculiar to microorganisms. The pattern recognition molecules are the complements, lectins, Toll-like receptors, etc., which are encoded in the genome (Hoshino et al., 1999; Pandey et al., 2014). Innate immunity does not have a complex and specific immune memory, like adaptive immunity, and responds immediately to specific components of pathogens. However, it could be considered that there is a different form of the memory system in innate immunity and distinct microglia/macrophage responses, as seen in the disease-associated microglia (DAM), which are thought as the microglia population that acquired pro-inflammatory activity by altering the expression of genes involved in the lysosomal/phagocytic pathway, endocytosis and the regulation of immune responses in response to central nervous system (CNS) pathologies (Deczkowska et al., 2018). Whereas disorders of acquired immunity are common in autoimmune diseases, innate immune system anomalies are often noted in many psychiatric disorders. At the evolutionary origin, acquired immunity appears in vertebrate lampreys and hagfishes (Cyclostomatas). On the other hand, innate immunity existed long before the advent of vertebrates. The complement system has been found in ascidians and sea urchins, and Toll-like receptors are already present in cnidarians (Nonaka and Yoshizaki, 2004; Brennan and Gilmore, 2018; Liu et al., 2020).
Inflammatory mediators, such as cytokines and chemokines, are molecules released from immune cells in response to the damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), and neurodegeneration-associated molecular patterns (NAMPs), and the mediators play distinct roles in both physiology and pathology in the CNS (Hopkins and Rothwell, 1995; Gruol and Nelson, 1997; Deczkowska et al., 2018). Regarding disease association with the brain immunity, excessive inflammatory cytokines, disruption of brain vasculature systems, the proliferation of activated immune cells in the parenchyma, and malformation of neurons are observed in the various animal models emerging the symptoms of psychiatric disorders (Hopkins and Rothwell, 1995; Gilmore et al., 2004; Choi et al., 2016; Menard et al., 2017; Greene et al., 2018; Nie et al., 2018). Previously, researchers had regarded the cerebellum as not an important brain site in the development of stress responses by aberrant immunity (Nguyen et al., 1998). The current view suggests that the cerebellum is vulnerable to aberrant immunity and infection in the early developmental period, which is the new region causing the emergence of psychiatric diseases including autism spectrum disorders, mood disorder, and schizophrenia (Schmahmann, 2004; Tsai et al., 2012, 2018; Piochon et al., 2014; Moberget et al., 2018; Yamamoto et al., 2019; Cook et al., 2021; Ozaki et al., 2021; Baek et al., 2022; Hwang et al., 2022). Typically, in autism spectrum disorder (ASD)-patient brains, the cerebellum presented various pathological impairments, including the uneven structure of the Purkinje cell layer and granular cell layer, marked activation of microglia and reactive Bergmann's astroglia, and accumulation of perivascular macrophages and microglia. The cytokine profiling also indicated that monocyte chemoattractant protein 1 [MCP-1; known as CCL2: chemokine (C-C motif) ligand 2], interleukin-6 (IL-6), and transforming growth factor-β1 (TGFβ1) were the most prevalent cytokines in ASD brain tissues. Cerebrospinal fluid from patients showed a unique pro-inflammatory profile of cytokines, including a marked increase in interferon-γ (IFN γ), TGFβ, MCP-1, IL-8, fibroblast growth factor (FGF)-9, and vascular endothelial growth factor (VEGF) (Vargas et al., 2005; Parker-Athill and Tan, 2010). In maternal immune activation (MIA) mice by administration of Poly(I:C) to the pregnant mothers, adult offspring show ASD-like behaviors and a deficit of Purkinje cells in the middle lobes (e.g., lobule VIa-VII) of the cerebellum (Shi et al., 2009; Naviaux et al., 2013), presumably reflecting the vulnerability of the vasculature and promoted access of the peripheral immunity in the developmental period. A spatially localized delay of granule-cell migration is also observed during the cerebellar development in the MIA exposed animals (Shi et al., 2009). Thus, the cerebellum is expected to be one of the brain regions implicated in developmental disorders with distinct neuroinflammation. Despite that, the mechanisms of how inflammatory mediators modulate neuronal activity as the synaptic transmission and the intrinsic excitability are not utterly elucidated. We speculate that the cerebellum is vulnerable to immune-cell invasion through the fourth ventricle, capillaries, and pial vasculatures between folia. For a better understanding of cerebellar inflammation and its relevance to psychiatric disorders, an overview of recent studies regarding immune-triggered plasticity across the brain is required. Such effort should help advance this field and find therapeutic targets.
The resident immune cells in the brain, microglia, are the most abundant immune cells in the brain. Microglia originate from erythromyeloid progenitors in the yolk sac and invade the brain before forming the blood-brain barrier (BBB) (Ling and Wong, 1993; Nakajima and Kohsaka, 1993; Ginhoux et al., 2010; Ginhoux and Prinz, 2015). Microglia account for 5–12% of the total number of cells present in the mouse brain (Lawson et al., 1990) and 0.5–16.6% of the total number of cells in the human brain (Mittelbronn et al., 2001), depending on the brain region. Microglial cell densities across mammalian species remain conserved (e.g., around 6% of all cells in the mammalian cortex) for over 200 million years (Dos Santos et al., 2020). Askew et al. (2017) demonstrated that the number of microglial cells remains steady from early postnatal stages until aging and is maintained by the regulated coupling of proliferation and apoptosis without a contribution from circulating progenitors (Askew et al., 2017).
Classically, microglia could be divided into two phenotypes: amoeboid microglia and ramified microglia (Ling and Wong, 1993). In the adult brain, resting-state microglia are dynamic and have many functions from the homeostasis of the brain-parenchymal milieu to the refinement of the neural circuit (Nimmerjahn et al., 2005). It is well-known that clearance of the protein aggregates, damaged organelles, redundant synapses, and dead cells are crucial for the maintenance of brain health and brain development (Colonna and Butovsky, 2017; Deczkowska et al., 2018). When microglia are reactivated via the innate immune system by DAMPs and PAMPs, they transform into activated microglia which change gene expression patterns and their morphology. They have the characteristic of the high ability of proliferation, migratory, and phagocytosis, and release various inflammatory mediators. Recent studies indicate microglia are heterogeneous across brain regions in terms of morphology, signaling pathways, and gene expression patterns (Grabert et al., 2016; Ayata et al., 2018; Silvin and Ginhoux, 2018; Süß et al., 2020). The phenotypes as transcripts and morphology of microglia differ from the localized region or age (De Biase et al., 2017; Masuda et al., 2019). Ablation and repopulation of microglia also alter phenotypes and localizations (De Biase et al., 2017; Masuda et al., 2019). Our current understanding of gene expression patterns is based on many transcriptome studies and morphological analyses. While we admit the importance of the transcriptome, such studies do not provide any information on functional protein expression. Upcoming multiplexed proteome studies, such as imaging masscytometry, should overcome the points (Fernández-Zapata et al., 2020). As abovementioned, there is no complex and specific immune memory in innate immunity, but the feature of heterogeneity might be a form of the memory in innate immunity.
Importantly, microglia are one of the major resources of inflammatory cytokine releases in response to peripheral inflammation, invaded microbes, bacterial endotoxin, and nucleic acids. For instance, transient exposure of brain slices to the Gram-negative bacterial endotoxin, lipopolysaccharide (LPS), activates microglia through Toll-like receptor 4 (TLR4), a pattern recognition receptor. Exposure to LPS facilitates the vesicular release at hippocampal excitatory presynaptic terminals (Pascual et al., 2012). It was also shown that under the hypoxia condition exposure to LPS could depress the post-synaptic efficacy of glutamate receptors by the superoxide and nitric oxide production (Zhang et al., 2014). The number of studies on such immune-triggered modulations of synaptic transmission in the CNS is increasing these days (Pascual et al., 2012; Parkhurst et al., 2013; Zhan et al., 2014; Zhang et al., 2014; Yamamoto et al., 2019; Di Filippo et al., 2021). Therefore, brain immunity is the triggering factor of synaptic plasticity via immune receptors, such as pattern-recognition receptors and cytokine receptors, expressed in the neurons. In contrast, activated microglia also modulate the non-synaptic membrane excitability of neurons in the neocortex, hippocampus, amygdala, and cerebellum (Gao et al., 2014; Klapal et al., 2016; Tzour et al., 2017; Duan et al., 2018; Luo et al., 2019; Yamamoto et al., 2019; Zheng et al., 2021; Yamawaki et al., 2022). In a study by Zhang et al. (2014), changes in waveforms of field EPSPs after microglia activation by exposure to LPS were shown, and the result not only indicated the long-term depression (LTD) of the synaptic transmission but also implied the modulation of the late component of evoked synaptic transmission as an ion-channel modulation. Nevertheless, the cellular mechanism for inducing the long-term plasticity of intrinsic excitability is known in a limited number of cell types. The neuromodulation of the microglial functions is not only for the synaptic plasticity (i.e., synaptic efficacy and synaptic structure) but also for the excitability of neurons and even their dendrites (Tables 1, 2). And the involved immune cell types including microglia and the released inflammatory mediators are just started to be identified.
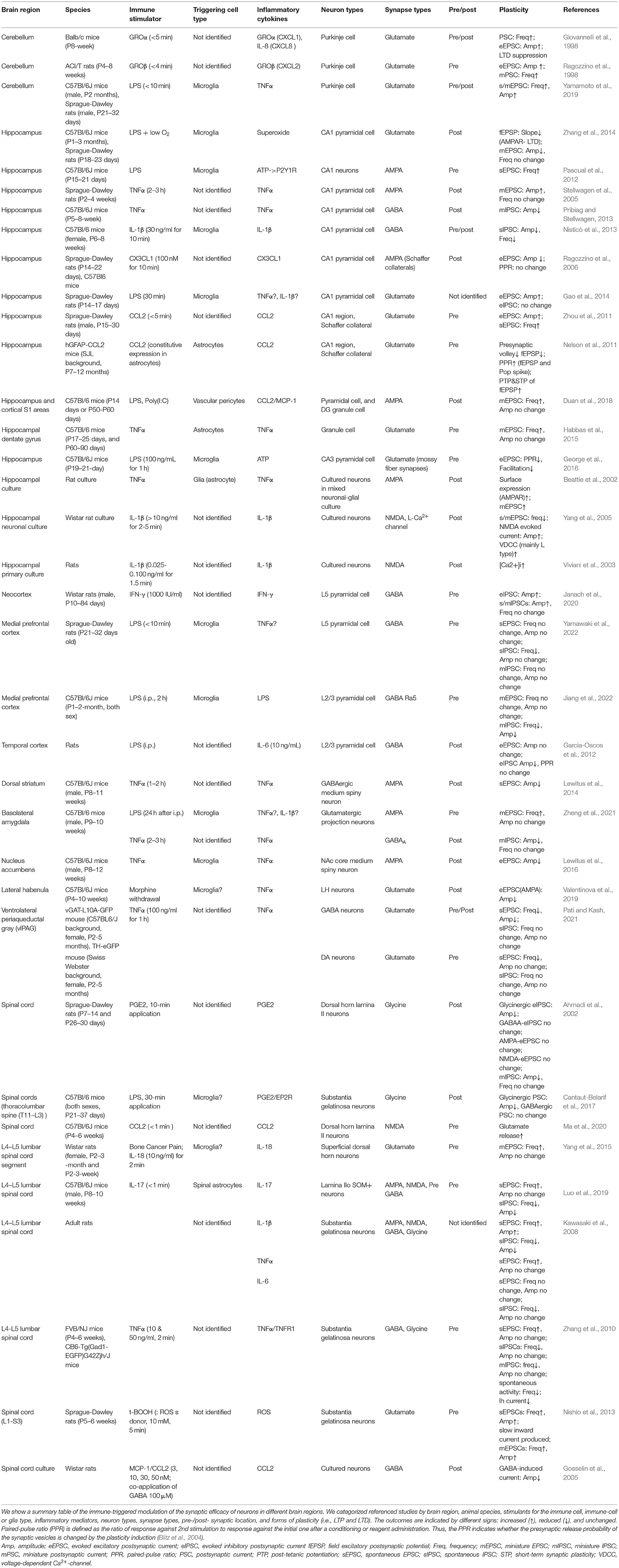
Table 1. Immune-triggered modulation of the synaptic transmission of neurons.
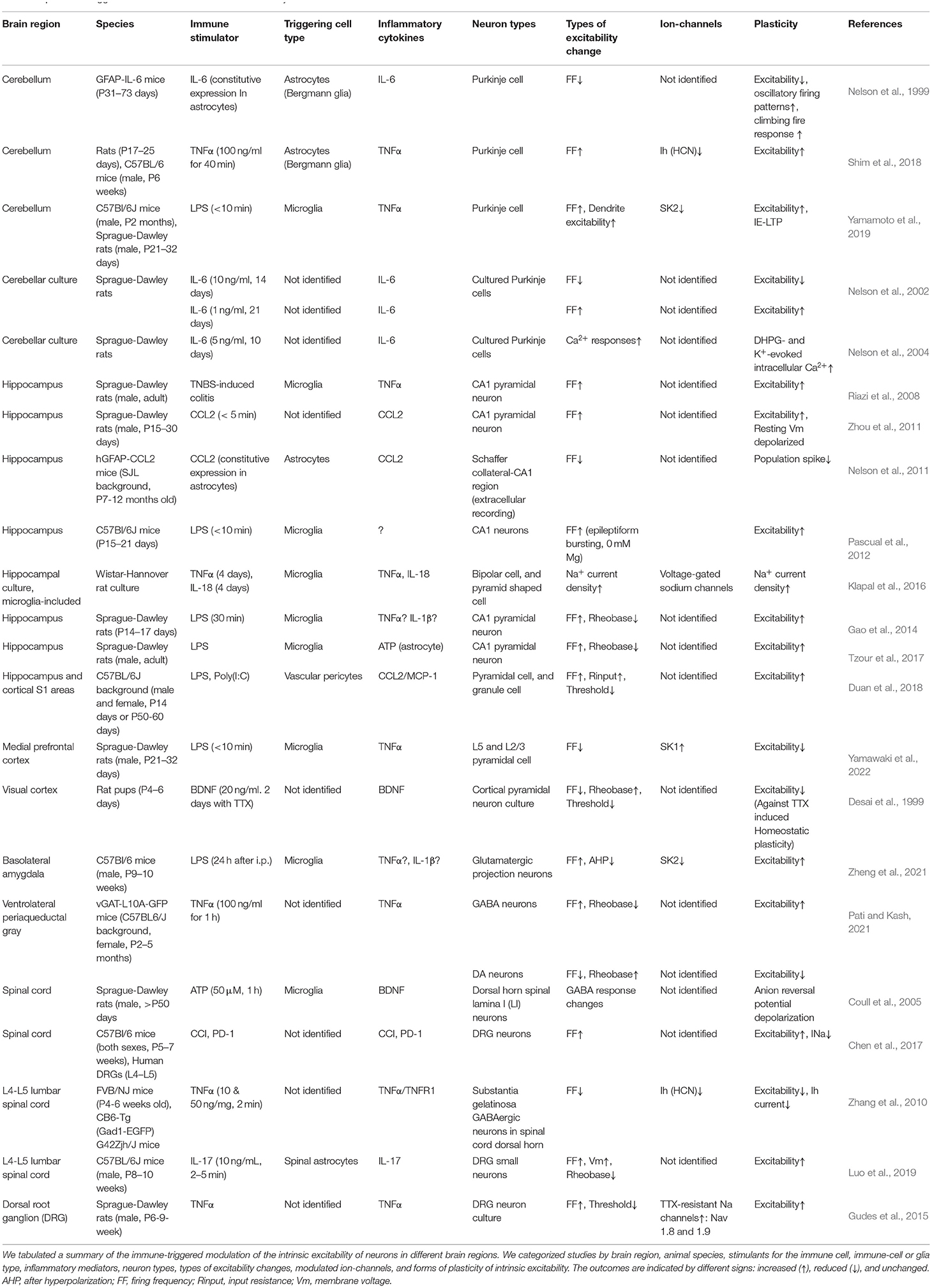
Table 2. Immune-triggered modulation of the intrinsic excitability of neurons.
Considering microglia as the most abundant immune cells in the brain parenchyma, they should play the most responsible roles for brain homeostasis and disease state. For the induction of immune-triggered plasticity, however, the mechanisms for induction are utterly the frontier in the present neuroscience field. In the following sections, we review recent advances in our understanding of the synaptic and non-synaptic plasticity induction by microglia and other immune cells, their contribution to the homeostasis of network dynamics throughout the CNS regions, and their relevance to the emergence of psychiatric diseases. We also highlight the ion channels and molecular signaling implicating the immune-triggered plasticity.
In the cerebellar cortex, a study indicated that activated microglia by LPS-exposure induce long-term potentiation (LTP) of intrinsic excitability, as the increase in the firing frequency of action potentials, and facilitate synaptic transmission in Purkinje neurons of juvenile rodents (Yamamoto et al., 2019; Figure 1). The resultant hyperexcitability in the cerebellar cortex by acute inflammation causes an abnormality in psychomotor behaviors of rodents via the pathways to the cerebello-frontal cortex (Yamamoto et al., 2019; Ohtsuki et al., 2020; Ozaki et al., 2021). Depletion of microglia made the animals resistant to the LPS-exposure, and immune-triggered hyperexcitability was not induced in the microglia-depleted cerebellum (Yamamoto et al., 2019). After exposure to the endotoxin, microglia secrete tumor necrosis factor (TNF)-α rapidly through a protein-synthesis-independent manner and a non-constitutive pathway. TLR4 on the surface of microglia recognizes the endotoxin, primary response gene 88 (MyD88), and Toll/IL-1 receptor (TIR) domain-containing adaptor-inducing interferon-β (TRIF) play the downstream pathway (Yamamoto et al., 2019). The released mediator TNF-α increases in action potential firing in a translation-independent manner. Induction of intrinsic plasticity, as LTP of the intrinsic excitability of Purkinje cells, was humbled by administration of LPS, heat-killed Gram-negative bacteria, and TNF-α in advance, suggesting crosstalk of the induction mechanism between naïve form of intrinsic plasticity and immune-triggered plasticity. Pharmacological suppression of the cerebellar astrocytes, such as Bergmann glia, did not diminish from inducing hyperexcitability plasticity of Purkinje neurons (Yamamoto et al., 2019). After activating TNF receptors expressing on the neurons, activation of the protein phosphatases elicits the trafficking of ion channels. Cerebellar microglia induce phosphatase activation and downregulation of small-conductance Ca2+-activated K+ channels (SK channels), which intensifies the intrinsic excitability of Purkinje cells (Yamamoto et al., 2019; Figure 1). In Purkinje cells, SK2 channels are specifically expressed, which is also shown to augment the excitability of dendrites (Belmeguenai et al., 2010; Ohtsuki et al., 2012; Ohtsuki and Hansel, 2018; Yamamoto et al., 2019; Ohtsuki, 2020). While TNF-α does not solely target the neuronal membrane, secreted TNF-α also modulates presynaptic transmission via TNF receptors on astrocytes (Santello et al., 2011; Habbas et al., 2015). Another study showed that the interference of TNF receptor 1 in Bergmann glia suppressed the effect of the TNF-α on the excitability increase of Purkinje cells (Shim et al., 2018). In response to endotoxin, activated microglia lead to activating Bergmann glia. Through the activation, Bergmann glia may promote presynaptic release from excitatory parallel fibers and climbing fibers following TNF-α stimulation and ATP release (Yamamoto et al., 2019). Indeed, Yamamoto et al. (2019) showed an increase in both spontaneous excitatory postsynaptic current (EPSC) and miniature EPSC frequency, and their amplitude, suggesting the modulation of both an increase in the activity of presynaptic neurons or the local excitability of terminals (e.g., granule cells and climbing fibers), vesicular release from pre-synapses, and post-synaptic responsiveness (Ohtsuki and Hirano, 2008; Yamamoto et al., 2019). However, the machinery for synaptic plasticity is not completely characterized yet.
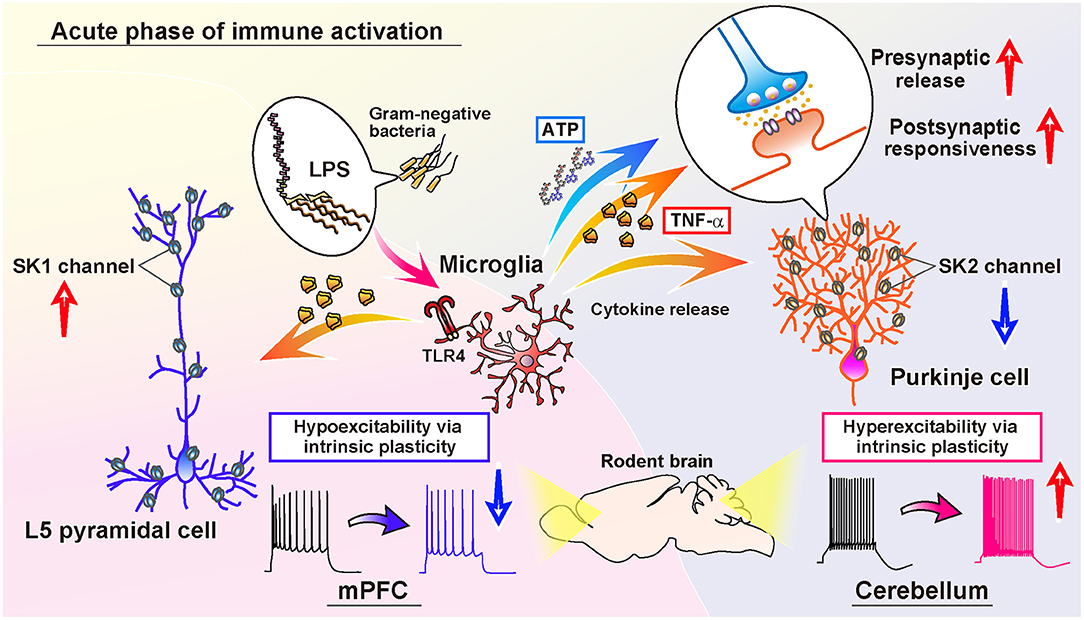
Figure 1. Comparison of the immune-triggered plasticity of intrinsic excitability of cerebellar Purkinje cells and mPFC L5 pyramidal cells. Exposure to LPS activates microglia in both the cerebellum and mPFC. In the cerebellum, activated microglia release inflammatory cytokines, including TNF-α, which induce downregulation of SK2 channels in the Purkinje cells. Activated microglia also contribute to ATP release in the cerebellar cortex, and both ATP and TNF-α promote the release of presynaptic vesicles of the glutamatergic terminals. Moreover, the postsynaptic responsiveness is also increased via intraneuronal TNF-signaling. As a result, both the amplitude and frequency of both spontaneous and miniature EPSC increases (Yamamoto et al., 2019). In the mPFC, TNF-α released from activated microglia causes the upregulation of SK1 channels of L5 pyramidal cells, in contrast to the cerebellum. Subsequently, the intrinsic excitability of L5 pyramidal cells is reduced long-lastingly. In addition, the frequency of spontaneous IPSC decreases, whereas the spontaneous EPSC does not change (Yamawaki et al., 2022).
It is noteworthy that, generally, in synapses, there are two sites for modulation: pre- and post-synapses. The modulation of presynaptic release is identified by the increase and decrease in the frequency of miniature EPSC and inhibitory postsynaptic current (IPSC) of the excitatory and inhibitory synapses, respectively. Miniature EPSC and IPSC are the synaptic events recorded under voltage-clamp in the presence of a blocker of voltage-sensitive Na+-channels (i.e., tetrodotoxin, TTX). In contrast, spontaneous events are recorded without TTX. When the amplitude of miniature EPSC and IPSC changes, it indicates changes in postsynaptic responsiveness.
In contrast to the immune-triggered plasticity in the cerebellum, transient LPS-exposure to the medial prefrontal cortex (mPFC) and the microglia activation reduces the intrinsic excitability of layer 5 (L5) and L2/3 pyramidal neurons (Yamawaki et al., 2022; Figure 1). Interestingly, the exposure to LPS did not decrease the firing frequency of fast-spiking interneurons, indicating cell-type specificity of the LPS-triggered plasticity. Such hypoexcitability plasticity—a sustained reduction of the intrinsic excitability of neurons—was prevented under the suppression of microglial activity, too. The hypoexcitability plasticity of pyramidal cells by LPS-exposure was suppressed in the preparations pre-incubated by colony-stimulating factor 1 receptor [CSF1R, also known as macrophage colony-stimulating factor receptor (M-CSFR), which is required for the maintenance of microglia (Yamawaki et al., 2022)]. Whereas the excitability of cerebellar Purkinje cells increases via downregulation of SK channels (Yamamoto et al., 2019), interestingly, the excitability of mPFC pyramidal neurons decreases via upregulation of SK1 channels in response to microglia activation and the release of inflammatory cytokine TNF-α (Yamawaki et al., 2022). The TNF-signaling to the intracellular cascade of pyramidal neurons is suggested via the activation of protein phosphatases in the mPFC L5 pyramidal neurons (Beattie et al., 2002; Santello et al., 2011; Pribiag and Stellwagen, 2013; Habbas et al., 2015; Yamawaki et al., 2022). This prominent comparison of the directionality of modulation of neurophysiological properties represents the inverted form of the plasticity in both cerebellar Purkinje cells and mPFC pyramidal cells. The inverted directionality of immune-triggered intrinsic excitability plasticity would be derived from both neuronal and microglial mechanisms (Yamawaki et al., 2022; Figure 1). In addition, exposure to LPS or interleukin-1β (IL-1β) did not change the spontaneous EPSC in the mPFC, whereas it decreased the frequency of spontaneous IPSC but not miniature IPSC, implying the reduction of the network activity through hypoexcitability of pyramidal cells after acute inflammation in the mPFC (Yamawaki et al., 2022). In the neocortical L5 pyramidal neurons, IFN-γ was also reported to increase amplitudes of spontaneous and miniature IPSCs without changes in frequency, suggesting postsynaptic modulation of γ-aminobutyric acid (GABA)-ergic transmission (Janach et al., 2020). Besides, IFN-γ diminished Ih current, suggesting involvement of protein kinase C (PKC), while IFN-γ did neither alter subthreshold nor suprathreshold neuronal excitability (Janach et al., 2020). Therefore, among the phases of inflammation in the CNS, the neuromodulatory effects are various, and it is also dependent on the brain region, cell type, inflammatory mediator, and related immune cells.
As the phenomenon of synaptic modulation during inflammation, the neural pain accompanied by hypersensitivity of neurons in the spinal cord is relatively well-studied, and it would be a good example to introduce here. In the early studies in the spinal cord, it has been shown that brain-derived neurotrophic factor (BDNF) released from microglia induces depolarization of anion reversal potential and modulates the driving force of inhibitory GABAergic channels via downregulation of the potassium-chloride cotransporter KCC2 (also known as: potassium-chloride transporter member 5 and SLC12A5) mediated by TrkB signaling, associated with the neural pain (Coull et al., 2003, 2005). Thus, inflammatory mediators released from immune cells, like microglia and T-cells, have various influences on the synaptic modulations depending on the type of synapses or their receptors associated with nociception and the hypersensitivity of neurons (Grace et al., 2014). In the excitatory presynaptic terminals of the spinal cord, TNF-α, IL-1β, CCL2/MCP-1, reactive oxygen species (ROS), and IFN γ increase glutamate release via transient receptor potential vanilloid 1 (TRPV1), transient receptor potential cation channel subfamily A member 1 (TRPA1), interleukin-1 receptor 1(IL-1R1), and presynaptic ionotropic glutamate receptor [N-methyl-D-aspartate receptor (NMDA receptor)] (Kawasaki et al., 2008; Medvedeva et al., 2008; Park et al., 2011a,b; Nishio et al., 2013; Ma et al., 2020; Donnelly et al., 2021). On the other hand, GABAergic synaptic transmission of the inhibitory interneurons is attenuated by TNF-α, IL-1β, IL-6, the pair of CLL2 and CCL2/MCP-1, and ROS (Gosselin et al., 2005; Vikman et al., 2007; Kawasaki et al., 2008; Zhang et al., 2010). CCR2 (C-C Motif Chemokine Receptor 2) is the receptor of CCL2 expressed in the neurons of brain-wide regions including the telencephalon, diencephalon, mesencephalon, cerebellum (cortex and nuclei), and brainstem (Banisadr et al., 2005a,b). Although IFN-γ was also implied to modulate the release of GABAergic transmission in the dorsal horn neurons (Vikman et al., 2007; Grace et al., 2014), bicuculline, which was used in the study, is not specific to GABAA receptor but it also affects small-conductance Ca2+-activated K+ channels (Khawaled et al., 1999), and thus further investigation would be required. In general, it is required to consider that many experiments using bicuculline for specifying excitatory synaptic transmission should modulate the neuronal excitability per se and do not necessarily exclude the effect of excitability modulation (Takeuchi et al., 2008). As mentioned, according to Janach et al. (2020), IFN-γ receptors express in the neocortical pyramidal neurons, and the administration of IFN-γ increased both amplitudes of spontaneous and miniature IPSCs without changes in their frequency, suggesting postsynaptic modulation of GABAergic transmission (Janach et al., 2020). In the spinal cord, again, BDNF from microglia changes the driving force of GABAergic conductance via downregulation of the potassium-chloride cotransporter KCC2 mediated by TrkB signaling (Coull et al., 2003, 2005). Glycinergic neurotransmission is also inhibited by IL-1β and IL-6 (Kawasaki et al., 2008). As for the postsynaptic responsiveness, various inflammatory cytokines, including TNF-α, IL-1β, IL-17, ROS, CXCL1, and CXCR2 may promote phosphorylation of NMDA receptors, potentiate the efficacy of NMDA receptors, and may maintain the neuropathic pain and mechanical allodynia (Gao et al., 2007; Kleibeuker et al., 2008; Meng et al., 2013; Zhang et al., 2013). However, we have to note again that the protein expression or quantification by merely western blotting and immunostaining does not always indicate modulation of synapse functions and localization. Therefore, we consider that current NMDA receptor trafficking in the spinal cord is based on indirect evidence yet without tests with the physiological approaches. Therefore, we just describe the possibility of the functional modulation of NMDA receptors and stress the necessity of further investigation regarding immune-triggered plasticity. Katsura et al. (2006) suggested that activation of Src-family tyrosine kinases in spinal cord microglia is crucial for mechanical hypersensitivity after peripheral nerve injury, but not in neurons or astrocytes. Their results indicate no change in phosphorylation of Src-family kinase in primary sensory neurons but OX42-positive microglia. Src-family kinase in microglia contributes to mechanical hypersensitivity presumably via the expression of TRPV1 and TRPA1 in the sensory neurons (Katsura et al., 2006). Prostaglandins (PGs) also have a crucial role in pain sensitization (Ahmadi et al., 2002; Harvey et al., 2004), but we will discuss this physiologically active lipid in the latter section. In sum, the modulations of synaptic transmission and intrinsic excitability in the spinal cord have been gradually revealed, and inflammation certainly links to the neuronal modulations in the spinal cord associated with the sense of pain. These forms of plasticity of neuronal activity by immunity would no wonder exist in CNS regions ubiquitously (Tables 1, 2).
The modulation of synaptic transmission and intrinsic excitability by immunity is expected to exist in CNS regions other than the cerebellum and cerebral cortex. Here, we show summary tables of the immune-triggered modulation of the synaptic efficacy and intrinsic excitability of neurons in different brain regions (Tables 1, 2). Table 1 shows our summary of the immune-triggered synaptic plasticity. We categorized studies by animal species, brain region, neuron type, synapse types, pre-/post- synaptic location, a form of plasticity (i.e., LTP and LTD), inflammatory mediators, immune-cell or glia type, a stimulant for the immune cell, and comments regarding the induction. Arbitrarily, we allowed classifying LTP or LTD if the changes last more than 20 min after administration of immune stimulant or induction molecule. We also made a summary table of the immune-triggered modulation of the intrinsic excitability (Table 2). In tables, we included astrocytes and pericytes studies to compare the microglia because the classification of the “brain immune cells” is now not simple: e.g., astrocytes also express the immunological molecules, like interleukins, which have been believed to express only in immune cells. As in these tables, immune-triggered plasticity is induced in various regions not only the cerebellum and cerebral cortex but hippocampus, amygdala, basal ganglia, ventral periaqueductal gray (PAG), and spinal cord. Now, it is no doubt to occur ubiquitous modulations of the synaptic efficacy and the intrinsic excitability of neurons by immune cells mainly via innate immune responses. The related inflammatory cytokines and the mechanism for induction have numerous variations. In the following sections, we discuss the roles of inflammatory mediators, cytokines, in synaptic and non-synaptic plasticity (Roles of Inflammatory Cytokines and Lipid Mediators in Synaptic and Non-synaptic Plasticity). And we describe modulations by chemokines (Modulation of Neuronal Activity by Chemokines). Lastly, we discuss the possible involvement of the immune-triggered plasticity in psychiatric diseases (Plasticity Triggered by Inflammatory Cytokines and Relevance to Psychiatric Diseases).
Tumor necrosis factor (TNF) was identified in 1975 as a factor causing hemorrhagic necrosis in tumors planted in mice (Carswell et al., 1975), now it is known as a multifunctional cytokine. The TNF family has three members: TNF-α, TNF-β/LT-α, and LT-β. TNF-α is a cytokine produced by an antigen-presenting cell (APC), and two isoforms can exist: 26-kDa membrane-tethered form (mTNF-α) and 16-kDa soluble form (sTNF-α; Kriegler et al., 1988). The mTNF-α is cleaved by the inducible TNF converting enzyme (TACE) to release the form of sTNF-α (Black et al., 1997; Moss et al., 1997), which exerts its effect through TNF receptors. In the hippocampus, TNF-α, but not IL-6 and IL-10, increases the surface expression of an α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subunit GluR1 and reduces the surface expression of GABAA receptors (Beattie et al., 2002; Stellwagen et al., 2005). TNF-α signaling is not required for the induction of CA1 hippocampal synaptic LTP or LTD (Stellwagen and Malenka, 2006). TNF-α activates phosphatase signaling via TNF receptor 1 (Pribiag and Stellwagen, 2013). As per the previous sections, TNF-α released by microglia induces plasticity in the CNS. In the cerebellar Purkinje cells, transient administration of TNF-α increases the excitatory synaptic transmission and the intrinsic excitability (Yamamoto et al., 2019). In the cerebral pyramidal cells, TNF-α decreases the intrinsic excitability, and both TNF-α and IL-1β did not alter the excitatory synaptic transmission (Yamawaki et al., 2022). In the ventral PAG, using female tyrosine hydroxylase-eGFP reporter mice, it was suggested that exposure to TNF-α decreased the excitability of dopaminergic neurons along with a modest reduction in glutamatergic synaptic transmission (Pati and Kash, 2021).
Activated microglia and other immune cells release not only TNF-α but also multiple types of interleukins. In this section, we discuss the roles of interleukins in plasticity induction. Interleukins are secreted in various brain regions, including the cerebellum, brainstem, hippocampus, and cerebrum, after injection of LPS to both the peripheral and central nervous systems (Quan et al., 1994). Here, we introduce the roles of IL-1, 6, and 17.
IL-1 is a protein with a molecular weight of 17 kDa, which is secreted from various cells in the body other than macrophages. Its function is not only for immune cells but also for connective tissue and CNS neurons. There are two types of molecular species, IL-1α and 1β. Another molecule with homology is the IL-1 receptor antagonist (IL-1Ra), which binds to the same receptor but does not convey a signal. IL-1 signal induces the expression of IL-8 and IL-6 by activating NFκB and AP-1 via MyD88. There are two types of their receptors: type IR transmits a signal, but type IIR has a deficit in the cytoplasmic region and does not transmit the signal. Similar signaling is found in IL-18R and TLR family molecules (Dinarello, 2018; Fields et al., 2019).
Among interleukins, IL-1β and IL-6 had been reported to prevent the induction of synaptic and non-synaptic plasticity in the hippocampal CA1 regions couple of decades ago (Bellinger et al., 1993; Li et al., 1997; Ross et al., 2003). These studies showed the impairment or the reduction of LTP of the excitatory synaptic transmission and the population spikes by administrating interleukins before applying the tetanus stimulation (i.e., LTP induction protocol) in the hippocampal slice preparations. In vivo, administration of LPS to the fourth ventricle impaired hippocampal memory tasks (Min et al., 2009). In hippocampal slices, it is reported that LTP induction was facilitated following 30 ng/ml IL-1β perfusion for 10 min, and LTD induction was prevented (Nisticò et al., 2013). In mPFC pyramidal neurons, IL-1β does not change the intrinsic excitability of action potential firing (Yamawaki et al., 2022). However, there is a difference in the modulation of synaptic transmission in primary culture. Yang et al. (2005) showed that more than 10 ng/ml IL-1β decreased the frequencies but not the amplitudes of spontaneous and miniature EPSC of neurons, indicating the suppression of presynaptic release. In the same study, IL-1β increased current mediated by both NMDA receptors and L-type Ca2+ channels (Yang et al., 2005). In the primary culture of hippocampal neurons, it was shown that IL-1β increases NMDA receptor function and Ca2+ influx through activation of Src-family tyrosine kinases and subsequent phosphorylation of NR2 subunits (Viviani et al., 2003). IL-1β also reduces the level of newly synthesized proteins in dendrites following chemically induced LTP (cLTP) by inhibiting Akt/mTOR signal transduction (Prieto et al., 2019a). Another inflammatory cytokine, IL-18, on the other hand, does not show a reduction of cLTP (Prieto et al., 2019b). Together, these studies suggest that IL-1β potentially modulates various forms of neuronal activity of hippocampal neurons: synaptic activity, Ca2+ signaling, and mTOR signaling required for spine stabilization and memory consolidation. Nevertheless, cultured neurons are immature and without an immunological milieu, so we cannot always consider the findings as to the biological representatives of cortical responses.
In the cerebellum, iontophoretic administration of IL-1β to the cerebellum increases the firing frequency of Purkinje cells in vivo (Motoki et al., 2009). In an EAE model, results suggest that the waveform of spontaneous EPSC was expanded by the activity of Bergman glia, EAAT, and IL-1β, but not by TNF-α, and such modulation was reverted by administration of IL-1Ra (Mandolesi et al., 2013).
A recent study by Willis et al. (2020) revealed repopulating microglia after traumatic brain injury promote brain repair in an IL-6-dependent manner (Willis et al., 2020). In general, IL-6 receptors (also known as CD126) include the membrane-bound IL-6 receptor (IL-6R) as well as the secretory soluble IL-6 receptor (sIL-6R) in the serum (Kishimoto, 2010). The secretory type has a structure in which the intracellular and transmembrane regions of the membrane-bound receptor are truncated, and the secretory receptor also exhibits IL-6 affinity similar to that of the membrane-bound receptor. IL-6R cannot function by itself because it does not have the ability for signal transduction. The IL-6R can transmit signals only by associating with gp130. gp130 is a protein widely expressed in various types of cells, and sIL-6R associated with IL-6 acquires reactivity by interacting with gp130. When the ligand binds to the IL-6R, the IL-6R associated with gp130 transmits a signal into the cell, mainly via two downward signaling pathways. One is the JAK-STAT pathway, in which activated JAK1/2 phosphorylates a tyrosine residue of gp130. The phosphorylated tyrosine of gp130 binds to the SH2 domain of the STAT1/3 molecule. When the transcription factor STAT1/3 is activated via the SH2 domain, the complex translocates into the nucleus, and binds to DNA sequences, inducing transcription of the target genes. The other is the MAPK pathway, in which Shp2 binds to gp130, it converts Ras, a small G protein, into an active form via the adapter protein Grb2. Then, Ras activates Raf-1, Raf-1 phosphorylates MEK, and MEK activates ERK, in turn. Activated ERK regulates gene expression after translocating into the nucleus. Surprisingly, IL-6 may alter the immune milieu of the brain parenchyma via peripheral stimulation. Exposure to chronic social stress deteriorates BBB integrity via loss of tight junction, promoting peripheral IL-6 passage across the BBB and depression behavior (Menard et al., 2017). In the rat slice preparations of the temporal cortex, IL-6 selectively reduced the amplitude of inhibitory postsynaptic currents without affecting excitatory postsynaptic currents (Garcia-Oscos et al., 2012). The IL-6-induced decrease in inhibitory postsynaptic currents is mediated by gp130 and via internalization of GABAA receptors. IL-6 shifts the balance between synaptic inhibition and excitation to increased excitability (Garcia-Oscos et al., 2012).
Since the discovery of IL-6 in 1986 (Kishimoto, 2010), the localization of the mRNA of IL-6 and IL-6A has been revealed in various regions of the rodent brain, including the cerebellum, medulla, neocortex, and hippocampus (Schöbitz et al., 1993; Gadient and Otten, 1994a,b). In the cerebellum, the IL-6R and gp130 express in neurons, including Purkinje cells and granule cells (Morikawa et al., 2000). And chronic administration of interleukins is neurotoxic and induces apoptosis of granule cells and Purkinje cells (Peng et al., 2005; Kaur et al., 2014). In the GFAP-IL6 transgenic mouse, IL6 is overexpressed in the brain under the control of the GFAP promoter, as a model of chronic brain inflammation (Campbell et al., 1993). According to Gyengesi et al. (2019), the motor performance of GFAP-IL6 mice showed an age-dependent difference: reduced performance on the rotarod, higher ataxia scores, and deficits in beam walking, later in life. GFAP-IL6 mice also showed an increased number of Iba1-positive microglia and high TNF-α levels, as well as a reduction of cerebellar volume, which starts at 6 months to a loss of about 50%. While synaptophysin levels as a presynaptic marker remained unchanged, PSD95 levels as a postsynaptic marker decreased in the aging GFAP-IL6 mice (Gyengesi et al., 2019). Purkinje cells of GFAP-IL6 mice show a relatively low firing rate of simple spikes, compared to wild-type. And characteristically, a significantly greater proportion of Purkinje neurons from GFAP-IL6 mice exhibited an oscillatory pattern of spontaneous firing (Nelson et al., 1999). The pause period after stimulation of a climbing fiber afferent (complex spike) of Purkinje neurons was significantly longer than control, indicating that IL-6 could act directly on Purkinje neurons to alter their physiological properties (Nelson et al., 1999). Purkinje cells that are chronically exposed to IL-6 also show elevated resting levels of intracellular calcium, an increase in both AMPA-induced and metabotropic glutamate receptor (mGluR)-activated intracellular Ca2+ signals, and release from intracellular Ca2+ stores (Nelson et al., 2002, 2004; Gruol, 2013). These studies imply the possible modulation or even disruption of the induction of various forms of plasticity of Purkinje cells (Ohtsuki and Hirano, 2008; Ohtsuki et al., 2009, 2020). Another study in cultured cerebellar granule neurons suggests chronic incubation with IL-6 at a concentration of 5 ng/ml alters NMDA receptor-mediated membrane responses, increases Ca2+ influx through the L-, N-, and P/Q-type voltage-gated calcium channels, and enhances neurotoxicity in a developmentally regulated manner (Qiu et al., 1995, 1998). In contrast, the following study suggests a reduction of the activity of L-type calcium channels by pretreatment of granule cell culture with 120 ng/ml IL-6 (Ma et al., 2012). Thus, we notice that modulation of the excitability of CNS neurons by inflammatory cytokines is dependent on the concentration. Rather, an increase in the levels of IL-6 and its receptors in the cerebellar cortex may correlate with the disruption of the cerebellar functions associated with the cerebellum-related psychiatric disease models.
IL-17A is a glycoprotein with 21 kDa molecular weight, which is secreted as a disulfide-linked homodimer. In 1993, Rouvier et al. cloned IL-17A from the cDNA library of a murine cytotoxic T lymphocyte hybridoma (Rouvier et al., 1993; Iwakura et al., 2011; McGeachy et al., 2019). Murine IL-17A is composed of 147 amino acid residues that share 63% amino acid sequence homology with human IL-17A of 155 amino acids (Iwakura et al., 2011). Five additional cytokines, IL-17B, IL-17C, IL-17D, IL-17E (also known as IL-25), and IL-17F, were identified, and they form the IL-17 family (Kolls and Lindén, 2004). IL-17 strongly induces inflammation by interacting with a wide range of cell types (epithelial cells, endothelial cells, fibroblasts, osteoblasts, macrophages, dendritic cells, etc.), by producing other cytokines (G-CSF, GM-CSF, IL-6, IL-1β, TGFβ, TNF-α, etc.), chemokines (CCL2/MCP-1, CXCL1/GROα, CXCL2/GROβ, CXCL5, CXCL8/IL-8, CXCL10, etc.) and prostaglandins (PGE2, etc.), and by driving migration of neutrophils and macrophages. Although IL-17A and IL-17F share the highest amino acid sequence homology and perform distinct functions, the functions of IL-17B, IL-17C, and IL-17D remain less understood. IL-17A is the signature cytokine of the recently identified T helper 17 (Th17) cell subset (Iwakura et al., 2011; McGeachy et al., 2019). The IL-17 receptor (IL-17R) family includes five members (IL-17RA to IL-17RE), containing conserved extracellular fibronectin-like domain, cytoplasmic SEF/IL-17R (SEFIR) domain, and cytoplasmic C/EBPβ activation domains (Iwakura et al., 2011; McGeachy et al., 2019). Both homo- and heterodimers are functional receptors for IL-17 family cytokines. IL-17A and IL-17F activate NF-κB, MAPK, and C/EBP after binding to the receptors (Iwakura et al., 2011; McGeachy et al., 2019).
According to Ribeiro et al. (2019), the extent of the hippocampal LTP and AMPA/NMDA ratio was reduced in the IL-17-deficient mice after the short-term memory training (i.e., Y-maze) but not long-term training (i.e., Morris water maze). They showed that IL-17 produced from meningeal γδ T cells modulated synaptic plasticity induction and IL-17 promoted BDNF production in astrocytes using GFAP-IL17R deletion mice. Administration of IL-17 or BDNF amended the synaptic plasticity and behavior. Therefore, the IL-17 derived from a fetal-derived meningeal-resident γδ-T-cell subset was suggested to promote cognitive behavior (Ribeiro et al., 2019). In an early study of IL-17 expression by Moore et al. (2002), mRNA of IL-17B expresses in the cerebral cortex, hippocampus, cerebellum, and spinal cord of mice. In the cerebellum, the expression is prominent in the Purkinje cells, although cells in the granule cell layer and the molecular layer were not. Monoclonal anti-IL-17B histochemistry displays its expression in neurons and glial cells in the cerebellum, hippocampus, and cerebral cortex (Moore et al., 2002). IL-17A is also studied in the mouse models of neuropathic pain and multiple sclerosis (Luo et al., 2019; Di Filippo et al., 2021). Luo et al. (2019) demonstrated that IL-17A is mainly released from spinal cord astrocytes, and its receptor IL-17R exists in somatostatin-expressing interneurons. IL-17A increases EPSCs and decreases IPSCs in lamina IIo somatostatin-expressing neurons. Administration of 10 ng/ml IL-17A induces hyperexcitability of neurons in dorsal root ganglia. IL-17R antibody decreases the excitability of neurons. Selective knockdown of IL-17R in spinal somatostatin-expressing interneurons reduces paclitaxel-induced hypersensitivity after paclitaxel and neuropathic pain (Luo et al., 2019). According to Di Filippo et al. (2021), the IL-17A receptor (IL-17RA) is highly expressed in neurons in the hippocampal CA1, and exposure to 20 ng/ml IL-17A disrupts hippocampal LTP through the activation of IL-17RA and p38 mitogen-activated protein kinase (MAPK). IL-17A overexpression mimics the disruption of LTP as neurons in the EAE. The loss of IL-17A ameliorates EAE-related cognitive deficits (Di Filippo et al., 2021). In contrast, the roles of cerebellar IL-17 are not well-understood yet, and an advance investigation is required.
TGFβ family members (: TGFβ1, TGFβ2, and TGFβ3) are synthesized by many cell types, including macrophages, as pre-pro-TGFβ (pre-propeptide), and the pre-propeptide is cleaved in the endoplasmic reticulum and secreted as an inactive dimer bound as a latent complex, tethered by the extracellular matrix. Serum proteinases and matrix metalloproteinases (MMPs) liberate the latent form, and TGFβ-binding to transforming growth factor beta receptors (TGFBRs) transduce the signaling in the SMAD-dependent and SMAD-independent manners (Spittau et al., 2020). TGFβ1 has been considered as an essential factor for development, differentiation, activation, reactivation, and homeostasis of both innate and adaptive immunity. Interestingly, TGFβ1 increases phosphorylation of cAMP response element-binding protein (CREB) and augments the maintenance of LTP (i.e., late-phase LTP; Caraci et al., 2015). Exogenous administration of both IL-10 and TGFβ1 facilitates induction of LTP in hippocampal CA1 neurons without any effect on short-term plasticity (Nenov et al., 2019). However, there is a possibility of secondary effect via regulating microglia functions (Spittau et al., 2020). Further, these studies lack an investigation of the membrane properties of neurons after exposure to TGFβ, like many studies on interleukins and endotoxin. As discussed in the latter paragraph: Meta-plasticity by brain immunity, membrane properties are crucial for inducing multiple forms of plasticity. Voltage- and Ca2+-dependent ion channels underlie the membrane properties, which determine the intrinsic excitability of the neurons, affect the Ca2+ influx through the voltage-gated and ligand-gated Ca2+ channels, and decide the consequence as the plasticity induction.
Prostaglandins (PGs) are the physically active lipid mediators, which are produced in almost all tissues and involved in the induction of plasticity in the brain (Akaneya and Tsumoto, 2006; Le et al., 2010). In the inflammatory pain sensitization, PGE2 activates protein kinase A (PKA), specifically phosphorylates glycine receptor subtype GlyR alpha3 and reduces the glycinergic transmission by inhibiting GlyR3 (Harvey et al., 2004). PGE2 specifically decreased fast inhibitory synaptic current through glycine receptors, but not GABAA, AMPA, and NMDA receptors in the rodent spinal cord dorsal horn. PGE2, but not PGF2 alpha, PGD2, or PGI2, reduced inhibitory glycinergic synaptic transmission (Ahmadi et al., 2002). In microglia, phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) drives PGE2 release, which activates PGE2 receptor E-prostanoid 2 (EP2) of neurons in the dorsal horn. Thus, PGE2 contributes to signaling between microglia and neurons in pain maintenance after injury (Zhao et al., 2007). PGE2 signaling could also regulate membrane excitability (i.e., action potentials and back-propagating action potential-associated dendritic Ca2+ influx) and LTP in hippocampal perforant path-dentate gyrus synapses (Chen et al., 2002). A recent study indicated that striatal microglia modulate the excitability of neurons via IL-6 and PGE2 signaling and induce negative mood (Klawonn et al., 2021). Peripheral inflammation was also suggested to induce PGE2-mediated modulation of the dopaminergic circuit for motivation in the striatum (Fritz et al., 2016). Considering the highly broad modulation by PGs (Furuyashiki and Narumiya, 2011), microglia-produced PGs could influence memory, learning, mood, and behaviors. PGs have been traditionally thought to function mostly as mediators of acute inflammation, even in the nervous system. However, a recent view suggests the involvement of PGs in chronic inflammation. PGs crosstalk with cytokines and both PGs and cytokines may synergistically activate NF-κB to induce the expression of inflammation-related genes (Yao and Narumiya, 2019), and thus, may generate the heterogeneity in brain inflammation.
Chemokines are classified into four main subfamilies: CXC, CC, CX3C, and C chemokines. Four cysteine residues are conserved in the amino acid sequence of a typical chemokine. All of these proteins bind with G protein-coupled receptors containing seven transmembrane domains called chemokine receptors: CXCRs, CCRs, CX3CR1, and XCR1. In mouse cerebellar slices, 50-nM chemokine (C-X-C motif) ligand 1 (CXCL1; also known as GROα and GRO1) suppressed the induction of the LTD of parallel fiber-Purkinje cell synapses (Giovannelli et al., 1998). CXCL1 and IL-8 as CXCL8 increased the number of spontaneous synaptic transmissions (Giovannelli et al., 1998). It is also reported that 60-nM CXCL2 (also known as GROβ and GRO2) increased the frequency of synaptic currents in rats (Ragozzino et al., 1998). In the hippocampus, CXCL10 (10 ng/ml) suppressed the extent of LTP (Vlkolinský et al., 2004). Acute application of CX3CL1 chemokines as fractalkine, which functions as an adhesion molecule, caused a sustained reduction of EPSC amplitude (Ragozzino et al., 2006). The CX3CL1-induced LTD is due to a decrease in postsynaptic responsiveness and is mediated by the CX3CL1 receptor (CX3CR1). CX3CL1-induced depression of EPSC was not observed in the absence of afferent fiber stimulation or AMPA receptor activation, respectively, indicating the requirement of sustained CX3CR1 activation with AMPA receptor activation for its development. Ca2+-, cAMP-, and Ser845 GluR1 phosphorylation-dependent process is likely to modulate CX3CL1-induced LTD. Therefore, different chemokines modulate the function of synapses via distinct cell signaling by influencing the balance between kinase and phosphatase activity (Ragozzino et al., 2006).
As stated previously, transient exposure to a chemokine CCL2/MCP-1 increases the presynaptic release of the spinal cord (Ma et al., 2020). In hippocampal CA1 pyramidal neurons, acute administration of CCL2 increased excitability and presynaptic transmission (Zhou et al., 2011; Duan et al., 2018). On the other hand, a transgenic mouse expressing CCL2 under regulation by GFAP promoter decreased the excitability of neurons and weakened plasticity extent, which implies that chronic exposure to the chemokine may change the homeostasis of the activity in the hippocampal circuit (Nelson et al., 2011; Bray et al., 2013). Nevertheless, not only the identification of leukocytes releasing those molecules in the brain but the effects on cerebellar synaptic transmission and intrinsic excitability of neurons are unsolved.
Recently, IL-17 has been shown involved in the disruption of the cortical neural circuit and aberrant behavioral phenotypes (Choi et al., 2016; Reed et al., 2020), which attracted researchers. Viral infection during pregnancy has been considered correlated with increased frequency of autism spectrum disorder, ASD, in offspring. This observation has been modeled in rodents subjected to the maternal immune activation, MIA. The Th17 and IL-17 in mothers are involved in MIA-induced behavioral anomalies of offspring, representing an increase in the pup ultrasonic vocalization (USV) responses, a decrease in the social interaction, and an increase in the marble burying. MIA also induces an abnormal cortical layer formation, dependent on maternal IL-17 in the fetal brain. And the administration of anti-IL-17a antibody to the pregnant mother ameliorates both behavioral and histological phenotypes (Choi et al., 2016). Since some children with ASD show an improvement in their behavioral symptoms during a fever, a sign of systemic inflammation, the social behavior deficits in the offspring exposed to MIA can be temporarily rescued by the administration of LPS. This was accompanied by a reduction in neuronal activity in the primary somatosensory cortex dysgranular zone (S1DZ) and its hyperactivity in the behavioral phenotypes of the offspring exposed to MIA. It was also due to an increase in IL-17 in S1DZ (Reed et al., 2020). However, the study did not answer why the administration of LPS rescued the deficits of sociability in the MIA offspring and how neuronal hyperexcitability was attenuated. Further, such studies did not see the activity in the different regions of the MIA-offspring brain nor the brain-wide connectivity, so the effect of MIA was merely investigated in the studied regions.
As seen above, the immune cell activation and inflammatory mediators modulate the intrinsic excitability of neurons via such as SK channels and voltage-gated Ca2+ channels. Modulation of the intrinsic excitability of neurons could determine the induction of synaptic plasticity. The modulation of intrinsic excitability of neurons, in turn, changes the membrane voltage of neurons, which affects the driving force of ion current through channels, and the Ca2+ influx via voltage-gated Ca2+ channels. The membrane properties determine the induction of synaptic plasticity. It is because the induction of activity-dependent synaptic plasticity relies on the activation of the Ca2+ influx and downward signaling of the kinases and phosphatases, which drive exocytosis and endocytosis of neurotransmitter channels, including AMPAR, NMDAR, mGluR, GABAAR, GABABR, glycine receptor, and so on. Indeed, the exposure to TNF-α to cerebellar Purkinje cells increases their excitability, suppresses the induction of further intrinsic plasticity, and potentiates both pre- and post-synaptic efficacy. It could imply that further induction of plasticity at granule cell synapses is occluded since the cellular signaling of intrinsic plasticity and microglia-triggered intrinsic plasticity share the intracellular signaling (Yamamoto et al., 2019). Hyperexcitability of the cerebellar cortex after LPS injection lasts at least 6 h (Yamamoto et al., 2019) so that further induction of plasticity and learning could be hampered for the duration. In the mPFC, it is speculated that further induction of synaptic plasticity is suppressed after activation of microglia and exposure to TNF-α (Yamawaki et al., 2022). Akin to this, the concept of “meta-plasticity” as the plasticity of synaptic plasticity is coined by Drs. Wickliffe C. Abraham and Mark F. Bear (Abraham and Bear, 1996; Abraham, 2008; Hulme et al., 2013). Brain immunity should give another field of research, again. It is also elusive how spike-timing-dependent plasticity (Zhang et al., 1998; Wang et al., 2000; Dan and Poo, 2004; Jörntell and Hansel, 2006; Suvrathan et al., 2016; Sgritta et al., 2017; Ohtsuki et al., 2020) is modulated by aberrant immunity or in the animal models with brain inflammation. Chronic administration of excess immunity during development may modulate the neuronal activity long-lastingly. In dendrites, the intrinsic excitability influences the directionality of the LTP/LTD induction (Wang et al., 2003; Sjöström and Häusser, 2006; Sjöström et al., 2008) and the conductivity of synaptic transmission along dendrites (Ohtsuki et al., 2012; Ohtsuki and Hansel, 2018; Ohtsuki, 2020). Considering those, many previous studies just showing the correlation between the impairment of synaptic plasticity and disruption of memory by immune stimulants would be required reinvestigation. New findings are expected to come from such efforts. Concurrently, it would be required to prove the identification of not only inflammatory mediators but also source immune cells by abolishing related cells (by depletion of microglia and T cells pharmacologically or genetically) and by regulating the activity of specific immune cells with gene manipulation.
Despite the difficulty in interpreting the biological significance of behaviors, learning, and cognition, the interaction of immune cells with neurons is a quite important feature of brain functioning. The immune-triggered plasticity and related meta-plasticity are expected to be involved in psychiatric disorders evoked by aberrant immunity, specifically at the stage of neurodevelopment and maturation of brain vasculatures (Hopkins and Rothwell, 1995; Gilmore et al., 2004; Choi et al., 2016; Menard et al., 2017; Greene et al., 2018; Nie et al., 2018; Segawa et al., 2021; Parvez and Ohtsuki, 2022). It is also plausible that brain immunity, such as microglia or disease-associated macrophages in the brain, may have an “immune-memory” in the ways of responses against immune stimulants via an inflammatory mediator signaling and plasticity. Subsequently, the myelination of neuronal axons might also become impaired, which is seen in autism patients (Ben Bashat et al., 2007) and schizophrenia model animals (Hirahara et al., 2013). Questions are the timing when brain functions are severely affected; which brain regions are vulnerable; what specificity and localization the brain immune cells have; how long immune reactivity maintains; what the molecular or genetic mechanisms in immune cells and neurons are; and whether we can suppress or revert the reactivity. In the disease brains, resultant anomalies in the brain-wide functional connectivity caused by immune-triggered plasticity have been found recently (Yamamoto et al., 2019; Tsurugizawa et al., 2020), and the pathways for diseases would be clarified (Ohtsuki et al., 2020; Wang et al., 2021). Those efforts should help us to clarify the target brain regions, immune-cell targets, or pathways for emerging the disease and to ameliorate symptoms with the drug delivery. Highly multiplexed special expression pattern of distinct marker proteins with multi-dimensional data analyses must enable us to interpret the whole embodied schema of immune-triggered plasticity and disease association in the brain microenvironment and tissue architecture, as done in the field of cancer (Fernández-Zapata et al., 2020; Chen et al., 2021; van Maldegem et al., 2021; Kuett et al., 2022). Additionally, ion channels significant for the intrinsic excitability plasticity of neurons, such as Ca2+-activated K+-channels (i.e., SK-, IK-, and BK-channels) or HCN channels, would be another potential target for the drug administration, while we should note that inflammatory mediators could be modulators of not only fast synaptic transmissions and the intrinsic excitability but also metabotropic receptors of monoamine, mGluRs, GABABR, and other G protein-coupled receptors, which are also involved in the plasticity induction.
In this review, we introduced the induction of immune-triggered plasticity of neurons across different brain regions. Typically, in the cerebellum, the Purkinje cells showed hyperexcitability plasticity of action potential firing, as well as an increase in excitatory synaptic transmission. In contrast, the mPFC pyramidal cells showed hypoexcitability plasticity as the reduction of action potential firing (Figure 1). In other brain regions, such as the spinal cord, midbrain, hippocampus, amygdala, and basal ganglia, neurons show different forms of plasticity, and it is obvious that inflammatory mediators such as inflammatory cytokines, chemokines, and neurotrophic factors change the activity of neurons in different ways in the different brain immune milieu. Further, the exposure to inflammatory cytokines influences the induction of the following plasticity in the ways of reduction or facilitation of the induction of plasticity, as an immune-mediated meta-plasticity. However, it is more complex than easily considered because the modulation of the intrinsic excitability of neurons by transient and chronic activation of the brain immunity also alters the induction of synaptic plasticity and disease association. Therefore, this field is quite broad and full of phenomena highly of biological significance.
GO designed the research. MH, TM, TK, and GO wrote the original manuscript. YW, TS, and GO edited the manuscript. MH, TK, and GO revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Mitsubishi Foundation, the Takeda Science Foundation, and JSPS Kakenhi Grant-in-Aid for Scientific Research (B). The funders had no role in the study design, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to present our deep appreciation to Shu Narumiya, Md Sorwer Alam Parvez, and Yuki Yamawaki for their invaluable comments and support of the research. We also appreciate Sumitomo Dainippon Pharma Co., Ltd., Ono Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation., and KYORIN Pharmaceutical Co., Ltd. as the sponsors for the Department of Drug Discovery Medicine.
Abraham, W. C.. (2008). Metaplasticity: tuning synapses and networks for plasticity. Nat. Rev. Neurosci. 9, 387. doi: 10.1038/nrn2356
Abraham, W. C., and Bear, M. F. (1996). Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 19, 126–130
Ahmadi, S., Lippross, S., Neuhuber, W. L., and Zeilhofer, H. U. (2002). PGE(2) selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nat. Neurosci. 5, 34–40. doi: 10.1038/nn778
Akaneya, Y., and Tsumoto, T. (2006). Bidirectional trafficking of prostaglandin E2 receptors involved in long-term potentiation in visual cortex. J. Neurosci. 26, 10209–10221. doi: 10.1523/JNEUROSCI.3028-06.2006
Askew, K., Li, K., Olmos-Alonso, A., Garcia-Moreno, F., Liang, Y., Richardson, P., et al. (2017). Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. 18, 391–405. doi: 10.1016/j.celrep.2016.12.041
Ayata, P., Badimon, A., Strasburger, H. J., Duff, M. K., Montgomery, S. E., Loh, Y. E., et al. (2018). Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci. 21, 1049–1060. doi: 10.1038/s41593-018-0192-3
Baek, S. J., Park, J. S., Kim, J., Yamamoto, Y., and Tanaka-Yamamoto, K. (2022). VTA-projecting cerebellar neurons mediate stress-dependent depression-like behaviors. Elife 11:e72981. doi: 10.7554/eLife.72981
Banisadr, G., Gosselin, R. D., Mechighel, P., Kitabgi, P., Rostène, W., and Melik-Parsadaniantz, S. (2005b). Highly regionalized neuronal expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) in rat brain: evidence for its colocalization with neurotransmitters and neuropeptides. J. Comput. Neurol. 489, 275–292. doi: 10.1002/cne.20598
Banisadr, G., Gosselin, R. D., Mechighel, P., Rostène, W., Kitabgi, P., and Mélik Parsadaniantz, S. (2005a). Constitutive neuronal expression of CCR2 chemokine receptor and its colocalization with neurotransmitters in normal rat brain: functional effect of MCP-1/CCL2 on calcium mobilization in primary cultured neurons. J. Comput. Neurol. 492, 178–192. doi: 10.1002/cne.20729
Barichello, T., Sayana, P., Giridharan, V. V., Arumanayagam, A. S., Narendran, B., Giustina, A. D., et al. (2019). Long-term cognitive outcomes after sepsis: a translational systematic review. Mol. Neurobiol. 56, 186–251. doi: 10.1007/s12035-018-1048-2
Beattie, E. C., Stellwagen, D., Morishita, W., Bresnahan, J. C., Ha, B. K., Von Zastrow, M., et al. (2002). Control of synaptic strength by glial TNFalpha. Science 295, 2282–2285 doi: 10.1126/science.1067859
Bellinger, F. P., Madamba, S., and Siggins, G. R. (1993). Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 628, 227–234.
Belmeguenai, A., Hosy, E., Bengtsson, F., Pedroarena, C. M., Piochon, C., Teuling, E., et al. (2010). Intrinsic plasticity complements long-term potentiation in parallel fiber input gain control in cerebellar Purkinje cells. J. Neurosci. 30, 13630–13643. doi: 10.1523/JNEUROSCI.3226-10.2010
Ben Bashat, D., Kronfeld-Duenias, V., Zachor, D. A., Ekstein, P. M., Hendler, T., Tarrasch, R., et al. (2007). Accelerated maturation of white matter in young children with autism: a high b value DWI study. Neuroimage 37, 40–47. doi: 10.1016/j.neuroimage.2007.04.060
Black, R. A., Rauch, C. T., Kozlosky, C. J., Peschon, J. J., Slack, J. L., Wolfson, M. F., et al. (1997). A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385, 729–733.
Blitz, D. M., Foster, K. A., and Regehr, W. G. (2004). Short-term synaptic plasticity: a comparison of two synapses. Nat. Rev. Neurosci. 5, 630–640. doi: 10.1038/nrn1475
Bray, J. G., Reyes, K. C., Roberts, A. J., Ransohoff, R. M., and Gruol, D. L. (2013). Synaptic plasticity in the hippocampus shows resistance to acute ethanol exposure in transgenic mice with astrocyte-targeted enhanced CCL2 expression. Neuropharmacology 67, 115–125. doi: 10.1016/j.neuropharm.2012.11.007
Brennan, J. J., and Gilmore, T. D. (2018). Evolutionary origins of toll-like receptor signaling. Mol. Biol. Evol. 35, 1576–1587. doi: 10.1093/molbev/msy050
Campbell, I. L., Abraham, C. R., Masliah, E., Kemper, P., Inglis, J. D., Oldstone, M. B., et al. (1993). Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. U. S. A. 90, 10061–10065.
Cantaut-Belarif, Y., Antri, M., Pizzarelli, R., Colasse, S., Vaccari, I., Soares, S., et al. (2017). Microglia control the glycinergic but not the GABAergic synapses via prostaglandin E2 in the spinal cord. J. Cell Biol. 216, 2979–2989. doi: 10.1083/jcb.201607048
Caraci, F., Gulisano, W., Guida, C. A., Impellizzeri, A. A., Drago, F., Puzzo, D., et al. (2015). A key role for TGF-β1 in hippocampal synaptic plasticity and memory. Sci. Rep. 5:11252. doi: 10.1038/srep11252
Carswell, E. A., Old, L. J., Kassel, R. L., Green, S., Fiore, N., and Williamson, B. (1975). An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. U. S. A. 72, 3666–3670.
Chen, C., Magee, J. C., and Bazan, N. G. (2002). Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. J. Neurophysiol. 87, 2851–2857. doi: 10.1152/jn.2002.87.6.2851
Chen, C., McDonald, D., Blain, A., Sachdeva, A., Bone, L., Smith, A. L. M., et al. (2021). Imaging mass cytometry reveals generalised deficiency in OXPHOS complexes in Parkinson's disease. NPJ Parkinsons Dis. 7:39. doi: 10.1038/s41531-021-00182-x
Chen, G., Kim, Y. H., Li, H., Luo, H., Liu, D. L., Zhang, Z. J., et al. (2017). PD-L1 inhibits acute and chronic pain by suppressing nociceptive neuron activity via PD-1. Nat. Neurosci. 20, 917–926. doi: 10.1038/nn.4571
Choi, G. B., Yim, Y. S., Wong, H., Kim, S., Kim, H., Kim, S. V., et al. (2016). The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939. doi: 10.1126/science.aad0314
Colonna, M., and Butovsky, O. (2017). Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 35, 441–468. doi: 10.1146/annurev-immunol-051116-052358
Cook, A. A., Fields, E., and Watt, A. J. (2021). Losing the beat: contribution of purkinje cell firing dysfunction to disease, and its reversal. Neuroscience 462, 247–261. doi: 10.1016/j.neuroscience.2020.06.008
Coull, J. A., Beggs, S., Boudreau, D., Boivin, D., Tsuda, M., Inoue, K., et al. (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021. doi: 10.1038/nature04223
Coull, J. A., Boudreau, D., Bachand, K., Prescott, S. A., Nault, F., Sík, A., et al. (2003). Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424, 938–942. doi: 10.1038/nature01868
Dan, Y., and Poo, M. M. (2004). Spike timing-dependent plasticity of neural circuits. Neuron 44, 23–30. doi: 10.1016/j.neuron.2004.09.007
De Biase, L. M., Schuebel, K. E., Fusfeld, Z. H., Jair, K., Hawes, I. A., Cimbro, R., et al. (2017). Local cues establish and maintain region-specific phenotypes of Basal Ganglia Microglia. Neuron 95, 341–356.e6. doi: 10.1016/j.neuron.2017.06.020
Deczkowska, A., Keren-Shaul, H., Assaf Weiner, A., Colonna, M., Schwartz, M., and Amit, I. (2018). Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 173, 1073–1081. doi: 10.1016/j.cell.2018.05.003
Desai, N. S., Rutherford, L. C., and Turrigiano, G. G. (1999). BDNF regulates the intrinsic excitability of cortical neurons. Learn. Mem. 6, 284–291.
Di Filippo, M., Mancini, A., Bellingacci, L., Gaetani, L., Mazzocchetti, P., Zelante, T., et al. (2021). Interleukin-17 affects synaptic plasticity and cognition in an experimental model of multiple sclerosis. Cell Rep. 37:110094. doi: 10.1016/j.celrep.2021.110094
Dinarello, C. A.. (2018). Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 281, 8–27. doi: 10.1111/imr.12621
Donnelly, C. R., Jiang, C., Andriessen, A. S., Wang, K., Wang, Z., Ding, H., et al. (2021). STING controls nociception via type I interferon signalling in sensory neurons. Nature 591, 275–280. doi: 10.1038/s41586-020-03151-1
Dos Santos, S. E., Medeiros, M., Porfirio, J., Tavares, W., Pessôa, L., Grinberg, L., et al. (2020). Similar microglial cell densities across brain structures and mammalian species: implications for brain tissue function. J. Neurosci. 40, 4622–4643. doi: 10.1523/JNEUROSCI.2339-19.2020
Duan, L., Zhang, X. D., Miao, W. Y., Sun, Y. J., Xiong, G., Wu, Q., et al. (2018). PDGFRβ cells rapidly relay inflammatory signal from the circulatory system to neurons via chemokine CCL2. Neuron 100, 183–200.e8. doi: 10.1016/j.neuron.2018.08.030
Fernández-Zapata, C., Leman, J. K. H., Priller, J., and Böttcher, C. (2020). The use and limitations of single-cell mass cytometry for studying human microglia function. Brain Pathol. 30, 1178–1191. doi: 10.1111/bpa.12909
Fields, J. K., Günther, S., and Sundberg, E. J. (2019). Structural basis of IL-1 family cytokine signaling. Front. Immunol. 10:1412. doi: 10.3389/fimmu.2019.01412
Fritz, M., Klawonn, A. M., Nilsson, A., Singh, A. K., Zajdel, J., Wilhelms, D. B., et al. (2016). Prostaglandin-dependent modulation of dopaminergic neurotransmission elicits inflammation-induced aversion in mice. J. Clin. Invest. 126, 695–705. doi: 10.1172/JCI83844
Furuyashiki, T., and Narumiya, S. (2011). Stress responses: the contribution of prostaglandin E(2) and its receptors. Nat. Rev. Endocrinol. 7, 163–175. doi: 10.1038/nrendo.2010.194
Gadient, R. A., and Otten, U. (1994a). Identification of interleukin-6 (IL-6)-expressing neurons in the cerebellum and hippocampus of normal adult rats. Neurosci. Lett. 182, 243–246.
Gadient, R. A., and Otten, U. (1994b). Expression of interleukin-6 (IL-6) and interleukin-6 receptor (IL-6R) mRNAs in rat brain during postnatal development. Brain. Res. 637, 10–14.
Gao, F., Liu, Z., Ren, W., and Jiang, W. (2014). Acute lipopolysaccharide exposure facilitates epileptiform activity via enhanced excitatory synaptic transmission and neuronal excitability in vitro. Neuropsychiatr. Dis. Treat. 10, 1489–1495. doi: 10.2147/NDT.S65695
Gao, X., Kim, H. K., Mo Chung, J., and Chung, K. (2007). Reactive oxygen species (ROS) are involved in enhancement of NMDA-receptor phosphorylation in animal models of pain. Pain 131, 262–271. doi: 10.1016/j.pain.2007.01.011
Garcia-Oscos, F., Salgado, H., Hall, S., Thomas, F., Farmer, G. E., Bermeo, J., et al. (2012). The stress-induced cytokine interleukin-6 decreases the inhibition/excitation ratio in the rat temporal cortex via trans-signaling. Biol. Psychiatry 71, 574–582. doi: 10.1016/j.biopsych.2011.11.018
George, J., Cunha, R. A., Mulle, C., and Amédée, T. (2016). Microglia-derived purines modulate mossy fibre synaptic transmission and plasticity through P2X4 and A1 receptors. Eur. J. Neurosci. 43, 1366–1378. doi: 10.1111/ejn.13191
Gilmore, J. H., Fredrik Jarskog, L., Vadlamudi, S., and Lauder, J. M. (2004). Prenatal infection and risk for schizophrenia: IL-1beta, IL-6, and TNFalpha inhibit cortical neuron dendrite development. Neuropsychopharmacology 29, 1221–1229. doi: 10.1038/sj.npp.1300446
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Ginhoux, F., and Prinz, M. (2015). Origin of microglia: current concepts and past controversies. Cold Spring Harb. Perspect. Biol. 7 :a020537. doi: 10.1101/cshperspect.a020537
Giovannelli, A., Limatola, C., Ragozzino, D., Mileo, A. M., Ruggieri, A., Ciotti, M. T., et al. (1998). CXC chemokines interleukin-8 (IL-8) and growth-related gene product alpha (GROalpha) modulate Purkinje neuron activity in mouse cerebellum. J. Neuroimmunol. 92, 122–132.
Gosselin, R. D., Varela, C., Banisadr, G., Mechighel, P., Rostene, W., Kitabgi, P., et al. (2005). Constitutive expression of CCR2 chemokine receptor and inhibition by MCP-1/CCL2 of GABA-induced currents in spinal cord neurones. J. Neurochem. 95, 1023–1034. doi: 10.1111/j.1471-4159.2005.03431.x
Grabert, K., Michoel, T., Karavolos, M. H., Clohisey, S., Baillie, J. K., Stevens, M. P., et al. (2016). Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 19, 504–516. doi: 10.1038/nn.4222
Grace, P. M., Hutchinson, M. R., Maier, S. F., and Watkins, L. R. (2014). Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 14, 217–231. doi: 10.1038/nri3621
Greene, C., Kealy, J., Humphries, M. M., Gong, Y., Hou, J., Hudson, N., et al. (2018). Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol. Psychiatry 23, 2156–2166. doi: 10.1038/mp.2017.156
Gruol, D. L.. (2013). Neuroimmune regulation of neurophysiology in the cerebellum. Cerebellum 12, 307–309. doi: 10.1007/s12311-012-0445-8
Gruol, D. L., and Nelson, T. E. (1997). Physiological and pathological roles of interleukin-6 in the central nervous system. Mol. Neurobiol. 15, 307–339.
Gudes, S., Barkai, O., Caspi, Y., Katz, B., Lev, S., and Binshtok, A. M. (2015). The role of slow and persistent TTX-resistant sodium currents in acute tumor necrosis factor-α-mediated increase in nociceptors excitability. J. Neurophysiol. 113, 601–619. doi: 10.1152/jn.00652.2014
Gyengesi, E., Rangel, A., Ullah, F., Liang, H., Niedermayer, G., Asgarov, R., et al. (2019). Chronic microglial activation in the GFAP-IL6 mouse contributes to age-dependent cerebellar volume loss and impairment in motor function. Front. Neurosci. 13, 303. doi: 10.3389/fnins.2019.00303
Habbas, S., Santello, M., Becker, D., Stubbe, H., Zappia, G., Liaudet, N., et al. (2015). Neuroinflammatory TNFα impairs memory via astrocyte signaling. Cell 163, 1730–1741. doi: 10.1016/j.cell.2015.11.023
Harvey, R. J., Depner, U. B., Wässle, H., Ahmadi, S., Heindl, C., Reinold, H., et al. (2004). GlyR alpha3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science 304, 884–887. doi: 10.1126/science.1094925
Hirahara, Y., Matsuda, K. I., Yamada, H., Saitou, A., Morisaki, S., Takanami, K., et al. (2013). G protein-coupled receptor 30 contributes to improved remyelination after cuprizone-induced demyelination. Glia 61, 420–431. doi: 10.1002/glia.22445
Hopkins, S. J., and Rothwell, N. J. (1995). Cytokines and the nervous system. I: expression and recognition. Trends Neurosci. 18, 83–88.
Hoshino, K., Takeuchi, O., Kawai, T., Sanjo, H., Ogawa, T., Takeda, Y., et al. (1999). Cutting edge: toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162, 3749–3752
Hulme, S. R., Jones, O. D., and Abraham, W. C. (2013). Emerging roles of metaplasticity in behaviour and disease. Trends Neurosci. 36, 353–362. doi: 10.1016/j.tins.2013.03.007
Hwang, I., Kim, B. S., Ko, H. R., Cho, S., Lee, H. Y., Cho, S. W., et al. (2022). Cerebellar dysfunction and schizophrenia-like behavior in Ebp1-deficient mice. Mol. Psychiatry 27, 2030–2041. doi: 10.1038/s41380-022-01458-1
Iwakura, Y., Ishigame, H., Saijo, S., and Nakae, S. (2011). Functional specialization of interleukin-17 family members. Immunity 34, 149–162. doi: 10.1016/j.immuni.2011.02.012
Janach, G. M. S., Reetz, O., Döhne, N., Stadler, K., Grosser, S., Byvaltcev, E., et al. (2020). Interferon-γ acutely augments inhibition of neocortical layer 5 pyramidal neurons. J. Neuroinflamm. 17:69. doi: 10.1186/s12974-020-1722-y
Jiang, J., Tang, B., Wang, L., Huo, Q., Tan, S., Misrani, A., et al. (2022). Systemic LPS-induced microglial activation results in increased GABAergic tone: a mechanism of protection against neuroinflammation in the medial prefrontal cortex in mice. Brain Behav. Immun. 99, 53–69. doi: 10.1016/j.bbi.2021.09.017
Jörntell, H., and Hansel, C. (2006). Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron 52, 227–238. doi: 10.1016/j.neuron.2006.09.032
Katsura, H., Obata, K., Mizushima, T., Sakurai, J., Kobayashi, K., Yamanaka, H., et al. (2006). Activation of Src-family kinases in spinal microglia contributes to mechanical hypersensitivity after nerve injury. J. Neurosci. 26, 8680–8690. doi: 10.1523/JNEUROSCI.1771-06.2006
Kaur, C., Sivakumar, V., Zou, Z., and Ling, E. A. (2014). Microglia-derived proinflammatory cytokines tumor necrosis factor-alpha and interleukin-1beta induce Purkinje neuronal apoptosis via their receptors in hypoxic neonatal rat brain. Brain Struct. Funct. 219, 151–170. doi: 10.1007/s00429-012-0491-5
Kawasaki, Y., Zhang, L., Cheng, J. K., and Ji, R. R. (2008). Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci. 28, 5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008
Khandaker, G. M., Cousins, L., Deakin, J., Lennox, B. R., Yolken, R., and Jones, P. B. (2015). Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry 2, 258–270. doi: 10.1016/S2215-0366(14)00122-9
Khawaled, R., Bruening-Wright, A., Adelman, J. P., and Maylie, J. (1999). Bicuculline block of small-conductance calcium-activated potassium channels. Pflugers Arch. 438, 314–321.
Kishimoto, T.. (2010). IL-6: from its discovery to clinical applications. Int. Immunol. 22, 347–352. doi: 10.1093/intimm/dxq030
Klapal, L., Igelhorst, B. A., and Dietzel-Meyer, I. D. (2016). Changes in neuronal excitability by activated microglia: differential Na(+) current upregulation in pyramid-shaped and bipolar neurons by TNF-α and IL-18. Front. Neurol. 7:44. doi: 10.3389/fneur.2016.00044
Klawonn, A. M., Fritz, M., Castany, S., Pignatelli, M., Canal, C., Similä, F., et al. (2021). Microglial activation elicits a negative affective state through prostaglandin-mediated modulation of striatal neurons. Immunity 54, 225–234.e6. doi: 10.1016/j.immuni.2020.12.016
Kleibeuker, W., Gabay, E., Kavelaars, A., Zijlstra, J., Wolf, G., Ziv, N., et al. (2008). IL-1 beta signaling is required for mechanical allodynia induced by nerve injury and for the ensuing reduction in spinal cord neuronal GRK2. Brain Behav. Immun. 22, 200–208. doi: 10.1016/j.bbi.2007.07.009
Kolls, J. K., and Lindén, A. (2004). Interleukin-17 family members and inflammation. Immunity 21, 467–476. doi: 10.1016/j.immuni.2004.08.018
Kriegler, M., Perez, C., DeFay, K., Albert, I., and Lu, S. D. (1988). A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell 53, 45–53.
Kuett, L., Catena, R., Özcan, A., Plüss, A., Cancer Grand Challenges IMAXT Consortium, Schraml, P., et al. (2022). Three-dimensional imaging mass cytometry for highly multiplexed molecular and cellular mapping of tissues and the tumor microenvironment. Nat. Cancer 3, 122–133. doi: 10.1038/s43018-021-00301-w
Lawson, L. J., Perry, V. H., Dri, P., and Gordon, S. (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170.
Le, T. D., Shirai, Y., Okamoto, T., Tatsukawa, T., Nagao, S., Shimizu, T., et al. (2010). Lipid signaling in cytosolic phospholipase A2alpha-cyclooxygenase-2 cascade mediates cerebellar long-term depression and motor learning. Proc. Natl. Acad. Sci. U. S. A. 107, 3198–3203. doi: 10.1073/pnas.0915020107
Lewitus, G. M., Konefal, S. C., Greenhalgh, A. D., Pribiag, H., Augereau, K., and Stellwagen, D. (2016). Microglial TNF-α suppresses cocaine-induced plasticity and behavioral sensitization. Neuron 90, 483–491. doi: 10.1016/j.neuron.2016.03.030
Lewitus, G. M., Pribiag, H., Duseja, R., St-Hilaire, M., and Stellwagen, D. (2014). An adaptive role of TNFα in the regulation of striatal synapses. J. Neurosci. 34, 6146–6155. doi: 10.1523/JNEUROSCI.3481-13.2014
Li, A. J., Katafuchi, T., Oda, S., Hori, T., and Oomura, Y. (1997). Interleukin-6 inhibits long-term potentiation in rat hippocampal slices. Brain Res. 748, 30–38.
Ling, E. A., and Wong, W. C. (1993). The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia 7, 9–18.
Liu, G., Zhang, H., Zhao, C., and Zhang, H. (2020). Evolutionary history of the toll-like receptor gene family across vertebrates. Genome Biol. Evol. 12, 3615–3634. doi: 10.1093/gbe/evz266
Luo, H., Liu, H. Z., Zhang, W. W., Matsuda, M., Lv, N., Chen, G., et al. (2019). Interleukin-17 regulates neuron-glial communications, synaptic transmission, and neuropathic pain after chemotherapy. Cell Rep. 29, 2384–2397.e5. doi: 10.1016/j.celrep.2019.10.085
Ma, S. B., Xian, H., Wu, W. B., Ma, S. Y., Liu, Y. K., Liang, Y. T., et al. (2020). CCL2 facilitates spinal synaptic transmission and pain via interaction with presynaptic CCR2 in spinal nociceptor terminals. Mol. Brain 13:161. doi: 10.1186/s13041-020-00701-6
Ma, S. H., Li, B., Huang, H. W., Peng, Y. P., and Qiu, Y. H. (2012). Interleukin-6 inhibits L-type calcium channel activity of cultured cerebellar granule neurons. J. Physiol. Sci. 62, 385–392. doi: 10.1007/s12576-012-0215-x
Mandolesi, G., Musella, A., Gentile, A., Grasselli, G., Haji, N., Sepman, H., et al. (2013). Interleukin-1β alters glutamate transmission at purkinje cell synapses in a mouse model of multiple sclerosis. J. Neurosci. 33, 12105–12121. doi: 10.1523/JNEUROSCI.5369-12.2013
Masuda, T., Sankowski, R., Staszewski, O., Böttcher, C., Amann, L., Sagar Scheiwe, C., et al. (2019). Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392. doi: 10.1038/s41586-019-0924-x
McGeachy, M. J., Cua, D. J., and Gaffen, S. L. (2019). The IL-17 family of cytokines in health and disease. Immunity 50, 892–906. doi: 10.1016/j.immuni.2019.03.021
Medvedeva, Y. V., Kim, M. S., and Usachev, Y. M. (2008). Mechanisms of prolonged presynaptic Ca2+ signaling and glutamate release induced by TRPV1 activation in rat sensory neurons. J. Neurosci. 28, 5295–5311. doi: 10.1523/JNEUROSCI.4810-07.2008
Medzhitov, R., and Janeway, C. Jr. (2000). Innate immunity. N. Engl. J. Med. 343, 338–344. doi: 10.1056/NEJM200008033430506
Menard, C., Pfau, M. L., Hodes, G. E., Kana, V., Wang, V. X., et al. (2017). Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 20, 1752–1760. doi: 10.1038/s41593-017-0010-3
Meng, X., Zhang, Y., Lao, L., Saito, R., Li, A., Bäckman, C. M., et al. (2013). Spinal interleukin-17 promotes thermal hyperalgesia and NMDA NR1 phosphorylation in an inflammatory pain rat model. Pain 154, 294–305. doi: 10.1016/j.pain.2012.10.022
Min, S. S., Quan, H. Y., Ma, J., Lee, K. H., Back, S. K., Na, H. S., et al. (2009). Impairment of long-term depression induced by chronic brain inflammation in rats. Biochem. Biophys. Res. Commun. 383, 93–97. doi: 10.1016/j.bbrc.2009.03.133
Mittelbronn, M., Dietz, K., Schluesener, H. J., and Meyermann, R. (2001). Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 101, 249–255. doi: 10.1007/s004010000284
Moberget, T., Doan, N. T., Alnæs, D., Kaufmann, T., Córdova-Palomera, A., Lagerberg, T. V., et al. (2018). Cerebellar volume and cerebellocerebral structural covariance in schizophrenia: a multisite mega-analysis of 983 patients and 1349 healthy controls. Mol. Psychiatry 23, 1512–1520. doi: 10.1038/mp.2017.106
Moore, E. E., Presnell, S., Garrigues, U., Guilbot, A., LeGuern, E., Smith, D., et al. (2002). Expression of IL-17B in neurons and evaluation of its possible role in the chromosome 5q-linked form of Charcot-Marie-Tooth disease. Neuromuscul. Disord. 12, 141–150. doi: 10.1016/S0960-8966(01)00250-4
Morikawa, Y., Tohya, K., Tamura, S., Ichihara, M., Miyajima, A., and Senba, E. (2000). Expression of interleukin-6 receptor, leukemia inhibitory factor receptor and glycoprotein 130 in the murine cerebellum and neuropathological effect of leukemia inhibitory factor on cerebellar Purkinje cells. Neuroscience 100, 841–848. doi: 10.1016/S0306-4522(00)00302-X
Moss, M. L., Jin, S. L., Milla, M. E., Bickett, D. M., Burkhart, W., Carter, H. L., et al. (1997). Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 385, 733–736.
Motoki, K., Kishi, H., Hori, E., Tajiri, K., Nishijo, H., and Muraguchi, A. (2009). The direct excitatory effect of IL-1beta on cerebellar Purkinje cell. Biochem. Biophys. Res. Commun. 379, 665–668. doi: 10.1016/j.bbrc.2008.12.023
Nakajima, K., and Kohsaka, S. (1993). Functional roles of microglia in the brain. Neurosci. Res. 17, 187–203.
Naviaux, R. K., Zolkipli, Z., Wang, L., Nakayama, T., Naviaux, J. C., Le, T. P., et al. (2013). Antipurinergic therapy corrects the autism-like features in the poly(IC) mouse model. PLoS ONE 8:e57380. doi: 10.1371/journal.pone.0057380
Nelson, T. E., Campbell, I. L., and Gruol, D. L. (1999). Altered physiology of Purkinje neurons in cerebellar slices from transgenic mice with chronic central nervous system expression of interleukin-6. Neuroscience 89, 127–136.
Nelson, T. E., Hao, C., Manos, J., Ransohoff, R. M., and Gruol, D. L. (2011). Altered hippocampal synaptic transmission in transgenic mice with astrocyte-targeted enhanced CCL2 expression. Brain Behav. Immun. 25(Suppl 1), S106–S119. doi: 10.1016/j.bbi.2011.02.013
Nelson, T. E., Netzeband, J. G., and Gruol, D. L. (2004). Chronic interleukin-6 exposure alters metabotropic glutamate receptor-activated calcium signalling in cerebellar Purkinje neurons. Eur. J. Neurosci. 20, 2387–2400. doi: 10.1111/j.1460-9568.2004.03706.x
Nelson, T. E., Ur, C. L., and Gruol, D. L. (2002). Chronic interleukin-6 exposure alters electrophysiological properties and calcium signaling in developing cerebellar purkinje neurons in culture. J. Neurophysiol. 88, 475–486. doi: 10.1152/jn.2002.88.1.475
Nenov, M. N., Malkov, A. E., Konakov, M. V., and Levin, S. G. (2019). Interleukin-10 and transforming growth factor-β1 facilitate long-term potentiation in CA1 region of hippocampus. Biochem. Biophys. Res. Commun. 518, 486–491. doi: 10.1016/j.bbrc.2019.08.072
Nguyen, K. T., Deak, T., Owens, S. M., Kohno, T., Fleshner, M., Watkins, L. R., et al. (1998). Exposure to acute stress induces brain interleukin-1beta protein in the rat. J. Neurosci. 18, 2239–2246.
Nie, X., Kitaoka, S., Tanaka, K., Segi-Nishida, E., Imoto, Y., Ogawa, A., et al. (2018). The innate immune receptors TLR2/4 mediate repeated social defeat stress-induced social avoidance through prefrontal microglial activation. Neuron 99, 464–479. doi: 10.1016/j.neuron.2018.06.035
Nimgaonkar, V. L., Prasad, K. M., Chowdari, K. V., Severance, E. G., and Yolken, R. H. (2017). The complement system: a gateway to gene-environment interactions in schizophrenia pathogenesis. Mol. Psychiatry 22, 1554–1561. doi: 10.1038/mp.2017.151
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Nishio, N., Taniguchi, W., Sugimura, Y. K., Takiguchi, N., Yamanaka, M., Kiyoyuki, Y., et al. (2013). Reactive oxygen species enhance excitatory synaptic transmission in rat spinal dorsal horn neurons by activating TRPA1 and TRPV1 channels. Neuroscience 247, 201–212. doi: 10.1016/j.neuroscience.2013.05.023
Nisticò, R., Mango, D., Mandolesi, G., Piccinin, S., Berretta, N., Pignatelli, M., et al. (2013). Inflammation subverts hippocampal synaptic plasticity in experimental multiple sclerosis. PLoS ONE 8:e54666. doi: 10.1371/journal.pone.0054666
Nonaka, M., and Yoshizaki, F. (2004). Primitive complement system of invertebrates. Immunol. Rev. 198, 203–215. doi: 10.1111/j.0105-2896.2004.00118.x
Ohtsuki, G.. (2020). Modification of synaptic-input clustering by intrinsic excitability plasticity on cerebellar Purkinje cell dendrites. J. Neurosci. 40, 267–282. doi: 10.1523/JNEUROSCI.3211-18.2019
Ohtsuki, G., and Hansel, C. (2018). Synaptic potential and plasticity of an SK2 channel gate regulate spike burst activity in cerebellar Purkinje cells. iScience 1, 49–54. doi: 10.1016/j.isci.2018.02.001
Ohtsuki, G., and Hirano, T. (2008). Bidirectional plasticity at developing climbing fiber-Purkinje neuron synapses. Eur. J. Neurosci. 28, 2393–2400. doi: 10.1111/j.1460-9568.2008.06539.x
Ohtsuki, G., Piochon, C., Adelman, J. P., and Hansel, C. (2012). SK2 channel modulation contributes to compartment-specific dendritic plasticity in cerebellar Purkinje cells. Neuron 75, 108–120. doi: 10.1016/j.neuron.2012.05.025
Ohtsuki, G., Piochon, C., and Hansel, C. (2009). Climbing fiber signaling and cerebellar gain control. Front. Cell Neurosci. 3:4. doi: 10.3389/neuro.03.004.2009
Ohtsuki, G., Shishikura, M., and Ozaki, A. (2020). Synergistic excitability plasticity in cerebellar functioning. FEBS J. 287, 4557–4593. doi: 10.1111/febs.15355
Ozaki, A., Yamawaki, Y., and Ohtsuki, G. (2021). Psychosis symptoms following aberrant immunity in the brain. Neural. Regen. Res. 16, 512–513. doi: 10.4103/1673-5374.293148
Pandey, S., Kawai, T., and Akira, S. (2014). Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect Biol. 7:a016246. doi: 10.1101/cshperspect.a016246
Pape, K., Tamouza, R., Leboyer, M., and Zipp, F. (2019). Immunoneuropsychiatry: novel perspectives on brain disorders. Nat. Rev. Neurol. 15, 317–328. doi: 10.1038/s41582-019-0174-4
Park, C. K., Lü, N., Xu, Z. Z., Liu, T., Serhan, C. N., and Ji, R. R. (2011a). Resolving TRPV1- and TNF-α-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J. Neurosci. 31, 15072–15085. doi: 10.1523/JNEUROSCI.2443-11.2011
Park, C. K., Xu, Z. Z., Liu, T., Lü, N., Serhan, C. N., and Ji, R. R. (2011b). Resolvin D2 is a potent endogenous inhibitor for transient receptor potential subtype V1/A1, inflammatory pain, and spinal cord synaptic plasticity in mice: distinct roles of resolvin D1, D2, and E1. J. Neurosci. 31, 18433–18438. doi: 10.1523/JNEUROSCI.4192-11.2011
Parker-Athill, E. C., and Tan, J. (2010). Maternal immune activation and autism spectrum disorder: interleukin-6 signaling as a key mechanistic pathway. Neurosignals 18, 113–128. doi: 10.1159/000319828
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R., Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Parvez, M. S. A., and Ohtsuki, G. (2022). Acute cerebellar inflammation and related ataxia: mechanisms and pathophysiology. Brain Sci. 12:367. doi: 10.3390/brainsci12030367
Pascual, O., Ben Achour, S., Rostaing, P., Triller, A., and Bessis, A. (2012). Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. U. S. A. 109, E197–205. doi: 10.1073/pnas.1111098109
Pati, D., and Kash, T. L. (2021). Tumor necrosis factor-α modulates GABAergic and dopaminergic neurons in the ventrolateral periaqueductal gray of female mice. J. Neurophysiol. 126, 2119–2129. doi: 10.1152/jn.00251.2021
Peng, Y. P., Qiu, Y. H., Lu, J. H., and Wang, J. J. (2005). Interleukin-6 protects cultured cerebellar granule neurons against glutamate-induced neurotoxicity. Neurosci. Lett. 374, 192–196. doi: 10.1016/j.neulet.2004.10.069
Piochon, C., Kloth, A. D., Grasselli, G., Titley, H. K., Nakayama, H., Hashimoto, K., et al. (2014). Cerebellar plasticity and motor learning deficits in a copy-number variation mouse model of autism. Nat. Commun. 5:5586. doi: 10.1038/ncomms6586
Pribiag, H., and Stellwagen, D. (2013). TNF-α downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABA(A) receptors. J. Neurosci. 33, 15879–15893 doi: 10.1523/JNEUROSCI.0530-13.2013
Prieto, G. A., Smith, E. D., Tong, L., Nguyen, M., and Cotman, C. W. (2019a). Inhibition of LTP-induced translation by IL-1β reduces the level of newly synthesized proteins in hippocampal dendrites. ACS Chem. Neurosci. 10, 1197–1203. doi: 10.1021/acschemneuro.8b00511
Prieto, G. A., Tong, L., Smith, E. D., and Cotman, C. W. (2019b). TNFα and IL-1β but not IL-18 suppresses hippocampal long-term potentiation directly at the synapse. Neurochem. Res. 44, 49–60. doi: 10.1007/s11064-018-2517-8
Qiu, Z., Parsons, K. L., and Gruol, D. L. (1995). Interleukin-6 selectively enhances the intracellular calcium response to NMDA in developing CNS neurons. J. Neurosci. 15, 6688–6699.
Qiu, Z., Sweeney, D. D., Netzeband, J. G., and Gruol, D. L. (1998). Chronic interleukin-6 alters NMDA receptor-mediated membrane responses and enhances neurotoxicity in developing CNS neurons. J. Neurosci. 18, 10445–10456.
Quan, N., Sundar, S. K., and Weiss, J. M. (1994). Induction of interleukin-1 in various brain regions after peripheral and central injections of lipopolysaccharide. J. Neuroimmunol. 49, 125–134.
Ragozzino, D., Di Angelantonio, S., Trettel, F., Bertollini, C., Maggi, L., Gross, C., et al. (2006). Chemokine fractalkine/CX3CL1 negatively modulates active glutamatergic synapses in rat hippocampal neurons. J. Neurosci. 26, 10488–10498. doi: 10.1523/JNEUROSCI.3192-06.2006
Ragozzino, D., Giovannelli, A., Mileo, A. M., Limatola, C., Santoni, A., and Eusebi, F. (1998). Modulation of the neurotransmitter release in rat cerebellar neurons by GRO beta. Neuroreport 9, 3601–3606.
Reed, M. D., Yim, Y. S., Wimmer, R. D., Kim, H., Ryu, C., Welch, G. M., et al. (2020). IL-17a promotes sociability in mouse models of neurodevelopmental disorders. Nature 577, 249–253. doi: 10.1038/s41586-019-1843-6
Riazi, K., Galic, M. A., Kuzmiski, J. B., Ho, W., Sharkey, K. A., and Pittman, Q. J. (2008). Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc. Natl. Acad. Sci. U. S. A. 105, 17151–17156. doi: 10.1073/pnas.0806682105
Ribeiro, M., Brigas, H. C., Temido-Ferreira, M., Pousinha, P. A., Regen, T., Santa, C., et al. (2019). Meningeal γδ T cell-derived IL-17 controls synaptic plasticity and short-term memory. Sci. Immunol. 4:eaay5199. doi: 10.1126/sciimmunol.aay5199
Ross, F. M., Allan, S. M., Rothwell, N. J., and Verkhratsky, A. (2003). A dual role for interleukin-1 in LTP in mouse hippocampal slices. J. Neuroimmunol. 144, 61–67. doi: 10.1016/j.jneuroim.2003.08.030
Rouvier, E., Luciani, M. F., Mattéi, M. G., Denizot, F., and Golstein, P. (1993). CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J. Immunol. 150, 5445–5456.
Santello, M., Bezzi, P., and Volterra, A. (2011). TNFα controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 69, 988–1001. doi: 10.1016/j.neuron.2011.02.003
Schmahmann, J. D.. (2004). Disorders of the cerebellum: ataxia, dysmetria of thought, and the cerebellar cognitive affective syndrome. J. Neuropsychiatry Clin. Neurosci. 16, 367–378. doi: 10.1176/jnp.16.3.367
Schöbitz, B., de Kloet, E. R., Sutanto, W., and Holsboer, F. (1993). Cellular localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Eur. J. Neurosci. 5, 1426–1435.
Segawa, K., Blumenthal, Y., Yamawaki, Y., and Ohtsuki, G. (2021). A destruction model of the vascular and lymphatic systems in the emergence of psychiatric symptoms. Biology 10:34. doi: 10.3390/biology10010034
Sgritta, M., Locatelli, F., Soda, T., Prestori, F., and D'Angelo, E. U. (2017). Hebbian spike-timing dependent plasticity at the cerebellar input stage. J. Neurosci. 37, 2809–2823. doi: 10.1523/JNEUROSCI.2079-16.2016
Shi, L., Smith, S. E., Malkova, N., Tse, D., Su, Y., and Patterson, P. H. (2009). Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav. Immun. 23, 116–123. doi: 10.1016/j.bbi.2008.07.012
Shim, H. G., Jang, S. S., Kim, S. H., Hwang, E. M., Min, J. O., Kim, H. Y., et al. (2018). TNF-α increases the intrinsic excitability of cerebellar Purkinje cells through elevating glutamate release in Bergmann Glia. Sci. Rep. 8:11589. doi: 10.1038/s41598-018-29786-9
Silvin, A., and Ginhoux, F. (2018). Microglia heterogeneity along a spatio-temporal axis: more questions than answers. Glia 66, 2045–2057. doi: 10.1002/glia.23458
Sjöström, P. J., and Häusser, M. (2006). A cooperative switch determines the sign of synaptic plasticity in distal dendrites of neocortical pyramidal neurons. Neuron 51, 227–238. doi: 10.1016/j.neuron.2006.06.017
Sjöström, P. J., Rancz, E. A., Roth, A., and Häusser, M. (2008). Dendritic excitability and synaptic plasticity. Physiol. Rev. 88, 769–840. doi: 10.1152/physrev.00016.2007
Spittau, B., Dokalis, N., and Prinz, M. (2020). The role of TGFβ signaling in microglia maturation and activation. Trends Immunol. 41, 836–848. doi: 10.1016/j.it.2020.07.003
Stellwagen, D., Beattie, E. C., Seo, J. Y., and Malenka, R. C. (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 25, 3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005
Stellwagen, D., and Malenka, R. C. (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440, 1054–1059. doi: 10.1038/nature04671
Strecker, J. K., Schmidt, A., Schäbitz, W. R., and Minnerup, J. (2017). Neutrophil granulocytes in cerebral ischemia: evolution from killers to key players. Neurochem. Int. 107, 117–126. doi: 10.1016/j.neuint.2016.11.006
Süß, P., Hoffmann, A., Rothe, T., Ouyang, Z., Baum, W., Staszewski, O., et al. (2020). Chronic peripheral inflammation causes a region-specific myeloid response in the central nervous system. Cell Rep. 30, 4082–4095.e6. doi: 10.1016/j.celrep.2020.02.109
Suvrathan, A., Payne, H. L., and Raymond, J. L. (2016). Timing rules for synaptic plasticity matched to behavioral function. Neuron 92, 959–967. doi: 10.1016/j.neuron.2016.10.022
Takeuchi, T., Ohtsuki, G., Yoshida, T., Fukaya, M., Wainai, T., Yamashita, M., et al. (2008). Enhancement of both long-term depression induction and optokinetic response adaptation in mice lacking delphilin. PLoS ONE 3:e2297. doi: 10.1371/journal.pone.0002297
Tsai, P. T., Hull, C., Chu, Y., Greene-Colozzi, E., Sadowski, A. R., Leech, J. M., et al. (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. doi: 10.1038/nature11310
Tsai, P. T., Rudolph, S., Guo, C., Ellegood, J., Gibson, J. M., Schaeffer, S. M., et al. (2018). Sensitive periods for cerebellar-mediated autistic-like behaviors. Cell Rep. 25, 357–367. doi: 10.1016/j.celrep.2018.09.039
Tsurugizawa, T., Tamada, K., Ono, N., Karakawa, S., Kodama, Y., Debacker, C., et al. (2020). Awake functional MRI detects neural circuit dysfunction in a mouse model of autism. Sci. Adv. 6:eaav4520. doi: 10.1126/sciadv.aav4520
Tzour, A., Leibovich, H., Barkai, O., Biala, Y., Lev, S., Yaari, Y., et al. (2017). KV 7/M channels as targets for lipopolysaccharide-induced inflammatory neuronal hyperexcitability. J. Physiol. 595, 713–738. doi: 10.1113/JP272547
Valentinova, K., Tchenio, A., Trusel, M., Clerke, J. A., Lalive, A. L., Tzanoulinou, S., et al. (2019). Morphine withdrawal recruits lateral habenula cytokine signaling to reduce synaptic excitation and sociability. Nat. Neurosci. 22, 1053–1056. doi: 10.1038/s41593-019-0421-4
van Maldegem, F., Valand, K., Cole, M., Patel, H., Angelova, M., Rana, S., et al. (2021). Characterisation of tumour microenvironment remodelling following oncogene inhibition in preclinical studies with imaging mass cytometry. Nat. Commun. 12:5906. doi: 10.1038/s41467-021-26214-x
Vargas, D. L., Nascimbene, C., Krishnan, C., Zimmerman, A. W., and Pardo, C. A. (2005). Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 57, 67–81. doi: 10.1002/ana.20315
Vikman, K. S., Duggan, A. W., and Siddall, P. J. (2007). Interferon-gamma induced disruption of GABAergic inhibition in the spinal dorsal horn in vivo. Pain 133, 18–28. doi: 10.1016/j.pain.2007.02.010
Viviani, B., Bartesaghi, S., Gardoni, F., Vezzani, A., Behrens, M. M., Bartfai, T., et al. (2003). Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 23, 8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003
Vlkolinský, R., Siggins, G. R., Campbell, I. L., and Krucker, T. (2004). Acute exposure to CXC chemokine ligand 10, but not its chronic astroglial production, alters synaptic plasticity in mouse hippocampal slices. J. Neuroimmunol. 150, 37–47. doi: 10.1016/j.jneuroim.2004.01.011
Wang, S. S., Denk, W., and Häusser, M. (2000). Coincidence detection in single dendritic spines mediated by calcium release. Nat. Neurosci. 3, 1266–1273. doi: 10.1038/81792
Wang, X., Novello, M., Gao, Z., Ruigrok, T. J. H., and De Zeeuw, C. I. (2021). Input and output organization of the mesodiencephalic junction for cerebro-cerebellar communication. J. Neurosci. Res. 100, 620–637. doi: 10.1101/2021.02.23.432495
Wang, Z., Xu, N. L., Wu, C. P., Duan, S., and Poo, M. M. (2003). Bidirectional changes in spatial dendritic integration accompanying long-term synaptic modifications. Neuron 37, 463–472. doi: 10.1016/S0896-6273(02)01189-3
Willis, E. F., MacDonald, K. P. A., Nguyen, Q. H., Garrido, A. L., Gillespie, E. R., Harley, S. B. R., et al. (2020). Repopulating microglia promote brain repair in an IL-6-dependent manner. Cell 180, 833–846.e16. doi: 10.1016/j.cell.2020.02.013
Yamamoto, M., Kim, M., Imai, H., Itakura, Y., and Ohtsuki, G. (2019). Microglia-triggered plasticity of intrinsic excitability modulates psychomotor behaviors in acute cerebellar inflammation. Cell Rep. 28, 2923–2938.e8. doi: 10.1016/j.celrep.2019.07.078
Yamawaki, Y., Wada, Y., Matsui, S., and Ohtsuki, G. (2022). Microglia-triggered hypoexcitability plasticity of pyramidal neurons in the rat medial prefrontal cortex. Curr. Res. Neurobiol. 3:100028. doi: 10.1016/j.crneur.2022.100028
Yang, S., Liu, Z. W., Wen, L., Qiao, H. F., Zhou, W. X., and Zhang, Y. X. (2005). Interleukin-1beta enhances NMDA receptor-mediated current but inhibits excitatory synaptic transmission. Brain Res. 1034, 172–179. doi: 10.1016/j.brainres.2004.11.018
Yang, Y., Li, H., Li, T. T., Luo, H., Gu, X. Y., Lü, N., et al. (2015). Delayed activation of spinal microglia contributes to the maintenance of bone cancer pain in female Wistar rats via P2X7 receptor and IL-18. J. Neurosci. 35, 7950–7963. doi: 10.1523/JNEUROSCI.5250-14.2015
Yao, C., and Narumiya, S. (2019). Prostaglandin-cytokine crosstalk in chronic inflammation. Br. J. Pharmacol. 176, 337–354. doi: 10.1111/bph.14530
Yolken, R. H., and Torrey, E. F. (1995). Viruses, schizophrenia, and bipolar disorder. Clin. Microbiol. Rev. 8, 131–145.
Zhan, Y., Paolicelli, R. C., Sforazzini, F., Weinhard, L., Bolasco, G., Pagani, F., et al. (2014). Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 17, 400–406. doi: 10.1038/nn.3641
Zhang, H., Nei, H., and Dougherty, P. M. (2010). A p38 mitogen-activated protein kinase-dependent mechanism of disinhibition in spinal synaptic transmission induced by tumor necrosis factor-alpha. J. Neurosci. 30, 12844–12855. doi: 10.1523/JNEUROSCI.2437-10.2010
Zhang, J., Malik, A., Choi, H. B., Ko, R. W. Y., Dissing-Olesen, L., and MacVicar, B. A. (2014). Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron 82, 195–207. doi: 10.1016/j.neuron.2014.01.043
Zhang, L. I., Tao, H. W., Holt, C. E., Harris, W. A., and Poo, M. (1998). A critical window for cooperation and competition among developing retinotectal synapses. Nature 395, 37–44.
Zhang, Z. J., Cao, D. L., Zhang, X., Ji, R. R., and Gao, Y. J. (2013). Chemokine contribution to neuropathic pain: respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain 154, 2185–2197. doi: 10.1016/j.pain.2013.07.002
Zhao, P., Waxman, S. G., and Hains, B. C. (2007). Extracellular signal-regulated kinase-regulated microglia-neuron signaling by prostaglandin E2 contributes to pain after spinal cord injury. J. Neurosci. 27, 2357–2368. doi: 10.1523/JNEUROSCI.0138-07.2007
Zheng, Z. H., Tu, J. L., Li, X. H., Hua, Q., Liu, W. Z., Liu, Y., et al. (2021). Neuroinflammation induces anxiety- and depressive-like behavior by modulating neuronal plasticity in the basolateral amygdala. Brain Behav. Immun. 91, 505–518. doi: 10.1016/j.bbi.2020.11.007
Keywords: immune-triggered plasticity, brain immune cells, inflammatory mediators, synaptic transmission, intrinsic excitability, cerebellum, medial PFC (mPFC), psychiatric diseases
Citation: Hikosaka M, Kawano T, Wada Y, Maeda T, Sakurai T and Ohtsuki G (2022) Immune-Triggered Forms of Plasticity Across Brain Regions. Front. Cell. Neurosci. 16:925493. doi: 10.3389/fncel.2022.925493
Received: 21 April 2022; Accepted: 16 June 2022;
Published: 22 July 2022.
Edited by:
Haitao Wang, Southern Medical University, ChinaReviewed by:
Marco Pignatelli, Washington University in St. Louis, United StatesCopyright © 2022 Hikosaka, Kawano, Wada, Maeda, Sakurai and Ohtsuki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gen Ohtsuki, b2h0c3VraS5nZW4uN3dAa3lvdG8tdS5hYy5qcA==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.