 Jianjun Xu
Jianjun Xu Etsuko Minobe
Etsuko Minobe Masaki Kameyama
Masaki Kameyama
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 14 April 2022
Sec. Cellular Neurophysiology
Volume 16 - 2022 | https://doi.org/10.3389/fncel.2022.867385
This article is part of the Research Topic Role of Metal Ions in Central Nervous System: Physiology and Pathophysiology View all 7 articles
Parkinson’s disease (PD), a common neurodegenerative disease characterized by motor dysfunction, results from the death of dopaminergic neurons in the substantia nigra pars compacta (SNc). Although the precise causes of PD are still unknown, several risk factors for PD have been determined, including aging, genetic mutations, environmental factors, and gender. Currently, the molecular mechanisms underlying risk factor-related neurodegeneration in PD remain elusive. Endoplasmic reticulum stress, excessive reactive oxygen species production, and impaired autophagy have been implicated in neuronal death in the SNc in PD. Considering that these pathological processes are tightly associated with intracellular Ca2+, it is reasonable to hypothesize that dysregulation of Ca2+ handling may mediate risk factors-related PD pathogenesis. We review the recent findings on how risk factors cause Ca2+ dyshomeostasis and how aberrant Ca2+ handling triggers dopaminergic neurodegeneration in the SNc in PD, thus putting forward the possibility that manipulation of specific Ca2+ handling proteins and subcellular Ca2+ homeostasis may lead to new promising strategies for PD treatment.
Parkinson’s disease (PD) is the second most common progressive neurodegenerative disorder in humans (Willis et al., 2010). It is characterized by motor dysfunction, cardinally manifested with resting tremor, slowness of movement, and muscle rigidity (Jankovic, 2008). Pathological analysis and clinical studies indicate that these motor PD symptoms result from the degeneration and loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc) (Kalia and Lang, 2016; Surmeier et al., 2017), a region in the midbrain involved in the regulation of movement and coordination of the human body. Unfortunately, since the mechanisms underlying PD pathogenesis remain elusive, there is currently no available cure for PD.
The precise causes of PD are largely unknown, and several etiologic factors, including aging, genetic and environmental factors, and gender (Ascherio and Schwarzschild, 2016), have been shown to increase the risk of PD development. One risk factor, or a combination of two or more factors, can distinctly contribute to PD pathogenesis (Pang et al., 2019; Vasta et al., 2021). Among these factors, aging is considered as the number one risk factor for PD development and progression (Hindle, 2010). Over 1% of the population worldwide suffers from PD at the age of 60, and this incidence rate increases to 5% in individuals over 85 years old (de Lau and Breteler, 2006), highlighting the impact of aging on the risk of PD. Genetic factors constitute another major risk factor for PD. Approximately 10–15% of all PD cases are caused by genetic mutations (Deng et al., 2018). Many mutations in genes associated with PD, including GBA, LRRK2, PRKN, PINK1, and SNCA (Kitada et al., 1998; Valente et al., 2001; Paisán-Ruíz et al., 2004; Lunati et al., 2018), have been identified. Moreover, several gene mutations have been shown to be the major cause of early onset PD (diagnosed in individuals younger than 45 years old) (Schrag and Schott, 2006), implying a superimposed effect of genetic factors on age-related PD pathogenesis. It is notable that PD is induced by a combination of aging with environmental and/or genetic factors (Pang et al., 2019; Vasta et al., 2021). Environmental factors include exposure to toxins and chemicals, which are associated with an increased risk of PD (Chen, 2018). Studies on the gender-based incidence of PD show that men are approximately 1.5∼2 times more likely than women to develop PD (de Lau et al., 2004), suggesting that gender is associated with PD incidence. The possible reasons for the higher risk of PD in men include lower neuroprotection by estrogen and higher chances of exposure to PD-related environmental factors. Moreover, other factors, such as head trauma and some medications have also been determined as the PD risk factors.
Although a number of PD risk factors have been identified, the molecular pathways of risk factor-related progressive neurodegeneration in PD require further elucidation. Dysfunction of the endoplasmic reticulum (ER), mitochondria, and lysosomes has been proposed to be the primary cause of neuronal death in PD patients (Michel et al., 2016; Sunanda et al., 2021). Dysfunction of the ER causes accumulation of unfolded proteins, which in turn triggers ER stress, resulting in an unfolded protein response (UPR). Impaired protein quality control and cellular Ca2+ dyshomeostasis have been implicated in the pathogenesis of PD (Xu et al., 2005; Lindholm et al., 2006; Kim et al., 2008). Mitochondrial dysfunction in pathological conditions leads to decreased ATP production, oxidative stress, and reduced calcium handling capacity, all of which are associated with neuronal damage in PD (Abou-Sleiman et al., 2006; Henchcliffe and Beal, 2008). Maintenance of a constant balance between synthesis and degradation of cellular constituents by the lysosomal system is essential for cellular homeostasis. Dysfunction of lysosomes results in the accumulation of aberrant proteins, lipids, and damaged organelles, especially mitochondria, leading to increased formation of toxic protein aggregates and excessive generation of reactive oxygen species (ROS), which are also crucially associated with neurodegeneration and neuronal death (Lin et al., 2019; Ray et al., 2021). Furthermore, recent studies have highlighted the essential role of the interactions between the ER, mitochondria, and lysosomes in the maintenance of normal cellular homeostasis (Raffaello et al., 2016; Rossini et al., 2021). However, dysfunction of one organelle has been shown to cause failure of the other two organelles, suggesting a switch-like mechanism or molecule in the cells. Once this mechanism or molecule is activated or reaches a threshold, virtuous interactions between organelles become vicious interplays, driving a vicious cycle between the ER, mitochondria, and lysosomes. Consistent with this hypothesis, dysfunction of these three important organelles has been shown to be a common feature in PD pathology (Perić et al., 2016; Ray et al., 2021). Ca2+, as the most important signaling molecule in neuronal cells, is crucial for the regulation of ER and mitochondrial and lysosomal functions and mediates the interactions between these organelles (Raffaello et al., 2016). Recently, dysregulation of intracellular Ca2+ handling, together with ER, mitochondrial, and lysosomal dysfunction, has been suggested as a common pathway in PD pathogenesis (Wojda et al., 2008; Michel et al., 2016; Zaichick et al., 2017). Therefore, it is worth considering whether Ca2+ dyshomeostasis links PD risk factors to neurodegeneration by triggering detrimental interactions between subcellular organelles.
We review recent findings and discuss how intracellular Ca2+ dyshomeostasis is induced by PD risk factors, which eventually leads to neurodegeneration and death of SNc DA neurons in PD.
As the most ubiquitous and versatile second messenger in signaling pathways in neurons, Ca2+ is fundamental for neuronal development and survival by controlling diverse cellular processes including gene expression, synaptogenesis, synaptic transmission, energy production, membrane excitability, and neuronal plasticity (Kawamoto et al., 2012). Neuronal Ca2+ signals are induced after transient and reversible elevation of cytosolic Ca2+ concentration via Ca2+ influx through plasma membrane Ca2+-permeable channels and Ca2+ release from subcellular organelles (Brini et al., 2014) (Figure 1). The Ca2+-permeable channels in plasma membrane which mediate Ca2+ influx upon activation are categorized into three major groups (Brini et al., 2014): (1) voltage-gated Ca2+ channels (VGCCs). Neuronal VGCCs include L-type (Cav1.2-1.4), P/Q type (Cav2.1), N type (Cav2.2), R type (Cav2.3), and T type (Cav3.1-3.3) VGCCs. Cav1.2 and Cav1.3 are the main VGCC types in neurons. (2) Receptor-operated Ca2+ channels (ROCs). The main ROCs in neurons are glutamate and purinergic receptors. They are further divided into ionotropic and metabotropic receptors. Alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid-sensitive receptors (AMPARs) and N-methyl-d-aspartate sensitive receptors (NMDARs) are the principal types of ionotropic glutamate receptors. Ionotropic purinergic receptors (P2XRs) mediate Ca2+ influx and membrane depolarization, while metabotropic purinergic receptors (P2YRs) activate the Gq/11 signaling pathway upon activation by ATP binding. (3) Store-operated Ca2+ entry channels (SOCs) activated by depletion of ER Ca2+ stores. SOCs communicate with the ER through interactions between STIM (the transmembrane stromal interaction molecules within the ER) and the ORAI subunit of SOC (Yuan et al., 2009).
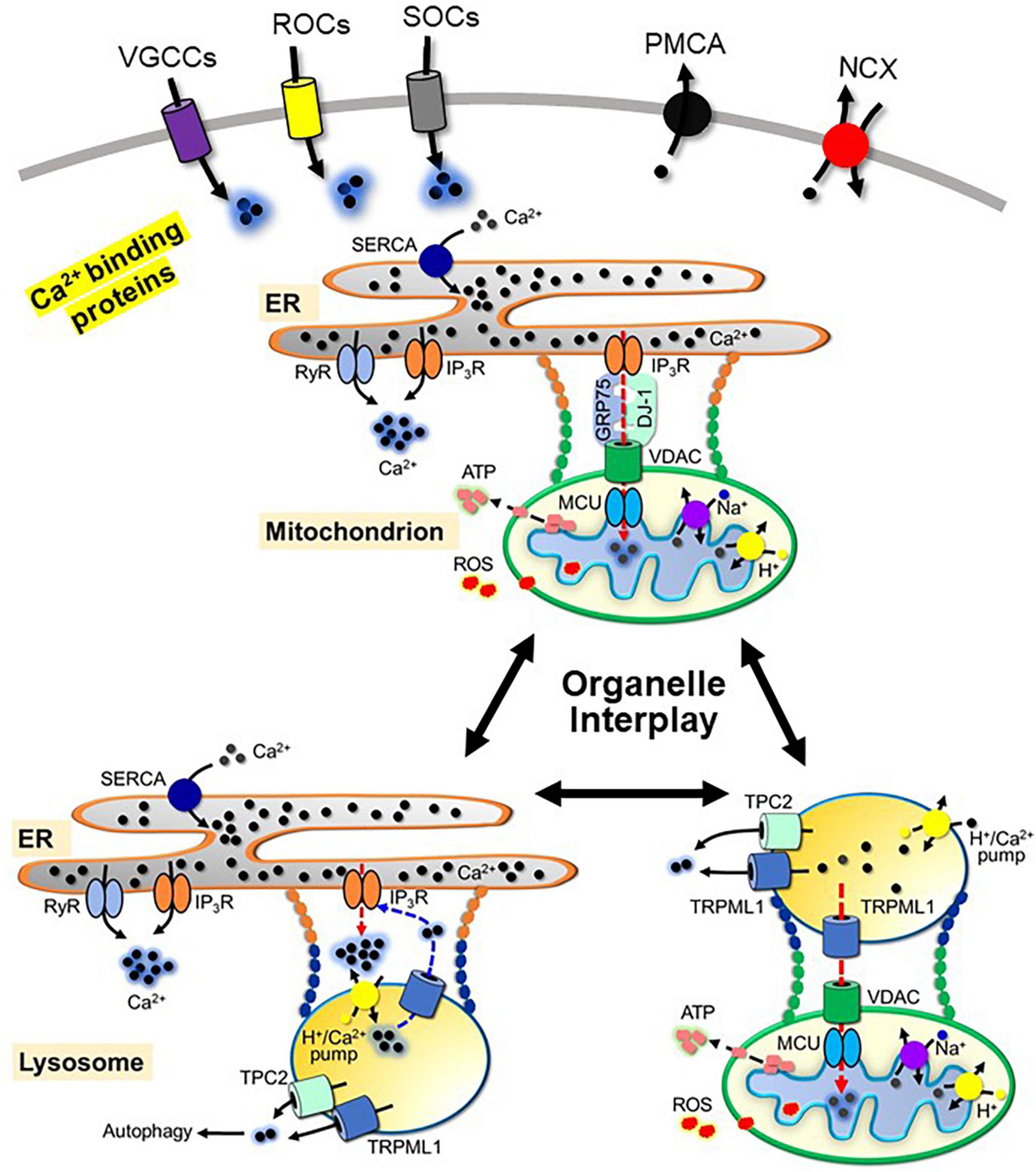
Figure 1. Schematic view of neuronal Ca2+ handling and the interplay between the ER, mitochondria and lysosomes. Ca2+ influx through voltage-gated Ca2+ channels (VGCCs), receptor-operated Ca2+ channels (ROCs) and store-operated Ca2+ entry channels (SOCs), and Ca2+ release from the ER through Ryanodine receptor (RyR) and inositol 1,4,5-triphosphate receptor (IP3R), from the mitochondria through Na+-Ca2+ exchanger and H+-Ca2+ exchanger, and from the lysosomes through the transient receptor potential channel mucolipin 1 (TRPML1) and two-pore channel 2 (TPC2) contribute to Ca2+ signaling. Mechanisms to regulate Ca2+ signaling include three pathways: extrusion via plasma membrane Ca2+-ATPase (PMCA) and Na+-Ca2+ exchanger (NCX); sequestration by Ca2+-binding proteins; uptake by the ER through sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), uptake by the mitochondria through voltage-dependent anion channels (VDAC) and mitochondrial Ca2+ uniporter complex (MCU) and uptake by the lysosomes through lysosomal H+/Ca2+-exchanger. Organelle contacts between the ER, mitochondria and lysosomes include physical tethering and functional interactions. For simplicity, the components of each physical tether mentioned in the text are omitted. Ca2+ mediates functional interactions between organelles. Functional interactions between the ER and mitochondria are formed by the tetramer complex of VDAC, IP3R, deglycase 1 (DJ-1) and glucose-related protein 75 (GRP75). The interaction between the ER and lysosomes is mediated by IP3R and TRPML1. The interaction between the mitochondria and lysosomes is mediated by TRPML1, VDAC and MCU.
In addition to Ca2+ influx, Ca2+ release from subcellular organelles contributes to Ca2+ signaling. ER and mitochondria are major intracellular Ca2+ stores in neurons, while lysosomes are also involved in the control of cellular Ca2+ homeostasis (Yang et al., 2019). Ca2+ release from the ER is mediated by the ryanodine receptor (RyR), which is activated by Ca2+ influx, and inositol 1,4,5-trisphosphate receptor (IP3R), which is activated by IP3 from Gq/11 signaling pathways. Both RyR and IP3R are sensitive to cytosolic Ca2+ and oxidation states (Mak et al., 2001; Aracena-Parks et al., 2006; Xiong et al., 2006; Joseph et al., 2018). The mitochondrial system mainly consists of a Na+-Ca2+ exchanger (mNCX) and a H+–Ca2+ exchanger (Austin et al., 2017) responsible for Ca2+ release from the mitochondria. Ca2+ release from the lysosomes is mainly mediated through the transient receptor potential channel mucolipin 1 (TRPML1), which belongs to the mucolipin subgroup of the TRP ion channel family (Ramsey et al., 2006), and two-pore channel 2 (TPC2) (Calcraft et al., 2009).
Termination of cytosolic Ca2+ signals is mediated through three mechanisms: extracellular extrusion, uptake into intracellular organelles, and sequestration by Ca2+ binding proteins. Calcium efflux from the cytoplasm to the extracellular space is mainly mediated through the action of the plasma membrane Ca2+-ATPase (PMCA) and the Na+-Ca2+ exchanger (NCX) by using the energy stored in ATP and the chemical energy of the Na+ gradient, respectively. Uptake of Ca2+ into the ER is mediated by the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) in an ATP-dependent manner. The mechanism of Ca2+ uptake by the lysosomes is not completely understood. Melchionda et al. (2016) characterized the lysosomal H+/Ca2+-exchanger involved in lysosomal Ca2+ refilling in non-placental mammals. SERCA, PMCA, and ion exchangers are sensitive to the cellular oxidative status (Görlach et al., 2015). Increased ROS production inhibits the activity of these proteins. Mitochondria take up Ca2+ from the cytoplasm through voltage-dependent anion channels (VDAC) and mitochondrial Ca2+ uniporter complex (MCU), which is crucially involved in the sequestration of Ca2+ signaling (Shoshan-Barmatz and De, 2017). Ca2+ efflux and uptake into organelles by Ca2+ pumps and exchangers is a slow Ca2+ buffering process, while rapid Ca2+ buffering is mediated by intracellular calcium binding proteins (CaBPs). CaBPs are classified into Ca2+ sensors and Ca2+ buffer proteins (Burgoyne, 2007). Ca2+ sensor proteins, such as calmodulin, change their conformation upon Ca2+ binding and activate downstream signaling molecules. In contrast to Ca2+ sensor proteins, Ca2+ buffer proteins, such as calretinin, calbindin, or parvalbumin, quickly bind Ca2+ with different affinities when Ca2+ signals are generated in neurons and act as negative modulators of cytosolic calcium levels.
Ca2+ homeostasis is strictly controlled in healthy neurons. Disturbance in Ca2+ homeostasis leads to profound alterations in neuronal function. Especially, synapses, as the primary target of Ca2+, is highly vulnerable to Ca2+ dyshomeostasis. The axonal terminal of SNc DA neurons have high-energy requirement due to their extensive axonal arborization. Ca2+ dyshomeostasis in PD may initially attenuate nigrostriatal synaptic plasticity and DA release, and subsequently cause deconstruction of axons and loss of neuronal connectivity, preceding to the death of DA neurons and occurrence of gross PD motor symptom (Kouroupi et al., 2017; González-Rodríguez et al., 2021).
In contrast to other neurons, SNc DA neurons show two key features in cytosolic Ca2+ handling: (1) low Ca2+ buffering capacity. SNc DA neurons lack expression of the Ca2+ buffer protein calbindin (Dopeso-Reyes et al., 2014), which binds Ca2+ with rapid to intermediate kinetics. Calbindin is important for rapid restoration of basal Ca2+ levels within a cell soon after Ca2+ signal occurs. (2) SNc DA neurons are constitutively active in the absence of external input, generating slow, broad spikes that lead to a significant increase in resting cytosolic Ca2+ concentration. Although the mechanism underlying the pacemaking activity of SNc DA neurons is not completely clear, Cav1.3 channels have been shown to support pacemaking (Chan et al., 2007; Guzman et al., 2009). The combination of these two features in Ca2+ handling is shared by neurons at risk of neurodegeneration (Surmeier and Schumacker, 2013).
Ca2+ dyshomeostasis results from the disrupted balance between induction and termination of Ca2+ signaling, which is dynamically controlled by the Ca2+-handling molecules mentioned earlier under physiological conditions. Alterations in the activities or expression levels of Ca2+-handling proteins in pathological conditions causes dysregulation of Ca2+ handling, which has been extensively reported to contribute to neurodegenerative diseases (Mattson, 2004, 2007; Surmeier, 2007; Wojda et al., 2008; Marambaud et al., 2009). Considering the two features of Ca2+ handling (low Ca2+ buffering capacity and higher basal cytosolic Ca2+ level), SNc DA neurons are extremely susceptible to the increase in cytosolic Ca2+ induced by PD risk factors, such as aging, and genetic and environmental factors. Sustained Ca2+ elevation leads to neuronal damage through both direct and indirect pathways.
Excessive cytosolic Ca2+ can directly activate a variety of Ca2+-dependent enzymes, including phospholipases, proteases, endonucleases, and nitric oxide synthase (NOS). These activated enzymes contribute to neurodegeneration by damaging various cell structures, such as the membrane, cytoskeleton, and DNA, as well as through NO-induced oxidative stress (Tymianski and Tator, 1996).
In healthy neurons, cytosolic Ca2+ is tightly controlled within a physiological range by Ca2+-handling proteins, and Ca2+, in turn, regulates these Ca2+-handling proteins. In addition, Ca2+ plays a crucial role in the regulation of organelle functions, especially the ER, mitochondria, and lysosomes. Recently, increasing evidence has shed light on the central role of Ca2+ signaling in the crosstalk between the ER, mitochondria, and lysosomes (Raffaello et al., 2016). Prolonged Ca2+ dyshomeostasis under pathological conditions may feed a vicious interplay between ER, mitochondria, and lysosomes, which are hypothesized to participate in common pathways in neurodegeneration in PD.
Each subcellular organelle is physically separated from the cytoplasm, with its own unique role in cell function. Originally, subcellular organelles were considered as independent entities within a cell, however, recent discoveries have shown that there are dynamic interactions between organelles. Organelles are in close proximity, and they dynamically come in contact, with each other through membrane contact sites, which are enriched in signaling molecules and ion channels (Rowland and Voeltz, 2012; Prinz, 2014; Phillips and Voeltz, 2016; Muallem et al., 2017; Cohen et al., 2018; Lee et al., 2020; Rossini et al., 2021) (Figure 1). Membrane contact sites serve as “hotspots” for the transfer of signaling molecules and crosstalk between organelles, which are essential for cellular homeostasis. Disruption of the coordinated interactions between organelles results in the pathogenesis of various diseases, including PD.
The most well-documented membrane contact site is the mitochondria-ER contact site (MERCS). In MERCS, the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane (OMM) is bridged with the ER membrane IP3R with deglycase 1 (DJ-1) and glucose-related protein 75 (GRP75). DJ-1 interacts with VDAC, IP3R, and GRP75. GRP75 links IP3R to VDAC. Together, IP3R, VDAC, GRP75, and DJ-1 form a functional tetramer complex (IP3R/GRP75/DJ-1/VDAC complex) at MERCS (Liu et al., 2019), which mediates Ca2+ transfer from the ER to the mitochondrial matrix via the inner membrane mitochondrial calcium uniporter (MCU) and likely ROS transfer from the mitochondria to the ER. MERCSs are regulated by multiple proteins in the ER and mitochondria membrane, which contribute to dynamic physical tethering between the organelles, and regulate the integrity, stability, size, and length of the MERCS (Csordás et al., 2018; Wilson and Metzakopian, 2021). Ca2+ dyshomeostasis may strengthen MERCS, by activation of Ca2+-dependent proteins in MERCS (Hajnóczky et al., 2014), which promotes physical and functional interaction between the ER and mitochondria. Enhanced MERCS has been reported to facilitate mitochondrial Ca2+ uptake, causing Ca2+ overload and mitochondrial dysfunction. Consistent with this hypothesis, there are increased ER–mitochondria contacts and upregulated MERCS-associated proteins, including IP3Rs, RyRs, and VDACs (Hedskog et al., 2013), and Ca2+ dyshomeostasis in neurodegenerative diseases (Wojda et al., 2008). Therefore, dynamic contacts between the ER and mitochondrial are beneficial for normal function of the ER and mitochondria. Deficits in MERCS lead to mitochondrial Ca2+ dyshomeostasis and dysfunction, and subsequent dopaminergic neuronal death in PD (Gómez-Suaga et al., 2018; Ray et al., 2021; Wilson and Metzakopian, 2021).
The ER also dynamically contacts endolysosomal organelles. In the ER-lysosome contact sites, IP3Rs are highly clustered in the ER membrane (Atakpa et al., 2018). IP3R, together with lysosomal Ca2+-handling proteins such as the H+/Ca2+-exchanger, Ca2+ release channel TRPML1, and TPC2 form a functional unit in ER-lysosome contact sites, which contributes to ER-dependent lysosomal Ca2+ refilling. It has been reported that Ca2+ release from lysosomes via TRPML1 results in increased cytosolic Ca2+ concentration within the ER-lysosome contact sites, which subsequently activates IP3R to trigger massive Ca2+ release from the ER through the Ca2+-induced Ca2+ release (CICR) mechanism (Galione, 2015). Thus, ER and lysosomal Ca2+ release channels form an amplifying loop that controls lysosomal Ca2+ refilling. Both lysosomal Ca2+ refilling and Ca2+ release play essential roles in lysosomal functions, including late endosome-lysosome fusion (Pryor et al., 2000), lysosomal exocytosis, phagocytosis, membrane repair, and signal transduction (Reddy et al., 2001; Kinnear et al., 2004). The ER-lysosome contact is regulated by many proteins in the ER and endo-lysosomal membranes, which constitute dynamic physical interactions in the contact site (van der Kant and Neefjes, 2014). The ER-lysosome contact generates microdomain for lipid transport and Ca2+ transfer between the two organelles. Interestingly, the ER-endolysosome contact seems to be a Ca2+-sensitive structure. Epidermal growth factor stimulation triggers a spike in cytosolic Ca2+, and results in increased ER-endosome contacts formation, likely through the recruitment of some Ca2+-sensitive tethering proteins (Eden et al., 2010). Thus, the role of Ca2+ dyshomeostasis-induced increased ER-endolysosome contacts in PD pathogenesis warrants further investigation.
Recently, several laboratories have observed physical dynamic contacts between lysosomes and mitochondria that are independent of mitophagy (Han et al., 2017; Valm et al., 2017; Wong et al., 2018). Lysosome-mitochondria contact has been shown to be regulated by the small GTPase Rab7 through the modulation of the contact site tethering and untethering dynamics (Wong et al., 2018). Rab7 exists in two forms in the cell: an active GTP-bound and an inactive GDP-bound form (Rab7-GTP and Rab7-GDP). Rab7-GTP at the lysosomal membrane promotes lysosome-mitochondria contact by interacting with unidentified effector proteins at the mitochondrial membrane (Wong et al., 2019). Rab7-GTP is inactivated by hydrolysis of its bound GTP into GDP upon activation of Rab7 GTPase, which causes lysosome-mitochondria untethering. Thus, lysosome-mitochondria contact depends on nucleotide binding to Rab7, which can be regulated by several cytoplasmic factors, such as guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs). The contact site offers a platform for proteins at the mitochondrial and lysosomal membranes that regulates their interplay, such as metabolic exchange, and Ca2+ and ROS transfer between the two organelles. The lysosomal Ca2+ release channels TRPML1 and TPC2 together with mitochondrial VDAC in the outer membrane and MCU in the inner membrane form a functional unit that mediates Ca2+ transfer from the lysosome to the mitochondria (Peng et al., 2020). Dysregulation of mitochondria-lysosome contacts results in dysfunction of both the mitochondria and lysosomes and contributes to PD pathogenesis (Peng et al., 2020). Recent study shows prolonged lysosome-mitochondria contacts in neurons modeling PD, due to defect in GTP hydrolysis (Kim et al., 2021). Whether Ca2+ dyshomeostasis in PD is involved in facilitation of lysosome-mitochondria contacts remains to be elucidated.
The main functions of mitochondria include ATP production, cytosolic Ca2+ buffering, and apoptosis control. Mitochondria synthesize ATP through the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (Zhao et al., 2019). During oxidative phosphorylation, electrons are transported down the electron transport chain (ETC) and provide energy for the activity of the downstream respiratory H+ pump chain (respiratory complex I–IV). Respiratory complexes pump H+ out of the mitochondrial matrix and thus establish an H+ gradient and a mitochondrial membrane potential across the mitochondrial inner membrane. Mitochondrial membrane potential promotes ATP synthesis by complex V (Zorova et al., 2018) and supports MCU activity for Ca2+ uptake (Vais et al., 2020). Mitochondria are the major source of cellular ROS. ROS is produced in the mitochondria when electrons in the ETC are captured by oxygen, thus producing superoxide anion radicals. A deficit in ETC leads to excessive ROS production, which is known as mitochondrial oxidative stress (Bhatti et al., 2017). Ca2+ is a key regulator of mitochondrial function. The transient increase in cytosolic Ca2+ rapidly stimulates mitochondrial uptake, which helps to prevent Ca2+ overload in the cytosol and enhances ATP production to meet the increased metabolic demands by activation of Ca2+-dependent enzymes of both the TCA cycle and oxidative phosphorylation (Glancy and Balaban, 2012). However, a prolonged increase in cytosolic Ca2+ causes mitochondrial Ca2+ overload, resulting in the collapse of mitochondrial membrane potential, which reduces ATP production. Importantly, mitochondrial Ca2+ overload triggers mitochondrial oxidative stress by enhancing ROS production. Severe oxidative stress promotes the opening of the mitochondrial permeability transition pore (mPTP) and leads to the release of cytochrome c, triggering apoptosis (Peng and Jou, 2010). The mitochondria also release ROS to the cytoplasm via the mPTP.
The ER is a major intracellular Ca2+ storage site in neurons involved in the generation and termination of cellular Ca2+ signaling. Three types of proteins are highly expressed in the ER with different functions in the control of cellular Ca2+ homeostasis: Ca2+ release channels (IP3R and RyR) contribute to cellular Ca2+ signaling generation by transporting Ca2+ from the ER lumen to the cytoplasm; ER resident Ca2+ binding proteins determine the ER Ca2+ buffering capacity; Ca2+ pumps (SERCA) contribute to sequestration of elevated cytosolic Ca2+ by pumping Ca2+ into the ER lumen. Both Ca2+ release channels and pumps are sensitive to cytosolic and ER luminal Ca2+ and ROS. Luminal Ca2+-binding proteins, such as calreticulin, calnexin, protein disulfide isomerases (PDI), and glucose-regulated protein 94 kDa (GRP94), regulate ER function not only by directly buffering luminal Ca2+, but also by regulating Ca2+ release channels, Ca2+ pumps, or ER proteins involved in protein synthesis and folding (Prins and Michalak, 2011).
The ER is also a “protein synthesis factory” responsible for the synthesis, folding, maturation, degradation, and translocation of secretory and transmembrane proteins (Braakman and Bulleid, 2011). Once translocated to the ER from ribosomes, the newly synthesized proteins undergo several post-translational modifications. Proper folding of proteins into their tertiary and quaternary structures is assisted by the coordinated work of chaperones, foldases, and cofactors (Yoshida et al., 2001). The optimal concentrations of ATP, Ca2+, and ROS regulate protein folding through disulfide bond formation (Hetz et al., 2011). Transportation of properly folded proteins inside vesicles to the surface of the cell membrane or extracellular space is strictly monitored by the ER protein quality control system (ERQC). If a protein is unfolded or misfolded, the protein is targeted for ER retention, which subsequently stimulates ER-associated degradation (ERAD) through proteolysis (Benyair et al., 2011). Depletion of ER Ca2+ stores, a decrease in ATP levels and an increase in the oxidative state lead to reduced protein-folding capacity of the ER, resulting in the accumulation and aggregation of unfolded or misfolded proteins, a condition termed ER stress (Bravo et al., 2013). ER stress triggers an adaptive response, called the unfolded protein response (UPR). UPR is a complex signaling pathway that controls adaptive processes through both transcriptional and non-transcriptional responses, regulating each aspect of the protein synthesis pathway, including protein folding, ER biogenesis, ERAD, protein entry to the ER, autophagy, and secretion, thus, UPR is a pro-survival response that is beneficial for immediate removal of unfolded or misfolded proteins and restoration of the normal function of the ER (Oslowski and Urano, 2011). However, prolonged ER stress leads to sustained UPR, converting a pro-survival response to pro-apoptotic signaling (Read and Schröder, 2021). Dyshomeostasis of cellular Ca2+ may induce depletion of ER Ca2+ stores by increasing Ca2+ release via RyR and IP3R, and cause Ca2+ overload in mitochondria, leading to reduced ATP production and oxidative stress, all of which contribute to ER stress and impaired UPR, thus initiating apoptosis.
Lysosomes are small organelles that function as the “recycling plant” of the cells, responsible for degradation of macromolecules (proteins, nucleic acids, lipids, and carbohydrates), damaged organelles and microorganisms (Trivedi et al., 2020). Lysosomes contain more than 100 membrane proteins that are involved in ion transport, autophagy, and membrane fusion and repair (Schwake et al., 2013). The acidic lumen of the lysosome is also abundant in hydrolases, including nucleases, proteases, lipases, glycosidases, sulfatases, and phospholipases (Luzio et al., 2014). These lysosomal hydrolases are responsible for breaking large molecules down to small molecules for recycling into the cytosol to form new macromolecules. They are also involved in the degradation of dysfunctional organelles, invaders, toxins, and pathogens that enter the cell via endocytic mechanisms. Autophagy is a cellular process through which cytosolic misfolded proteins or damaged organelles are directed to lysosomes for degradation. This process is important for the maintenance of normal cellular homeostasis by the clearance of dysfunctional proteins and organelles. Dysfunction of autophagy results in the accumulation and aggregation of aberrant proteins, such as α-synuclein, which is the major component of the Lewy bodies in PD neurons (Spillantini et al., 1997). Autophagy is highly regulated by Ca2+. Recent studies have shown that lysosomal Ca2+ release via TRPML1 is crucial for the regulation of autophagy. Ca2+ release through TRPML1 induces dephosphorylation and subsequent nuclear translocation of transcription factor EB (TFEB) by activation of Ca2+/calmodulin (CaM)-dependent phosphatase calcineurin. TFEB is a lysosomal master gene regulator that enters the nucleus and promotes expression of several genes associated with the regulation of the autophagic pathway (Medina et al., 2015). TRPML1 is a target of lysosomal membrane-resident mammalian target of rapamycin (mTOR). mTOR-mediated phosphorylation of TRPML1 inhibits TRPML1 activity, while mTOR function depends on Ca2+-CaM (Li et al., 2016). Thus, Ca2+ release through TRPML1 mediates a negative feedback regulation between mTOR and TRPML1. TRPML1 is also a ROS sensor whose activity is upregulated by ROS (Zhang et al., 2016). Once oxidative stress occurs, ROS induces Ca2+ release through TRPML1, which in turn enhances autophagy through the calcineurin-TFEB signaling pathway. Augmented autophagy promotes elimination of damaged mitochondria that produce excessive ROS and restores normal redox homeostasis. Mutations and dysfunction of TRPML1 have been implicated in the development of PD (Tsunemi et al., 2019). Dyshomeostasis of cytosolic Ca2+ may cause dysregulation of lysosomal Ca2+ homeostasis by triggering mitochondria-derived oxidative stress and ER stress. Excessive ROS stimulates lysosomal release via TRPML1, while ER stress-induced depletion of Ca2+ stores reduces lysosomal refilling, both of which may contribute to impaired lysosomal Ca2+ homeostasis, lysosomal dysfunction, and autophagy deregulation.
Under physiological conditions, Ca2+ influx, via the plasma membrane Ca2+-permeable channel and subsequent Ca2+ release from subcellular organelles, induces Ca2+ signaling within a cell in response to various stimulations. Cellular Ca2+ signaling is a transient elevation of cytosolic Ca2+, which can be quickly terminated by extracellular extrusion, uptake into intracellular organelles, and sequestration by Ca2+-binding proteins. Transient elevation of cytosolic Ca2+ enhances the functions of the ER, mitochondria, and lysosomes. Elevated cytosolic Ca2+ moderately increases the Ca2+ load in the mitochondria, which promotes ATP production and ROS release from mitochondria (Glancy and Balaban, 2012). Ca2+ release from the ER and ROS release from the mitochondria in turn enhance lysosomal Ca2+ refilling and Ca2+ release via TRPML1, which augments autophagy signaling. All these processes are beneficial for cellular homeostasis. However, in pathological conditions, prolonged elevation of cytosolic Ca2+ converts the virtuous interplay between the ER, mitochondria, and lysosomes to a vicious cycle (Figure 2). Excessive cytosolic Ca2+ increases Ca2+ release from the ER and is transported to the mitochondria, causing mitochondrial Ca2+ overload (Santulli et al., 2015). Ca2+ overload leads to mitochondrial dysfunction, resulting in reduced ATP production and oxidative stress (Peng and Jou, 2010). Low ATP supply decreases ER Ca2+ uptake via SERCA, which requires ATP, while excessive ROS facilitates Ca2+ release from the ER through upregulation of RyR and IP3R, thus resulting in depletion of ER Ca2+ stores (Zhang et al., 2006). Reduced Ca2+ stores together with low ATP and higher levels of ROS cause ER dysfunction and impaired UPR, which enables accumulation of unfolded and misfolded proteins in the cells (Bahar et al., 2016). Depletion of ER Ca2+ stores attenuates lysosomal refilling and subsequent Ca2+ release via TRPML1, which impairs the autophagy signaling pathway, leading to accumulation of abnormal protein aggregates and damaged mitochondria within the cell (Shulman et al., 2011). Protein aggregates, such as α-synuclein aggregates, and accumulation of damaged mitochondria further enhance cellular oxidative stress (Reeve et al., 2015). Therefore, sustained cytosolic Ca2+ dyshomeostasis mediates the vicious interplay between ER stress, oxidative stress, and impaired autophagy. In contrast, dysfunction of the ER, mitochondria, and lysosomes further exacerbates Ca2+ dyshomeostasis. For example, depletion of ER Ca2+ stores activates SOCs, resulting in increased Ca2+ influx (Yuan et al., 2009). Toxic α-synuclein oligomers may integrate into the cell membrane to form a pore that allows Ca2+ influx (Kim et al., 2009). Excessive ROS has opposite effects on the activities of Ca2+-handling proteins responsible for the elevation and sequestration of cytosolic Ca2+. ROS facilitates plasma membrane Ca2+-permeable channels and organelle membrane Ca2+ release channels, however, it inhibits Ca2+-ATPase and Ca2+-transporters, which are responsible for pumping Ca2+ out of the cell or uptake by organelles, respectively (Görlach et al., 2015). Recently, accumulating evidence has shown that Ca2+ dyshomeostasis, ER stress, mitochondrial oxidative stress, and impaired autophagy typically appear in SNc DA neurons in PD (Abou-Sleiman et al., 2006; Calì et al., 2011; Michel et al., 2016; Sunanda et al., 2021), suggesting that dyshomeostasis is likely a common pathway in PD pathogenesis.
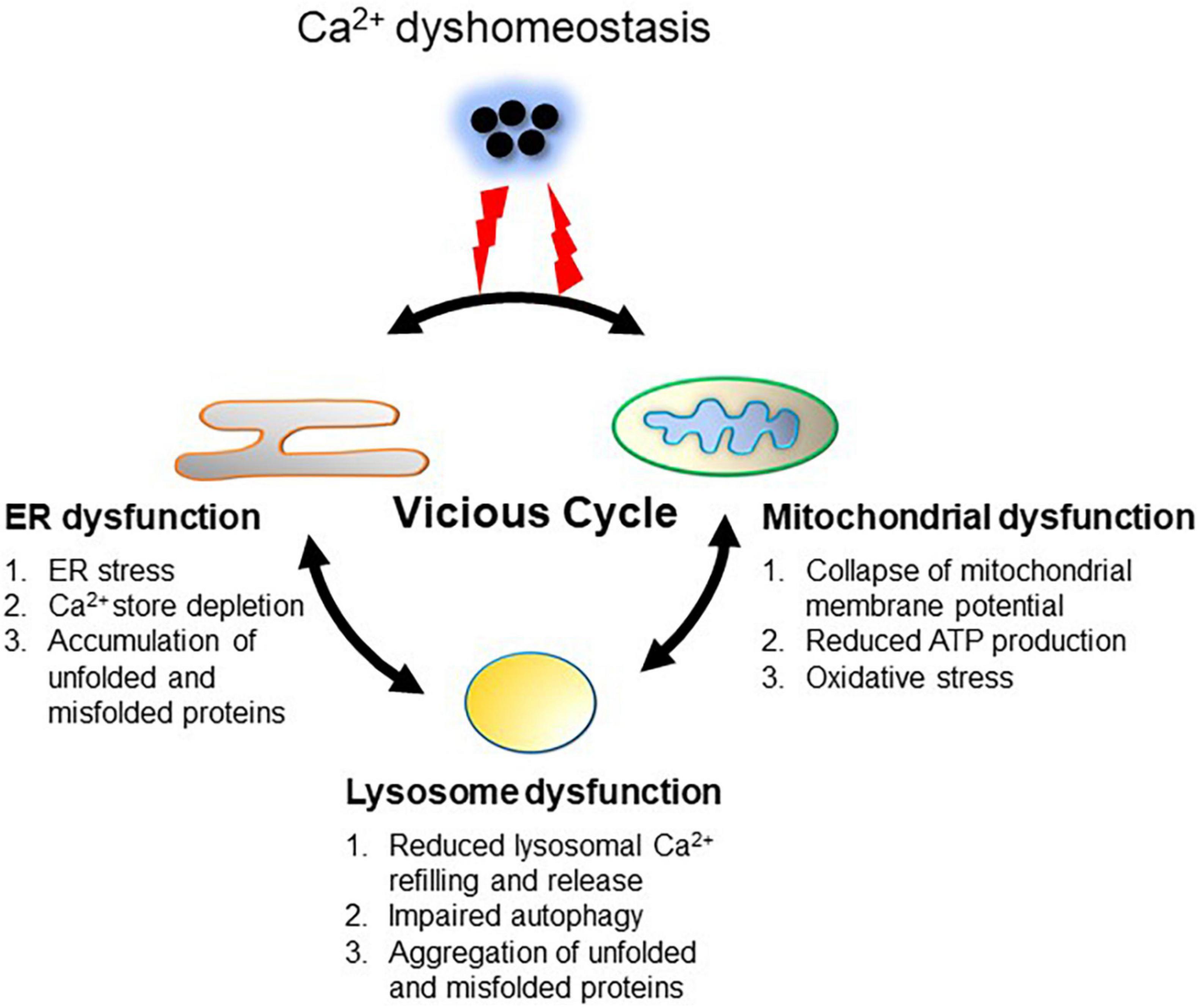
Figure 2. Ca2+ dyshomeostasis triggers a vicious cycle between the ER, mitochondria and lysosomes. Dysregulation of Ca2+ handling leads to intracellular Ca2+ overload. Prolonged excessive Ca2+ induces Ca2+ overload in the mitochondria through increasing mitochondrial uptake from the ER and cytoplasm, resulting in mitochondrial dysfunction (oxidative stress and reduced ATP production). ER dysfunction is induced by intracellular Ca2+ overload as well as increased ROS and reduced ATP from the mitochondria, resulting in ER stress, Ca2+ store depletion, and accumulation of unfolded or misfolded proteins. Dysfunction of the ER and mitochondria impairs lysosome functions through reduction of lysosomal Ca2+ refilling and subsequent Ca2+ release via TRPML1, which is tightly associated with impaired autophagy, leading to aggregation of abnormal proteins such as α-synuclein. Aggregation of α-synuclein in turn deteriorates ER and mitochondrial function. This vicious cycle between the ER, mitochondria and lysosomes perpetuates cellular Ca2+ dyshomeostasis.
Aging is an inevitable and irreversible biological process. The underlying mechanisms of aging remain unclear. Aging is a stochastic event with no defined triggering mechanisms and clear starting age, suggesting that advanced age itself may not be a problem, however, it increases the possibilities of exposure to environmental detrimental factors and gene mutations, which cause accumulation of cellular pathologies over time due to constant disruption of the balance between cellular damage and repair. Therefore body cells may become unable to maintain their homeostasis, in particular Ca2+ and redox homeostasis. Long-term exposure to toxic chemicals and ultraviolet light, chronic infections and inflammation, hormonal imbalance, psychological stress, and even bad lifestyle (dietary habits and sleep) may contribute to aging (Pyo et al., 2020).
Aging is now considered the greatest risk factor for PD. However, the mechanism linking aging to the loss of SNc DA neurons in PD is largely unknown. Ca2+ is crucial for the regulation of neuronal development, survival, and death, even slight alterations in cellular Ca2+ levels may have deleterious consequences on neurons. Recently, increasing evidence has shown that Ca2+ dyshomeostasis is involved in both aging and PD (Cortes et al., 2020), suggesting that dysregulation of Ca2+ handling is a common feature of aging and PD.
It has been widely reported that neuronal Ca2+ handling is dysregulated with age as a result of changes in the activity or cellular expression of proteins involved in the control of cellular Ca2+ homeostasis. Increased Ca2+ influx has been reported in aging neurons. AMPAR mediates Na+ influx in the presence of the GluR2 subunit in developing neurons, whereas AMPAR without GluR2 in aging neurons allows Ca2+ entry (Pandey et al., 2015). Upregulation of the expression level and activities of Cav1.3 channels in neurons has been reported in aging and PD (Veng and Browning, 2002; Hurley et al., 2013). α-synuclein aggregates in neurons, the hallmark of PD, are usually observed in aging too (Bobela et al., 2015). α-synuclein oligomers have been reported to form Ca2+-permeable channels, which promote an increase in cytosolic Ca2+ levels (Kim et al., 2009). Ca2+ release from the ER is increased, presumably due to the activation of IP3R and RyR by dysfunctional mitochondria-related ROS in aged neurons (Keller et al., 2004). In contrast, increased ROS and decreased ATP levels due to mitochondrial dysfunction in aging and PD inhibit SERCA, PMCA, and NCX, leading to impaired Ca2+ sequestration. Additionally, during brain aging, the expression levels of cellular Ca2+-binding proteins, such as calbindin and calretinin, are decreased in SNc DA neurons (Hurley et al., 2013), leading to lower Ca2+-buffering capacity. Therefore, with age, any detrimental factors or gene mutations that can directly impact neuronal Ca2+ handling or indirectly by affecting organelle functions may lead to Ca2+ dyshomeostasis and contribute to neurodegeneration and loss of neurons in PD.
Since the first reports showing that SNCA was the primary causative gene involved in the early onset of familial PD (Polymeropoulos et al., 1997), an increasing number of genes have been reported as direct contributors or risk factors for PD. It is notable that the majority of PD cases are sporadic, with only 10-15% of cases having familial history (Deng et al., 2018). Therefore, there is now a general consensus that interactions between genetic factors and environmental exposure are major contributors to the etiology of PD, especially early onset PD (Cannon and Greenamyre, 2013). Moreover, accumulating clinical and pathological evidence shows that familial and sporadic forms of PD are often highly similar, suggesting that they may share common pathogenetic mechanisms (Pang et al., 2019). Indeed, similar to sporadic forms of PD, familial forms of PD are associated with genetic mutations that lead to mitochondrial dysfunction, oxidative stress, impaired autophagy, and accumulation of α-synuclein aggregates (Pang et al., 2019). As mentioned earlier, these pathological processes are regulated and triggered by Ca2+, which in turn exacerbates cellular Ca2+ dyshomeostasis, forming a vicious cycle in PD pathogenesis.
To date, there have been numerous genes whose mutations have been known to increase the risk of PD. Here, we focus on several reported genetic mutations that are well documented to be associated with PD pathogenesis and discuss how these mutations induce dysregulation of Ca2+ handling during PD progression.
SNCA is the first gene identified with mutations that contribute to early onset of familial PD. The SCNA gene encodes α-synuclein, a 140-amino acid protein. Most missense mutations, such as A30P, A53T, E46K, H50Q, and G51D are located at the amino terminus of α-synuclein and may cause familial PD (Klein and Westenberger, 2012). α-synuclein is natively unfolded and adopts an α-helical conformation when it binds to the cell membrane with its amino terminus (Burré et al., 2013), while the pathogenic missense SNCA variants tend to form stable β sheets that promote aggregation of α-synuclein, which is toxic to neurons (Bertoncini et al., 2005). Previous studies have shown that natively unfolded α-synuclein is selectively translocated into lysosomes for degradation (Cuervo et al., 2004). Decreasing lysosomal degradation or increasing the expression of unfolded and misfolded α-synuclein leads to aggregation.
The GBA gene encodes for a lysosomal glucocerebrosidase (GCase) that catalyzes the conversion of the sphingolipid glucosylceramide to ceramide and glucose. GBA mutations, such as E326K, are now considered as the single largest risk factor for idiopathic PD. GBA mutations cause decreased expression and activity of GCase, resulting in accumulation of glucosylceramide, which enhances α-synuclein aggregation by stabilizing soluble α-synuclein oligomers (Muñoz et al., 2021). Both GBA mutations and sporadic PD showed decreased GCase levels and increased α-synuclein aggregation.
Accumulation of α-synuclein aggregates has been reported in both familial and sporadic PD patients (Stefanis, 2012), suggesting a central role of this protein in PD pathogenesis. Aggregation of α-synuclein exacerbates cellular Ca2+ dyshomeostasis directly or indirectly, through the following mechanisms: (1) α-synuclein oligomers form Ca2+-permeable pores in the cell membrane that increase Ca2+ influx; (2) aggregated or misfolded α-synuclein interacts with and impairs mitochondrial complex I, causing mitochondrial dysfunction and oxidative stress in cells (Reeve et al., 2015), which indirectly causes Ca2+ dysregulation. When Ca2+ dyshomeostasis surpasses a certain threshold, it accelerates PD progression through perpetuating a vicious cycle between the mitochondria, ER, and lysosomes. Therefore, Ca2+ dyshomeostasis may link SNCA and GBA mutations to neurodegeneration and ultimately to the loss of neurons in PD.
LRRK2 encodes for leucine-rich repeat kinase 2 (LRRK2), a protein containing a GTPase and a kinase domain. LRRK2 mutations are typically associated with late-onset familial PD (Tolosa et al., 2020). The G2019S variant has been reported to increase LRRK2 kinase activity (West et al., 2005). Although the exact mechanism underlying LRRK2-related PD pathogenesis is not fully clear, the G2019S variant has been shown to impair lysosomal functions through phosphorylation of its substrates, especially Rab proteins, which are required for all vesicular trafficking steps (MacLeod et al., 2006) as well as the interaction between mitochondria and lysosomes (Wong et al., 2018). Inactivation of Rab proteins by LRRK2-mediated phosphorylation is associated with lysosomal dysfunction in LRRK2-related PD. Interestingly, mutant LRRK2-mediated autophagic deficits can be reversed by inhibition of the endolysosomal Ca2+ release channel TPC2 (Hockey et al., 2015), suggesting that dysregulation of lysosomal Ca2+ handling is associated with LRRK2-related pathogenic processes.
The PRKN (PARK2) gene encodes for the E3 ubiquitin ligase parkin, and mutations in parkin constitute the most common cause of early onset PD (Klein and Lohmann-Hedrich, 2007). Parkin contains a cysteine in its catalytic site that forms an intermediate thioester bond with ubiquitin, accelerating the covalent interaction between ubiquitin and the lysine residue of a substrate protein. Activation of parkin is mainly triggered by the binding of phosphorylated ubiquitin (pUbS65) or direct phosphorylation of parkin at serine 65 (pParkinS65). The mitochondria-resident protein, PTEN-induced putative kinase protein 1 (PINK1), mediates phosphorylation and activation of parkin; therefore, loss-of-function mutations in PINK1 lead to impaired parkin activity and are associated with a PD phenotype similar to that of mutated PRKN-related PD (Kamienieva et al., 2021). Parkin directs ubiquitin to its target substrates for proteasomal degradation. Parkin targets the mitochondria through interactions with PINK1. Once inactivated by PINK1, parkin ubiquitinates various mitochondrial regulatory proteins at the membrane surface for degradation, thus regulating mitochondrial functions, including mitochondrial fission and fusion, biogenesis, mitophagy, and ER-mitochondrial contacts. Mutations in PRKN lead to reduced parkin activity and subsequent mitochondrial dysfunction (Kamienieva et al., 2021).
Although exposure to environmental factors (chemicals and toxins) has been associated with increased risk of PD, it is difficult to determine whether an environmental factor is the definitive cause of PD. In other words, each environmental factor may contribute to increased risk, but may not be sufficient to cause PD. It is widely accepted that PD results from complex interactions between two or more environmental factors, possibly independent of gene mutations (Ball et al., 2019). Curiously, few environmental factors directly affect Ca2+-handling proteins, suggesting that Ca2+ dyshomeostasis is indirectly triggered by cumulative cellular dyshomeostasis initiated by environmental factors, particularly mitochondrial dysfunction and oxidative stress. Once Ca2+ dysregulation reaches a threshold, it drives vicious interactions between the ER, mitochondria, and lysosomes, thus causing PD. In this review, some environmental factors that selectively target substantia nigra neurons are briefly discussed, including pesticides, heavy metals, and solvents.
Pesticides are used in the agricultural industry to control pests and destroy weeds. Exposure to pesticides has been linked to increased risk of PD. Organochlorines and organophosphates are well-documented pesticides. One of the organochloride pesticides, dieldrin, preferentially targets SNc DA neurons and causes neurotoxic damage through the induction of oxidative stress and mitochondrial dysfunction (Kanthasamy et al., 2005). Application of the antioxidant N-acetyl cysteine protects from dieldrin-induced neurotoxicity by restoring normal ROS levels and mitochondrial membrane potential (Sharma et al., 2010). Rotenone is the main organophosphate component involved in increased PD risk through promotion of mitochondrial dysfunction by inhibiting mitochondrial complex I and ROS-activated aggregation of α-synuclein (Betarbet et al., 2000).
There is a link between exposure to various metals and increased risk of PD. Occupational exposure to high-dose manganese during welding is associated with a form of parkinsonism, known as manganism, with symptoms such as tremors, gait abnormalities, and facial muscle spasms. Promotion of aggregation and cell-to-cell transmission of α-synuclein by manganese has recently been suggested to cause neurotoxicity in SNc DA neurons (Harischandra et al., 2019). Accumulation of either iron or copper in the midbrain has been linked to increased PD risk. Iron and copper, as redox metal ions, can elevate the oxidative state of DA neurons by contributing to the conversion of the superoxide anion and hydrogen peroxide into the hydroxyl radical through the Fenton and Haber-Weiss reactions (Collin, 2019). Long-term exposure to lead in the environment is also a risk factor for PD. Lead has been reported to decrease DA release by disrupting the dopaminergic system. In addition, lead is a pro-oxidant that can cause oxidative stress by enhancing ROS production, leading to cellular dyshomeostasis and subsequent neuronal damage (Neal and Guilarte, 2013).
Exposure to organic solvents may also play an etiologic role in PD development. Organic solvents are commonly used in metal degreasing, dry cleaning, and as detergents. Trichloroethylene (TCE) is an industrial solvent, and exposure to TCE has been implicated as an environmental risk factor for PD. Although the exact mechanism underlying the effect of TCE on PD risk is not completely clear, TCE-associated mitochondrial dysfunction and deficits in complex I contribute to PD pathogenesis (Lock et al., 2013).
Epidemiological studies have shown that PD is 1.5–2 times more frequent in men than in women (de Lau et al., 2004). The precise reasons for the gender difference in the incidence of PD are not fully understood. One hypothesis is that men are more likely to be exposed to PD-related environmental factors than women. However, meta-analysis reveals a similar incidence of PD in men and post-menopausal women, suggesting that female sex hormones may be involved in the protective effect in PD (Moisan et al., 2016). Increasing evidence suggests that estrogen may elicit a beneficial effect on SNc DA neurons in PD, although the underlying mechanism of estrogen actions remains to be clarified. Several aspects of estrogen action have been reported to help protect women from PD through genomic and non-genomic mechanisms. Estrogen enhances nigrostriatal DA synthesis and release (Jacobs and D’Esposito, 2011), which are tightly associated with Ca2+ signals. Whether the increased DA release is due to a direct action of estrogen or an indirect effect through an estrogen-induced increase in cytosolic Ca2+ requires further investigation. Recently, estrogenic control of mitochondrial function and biogenesis has drawn more attention. It has been extensively reported that estrogen enhances electron transport chain activity, maintains the mitochondrial membrane potential, increases ATP production, inhibits excessive ROS production, and prevents mitochondrial Ca2+ overload (Klinge, 2008, 2020; Lejri et al., 2018; Ventura-Clapier et al., 2019), which help maintain Ca2+ and cellular homeostasis. Furthermore, estrogen has been reported to support stable, helically folded α-synuclein tetramer that resists aggregation (Rajsombath et al., 2019), consistent with the findings that females show less accumulation of α-synuclein than males. However, it is notable that the beneficial effects of estrogen are related to healthy status of the neurons. Although estrogen protects healthy neurons against PD development, once neurodegeneration is induced, estrogen has little effect or even exacerbates disease (Morrison et al., 2006; Miller and Harman, 2017). Therefore, the precise mechanism of estrogen effect on SNc DA neurons remains to be further elucidated.
There are also other risk factors for PD, such as some medications which block the action of dopamine, and head trauma. In this review, the possible effect of head trauma on PD development is briefly discussed.
Growing evidence shows a clear link between head trauma and an increased risk of PD. Head injuries are common and can be caused by unexpected incidents, such as traffic accidents and falls, or some specific sport activities. The risk of PD due to head injuries is higher with more severe or recurrent injuries and loss of consciousness (Mckee and Daneshvar, 2015). Recent studies have shown that even mild traumatic brain injury (TBI) with loss of consciousness for more than 1 h may increase the risk of subsequent onset of PD by 1.5-fold, compared to controls (Gardner et al., 2018). The exact mechanism by which TBI increases the risk of PD is not clear. TBI-induced inflammation, metabolic dysregulation, and protein aggregation have been suggested as underlying mechanisms involved in the pathogenesis of PD (Delic et al., 2020). These studies showed that TBI accelerated α-synuclein aggregation in DA neurons (Acosta et al., 2015). Moreover, upregulation of LRRK2 has been found in the brains of mice following TBI, and inhibition of LRRK2 was neuroprotective in PD and TBI models (Bae et al., 2018). Rab is a substrate of LRRK2, which has been associated with PD progression; therefore, further studies are required to determine whether the contribution of TBI-induced LRRK2 upregulation to PD pathogenesis is mediated through Rab signaling.
Parkinson’s disease is a chronic and progressive neurodegenerative disorder initiated by a number of risk factors, suggesting that risk factor-induced cellular dyshomeostasis accumulates over time. When cellular repair and compensatory systems cannot overcome sustained pathological injuries within cells, cell damage occurs. The ER, mitochondria, and lysosomes are crucial for the maintenance of cellular homeostasis, they interact with each other, and change their functions in response to signal stimulation. Ca2+ is the most important signaling molecule that participates in all cellular processes. Ca2+ transfer between subcellular organelles mediates coordinated and concerted interactions involved in cellular homeostasis. Under physiological conditions, cellular Ca2+ homeostasis is tightly controlled by Ca2+-handling proteins. However, in pathological conditions, prolonged impairment of cellular functions may directly or indirectly cause dysregulation of Ca2+ handling, resulting in cellular Ca2+ dyshomeostasis. SNc DA neurons are highly susceptible to an additional increase in cellular Ca2+, based on their features in Ca2+ handling. Sustained Ca2+ overload in SNc DA neurons may convert Ca2+ from a physiological regulatory factor to a pathological trigger. Indeed, growing evidence suggests that long-term excessive cytosolic Ca2+ contributes to ER, mitochondria, and lysosomal dysfunction in PD pathogenesis. Furthermore, recent studies support the hypothesis that Ca2+ dyshomeostasis is central in perpetuating the vicious cycle between the ER, mitochondria, and lysosomes, because increasing Ca2+ buffering capacity by overexpression of Ca2+-binding proteins or manipulation of subcellular Ca2+ handling can reduce oxidative stress, ameliorate autophagy, and decrease protein aggregation (Liu et al., 1997; Jung et al., 2019; Kang et al., 2021). Therefore, elimination of cellular Ca2+ overload and Ca2+ dyshomeostasis in subcellular organelles by application or expression of cell-specific and organelle-specific Ca2+-handling molecules may represent a promising strategy for PD treatment.
JX and MK wrote the manuscript. EM participated in discussion and collection of the manuscript. All authors approved the manuscript for publication.
This work was supported by the JSPS KAKENHI Grant (numbers 19k07285 to JX, 20K07263 to MK, and 19K07303 to EM) and a Grant from the Kodama Memorial Foundation for Medical Research to JX.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Editage (www.editage.com) for English language editing. JX thanks Wei-Jun Fu and Ke Xu for encouragement.
Abou-Sleiman, P. M., Muqit, M. M., and Wood, N. W. (2006). Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 7, 207–219. doi: 10.1038/nrn1868
Acosta, S. A., Tajiri, N., de la Pena, I., Bastawrous, M., Sanberg, P. R., Kaneko, Y., et al. (2015). Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson’s disease. J. Cell Physiol. 230, 1024–1032. doi: 10.1002/jcp.24830
Aracena-Parks, P., Goonasekera, S. A., Gilman, C. P., Dirksen, R. T., Hidalgo, C., and Hamilton, S. L. (2006). Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J. Biol. Chem. 281, 40354–40368. doi: 10.1074/jbc.M600876200
Ascherio, A., and Schwarzschild, M. A. (2016). The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol. 15, 1257–1272. doi: 10.1016/S1474-4422(16)30230-7
Atakpa, P., Thillaiappan, N. B., Mataragka, S., Prole, D. L., and Taylor, C. W. (2018). IP3 receptors preferentially associate with ER-lysosome contact sites and selectively deliver Ca2+ to lysosomes. Cell Rep. 25, 3180.e–3193.e. doi: 10.1016/j.celrep.2018.11.064
Austin, S., Tavakoli, M., Pfeiffer, C., Seifert, J., Mattarei, A., De Stefani, D., et al. (2017). LETM1-mediated K+ and Na+ homeostasis regulates mitochondrial Ca2+ efflux. Front. Physiol. 8:839. doi: 10.3389/fphys.2017.00839
Bae, Y. H., Joo, H., Bae, J., Hyeon, S. J., Her, S., Ko, E., et al. (2018). Brain injury induces HIF-1α-dependent transcriptional activation of LRRK2 that exacerbates brain damage. Cell Death Dis. 9:1125. doi: 10.1038/s41419-018-1180-y
Bahar, E., Kim, H., and Yoon, H. (2016). ER stress-mediated signaling: action potential and Ca2+ as key players. Int. J. Mol. Sci. 17:1558. doi: 10.3390/ijms17091558
Ball, N., Teo, W. P., Chandra, S., and Chapman, J. (2019). Parkinson’s disease and the environment. Front. Neurol. 10:218. doi: 10.3389/fneur.2019.00218
Benyair, R., Ron, E., and Lederkremer, G. Z. (2011). Protein quality control, retention, and degradation at the endoplasmic reticulum. Int. Rev. Cell. Mol. Biol. 292, 197–280. doi: 10.1016/B978-0-12-386033-0.00005-0
Bertoncini, C. W., Fernandez, C. O., Griesinger, C., Jovin, T. M., and Zweckstetter, M. (2005). Familial mutants of alpha-synuclein with increased neurotoxicity have a destabilized conformation. J. Biol. Chem. 280, 30649–30652. doi: 10.1074/jbc.C500288200
Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V., and Greenamyre, J. T. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 3, 1301–1306. doi: 10.1038/81834
Bhatti, J. S., Bhatti, G. K., and Reddy, P. H. (2017). Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1066–1077. doi: 10.1016/j.bbadis.2016.11.010
Bobela, W., Aebischer, P., and Schneider, B. L. (2015). A lpha-synuclein as a mediator in the interplay between aging and Parkinson’s disease. Biomolecules 5, 2675–2700. doi: 10.3390/biom5042675
Braakman, I., and Bulleid, N. J. (2011). Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 80, 71–99. doi: 10.1146/annurev-biochem-062209-093836
Bravo, R., Parra, V., Gatica, D., Rodriguez, A. E., Torrealba, N., Paredes, F., et al. (2013). Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic inte gration. Int. Rev. Cell Mol. Biol. 301, 215–290. doi: 10.1016/B978-0-12-407704-1.00005-1
Brini, M., Calì, T., Ottolini, D., and Carafoli, E. (2014). Neuronal calcium signaling: function and dysfunction. Cell. Mol. Life Sci. 71, 2787–2814. doi: 10.1007/s00018-013-1550-7
Burgoyne, R. D. (2007). Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 8, 182–193. doi: 10.1038/nrn2093
Burré, J., Vivona, S., Diao, J., Sharma, M., Brunger, A. T., and Südhof, T. C. (2013). Properties of native brain α-synuclein. Nature 498, E4–E6.
Calcraft, P. J., Ruas, M., Pan, Z., Cheng, X., Arredouani, A., Hao, X., et al. (2009). NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459, 596–600. doi: 10.1038/nature08030
Calì, T., Ottolini, D., and Brini, M. (2011). Mitochondria, calcium, and endoplasmic reticulum stress in Parkinson’s disease. Biofactors 37, 228–240. doi: 10.1002/biof.159
Cannon, J. R., and Greenamyre, J. T. (2013). Gene-environment interactions in Parkinson’s disease: specific evidence in humans and mammalian models. Neurobiol. Dis. 57, 38–46. doi: 10.1016/j.nbd.2012.06.025
Chan, C. S., Guzman, J. N., Ilijic, E., Mercer, J. N., Rick, C., Tkatch, T., et al. (2007). ‘Rejuve nation’ protects neurons in mouse models of Parkinson’s disease. Nature 447, 1081–1086. doi: 10.1038/nature05865
Chen, H. (2018). The changing landscape of Parkinson epidemiologic research. J. Parkinsons. Dis. 8, 1–12. doi: 10.3233/JPD-171238
Cohen, S., Valm, A. M., and Lippincott-Schwartz, J. (2018). Interacting organelles. Curr. Opin. Cell Biol. 53, 84–91. doi: 10.1016/j.ceb.2018.06.003
Collin, F. (2019). Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int. J. Mol. Sci. 20:2407. doi: 10.3390/ijms20102407
Cortes, L., Malva, J., Rego, A. C., and Pereira, C. F. (2020). Calcium signaling in aging and neurodegenerative diseases 2019. Int. J. Mol. Sci. 21:1125. doi: 10.3390/ijms21031125
Csordás, G., Weaver, D., and Hajnóczky, G. (2018). Endoplasmic reticulum-mitochondrial contactology: Structure and signaling functions. Trends Cell Biol. 28, 523–540. doi: 10.1016/j.tcb.2018.02.009
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., and Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. doi: 10.1126/science.1101738
de Lau, L. M., Giesbergen, P. C., de Rijk, M. C., Hofman, A., Koudstaal, P. J., and Breteler, M. M. (2004). Incidence of parkinsonism and Parkinson disease in a general population: the rotterdam study. Neurology 63, 1240–1244. doi: 10.1212/01.wnl.0000140706.52798.be
de Lau, L. M. L., and Breteler, M. M. B. (2006). Epidemiology of Parkinson’s disease. Lancet Neurol. 5, 525–535. doi: 10.1016/S1474-4422(06)70471-9
Delic, V., Beck, K. D., Pang, K. C. H., and Citron, B. A. (2020). Biological links between traumatic brain injury and Parkinson’s disease. Acta Neuropathol. Commun. 8:45. doi: 10.1186/s40478-020-00924-7
Deng, H., Wang, P., and Jankovic, J. (2018). The genetics of Parkinson disease. Ageing Res. Rev. 42, 72–85. doi: 10.1016/j.arr.2017.12.007
Dopeso-Reyes, I. G., Rico, A. J., Roda, E., Sierra, S., Pignataro, D., Lanz, M., et al. (2014). Calbindin content and differential vulnerability of midbrain efferent dopaminergic neurons in macaques. Front. Neuroanat. 8:146. doi: 10.3389/fnana.2014.00146
Eden, E. R., White, I. J., Tsapara, A., and Futter, C. E. (2010). Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat. Cell Biol. 12, 267–272. doi: 10.1038/ncb2026
Galione, A. (2015). A primer of NAADP-mediated Ca2+ signalling: From sea urchin eggs to mammalian cells. Cell Calcium. 58, 27–47. doi: 10.1016/j.ceca.2014.09.010
Gardner, R. C., Byers, A. L., Barnes, D. E., Li, Y., Boscardin, J., and Yaffe, K. (2018). Mild TBI and risk of Parkinson disease: A chronic effects of neurotrauma consortium study. Neurology 90, e1771–e1779. doi: 10.1212/WNL.0000000000005522
Glancy, B., and Balaban, R. S. (2012). Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51, 2959–2973. doi: 10.1021/bi2018909
Gómez-Suaga, P., Bravo-San Pedro, J. M., González-Polo, R. A., Fuentes, J. M., and Niso-Santano, M. (2018). ER-mitochondria signaling in Parkinson’s disease. Cell Death Dis. 9:337. doi: 10.1038/s41419-017-0079-3
González-Rodríguez, P., Zampese, E., Stout, K. A., Guzman, J. N., Ilijic, E., Yang, B., et al. (2021). Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 599, 650–656. doi: 10.1038/s41586-021-04059-0
Görlach, A., Bertram, K., Hudecova, S., and Krizanova, O. (2015). Calcium and ROS: A mutual interplay. Redox Biol. 6, 260–271. doi: 10.1016/j.redox.2015.08.010
Guzman, J. N., Sánchez-Padilla, J., Chan, C. S., and Surmeier, D. J. (2009). Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 29, 11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009
Hajnóczky, G., Booth, D., Csordás, G., Debattisti, V., Golenár, T., Naghdi, S., et al. (2014). Reliance of ER-mitochondrial calcium signaling on mitochondrial EF-hand Ca2+ binding proteins: Miros, MICUs, LETM1 and solute carriers. Curr. Opin. Cell. Biol. 29, 133–141. doi: 10.1016/j.ceb.2014.06.002
Han, Y., Li, M., Qiu, F., Zhang, M., and Zhang, Y. H. (2017). Cell-permeable organic fluorescent probes for live-cell long-term super-resolution imaging reveal lysosome-mitochondrion interactions. Nat. Commun. 8:1307. doi: 10.1038/s41467-017-01503-6
Harischandra, D. S., Rokad, D., Neal, M. L., Ghaisas, S., Manne, S., Sarkar, S., et al. (2019). Manganese promotes the aggregation and prion-like cell-to-cell exosomal transmission of α-synuclein. Sci. Signal. 12:eaau4543. doi: 10.1126/scisignal.aau4543
Hedskog, L., Pinho, C. M., Filadi, R., Ronnback, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the endoplasmic reticulum–mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. U.S.A. 110, 7916–7921. doi: 10.1073/pnas.1300677110
Henchcliffe, C., and Beal, M. F. (2008). Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 4, 600–609. doi: 10.1038/ncpneuro0924
Hetz, C., Martinon, F., Rodriguez, D., and Glimcher, L. H. (2011). The unfolded protein response: integrating stress signals through the stress sensor IRE1α. Physiol. Rev. 91, 1219–1243. doi: 10.1152/physrev.00001.2011
Hindle, J. V. (2010). Ageing, neurodegeneration and Parkinson’s disease. Age Ageing. 39, 156–161. doi: 10.1093/ageing/afp223
Hockey, L. N., Kilpatrick, B. S., Eden, E. R., Lin-Moshier, Y., Brailoiu, G. C., Brailoiu, E., et al. (2015). Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J. Cell Sci. 128, 232–238. doi: 10.1242/jcs.164152
Hurley, M. J., Brandon, B., Gentleman, S. M., and Dexter, D. T. (2013). Parkinson’s disease is associated with altered expression of Cav1 channels and calcium-binding proteins. Brain. 136, 2077–2097. doi: 10.1093/brain/awt134
Jacobs, E., and D’Esposito, M. (2011). Estrogen shapes dopamine-dependent cognitive processes: implications for women’s health. J. Neurosci. 31, 5286–5293. doi: 10.1523/JNEUROSCI.6394-10.2011
Jankovic, J. (2008). Parkinson’s disease clinical features and diagnosis. J. Neurol. Neurosurg. Psychiat. 79, 368–376. doi: 10.1136/jnnp.2007.131045
Joseph, S. K., Young, M. P., Alzayady, K., Yule, D. I., Ali, M., Booth, D. M., et al. (2018). Redox regulation of type-I inositol trisphosphate receptors in intact mammalian cells. J. Biol. Chem. 293, 17464–17476. doi: 10.1074/jbc.RA118.005624
Jung, S., Chung, Y., Lee, Y., Lee, Y., Cho, J. W., Shin, E. J., et al. (2019). Buffering of cytosolic calcium plays a neuroprotective role by preserving the autophagy-lysosome pathway during MPP+-induced neuronal death. Cell Death Discov. 5:130. doi: 10.1038/s41420-019-0210-6
Kalia, L. V., and Lang, A. E. (2016). Parkinson’s disease in 2015: evolving basic, pathological and clinical concepts in PD. Nat. Rev. Neurol. 12, 65–66. doi: 10.1038/nrneurol.2015
Kamienieva, I., Duszyński, J., and Szczepanowska, J. (2021). Multitasking guardian of mitochondrial quality: Parkin function and Parkinson’s disease. Transl. Neurodegener. 10:5. doi: 10.1186/s40035-020-00229-8
Kang, K. R., Kim, J., Ryu, B., Lee, S. G., Oh, M. S., Baek, J., et al. (2021). BAPTA, a calcium chelator, neuroprotects injured neurons in vitro and promotes motor recovery after spinal cord transection in vivo. CNS Neurosci. Ther. 27, 919–929. doi: 10.1111/cns.13651
Kanthasamy, A. G., Kitazawa, M., Kanthasamy, A., and Anantharam, V. (2005). Dieldrin-induced neurotoxicity: relevance to Parkinson’s disease pathogenesis. Neurotoxicology 26, 701–719. doi: 10.1016/j.neuro.2004.07.010
Kawamoto, E. M., Vivar, C., and Camandola, S. (2012). Physiology and pathology of calcium signaling in the brain. Front. Pharmacol. 3:61. doi: 10.3389/fphar.2012.00061
Keller, J. N., Dimayuga, E., Chen, Q., Thorpe, J., Gee, J., and Ding, Q. (2004). Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int. J. Biochem. Cell Biol. 36, 2376–2391. doi: 10.1016/j.biocel.2004.05.003
Kim, H. Y., Cho, M. K., Kumar, A., Maier, E., Siebenhaar, C., Becker, S., et al. (2009). Structural properties of pore-forming oligomers of alpha-synuclein. J. Am. Chem. Soc. 131, 17482–17489. doi: 10.1021/ja9077599
Kim, I., Xu, W., and Reed, J. C. (2008). Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat. Rev. Drug. Discov. 7, 1013–1030. doi: 10.1038/nrd2755
Kim, S., Wong, Y. C., Gao, F., and Krainc, D. (2021). Dysregulation of mitochondria-lysosome contacts by GBA1 dysfunction in dopaminergic neuronal models of Parkinson’s disease. Nat. Commun. 12:1807. doi: 10.1038/s41467-021-22113-3
Kinnear, N. P., Boittin, F. X., Thomas, J. M., Galione, A., and Evans, A. M. (2004). Lysosome-sarcoplasmic reticulum junctions. A trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J. Biol. Chem. 279, 54319–54326. doi: 10.1074/jbc.M406132200
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Klein, C., and Lohmann-Hedrich, K. (2007). Impact of recent genetic findings in Parkinson’s disease. Curr. Opin. Neurol. 20, 453–464. doi: 10.1097/WCO.0b013e3281e6692b
Klein, C., and Westenberger, A. (2012). Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a008888. doi: 10.1101/cshperspect.a008888
Klinge, C. M. (2008). Estrogenic control of mitochondrial function and biogenesis. J. Cell. Biochem. 105, 1342–1351. doi: 10.1002/jcb.21936
Klinge, C. M. (2020). Estrogenic control of mitochondrial function. Redox Biol. 31:101435. doi: 10.1016/j.redox.2020.101435
Kouroupi, G., Taoufik, E., Vlachos, I. S., Tsioras, K., Antoniou, N., Papastefanaki, F., et al. (2017). Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 114, E3679–E3688. doi: 10.1073/pnas.1617259114
Lee, J. E., Cathey, P. I., Wu, H., Parker, R., and Voeltz, G. K. (2020). Endoplasmic reticulum contact sites regulate the dynamics of membraneless organelles. Science 367:eaay7108. doi: 10.1126/science.aay7108
Lejri, I., Grimm, A., and Eckert, A. (2018). Mitochondria, estrogen and female brain aging. Front. Aging Neurosci. 10:124. doi: 10.3389/fnagi.2018.00124
Li, R. J., Xu, J., Fu, C., Zhang, J., Zheng, Y. G., Jia, H., et al. (2016). Regulation of mTORC1 by lysosomal calcium and calmodulin. Elife. 5:e19360. doi: 10.7554/eLife.19360
Lin, K. J., Lin, K. L., Chen, S. D., Liou, C. W., Chuang, Y. C., Lin, H. Y., et al. (2019). The overcrowded crossroads: Mitochondria, alpha-synuclein, and the endo-lysosomal system interaction in Parkinson’s disease. Int. J. Mol. Sci. 20:5312. doi: 10.3390/ijms20215312
Lindholm, D., Wootz, H., and Korhonen, L. (2006). ER stress and neurodegenerative diseases. Cell Death Differ. 13, 385–392. doi: 10.1038/sj.cdd.4401778
Liu, H., Bowes, R. C. III, van de Water, B., Sillence, C., Nagelkerke, J. F., and Stevens, J. L. (1997). Endoplasmic reticulum chaperones GRP78 and calreticulin prevent oxidative stress, Ca2+ disturbances, and cell death in renal epithelial cells. J. Biol. Chem. 272, 21751–21759. doi: 10.1074/jbc.272.35.21751
Liu, Y., Ma, X., Fujioka, H., Liu, J., Chen, S., and Zhu, X. (2019). DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. U.S.A. 116, 25322–25328. doi: 10.1073/pnas.1906565116
Lock, E. A., Zhang, J., and Checkoway, H. (2013). Solvents and Parkinson disease: a systematic review of toxicological and epidemiological evidence. Toxicol. Appl. Pharmacol. 266, 345–355. doi: 10.1016/j.taap.2012.11.016
Lunati, A., Lesage, S., and Brice, A. (2018). The genetic landscape of Parkinson’s disease. Rev. Neurol. 174, 628–643. doi: 10.1016/j.neurol.2018.08.004
Luzio, J. P., Hackmann, Y., Dieckmann, N. M., and Griffiths, G. M. (2014). The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb Perspect. Biol. 6:a016840. doi: 10.1101/cshperspect.a016840
MacLeod, D., Dowman, J., Hammond, R., Leete, T., Inoue, K., and Abeliovich, A. (2006). The Familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52, 587–593. doi: 10.1016/j.neuron.2006.10.008
Mak, D. O., McBride, S., and Foskett, J. K. (2001). Regulation by Ca2+ and inositol 1,4,5- trisphosphate (InsP3) of single recombinant type 3 InsP3 receptor channels. Ca2+ activation uniquely distinguishes types 1 and 3 insp3 receptors. J. Gen. Physiol. 117, 435–446. doi: 10.1085/jgp.117.5.435
Marambaud, P., Dreses-Werringloer, U., and Vingtdeux, V. (2009). Calcium signaling in neurodegeneration. Mol. Neurodegener. 4:20. doi: 10.1186/1750-1326-4-20
Mattson, M. P. (2004). Pathways towards and away from Alzheimer’s disease. Nature 430, 631–639. doi: 10.1038/nature02621
Mattson, M. P. (2007). Calcium and neurodegeneration. Aging Cell. 6, 337–350. doi: 10.1111/j.1474-9726.2007.00275.x
Mckee, A. C., and Daneshvar, D. H. (2015). The neuropathology of traumatic brain injury. Handb. Clin. Neurol. 127, 45–66. doi: 10.1016/B978-0-444-52892-6.00004-0
Medina, D. L., Di Paola, S., Peluso, I., Armani, A., De Stefani, D., Venditti, R., et al. (2015). Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299. doi: 10.1038/ncb3114
Melchionda, M., Pittman, J. K., Mayor, R., and Patel, S. (2016). Ca2+/H+ exchange by acidic organelles regulates cell migration in vivo. J. Cell Biol. 212, 803–813. doi: 10.1083/jcb.201510019
Michel, P. P., Hirsch, E. C., and Hunot, S. (2016). Understanding dopaminergic cell death path- ways in Parkinson disease. Neuron 90, 675–691. doi: 10.1016/j.neuron.2016.03.038
Miller, V. M., and Harman, S. M. (2017). An update on hormone therapy in postmenopausal women: mini-review for the basic scientist. Am. J. Physiol. Heart Circ. Physiol. 313, H1013–H1021. doi: 10.1152/ajpheart.00383.2017
Moisan, F., Kab, S., Mohamed, F., Canonico, M., Le Guern, M., Quintin, C., et al. (2016). Parkinson disease male-to-female ratios increase with age: French nationwide study and meta-analysis. J. Neurol. Neurosurg. Psychiat. 87, 952–957. doi: 10.1136/jnnp-2015-312283
Morrison, J. H., Brinton, R. D., Schmidt, P. J., and Gore, A. C. (2006). Estrogen, menopause, and the aging brain: how basic neuroscience can inform hormone therapy in women. J. Neurosci. 26, 10332–10348. doi: 10.1523/JNEUROSCI.3369-06.2006
Muallem, S., Chung, W. Y., Jha, A., and Ahuja, M. (2017). Lipids at membrane contact sites: cell signaling and ion transport. EMBO Rep. 18, 1893–1904. doi: 10.15252/embr.201744331
Muñoz, S. S., Petersen, D., Marlet, F. R., Kücükköse, E., and Galvagnion, C. (2021). The interplay between Glucocerebrosidase, α-synuclein and lipids in human models of Parkinson’s disease. Biophys. Chem. 273, 106534. doi: 10.1016/j.bpc.2020.106534
Neal, A. P., and Guilarte, T. R. (2013). Mechanisms of lead and manganese neurotoxicity. Toxicol. Res. 2, 99–114. doi: 10.1039/C2TX20064C
Oslowski, C. M., and Urano, F. (2011). Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 490, 71–92. doi: 10.1016/B978-0-12-385114-7.00004-0
Paisán-Ruíz, C., Jain, S., Evans, E. W., Gilks, W. P., Simón, J., van der Brug, M., et al. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. doi: 10.1016/j.neuron.2004.10.023
Pandey, S. P., Rai, R., Gaur, P., and Prasad, S. (2015). Development- and age- related alterations in the expression of AMPA receptor subunit GluR2 and its trafficking proteins in the hippocampus of male mouse brain. Biogerontology 16, 317–328. doi: 10.1007/s10522-014-9548-6
Pang, S. Y., Ho, P. W., Liu, H. F., Leung, C. T., Li, L., Chang, E. E. S., et al. (2019). The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. 8:23. doi: 10.1186/s40035-019-0165-9
Peng, T. I., and Jou, M. J. (2010). Oxidative stress caused by mitochondrial calcium overload. Ann. N.Y. Acad. Sci. 1201, 183–188. doi: 10.1111/j.1749-6632.2010.05634.x
Peng, W., Wong, Y. C., and Krainc, D. (2020). Mitochondria-lysosome contacts regulate mitochondrial Ca2+ dynamics via lysosomal TRPML1. Proc. Natl. Acad. Sci. U.S.A. 117, 19266–19275. doi: 10.1073/pnas.2003236117
Perić, M., Bou Dib, P., Dennerlein, S., Musa, M., Rudan, M., Lovrić, A., et al. (2016). Crosstalk between cellular compartments protects against proteotoxicity and extends lifespan. Sci. Rep. 6:28751. doi: 10.1038/srep28751
Phillips, M. J., and Voeltz, G. K. (2016). Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 17, 69–82. doi: 10.1038/nrm.2015.8
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Prins, D., and Michalak, M. (2011). Organellar calcium buffers. Cold Spring Harb Perspect. Biol. 3:a004069. doi: 10.1101/cshperspect.a004069
Prinz, W. A. (2014). Bridging the gap: membrane contact sites in signaling, metabolism, and organelle dynamics. J. Cell Biol. 205, 759–769. doi: 10.1083/jcb.201401126
Pryor, P. R., Mullock, B. M., Bright, N. A., Gray, S. R., and Luzio, J. P. (2000). The role of intraorganellar Ca2+ in late endosome-lysosome heterotypic fusion and in the reformation of lysosomes from hybrid organelles. J. Cell Biol. 149, 1053–1062. doi: 10.1083/jcb.149.5.1053
Pyo, I. S., Yun, S., Yoon, Y. E., Choi, J. W., and Lee, S. J. (2020). Mechanisms of aging and the preventive effects of resveratrol on age-related diseases. Molecules 25:4649. doi: 10.3390/molecules25204649
Raffaello, A., Mammucari, C., Gherardi, G., and Rizzuto, R. (2016). Calcium at the center of cell signaling: Interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci. 41, 1035–1049. doi: 10.1016/j.tibs.2016.09.001
Rajsombath, M. M., Nam, A. Y., Ericsson, M., and Nuber, S. (2019). Female sex and brain-selective estrogen benefit α-synuclein tetramerization and the PD-like motor syndrome in 3K transgenic mice. J. Neurosci. 39, 7628–7640. doi: 10.1523/JNEUROSCI.0313-19.2019
Ramsey, I. S., Markus Delling, M., and Clapham, D. E. (2006). An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647. doi: 10.1146/annurev.physiol.68.040204.100431
Ray, B., Bhat, A., Mahalakshmi, A. M., Tuladhar, S., Bishir, M., Mohan, S. K., et al. (2021). Mitochondrial and organellar crosstalk in Parkinson’s disease. ASN Neuro. 13:17590914211028364. doi: 10.1177/17590914211028364
Read, A., and Schröder, M. (2021). The unfolded protein response: An overview. Biology 10:384. doi: 10.3390/biology10050384
Reddy, A., Caler, E. V., and Andrews, N. W. (2001). Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 106, 157–169. doi: 10.1016/s0092-8674(01)00421-4
Reeve, A. K., Ludtmann, M. H., Angelova, P. R., Simcox, E. M., Horrocks, M. H., Klenerman, D., et al. (2015). Aggregated alpha-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons. Cell Death Dis. 6:e1820. doi: 10.1038/cddis.2015.166
Rossini, M., Pizzo, P., and Filadi, R. (2021). Better to keep in touch: investigating inter-organelle cross-talk. FEBS J. 288, 740–755. doi: 10.1111/febs.15451
Rowland, A. A., and Voeltz, G. K. (2012). Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat. Rev. Mol. Cell Biol. 13, 607–625. doi: 10.1038/nrm3440
Santulli, G., Xie, W., Reiken, S. R., and Marks, A. R. (2015). Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. U.S.A. 112, 11389–11394. doi: 10.1073/pnas.1513047112
Schrag, A., and Schott, J. M. (2006). Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet Neurol. 5, 355–363. doi: 10.1016/S1474-4422(06)70411-2
Schwake, M., Schröder, B., and Saftig, P. (2013). Lysosomal membrane proteins and their central role in physiology. Traffic 14, 739–748. doi: 10.1111/tra.12056
Sharma, H., Zhang, P., Barber, D. S., and Liu, B. (2010). Organochlorine pesticides dieldrin and lindane induce cooperative toxicity in dopaminergic neurons: role of oxidative stress. Neurotoxicology 31, 215–222. doi: 10.1016/j.neuro.2009.12.007
Shoshan-Barmatz, V., and De, S. (2017). Mitochondrial VDAC, the Na+ /Ca2+ exchanger, and the Ca2+ uniporter in Ca2+ dynamics and signaling. Adv. Exp. Med. Biol. 981, 323–347. doi: 10.1007/978-3-319-55858-5_13
Shulman, J. M., De Jager, P. L., and Feany, M. B. (2011). Parkinson’s disease: genetics and pathogenesis. Annu. Rev. Pathol. 6, 193–222. doi: 10.1146/annurev-pathol-011110-130242
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840.
Stefanis, L. (2012). α-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect. Med. 2:a009399. doi: 10.1101/cshperspect.a009399
Sunanda, T., Ray, B., Mahalakshmi, A. M., Bhat, A., Rashan, L., Rungratanawanich, W., et al. (2021). Mitochondria-endoplasmic reticulum crosstalk in Parkinson’s disease: The role of brain renin angiotensin system components. Biomolecules 11:1669. doi: 10.3390/biom11111669
Surmeier, D. J. (2007). Calcium, ageing, and neuronal vulnerability in Parkinson’s disease. Lancet Neurol. 6, 933–938. doi: 10.1016/S1474-4422(07)70246-6
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017). Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 18, 101–113. doi: 10.1038/nrn.2016.178
Surmeier, D. J., and Schumacker, P. T. (2013). Calcium, bioenergetics, and neuronal vulnerability in Parkinson’s disease. J. Biol. Chem. 288, 10736–10741. doi: 10.1074/jbc.R112
Tolosa, E., Vila, M., Klein, C., and Rascol, O. (2020). LRRK2 in Parkinson disease: challenges of clinical trials. Nat. Rev. Neurol. 16, 97–107. doi: 10.1038/s41582-019-0301-2
Trivedi, P. C., Bartlett, J. J., and Pulinilkunnil, T. (2020). Lysosomal biology and function: Modern view of cellular debris Bin. Cells 9:1131. doi: 10.3390/cells9051131
Tsunemi, T., Perez-Rosello, T., Ishiguro, Y., Yoroisaka, A., Jeon, S., Hamada, K., et al. (2019). Increased lysosomal exocytosis induced by lysosomal Ca2+ channel agonists protects human dopaminergic neurons from α-Synuclein toxicity. J. Neurosci. 39, 5760-5772. doi: 10.1523/JNEUROSCI.3085-18.2019
Tymianski, M., and Tator, C. H. (1996). Normal and abnormal calcium homeostasis in neurons: a basic for the pathophysiology of traumatic and ischemic central nervous system injury. Neurosurgery 38, 1176–1195. doi: 10.1097/00006123-199606000-00028
Vais, H., Payne, R., Paudel, U., Li, C., and Foskett, J. K. (2020). Coupled transmembrane mechanisms control MCU-mediated mitochondrial Ca2+ uptake. Proc. Natl. Acad. Sci. U.S.A. 117, 21731–21739. doi: 10.1073/pnas.2005976117
Valente, E. M., Bentivoglio, A. R., Dixon, P. H., Ferraris, A., Ialongo, T., Frontali, M., et al. (2001). Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am. J. Hum. Genet. 68, 895–900. doi: 10.1086/319522
Valm, A. M., Cohen, S., Legant, W. R., Melunis, J., Hershberg, U., Wait, E., et al. (2017). Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546, 162–167. doi: 10.1038/nature22369
van der Kant, R., and Neefjes, J. (2014). Small regulators, major consequences - Ca2 + and cholesterol at the endosome-ER interface. J. Cell Sci. 127, 929–938. doi: 10.1242/jcs.137539
Vasta, R., Chia, R., Traynor, B. J., and Chiò, A. (2021). Unraveling the complex interplay between genes, environment, and climate in ALS. EBioMedicine 75:103795. doi: 10.1016/j.ebiom.2021.103795
Veng, L. M., and Browning, M. D. (2002). Regionally selective alterations in expression of the α1D subunit Cav1.3 of L-type calcium channels in the hippocampus of aged rats. Brain Res. Mol. Brain Res. 107, 120–127. doi: 10.1016/s0169-328x(02)00453-9
Ventura-Clapier, R., Piquereau, J., Veksler, V., and Garnier, A. (2019). Estrogens, estrogen receptors effects on cardiac and skeletal muscle mitochondria. Front. Endocrinol. 10:557. doi: 10.3389/fendo.2019.00557
West, A. B., Moore, D. J., Biskup, S., Bugayenko, A., Smith, W. W., Ross, C. A., et al. (2005). Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U. S. A. 102, 16842–16847. doi: 10.1073/pnas.0507360102
Willis, A. W., Evanoff, B. A., Lian, M., Criswell, S. R., and Racette, B. A. (2010). Geographic and ethnic variation in Parkinson disease: a population-based study of US Medicare beneficiaries. Neuroepidemiology 34, 143–151. doi: 10.1159/000275491
Wilson, E. L., and Metzakopian, E. (2021). ER-mitochondria contact sites in neurodegeneration: genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ. 28, 1804–1821. doi: 10.1038/s41418-020-00705-8
Wojda, U., Salinska, E., and Kuznicki, J. (2008). Calcium ions in neuronal degeneration. IUBMB Life 60, 575–590. doi: 10.1002/iub.91
Wong, Y. C., Kim, S., Peng, W., and Krainc, D. (2019). Regulation and function of mitochondria-lysosome membrane contact sites in cellular homeostasis. Trends Cell Biol. 29, 500–513. doi: 10.1016/j.tcb.2019.02.004
Wong, Y. C., Ysselstein, D., and Krainc, D. (2018). Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554, 382–386. doi: 10.1038/nature25486
Xiong, L., Zhang, J., He, R., and Hamilton, S. L. (2006). A Ca2+-binding domain in RyR1 that interacts with the calmodulin binding site and modulates channel activity. Biophys. J. 90, 173–182. doi: 10.1529/biophysj.105.066092
Xu, C., Bailly-Maitre, B., and Reed, J. C. (2005). Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Invest. 115, 2656–2664. doi: 10.1172/JCI26373
Yang, J., Zhao, Z., Gu, M., Feng, X., and Xu, H. (2019). Release and uptake mechanisms of vesicular Ca2+ store. Protein Cell 10, 8–19. doi: 10.1007/s13238-018-0523-x
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. doi: 10.1016/s0092-8674(01)00611-0
Yuan, J. P., Zeng, W., Dorwart, M. R., Choi, Y. J., Worley, P. F., and Muallem, S. (2009). SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 11, 337–343. doi: 10.1038/ncb1842
Zaichick, S. V., McGrath, K. M., and Caraveo, G. (2017). The role of Ca2+ signaling in Parkinson’s disease. Dis. Model Mech. 10, 519–535. doi: 10.1242/dmm.028738
Zhang, X., Cheng, X., Yu, L., Yang, J., Calvo, R., Patnaik, S., et al. (2016). MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 7:12109. doi: 10.1038/ncomms12109
Zhang, Y., Soboloff, J., Zhu, Z., and Berger, S. A. (2006). Inhibition of Ca2+ influx is required for mitochondrial reactive oxygen species-induced endoplasmic reticulum Ca2+ depletion and cell death in leukemia cells. Mol. Pharmacol. 70, 1424–1434. doi: 10.1124/mol.106.024323
Zhao, R. Z., Jiang, S., Zhang, L., and Yu, Z. B. (2019). Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 44, 3–15. doi: 10.3892/ijmm.2019.4188
Keywords: Parkinson’s disease, Ca2+ homeostasis, oxidative stress, ER stress, autophagy
Citation: Xu J, Minobe E and Kameyama M (2022) Ca2+ Dyshomeostasis Links Risk Factors to Neurodegeneration in Parkinson’s Disease. Front. Cell. Neurosci. 16:867385. doi: 10.3389/fncel.2022.867385
Received: 01 February 2022; Accepted: 23 March 2022;
Published: 14 April 2022.
Edited by:
Zhiyi Yu, Shandong University, ChinaReviewed by:
Yan Zhang, University of Science and Technology of China, ChinaCopyright © 2022 Xu, Minobe and Kameyama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianjun Xu, amp4dWtAbTMua3VmbS5rYWdvc2hpbWEtdS5hYy5qcA==; Masaki Kameyama, bWthbWUwMDlAeWFob28uY28uanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.