 Ario Mirian1†
Ario Mirian1† Alexander Moszczynski2†
Alexander Moszczynski2† Serena Soleimani3
Serena Soleimani3 Isabelle Aubert4,5
Isabelle Aubert4,5 Lorne Zinman6,7,8‡
Lorne Zinman6,7,8‡ Agessandro Abrahao6,7,8,9,*,‡
Agessandro Abrahao6,7,8,9,*,‡- 1Clinical Neurological Sciences, Western University, London Health Sciences, London, ON, Canada
- 2School of Medicine, Queen's University, Kingston, ON, Canada
- 3College of Osteopathic Medicine, Michigan State University, East Lansing, MI, United States
- 4Biological Sciences, Hurvitz Brain Sciences Research Program, Sunnybrook Research Institute, Sunnybrook Health Sciences Centre, Toronto, ON, Canada
- 5Department of Laboratory Medicine and Pathobiology, Temerty Faculty of Medicine, University of Toronto, Toronto, ON, Canada
- 6Division of Neurology, Department of Medicine, Temerty Faculty of Medicine, University of Toronto, Toronto, ON, Canada
- 7Division of Neurology, Department of Medicine, Sunnybrook Health Science Centre, Toronto, ON, Canada
- 8Evaluative Clinical Sciences, Hurvitz Brain Sciences Research Program, Sunnybrook Research Institute, Sunnybrook Health Sciences Centre, Toronto, ON, Canada
- 9Harquail Centre for Neuromodulation, Sunnybrook Research Institute, Toronto, ON, Canada
Introduction: Recent studies have implicated changes in the blood-central nervous system barriers (BCNSB) in amyotrophic lateral sclerosis (ALS). The objective of this scoping review is to synthesize the current evidence for BCNSB structure and functional abnormalities in ALS studies and propose how BCNSB pathology may impact therapeutic development.
Methods: A literature search was conducted using Ovid Medline, EMBASE, and Web of Science, from inception to November 2021 and limited to entries in English language. Simplified search strategy included the terms ALS/motor neuron disease and [BCNSB or blood-brain barrier (BBB) or blood-spinal cord barrier (BSCB)]. Henceforth, BCNSB is used as a term that is inclusive of the BBB and BSCB. Four independent reviewers conducted a title and abstract screening, hand-searched the reference lists of review papers, and performed a full text review of eligible studies. Included studies were original peer-reviewed full text publications, evaluating the structure and function of the BCNSB in preclinical models of ALS, clinical ALS, or postmortem human ALS tissue. There was no restriction on study design. The four reviewers independently extracted the data.
Results: The search retrieved 2,221 non-duplicated articles and 48 original studies were included in the synthesis. There was evidence that the integrity of the BCNSB is disrupted throughout the course of the disease in rodent models, beginning prior to symptom onset and detectable neurodegeneration. Increased permeability, pharmacoresistance with upregulated efflux transporters, and morphological changes in the supporting cells of the BCNSB, including pericytes, astrocytes, and endothelial cells were observed in animal models. BCNSB abnormalities were also demonstrated in postmortem studies of ALS patients. Therapeutic interventions targeting BCNSB dysfunction were associated with improved motor neuron survival in animal models of ALS.
Conclusion: BCNSB structural and functional abnormalities are likely implicated in ALS pathophysiology and may occur upstream to neurodegeneration. Promising therapeutic strategies targeting BCNSB dysfunction have been tested in animals and can be translated into ALS clinical trials.
Introduction
Amyotrophic lateral sclerosis (ALS) is a terminal neurodegenerative disease that results in progressive wasting and paralysis of voluntary muscles. Only two medications, riluzole and edaravone, are approved with a marginal effect of slowing disease progression. ALS pathophysiology remains poorly understood, but multiple preclinical and postmortem studies, along with in vivo neuroimaging, and serum, CSF, and neurophysiological biomarkers, have been utilized to investigate this complex disease. These studies have demonstrated that ALS pathophysiology includes: hyperexcitability and degeneration of the motor network in the motor cortex, brainstem, and spinal cord; protein misfolding; impaired astrocytic and microglial functions; neuroinflammation; free radical toxicity; and mitochondrial and RNA metabolism dysfunction (Taylor et al., 2016; Eisen, 2021).
Studies led by Garbuzova-Davis et al. have implicated changes in the neurovascular unit in ALS pathology (Garbuzova-Davis et al., 2007a,b, 2012). The neurovascular unit comprises the capillary and surrounding neurons, astrocyte end-feet, microglia, and pericytes. In contrast to fenestrated and sinusoidal capillaries in other organs, the CNS capillaries are continuous and composed of a protective layer of tightly joined endothelial cells and basal membrane, known as the blood-brain barrier (BBB) and blood-spinal cord barrier (BSCB), hereafter collectively referred to as blood-central nervous system barriers (BCNSB) (Supplementary Figure 1).
The objective of this scoping review is to synthesize the current evidence for BCNSB structure and functional abnormalities described in ALS animal and human studies and propose mechanisms by which BCNSB-associated pathology may impact therapeutic development.
Methods
Literature Search
Literature search was conducted in November 2021 on the Ovid MEDLINE, EMBASE, and Web of Science databases for entries in English from inception to search date. Briefly, the search strategy (Supplementary Table 1) used the keywords: (“motor neuron disease” or “motor neurone disease” or “amyotrophic lateral sclerosis” or “ALS” or “primary lateral sclerosis” or “PLS” or “progressive muscle atrophy” or “PMA” or “Lou Gehrig Disease”) AND (“blood brain barrier” or “BBB” or “blood spinal cord barrier” or “BSCB” or “blood cerebral spinal fluid barrier” or “blood cerebrospinal fluid barrier” or “BCSFB” or “blood central nervous system barrier” or “BCNSB”).
Eligibility Criteria, Data Extraction, and Synthesis
Four independent reviewers (Abrahao, Mirian, Moszczynski, and Soleimani) performed a primary title and abstract screening and extracted full data from eligible articles following the PRISMA extension for scoping reviews (PRISMA-ScR) guidelines (Tricco et al., 2018) and a pre-specified protocol registered with PROSPERO (CRD42017065405) to mitigate selection bias.
Included studies were original, peer-reviewed, full-text published or accepted articles assessing the following research questions: (1) in the context of ALS pathogenesis, what is known from the literature about the structure and function of the BBB and BSCB, as well as therapeutic interventions targeting these barriers, in animal and human ALS studies? (2) in the context of ALS treatment development, how does the BBB and BSCB impact therapeutic access to the CNS in animal and human ALS studies? Exclusion criteria included non-peer-reviewed articles; publications in the format of poster abstracts, editorial letters, conference papers; and articles without extractable data on ALS or its variants, primary lateral sclerosis (PLS) or progressive muscular atrophy (PMA). Studies cited in peer-reviewed review papers were also hand-searched for broader inclusion. Additional studies that fell outside the scope of the search strategy but were considered relevant were also included for the discussion of the clinical context of the BCNSB pathological findings and therapeutic delivery.
Data charting from each study was performed in duplicate by independent reviewers. Data items included the studied structure (BSCB and BBB), the research questions, interventions, experimental groups or population, sample size, animal disease stage (pre vs. symptomatic), ALS model description, BCNSB functional or structural measures, and descriptive findings. The synthesis and interpretation of the extracted data were presented in a descriptive manner without statistical inferences or meta-analyses given the largely qualitative nature of the studies in this review. Discrepancies in data extraction and synthesis were resolved by consensus decision of all reviewers. Supplementary Table 2 reports the PRISMA-ScR checklist.
Results
The search yielded 2,221 non-duplicated entries. After the primary and full-text screening, data from 48 studies investigating BCNSB pathology in ALS were extracted (Supplementary Figure 2). Tables 1–3 summarize these studies according to preclinical, postmortem tissue, and clinical evidence, respectively.

Table 1. Preclinical studies investigating BCNSB integrity and function in ALS.
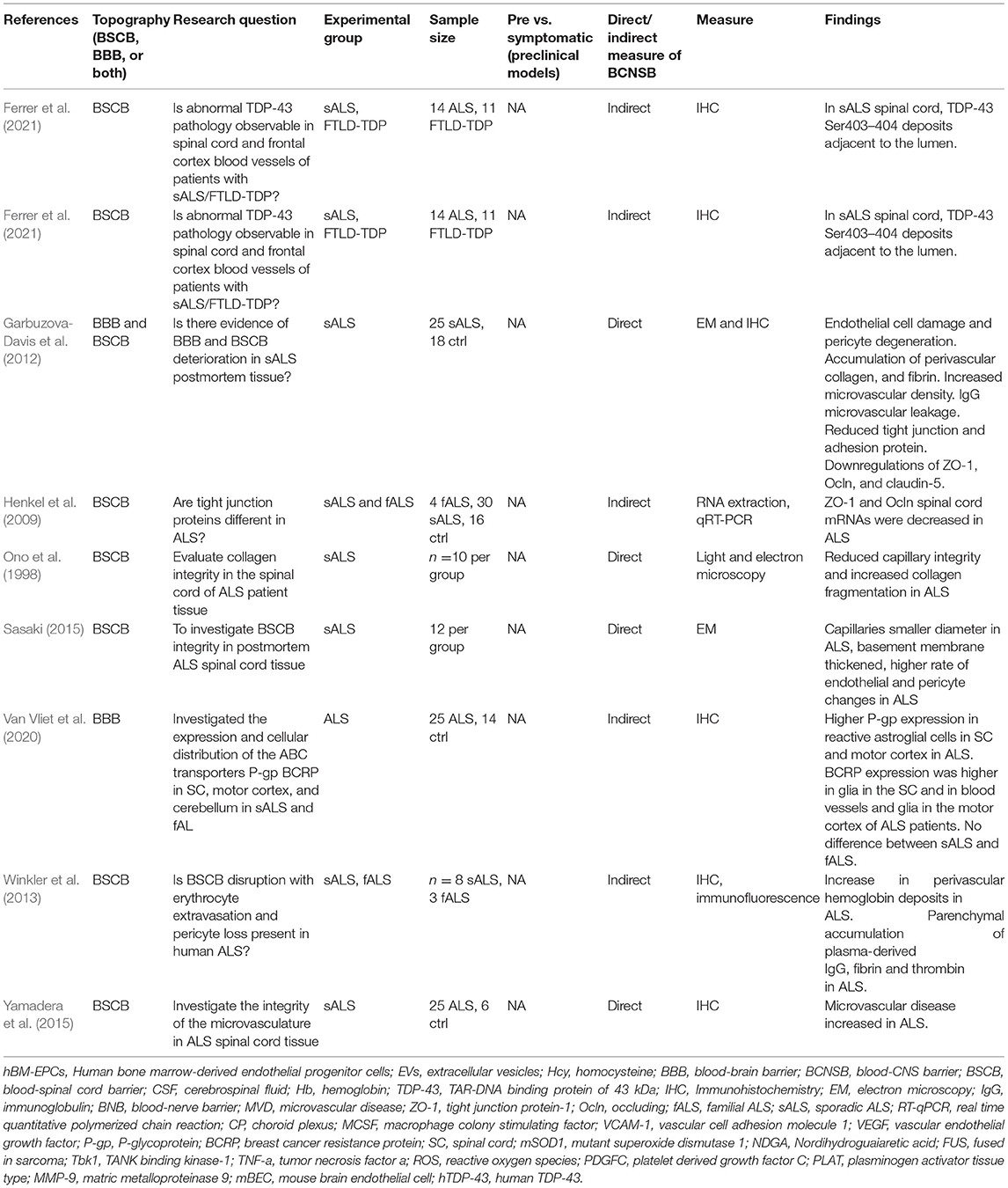
Table 2. Human post-mortem studies investigating BCNSB integrity and function in ALS.
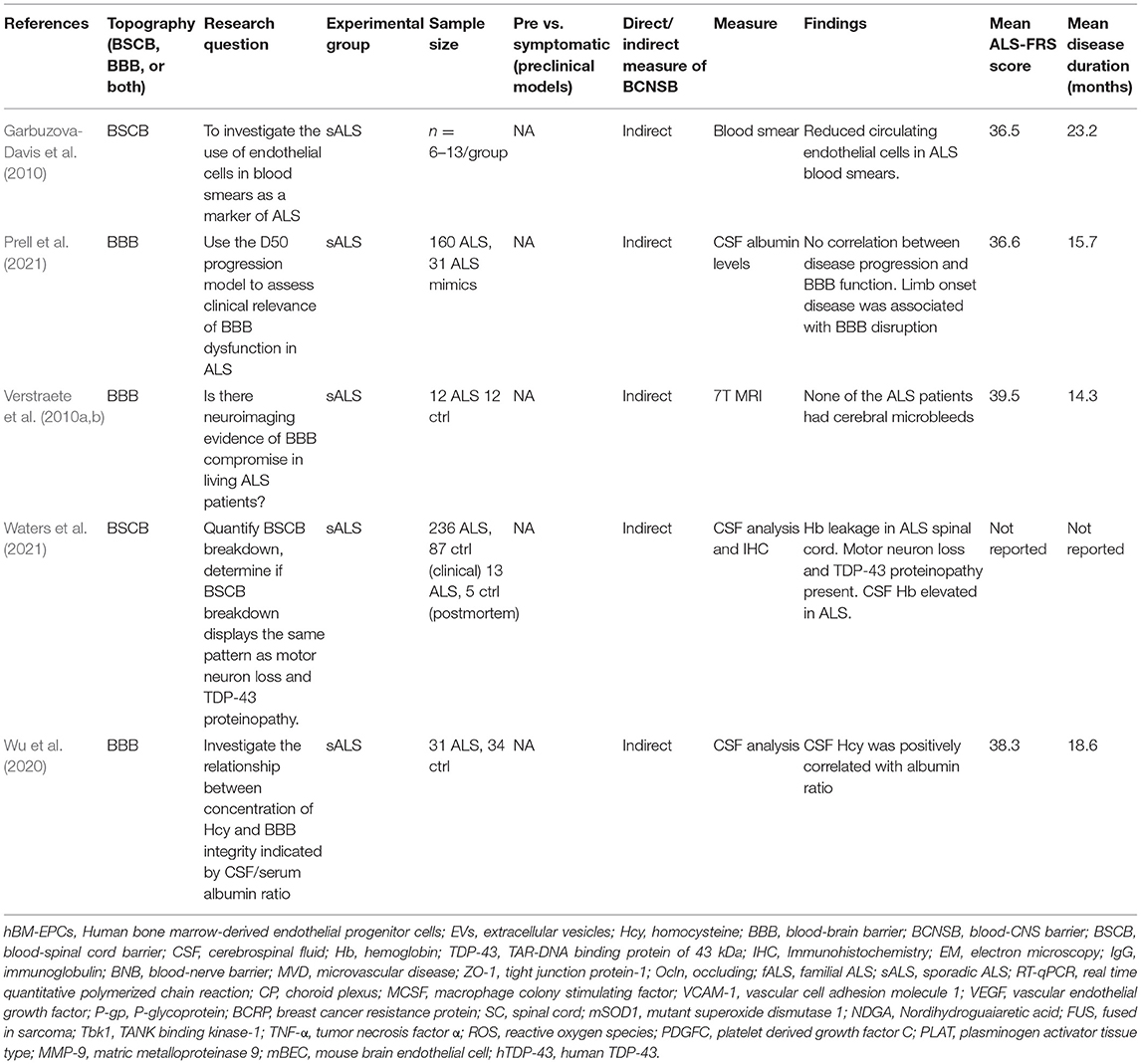
Table 3. Clinical studies investigating BCNSB integrity and function in ALS.
BCNSB Structure and Function in ALS Preclinical Models
In transgenic superoxide dismutase-1 (SOD1) rodent models of ALS, studies have demonstrated abnormal neurovascular unit and BCNSB ultrastructure (Garbuzova-Davis et al., 2007a,b) with upregulated pharmacoresistance mechanisms (Jablonski et al., 2012). Neurovascular unit changes include dysfunction of glia, endothelial cells, and pericytes, downregulation of tight junction expression, upregulation of drug efflux proteins, and increased paracellular permeability. TAR-DNA binding protein of 43 kDa (TDP-43), TANK-binding kinase 1 (TBK1), and Fused In Sarcoma (FUS) rodent models of ALS have also shown impairment of the BCNSB manifesting as ZO-1 adherens protein dysfunction, reduced vascular density, decreased expression of AQP4 and increased albumin levels in the CSF, among other changes (Jablonski et al., 2012; Sasaki et al., 2015; Ouali Alami et al., 2020).
Early morphological changes in the BSCB preceded motor neuron degeneration in SOD1 rodent models, including reduced capillary density and premature dissociation of the astrocyte end-foot and endothelial cells (Figure 1; Zhong et al., 2008; Miyazaki et al., 2011). Blood flow was reduced by 30–45% in the lower spinal cord prior to symptom onset in SOD1 models compared to non-diseased animals (Zhong et al., 2008). During the presymptomatic stage, activation of the platelet-derived growth factor C (PDGF-CC) pathway may contribute to BSCB leakage (Lewandowski et al., 2016). Deposits of hemosiderin, a hemoglobin degradation marker and indicator of microhemorrhage, were observed in the anterior horn of SOD1 mice (Zhong et al., 2008) and SOD1 rats (Nicaise et al., 2009a). Through BSCB dysfunction, it was proposed that iron from hemoglobin metabolism, endogenous immune or inflammatory mediators (Garbuzova-Davis et al., 2019) and reactive oxidative stress can contribute to early motor neuron damage or exacerbate ongoing motor neuron damage (Winkler et al., 2014). Furthermore, electron microscopy demonstrated focal accumulation of extracellular fluid between vessel walls and adjacent parenchyma, along with swollen astrocyte foot processes in SOD1 rodents (Garbuzova-Davis et al., 2007a).
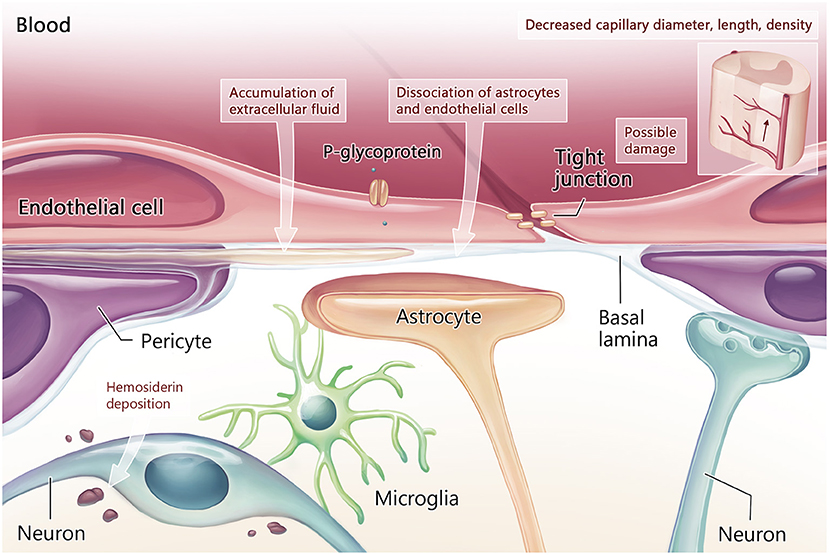
Figure 1. Blood-CNS barrier structure and function in the early stage of ALS. Early in the disease process, damage to tight junctions is associated with vascular pathology including reduction in capillary diameter, length, and density. Other structural damage includes dissociation of the astrocytes and endothelial cells of the neurovascular unit, which have been noted prior to motor neuron degeneration. Loss of neurovascular unit integrity leads to accumulation of extracellular fluid and hemosiderin deposition around motor neurons.
Ultrastructural abnormalities accumulated over time in the BSCB of symptomatic SOD1 animal models as neuronal loss progressed (Garbuzova-Davis et al., 2007a). Endothelial cells showed cytoplasmic disorganization, vacuolated mitochondria, and impaired basal membrane with reduced expression of tight junction proteins claudin-5, occludin, ZO-1 and proteoglycan agrin (Nicaise et al., 2009a; Garbuzova-Davis et al., 2012; Meister et al., 2015; Ouali Alami et al., 2020; Tang et al., 2021).
Structural changes were associated with progressive functional impairment of the BSCB in ALS animals. IgG, an immunoglobulin typically unable to cross the BSCB, had higher deposition in the extra- and intra-neuronal spaces within the spinal cord of pre-symptomatic and symptomatic SOD1 rodents (Zhong et al., 2008; Nicaise et al., 2009a; Garbuzova-Davis et al., 2012; Boswell et al., 2013; Rabinovich-Nikitin et al., 2016). The mechanism of IgG deposition remains unknown as it is unclear whether immunoglobulins have extravasated from the bloodstream through a disrupted BSCB, by transcytosis across endothelial cells (Poduslo et al., 1994), or have originated from a non-vascular mechanism of retrograde axonal transport by the motor neurons (Fabian and Petroff, 1987; Fratantoni et al., 1996). BSCB permeability increased with disease progression in SOD1 rodents, as suggested by the extravasation of intravenous Evans Blue dye into the spinal cord of symptomatic animals, in contrast to the lack of Evans Blue leakage in presymptomatic animals (Garbuzova-Davis et al., 2007a; Zhong et al., 2008; Nicaise et al., 2009b; Miyazaki et al., 2011).
Compared to the number of animal studies focusing on the spinal cord, fewer reports have demonstrated neurovascular unit changes in the brain and brainstem. BBB disruption in the midbrain of symptomatic SOD1 ALS rats was detected by the extravasation of gadolinium from the intravascular compartment into the midbrain on MRI. Gadolinium enhancement was correlated with activation of microglia and immune cell infiltration as detected by immunohistochemistry (Andjus et al., 2009; Bataveljic et al., 2011). Downregulation of potassium channel Kir4.1 and upregulation of water channel aquaporin-4 (AQP4) in astrocytic end-feet were associated with BBB dysfunction within the motor cortex and brainstem of mutant SOD1 rats (Bataveljic et al., 2012). This astrocyte-induced impairment of water and potassium homeostasis was indicated as the driving factor for increased BBB permeability and leakage of intravascular toxic mediators into the motor neuron microenvironment. Additional toxic effects occur with the accumulation of extracellular ions, such as potassium, which overcomes physiological buffer mechanisms in the ALS disease state (Bataveljic et al., 2012, 2014).
AQP4 upregulation has also been demonstrated in mouse models of ALS carrying the SOD1G93A, SOD1G85R, and LoxSOD1G37R mutations (Watanabe-Matsumoto et al., 2018). In these studies, AQP4 mislocalization along astrocyte end-feet was unique to ALS pathology and was not seen in other nerve damage models, such as sciatic nerve axotomy. While these studies noted upregulation of AQP4, other studies have demonstrated downregulation (Ouali Alami et al., 2020). This discrepancy may be due to variability in models or timepoints, as the response of regulation of the protein may go through different phases of dysregulation after its equilibrium has shifted, downregulating and then upregulating to compensate or vice versa. Future research may focus on this phenomenon. Furthermore, SOD1 ALS mice with knocked out AQP4 gene did not develop BBB dysfunction to the same extent as their AQP4 expressing counterparts. Despite improved BBB integrity, SOD1 ALS mice lacking AQP4 demonstrated accelerated disease progression and shortened survival (Watanabe-Matsumoto et al., 2018). This seemingly opposite effect whereby a lack of AQP4 improved BBB integrity but worsened disease phenotype may be due to independent roles of AQP4 in BBB-mediated CNS homeostasis and ALS pathogenesis.
Postmortem Evidence of BCNSB Dysfunction in ALS
The BCNSB findings observed in animal studies have also been corroborated in postmortem tissue from ALS patients (Garbuzova-Davis and Sanberg, 2014). In comparison to non-ALS human controls, there was disorganization of the microvascular architecture in the anterior horn, along with reduced capillary diameter (Sasaki, 2015) and density (Yamadera et al., 2015). Pericytes and endothelial cells within the spinal cord and brainstem exhibited degeneration, along with dissociation of astrocyte end-feet (Garbuzova-Davis et al., 2012; Winkler et al., 2013; Sasaki, 2015) and downregulation of tight junction proteins (Henkel et al., 2009). Disruptions in the endothelial lining indicating BCNSB breakage were also identified, with deposition of IgG, fibrin, thrombin, hemoglobin, and erythrocytes in the anterior horn tissue samples of patients with ALS (Miyazaki et al., 2011; Winkler et al., 2013; Sasaki, 2015). In the interstitial component of the BSCB, there was evidence for reduced and fragmented collagen bundles in the anterior horn of patients with ALS (Ono et al., 1998; Garbuzova-Davis and Sanberg, 2014). There was significant accumulation of perivascular collagen IV in the spinal cord and medulla (Garbuzova-Davis et al., 2012), a factor that can limit effective drug influx to the CNS (Garbuzova-Davis et al., 2016). The morphological and functional changes in the BCNSB in the late stages of ALS are schematized in Figures 2, 3.
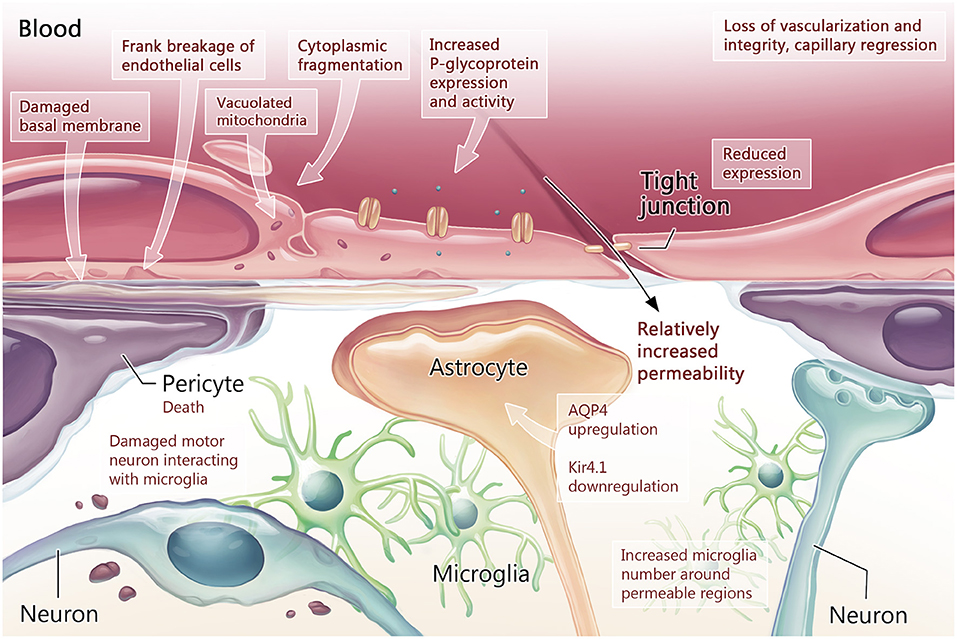
Figure 2. Blood-CNS barrier structure and function in the symptomatic stage of ALS. Once measurable behavioral changes have occurred, the early loss of neurovascular unit integrity has progressed to loss of vascularization through capillary regression. Tight junction protein expression is measurably reduced in endothelial cells, while export proteins such as P-glycoprotein (P-gp) demonstrate increased expression and activity. Within endothelial cells, signs of damage manifest as vacuolated mitochondria and cytoplasmic fragmentation. Damage to the basement membrane is also present. These changes culminate as breakage of the endothelial cell lining. With increased blood-CNS-barrier (BCNSB) permeability, changes in the cellular- molecular environment of the CNS can be measured including pericyte death and increased neuroinflammation as microglia satellite to damaged motor neurons and permeable regions. Astrocytes manifest molecular changes in the form of aquaporin 4 (AQP4) upregulation and potassium channel (Kir4.1) downregulation, leading to dysregulation the perineuronal environment.
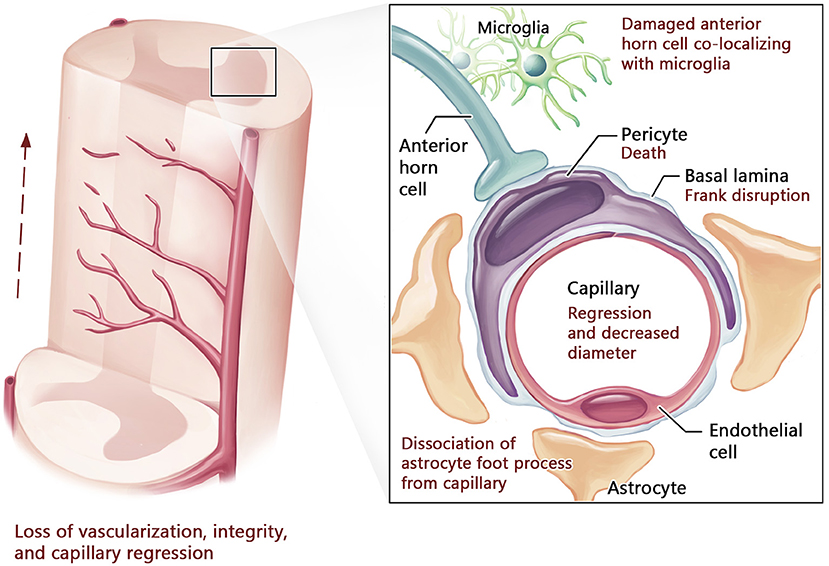
Figure 3. Blood-spinal cord barrier structure in advanced stage of ALS. In advanced ALS, neurovascular compromise progresses with further capillary regression and decreased diameter leading to reduced vascularization, particularly in the anterior horn of the spinal cord. This is accompanied by pericyte death, basal lamina disruption, collagen IV deposition, and marked dissociation of astrocyte foot processes from capillaries. Damaged cells within the anterior horn co-localize with microglia, indicating neuroinflammation.
TDP-43 deposition in endothelial cells has been observed in both ALS and frontotemporal lobar dementia with TDP-43 pathology (FTLD-TDP) in postmortem tissue (Ferrer et al., 2021). This has led to speculation over whether the pathological protein is being taken up by endothelial cells from neurons, or whether the observed deposits are from endogenous TDP-43 in the endothelial cells themselves. Both cases indicate that a vasculopathy is present in TDP-43 proteinopathies.
Whether the post-mortem BCNSB changes described in the vicinity of lower motor neurons (LMNs) in the spinal cord and brainstem also apply to the BBB surrounding the upper motor neurons (UMNs) in primary motor cortex is undetermined. Historically, studies in ALS have primarily investigated the LMNs and their connectivity with muscles at the neuromuscular junction, with less focus on the motor cortex. Given the robust data supporting a key pathological role of UMN dysfunction in ALS (Eisen, 2021), future preclinical, clinical and post-mortem studies on the interplay of astrocytes, UMNs, and motor cortex capillaries are warranted.
Clinical Evidence of BCNSB Dysfunction in Patients Living With ALS
This scoping review did not identify validated imaging, serum, or CSF biomarkers to measure BCNSB integrity and function directly and reliably in patients living with ALS. Therefore, our understanding of BCNSB pathology throughout early to late stages of ALS in humans is limited. However, a small number of studies attempted to evaluate the BCNSB function in patients with ALS using pharmacokinetic assumptions and indirect markers.
Among these indirect measures, increased cerebrospinal fluid (CSF) total protein or albumin levels and CSF/serum concentration ratio of any given therapeutic may correlate with BCNSB permeability. Elevated CSF protein has been reported in some case series of ALS (Chelstowska and Kuzma-Kozakiewicz, 2014). However, protein may also leak into the CSF from the degenerating motor roots, limiting the utility of this measure as a BCNSB functional biomarker. For instance, breakdown of the blood-peripheral nerve barrier has been demonstrated by gadolinium enhancement on MRI of lumbosacral roots and leptomeninges of patients with ALS (Luigetti et al., 2010; Young et al., 2010). Circulating endothelial cells in peripheral blood was not a reliable indicator of BCNSB endothelial damage in patients with ALS (Garbuzova-Davis et al., 2010).
Measuring BCNSB permeability using CSF/serum therapeutic concentrations has limitations. It relies on the assumption of therapeutic extravasation from the vascular compartment to the CNS parenchyma and then to the subarachnoid space without direct CSF excretion at the choroid plexus (i.e., via the BCSFB) or complete therapeutic metabolism within neural tissue. Pharmacokinetic studies investigating CSF/serum concentration ratios of multiple ALS therapeutics have typically demonstrated low CSF/serum ratios suggesting limited therapeutic access across the BCNSB (Sussmuth et al., 2010; Wu et al., 2020; Prell et al., 2021; Waters et al., 2021).
Current structural and functional neuroimaging techniques lack the spatial resolution and accuracy required to directly assess the structure and function of BCNSB in patients with ALS. Radiologically, the BCNSB appears intact in ALS patients on standard imaging techniques, in contrast to multiple sclerosis and other CNS inflammatory conditions whereby BCNSB disruption is typically demonstrated by gadolinium extravasation on MRI. While early clinical ALS studies using novel 7T MRI scanners have largely explored the CNS anatomy and connectivity (Verstraete et al., 2010b; Cosottini et al., 2016; Barry et al., 2021), BCNSB leakage has not been extensively investigated with this technique. Of note, indirect measures of BBB integrity such as microbleeds in ALS were not increased as measured by 7T MRI (Verstraete et al., 2010a).
Implications for Therapeutic Development in ALS
Despite increased permeability in preclinical and postmortem models, therapeutics with high molecular weight still have limited access to the CNS (Garbuzova-Davis et al., 2016). While lipid soluble and low molecular weight molecules can diffuse across the endothelial cell membranes, most of these compounds do not effectively enter the CNS and are transported back to the bloodstream by BCNSB-driven efflux or pharmacoresistance systems.
These pharmacoresistance proteins include specialized ATP binding cassette (ABC) efflux transporters that actively pump a number of endogenous and exogenous substrates out of the BCNSB (Mohamed et al., 2017). Relevant to ALS, the upregulation of key pharmacoresistance efflux proteins, P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), has been shown in multiple animal models (Milane et al., 2010; Jablonski et al., 2012; Chan et al., 2017), a finding also supported by postmortem human tissue studies (Jablonski et al., 2012; Qosa et al., 2016; Van Vliet et al., 2020).
Increased expression and activity of P-gp began after symptom onset in the BSCB and BBB of mutated SOD1 models (Boston-Howes et al., 2008; Milane et al., 2010; Chan et al., 2017). TDP-43A315T mutant mice also demonstrated P-gp and BCRP overexpression (Jablonski et al., 2012). In vitro, the P-gp upregulation in the endothelial cells was mediated by nuclear factor κB (NF-κB) activation and was induced when these cells were co-cultured with ALS-derived astrocytes (Qosa et al., 2016; Mohamed et al., 2019). In postmortem tissue, P-gp and BCRP upregulation was observed in the spinal cord and motor cortex of both sporadic and familial ALS cases (Van Vliet et al., 2020). It has been suggested that P-gp evolved for the purpose of responding to harmful substances (Broeks et al., 1995). Therefore, in ALS it may be responding to the toxic sequelae of aberrant protein in the CNS or other circulating toxins. Upregulation may be the result of a physiological attempt to dispense of harmful substrates. A consequence, however, may be decreased drug efficacy.
Of clinical relevance, riluzole, the first approved ALS drug, is a substrate of these efflux proteins (Milane et al., 2009b, 2010). The P-gp overexpression over the course of the disease may explain the poor bioavailability of riluzole within CNS parenchyma thereby limiting its therapeutic efficacy (Milane et al., 2010; Jablonski et al., 2014). In contrast, edavarone, a free radical scavenger approved for ALS treatment, has negligible P-gp binding and is potentially less influenced by BCNSB efflux activity (Dash et al., 2018; Hyung et al., 2018).
Reversing BCNSB pharmacoresistance in ALS is a long-sought approach to enhance therapeutic effect via inhibition of efflux transporters (Milane et al., 2009a; Jablonski et al., 2014). There was a significant increase in the parenchymal concentration of riluzole in rodents co-treated with efflux transporter inhibitors, such as minocycline (Milane et al., 2009a), verapamil liposomes (Yang et al., 2018) and elacridar (Jablonski et al., 2014). For instance, riluzole plus elacridar, a third-generation inhibitor BCRP and P-gp, improved muscle function, disease progression and survival in ALS mice (Jablonski et al., 2014).
While inhibition of efflux transporters is a promising strategy for clinical trials there are some challenges with current P-gp inhibitors. This includes the high dose needed to block P-gp which may lead to adverse reactions and systemic toxicity (Milane et al., 2007; Kalvass et al., 2013), as well as the need for selectivity and specificity of P-gp only at the BBB to prevent adverse effects on other organs (Amin, 2013).
BCNSB Pathology as a Therapeutic Target to Ameliorate ALS Disease Progression
Repair of the BCNSB pathology in ALS via stem cell transplantation has provided some evidence of symptomatic improvement in SOD1G93A mice. The intravenous transplantation of human bone marrow-derived CD34+ cells (hBM34+) and endothelial progenitor cells (hBM-EPCs) enhanced the replacement of damaged endothelial cells in the CNS capillaries (Garbuzova-Davis et al., 2017, 2018, 2019, 2021; Eve et al., 2018). One mechanism of hBM-EPCs on the endothelium includes excretion of extracellular vesicles that transfer biomolecules to facilitate the repair of damaged microvascular endothelium in ALS (Garbuzova-Davis et al., 2020). Additionally, mice receiving hBM-EPCs have increased tight junction protein levels, capillary pericyte coverage, and basement membrane laminin expression, all of which maintain capillary endothelium integrity (Garbuzova-Davis et al., 2021). Ultimately, these mechanisms may prevent the entry of immune or inflammatory mediators which can contribute to motor neuron dysfunction (Garbuzova-Davis et al., 2019). At the symptomatic disease stage, hBM-EPC-treated mice have shown an improvement of behavioral outcomes and motor neuron survival, making it a promising therapeutic strategy for future translational clinical trials (Garbuzova-Davis et al., 2019).
Other therapeutics, such as an antagonist of the chemokine receptor CXCR4 (Rabinovich-Nikitin et al., 2016) and the myelosuppressive dealkylating agent busulfan (Peake et al., 2017), demonstrated improved BCNSB function in ALS models. Edaravone has been previously shown to mediate BBB repair in models of ischemic stroke, in addition to its primary mechanism of action as a free radical scavenger (Miyamoto et al., 2014; Tóth et al., 2014; Watanabe et al., 2015). Yet, BCNSB repair largely remains only a theoretical consideration in edavarone's mechanism of action in ALS.
Discussion
ALS pathophysiology remains poorly understood and has typically focused on the interplay between degenerating motor neurons and activated glia. Recent evidence supported a key role of neurovascular unit pathology contributing to motor neuron degeneration in ALS. In this scoping review, rodent models demonstrated progressive BCNSB disruption throughout the course of the disease, beginning prior to detectable motor neuron loss. Morphological changes in the supporting cells of the BCNSB, including pericytes, astrocytes, and endothelial cells, were observed in animal models and postmortem studies of ALS patients. Progressive BCNSB changes have also been described in other neurodegenerative disorders, such as Alzheimer's disease (Zipser et al., 2007) and Parkinson's disease (Pan and Nicolazzo, 2018), suggesting commonalities in vascular dysfunction and neurodegeneration in the context of different proteinopathies.
BCNSB leakage may be driven by activated astrocytes, imbalance of extracellular ion and water channel homeostasis (Watanabe-Matsumoto et al., 2018), and oxidative stress (Garbuzova-Davis et al., 2016). Although it remains unclear whether the neurovascular unit insult is the driving causative mechanism in ALS pathophysiology, the studies in this scoping review indicate that it is likely to contribute to motor neuron degeneration as an early or upstream mechanism. Disrupted barriers can allow endogenous immune or inflammatory mediators to enter the motor neuron microenvironment (Garbuzova-Davis et al., 2008, 2019). Activated astrocytes lose their ability to provide trophic and metabolic support and become toxic to motor neurons (Nagai et al., 2007; Haidet-Phillips et al., 2011).
Evidence of BCNSB abnormalities in animal models and human postmortem tissue need further in vivo investigation in patients with ALS. Part of the endothelial cell injury in SOD1 rodents relates to a direct toxic effect of the mutated SOD1 protein and oxidative stress (Garbuzova-Davis et al., 2016), which may not be translatable to the majority sporadic ALS cases. Progressive respiratory failure and aspiration pneumonia, common causes of death among patients (Corcia et al., 2008), along with disrupted blood-gas homeostasis and a systemic inflammatory response, may account for some of the BCNSB abnormalities in end-stage ALS. These factors may increase tissue susceptibility to anoxia from the time of death to histological fixation as compared to non-ALS controls. Novel non-invasive imaging, serum and CSF biomarkers are warranted to ascertain BCNSB changes throughout the stages of human ALS.
Despite the evidence of BCNSB leakage in ALS models, therapeutic access to the CNS remains a challenge as these barriers still limit the passage of most therapeutics (e.g., antibodies, proteins, gene carriers and cells) from the vascular compartment to the CNS parenchyma. Limited therapeutic access to the CNS may account, at least in part, for the discrepancies between large therapeutic effect sizes observed in SOD1 mouse trials compared to lack of benefit in ALS clinical trials (Garbuzova-Davis et al., 2016). An important distinction between mutant SOD1 animal models and sporadic ALS patients is the degree of pericyte degeneration and perivascular collagen-IV accumulation in the latter (Garbuzova-Davis et al., 2016). Pericyte degeneration has been shown to reduce capillary blood flow which in turn may limit therapeutic delivery (Garbuzova-Davis and Sanberg, 2014; Garbuzova-Davis et al., 2016). Collagen-IV build up in the brain and spinal cord vessels may be a compensatory mechanism to BCNSB dysfunction and can further limit the diffusion of therapeutics across the BCNSB (Garbuzova-Davis et al., 2012, 2016). In addition, the upregulation of transmembrane efflux transporters, such as P-gp, further reduces drug bioavailability to the motor neuron network. Contrasting to SOD1 mice, the scarceness of in vivo proof of BCNSB disruption in patients with ALS may reflect the human disease heterogeneity and the lack of optimal tools to elucidate BCNSB function in patients.
Multiple strategies to enhance therapeutic delivery to the brain and spinal cord across the BCNSB have been reported in ALS preclinical and clinical studies. In addition to safety, ideally, BCNSB-modifying techniques should be temporary and reversible to avoid worsening of chronic BCNSB pathology in ALS. These approaches included the co-administration of mannitol for increased BCNSB permeability (Chi et al., 2011) or elacridar for P-gp inhibition (Jablonski et al., 2014), and direct tissue injections via open surgery. In humans, interventions involving invasive injection of therapeutics into the motor cortex (Martínez et al., 2009) and spinal cord (Feldman et al., 2014) have been performed. However, in addition to uncertain efficacy, the generalizability of surgical approaches, particularly for repeated procedures, is limited due to morbidity and tolerability in ALS patients with respiratory impairment.
As a less invasive alternative to bypass the BCNSB, repeated intrathecal injections of antisense oligonucleotides targeting SOD1 (Miller et al., 2013, 2020), gene therapy with adeno-associated virus rh10 containing an anti-SOD1 microRNA (AAV-miR-SOD1) (Mueller et al., 2020), and mesenchymal stem cells (Petrou et al., 2016; Cudkowicz et al., 2021) have been safely tested in humans with ALS. Non-invasive magnetic resonance-guided focused ultrasound (MRgFUS) has emerged as a technique to open the BBB safely and temporarily for targeted drug delivery to the motor cortex in patients with ALS (Abrahao et al., 2019). MRgFUS combines transcranial acoustic energy and intravenous microbubbles to disrupt the tight junctions of the targeted capillaries, allowing large therapeutics such as antibodies to gain access to the human human brain (Meng et al., 2021). Also, reversible MRgFUS-induced BBB permeability was safely performed in patients with Alzheimer's disease (Lipsman et al., 2018; Rezai et al., 2020; Park et al., 2021), another neurodegenerative condition with chronic BCNSB leakage (Zipser et al., 2007).
While the search strategy employed broad terms, it is possible that some relevant studies were missed and thus not reflected here. This scoping review aimed to summarize qualitative and quantitative BCNSB measures from heterogenous ALS study models but did not attempt to conduct meta-analyses or a critical appraisal of individual studies. To mitigate bias, four independent reviewers analyzed the extracted data and achieved consensus on data synthesis. Therefore, despite all the efforts to mitigate selection bias, reporting bias is still a possibility. The generalizability of the findings is also limited given the majority of studies investigated BCNSB changes in mutated SOD1 animal models with a relative paucity of studies in other models of motor neuron disease (e.g., FUS or TDP-43) and clinical data. Lastly, this review did not focus on the BCSFB at the choroid plexus. Given the structural differences in relation to the BBB and BSCB and recent reports of BCSFB disruption (Saul et al., 2020) and potential therapeutic target in ALS (Kunis et al., 2015), a dedicated review on this topic is suggested.
This synthesis of the literature demonstrates evidence that BCNSB structural and functional abnormalities are likely implicated in ALS pathophysiology. These BCNSB changes in preclinical models may represent an upstream process in relation to motor neuron degeneration. Therefore, more studies are needed to validate these findings in vivo in humans with ALS and to further elucidate the potential role of BCNSB disruption as a component of the disease pathophysiology. Promising therapeutic strategies targeting these BCNSB changes in animal models can be translated into future ALS clinical trials.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
AMi, AMo, SS, IA, and LZ: acquisition of data, analysis and interpretation of data, and manuscript writing. AA: study conceptualization, acquisition of data, analysis and interpretation of data, and manuscript writing. All authors contributed to the article and approved the submitted version.
Funding
This research was undertaken, in part, thanks to funding from the Canada Research Chairs Program (to IA; CRC Tier 1 in Brain Repair and Regeneration), ALS Society of Canada and Brain Canada (to AA and LZ), and generosity of philanthropic gifts to the Sunnybrook Foundation for ALS research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Mr. Henry Lam from Sunnybrook Library for his assistance on the literature search strategy. Hang Yu Lin, from IA Laboratory at Sunnybrook Research Institute, for creating customized, original illustrations for this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2022.851563/full#supplementary-material
References
Abrahao, A., Meng, Y., Llinas, M., Huang, Y., Hamani, C., Mainprize, T., et al. (2019). First-in-human trial of blood-brain barrier opening in amyotrophic lateral sclerosis using MR-guided focused ultrasound. Nat. Commun. 10, 4373. doi: 10.1038/s41467-019-12426-9
Amin, M. L. (2013). P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 7, 27–34. doi: 10.4137/DTI.S12519
Andjus, P. R., Bataveljic, D., Vanhoutte, G., Mitrecic, D., Pizzolante, F., Djogo, N., et al. (2009). In vivo morphological changes in animal models of amyotrophic lateral sclerosis and Alzheimer's-like disease: MRI approach. Anat. Rec. (Hoboken) 292, 1882–1892. doi: 10.1002/ar.20995
Barry, R. L., Babu, S., Anteraper, S. A., Triantafyllou, C., Keil, B., Rowe, O. E., et al. (2021). Ultra-high field (7T) functional magnetic resonance imaging in amyotrophic lateral sclerosis: a pilot study. Neuroimage Clin. 30, 102648. doi: 10.1016/j.nicl.2021.102648
Bataveljic, D., Milosevic, M., Radenovic, L., and Andjus, P. (2014). Novel molecular biomarkers at the blood-brain barrier in ALS. Biomed. Res. Int. 2014, 907545. doi: 10.1155/2014/907545
Bataveljic, D., Nikolic, L., Milosevic, M., Todorovic, N., and Andjus, P. R. (2012). Changes in the astrocytic aquaporin-4 and inwardly rectifying potassium channel expression in the brain of the amyotrophic lateral sclerosis SOD1(G93A) rat model. Glia 60, 1991–2003. doi: 10.1002/glia.22414
Bataveljic, D., Stamenkovic, S., Bacic, G., and Andjus, P. R. (2011). Imaging cellular markers of neuroinflammation in the brain of the rat model of amyotrophic lateral sclerosis. Acta Physiol. Hung. 98, 27–31. doi: 10.1556/APhysiol.98.2011.1.4
Boston-Howes, W., Williams, E. O., Bogush, A., Scolere, M., Pasinelli, P., and Trotti, D. (2008). Nordihydroguaiaretic acid increases glutamate uptake in vitro and in vivo: therapeutic implications for amyotrophic lateral sclerosis. Exp. Neurol. 213, 229–237. doi: 10.1016/j.expneurol.2008.06.010
Boswell, C. A., Mundo, E. E., Johnstone, B., Ulufatu, S., Schweiger, M. G., Bumbaca, D., et al. (2013). Vascular physiology and protein disposition in a preclinical model of neurodegeneration. Mol. Pharm. 10, 1514–1521. doi: 10.1021/mp3004786
Broeks, A., Janssen, H. W., Calafat, J., and Plasterk, R. H. (1995). A P-glycoprotein protects Caenorhabditis elegans against natural toxins. EMBO J. 14, 1858–1866. doi: 10.1002/j.1460-2075.1995.tb07178.x
Chan, G. N., Evans, R. A., Banks, D. B., Mesev, E. V., Miller, D. S., and Cannon, R. E. (2017). Selective induction of P-glycoprotein at the CNS barriers during symptomatic stage of an ALS animal model. Neurosci. Lett. 639, 103–113. doi: 10.1016/j.neulet.2016.12.049
Chelstowska, B., and Kuzma-Kozakiewicz, M. (2014). Is cerebrospinal fluid obtained for diagnostic purpose a good material for biomarker studies in amyotrophic lateral sclerosis? Biomarkers 19, 571–577. doi: 10.3109/1354750X.2014.949867
Chi,ò, A., Mora, G., Bella, V. L., Caponnetto, C., Mancardi, G., Sabatelli, M., et al. (2011). Repeated courses of granulocyte colony-stimulating factor in amyotrophic lateral sclerosis: clinical and biological results from a prospective multicenter study. Muscle Nerve 43, 189–195. doi: 10.1002/mus.21851
Corcia, P., Pradat, P. F., Salachas, F., Bruneteau, G., Forestier, N., Seilhean, D., et al. (2008). Causes of death in a post-mortem series of ALS patients. Amyotroph. Lateral Scler. 9, 59–62. doi: 10.1080/17482960701656940
Cosottini, M., Donatelli, G., Costagli, M., Caldarazzo Ienco, E., Frosini, D., Pesaresi, I., et al. (2016). High-resolution 7T MR imaging of the motor cortex in amyotrophic lateral sclerosis. AJNR Am. J. Neuroradiol. 37, 455–461. doi: 10.3174/ajnr.A4562
Cudkowicz, M. E., Lindborg, S. R., Goyal, N. A., Miller, R. G., Burford, M. J., Berry, J. D., et al. (2021). A randomized placebo-controlled phase 3 study of mesenchymal stem cells induced to secrete high levels of neurotrophic factors in amyotrophic lateral sclerosis. Muscle Nerve. 65, 291–302. doi: 10.1002/mus.27472
Dash, R. P., Babu, R. J., and Srinivas, N. R. (2018). Two decades-long journey from riluzole to edaravone: revisiting the clinical pharmacokinetics of the only two amyotrophic lateral sclerosis therapeutics. Clin. Pharmacokinet. 57, 1385–1398. doi: 10.1007/s40262-018-0655-4
Eisen, A. (2021). The dying forward hypothesis of ALS: tracing its history. Brain Sci. 11, 300. doi: 10.3390/brainsci11030300
Evans, M. C., Serres, S., Khrapitchev, A. A., Stolp, H. B., Anthony, D. C., and Talbot, K.. (2014). T2-weighted MRI detects presymptomatic pathology in the SOD1 mouse model of ALS. J Cereb. Blood Flow Metab. 34, 785–793.
Eve, D. J., Steiner, G., Mahendrasah, A., Sanberg, P. R., Kurien, C., Thomson, A., et al. (2018). Reduction of microhemorrhages in the spinal cord of symptomatic ALS mice after intravenous human bone marrow stem cell transplantation accompanies repair of the blood-spinal cord barrier. Oncotarget 9, 10621–10634. doi: 10.18632/oncotarget.24360
Fabian, R. H., and Petroff, G. (1987). Intraneuronal IgG in the central nervous system: uptake by retrograde axonal transport. Neurology 37, 1780–1784. doi: 10.1212/WNL.37.11.1780
Feldman, E. L., Boulis, N. M., Hur, J., Johe, K., Rutkove, S. B., Federici, T., et al. (2014). Intraspinal neural stem cell transplantation in amyotrophic lateral sclerosis: phase 1 trial outcomes. Ann. Neurol. 75, 363–373. doi: 10.1002/ana.24113
Ferrer, I., Andres-Benito, P., Carmona, M., Assialioui, A., and Povedano, M. (2021). TDP-43 vasculopathy in the spinal cord in sporadic amyotrophic lateral sclerosis (sALS) and frontal cortex in sALS/FTLD-TDP. J. Neuropathol. Exp. Neurol. 80, 229–239. doi: 10.1093/jnen/nlaa162
Fratantoni, S. A., Dubrovsky, A. L., and Uchitel, O. D. (1996). Uptake of immunoglobulin G from amyotrophic lateral sclerosis patients by motor nerve terminals in mice. J. Neurol. Sci. 137, 97–102. doi: 10.1016/0022-510X(95)00345-3
Garbuzova-Davis, S., Boccio, K. J., Llauget, A., Shell, R., Hailu, S., Mustafa, H., et al. (2021). Beneficial effects of transplanted human bone marrow endothelial progenitors on functional and cellular components of blood-spinal cord barrier in ALS mice. eNeuro 8, ENEURO.0314-0321.2021. doi: 10.1523/ENEURO.0314-21.2021
Garbuzova-Davis, S., Haller, E., Navarro, S., Besong, T. E., Boccio, K. J., Hailu, S., et al. (2018). Transplantation of human bone marrow stem cells into symptomatic ALS mice enhances structural and functional blood-spinal cord barrier repair. Exp. Neurol. 310, 33–47. doi: 10.1016/j.expneurol.2018.08.012
Garbuzova-Davis, S., Haller, E., Saporta, S., Kolomey, I., Nicosia, S. V., and Sanberg, P. R. (2007a). Ultrastructure of blood–brain barrier and blood–spinal cord barrier in SOD1 mice modeling ALS. Brain Res. 1157, 126–137. doi: 10.1016/j.brainres.2007.04.044
Garbuzova-Davis, S., Hernandez-Ontiveros, D. G., Rodrigues, M. C., Haller, E., Frisina-Deyo, A., Mirtyl, S., et al. (2012). Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 1469, 114–128. doi: 10.1016/j.brainres.2012.05.056
Garbuzova-Davis, S., Kurien, C., Haller, E., Eve, D. J., Navarro, S., Steiner, G., et al. (2019). Human bone marrow endothelial progenitor cell transplantation into symptomatic ALS mice delays disease progression and increases motor neuron survival by repairing blood-spinal cord barrier. Sci. Rep. 9, 5280. doi: 10.1038/s41598-019-41747-4
Garbuzova-Davis, S., Kurien, C., Thomson, A., Falco, D., Ahmad, S., Staffetti, J., et al. (2017). Endothelial and astrocytic support by human bone marrow stem cell grafts into symptomatic ALS mice towards blood-spinal cord barrier repair. Sci. Rep. 7, 884. doi: 10.1038/s41598-017-00993-0
Garbuzova-Davis, S., and Sanberg, P. R. (2014). Blood-CNS Barrier Impairment in ALS patients versus an animal model. Front. Cell. Neurosci. 8, 21. doi: 10.3389/fncel.2014.00021
Garbuzova-Davis, S., Saporta, S., Haller, E., Kolomey, I., Bennett, S. P., Potter, H., et al. (2007b). Evidence of compromised blood-spinal cord barrier in early and late symptomatic SOD1 mice modeling ALS. PLoS ONE 2, e1205. doi: 10.1371/journal.pone.0001205
Garbuzova-Davis, S., Saporta, S., and Sanberg, P. R. (2008). Implications of blood-brain barrier disruption in ALS. Amyotroph. Lateral Scler. 9, 375–376. doi: 10.1080/17482960802160990
Garbuzova-Davis, S., Thomson, A., Kurien, C., Shytle, R. D., and Sanberg, P. R. (2016). Potential new complication in drug therapy development for amyotrophic lateral sclerosis. Expert Rev. Neurother. 16, 1397–1405. doi: 10.1080/14737175.2016.1207530
Garbuzova-Davis, S., Willing, A. E., Ehrhart, J., Wang, L., Sanberg, P. R., and Borlongan, C. V. (2020). Cell-free extracellular vesicles derived from human bone marrow endothelial progenitor cells as potential therapeutics for microvascular endothelium restoration in ALS. Neuromolecular Med. 22, 503–516. doi: 10.1007/s12017-020-08607-1
Garbuzova-Davis, S., Woods, R. L. 3rd, Louis, M. K., Zesiewicz, T. A., Kuzmin-Nichols, N., Sullivan, K. L., Miller, A. M., et al. (2010). Reduction of circulating endothelial cells in peripheral blood of ALS patients. PLoS ONE 5, e10614. doi: 10.1371/journal.pone.0010614
Haidet-Phillips, A. M., Hester, M. E., Miranda, C. J., Meyer, K., Braun, L., Frakes, A., et al. (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828. doi: 10.1038/nbt.1957
Henkel, J. S., Beers, D. R., Wen, S., Bowser, R., and Appel, S. H. (2009). Decreased mRNA expression of tight junction proteins in lumbar spinal cords of patients with ALS. Neurology 72, 1614–1616. doi: 10.1212/WNL.0b013e3181a41228
Hyung, S., Jeong, Y. S., Yeo, J., Song, Y. K., Kim, M. S., Im, Y. J., et al. (2018). Identification of the primary determining factor(s) governing the oral absorption of edaravone in rats. Eur. J. Pharm. Sci. 123, 312–320. doi: 10.1016/j.ejps.2018.07.052
Jablonski, M. R., Jacob, D. A., Campos, C., Miller, D. S., Maragakis, N. J., Pasinelli, P., et al. (2012). Selective increase of two ABC drug efflux transporters at the blood-spinal cord barrier suggests induced pharmacoresistance in ALS. Neurobiol. Dis. 47, 194–200. doi: 10.1016/j.nbd.2012.03.040
Jablonski, M. R., Markandaiah, S. S., Jacob, D., Meng, N. J., Li, K., Gennaro, V., et al. (2014). Inhibiting drug efflux transporters improves efficacy of ALS therapeutics. Ann Clin Transl Neurol 1, 996–1005. doi: 10.1002/acn3.141
Kalvass, J. C., Polli, J. W., Bourdet, D. L., Feng, B., Huang, S. M., Liu, X., et al. (2013). Why clinical modulation of efflux transport at the human blood-brain barrier is unlikely: the ITC evidence-based position. Clin. Pharmacol. Ther. 94, 80–94. doi: 10.1038/clpt.2013.34
Kunis, G., Baruch, K., Miller, O., and Schwartz, M. (2015). Immunization with a myelin-derived antigen activates the brain's choroid plexus for recruitment of immunoregulatory cells to the CNS and attenuates disease progression in a mouse model of ALS. J. Neurosci. 35, 6381–6393. doi: 10.1523/JNEUROSCI.3644-14.2015
Lewandowski, S. A., Nilsson, I., Fredriksson, L., Lonnerberg, P., Muhl, L., Zeitelhofer, M., et al. (2016). Presymptomatic activation of the PDGF-CC pathway accelerates onset of ALS neurodegeneration. Acta Neuropathol. 131, 453–464. doi: 10.1007/s00401-015-1520-2
Lipsman, N., Meng, Y., Bethune, A. J., Huang, Y., Lam, B., Masellis, M., et al. (2018). Blood-brain barrier opening in Alzheimer's disease using MR-guided focused ultrasound. Nat. Commun. 9, 2336. doi: 10.1038/s41467-018-04529-6
Luigetti, M., Cianfoni, A., Conte, A., and Sabatelli, M. (2010). Gadolinium enhancement of the lumbar leptomeninges and roots in a case of ALS. Amyotroph. Lateral Scler. 11, 412–413. doi: 10.3109/17482960903234728
Martínez, H. R., Gonzalez-Garza, M. T., Moreno-Cuevas, J. E., Caro, E., Gutierrez-Jimenez, E., and Segura, J. J. (2009). Stem-cell transplantation into the frontal motor cortex in amyotrophic lateral sclerosis patients. Cytotherapy 11, 26–34. doi: 10.1080/14653240802644651
Meister, S., Storck, S. E., Hameister, E., Behl, C., Weggen, S., Clement, A. M., et al. (2015). Expression of the ALS-causing variant hSOD1(G93A) leads to an impaired integrity and altered regulation of claudin-5 expression in an in vitro blood-spinal cord barrier model. J. Cereb. Blood Flow Metab. 35, 1112–1121. doi: 10.1038/jcbfm.2015.57
Meng, Y., Reilly, R. M., Pezo, R. C., Trudeau, M., Sahgal, A., Singnurkar, A., et al. (2021). MR-guided focused ultrasound enhances delivery of trastuzumab to Her2-positive brain metastases. Sci Transl Med 13, eabj4011. doi: 10.1126/scitranslmed.abj4011
Milane, A., Fernandez, C., Dupuis, L., Buyse, M., Loeffler, J. P., Farinotti, R., et al. (2010). P-glycoprotein expression and function are increased in an animal model of amyotrophic lateral sclerosis. Neurosci. Lett. 472, 166–170. doi: 10.1016/j.neulet.2010.01.078
Milane, A., Fernandez, C., Vautier, S., Bensimon, G., Meininger, V., and Farinotti, R. (2007). Minocycline and riluzole brain disposition: interactions with p-glycoprotein at the blood-brain barrier. J. Neurochem. 103, 164–173. doi: 10.1111/j.1471-4159.2007.04772.x
Milane, A., Tortolano, L., Fernandez, C., Bensimon, G., Meininger, V., and Farinotti, R. (2009a). Brain and plasma riluzole pharmacokinetics: effect of minocycline combination. J. Pharm. Pharm. Sci. 12, 209–217. doi: 10.18433/J36C78
Milane, A., Vautier, S., Chacun, H., Meininger, V., Bensimon, G., Farinotti, R., et al. (2009b). Interactions between riluzole and ABCG2/BCRP transporter. Neurosci. Lett. 452, 12–16. doi: 10.1016/j.neulet.2008.12.061
Miller, T., Cudkowicz, M., Shaw, P. J., Andersen, P. M., Atassi, N., Bucelli, R. C., et al. (2020). Phase 1-2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N. Engl. J. Med. 383, 109–119. doi: 10.1056/NEJMoa2003715
Miller, T. M., Pestronk, A., David, W., Rothstein, J., Simpson, E., Appel, S. H., et al. (2013). An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 12, 435–442. doi: 10.1016/S1474-4422(13)70061-9
Miyamoto, N., Pham, L. D., Maki, T., Liang, A. C., and Arai, K. (2014). A radical scavenger edaravone inhibits matrix metalloproteinase-9 upregulation and blood-brain barrier breakdown in a mouse model of prolonged cerebral hypoperfusion. Neurosci. Lett. 573, 40–45. doi: 10.1016/j.neulet.2014.05.005
Miyazaki, K., Ohta, Y., Nagai, M., Morimoto, N., Kurata, T., Takehisa, Y., et al. (2011). Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 89, 718–728. doi: 10.1002/jnr.22594
Mohamed, L. A., Markandaiah, S., Bonanno, S., Pasinelli, P., and Trotti, D. (2017). Blood–brain barrier driven pharmacoresistance in amyotrophic lateral sclerosis and challenges for effective drug therapies. AAPS J. 19, 1–15. doi: 10.1208/s12248-017-0120-6
Mohamed, L. A., Markandaiah, S. S., Bonanno, S., Pasinelli, P., and Trotti, D. (2019). Excess glutamate secreted from astrocytes drives upregulation of P-glycoprotein in endothelial cells in amyotrophic lateral sclerosis. Exp. Neurol. 316, 27–38. doi: 10.1016/j.expneurol.2019.04.002
Mueller, C., Berry, J. D., Mckenna-Yasek, D. M., Gernoux, G., Owegi, M. A., Pothier, L. M., et al. (2020). SOD1 suppression with adeno-associated virus and MicroRNA in familial ALS. N. Engl. J. Med. 383, 151–158. doi: 10.1056/NEJMoa2005056
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Nicaise, C., Mitrecic, D., Demetter, P., De Decker, R., Authelet, M., Boom, A., et al. (2009a). Impaired blood-brain and blood-spinal cord barriers in mutant SOD1-linked ALS rat. Brain Res. 1301, 152–162. doi: 10.1016/j.brainres.2009.09.018
Nicaise, C., Soyfoo, M. S., Authelet, M., De Decker, R., Bataveljic, D., Delporte, C., et al. (2009b). Aquaporin-4 overexpression in rat ALS model. Anat. Rec. (Hoboken) 292, 207–213. doi: 10.1002/ar.20838
Ono, S., Imai, T., Munakata, S., Takahashi, K., Kanda, F., Hashimoto, K., et al. (1998). Collagen abnormalities in the spinal cord from patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 160, 140–147. doi: 10.1016/S0022-510X(98)00223-8
Ouali Alami, N., Tang, L., Wiesner, D., Commisso, B., Bayer, D., Weishaupt, J., et al. (2020). Multiplexed chemogenetics in astrocytes and motoneurons restore blood-spinal cord barrier in ALS. Life Sci Alliance 3, 571. doi: 10.26508/lsa.201900571
Pan, Y., and Nicolazzo, J. A. (2018). Impact of aging, Alzheimer's disease and Parkinson's disease on the blood-brain barrier transport of therapeutics. Adv. Drug Deliv. Rev. 135, 62–74. doi: 10.1016/j.addr.2018.04.009
Park, S. H., Baik, K., Jeon, S., Chang, W. S., Ye, B. S., and Chang, J. W. (2021). Extensive frontal focused ultrasound mediated blood-brain barrier opening for the treatment of Alzheimer's disease: a proof-of-concept study. Transl. Neurodegener. 10, 44. doi: 10.1186/s40035-021-00269-8
Peake, K., Manning, J., Lewis, C. A., Tran, K., Rossi, F., and Krieger, C. (2017). Bone marrow-derived cell accumulation in the spinal cord is independent of peripheral mobilization in a mouse model of amyotrophic lateral sclerosis. Front. Neurol. 8, 75. doi: 10.3389/fneur.2017.00075
Petrou, P., Gothelf, Y., Argov, Z., Gotkine, M., Levy, Y. S., Kassis, I., et al. (2016). Safety and clinical effects of mesenchymal stem cells secreting neurotrophic factor transplantation in patients with amyotrophic lateral sclerosis. JAMA Neurol. 73, 337–338. doi: 10.1001/jamaneurol.2015.4321
Poduslo, J. F., Curran, G. L., and Berg, C. T. (1994). Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc. Natl. Acad. Sci. U.S.A. 91, 5705–5709. doi: 10.1073/pnas.91.12.5705
Prell, T., Vlad, B., Gaur, N., Stubendorff, B., and Grosskreutz, J. (2021). Blood-brain barrier disruption is not associated with disease aggressiveness in amyotrophic lateral sclerosis. Front. Neurosci. 15, 656456. doi: 10.3389/fnins.2021.656456
Qosa, H., Lichter, J., Sarlo, M., Markandaiah, S. S., Mcavoy, K., Richard, J. P., et al. (2016). Astrocytes drive upregulation of the multidrug resistance transporter ABCB1 (P-Glycoprotein) in endothelial cells of the blood-brain barrier in mutant superoxide dismutase 1-linked amyotrophic lateral sclerosis. Glia 64, 1298–1313. doi: 10.1002/glia.23003
Rabinovich-Nikitin, I., Ezra, A., Barbiro, B., Rabinovich-Toidman, P., and Solomon, B. (2016). Chronic administration of AMD3100 increases survival and alleviates pathology in SOD1(G93A) mice model of ALS. J. Neuroinflammation 13, 123. doi: 10.1186/s12974-016-0587-6
Rezai, A. R., Ranjan, M., D'haese, P. F., Haut, M. W., Carpenter, J., Najib, U., et al. (2020). Noninvasive hippocampal blood-brain barrier opening in Alzheimer's disease with focused ultrasound. Proc. Natl. Acad. Sci. U.S.A. 117, 9180–9182. doi: 10.1073/pnas.2002571117
Sasaki, S. (2015). Alterations of the blood-spinal cord barrier in sporadic amyotrophic lateral sclerosis. Neuropathology 35, 518–528. doi: 10.1111/neup.12221
Sasaki, S., Iguchi, Y., Katsuno, M., and Sobue, G. (2015). Alterations in the blood-spinal cord barrier in TDP-43 conditional knockout mice. Neurosci. Lett. 598, 1–5. doi: 10.1016/j.neulet.2015.05.005
Saul, J., Hutchins, E., Reiman, R., Saul, M., Ostrow, L. W., Harris, B. T., et al. (2020). Global alterations to the choroid plexus blood-CSF barrier in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 8, 92. doi: 10.1186/s40478-020-00968-9
Stamenković, S., Pavićević, A., Mojović, M., Popović-Bijelić, A., Selaković, V., Andjus, P., et al. (2017). In vivo EPR pharmacokinetic evaluation of the redox status and the blood brain barrier permeability in the SOD1G93A ALS rat model. Free Radic. Biol. Med. 108, 258–269. doi: 10.1016/j.freeradbiomed.2017.03.034
Sussmuth, S. D., Sperfeld, A. D., Ludolph, A. C., and Tumani, H. (2010). Hypercapnia is a possible determinant of the function of the blood-cerebrospinal fluid barrier in amyotrophic lateral sclerosis. Neurochem. Res. 35, 1071–1074. doi: 10.1007/s11064-010-0156-9
Tang, J., Kang, Y., Zhou, Y., Li, X., Lan, J., Wu, L., et al. (2021). ALS-causing SOD1 mutants regulate occludin phosphorylation/ubiquitination and endocytic trafficking via the ITCH/Eps15/Rab5 axis. Neurobiol. Dis. 153, 105315. doi: 10.1016/j.nbd.2021.105315
Taylor, J. P., Brown, R. H., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Tóth, A. E., Walter, F. R., Bocsik, A., Sántha, P., Veszelka, S., Nagy, L., et al. (2014). Edaravone protects against methylglyoxal-induced barrier damage in human brain endothelial cells. PLoS ONE 9, e100152. doi: 10.1371/journal.pone.0100152
Tricco, A. C., Lillie, E., Zarin, W., O'brien, K. K., Colquhoun, H., Levac, D., et al. (2018). PRISMA extension for scoping reviews (PRISMA-ScR): checklist and explanation. Ann. Intern. Med. 169, 467–473. doi: 10.7326/M18-0850
Van Vliet, E. A., Iyer, A. M., Mesarosova, L., Colakoglu, H., Anink, J. J., Van Tellingen, O., et al. (2020). Expression and cellular distribution of P-glycoprotein and breast cancer resistance protein in amyotrophic lateral sclerosis patients. J. Neuropathol. Exp. Neurol. 79, 266–276. doi: 10.1093/jnen/nlz142
Verstraete, E., Biessels, G. J., Van Den Heuvel, M. P., Visser, F., Luijten, P. R., and Van Den Berg, L. H. (2010a). No evidence of microbleeds in ALS patients at 7 Tesla MRI. Amyotroph. Lateral Scler. 11, 555–557. doi: 10.3109/17482968.2010.513053
Verstraete, E., Van Den Heuvel, M. P., Veldink, J. H., Blanken, N., Mandl, R. C., Hulshoff Pol, H. E., et al. (2010b). Motor network degeneration in amyotrophic lateral sclerosis: a structural and functional connectivity study. PLoS ONE 5, e13664. doi: 10.1371/journal.pone.0013664
Watanabe, Y., Kazuki, Y., Kazuki, K., Ebiki, M., Nakanishi, M., Nakamura, K., et al. (2015). Use of a human artificial chromosome for delivering trophic factors in a rodent model of amyotrophic lateral sclerosis. Mol. Ther. Nucleic Acids 4, e253. doi: 10.1038/mtna.2015.28
Watanabe-Matsumoto, S., Moriwaki, Y., Okuda, T., Misawa, H., Ohara, S., Yamanaka, K., et al. (2018). Dissociation of blood-brain barrier disruption and disease manifestation in an aquaporin-4-deficient mouse model of amyotrophic lateral sclerosis. Neurosci. Res. 133, 48–57. doi: 10.1016/j.neures.2017.11.001
Waters, S., Swanson, M. E. V., Dieriks, B. V., Zhang, Y. B., Grimsey, N. L., Murray, H. C., et al. (2021). Blood-spinal cord barrier leakage is independent of motor neuron pathology in ALS. Acta Neuropathol Commun 9, 144. doi: 10.1186/s40478-021-01244-0
Winkler, E. A., Sengillo, J. D., Sagare, A. P., Zhao, Z., Ma, Q., Zuniga, E., et al. (2014). Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc. Natl. Acad. Sci. U.S.A. 111, E1035–1042. doi: 10.1073/pnas.1401595111
Winkler, E. A., Sengillo, J. D., Sullivan, J. S., Henkel, J. S., Appel, S. H., and Zlokovic, B. V. (2013). Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 111–120. doi: 10.1007/s00401-012-1039-8
Wu, Y., Yang, X., Li, X., Wang, H., and Wang, T. (2020). Elevated cerebrospinal fluid homocysteine is associated with blood-brain barrier disruption in amyotrophic lateral sclerosis patients. Neurol. Sci. 41, 1865–1872. doi: 10.1007/s10072-020-04292-x
Yamadera, M., Fujimura, H., Inoue, K., Toyooka, K., Mori, C., Hirano, H., et al. (2015). Microvascular disturbance with decreased pericyte coverage is prominent in the ventral horn of patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 16, 393–401. doi: 10.3109/21678421.2015.1011663
Yang, T., Ferrill, L., Gallant, L., Mcgillicuddy, S., Fernandes, T., Schields, N., et al. (2018). Verapamil and riluzole cocktail liposomes overcome pharmacoresistance by inhibiting P-glycoprotein in brain endothelial and astrocyte cells: a potent approach to treat amyotrophic lateral sclerosis. Eur. J. Pharm. Sci. 120, 30–39. doi: 10.1016/j.ejps.2018.04.026
Young, N. P., Laughlin, R. S., and Sorenson, E. J. (2010). Gadolinium enhancement of the lumbar roots in a case of ALS. Amyotroph. Lateral Scler. 11, 207–209. doi: 10.3109/17482960802642161
Zhong, Z., Deane, R., Ali, Z., Parisi, M., Shapovalov, Y., O'banion, M. K., et al. (2008). ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 11, 420–422. doi: 10.1038/nn2073
Keywords: blood-brain barrier, blood-spinal cord barrier (BSCB), motor neuron disease (MND), amyotrophic lateral sclerosis, neurovascular unit, pericyte
Citation: Mirian A, Moszczynski A, Soleimani S, Aubert I, Zinman L and Abrahao A (2022) Breached Barriers: A Scoping Review of Blood-Central Nervous System Barrier Pathology in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 16:851563. doi: 10.3389/fncel.2022.851563
Received: 10 January 2022; Accepted: 07 March 2022;
Published: 31 March 2022.
Edited by:
James P. Clement, Jawaharlal Nehru Centre for Advanced Scientific Research, IndiaReviewed by:
Jorge Matias-Guiu, Complutense University of Madrid, SpainPavle R. Andjus, University of Belgrade, Serbia
Copyright © 2022 Mirian, Moszczynski, Soleimani, Aubert, Zinman and Abrahao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agessandro Abrahao, YWdlc3NhbmRyby5hYnJhaGFvQHV0b3JvbnRvLmNh
†These authors have contributed equally to this work and share first authorship
‡These authors share senior authorship