 Subrata Pradhan
Subrata Pradhan Rui Gao1
Rui Gao1 Nan Zhang
Nan Zhang Yogesh P. Wairkar
Yogesh P. Wairkar Partha S. Sarkar
Partha S. Sarkar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 04 April 2022
Sec. Cellular Neuropathology
Volume 16 - 2022 | https://doi.org/10.3389/fncel.2022.837576
This article is part of the Research TopicDefective DNA Damage Response – Repair Axis in Post-Mitotic Neurons in Human Health and Neurodegenerative DiseasesView all 5 articles
Emerging evidence suggests that DNA repair deficiency and genome instability may be the impending signs of many neurological diseases. Genome-wide association (GWAS) studies have established a strong correlation between genes that play a role in DNA damage repair and many neurodegenerative diseases, including Huntington’s disease (HD), and several other trinucleotides repeat expansion-related hereditary ataxias. Recently, many reports have documented a significant role played by the DNA repair processes in aging and in modifying many neurodegenerative diseases, early during their progression. Studies from our lab and others have now begun to understand the mechanisms that cause defective DNA repair in HD and surprisingly, many proteins that have a strong link to known neurodegenerative diseases seem to be important players in these cellular pathways. Mutations in huntingtin (HTT) gene that lead to polyglutamine repeat expansion at the N-terminal of HTT protein has been shown to disrupt transcription-coupled DNA repair process, a specialized DNA repair process associated with transcription. Due to the recent progress made in understanding the mechanisms of DNA repair in relation to HD, in this review, we will mainly focus on the mechanisms by which the wild-type huntingtin (HTT) protein helps in DNA repair during transcription, and the how polyglutamine expansions in HTT impedes this process in HD. Further studies that identify new players in DNA repair will help in our understanding of this process in neurons. Furthermore, it should help us understand how various DNA repair mechanism(s) coordinate to maintain the normal physiology of neurons, and provide insights for the development of novel drugs at prodromal stages of these neurodegenerative diseases.
Understanding of most neurodegenerative disorders suffer from the lack of the knowledge of their etiology. This creates a huge and significant challenge for designing and targeting effective therapies for the patients suffering from these untreatable, and terminal conditions. For example, although pathological features of Alzheimer’s disease (AD) are the deposition of Aβ peptides and tau aggregates, the therapies that have been directed toward these proteins have not had a remarkable success. Part of the problem could be that we are not targeting early etiological causes and missing opportunities to dramatically improve the patient outcomes by not targeting early causes of the disease. Recent studies have shown that substantial DNA damage occurs early in many neurodegenerative diseases (Rolig and McKinnon, 2000; McKinnon and Caldecott, 2007; McKinnon, 2009; Madabhushi et al., 2014), including AD (Shackelford, 2006; Shanbhag et al., 2019), Parkinson’s disease (PD; Milanese et al., 2018; Schaser et al., 2019; Martín-Jiménez et al., 2020; Gonzalez-Hunt and Sanders, 2021), Huntington’s disease (HD; Browne et al., 1997; Bogdanov et al., 2001; Giuliano et al., 2003; Illuzzi et al., 2009; Enokido et al., 2010; Bertoni et al., 2011; Tamura et al., 2011; Askeland et al., 2018; Massey and Jones, 2018), spinocerebellar ataxia type 3 (SCA3; Chatterjee et al., 2015; Gao et al., 2015; Chakraborty et al., 2020), and amyotrophic lateral sclerosis (ALS; Konopka and Atkin, 2018; Mitra et al., 2019; Konopka et al., 2020); while it is becoming increasingly clear that DNA damage may occur early and maybe an important early contributor to these diseases, it is unclear how DNA repair processes contribute to the dysfunction of neurons. Because our lab along with others, have made significant contributions toward understanding transcription-coupled DNA repair (TCR) processes in HD and other polyglutamine (polyQ) diseases, for the sake of this review, we will largely focus on DNA repair mechanisms and their relationship with TCR in HD.
Huntington’s disease is a rare genetic disorder with a prevalence of 5–10 individuals per 100,000 in the Caucasian population and many more people are at risk of developing the disease (Morrison, 2012; Baig et al., 2016; Rawlins et al., 2016). Some juvenile forms of the disease also exist but are rare, accounting for about 5% of the cases (Nance and Myers, 2001). The term “Juvenile HD” is applied to the cases of HD with disease onset before the age of 20. While HD symptoms that appear in adults primarily manifest with pure movement disorder or chorea (No authors listed, 1993; Vonsattel and DiFiglia, 1998; Rosenblatt et al., 2003; Ross and Tabrizi, 2011), patients with Juvenile HD develop symptoms of mental disturbance and rigidity rather than chorea (Nance and Myers, 2001; Geevasinga et al., 2006).
Huntington’s disease is a complex disease, and the disease symptoms vary between individuals but is typically manifested with three distinct sets of symptoms known as classical triad consisting of: (1) involuntary choreatic movements and motor coordination defects, (2) mild to moderate cognitive decline, and (3) psychiatric and behavioral abnormalities. At the onset, all the symptoms may not appear simultaneously, and symptoms often vary between individuals and the affected members of a family, however, the symptoms progress predictably with age (Ross et al., 1997; Walker, 2007). Typically, the phenotypes manifest in the middle-age (Ross et al., 1997; Walker, 2007) when they present with psychiatric and cognitive symptoms, although there are cases of juvenile onset at ages as young as 2 years (Nance and Myers, 2001). The motor coordination defects in adult-onset HD can be divided into the choreiform movements with gait disturbances that usually appear at the early stages of disease progression, and motor impairments such as bradykinesia and rigidity that are usually observed in the later stages of the disease (No authors listed, 1993; Vonsattel and DiFiglia, 1998; Rosenblatt et al., 2003; Ross and Tabrizi, 2011). Importantly, there is mounting evidence that cognitive deficits become apparent in mutant carriers several years prior to the onset of clinical symptoms (Jason et al., 1988; Hahn-Barma et al., 1998; Lawrence et al., 1998; Marder et al., 2000; Berrios et al., 2002). Cognitive impairment starts with subtle disturbances but progressively leads to detectable cognitive decline. In addition, HD patients cannot organize or plan simple tasks in daily life and lose flexibility of mind and often fail to make simple adjustments. Lastly, the HD patients can also be diagnosed with frequent depression, excessive mood swings and anxiety, and signs of apathy, irritability, impulsivity, and social disinhibition. Thus, HD is a complex disease, but subtle molecular changes likely occur well before the clinical manifestation of the disease, providing a window of opportunity for therapeutic interventions.
In a seminal discovery, the unique pathological mutation in HD was discovered in 1993 (No authors listed, 1993). This ground-breaking discovery was not only important in identifying a unique genetic mutation that causes human genetic disease and in uncovering the complex HD pathogenic mechanism(s) but also led the way in unraveling the underlying pathomechanism of several other polyglutamine (polyQ)-expansion related hereditary ataxias. The mutation associated with HD was found to be an expansion of a polyQ tract at the N-terminal coding region of the huntingtin (HTT) protein due to an expansion of a polymorphic CAG trinucleotide repeat sequences in the mutant huntingtin (mHTT) gene, which leads to progressive deterioration of cognitive and motor functions in patients with HD (No authors listed, 1993; Vonsattel and DiFiglia, 1998; Ross and Tabrizi, 2011). In normal population, the polymorphic CAG repeat sequences are in the range of 5–35 repeats, with a median length of 17–20 CAG repeats. Full penetrance is observed for mHTT alleles containing more than 40 CAG repeats (No authors listed, 1993). CAG repeat length of 36 or more at the N-terminal of HTT gene was found to be associated with adult-onset HD (No authors listed, 1993). An inverse correlation was observed between the length of the CAG repeats and the age of onset, determined by the first motor manifestation (No authors listed, 1993; Rosenblatt et al., 2006). In another interesting report, the length of uninterrupted CAG repeats in DNA, rather than the polyQ length at the N-terminal of HTT was found to be a critical contributing factor in HD disease onset (Genetic Modifiers of Huntington’s Disease [GeM-HD] Consortium, 2019).
Clinical studies and studies in animal models of HD support the idea that mHTT protein carrying the extended stretch (more than 35 repeats) of polyQ sequences at the N-terminal region results in progressive degeneration of the gamma-aminobutyric acid (GABA)-releasing striatal neurons and glutamatergic cortical neurons in the basal ganglia. Additionally, neuronal dysfunction and tissue atrophy in other brain regions has also been reported in HD (Vonsattel and DiFiglia, 1998; Ross and Tabrizi, 2011).
Microscopic and molecular analyses of postmortem HD patient brains reveal the presence of aggregated form of mHTT (Davies et al., 1997; DiFiglia et al., 1997; Hoffner et al., 2007; Bauer and Nukina, 2009; Legleiter et al., 2010; Sontag et al., 2012). Based on these observations, it was hypothesized that the mHTT protein carrying extended stretch of polyQ sequences adopts unusual structural conformations, which facilitate aberrant protein–protein interactions. These result in the formation of large insoluble protein aggregates in the HD neurons. How do the aggregates affect the neuronal health? Many possibilities exist. First, the protein aggregates could negatively impact transcription of several neuronal genes because the aggregated HTT can interact and sequester key transcription factors and co-activators that regulate transcription of these genes (Luthi-Carter and Cha, 2003; Cha, 2007; Ross and Tabrizi, 2011; Kumar et al., 2014). Second, protein aggregates could physically interfere with the trafficking of organelles (Chang et al., 2006; Orr et al., 2008), and vesicles (Truant et al., 2006; Caviston and Holzbaur, 2009) in the affected neurons and glial cells (Figure 1). Third, HTT aggregates can interact with important signaling pathways (Davies et al., 1997; DiFiglia et al., 1997; Hoffner et al., 2007), can cause mitochondrial dysfunction and energy dyshomeostasis (Orr et al., 2008; Shirendeb et al., 2011; Siddiqui et al., 2012), and directly impact synaptic function (Morton et al., 2001; Li J. Y. et al., 2003; Joshi et al., 2009; Raymond et al., 2011). Finally, the aggregates could also lead to a disruption in DNA damage repair mechanism(s) leading to the excessive accumulation of DNA damage/strand breaks, leading to the chronic activation of the ataxia telangiectasia-mutated (ATM) and p53 signaling pathways (Bogdanov et al., 2001; Giuliano et al., 2003; Bae et al., 2005; Illuzzi et al., 2009; Bertoni et al., 2011; Lu et al., 2014; Askeland et al., 2018). Of interest, a recent study highlighting the importance of ATM pathway has demonstrated that either genetic or pharmacological ablation of ATM kinase activity significantly reduces the neurotoxicity in HD animal models (Lu et al., 2014). This study supports the emerging theme that persistent accumulation of DNA damage in neuronal DNA, and subsequent chronic activation of DNA damage-response (DDR) ATM pathway might be one of the important contributing factors to early pathogenesis of HD. However, whether accumulation of DNA damage is due to defective or impaired DNA repair mechanism(s), or chronic oxidative stress that stem from HTT protein aggregates remains a debatable.
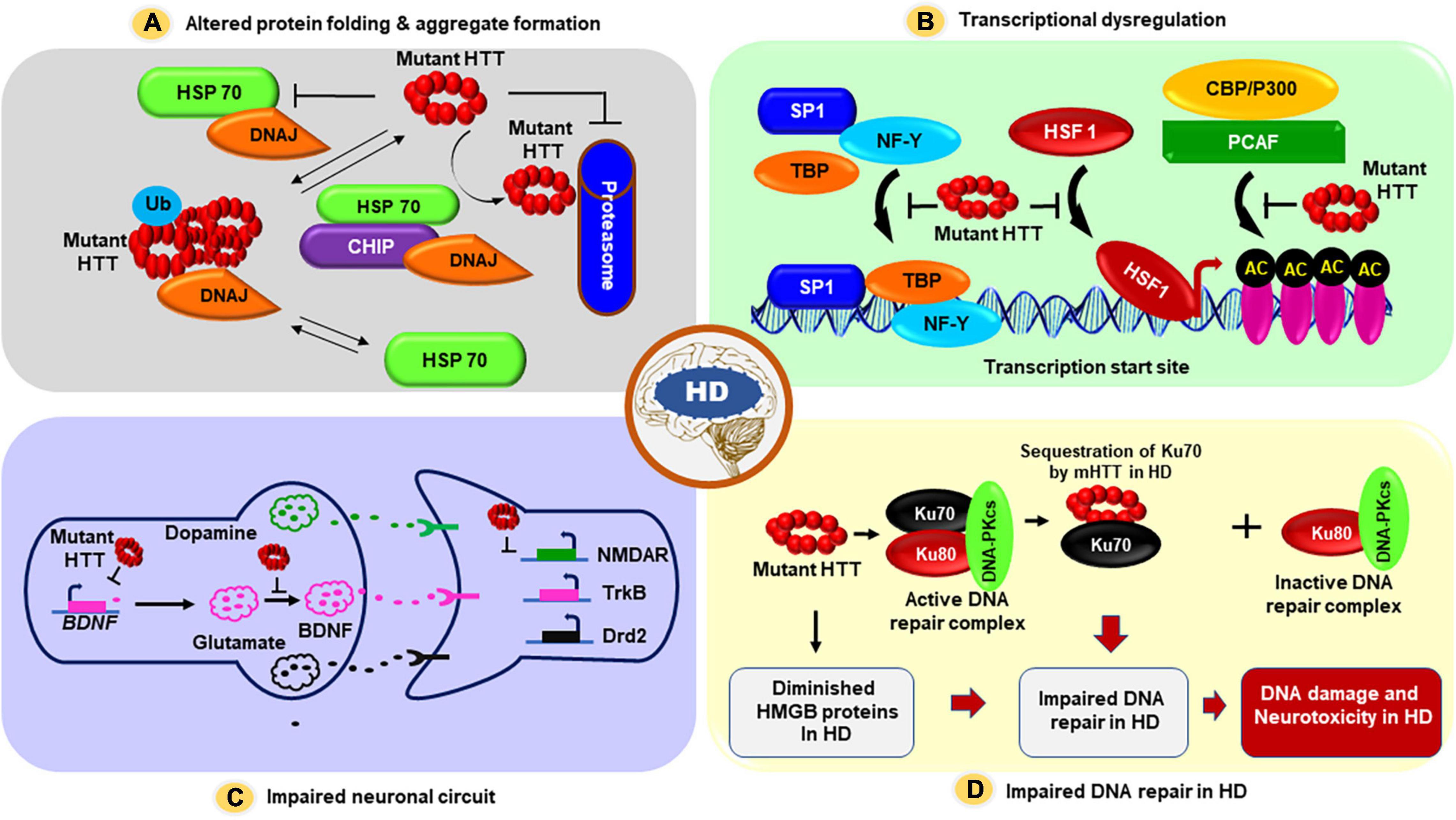
Figure 1. The proposed mechanism(s) by which mHTT triggers neuronal dysfunction and neurotoxicity in HD. Several possible pathogenic mechanism(s) for HD have been proposed: (A) First, mHTT can adopt altered structural conformations, and these inappropriately folded structures may aberrantly interact with many neuronal proteins, and these unusual protein-protein interactions may result in protein aggregate formation, and these aggregates may physically interfere with the vesicle and organelle transport within the cells, triggering neurotoxicity in HD. These aggregates may also fully or partially inactivate numerous proteins that are sequestered into HTT aggregates in HD neurons, contributing to neurotoxicity and HD pathogenesis. (B) mHTT carrying expanded polyQ sequences cause transcriptional dysregulation in HD: The mHTT protein translocate into the nucleus, interacts with various transcription factors and cofactors, and interferes with their normal functions, resulting in aberrant transcription of many neuronal genes that regulate neuronal function, survival, synaptic function, and vesicle transport. mHTT-mediated transcriptional dysregulation ultimately results in neuronal dysfunction, neurotoxicity, and neurodegeneration at early stages of disease progression in HD; (C) Evidence also suggests that in addition to overt transcription dysregulation, mHTT may also impair neuronal circuits and interfere with neurotransmission and synaptic function causing early neurotoxicity in HD. Hypothesized model illustrating how changes to the cortical-striatal synapse during early-stages of HD. In response to mHTT expression expression of BDNF is reduced in HD. Glial glutamate uptake by glutamate transporter 1 (GLT-1) is reduced, which probably enhances synaptic and/or extrasynaptic glutamate levels. Postsynaptic N-methyl-D-aspartate receptors (NMDAR) are increased outside the synapses, and gamma-aminobutyric acid (GABA)-argic inhibitory input is enhanced and tyrosine kinase B (TrkB) signaling is reduced. (D) Evidence also suggests that mHTT aberrantly interacts with and sequesters Ku70, an essential factor in NHEJ-mediated DNA double-strand break repair in HD. Sequestration of Ku70 by mHTT results in impaired DNA double-strand break repair in HD. Moreover, mHTT can also repress the expression of high mobility group proteins B1 and B2 (HMGB1/2), and mHTT-mediated downregulation of HMGB1/2 proteins can also result in defective DNA repair and DNA damage accumulation in HD. Abbreviations: SP1, Specificity protein 1; TBP, TATA binding protein; NF-Y, Nuclear factor Y; HSF1, Heat shock transcription factor 1; CBP, CREB-binding protein; AC, Acetylation; HSP70, Heat shock protein 70; DNAJ, Chaperone protein; Ub, Ubiquitination; BDNF, Brain-derived neurotrophic factor; NMDAR, N-methyl-D-aspartate receptors; TrkB, Tropomyosin receptor kinase B; Drd2, Dopamine Receptor D2.
In this review, we will briefly discuss some of the major pathogenic mechanisms that have been proposed for HD, with the focus on how HTT helps in DNA repair and genome maintenance. We will also comment upon the emerging view of and how mutant HTT (mHTT) protein impairs TCR, a specialized DNA repair mechanism in non-dividing postmitotic cells like neurons.
While the toxicity arising from the aggregates adopted by mHTT is largely hypothesized to be the cause and progression of the disease (Davies et al., 1997; DiFiglia et al., 1997; Benn et al., 2005; Hoffner et al., 2007; Sawada et al., 2007; Bauer and Nukina, 2009; Legleiter et al., 2010; Sontag et al., 2012), it is still a subject of intense debate whether the aggregates (Legleiter et al., 2010; Sontag et al., 2012) play any toxic role in HD pathogenic mechanisms or whether they are actually neuro protective. It also remains to be established whether the aggregates or oligomeric forms of the mutant protein are toxic for the HD neurons (Hoffner et al., 2007; Shirendeb et al., 2011). Arguments in favor of the aggregate hypothesis include interactions of HTT aggregates with various chaperone proteins, including heat shock proteins HSP40 and HSP70, and components of the neuronal proteasome system, resulting in sequestration of the proteins involved in protein clearance system. This might affect the protein folding machinery as well as clearance of the misfolded proteins in HD (Davies et al., 1997; DiFiglia et al., 1997; Muchowski et al., 2000). In fact, the HDJ-2 and HSP70 chaperones were found to co-localize with HTT aggregates in the brain of R6/2 HD transgenic mice expressing the N-terminal truncated fragment of mHTT (Mangiarini et al., 1996), and exogenous expression of chaperons HDJ-1 and HSP70 reduced the amount aggregates formed by mHTT and ameliorated the toxicity in cell models of HD (Jana et al., 2000). This unusual protein interactions might result in the inactivation of other key neuronal proteins adding to the complexity of the disease pathology. Importantly, increasing the levels of chaperons facilitates the clearance of the aggregates and reduces the neurotoxicity in HD models (Figure 1), suggesting that protein folding machinery and/or protein clearance systems are compromised in HD. However, the precise mechanisms of how mHTT disrupts these fundamental cellular processes in HD remains to be established.
Over the last two decades it has become clear that both HTT and mHTT are present in the nucleus, directly interact with various transcription factors, cofactors, and RNA polymerase, and regulate transcription of neuronal genes (Cha, 2007; Benn et al., 2008; Ross and Tabrizi, 2011; Valor, 2015; Jimenez-Sanchez et al., 2017). Both HTT and mHTT interact with several nuclear transcription factors and cofactors, including cyclic AMP-response element-binding protein (CBP; McCampbell et al., 2000; Steffan et al., 2000; Nucifora et al., 2001), TATA-binding protein (TBP; Huang et al., 1998), the tumor suppressor transcription factor p53 (Steffan et al., 2000; Bae et al., 2005), the general transcription factors TFIID and TFIIF (Zhai et al., 2005), and specificity protein 1 (SP1; Dunah et al., 2002). The RNA polymerase large subunit A (POLR2A) also interacts with HTT and is detected in nuclear inclusions in the HD brain (Huang et al., 1998; Suhr et al., 2001). Additionally, the transcription cofactor TAFII130 (a cofactor for CREB-dependent transcription) has also been shown to bind to expanded polyQ sequences to interfere with CREB-dependent transcription in HD (Shimohata et al., 2000a). Moreover, peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α (PGC-1α), a transcriptional coactivator (Lin et al., 2002, 2005) that works in combination with other transcription factors like PPARγ may play an important role in HD pathogenesis (Cui et al., 2006; Weydt et al., 2006). mHTT has been shown to interfere with the binding of the CREB/TAF4 complex on PGC-1α promoter, repressing its expression, and evidence suggest that reduced PGC-1α level/activity might contribute to neurotoxicity in HD (Cui et al., 2006; Weydt et al., 2006). Moreover, the repressor element 1 (RE1)-silencing transcription (REST) is a master regulator of neuronal gene transcription, repressing their transcription (Huang et al., 1999). Several REST target genes e.g., BDNF (brain-derived neurotrophic factor) are known to be downregulated in HD, and reduced BDNF activity contribute to HD pathogenesis (Zuccato et al., 2001; Canals et al., 2004). Overexpression of BDNF in HD transgenic mouse brain can rescue HD-like phenotypes (Xie et al., 2010). Evidence also suggest that REST regulates transcription of microRNAs, many of which regulate neuronal gene expression and are dysregulated in HD (Johnson et al., 2008; Johnson and Buckley, 2009). Further, HTT has been shown to bind REST in the cytoplasm and thus prevents REST-mediated suppression of BDNF (Zuccato et al., 2003, 2007). The presence of mHTT results in reduced interaction between mHTT and REST, and consequently nuclear levels of REST increases, repressing BDNF expression (Zuccato et al., 2003, 2007). Based on these observations, it is hypothesized that HTT probably assists in the assembly of transcription factor and co-activator into multi-protein active transcription complexes, which regulate target gene expression in neurons, and that mHTT perturbs either the assembly or the function of these transcription complexes (Luthi-Carter and Cha, 2003; Ross and Tabrizi, 2011; Kumar et al., 2014; Figure 1).
There is ample evidence to suggest that subtle alterations of synapses occur before overt neuronal death, and that these changes are predictive of the onset of behavioral problems associated with HD (Morton et al., 2001; Li H. et al., 2003; Joshi et al., 2009; Raymond et al., 2011). Huntingtin is highly expressed in the presynaptic terminals of nerve cells (DiFiglia et al., 1995), and various proteins that regulate both the presynaptic and postsynaptic function have been shown to interact with HTT (Li et al., 2002; Li J. Y. et al., 2003; Barron et al., 2021). These proteins affect a range of pre and postsynaptic functions, including endocytosis, synaptic homeostasis, and axonal transport on the presynaptic side, and postsynaptic receptor localization and dendritic protein transport at the post-synapse.
At the presynaptic terminal, HTT can regulate the normal exocytosis, endocytosis, and axonal transport. For example, Complexin II – a protein involved in neurotransmitter release (Rizo and Südhof, 2002), is decreased in HD patients (Morton and Edwardson, 2001), R6/2 transgenic mice (Morton et al., 2001) and neuronal cell model of HD (Edwardson et al., 2003). Another protein involved in docking of synaptic vesicles to the membrane is rabphilin 3A, the level of which is progressively decreased in R6/1 mouse model of HD (Smith et al., 2005). Also, reduction of HTT-interacting proteins involved in endocytosis might impair endocytic and intracellular trafficking in HD (Kalchman et al., 1997; Li et al., 1998; Sittler et al., 1998; Modregger et al., 2002; Singaraja et al., 2002; Trushina et al., 2004). HTT-interacting protein 1 (HIP1) is closely related HIP12, and these proteins are orthologs of the yeast Sla2p, which is involved in endocytosis (Engqvist-Goldstein et al., 1999). HIP1 interacts with clathrin and AP-2, whereas HIP12 interacts with F-actin and clathrin light chain (Legendre-Guillemin et al., 2002). The most studied of these proteins is HTT-associated protein 1 (HAP1) (Li et al., 1995) that interacts with the p150 Glued subunit of dynactin (Engelender et al., 1997; Li et al., 1998), which in turn, interacts with dynein, the motor protein involved in retrograde transport. These two proteins interact to facilitate efficient vesicular transport along the microtubules (Gill et al., 1991). Studies have shown that the binding of HAP1 with HTT is increased in HD (Li et al., 1995), and increased binding of mHTT with HAP1 depletes HAP1 from its normal functional site at the dynein/dynactin complex, negatively impacting axonal transport in HD (Gauthier et al., 2004).
These studies highlight the importance of HTT in normal synaptic function. It is no surprise then that various genetic therapies directed toward lowering mutant copy of HTT, which also reduced wild-type HTT level (Tabrizi et al., 2019), led to devastating consequences manifesting with motor and memory deficits (Kwon, 2021). It is interesting to note that both DNA damage and synaptic dysfunction are early signs of HD. However, there is no evidence that these two phenomena are linked or whether these processes are disrupted independent of one another in HD. One possibility is that synapses are disrupted because their energy demands are not met in HD. Most of the energy needed for synaptic function is supplied by mitochondria that localize to synapses and to the nodes of Ranvier in the motor neurons. There is plenty of evidence to suggest that mitochondrial function is disrupted in HD (Orr et al., 2008; Shirendeb et al., 2011; Siddiqui et al., 2012), and this could directly affect various synaptic processes. Second possibility is that in HD, there might a decrease in the interactions of HTT with its synaptic partners, much like we observed in DNA damage repair where mHTT can bind to the native DNA repair complex partners and decrease the efficacy of the complex to repair DNA damages (Gao et al., 2019). Finally, we know that inefficiency of DNA repair can disrupt transcription (Gao et al., 2019), which might affect the synaptic protein turnover leading to decreased synaptic function. However, to our knowledge, no study has yet demonstrated a direct link between DNA repair deficiency and synaptic dysfunction. Intriguingly, mutations that affect DNA repair have implications for many neurological diseases including ALS, AD, PD, and HD (Browne et al., 1997; Rolig and McKinnon, 2000; Bogdanov et al., 2001; Giuliano et al., 2003; Illuzzi et al., 2009; Bertoni et al., 2011; Madabhushi et al., 2014; Maynard et al., 2015; Askeland et al., 2018; Massey and Jones, 2018; Mitra et al., 2019; Shanbhag et al., 2019; Konopka et al., 2020). Thus, it would be interesting to investigate whether there is a direct link between synaptic demise and DNA repair and whether DNA repair is upstream of synaptic dysfunction in HD and related neurodegenerative diseases (Figure 1).
It is often debated whether persistent accumulation of DNA damage that is consistently observed in HD brain is the cause or a consequence of HD pathophysiology. Emerging evidence indicates that mHTT interferes with the DNA repair process in the post-mitotic neurons, leading to the accumulation of DNA damage (Qi et al., 2007; Enokido et al., 2010; Tamura et al., 2011). The wide-spread occurrence of unresolved DNA damages/lesions in postmortem HD patient brains, and especially in the pre-symptomatic HD transgenic mouse brain early during the disease progression suggest that accumulation of DNA damage/lesions in HD could be a cause rather than a symptom of HD (Enokido et al., 2010; Tamura et al., 2011; Gao et al., 2019). However, these observations do not rule out the possibility that DNA damages accumulate independently due to other possible causes of neurodegeneration in HD. Indeed, both could contribute to the HD disease pathology but recent evidence from genome-wide association (GWAS) studies of HD have found many genes that are involved in DNA damage repair in neurons (e.g., FAN1, LIG1, MLH1, MSH3, PMS1, and PMS2) as strong modifiers of age at onset and disease severity (Genetic Modifiers of Huntington’s Disease [GeM-HD] Consortium, 2015; Maiuri et al., 2019; Tabrizi et al., 2020), suggesting that impaired or defective DNA repair mechanism is mechanistically linked to HD pathomechanism. Second, many recent studies support the idea that presence of DNA damage/lesions is one of the early pathological hallmarks in HD brain (Browne et al., 1997; Bogdanov et al., 2001; Giuliano et al., 2003; Illuzzi et al., 2009; Bertoni et al., 2011; Askeland et al., 2018; Massey and Jones, 2018), indicating that DNA damage occur early in the disease progression and persistence of these lesions might trigger a cascade of pro-degenerative pathways in HD. Third, the presence of substantially higher oxidized bases in genomic DNA, in cell and animal models of HD, and in human postmortem brains (Browne et al., 1997; Bogdanov et al., 2001; Giuliano et al., 2003; Illuzzi et al., 2009; Bertoni et al., 2011; Askeland et al., 2018; Massey and Jones, 2018) also indicate problems with DNA repair system(s) in HD. Importantly, accumulation of oxidized bases in DNA, with frequent DNA deletions in HD postmortem brains strongly support the idea (Horton et al., 1995; Browne et al., 1997; Polidori et al., 1999; Siddiqui et al., 2012). Fourth, transgenic expression of mHTT in mouse brain also induces DNA damage early in the disease progression (Bogdanov et al., 2001; Gao et al., 2019), suggesting that DNA repair system(s) that maintains neuronal genome integrity might be affected early in the disease progression. Finally, many studies have implicated HTT in double strand break (DSB) repair mechanism, and mHTT has been shown to interact with Ku70, an essential DSB repair protein and a regulatory component of the DNA-dependent protein kinase (DNA-PK) (Enokido et al., 2010). Interaction and sequestration of Ku70 by mHTT has been suggested to impair non-homologous end-joining (NHEJ)-mediated DNA DSB repair in HD neurons (Enokido et al., 2010). DSBs are the most lethal form of DNA lesions in postmitotic cells like neurons and unlike other cells, neurons do not use homologous recombination. Instead, NHEJ pathway is used in neurons to repair these potentially lethal lesions (Rass et al., 2007; McKinnon, 2009; Lieber, 2010). Importantly, stimulating DSB repair activity by overexpressing Ku70 in mouse or Drosophila model of HD can rescue neurodegeneration (Enokido et al., 2010; Tamura et al., 2011), suggesting that mHTT impairs NHEJ-mediated DSB repair in HD. These findings strongly support the idea that failure of the neuronal DNA repair mechanisms due to polyQ expansion in HTT could be an important early contributing factor to the downstream degenerative events commonly observed in HD. Why would the DNA repair process be so important and effect the neurons disproportionately? We know that most of the mature neurons in adult brains are postmitotic and these repair processes maybe more important in neurons than other cells that can undergo apoptosis in case of heavy damage to their DNA. Also, we know that neurons are metabolically highly active, even at resting state, generating excessive levels of potentially damaging reactive oxygen species (ROS), and these damaging free radicals have been demonstrated to induce various types of lesions in neuronal DNA, including DNA single-strand breaks, oxidative damages, base deletions, DSBs (Lindahl and Barnes, 2000). Therefore, if not efficiently resolved, these potentially lethal and damaging lesions can accumulate during routine neuronal activities and become highly toxic if left unresolved. Thus, while DNA damage might contribute to HD pathology, and that HTT may play a critical role in DNA repair processes in neurons, until recently, it was not clear how the native HTT protein participates in the DNA repair processes in normal neurons, and how polyQ expansions in mHTT disrupts this vital genome maintenance process in neurons.
We know that the damaging, and potentially lethal lesions in neurons are generally faithfully and accurately resolved by a repertoire of DNA repair systems that strictly protect the genome integrity and health of mature neurons (Sancar et al., 2004; McKinnon, 2017). Oxidized DNA bases, abasic sites, single-strand breaks (SSBs), and DSBs are primarily repaired by the base-excision repair (BER) pathway in the central nervous system (Seeberg et al., 1995), and evidence to support the existence of nucleotide excision repair (NER), and mismatch repair (MMR) pathways in postmitotic neurons (Madabhushi et al., 2014; McKinnon, 2017). The plausible DNA damage repair pathways in mammalian cells are illustrated in Figure 2. However, since neurons are more vulnerable due to their high metabolic and transcriptional activities, and their postmitotic nature, there must be additional mechanisms that are important in protecting the neuronal genome of neurons. Indeed, emerging evidence including our own recent studies (Gao et al., 2015, 2019; Chakraborty et al., 2020) suggest that neurons have an additional and specialized repair mechanism in which the transcriptionally active genome is repaired more efficiently than the non-transcribing region to accurately maintain genomic integrity such that they do not produce defective or inactive proteins. How do long-lived cells like neurons accurately maintain their genome integrity despite constant exposure to endogenous genotoxic agents? Increasing evidence suggest that postmitotic cells like neurons have an additional layer of repair mechanism to protect their genome. It is hypothesized that the translocating RNA polymerase recruits specific DNA repair proteins to resolve the lesions during transcription elongation (Hanawalt, 1994; Hanawalt and Spivak, 2008). However, how in response to DNA damage, several DNA repair proteins are quickly assembled to accurately resolve the DNA lesions. However, how this process unfolds remains poorly understood, and needs further investigation. We have recently demonstrated that HTT plays a critical role in assembling a multi-protein transcription-coupled DNA single-strand break repair (TCR) complex, and that this novel multiprotein complex sense the DNA strand breaks/damages in the template DNA strand and resolves the DNA damages/lesions during transcription elongation (Gao et al., 2019). Our findings strongly suggest that the HTT-assembled macromolecular DNA repair-transcription complex provides an additional layer of protective mechanism to strictly maintain the sequence integrity of the protein-coding regions of neuronal genome. This specialized DNA repair system probably is an additional layer of protection evolved for postmitotic cells like neurons.
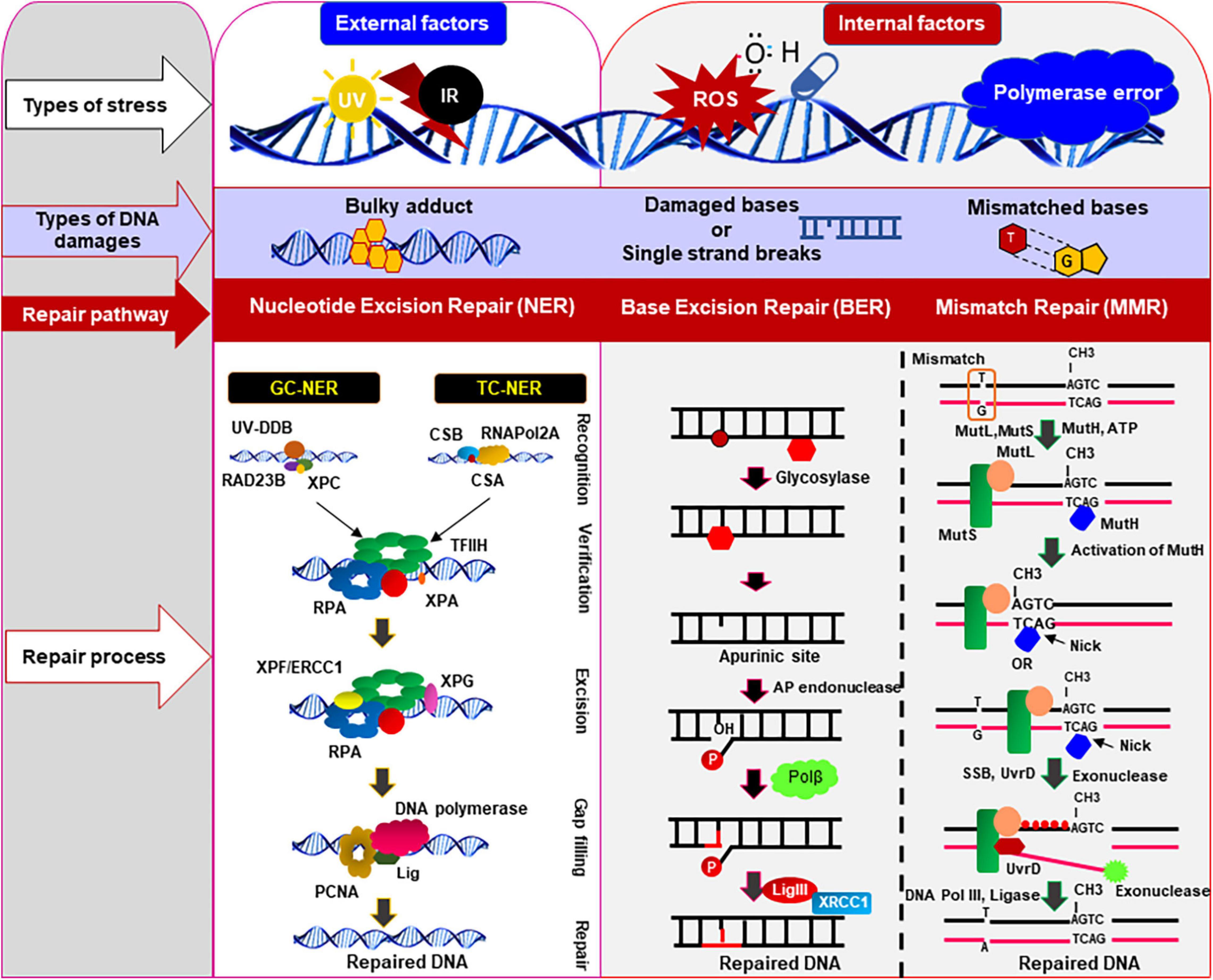
Figure 2. DNA damages and repair pathways in mammalian cells. Various types of DNA damages/lesions are routinely induced in genomic DNA by both internal genotoxic agents (e.g., reactive oxygen species: ROS, DNA polymerase error) as well as by external agents (e.g., ultra-violet radiation, infrared radiation, etc.). These potentially lethal damages are efficiently repaired by specialized DNA repair pathways, depending on types of DNA damages and cell types. This figure illustrates the common and simplified graphical presentation of the different types of DNA repair pathways involved to resolve various types of damages/lesions in mammalian cells in an error-free way. The set of repair proteins may vary between different pathways, and there might be some overlapping steps in different repair pathways. Nucleotide excision repair (NER), base excision repair (BER), and mismatch repair (MMR) pathways are the main repair pathways that protect genome from the internal or external genotoxic agents. These repair pathways play important role in repairing damages that routinely occur in genomic DNA of the post-mitotic neurons to maintain the sequence integrity and health of neurons. CSA/CSB, cockayne syndrome protein A/B; ERCC1, excision-repair cross-complementing 1; TFIIH, transcription factor IIH; RPA, replication protein A; GG-NER, global genomic nucleotide excision repair; TC-NER, transcription-coupled nucleotide excision repair; Lig, DNA ligase; RNAPol2A, RNA polymerase subunit 2A; XPA/XPC/XPF/XPG, proteins involved in different xeroderma pigmentosum (XP); PCNA, proliferating cell nuclear antigen; Polβ, DNA polymerase β; MLH, MutL protein homolog; MSH, MutS protein homolog; UV, ultraviolet; XRCC1, X-ray repair cross-complementing 1.
How does HTT help in the TCR process? HTT is a large (∼350 kDa) protein of 3,144 amino acids and many studies have shown that HTT is indispensable for neuronal development and survival (Duyao et al., 1995; Nasir et al., 1995; Zeitlin et al., 1995; Saudou and Humbert, 2016). While HTT is predominantly a cytosolic protein, it is also found to be present in the nucleus and mitochondria. HTT is enriched in the neurons, specifically in striatum, cerebral cortex, and cerebellum (Li et al., 1993; Sharp et al., 1995) and contains several copies of HEAT [huntingtin, elongation factor 3, protein phosphatase 2A (PP2A), and TOR1] motifs, which may adopt a tertiary structure that likely acts as a scaffold for the assembly of multiprotein complexes (Ochaba et al., 2014; Maiuri et al., 2017). In the nucleus, HTT also interacts with RNA polymerase and various transcription factors (Giuliano et al., 2003; Illuzzi et al., 2009; Bertoni et al., 2011; Ross and Tabrizi, 2011; Lu et al., 2014; Valor, 2015; Jimenez-Sanchez et al., 2017). Previous studies have also implicated HTT in DNA repair due to its interaction with ku70, an essential DNA repair protein (Enokido et al., 2010). Importantly, restoring DNA repair by overexpressing Ku70 in mouse or Drosophila model of HD was found to rescue neurodegeneration (Enokido et al., 2010; Tamura et al., 2011), suggesting that mHTT impairs DNA repair in HD and that DNA damage may be somehow linked to the demise of the neurons.
Our recent studies have shown that HTT organizes a multi-protein TCR complex with RNA polymerase, transcription factors and cofactors (e.g., TBP, CBP, TAF4, etc.), ataxin-3, a deubiquitinating enzyme and DNA repair enzymes that include DNA ligase 3 and polynucleotide kinase 3′-phosphatase (PNKP) (Gao et al., 2019). Interestingly, role of PNKP was implicated in DNA repair deficiency in polyQ expansion disease(s) pathology previously (Barclay et al., 2014). Our data indicate that the multiprotein TCR complex assembled by HTT stimulates DNA strand break repair in neurons by stimulating the end-processing activity of the DNA strand break repair enzyme PNKP (Gao et al., 2019). By contrast, our study has shown that the presence of mHTT in the TCR complex dramatically decreases the activity of PNKP, leading to persistent accumulation of DNA damages in HD (Gao et al., 2019). mHTT-mediated inactivation of the TCR complex in neurons results in preferential accumulation of DNA strand-breaks/damages within the transcriptionally active genome and the number of damages is significantly lower in the genomic regions that are not transcriptionally active in the neurons (Hanawalt, 1994; Hanawalt and Spivak, 2008; Chakraborty et al., 2016, 2020; Gao et al., 2019).
Moreover, the chromatin immunoprecipitation (ChIP) experiments showed that HTT and associated TCR complex components associate with the transcriptionally active genome compared with the inactive genome and the former accumulates more strand breaks/damages in the HD or SCA3 brains (Gao et al., 2019; Chakraborty et al., 2020). ChIP analysis revealed a significantly higher occupancy of HTT on the actively transcribing genome in the brain [e.g., neuronal differentiation factor 1 and 2 (Neurod1 and Neurod2), neurogenic basic-helix-loop-helix protein neurogenin 1 (Neurog1), tubulin beta 3 class III (Tubb3), neuron-specific enolase 2 (Eno2γ), and DNA polymerase beta (Pol b)] over genes that are not transcribed in the brain but actively transcribed in skeletal or cardiac muscle [e.g., myogenic differentiation factor 1 (Myod1); myogenic factor 4; myogenin (Myog); and myosin heavy chain 2, 4, 6, or 7 (Myh2, Myh4, Myh6, or Myh7); Gao et al., 2019]. Increased association between HTT with the transcriptionally active genome and mHTT-mediated abrogation of TCR complex activity suggest that HTT-assembled TCR complex predominantly repairs the DNA lesions during transcription elongation, but polyQ expansion in HTT might impair the TCR activity, resulting in DNA damage accumulation predominantly within the actively transcribing regions of genome in HD. Indeed, analysis of genomic DNA revealed 60–70% lower PCR-amplification of actively transcribing genes in asymptomatic (7 weeks old) transgenic zQ175 mouse brains compared to age-matched controls (Gao et al., 2019). In contrast, the amplification efficacy for non-transcribing genes in the zQ175 brains was only marginally (10–15%) reduced (Gao et al., 2019), indicating significantly less DNA strand break accumulation. These data suggest that HTT-TCR complex repairs the strand breaks that routinely accumulate during transcription, and that this function is impaired by polyQ expansion in HTT, resulting in persistent damage accumulation predominantly affecting the actively transcribing genes in neurons.
Persistence of DNA lesions/damages within the actively transcribing genes or promoter regions may impede transcription of a variety of neuronal genes, including the genes regulating synaptic function, vesicular transport, and calcium homeostasis, impacting overall neuronal health and function, contributing to neurotoxicity at the early stages in HD disease progression. It is tempting to speculate that inefficient TCR is an important contributor to the accumulation of DNA lesions, and transcriptional dysregulation, hallmark features of the HD (Browne et al., 1997; Ayala-Peña, 2013). Therefore, based on our data (Gao et al., 2019), we hypothesize that the combination of reduced HTT levels, and the presence of mHTT in the TCR complex causes DNA repair deficiency resulting in persistent accumulation of DNA damages in the neuronal genome, triggering early neurotoxicity and neurodegeneration in HD. Importantly, this raises the possibility that HTT may act as an interchangeable scaffold to organize components for different DNA repair processes as previously suggested (Qi et al., 2007). Also, we refer the readers to recent excellent reviews on the topic of various types of neuron-specific DNA repair mechanism and neurodegeneration (Jonson et al., 2013; Jones et al., 2017; Ross and Truant, 2017). Taken together, these studies provide compelling evidence to indicate that HTT-assembled TCR complex helps to resolve the DNA lesions during active transcription, and this mechanism might provide an additional protective mechanism to preserve the sequence integrity of the protein-coding regions of the neuronal genome.
Based on the data and what is already known about DNA damage repair and HTT, we propose a model in which RNA polymerase-mediated transcription pausing at DNA damage sites leads to phosphorylation-dependent activation of the DDR kinase ATM and DNA-PK, two of the important serine-threonine kinase that are phosphorylated and activated in response to DNA damage (Kurz and Lees-Miller, 2004; Guo et al., 2010; Yue et al., 2020). Phosphorylation of ATM and/or DNA-PK enhances phosphorylation-dependent recruitment of PNKP (Segal-Raz et al., 2011; Zolner et al., 2011) at the damaged sites, facilitating DNA damage repair. The presence of DNA lesions thus converts the translocating HTT-POLR2A transcription complex into an active TCR complex assisted by HTT. After repair, specific repair factor(s) are dephosphorylated by protein phosphatases, and the transcription complex restarts the paused transcription (Figure 3). When TCR activity is disrupted in neurodegenerative diseases like HD, the complex stalls at DNA lesions, failing or delaying the initiation of repair, compromising the genome integrity and transcription resulting in neuronal dysfunction, and premature death of neurons.
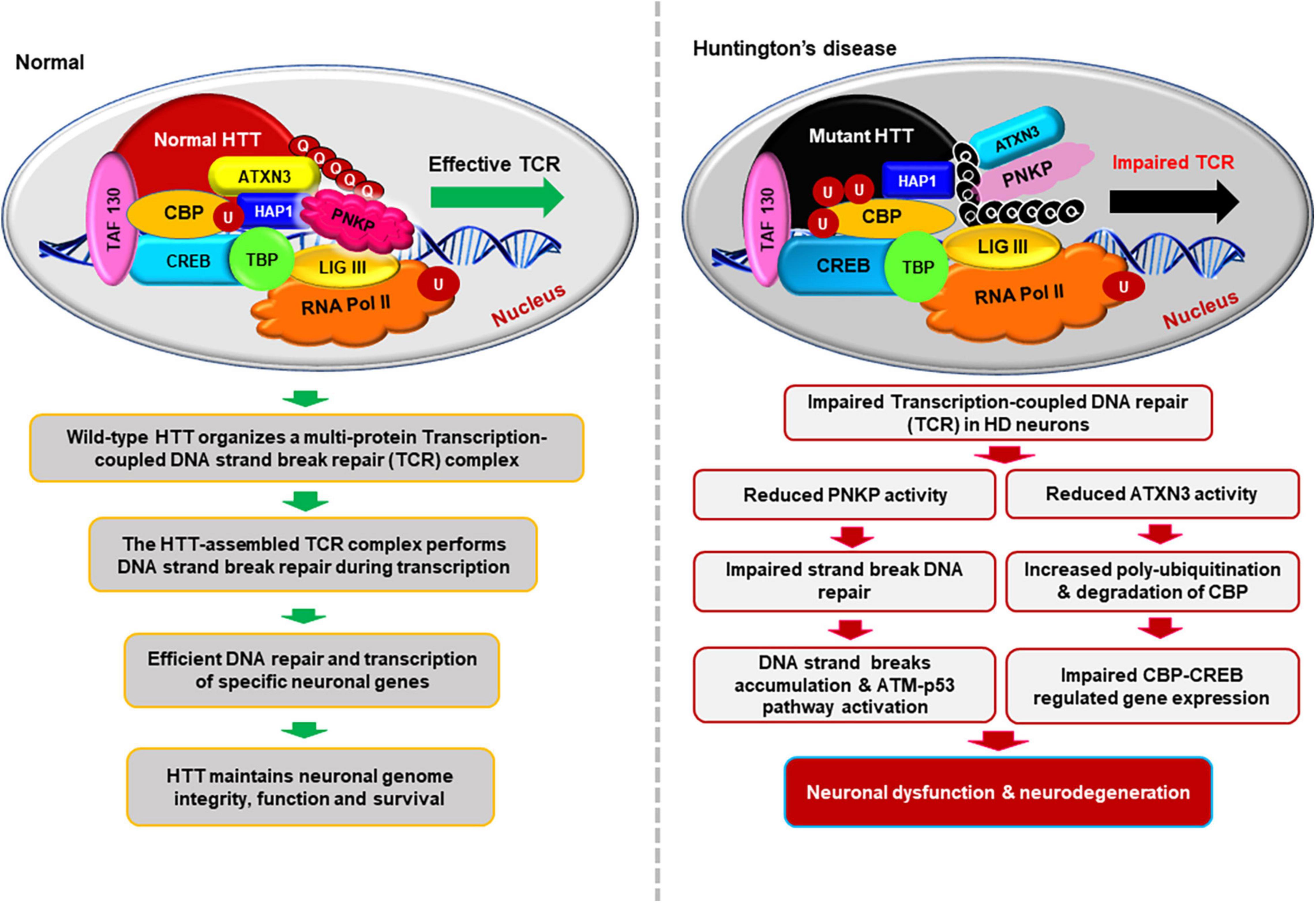
Figure 3. Schematics illustrating how HTT helps to maintain neuronal genome integrity and how mHTT synchronously impairs DNA repair and transcription in HD. This hypothesized multi-protein transcription-coupled DNA repair (TCR) complex assembled by HTT senses DNA strand breaks/lesions during transcription elongation and orchestrates their faithful repair to maintain the sequence integrity of neuronal genome. HTT thus plays a pivotal role in maintaining neuronal health, function, and survival. This novel complex thus provides an additional layer of protective mechanism to strictly maintain the sequence integrity of the transcriptionally active region of genome to protect the protein-coding regions of neuronal genome. By contrast, the presence of mHTT in the TCR complex impairs the activity of the complex, resulting in persistent accumulation of DNA lesions/damages in neuronal genome. Persistence of DNA strand breaks/lesions in genome can interfere with the translocation of transcribing RNA polymerase over the template DNA, impeding adequate transcription of many neuronal genes in HD. Inadequate expression of neuronal genes, may adversely impact neuronal health, neurotransmission, calcium homeostasis, and synaptic function, causing early neurotoxicity in HD. Additionally, persistence of unresolved DNA damage in neuronal genome may also result in chronic activation of the DNA damage-response (DDR) ATM-p53-dependent pro-apoptotic signaling pathways facilitating premature demise of neurons in HD. Moreover, mHTT also can inactivate ATXN3’s deubiquitinating activity, facilitating poly-ubiquitination and subsequent degradation of cyclic AMP-response element-binding protein (CBP), impairing CBP-CREB-regulated gene transcription, further amplifying the pro-degenerative output in the HD neurons.
A large number of studies have demonstrated that both wild-type HTT as well as mHTT carrying extended polyQ are present within the nuclei, and interact with several nuclear transcription factors and cofactors (Huang et al., 1998; McCampbell et al., 2000; Shimohata et al., 2000b; Steffan et al., 2000; Nucifora et al., 2001; Suhr et al., 2001; Dunah et al., 2002; Bae et al., 2005; Zhai et al., 2005; Cha, 2007; Benn et al., 2008; Ross and Tabrizi, 2011; Valor, 2015; Jimenez-Sanchez et al., 2017). Interactions of mHTT with the nuclear transcription factors have been implicated in extensive transcription dysregulation observed in HD (Ross and Tabrizi, 2011; Valor, 2015; Jimenez-Sanchez et al., 2017). In addition to these transcription factors, high mobility group proteins B1 and B2 (HMGB1/2) were also found to interact with the mutant form of HTT and ataxin-1, another polyQ-containing protein (Qi et al., 2007). HMGB proteins are evolutionarily conserved non-histone chromatin-associated proteins that play key roles in maintaining nuclear homeostasis. However, the function of HMGBs in the nuclei of brain cells is poorly understood. Interestingly, HMGB proteins are significantly reduced in the nuclear region outside of inclusion bodies in the affected neurons in HD and spinocerebellar ataxia type 1 (SCA1) (Qi et al., 2007). Furthermore, expression of HMGB proteins was found to significantly ameliorate polyQ-induced pathology in primary neurons as well as in Drosophila models of polyQ diseases (Qi et al., 2007), suggesting that HMGB proteins may be critical regulators of polyQ-mediated neurotoxicity. Furthermore, it was also demonstrated that reduction of nuclear HMGB protein level in the nucleus strongly correlated with DSB-mediated neuronal damage in HD (Qi et al., 2007). Moreover, HMGB1 protein was found to localize to neuronal nuclei, and its protein levels changed in various brain regions in an age-dependent manner (Enokido et al., 2008). Intriguingly, reduced expression of HMGB proteins correlated with the increased incidence of DSBs in neurons as compared with the astrocytes. These findings indicate that HMGB expression levels during aging might be an important indicator for DNA DSBs in neurons (Figure 1).
Finally, while there is overwhelming evidence that suggests that DNA repair mechanism(s) fail in HD and many other microsatellite repeat expansion related neurodegenerative disorders, there is some evidence in R6/1 mouse model of HD where it was shown that DNA repair system per se is not affected (Kovtun et al., 2007). These results while intriguing, need further evaluation in other models of HD and other related diseases.
Emerging evidence suggest that the mHTT transcripts carrying the expanded CAG RNA sequences can also contribute to genotoxicity in HD (Bañez-Coronel et al., 2012; Martí, 2016; Peng et al., 2021). A recent study by Peng et al. (2021) has raised the possibility that small RNA molecules encoding CAG sequences, generated from the cleavage of the mHTT transcripts, can contribute to DNA damage in HD. In this study, the expression of the nudix hydrolase 16 (NUDT16) gene was found to be downregulated in CAG RNA expressing cells and in mouse model of HD (Peng et al., 2021). Amongst its many functions, NUDT16 is an RNA de-capping enzyme that catalyzes the cleavage of the cap structure of small nucleolar RNAs (snoRNAs) and mRNAs. It also has a diphosphatase activity that removes m7G and/or m227G caps from U8 snoRNA and leaves a 5′monophosphate on the RNA (Iyama et al., 2010; Trésaugues et al., 2015). Importantly, Peng et al. (2021) have shown that loss of NUDT16 function in cell and animal model of HD results in misincorporation of damaging nucleotides into DNAs leading to DNA damage accumulation. These findings suggest that small CAG RNAs can hybridize with CUG-containing NUDT16 transcripts and form a CAG-CUG RNA heteroduplex, resulting in silencing of NUDT16 gene, which may lead to the DNA damage accumulation and neuronal apoptosis in HD (Peng et al., 2021).
A recent study highlighting the importance of DNA damage in neurodegenerative disorders has shown that either genetic or pharmacological ablation of DDR kinase ATM activity significantly ameliorates neurotoxicity in HD animal models (Lu et al., 2014). This report strongly supports the emerging view that persistent accumulation of unresolved DNA damage in neuronal genome and the subsequent chronic activation of the DDR ATM signaling pathway probably is one of the major contributing factors in HD pathogenic mechanism(s). Our recent publication also supports this idea because the presence of mHTT or mutant ataxin-3 in the transcription-linked DNA repair complex dramatically impairs its DNA repair activities, resulting in DNA damage/strand break accumulation and chronic activation of the DDR pathway in HD (Gao et al., 2019) as well as in SCA3 (Gao et al., 2015) respectively. Furthermore, our study suggests that HTT may help stimulate TCR complex activity and DNA repair, while mHTT reduces DNA repair activity thereby, impairing DNA damage repair and enhancing the DNA strand breaks in genome.
Persistent and cumulative accumulation of DNA damage/strand breaks can result in chronic activation of the DDR serine-threonine kinase ATM as well as DNA-PK, which in turn phosphorylate the tumor suppressor protein p53 (Kurz and Lees-Miller, 2004; Guo et al., 2010). The transcription factor p53 is the primary target of DDR pathway, and many of the functions of ATM/DNA-PK are p53-dependent and activated p53 regulates a wide variety of cellular processes such as transcription, cell-cycle regulation, DDR and DNA repair, and cell death (Miller et al., 2000; Kurz and Lees-Miller, 2004; Culmsee and Mattson, 2005; Chang et al., 2012). Aberrant activation of p53 pathway has also been reported in several other polyQ-expansion-associated hereditary ataxias including HD (Bae et al., 2005; Wang et al., 2010; Chou et al., 2011). The activated p53 (phosphorylated p53) can transactivate expression of many proapoptotic genes such as BAX, Bcl2L2 (encoding BIM), PMA1P1 (encoding NOXA), BBC3 (encoding PUMA), triggering apoptotic pathways (Oda et al., 2000; Nakano and Vousden, 2001; Shibue et al., 2003; Chipuk et al., 2004, 2005; Cregan et al., 2004; Kuwana et al., 2005; Gao et al., 2015). Persistence of unrepaired DNA damage in genome thus can facilitate p53-depndent activation of pro-degenerative pathways in HD as described earlier (Bae et al., 2005). In response to DNA strand breaks, ATM directly phosphorylates p53, which in turn activates transcription of many pro-apoptotic genes (Oda et al., 2000; Chipuk et al., 2004). Expression of mHTT has been shown to increase p53 target gene expression, whereas deleting p53 in the HD transgenic mouse brain rescues the behavioral abnormalities (Bae et al., 2005) highlighting the fact that mHTT-mediated chronic activation of DDR-p53 pathway contributes to neurotoxicity in HD. These data indicate that HTT is important in DNA strand break repair, whereas mHTT impairs DNA repair activity, resulting in DNA strand break/damage accumulation, and cumulative accumulation of DNA damage results in chronic activation of ATM-dependent p53 signaling pathway to triggers neurotoxicity in HD.
Furthermore, persistence of DNA damage can also trigger pro-apoptotic signaling in another ATM and/or DNA-PK-dependent but p53-independent pathway in HD. There is ample evidence suggesting that persistent accumulation of DNA damages and subsequent phosphorylation-dependent chronic activation of the DDR kinase ATM and/or DNA-PK can also result in the phosphorylation of another downstream tyrosine kinase c-Abl (encoded by the mammalian homolog of the v-Abl oncogene from the Abelson murine leukemia virus) (Baskaran et al., 1997; Kharbanda et al., 1997; Shafman et al., 1997). Experimental evidence suggests that the activated c-Abl kinase (phosphorylated form of c-Abl) can constitutively associates with the protein kinase C delta (PKCδ), resulting in phosphorylation and subsequent nuclear translocation of PKCδ (Yuan et al., 1998; Yoshida, 2008; Adwan et al., 2011). Several studies have shown that cytosolic retention of PKCδ is required for cell survival and function while its phosphorylation and subsequent nuclear translocation can activate the apoptotic pathways (Basu et al., 2001; Yoshida, 2008; Adwan et al., 2011). However, it remains to be tested whether chronic activation of the DDR pathways and aberrant activation of the c-Abl→PKCδ pathway contributes to degeneration of neurons in HD. The presence of unrepaired DNA damages in neuronal genome and subsequent chronic activation of the DNA damage sensor ATM and/or DNA-PK kinases thus can concomitantly activate several pro-degenerative signaling pathways in neurons, and presence of chronic but low-grade DDR pathways in neurons can lead to neuronal dysfunction, and finally their premature demise in HD (Figure 4).
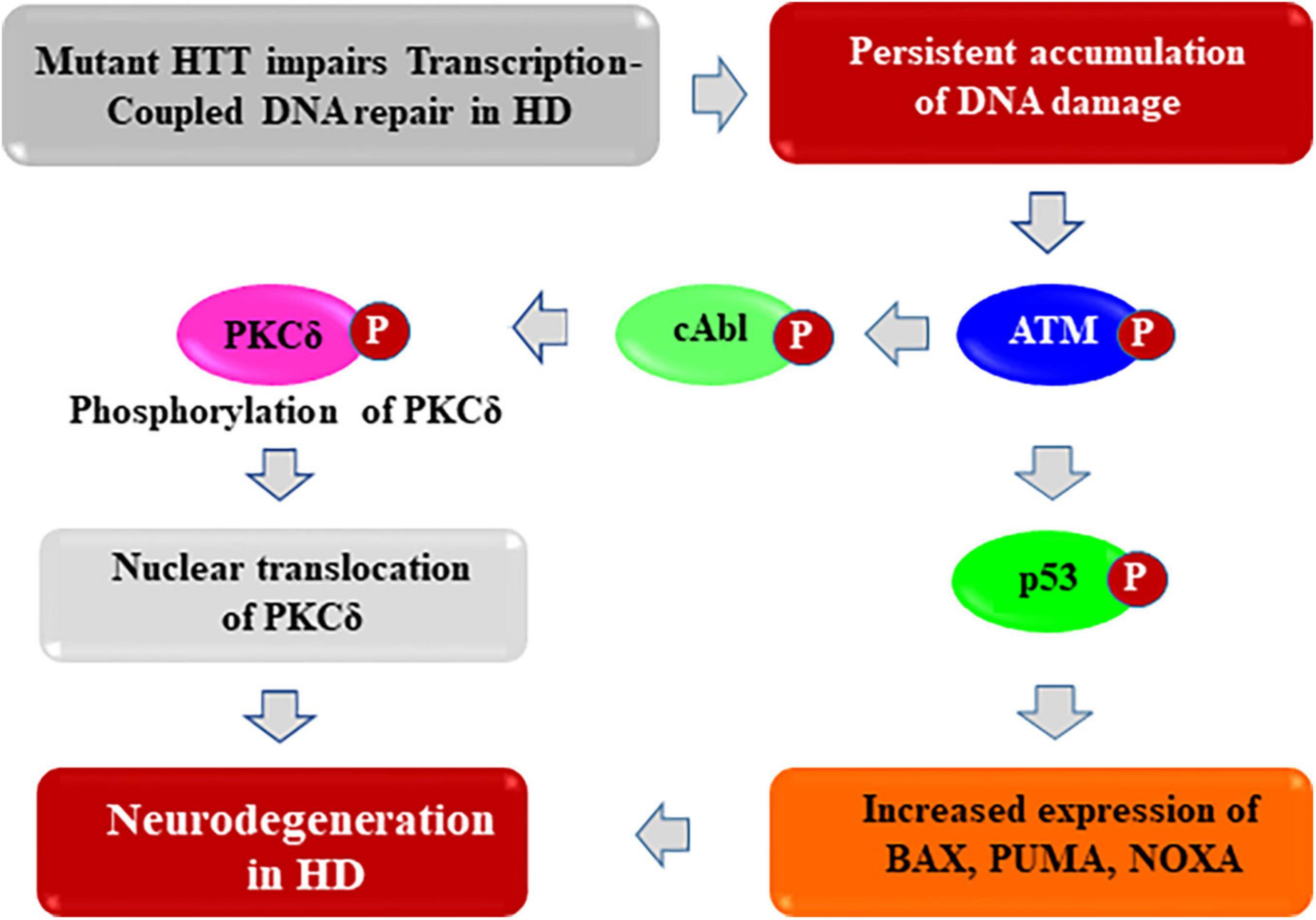
Figure 4. Proposed mechanism by which mHTT activates pro-apoptotic signaling to trigger neurotoxicity in HD. The huntingtin (HTT) protein probably acts as a structural scaffold to assemble a novel macromolecular transcription-coupled DNA repair (TCR) complex with RNA polymerase large subunit, various transcription factors and cofactors, and DNA repair enzymes including DNA ligase 3 (LIG 3) and DNA strand break repair enzyme PNKP (polynucleotide kinase 3′-phosphatase). This novel multi-protein complex senses the DNA strand breaks/lesions during transcription elongation and orchestrates their faithful repair to strictly maintain genome integrity. By contrast, the presence of mHTT in the TCR complex impairs its activity resulting damage accumulation in neuronal genome. This results in enhanced phosphorylation of DNA damage-response serine-threonine kinase ATM, and DNA-dependent protein kinase (DNA-PK), leading to chronic activation of ATM and/or DNA-PK-dependent p53 and c-Abl kinase signaling cascade in HD. This trigger pro-apoptotic signaling pathways by enhancing expression of various p53 target genes such as BAX, PUMA, and NOXA in HD. In parallel, activated ATM/DNA-PK can also phosphorylate the c-Abl kinase, which in turn phosphorylate PKCδ, facilitating nuclear translocation of PKCδ. Nuclear translocation of PKCδ may further amplify the pro-apoptotic signal in HD, ultimately leading to neuronal degeneration and development of complex neurological phenotypes in HD.
In response to DNA damage, several DDR kinases e.g., ATM, ATR, DNA-PK, p53, chk1 and 2 (checkpoint kinase 1 and 2), etc. are phosphorylated, and the phosphorylated form of these effector DDR kinases coordinately regulate DNA repair, neuronal survival, and their fate (Kurz and Lees-Miller, 2004; Guo et al., 2010). The important question that needs vigorous investigation is whether blocking ATM and/or DNA-PK with small molecule inhibitors can block neurodegeneration in HD. Therefore, significant efforts are being made to develop therapeutic approaches to block ATM or DNA-PK kinases (Lu et al., 2014). It is important to note that the DDR kinase also play important role in mediating DNA repair processes in the neurons by triggering phosphorylation-dependent activation of DNA damage repair proteins such as BRCA1, p53, PNKP, Ku70, ligase IV, etc. Therefore, one must consider the possibility that therapeutic approaches for HD aiming to block the DDR kinases with small molecule inhibitors may lead to impaired DNA repair in neurons, resulting in adverse consequences. Therefore, understanding the detailed molecular mechanism of DNA repair in neurons, and how polyQ expansion in HTT causes neurotoxicity affects these processes will provide more rational and targeted therapeutic strategies.
An important unanswered question is how mHTT impacts specific neuronal populations in HD despite its ubiquitous expression in the brain cells. Why do different neurons and/or brain regions show variable sensitivity to genotoxic insults? Neither the transcriptional signatures nor the activity patterns of any two classes of neurons are likely to be identical; therefore, the regions of robust and high-fidelity DNA repair, as well as those that are poorly maintained, likely vary from neuron to neuron. Although our findings suggest that the efficiency of TCR predominantly impacts the cortex and striatum with little effect on the cerebellar DNA, in HD, they do not explain why mHTT predominantly impairs TCR in specific brain regions in HD. How might inactivation of a potentially key DNA repair mechanism like TCR differentially affect various brain regions in HD? One possibility is that several factors specifically regulate the activity of this complex in various brain regions, particularly under genotoxic stress conditions. Inactivation of these region-specific regulators could potentially be impacted differentially causing the difference in the levels of DNA damage in different brain regions. Alternatively, specific brain regions might be differentially vulnerable to impaired TCR complex function. Therefore, complete, and detailed elucidation of the composition of this unique transcription-linked DNA repair complex, particularly in different brain regions and understanding how its activity is regulated by these factors may clarify the selective vulnerabilities of neurons in the HD brain.
While it is often argued that ineffective DNA repair and/or cumulative accumulation of unresolved DNA lesion/damages in neuronal genome are the consequences, and not the causes of neuronal dysfunction, toxicity and degeneration, we diverge and hypothesize that in fact, impaired or defective DNA repair might be an early pathology in not only in HD but also in several other terminal neurodegenerative diseases like ALS (Konopka and Atkin, 2018; Mitra et al., 2019; Konopka et al., 2020), AD (Shackelford, 2006; Shanbhag et al., 2019), PD (Milanese et al., 2018; Schaser et al., 2019; Martín-Jiménez et al., 2020; Gonzalez-Hunt and Sanders, 2021), SCA7 (Niss et al., 2021), and SCA3 (Chatterjee et al., 2015; Gao et al., 2015; Chakraborty et al., 2020). It is possible that once DNA damage occurs due to faulty or impaired repair system, the presence of highly damaging and potentially lethal lesions may become irreversible in adult post-mitotic neurons, and no interventions will rescue the damages or phenotype after that point. However, our data argue against this point. Our goal must therefore be to prevent neurons from reaching that critical tipping point and identify therapeutic targets within this window of opportunity to stop or reverse the degeneration for diseases like HD.
Studies showing that brain-specific knockout of HTT in postnatal mouse brain triggers neurodegeneration (O’Kusky et al., 1999; Dragatsis et al., 2000), suggesting that HTT plays important roles in neuronal development and survival. On the other hand, immortalized cell lines or primary neurons overexpressing HTT are resistant to degeneration following exposure to degenerative stimuli including expression of mHTT, and transgenic mice overexpressing HTT show tremendous resistance to degeneration when exposed to excitotoxic and ischemic injury (Rigamonti et al., 2000; Ho et al., 2001; Zhang et al., 2003, 2006; Leavitt et al., 2006). Although these reports suggest that increased HTT levels dramatically improve neuronal resistance to degeneration, the precise mechanism by which this protein provides neuroprotection remains poorly understood. Emerging evidence, including our studies, suggest that HTT plays a critical role in DNA strand break repair possibly by assembling the macromolecular TCR complex. Based on our recent data we hypothesize that this complex “senses” DNA damage/lesions in the template DNA and orchestrates their repair during transcription to maintain sequence integrity of neuronal genome. On the other hand, mHTT-mediated loss of DNA repair and deubiquitinating activities might be critical proximal events that impair DNA repair as well as transcription in HD. This mechanism would provide a mechanistic link between transcriptional dysregulation, cumulative DNA damage accumulations and inappropriate and chronic activation of the ATM/DNA-PK-dependent pro-apoptotic pathways, which trigger early neurotoxicity in HD. While the final biological output triggered by impaired TCR and persistence of unrepaired DNA damages in HD remains to be understood, our study points to a possible TCR mechanism that may be disrupted by mHTT in HD (Gao et al., 2019). The molecular strategies that interfere with the interaction of mHTT with the TCR complex might help slow neurotoxicity and the functional decline in HD. Alternatively, molecular approaches to stimulate PNKP activity for efficacious DNA repair may be another way to combat transcriptional dysregulation in HD. Collectively, the various studies on DNA repair deficiency in HD (Qi et al., 2007; Enokido et al., 2010; Tamura et al., 2011) and our recent findings (Gao et al., 2019) may help clarify how mHTT compromises genome integrity and neuronal function in HD.
SP, RG, NZ, and KB contributed to the experiments described in this review article. PS and YW wrote and edited the manuscript. All authors read and contributed to the editing of the manuscript and approved the submission of all versions of the review article.
This research was supported by the National Institutes of Health grants RO1 NSO79541-01 to PS and R01 EY026089-01A1 to PS, and Hereditary Disease Foundation grant to PS; National Institute of health grant R56 NS105681 to YW, and Alzheimer’s Association grant to YW.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adwan, T. S., Ohm, A. M., Jones, D. N. M., Humphries, M. J., and Reyland, M. E. (2011). Regulated binding of importin-α to protein kinase Cδ in response to apoptotic signals facilitates nuclear import. J. Biol. Chem. 286, 35716–35724. doi: 10.1074/jbc.M111.255950
Askeland, G., Dosoudilova, Z., Rodinova, M., Klempir, J., Liskova, I., Kuśnierczyk, A., et al. (2018). Increased nuclear DNA damage precedes mitochondrial dysfunction in peripheral blood mononuclear cells from Huntington’s disease patients. Sci. Rep. 8:9817. doi: 10.1038/s41598-018-27985-y
Ayala-Peña, S. (2013). Role of oxidative DNA damage in mitochondrial dysfunction and Huntington’s disease pathogenesis. Free Radic. Biol. Med. 62, 102–110. doi: 10.1016/j.freeradbiomed.2013.04.017
Bae, B. I., Xu, H., Igarashi, S., Fujimuro, M., Agrawal, N., Taya, Y., et al. (2005). p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 47, 29–41. doi: 10.1016/j.neuron.2005.06.005
Baig, S. S., Strong, M., and Quarrell, O. W. (2016). The global prevalence of Huntington’s disease: a systematic review and discussion. Neurodegener. Dis. Manag. 6, 331–343. doi: 10.2217/nmt-2016-0008
Bañez-Coronel, M., Porta, S., Kagerbauer, B., Mateu-Huertas, E., Pantano, L., Ferrer, I., et al. (2012). A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. 8:e1002481. doi: 10.1371/journal.pgen.1002481
Barclay, S. S., Tamura, T., Ito, H., Fujita, K., Tagawa, K., Shimamura, T., et al. (2014). Systems biology analysis of Drosophila in vivo screen data elucidates core networks for DNA damage repair in SCA1. Hum. Mol. Genet. 23, 1345–1364. doi: 10.1093/hmg/ddt524
Barron, J. C., Hurley, E. P., and Parsons, M. P. (2021). Huntingtin and the Synapse. Front. Cell Neurosci. 15:689332. doi: 10.3389/fncel.2021.689332
Baskaran, R., Wood, L. D., Whitaker, L. L., Canman, C. E., Morgan, S. E., Xu, Y., et al. (1997). Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature 387, 516–519. doi: 10.1038/387516a0
Basu, A., Woolard, M. D., and Johnson, C. L. (2001). Involvement of protein kinase C-delta in DNA damage-induced apoptosis. Cell Death Differ. 8, 899–908. doi: 10.1038/sj.cdd.4400885
Bauer, P. O., and Nukina, N. (2009). The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J. Neurochem. 110, 1737–1765. doi: 10.1111/j.1471-4159.2009.06302.x
Benn, C. L., Landles, C., Li, H., Strand, A. D., Woodman, B., Sathasivam, K., et al. (2005). Contribution of nuclear and extranuclear polyQ to neurological phenotypes in mouse models of Huntington’s disease. Hum. Mol. Genet. 14, 3065–3078. doi: 10.1093/hmg/ddi340
Benn, C. L., Sun, T., Sadri-Vakili, G., McFarland, K. N., DiRocco, D. P., Yohrling, G. J., et al. (2008). Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J. Neurosci. 28, 10720–10733. doi: 10.1523/JNEUROSCI.2126-08.2008
Berrios, G. E., Wagle, A. C., Marková, I. S., Wagle, S. A., Rosser, A., and Hodges, J. R. (2002). Psychiatric symptoms in neurologically asymptomatic Huntington’s disease gene carriers: a comparison with gene negative at risk subjects. Acta Psychiatr. Scand. 105, 224–230. doi: 10.1034/j.1600-0447.2002.0o456.x
Bertoni, A., Giuliano, P., Galgani, M., Rotoli, D., Ulianich, L., Adornetto, A., et al. (2011). Early and late events induced by polyQ-expanded proteins: identification of a common pathogenic property of polYQ-expanded proteins. J. Biol. Chem. 286, 4727–4741. doi: 10.1074/jbc.M110.156521
Bogdanov, M. B., Andreassen, O. A., Dedeoglu, A., Ferrante, R. J., and Beal, M. F. (2001). Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J. Neurochem. 79, 1246–1249. doi: 10.1046/j.1471-4159.2001.00689.x
Browne, S. E., Bowling, A. C., MacGarvey, U., Baik, M. J., Berger, S. C., Muqit, M. M., et al. (1997). Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann. Neurol. 41, 646–653. doi: 10.1002/ana.410410514
Canals, J. M., Pineda, J. R., Torres-Peraza, J. F., Bosch, M., Martín-Ibañez, R., Muñoz, M. T., et al. (2004). Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J Neurosci. 24, 7727–7739. doi: 10.1523/JNEUROSCI.1197-04.2004
Caviston, J. P., and Holzbaur, E. L. (2009). Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 19, 147–155. doi: 10.1016/j.tcb.2009.01.005
Cha, J. H. (2007). Transcriptional signatures in Huntington’s disease. Prog. Neurobiol. 83, 228–248. doi: 10.1016/j.pneurobio.2007.03.004
Chakraborty, A., Tapryal, N., Venkova, T., Horikoshi, N., Pandita, R. K., Sarker, A. H., et al. (2016). Classical non-homologous end-joining pathway utilizes nascent RNA for error-free double-strand break repair of transcribed genes. Nat. Commun. 7:13049. doi: 10.1038/ncomms13049
Chakraborty, A., Tapryal, N., Venkova, T., Mitra, J., Vasquez, V., Sarker, A. H., et al. (2020). Deficiency in classical nonhomologous end-joining-mediated repair of transcribed genes is linked to SCA3 pathogenesis. Proc. Natl. Acad. Sci. USA. 117, 8154–8165. doi: 10.1073/pnas.1917280117
Chang, D. T., Rintoul, G. L., Pandipati, S., and Reynolds, I. J. (2006). Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol. Dis. 22, 388–400. doi: 10.1016/j.nbd.2005.12.007
Chang, J. R., Ghafouri, M., Mukerjee, R., Bagashev, A., Chabrashvili, T., and Sawaya, B. E. (2012). Role of p53 in neurodegenerative diseases. Neurodegener. Dis. 9, 68–80. doi: 10.1159/000329999
Chatterjee, A., Saha, S., Chakraborty, A., Silva-Fernandes, A., Mandal, S. M., Neves-Carvalho, A., et al. (2015). The role of the mammalian DNA end-processing enzyme polynucleotide kinase 3’-phosphatase in spinocerebellar ataxia type 3 pathogenesis. PLoS Genet. 11:e1004749. doi: 10.1371/journal.pgen.1004749
Chipuk, J. E., Bouchier-Hayes, L., Kuwana, T., Newmeyer, D. D., and Green, D. R. (2005). PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 309, 1732–1735. doi: 10.1126/science.1114297
Chipuk, J. E., Kuwana, T., Bouchier-Hayes, L., Droin, N. M., Newmeyer, D. D., Schuler, M., et al. (2004). Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303, 1010–1014. doi: 10.1126/science.1092734
Chou, A. H., Lin, A. C., Hong, K. Y., Hu, S. H., Chen, Y. L., Chen, J. Y., et al. (2011). p53 activation mediates polyglutamine-expanded ataxin-3 upregulation of Bax expression in cerebellar and pontine nuclei neurons. Neurochem. Int. 58, 145–152. doi: 10.1016/j.neuint.2010.11.005
Cregan, S. P., Arbour, N. A., Maclaurin, J. G., Callaghan, S. M., Fortin, A., Cheung, E. C., et al. (2004). p53 activation domain 1 is essential for PUMA upregulation and p53-mediated neuronal cell death. J. Neurosci. 24, 10003–10012. doi: 10.1523/JNEUROSCI.2114-04.2004
Cui, L., Jeong, H., Borovecki, F., Parkhurst, C. N., Tanese, N., and Krainc, D. (2006). Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127, 59–69. doi: 10.1016/j.cell.2006.09.015
Culmsee, C., and Mattson, M. P. (2005). p53 in neuronal apoptosis. Biochem. Biophys. Res Commun. 331, 761–777. doi: 10.1016/j.bbrc.2005.03.149
Davies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp, A. H., Ross, C. A., et al. (1997). Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548. doi: 10.1016/s0092-8674(00)80513-9
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
DiFiglia, M., Sapp, E., Chase, K., Schwarz, C., Meloni, A., Young, C., et al. (1995). Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 14, 1075–1081. doi: 10.1016/0896-6273(95)90346-1
Dragatsis, I., Levine, M. S., and Zeitlin, S. (2000). Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 26, 300–306. doi: 10.1038/81593
Dunah, A. W., Jeong, H., Griffin, A., Kim, Y. M., Standaert, D. G., Hersch, S. M., et al. (2002). Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science 296, 2238–2243. doi: 10.1126/science.1072613
Duyao, M. P., Auerbach, A. B., Ryan, A., Persichetti, F., Barnes, G. T., McNeil, S. M., et al. (1995). Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 269, 407–410. doi: 10.1126/science.7618107
Edwardson, J. M., Wang, C. T., Gong, B., Wyttenbach, A., Bai, J., Jackson, M. B., et al. (2003). Expression of mutant Huntingtin blocks exocytosis in PC12 cells by depletion of complexin II. J. Biol Chem. 278, 30849–30853. doi: 10.1074/jbc.M304615200
Engelender, S., Sharp, A. H., Colomer, V., Tokito, M. K., Lanahan, A., Worley, P., et al. (1997). Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum. Mol. Genet. 6, 2205–2212. doi: 10.1093/hmg/6.13.2205
Engqvist-Goldstein, A. E., Kessels, M. M., Chopra, V. S., Hayden, M. R., and Drubin, D. G. (1999). An actin-binding protein of the Sla2/Huntingtin interacting protein 1 family is a novel component of clathrin-coated pits and vesicles. J. Cell Biol. 147, 1503–1518. doi: 10.1083/jcb.147.7.1503
Enokido, Y., Tamura, T., Ito, H., Arumughan, A., Komuro, A., Shiwaku, H., et al. (2010). Mutant Huntingtin impairs Ku70-mediated DNA repair. J. Cell Biol. 189, 425–443. doi: 10.1083/jcb.200905138
Enokido, Y., Yoshitake, A., Ito, H., and Okazawa, H. (2008). Age-dependent change of HMGB1 and DNA double-strand break accumulation in mouse brain. Biochem. Biophys. Res. Commun. 376, 128–133. doi: 10.1016/j.bbrc.2008.08.108
Gao, R., Chakraborty, A., Geater, C., Pradhan, S., Gordon, K. L., Snowden, J., et al. (2019). Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. Elife 8:e42988. doi: 10.7554/eLife.42988
Gao, R., Liu, Y., Silva-Fernandes, A., Fang, X., Paulucci-Holthauzen, A., Chatterjee, A., et al. (2015). Inactivation of PNKP by mutant ATXN3 triggers apoptosis by activating the DNA damage-response pathway in SCA3. PLoS Genet. 11:e1004834. doi: 10.1371/journal.pgen.1004834
Gauthier, L. R., Charrin, B. C., Borrell-Pagès, M., Dompierre, J. P., Rangone, H., Cordelières, F. P., et al. (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118, 127–138. doi: 10.1016/j.cell.2004.06.018
Geevasinga, N., Richards, F. H., Jones, K. J., and Ryan, M. M. (2006). Juvenile Huntington disease. J. Paediatr. Child Health 42, 552–554. doi: 10.1111/j.1440-1754.2006.00921.x
Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium (2015). Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162, 516–526. doi: 10.1016/j.cell.2015.07.003
Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium (2019). CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell 178, 887–900.e14. doi: 10.1016/j.cell.2019.06.036
Gill, S. R., Schroer, T. A., Szilak, I., Steuer, E. R., Sheetz, M. P., and Cleveland, D. W. (1991). Dynactin, a conserved, ubiquitously expressed component of an activator of vesicle motility mediated by cytoplasmic dynein. J. Cell Biol. 115, 1639–1650. doi: 10.1083/jcb.115.6.1639
Giuliano, P., De Cristofaro, T., Affaitati, A., Pizzulo, G. M., Feliciello, A., Criscuolo, C., et al. (2003). DNA damage induced by polyglutamine-expanded proteins. Hum. Mol. Genet. 12, 2301–2309. doi: 10.1093/hmg/ddg242
Gonzalez-Hunt, C. P., and Sanders, L. H. (2021). DNA damage and repair in Parkinson’s disease: recent advances and new opportunities. J. Neurosci. Res. 99, 180–189. doi: 10.1002/jnr.24592
Guo, Z., Kozlov, S., Lavin, M. F., Person, M. D., and Paull, T. T. (2010). ATM activation by oxidative stress. Science 330, 517–521. doi: 10.1126/science.1192912
Hahn-Barma, V., Deweer, B., Dürr, A., Dodé, C., Feingold, J., Pillon, B., et al. (1998). Are cognitive changes the first symptoms of Huntington’s disease? A study of gene carriers. J. Neurol. Neurosurg. Psychiatry. 64, 172–177. doi: 10.1136/jnnp.64.2.172
Hanawalt, P. C. (1994). Transcription-coupled repair and human disease. Science 266, 1957–1958. doi: 10.1126/science.7801121
Hanawalt, P. C., and Spivak, G. (2008). Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 9, 958–970. doi: 10.1038/nrm2549
Ho, L. W., Brown, R., Maxwell, M., Wyttenbach, A., and Rubinsztein, D. C. (2001). Wild type Huntingtin reduces the cellular toxicity of mutant Huntingtin in mammalian cell models of Huntington’s disease. J. Med. Genet. 38, 450–452. doi: 10.1136/jmg.38.7.450
Hoffner, G., Souès, S., and Djian, P. (2007). Aggregation of expanded Huntingtin in the brains of patients with Huntington disease. Prion 1, 26–31. doi: 10.4161/pri.1.1.4056
Horton, T. M., Graham, B. H., Corral-Debrinski, M., Shoffner, J. M., Kaufman, A. E., Beal, M. F., et al. (1995). Marked increase in mitochondrial DNA deletion levels in the cerebral cortex of Huntington’s disease patients. Neurology 45, 1879–1883. doi: 10.1212/wnl.45.10.1879
Huang, C. C., Faber, P. W., Persichetti, F., Mittal, V., Vonsattel, J. P., MacDonald, M. E., et al. (1998). Amyloid formation by mutant Huntingtin: threshold, progressivity and recruitment of normal polyglutamine proteins. Somat. Cell Mol. Genet. 24, 217–233. doi: 10.1023/b:scam.0000007124.19463.e5
Huang, Y., Myers, S. J., and Dingledine, R. (1999). Transcriptional repression by REST: recruitment of Sin3A and histone deacetylase to neuronal genes. Nat. Neurosci. 2, 867–872. doi: 10.1038/13165
Illuzzi, J., Yerkes, S., Parekh-Olmedo, H., and Kmiec, E. B. (2009). DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant Huntingtin aggregates. J. Neurosci. Res. 87, 733–747. doi: 10.1002/jnr.21881
Iyama, T., Abolhassani, N., Tsuchimoto, D., Nonaka, M., and Nakabeppu, Y. (2010). NUDT16 is a (deoxy)inosine diphosphatase, and its deficiency induces accumulation of single strand breaks in nuclear DNA and growth arrest. Nucleic Acids Res. 38, 4834–4843. doi: 10.1093/nar/gkq249
Jana, N. R., Tanaka, M., Wang, G. H., and Nukina, N. (2000). Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal Huntingtin: their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet. 9, 2009–2018. doi: 10.1093/hmg/9.13.2009
Jason, G. W., Pajurkova, E. M., Suchowersky, O., Hewitt, J., Hilbert, C., Reed, J., et al. (1988). Presymptomatic neuropsychological impairment in Huntington’s disease. Arch. Neurol. 45, 769–773. doi: 10.1001/archneur.1988.00520310079021
Jimenez-Sanchez, M., Licitra, F., Underwood, B. R., and Rubinsztein, D. C. (2017). Huntington’s disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb. Perspect. Med. 7:a024240. doi: 10.1101/cshperspect
Johnson, R., and Buckley, N. J. (2009). Gene dysregulation in Huntington’s disease: REST, microRNAs and beyond. Neuromol. Med. 11, 183–199. doi: 10.1007/s12017-009-8063-4
Johnson, R., Zuccato, C., Belyaev, N. D., Guest, D. J., Cattaneo, E., and Buckley, N. J. (2008). A microRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol. Dis. 29, 438–445. doi: 10.1016/j.nbd.2007.11.001
Jones, L., Houlden, H., and Tabrizi, S. J. (2017). DNA repair in the trinucleotide repeat disorders. Lancet Neurol. 16, 88–96. doi: 10.1016/S1474-4422(16)30350-7
Jonson, I., Ougland, R., and Larsen, E. (2013). DNA repair mechanisms in Huntington’s disease. Mol. Neurobiol. 47, 1093–1102. doi: 10.1007/s12035-013-8409-7
Joshi, P. R., Wu, N. P., André, V. M., Cummings, D. M., Cepeda, C., Joyce, J. A., et al. (2009). Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J. Neurosci. 29, 2414–2427. doi: 10.1523/JNEUROSCI.5687-08.2009
Kalchman, M. A., Koide, H. B., McCutcheon, K., Graham, R. K., Nichol, K., Nishiyama, K., et al. (1997). HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated Huntingtin in the brain. Nat. Genet. 16, 44–53. doi: 10.1038/ng0597-44
Kharbanda, S., Yuan, Z. M., Weichselbaum, R., and Kufe, D. (1997). Functional role for the c-Abl protein tyrosine kinase in the cellular response to genotoxic stress. Biochim. Biophys. Acta. 1333, O1–7. doi: 10.1016/s0304-419x(97)00020-6
Konopka, A., and Atkin, J. D. (2018). The emerging role of DNA damage in the pathogenesis of the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 19:3137. doi: 10.3390/ijms19103137
Konopka, A., Whelan, D. R., Jamali, M. S., Perri, E., Shahheydari, H., Toth, R. P., et al. (2020). Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Mol. Neurodegener. 15:51. doi: 10.1186/s13024-020-00386-4
Kovtun, I. V., Liu, Y., Bjoras, M., Klungland, A., Wilson, S. H., and McMurray, C. T. (2007). OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 447, 447–452. doi: 10.1038/nature05778
Kumar, A., Vaish, M., and Ratan, R. R. (2014). Transcriptional dysregulation in Huntington’s disease: a failure of adaptive transcriptional homeostasis. Drug Discov. Today 19, 956–962. doi: 10.1016/j.drudis.2014.03.016
Kurz, E. U., and Lees-Miller, S. P. (2004). DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst). 3, 889–900. doi: 10.1016/j.dnarep.2004.03.029
Kuwana, T., Bouchier-Hayes, L., Chipuk, J. E., Bonzon, C., Sullivan, B. A., Green, D. R., et al. (2005). BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell. 17, 525–535. doi: 10.1016/j.molcel.2005.02.003
Kwon, D. (2021). Failure of genetic therapies for Huntington’s devastates community. Nature 593:180. doi: 10.1038/d41586-021-01177-7
Lawrence, A. D., Hodges, J. R., Rosser, A. E., Kershaw, A., ffrench-Constant, C., Rubinsztein, D. C., et al. (1998). Evidence for specific cognitive deficits in preclinical Huntington’s disease. Brain 121, 1329–1341. doi: 10.1093/brain/121.7.1329
Leavitt, B. R., van Raamsdonk, J. M., Shehadeh, J., Fernandes, H., Murphy, Z., Graham, R. K., et al. (2006). Wild-type Huntingtin protects neurons from excitotoxicity. J. Neurochem. 96, 1121–1129. doi: 10.1111/j.1471-4159.2005.03605
Legendre-Guillemin, V., Metzler, M., Charbonneau, M., Gan, L., Chopra, V., Philie, J., et al. (2002). HIP1 and HIP12 display differential binding to F-actin, AP2, and clathrin. Identification of a novel interaction with clathrin light chain. J. Biol. Chem. 277, 19897–19904. doi: 10.1074/jbc.M112310200
Legleiter, J., Mitchell, E., Lotz, G. P., Sapp, E., Ng, C., DiFiglia, M., et al. (2010). Mutant Huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J. Biol. Chem. 285, 14777–14790. doi: 10.1074/jbc.M109.093708
Li, H., Wyman, T., Yu, Z. X., Li, S. H., and Li, X. J. (2003). Abnormal association of mutant Huntingtin with synaptic vesicles inhibits glutamate release. Hum. Mol. Genet. 12, 2021–2030. doi: 10.1093/hmg/ddg218
Li, J. Y., Plomann, M., and Brundin, P. (2003). Huntington’s disease: a synaptopathy? Trends Mol. Med. 9, 414–420. doi: 10.1016/j.molmed.2003.08.006
Li, S. H., Gutekunst, C. A., Hersch, S. M., and Li, X. J. (1998). Interaction of Huntingtin-associated protein with dynactin P150Glued. J. Neurosci. 18, 1261–1269. doi: 10.1523/JNEUROSCI.18-
Li, S. H., Schilling, G., Young, W. S., Li, X. J., Margolis, R. L., Stine, O. C., et al. (1993). Huntington’s disease gene (IT15) is widely expressed in human and rat tissues. Neuron. 11, 985–993. doi: 10.1016/0896-6273(93)90127-d
Li, X. J., Li, S. H., Sharp, A. H., Nucifora, F. C. Jr., Schilling, G., Lanahan, A., et al. (1995). A Huntingtin-associated protein enriched in brain with implications for pathology. Nature 378, 398–402. doi: 10.1038/378398a0
Li, Y., Chin, L. S., Levey, A. I., and Li, L. (2002). Huntingtin-associated protein 1 interacts with hepatocyte growth factor-regulated tyrosine kinase substrate and functions in endosomal trafficking. J. Biol. Chem. 277, 28212–28221. doi: 10.1074/jbc.M111612200
Lieber, M. R. (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211. doi: 10.1146/annurev.biochem.052308.093131
Lin, J., Handschin, C., and Spiegelman, B. M. (2005). Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370. doi: 10.1016/j.cmet.2005.05.004
Lin, J., Wu, H., Tarr, P. T., Zhang, C. Y., Wu, Z., Boss, O., et al. (2002). Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418, 797–801. doi: 10.1038/nature00904
Lindahl, T., and Barnes, D. E. (2000). Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 65, 127–133. doi: 10.1101/sqb.2000.65.127
Lu, X. H., Mattis, V. B., Wang, N., Al-Ramahi, I., van den Berg, N., Fratantoni, S. A., et al. (2014). Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci. Transl. Med. 6:268ra178. doi: 10.1126/scitranslmed.3010523
Luthi-Carter, R., and Cha, J. J. (2003). Mechanism of transcriptional dysregulation in Huntington’s disease. Clin. Neurosci. Res. 3, 165–177.
Madabhushi, R., Pan, L., and Tsai, L. H. (2014). DNA damage and its links to neurodegeneration. Neuron 83, 266–282. doi: 10.1016/j.neuron.2014.06.034
Maiuri, T., Mocle, A. J., Hung, C. L., Xia, J., van Roon-Mom, W. M., and Truant, R. (2017). Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum. Mol. Genet. 26, 395–406. doi: 10.1093/hmg/ddw395
Maiuri, T., Suart, C. E., Hung, C. L. K., Graham, K. J., Barba Bazan, C. A., and Truant, R. (2019). DNA Damage repair in Huntington’s disease and other neurodegenerative diseases. Neurotherapeutics 16, 948–956. doi: 10.1007/s13311-019-00768-7
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., et al. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506.
Marder, K., Zhao, H., Myers, R. H., Cudkowicz, M., Kayson, E., Kieburtz, K., et al. (2000). Rate of functional decline in Huntington’s disease. Huntington Study Group. Neurology 54, 452–458. doi: 10.1212/wnl.54.2.452
Martí, E. (2016). RNA toxicity induced by expanded CAG repeats in Huntington’s disease. Brain Pathol. 26, 779–786. doi: 10.1111/bpa.12427
Martín-Jiménez, R., Lurette, O., and Hebert-Chatelain, E. (2020). Damage in mitochondrial DNA associated with Parkinson’s Disease. DNA Cell Biol. 39, 1421–1430. doi: 10.1089/dna.2020.5398
Massey, T. H., and Jones, L. (2018). The central role of DNA damage and repair in CAG repeats diseases. Dis Model Mech. 11:dmm031930. doi: 10.1242/dmm.031930
Maynard, S., Fang, E. F., Scheibye-Knudsen, M., Croteau, D. L., and Bohr, V. A. (2015). DNA damage, DNA repair, aging, and neurodegeneration. Cold Spring Harb. Perspect. Med. 5:a025130. doi: 10.1101/cshperspect.a025130
McCampbell, A., Taylor, J. P., Taye, A. A., Robitschek, J., Li, M., Walcott, J., et al. (2000). CREB-binding protein sequestration by expanded polyglutamine. Hum. Mol. Genet. 9, 2197–2202. doi: 10.1093/hmg/9.14.2197
McKinnon, P. J. (2009). DNA repair deficiency and neurological disease. Nat. Rev. Neurosci. 10, 100–112. doi: 10.1038/nrn2559
McKinnon, P. J. (2017). Genome integrity and disease prevention in the nervous system. Genes Dev. 31, 1180–1194. doi: 10.1101/gad.301325.117
McKinnon, P. J., and Caldecott, K. W. (2007). DNA strand break repair and human genetic disease. Annu. Rev. Genomics Hum. Genet. 8, 37–55. doi: 10.1146/annurev.genom.7.080505.115648
Milanese, C., Cerri, S., Ulusoy, A., Gornati, S. V., Plat, A., Gabriels, S., et al. (2018). Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death Dis. 9:818. doi: 10.1038/s41419-018-0848-7
Miller, F. D., Pozniak, C. D., and Walsh, G. S. (2000). Neuronal life and death: an essential role for the p53 family. Cell Death Differ. 7, 880–888. doi: 10.1038/sj.cdd.4400736
Mitra, J., Guerrero, E. N., Hegde, P. M., Liachko, N. F., Wang, H., Vasquez, V., et al. (2019). Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. U. S. A. 116, 4696–4705. doi: 10.1073/pnas.1818415116
Modregger, J., DiProspero, N. A., Charles, V., Tagle, D. A., and Plomann, M. (2002). PACSIN 1 interacts with Huntingtin and is absent from synaptic varicosities in presymptomatic Huntington’s disease brains. Hum. Mol. Genet. 11, 2547–2558. doi: 10.1093/hmg/11.21.2547
Morrison, P. J. (2012). Prevalence estimates of Huntington disease in Caucasian populations are gross underestimates. Mov. Disord. 27, 1707–1708. doi: 10.1002/mds.25266
Morton, A. J., and Edwardson, J. M. (2001). Progressive depletion of complexin II in a transgenic mouse model of Huntington’s disease. J. Neurochem. 76, 166–172. doi: 10.1046/j.1471-4159.2001.00059.x
Morton, A. J., Faull, R. L., and Edwardson, J. M. (2001). Abnormalities in the synaptic vesicle fusion machinery in Huntington’s disease. Brain Res. Bull. 56, 111–117. doi: 10.1016/s0361-9230(01)00611-6
Muchowski, P. J., Schaffar, G., Sittler, A., Wanker, E. E., Hayer-Hartl, M. K., and Hartl, F. U. (2000). Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. U. S. A. 97, 7841–7846. doi: 10.1073/pnas.140202897
Nakano, K., and Vousden, K. H. (2001). PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7, 683–694. doi: 10.1016/s1097-2765(01)00214-3
Nance, M. A., and Myers, R. H. (2001). Juvenile onset Huntington’s disease–clinical and research perspectives. Ment. Retard. Dev. Disabil. Res. Rev. 7, 153–157. doi: 10.1002/mrdd.1022
Nasir, J., Floresco, S. B., O’Kusky, J. R., Diewert, V. M., Richman, J. M., Zeisler, J., et al. (1995). Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 81, 811–823. doi: 10.1016/0092-8674(95)90542-1
Niss, F., Zaidi, W., Hallberg, E., and Ström, A. L. (2021). Polyglutamine expanded Ataxin-7 induces DNA damage and alters FUS localization and function. Mol. Cell Neurosci. 110:103584. doi: 10.1016/j.mcn.2020.103584
No authors listed (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-e
Nucifora, F. C. Jr., Sasaki, M., Peters, M. F., Huang, H., Cooper, J. K., Yamada, M., et al. (2001). Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 291, 2423–2428. doi: 10.1126/science.1056784
Ochaba, J., Lukacsovich, T., Csikos, G., Zheng, S., Margulis, J., Salazar, L., et al. (2014). Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. U. S. A. 111, 16889–16894. doi: 10.1073/pnas.1420103111
Oda, E., Ohki, R., Murasawa, H., Nemoto, J., Shibue, T., Yamashita, T., et al. (2000). Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288, 1053–1058. doi: 10.1126/science.288.5468.1053
O’Kusky, J. R., Nasir, J., Cicchetti, F., Parent, A., and Hayden, M. R. (1999). Neuronal degeneration in the basal ganglia and loss of pallido-subthalamic synapses in mice with targeted disruption of the Huntington’s disease gene. Brain Res. 818, 468–479. doi: 10.1016/s0006-8993(98)01312-2
Orr, A. L., Li, S., Wang, C. E., Li, H., Wang, J., Rong, J., et al. (2008). N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 28, 2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008
Peng, S., Guo, P., Lin, X., An, Y., Sze, K. H., Lau, M. H. Y., et al. (2021). CAG RNAs induce DNA damage and apoptosis by silencing NUDT16 expression in polyglutamine degeneration. Proc. Natl. Acad. Sci. U. S. A. 118:e2022940118. doi: 10.1073/pnas.2022940118
Polidori, M. C., Mecocci, P., Browne, S. E., Senin, U., and Beal, M. F. (1999). Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci. Lett. 272, 53–56. doi: 10.1016/s0304-3940(99)00578-9
Qi, M. L., Tagawa, K., Enokido, Y., Yoshimura, N., Wada, Y., Watase, K., et al. (2007). Proteome analysis of soluble nuclear proteins reveals that HMGB1/2 suppress genotoxic stress in polyglutamine diseases. Nat. Cell Biol. 9, 402–414. doi: 10.1038/ncb1553
Rass, U., Ahel, I., and West, S. C. (2007). Defective DNA repair and neurodegenerative disease. Cell 130, 991–1004. doi: 10.1016/j.cell.2007.08.043
Rawlins, M. D., Wexler, N. S., Wexler, A. R., Tabrizi, S. J., Douglas, I., Evans, S. J., et al. (2016). The prevalence of Huntington’s disease. Neuroepidemiology 46, 144–153. doi: 10.1159/000443738
Raymond, L. A., André, V. M., Cepeda, C., Gladding, C. M., Milnerwood, A. J., and Levine, M. S. (2011). Pathophysiology of Huntington’s disease: time-dependent alterations in synaptic and receptor function. Neuroscience 198, 252–273. doi: 10.1016/j.neuroscience.2011.08.052
Rigamonti, D., Bauer, J. H., De-Fraja, C., Conti, L., Sipione, S., Sciorati, C., et al. (2000). Wildtype huntingtin protects from apoptosis upstream of caspase-3. J. Neurosci. 20, 3705–3713. doi: 10.1523/JNEUROSCI.20-10-03705.2000
Rizo, J., and Südhof, T. C. (2002). Snares and Munc18 in synaptic vesicle fusion. Nat. Rev. Neurosci. 3, 641–653. doi: 10.1038/nrn898
Rolig, R. L., and McKinnon, P. J. (2000). Linking DNA damage and neurodegeneration. Trends Neurosci. 23, 417–424. doi: 10.1016/s0166-2236(00)01625-8
Rosenblatt, A., Abbott, M. H., Gourley, L. M., Troncoso, J. C., Margolis, R. L., Brandt, J., et al. (2003). Predictors of neuropathological severity in 100 patients with Huntington’s disease. Ann. Neurol. 54, 488–493. doi: 10.1002/ana.10691
Rosenblatt, A., Liang, K. Y., Zhou, H., Abbott, M. H., Gourley, L. M., Margolis, R. L., et al. (2006). The association of CAG repeat length with clinical progression in Huntington disease. Neurology 66, 1016–1020. doi: 10.1212/01.wnl.0000204230.16619.d9
Ross, C. A., and Tabrizi, S. J. (2011). Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98. doi: 10.1016/S1474-4422(10)70245-3
Ross, C. A., and Truant, R. (2017). DNA repair: a unifying mechanism in neurodegeneration. Nature 541, 34–35. doi: 10.1038/nature21107
Ross, C. A., Margolis, R. L., Rosenblatt, A., Ranen, N. G., Becher, M. W., and Aylward, E. (1997). Huntington disease and the related disorder, dentatorubral-pallidoluysian atrophy (DRPLA). Medicine (Baltimore) 76, 305–338. doi: 10.1097/00005792-199709000-00001
Sancar, A., Lindsey-Boltz, L. A., Unsal-Kaçmaz, K., and Linn, S. (2004). Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85. doi: 10.1146/annurev.biochem.73.011303.073723
Saudou, F., and Humbert, S. (2016). The biology of Huntingtin. Neuron 89, 910–926. doi: 10.1016/j.neuron.2016.02.003
Sawada, H., Ishiguro, H., Nishii, K., Yamada, K., Tsuchida, K., Takahashi, H., et al. (2007). Characterization of neuron-specific Huntingtin aggregates in human Huntingtin knock-in mice. Neurosci. Res. 57, 559–573. doi: 10.1016/j.neures.2007.01.002
Schaser, A. J., Osterberg, V. R., Dent, S. E., Stackhouse, T. L., Wakeham, C. M., Boutros, S. W., et al. (2019). Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 9:10919. doi: 10.1038/s41598-019-47227-z
Seeberg, E., Eide, L., and Bjørås, M. (1995). The base excision repair pathway. Trends Biochem. Sci. 20, 391–397. doi: 10.1016/s0968-0004(00)89086-6
Segal-Raz, H., Mass, G., Baranes-Bachar, K., Lerenthal, Y., Wang, S. Y., Chung, Y. M., et al. (2011). ATM-mediated phosphorylation of polynucleotide kinase/phosphatase is required for effective DNA double-strand break repair. EMBO Rep. 12, 713–719. doi: 10.1038/embor.2011.96
Shackelford, D. A. (2006). DNA end joining activity is reduced in Alzheimer’s disease. Neurobiol. Aging. 27, 596–605. doi: 10.1016/j.neurobiolaging.2005.03.009
Shafman, T., Khanna, K. K., Kedar, P., Spring, K., Kozlov, S., Yen, T., et al. (1997). Interaction between ATM protein and c-Abl in response to DNA damage. Nature 387, 520–523. doi: 10.1038/387520a0
Shanbhag, N. M., Evans, M. D., Mao, W., Nana, A. L., Seeley, W. W., Adame, A., et al. (2019). Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 7:77. doi: 10.1186/s40478-019-0723-5
Sharp, A. H., Loev, S. J., Schilling, G., Li, S. H., Li, X. J., Bao, J., et al. (1995). Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron 14, 1065–1074. doi: 10.1016/0896-6273(95)90345-3
Shibue, T., Takeda, K., Oda, E., Tanaka, H., Murasawa, H., Takaoka, A., et al. (2003). Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 17, 2233–2238. doi: 10.1101/gad.1103603
Shimohata, T., Nakajima, T., Yamada, M., Uchida, C., Onodera, O., Naruse, S., et al. (2000a). Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat. Genet. 26, 29–36. doi: 10.1038/79139
Shimohata, T., Onodera, O., and Tsuji, S. (2000b). Interaction of expanded polyglutamine stretches with nuclear transcription factors leads to aberrant transcriptional regulation in polyglutamine diseases. Neuropathology 20, 326–333. doi: 10.1046/j.1440-1789.2000.00350.x
Shirendeb, U., Reddy, A. P., Manczak, M., Calkins, M. J., Mao, P., Tagle, D. A., et al. (2011). Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum. Mol. Genet. 20, 1438–1455. doi: 10.1093/hmg/ddr024
Siddiqui, A., Rivera-Sánchez, S., Castro Mdel, R., Acevedo-Torres, K., Rane, A., Torres-Ramos, C. A., et al. (2012). Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington’s disease. Free Radic. Biol. Med. 53, 1478–1488. doi: 10.1016/j.freeradbiomed.2012.06.008
Singaraja, R. R., Hadano, S., Metzler, M., Givan, S., Wellington, C. L., Warby, S., et al. (2002). HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum. Mol. Genet. 11, 2815–2828. doi: 10.1093/hmg/11.23.2815
Sittler, A., Wälter, S., Wedemeyer, N., Hasenbank, R., Scherzinger, E., Eickhoff, H., et al. (1998). SH3GL3 associates with the Huntingtin exon 1 protein and promotes the formation of polygln-containing protein aggregates. Mol. Cell 2, 427–436. doi: 10.1016/s1097-2765(00)80142-2
Smith, R., Petersén, A., Bates, G. P., Brundin, P., and Li, J. Y. (2005). Depletion of rabphilin 3A in a transgenic mouse model (R6/1) of Huntington’s disease, a possible culprit in synaptic dysfunction. Neurobiol. Dis. 20, 673–684. doi: 10.1016/j.nbd.2005.05.008
Sontag, E. M., Lotz, G. P., Yang, G., Sontag, C. J., Cummings, B. J., Glabe, C. G., et al. (2012). Detection of mutant Huntingtin aggregation conformers and modulation of SDS-soluble fibrillar oligomers by small molecules. J. Huntingtons Dis. 1, 119–132. doi: 10.3233/JHD-2012-129004
Steffan, J. S., Kazantsev, A., Spasic-Boskovic, O., Greenwald, M., Zhu, Y. Z., Gohler, H., et al. (2000). The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. U. S. A. 97, 6763–6768. doi: 10.1073/pnas.100110097
Suhr, S. T., Senut, M. C., Whitelegge, J. P., Faull, K. F., Cuizon, D. B., and Gage, F. H. (2001). Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J. Cell Biol. 153, 283–294. doi: 10.1083/jcb.153.2.283
Tabrizi, S. J., Flower, M. D., Ross, C. A., and Wild, E. J. (2020). Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 16, 529–546. doi: 10.1038/s41582-020-0389-4
Tabrizi, S. J., Ghosh, R., and Leavitt, B. R. (2019). Huntingtin lowering strategies for disease modification in Huntington’s disease. Neuron 101, 801–819. doi: 10.1016/j.neuron.2019.01.039
Tamura, T., Sone, M., Iwatsubo, T., Tagawa, K., Wanker, E. E., and Okazawa, H. (2011). Ku70 alleviates neurodegeneration in Drosophila models of Huntington’s disease. PLoS One 6:e27408. doi: 10.1371/journal.pone.0027408
Trésaugues, L., Lundbäck, T., Welin, M., Flodin, S., Nyman, T., Silvander, C., et al. (2015). Structural basis for the specificity of human NUDT16 and its regulation by inosine monophosphate. PLoS One 10:e0131507. doi: 10.1371/journal.pone.0131507
Truant, R., Atwal, R., and Burtnik, A. (2006). Hypothesis: huntingtin may function in membrane association and vesicular trafficking. Biochem. Cell Biol. 84, 912–917. doi: 10.1139/o06-181
Trushina, E., Dyer, R. B., Badger, J. D. II., Ure, D., Eide, L., Tran, D. D., et al. (2004). Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell Biol. 24, 8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004
Valor, L. M. (2015). Transcription, epigenetics and ameliorative strategies in Huntington’s disease: a genome-wide perspective. Mol. Neurobiol. 51, 406–423. doi: 10.1007/s12035-014-8715-8
Vonsattel, J. P., and DiFiglia, M. (1998). Huntington disease. J. Neuropathol. Exp. Neurol. 57, 369–384. doi: 10.1097/00005072-199805000-00001
Wang, H. L., Chou, A. H., Lin, A. C., Chen, S. Y., Weng, Y. H., and Yeh, T. H. (2010). Polyglutamine-expanded ataxin-7 upregulates Bax expression by activating p53 in cerebellar and inferior olivary neurons. Exp. Neurol. 224, 486–494. doi: 10.1016/j.expneurol.2010.05.011
Weydt, P., Pineda, V. V., Torrence, A. E., Libby, R. T., Satterfield, T. F., Lazarowski, E. R., et al. (2006). Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 4, 349–362. doi: 10.1016/j.cmet.2006.10.004
Xie, Y., Hayden, M. R., and Xu, B. (2010). BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. J. Neurosci. 30, 14708–14718. doi: 10.1523/JNEUROSCI.1637-10.2010
Yoshida, K. (2008). Nuclear trafficking of pro-apoptotic kinases in response to DNA damage. Trends Mol. Med. 14, 305–313. doi: 10.1016/j.molmed.2008.05.003
Yuan, Z. M., Utsugisawa, T., Ishiko, T., Nakada, S., Huang, Y., Kharbanda, S., et al. (1998). Activation of protein kinase C delta by the c-Abl tyrosine kinase in response to ionizing radiation. Oncogene 16, 1643–1648. doi: 10.1038/sj.onc.1201698
Yue, X., Bai, C., Xie, D., Ma, T., and Zhou, P. K. (2020). DNA-PKcs: a multi-faceted player in DNA damage response. Front. Genet. 11:607428. doi: 10.3389/fgene.2020.607428
Zeitlin, S., Liu, J. P., Chapman, D. L., Papaioannou, V. E., and Efstratiadis, A. (1995). Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 11, 155–163. doi: 10.1038/ng1095-155
Zhai, W., Jeong, H., Cui, L., Krainc, D., and Tjian, R. (2005). In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell 123, 1241–1253. doi: 10.1016/j.cell.2005.10.030
Zhang, Y., Leavitt, B. R., van Raamsdonk, J. M., Dragatsis, I., Goldowitz, D., MacDonald, M. E., et al. (2006). Huntingtin inhibits caspase-3 activation. EMBO J. 25, 5896–5906. doi: 10.1038/sj.emboj.7601445
Zhang, Y., Li, M., Drozda, M., Chen, M., Ren, S., Mejia Sanchez, R. O., et al. (2003). Depletion of wildtype huntingtin in mouse models of neurologic diseases. J. Neurochem. 87, 101–106. doi: 10.1046/j.1471-4159.2003.01980.x
Zolner, A. E., Abdou, I., Ye, R., Mani, R. S., Fanta, M., Yu, Y., et al. (2011). Phosphorylation of polynucleotide kinase/phosphatase by DNA-dependent protein kinase and ataxia-telangiectasia mutated regulates its association with sites of DNA damage. Nucleic Acids Res. 39, 9224–9237. doi: 10.1093/nar/gkr647
Zuccato, C., Belyaev, N., Conforti, P., Ooi, L., Tartari, M., Papadimou, E., et al. (2007). Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J. Neurosci. 27, 6972–6983. doi: 10.1523/JNEUROSCI.4278-06.2007
Zuccato, C., Ciammola, A., Rigamonti, D., Leavitt, B. R., Goffredo, D., Conti, L., et al. (2001). Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 293, 493–498. doi: 10.1126/science.1059581
Keywords: defective DNA damage repair, huntingtin, Huntington’s disease, polyglutamine repeat expansion, transcription-coupled DNA repair, microsatellite repeat expansion
Citation: Pradhan S, Gao R, Bush K, Zhang N, Wairkar YP and Sarkar PS (2022) Polyglutamine Expansion in Huntingtin and Mechanism of DNA Damage Repair Defects in Huntington’s Disease. Front. Cell. Neurosci. 16:837576. doi: 10.3389/fncel.2022.837576
Received: 16 December 2021; Accepted: 07 February 2022;
Published: 04 April 2022.
Edited by:
Anna Konopka, Macquarie University, AustraliaReviewed by:
Hitoshi Okazawa, Tokyo Medical and Dental University, JapanCopyright © 2022 Pradhan, Gao, Bush, Zhang, Wairkar and Sarkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Partha S. Sarkar, cHNzYXJrYXJAdXRtYi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.