 Anna Konopka
Anna Konopka Julie D. Atkin
Julie D. Atkin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 23 June 2022
Sec. Cellular Neurophysiology
Volume 16 - 2022 | https://doi.org/10.3389/fncel.2022.836885
This article is part of the Research TopicDefective DNA Damage Response – Repair Axis in Post-Mitotic Neurons in Human Health and Neurodegenerative DiseasesView all 5 articles
Damage to DNA is generally considered to be a harmful process associated with aging and aging-related disorders such as neurodegenerative diseases that involve the selective death of specific groups of neurons. However, recent studies have provided evidence that DNA damage and its subsequent repair are important processes in the physiology and normal function of neurons. Neurons are unique cells that form new neural connections throughout life by growth and re-organisation in response to various stimuli. This “plasticity” is essential for cognitive processes such as learning and memory as well as brain development, sensorial training, and recovery from brain lesions. Interestingly, recent evidence has suggested that the formation of double strand breaks (DSBs) in DNA, the most toxic form of damage, is a physiological process that modifies gene expression during normal brain activity. Together with subsequent DNA repair, this is thought to underlie neural plasticity and thus control neuronal function. Interestingly, neurodegenerative diseases such as Alzheimer’s disease, amyotrophic lateral sclerosis, frontotemporal dementia, and Huntington’s disease, manifest by a decline in cognitive functions, which are governed by plasticity. This suggests that DNA damage and DNA repair processes that normally function in neural plasticity may contribute to neurodegeneration. In this review, we summarize current understanding about the relationship between DNA damage and neural plasticity in physiological conditions, as well as in the pathophysiology of neurodegenerative diseases.
DNA damage is now widely implicated in aging and the pathophysiology of age-related neurodegenerative disorders, such as amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), Huntington’s disease (HD), and Parkinson’s disease (PD) (Madabhushi et al., 2014; Machiela and Southwell, 2020). However, emerging evidence suggests that DNA damage and DNA repair are not only induced by pathological conditions (Ju et al., 2006; Tiwari et al., 2012; Suberbielle et al., 2013; Madabhushi et al., 2015). The same processes involved in neurodegeneration as we age are also involved in fundamental physiological functions of neurons that are related to neural plasticity. Hence, DNA damage and repair are associated with neural plasticity, implying an important role for these processes in neuronal function. Furthermore, in neurodegenerative diseases the selective death of specific groups of neurons is present. This suggests that the unique properties of neurons may contribute to selective neurodegeneration in pathophysiology. There are no previous comprehensive reviews on the role of DNA damage and repair in neural plasticity in neurodegenerative disorders. A previous collection entitled “DNA Damage, Neurodegeneration, and Synaptic Plasticity” (consisting of three research articles and one review) covered only some aspects of this topic (Merlo et al., 2016). In this review, we summarize current knowledge regarding the link between neural plasticity and DNA damage in both physiological conditions and in the pathophysiology of neurodegenerative diseases.
DNA is subject to persistent assault from both endogenous and environmental sources, and specific molecular pathways detect and respond to specific types of damage (Jackson and Bartek, 2009). These mechanisms, collectively known as the “DNA damage response (DDR)”, are essential for neuronal viability, although they decline significantly during aging (Agathangelou et al., 2018). DNA can be damaged in several different ways. When the phosphodiester bonds break in one or both DNA strands, this results in single-stranded and double stranded breaks (SSBs and DSBs respectively). Whilst DSBs arise less frequently than SSBs, they are much more harmful (Chatterjee and Walker, 2017). Each type of damage requires a specific mechanism of DNA repair, although these processes can overlap (Ma and Dai, 2018).
Neurons are terminally differentiated, post-mitotic, non-replicating, long-lived cells, with high metabolic activity. Therefore, their ability to cope with DNA damage differs from mitotic or cycling cells because they need to withstand damage throughout their lifespan. Neurons rely mostly on base excision repair (BER) and nucleotide excision repair (NER) pathways to deal with SSBs, and non-homologous end joining (NHEJ) to repair DSBs (Fishel et al., 2007). Here we discuss those DNA repair mechanisms most relevant to neurons. The reader is directed to several excellent recent reviews for a comprehensive discussion of all DNA repair mechanisms1 (see DNA damage repair (Internet), 2021).
BER repairs minor base lesions that are not helix distorting, such as the formation of 8-oxo-guanine (8-oxoG) bases resulting from reactive oxygen species (ROS). Following damage, DNA glycosylases such as 8-oxoguanine DNA glycosylase (OGG1) or nei like DNA glycosylase 1 (NEIL1) remove the damaged base, leaving an abasic site. Then, a nick in the phosphodiesterase backbone is made by AP endonuclease 1 (APE1) or a bifunctional glycosylase with lyase activity, creating DNA SSBs. Poly (ADP-ribose) polymerase 1 (PARP1) then senses the SSBs, which undergo repair by short-patch or long-patch BER (Kim and Wilson, 2012).
NER repairs bulky lesions, including cyclobutane-pyrimidine dimers, 6-4 pyrimidine-pyrimidone photoproducts (6-4PPs), chemical adducts, intrastrand crosslinks, and ROS-generated cyclopurines (Horowitz et al., 2011; Gonzalez-Hunt and Sanders, 2021). These lesions are induced mostly by UV radiation and exposure to chemicals that cannot cross the blood-brain barrier. Therefore, the number of sources that can produce these lesions in the brain is limited, although dopamine neurons are vulnerable to oxidative DNA damage which can be repaired by NER (Horowitz et al., 2011; Gonzalez-Hunt and Sanders, 2021). In NER, after the detection of SSBs, an endonuclease complex cuts the damaged strand, followed by gap-filling synthesis and ligation (Chatterjee and Walker, 2017).
NHEJ is the primary DNA repair pathway for DSBs in neurons. DSBs are recognized by ataxia telangiectasia mutated kinase (ATM), which phosphorylates histone H2AX (γH2AX) over the surrounding mega-base region of DNA (Thompson and Limoli, 2000). The broken DNA ends are bound by the DNA protein kinase complex (DNA-PK), which is composed of Ku70/Ku80 heterodimers and the catalytic subunit of DNA-PK (DNA-PKcs; Ciccia and Elledge, 2010). These ends are then processed by the nuclease Artemis and DNA polymerases Polμ or Polλ (Lieber, 2010), and ligated by a DNA ligase complex, containing DNA ligase 4, X-ray cross-complementation group 4 (XRCC4), and XRCC4 like factor (XLF)/Cernunnos (Ahnesorg et al., 2006; Buck et al., 2006). However, when classical NHEJ repair is defective, DSBs can be repaired by alternative end joining repair, A-NHEJ. A subset of A-NHEJ, which relies on micro-homologous sequences on either side of the DSB, is termed microhomology-mediated end joining (MMEJ; Wang and Xu, 2017). MMEJ is an error-prone DSB repair mechanism that results in chromosome translocations and rearrangements (Wang and Xu, 2017). It begins with resection of the broken DNA ends, which facilitates exposure of the micro-homologous sequences that will be ligated. These sequences are then annealed prior to ligation to form an intermediate structure with a 3’-flap and gaps on both sides of the DSB. Subsequently, this intermediate is removed to allow DNA polymerase to fill in the gap and the broken ends are ligated by DNA ligase III/I (Wang and Xu, 2017).
As neurons are post-mitotic, and thus non-replicating, it is believed that DNA repair pathways associated with replicating cells, such as mismatch repair (MMR) and homologous recombination (HR), are absent in neurons (Fishel et al., 2007; Welty et al., 2018). MMR corrects spontaneous base-base mismatches and small insertion-deletion loops (indels) that are generated during DNA replication (Pecina-Slaus et al., 2020). HR requires undamaged DNA on the sister chromatid as a template for repair, therefore it operates mostly during the S and G2 phases of the cell cycle (Welty et al., 2018). However, recent studies have demonstrated that HR may be active in neurons because it can utilize nascent mRNA during active transcription as a template to repair DNA (Fishel et al., 2007; Welty et al., 2018), implying that this mechanism is present in non-dividing, differentiated neurons (Welty et al., 2018).
Neurons are structurally and functionally unique, with their unusual morphology and their ability to communicate electrically and chemically via intricate synaptic contacts. Synaptic plasticity refers specifically to the functional and structural alterations of synapses that modulate the strength and efficiency of communication between neurons. Neural plasticity has a broader meaning and describes the ability of the nervous system to change its activity in response to intrinsic or extrinsic stimuli, by reorganizing its structure, function, or connections. The phenomenon of neural and synaptic plasticity underlies the molecular mechanisms involved in cognitive processes such as learning and memory, but it is also important for brain development and homeostasis, sensorial training, and recovery from brain lesions (Mateos-Aparicio and Rodriguez-Moreno, 2019). Therefore, it is not surprising that disrupted plasticity leads to a decline in cognitive functions and is associated with age-related diseases, including Alzheimer’s disease, amyotrophic lateral sclerosis, Huntington’s disease and Parkinson’s disease (Phukan et al., 2007; Paulsen, 2011; Picconi et al., 2012; Zhao et al., 2014).
Neuronal electrical excitation modulates plasticity (Hogan et al., 2020) by three fundamental mechanisms: (a) long-term depression (LTD), an activity-dependent reduction in the efficacy of neuronal synapses (Ahn et al., 1999); (b) long-term potentiation (LTP), persistent strengthening of synapses that leads to a long-lasting increase in signal transmission between neurons (Kandel, 2001); and (c) activity-associated development of corticospinal circuitry for movement control (Martin, 2005). In parallel with activity-associated plasticity, structural modifications of axonal, dendritic branches, and spine morphology occurs, constituting structural synaptic plasticity (Mateos-Aparicio and Rodriguez-Moreno, 2019).
In the brain, glutamate is the major excitatory neurotransmitter, whereas gamma aminobutyric acid (GABA) is the principal inhibitory neurotransmitter. Both glutamatergic excitatory and GABAergic transmission are important molecular processes involved in synaptic plasticity and neuronal activation. Glutamate receptors mediate fast excitatory synaptic transmission in the CNS, and they regulate a broad spectrum of processes. Ionotropic glutamate receptors are named after their specific ligands: kainate, α-amino-3-hydroxy-5-methyl-isoxazole-4-propionate (AMPA), and N-methyl-D-aspartate (NMDA). The NMDA receptors (NMDARs) are crucial for activity-dependent synaptic changes and learning and memory (Baez et al., 2018). It is also increasingly recognized that NMDARs participate in dendritic synaptic integration and are critical for generating persistent activity of neural assemblies (Hunt and Castillo, 2012). GABAergic synapses, similar to glutamatergic synapses, adjust their strength depending on the pattern of neuronal activity. The plasticity of inhibitory synapses is largely mediated by modulation of the expression, localization, and function of GABA receptors (Mele et al., 2016).
Several studies have shown that neuronal activity generates DSBs in cultured neurons. Activation of ionotropic NMDA or AMPA/kainate glutamate receptors induced DNA damage in rat cortical neuronal cultures (Crowe et al., 2006). This was detected by the formation of γH2AX foci, implying that DSBs were generated by neuronal activation. Furthermore, this was associated with activation of the Mre11-dependent DNA repair pathway, which functions in DSB repair (Crowe et al., 2006). The induction of DNA damage was related to increases in intracellular calcium levels (Crowe et al., 2006), which is known to contribute to mitochondrial dysfunction and an increase in ROS (Krieger and Duchen, 2002). However, treatment with the antioxidant, Vitamin E, and intracellular calcium chelator, BAPTA-AM, did not protect neurons completely from DNA damage, suggesting that it is also generated by other sources not related to mitochondrial dysfunction and ROS (Crowe et al., 2006).
Similarly, another study showed that cerebral cortical neurons efficiently repair oxidative DNA lesions after transient activation of glutamate receptors (Yang et al., 2010). This was facilitated by BER involving DNA glycosylases OGG1 and NEIL1 and cAMP-response element-binding protein (CREB)-mediated APE1 expression (Stetler et al., 2010; Yang et al., 2010). Furthermore, glutamate also activates nuclear factor-kappa B (NF-kB) in neurons (Jiang et al., 2003), which promotes DNA repair (Wang et al., 2009). Interestingly, elevated levels of intracellular oxidative damage stimulate BER, resulting in an increase in cellular survival (Chen et al., 1998; Ramana et al., 1998; Cabelof et al., 2002). Higher concentrations of glutamate (100 μM) can be deleterious for neurons (Mattson et al., 1988). Interestingly, during learning and memory in hippocampal neurons or activation of motor system neurons during exercise, glutamate concentration in the synaptic cleft transiently increases above 100 μM and reaches millimolar values (Rao-Mirotznik et al., 1998; Choi et al., 2000). Therefore, activation of synaptic glutamate receptors may upregulate DNA repair systems, thereby increasing repair capability in neurons (Yang et al., 2011). However, with a compromised DNA repair system or increased oxidative stress, neurons become rapidly overwhelmed and prone to cell death (Yang et al., 2011).
A recent study concluded that DSBs are generated physiologically to resolve topological limitations to gene expression in neurons. Topoisomerase enzymes participate in the overwinding or underwinding of DNA and thus they manage DNA topological constraints (McKinnon, 2016). Neuronal activity produces DSBs at specific loci in vitro by topoisomerase IIβ (TopIIβ), in the promoters of early response genes (ERGs, also called immediate early genes, IEGs) that are crucial for experience-driven changes to synapses, learning, and memory (Ju et al., 2006; Tiwari et al., 2012; Madabhushi et al., 2015). Interestingly, the expression patterns of ERGs in response to neuronal stimulation correlated well with the formation and repair of activity-induced DSBs (Madabhushi et al., 2015), implying that generation of DSBs and their subsequent repair are essential steps for proper gene function. Furthermore, DSBs produced during neuronal excitation were repaired within 2 h of the initial stimulus, suggesting that this process employs rapid DNA repair mechanisms such as NHEJ (Madabhushi et al., 2015). Similarly, in a more recent study, physiological neuronal activity by exposure of mice to learning behaviors through contextual fear conditioning (CFC) induced the formation of DSBs. This was also associated with experience-driven transcriptional changes within neurons (Stott et al., 2021). Similarly, in another recent study, neurons were found to accumulate DNA SSBs at specific sites within the genome. These were repaired by PARP1 and XRCC1-dependent mechanisms and were located within enhancers at, or near, CpG dinucleotides or at sites of DNA demethylation (Wu et al., 2021). Hence, together these studies suggest that DNA damage may facilitate important physiological roles in neurons. This is highlighted by the specific location of the associated DNA breaks, the requirement for gene expression, and their generation as a result of normal physiological processes.
Whilst neuronal activation is associated with induction of DNA damage, conversely, DNA damage has been shown to modulate neuronal activity. Interestingly, DNA damage can alter the activity of AMPA glutamate receptors (Lu et al., 2001). Induction of SSBs in neurons with camptothecin, a topoisomerase I inhibitor, resulted in decreases in AMPA-induced current and calcium response to AMPA in whole cell patch clamp analyses. This effect was abolished by pharmacological inhibition of caspases, implying that caspases are involved in this process (Lu et al., 2001). A link between DNA damage and altered glutamatergic neurotransmission induced by stress and anxiety has also been suggested (Reus et al., 2015). Rats selectively bred for an anxiety phenotype, by either high or low contextual fear conditioning, displayed more SSBs and DSBs in the hippocampus, amygdala, and nucleus accumbens compared to control animals. This correlated with changes in expression of NMDA receptor subunits NR1, NR2A, and NR2B, and excitatory amino acid transporter 1 (EAAT1), which removes glutamate from the extracellular space. Expression of NMDA receptor subunits and EAAT1 underwent further alterations when animals were additionally stressed, further linking the anxious phenotype, DNA damage, and glutamatergic system (Reus et al., 2015).
The base-excision DNA repair (BER) pathway has also been linked to maintenance of synaptic plasticity (Yu et al., 2015). Epigenetic modifications play important roles in neuronal plasticity, learning, and memory, and in neurological disorders. Cytosine methylation is the major covalent modification of eukaryotic genomic DNA and demethylation is mediated by the ten-eleven translocation (Tet) family of proteins. Interestingly, Tet-initiated DNA demethylation is mediated through BER in neurons (Yu et al., 2015) and synaptic activity bi-directionally regulates neuronal expression of Tet3, and hence DNA demethylation through BER. Expression of Tet3 and Tet1 or inhibition of BER in hippocampal neurons correlated inversely with excitatory glutamatergic synaptic transmission. Furthermore, Tet3 regulated gene expression in response to global synaptic activity changes and determined glutamate receptor 1 (GluR1) levels at the neuronal surface (Yu et al., 2015). Hence, it was suggested that BER modulates synaptic plasticity and that Tet3 is a sensor to epigenetically regulate the homeostatic synaptic plasticity of neurons. Similarly, a role for the BER protein DNA polymerase β (Polβ) in the early development of postnatal hippocampal pyramidal neurons and learning and memory in mice was recently described in another study. Polβ was implicated as an epigenetic regulator that maintains genome stability in Tet-mediated active DNA demethylation (Uyeda et al., 2020). In this process, the modified base is replaced with cytosine by Polβ. Transgenic mice lacking Polβ expression in forebrain postmitotic excitatory neurons displayed extensive DSBs in hippocampal pyramidal neurons, and to a lesser extent in neocortical neurons. This process was related to the hydroxylation of 5-methylcytosine by Tet1 (Uyeda et al., 2020). Interestingly, hippocampal neurons displaying DNA damage exhibited aberrant gene expression profiles and dendrite formation, but not apoptosis. In adult animals, impaired spatial reference memory and contextual fear memory were detected (Uyeda et al., 2020). Thus, this study links physiological DNA damage to epigenetic regulation and subsequent learning and memory.
Increasing evidence suggests that the introduction and repair of DSBs is associated with long-term memory (LTM) processes. LTM requires the establishment, maintenance, and rearrangement of specific neural networks, within specific brain areas involved in learning and memory. This was recently shown to involve recombination-activating genes (RAGs) that encode components of the protein complex that mediates the rearrangement and recombination of genes of the adaptive immune system (Castro-Perez et al., 2016). V(D)J recombination is a somatic process that occurs in developing lymphocytes during the early stages of T and B cell maturation. It results in the highly diverse collection of immunoglobulins and T cell receptors (TCRs) found in B cells and T cells respectively. This is a tightly regulated process involving the generation of DNA DSBs, and activation of DNA ligases and DNA repair proteins for re-joining the new gene segments. Expression of RAG1 was induced in the amygdala of C57BL/6 mice after context fear conditioning, but not after context-only or shock-only conditioning (Castro-Perez et al., 2016). Furthermore, knock down of RAG1 using antisense oligonucleotides significantly impaired LTM. This study, therefore, implies the existence of a link between RAG1 and DSBs with associative learning processes and in the consolidation of LTMs (Castro-Perez et al., 2016).
DNA damage can modulate neural plasticity through the expression of ERGs, which encode transcription factors such as c-fos, c-myc or c-jun. These in turn govern the expression of late response genes such as bdnf and homer1 to regulate various cellular responses. In neurons, ERGs play important roles in cellular, circuitry and cognitive functions, including neurite outgrowth, synapse development and maturation, the balance between excitatory and inhibitory synapses, and learning and memory (Madabhushi et al., 2015). Furthermore, ERGs modulate and maintain neural connectivity in an activity-associated manner (Hogan et al., 2020). Interestingly, regulation of ERG expression is associated with the generation of DSBs (Madabhushi and Kim, 2018) and is triggered by neuronal activation (Madabhushi et al., 2015). Therefore, activity-induced DSBs can trigger transcriptional changes that alter a neuron’s overall generic expression and ultimately induce plasticity changes (Hogan et al., 2020). ERGs are also regulated by a member of the growth arrest and DNA damage (Gadd45) protein family, Gadd45γ, which performs critical roles in the repair of DSBs (Li et al., 2019). Gadd45γ temporally influences learning-induced ERG expression in the prelimbic prefrontal cortex of adult mice through its interaction with DSB-mediated changes in DNA methylation (Li et al., 2019). Another DNA repair protein, ataxia telangiectasia and Rad3-related (ATR), is involved in the regulation of neuronal activity (Kirtay et al., 2021). ATR deletion in neurons enhances intrinsic neuronal activity, resulting in aberrant firing and an increased epileptiform activity, and increased susceptibility to ataxia and epilepsy in mice. ATR-deleted neurons exhibit hyper-excitability, associated with changes in action potential conformation and presynaptic vesicle accumulation, albeit independently of DNA damage response signaling (Kirtay et al., 2021). Figure 1 illustrates a simplified model of the interplay between neuroplasticity and DNA damage and repair. Table 1 summarizes current evidence linking the DDR to neural plasticity.
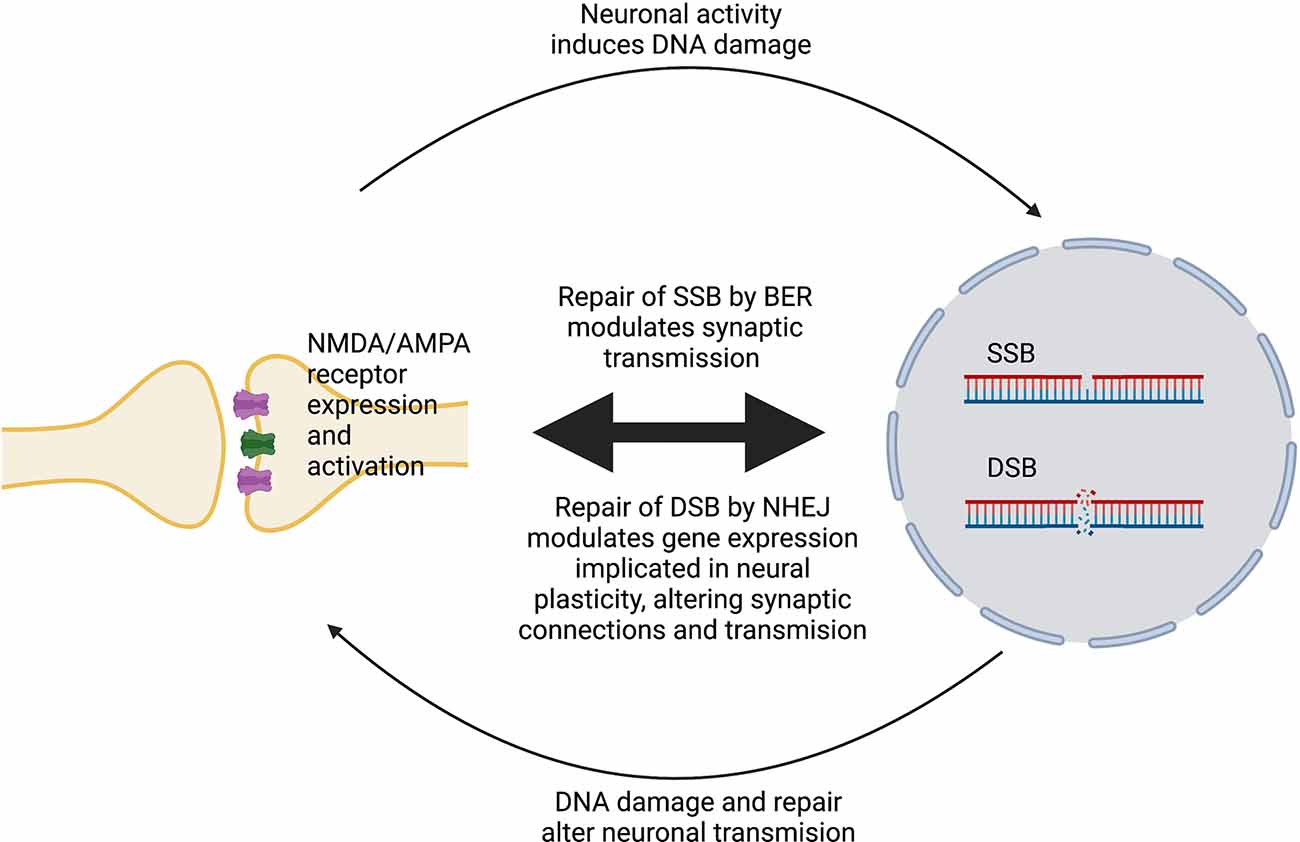
Figure 1. Simplified model of the interplay between neural plasticity and DNA damage and repair. Activation of NMDA/AMPA receptors at synapses induces SSBs or DSBs and promotes their repair by BER or NHEJ, respectively. In turn, DNA damage and repair alters the expression and activity of these receptors, which modulates neuronal gene expression, leading to changes in plasticity.
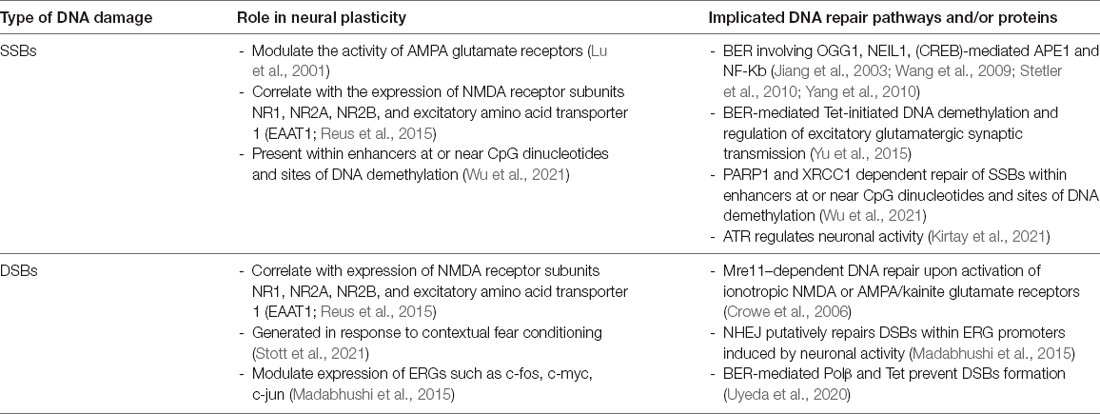
Table 1. Summary of the relationship between the DNA damage response and neural plasticity.
Physical activity induces neuronal stimulation, resulting in genetic alterations that regulate synaptic and brain plasticity (Vilela et al., 2020). Chronic exercise has a positive response on the brain, which is different compared with other organs (Liu et al., 2000). Aerobic and strength training improves spatial memory, reduces DNA damage, and increases expression of brain-derived neurotrophic factor (BDNF) and CREB in aging rats (Kim et al., 2010; Vilela et al., 2017) and mice (Yang et al., 2014). Physical exercise promotes DNA repair by activation of the enzymes involved in BER repair in human and animal models (Radak et al., 2009; Koltai et al., 2010; Siu et al., 2011). Regular exercise attenuates the age-associated increase in 8-hydroxy-2’-deoxyguanosine levels and increases DNA repair activity and neuronal resistance against oxidative stress (Radak et al., 2002). Similarly, an increase in the activity of 8-oxoG in human skeletal muscle was detected after a single-bout of physical exercise (Radak et al., 2003). In contrast, strenuous endurance exercise can result in muscle and lymphocyte DNA damage and the extent of this increases as running distance increases (Ryu et al., 2016).
The studies described above refer to the role of DNA damage in the normal physiological function of neurons. Below we describe the relationship between DNA damage and plasticity in relation to neurodegenerative diseases.
DSBs can alter gene expression, chromatin stability, and cellular functions, hence they can be an important driver of neurodegeneration and cognitive decline in neurodegenerative diseases. Aging is the major risk factor for neurodegenerative diseases. Interestingly, with aging the expression of genes related to synaptic plasticity and DNA repair is reduced (Lu et al., 2004). Moreover, DNA damage is increased in the promoters of genes that display reduced expression in the aged human cortex. Furthermore, these genes are selectively damaged by oxidative stress in cultured human neurons, resulting in reduced BER repair. These findings imply that DNA damage may reduce the expression of selectively vulnerable genes involved in learning and memory (Lu et al., 2004). Here we review the evidence linking dysfunction of the DDR with neural plasticity in the diseases described below.
Alzheimer’s disease (AD) is a neurodegenerative disorder resulting in brain atrophy and death of neurons located primarily in the hippocampus and cerebral neocortex. AD is the most common cause of dementia, which manifests as a continuous decline in thinking, behavioral and social skills, impairing person’s ability to function independently. Two hallmark pathologies are present in AD; both β-amyloid plaques and neurofibrillary tangles, containing hyperphosphorylated tau (Weller and Budson, 2018). The accumulation of DSBs is associated with loss of memory and neurons in AD (Lu et al., 2004; Shanbhag et al., 2019; Lin et al., 2020) and impairment of DNA repair processes is present prior to the onset of symptoms (Su et al., 1997; Sheng et al., 1998; Silva et al., 2014). More γH2AX foci, a widely used marker of DSBs, were present in neurons and astrocytes in the hippocampus and frontal cortex of AD patients compared to age-matched controls (Shanbhag et al., 2019). In this study some neurons and glia displayed diffuse pan-nuclear patterns of γH2AX immunoreactivity, which was not associated with DSBs, suggesting that DSBs accumulate selectively in vulnerable neuronal and glial cell populations (Shanbhag et al., 2019). Interestingly, β-amyloid inhibits DNA-PK-dependent NHEJ and contributes to the accumulation of DSBs in PC12 cells (Cardinale et al., 2012). Furthermore, whilst DSBs are generated in the dentate gyrus of wildtype mice in response to the exploration of a novel environment, they are repaired within 24 h (Suberbielle et al., 2013). In contrast, an AD transgenic mouse model expressing human amyloid precursor protein (hAPP) with aberrant neuronal activity, displayed more neuronal DSBs before and more severe and prolonged DSBs after, exploration of a novel environment. This finding suggests that excessive accumulation of DSBs in AD impairs spatial learning and memory. Interestingly, suppression of aberrant neuronal activity by ablating expression of tau, improved memory and led to the production of fewer DSBs in this model, suggesting that excessive DNA damage in AD is associated with synaptic dysfunction (Suberbielle et al., 2013). Furthermore, evidence for loss of function of ATM was detected in three different AD transgenic mouse models; R1.40, which expresses a single (full-length) APP transgene, PS/APP, expressing transgenes for both APP and presenilin-1 (PSEN1) and triple-transgenic animals (3xTg), which bear APP, PSEN1, and MAPT transgenes, together implying that defective repair of DSBs is present in AD (Shen et al., 2016). Excessive accumulation of DSBs in AD has also been linked to impairment of HR repair. In PS/APP mice, more persistent DSBs were present compared to control animals. This was accompanied by diminished HR repair in the subgranular zone of the dentate gyrus, the site of neurogenesis (Yu et al., 2018). In addition, disturbance in the cell cycle and DNA damage was present in neurons reprogrammed from induced pluripotent stem cells (iPSC) derived from familial AD patients. Specifically, an increase in a component of HR repair, a human tumor suppressor, BRACA1, was reported (Wezyk et al., 2018). BER repair is also linked to aberrant neural plasticity in AD. A significant decrease in NEIL DNA glycosylase was detected in AD brains compared to controls (Canugovi et al., 2014). Interestingly, NEIL1 null mice display defective memory retention in a water maze test (Canugovi et al., 2012). Furthermore, 3xTgAD mice with inefficient BER due to DNA polymerase-β (Polβ) haploinsufficiency displayed enhanced neurodegeneration, synaptic and cognitive deficits, and more DNA damage, compared to 3xTgAD mice with normal levels of Polβ (Sykora et al., 2015). Furthermore, olfactory deficits are present early in AD and these 3xTgAD/ POLβ+/− mice display greater degeneration of olfactory bulb neurons, in part via inhibiting the production of new neurons from neural progenitor cells (Misiak et al., 2017). Similarly, increased oxidative DNA damage and deficient BER was detected in post-mortem brain tissue samples derived from patients with mild cognitive impairment and AD, compared to age-matched neurologically normal subjects (Weissman et al., 2007). Consistent with this finding, reduced 8-oxoG activity was present in the hippocampal and para-hippocampal gyri, superior and middle temporal gyri, and inferior parietal lobule in AD patients compared to control individuals. This decrease was accompanied by altered DNA helicase activity, suggesting that this function may interfere with BER repair in AD (Lovell et al., 2000).
The deficiency of Polβ and decreased levels of NAD+, a cellular metabolite critical for neuronal resistance to stress and maintenance of DNA integrity, were found in AD patients’ brains (Hou et al., 2018). In an AD mouse model with deficiencies in DNA repair, NAD+ supplementation improved the DNA damage response and synaptic transmission, learning and memory, and motor function (Hou et al., 2018). Together these findings highlight the importance of efficient DNA repair processes in neuronal function related to neural plasticity in AD (Hou et al., 2018).
Amyotrophic lateral sclerosis (ALS) is a motor neuron disorder characterized by progressive loss of motor neurons in the cortex, brainstem, and spinal cord. This leads to muscle atrophy and weakness, eventually resulting in death. ALS overlaps significantly with frontotemporal dementia (FTD), the most common form of early-onset dementia (under 60 years of age) that primarily affects the frontal and temporal lobes of the brain (Shahheydari et al., 2017). DNA damage is increasingly implicated in the pathophysiology of ALS, and interestingly, the number of DNA repair proteins linked to ALS is steadily growing. Similarly, disruption of neurotransmission is associated with ALS pathogenesis. Cortical hyperexcitability, the enhanced response of a neuron to a stimulus, is an important pathophysiological process in ALS, contributing to the loss of motor neurons (Buskila et al., 2019; Brunet et al., 2020). Studies on ALS patients have revealed that this precedes clinical symptoms (Vucic and Kiernan, 2006), contributes to disease progression, and correlates with greater functional disabilities (Menon et al., 2020). Whilst the underlying mechanisms are unknown, hyperexcitability is related to the interplay between excitatory and inhibitory interneurons (Bae et al., 2013). Excitotoxicity involving the neurotransmitter glutamate, hypo-excitability, and loss of repetitive firing are also present in vulnerable motor neurons in vivo (Foran and Trotti, 2009; Martinez-Silva et al., 2018). Furthermore, frequent and strenuous physical activity is implicated as a risk factor for ALS (Julian et al., 2021). Importantly, this is known to induce DNA damage and is also related to changes in synaptic plasticity (Ryu et al., 2016; Vilela et al., 2020).
Interestingly, proteins that are central to ALS pathogenesis are implicated in both DNA damage and plasticity. Pathological forms of TAR DNA-binding protein 43 (TDP-43), a heterogenous nuclear ribonucleoprotein (hnRNP), is present in affected motor neurons in almost all ALS cases (97%). TDP-43 modulates RNA splicing and micro-RNA biogenesis and it also functions in NHEJ DNA repair (Mitra et al., 2019; Konopka et al., 2020). Its pathological mis-localization to the cytoplasm of motor neurons and the presence of ALS-associated mutations in TDP-43 are both known to impair DNA repair (Mitra et al., 2019; Konopka et al., 2020). TDP-43 also regulates synaptic functions. Transgenic animals expressing human A315T mutant TDP-43 exhibit reduced levels of the presynaptic protein synaptophysin in the brain, and display attenuated synaptic transmission, cognitive and motor deficits (Medina et al., 2014). TDP-43 depletion in transgenic rats enhances the acquisition of fear memory, decreases the short-term plasticity of intrinsic neuronal excitability, and affects the kinetics of AMPA receptors by slowing the decay time of AMPAR-mediated miniature excitatory postsynaptic currents (Koza et al., 2019). These findings imply that TDP-43 regulates activity-dependent neuronal plasticity, possibly by controlling the splicing of genes responsible for fast synaptic transmission and membrane potential (Koza et al., 2019). Interestingly, DNA damage modifies splicing proteins by modulating their activity or interaction with other proteins or RNA (Shkreta and Chabot, 2015), linking the role of TDP-43 in DNA damage with its role in activity-dependent neuronal plasticity.
Fused in sarcoma (FUS) is another DNA/RNA-binding protein implicated in both DNA repair and synaptic dysfunction in ALS, that displays structural and functional similarities to TDP-43. FUS interacts with histone deacetylase 1 (HDAC1) to facilitate DNA repair through NHEJ, while FUS-ALS associated mutants R514S and R521C display impaired DNA repair functions (Wang et al., 2013). FUS-ALS associated mutants R521C and P525L disrupt the formation of presynaptic active zones, subsequently reducing synaptic transmission with decreased quantal size (Machamer et al., 2014). Hence these studies imply a relationship between DNA repair and synaptic activity in ALS.
The DNA damage response is also induced by hexanucleotide (GGGGCC) repeat expansions in a non-coding region of C9orf72, the major genetic cause of ALS (Farg et al., 2017). In Drosophila, transgenic expression of the C9orf72 repeat expansion results in a dramatic reduction in synaptic arborization and the number of active zones at neuromuscular junctions (NMJs), and reduced neurotransmission. Hence synaptic dysfunction at NMJs is induced by C9orf72 ALS related pathology (Perry et al., 2017). Another prominent protein linked to familial forms of ALS, superoxide dismutase-1 (SOD1), is also implicated in both synaptic dysfunction and DNA damage. SOD1 localizes at the pre- and post-synapse, while the ALS-associated mutant G93A SOD1 shows mis-localization in pre-synaptic terminals as well as at the post-synapse, impairing axonal transport and contributing to neuronal cell death (Lee et al., 2015; Bae and Kim, 2016). Expression of mutant SOD1 G93A also decreases the formation of synaptophysin-positive presynaptic boutons (Zang et al., 2005). In ALS, over-activation of glutamate receptors in motor neurons is a well-described pathological event (Corona et al., 2007). Motor neurons from transgenic SOD1 G93A mice are sensitive to glutamate toxicity and this was associated with ROS production, resulting in oxidative DNA damage, elevation in intracellular calcium levels, and mitochondrial dysfunction (Kruman et al., 1999). Substantial increases in oxidative DNA damage have also been observed in nuclear DNA from the spinal cord, frontal cortex, striatum, and cerebellum from the transgenic G93A SOD1 mouse model (Aguirre et al., 2005). Therefore, taken together, these findings indicate that dysfunctional DNA repair and dysfunctional neural plasticity occur together in ALS.
Huntington’s disease (HD) is an inherited autosomal dominant neurodegenerative disorder characterized by motor dysfunction and cognitive deficits, due to neurodegeneration of specific brain regions such as the striatum and cerebral cortex. It is caused by mutation of the huntingtin protein, resulting in an abnormally high copy of polyglutamine (polyQ) repeats at its N-terminus. The expression of mutated huntingtin enhances NMDA activity and sensitizes type 1 inositol 1,4,5-trisphosphate receptors, causing a disturbance in calcium homeostasis (Bezprozvanny and Hayden, 2004; Zhang et al., 2008). In addition, expression of the presynaptic protein rabphilin 3A is decreased (Deak et al., 2006), while the levels of synaptic vesicle protein SCAMP5 are increased, in Caenorhabditis elegans neurons (Parker et al., 2007). This implies that alteration of expression of presynaptic proteins results in impairment of synaptic vesicle fusion or endocytosis. HD is also associated with DNA damage and defective DNA repair. Post-mortem brain samples of HD patients display higher levels of the oxidative DNA damage marker 8-Oxo-dG in both nuclei and mitochondria (Browne et al., 1997; Polidori et al., 1999). In addition, mutant huntingtin impairs NHEJ DNA repair by interaction with Ku70 protein, leading to the accumulation of DSBs in primary neurons (Enokido et al., 2010). Interestingly, expression of exogenous Ku70 rescues abnormal behavior and pathological phenotypes in the R6/2 mouse model of HD (Enokido et al., 2010). These findings imply that the huntingtin protein has a dual role in synaptic plasticity and DNA repair. For comprehensive reviews about the role of DNA damage in HD, we direct readers to the following articles about the role of mitochondrial DNA damage in HD (Yang et al., 2008), the role of MMR in HD (Iyer and Pluciennik, 2021), and the role of HR in HD (Jeon et al., 2012).
Parkinson’s disease (PD) is a neurodegenerative disorder associated with widespread degradation of dopaminergic neurons in the substantia nigra pars compacta (SNc), with subsequent loss of the neurons projecting to the striatum (Lang and Lozano, 1998). PD manifests principally by motor symptoms such as bradykinesia, rigidity, tremor at rest, postural instability, micrographia, and shuffling gait (Jankovic, 2008). Familial Parkinsonism is linked to mutations in a number of genes including α-synuclein, LRRK2, parkin, DJ-1, PINK1, UCHL-1, synphilin-1, and NR4A2 (Nussbaum and Polymeropoulos, 1997; Mizuno et al., 2001; Le et al., 2003; Marx et al., 2003; Healy et al., 2004; Maraganore et al., 2004; Valente et al., 2004; Rui et al., 2018).
Both DNA damage and dysfunction of synaptic plasticity have been reported in PD. Altered presynaptic plasticity is present in PD animal models, including α-synuclein, DJ-1, and PINK1 knock out mice (Abeliovich et al., 2000; Goldberg et al., 2005; Kitada et al., 2007). Similarly, altered vesicular transmission and defects in neurotransmission are present in experimental Parkinsonism (Abeliovich et al., 2000; Murphy et al., 2000; Li et al., 2010). Furthermore, altered composition of glutamatergic NMDA receptors contributes to clinical features of experimental Parkinsonism (Ulas and Cotman, 1996; Dunah and Standaert, 2001; see Picconi et al., 2012 for a comprehensive review about dysfunction to synaptic plasticity in PD). Similar to dysfunctional plasticity, DNA damage is present in the SNc of PD patients, including elevated levels of 8-oxo-guanine, abasic sites, and nuclear DNA strand breaks (Alam et al., 1997; Zhang et al., 1999; Hegde et al., 2006; Sanders et al., 2014; see Gonzalez-Hunt and Sanders, 2021 for a comprehensive review of DNA damage in PD). α-synuclein, which is known to function in synaptic plasticity, is also linked to DNA damage in PD. Overexpression of α-synuclein causes the formation of SSBs and DSBs, particularly under oxidative conditions (Vasquez et al., 2017) and it modulates repair of DSBs (Schaser et al., 2019). Depletion of Parkin results in impairment of dopamine release and synaptic plasticity in the striatum of Parkin knockout mice (Kitada et al., 2009). Interestingly, mutations in Parkin also result in the accumulation of BER factor, APE1 (Scott et al., 2017), suggesting that defective Parkin causes dysfunction to both DNA repair and synaptic plasticity. Mice with mutated excision repair cross-complementation group 1 (ERCC1) endonuclease, a competent of the NER repair pathway, display PD pathology, including impairment of striatal innervation, α-synuclein pathology, and the formation of γH2AX foci (Sepe et al., 2016). This implies an important role for DNA repair pathways in integrity of the nigrostriatal system in PD. Table 2 illustrates the potential and/or established roles of mutant proteins associated with neurodegeneration in DNA damage response mechanisms.
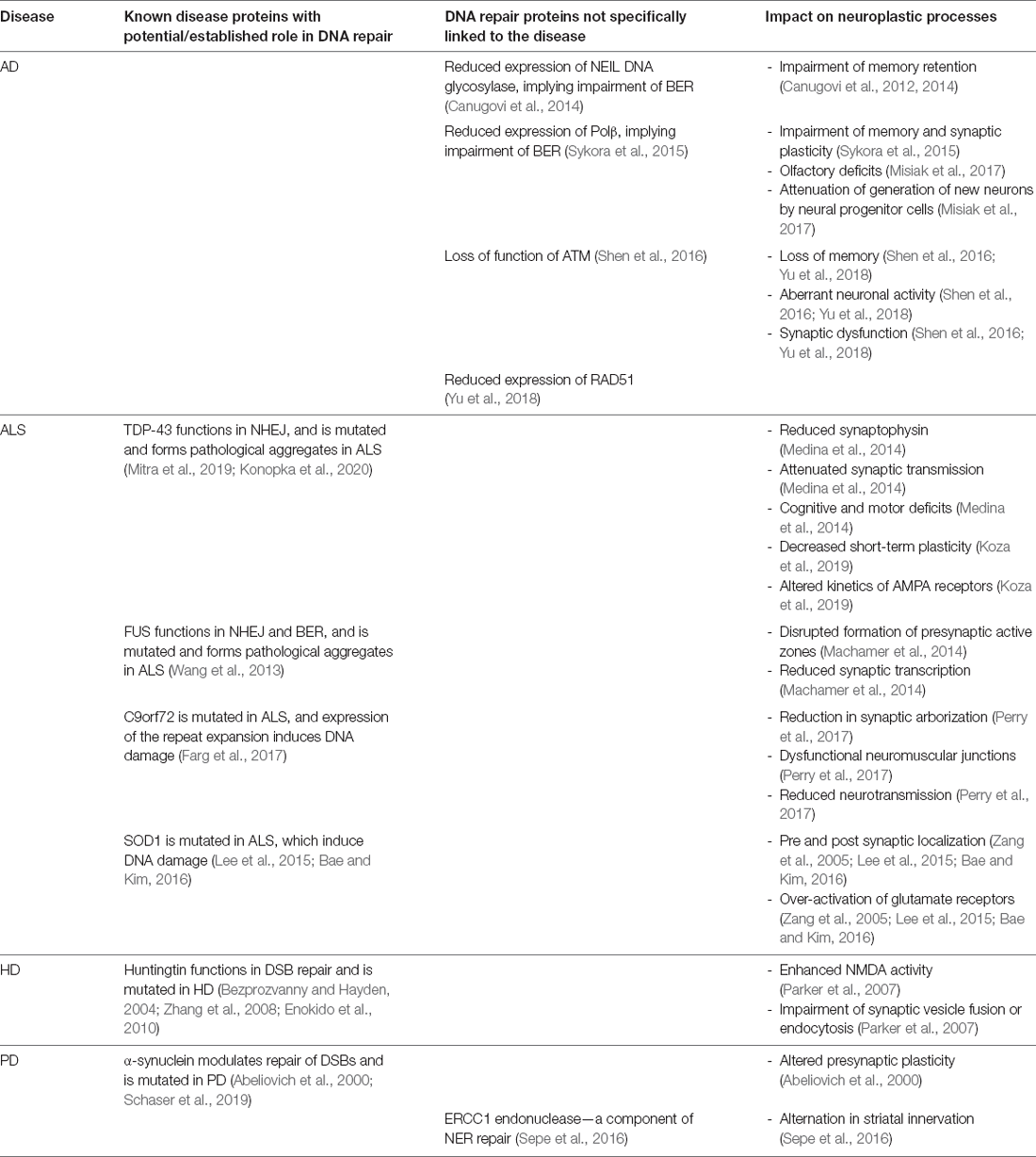
Table 2. Summary of the DNA damage and repair proteins associated with neural plasticity in neurodegenerative diseases.
Neurons, in contrast to other cell types, possess unique features, including their ability to undergo electrical excitation and their post-mitotic character. Recent findings imply that DNA damage is not limited to pathological conditions as previously thought, but it is also important for unique neuronal functions related to neural and synaptic plasticity under physiological conditions. However, dysfunction in these processes is also related to a decline in cognitive function and neuronal death in neurodegenerative diseases. However, human post-mortem tissues represent the end-point stage of the disease. Hence studies examining these tissues cannot be used to determine whether DNA damage has a primary or secondary role in pathogenesis. However, future studies on the relationship between plasticity and DNA damage may provide a better understanding of the cellular processes that contribute to higher order brain functions. Distinct groups of neurons are affected in different neurodegenerative diseases, such as motor neurons in ALS or neurons of the entorhinal cortex in AD, and these cells are specialized to perform specific functions. Given that DNA damage and repair are important for the unique functions of neurons, which in turn depend on their activation, it is possible that the interplay between DNA damage and neural plasticity is unique for specific groups of neurons. This could operate through the activation of specific genes by DNA damage, which would differ depending on the type of neurons involved and their associated functions. Therefore, a better understanding of the interplay between DNA damage and neural plasticity is required, as well as dysfunction in these processes in disease. In particular, the inclusion of specific neuronal types may reveal the causes of selective neuronal death in distinct neurodegenerative diseases. To date, no previous studies have examined therapeutic strategies directed at DNA damage and repair in relation to aberrant neural plasticity. However, this approach has the potential to identify novel treatments for impaired cognitive functions in neurodegenerative diseases associated with excessive DNA damage.
AK conceptualized and wrote the manuscript. AK provided resources for the figure preparation, and prepared the figure and tables. JA provided all other resources, and contributed with intellectual input and additional text. AK and JA edited the manuscript and approved it for publication. Both authors contributed to the article and approved the submitted version.
This work was supported by grants from the NHMRC Dementia Teams Research grant (1095215) and MND Research Australia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abeliovich, A., Schmitz, Y., Farinas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., et al. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. doi: 10.1016/s0896-6273(00)80886-7
Agathangelou, K., Apostolou, Z., and Garinis, G. A. (2018). Nuclear DNA damage and ageing. Subcell. Biochem. 90, 309–322. doi: 10.1007/978-981-13-2835-0_10
Aguirre, N., Beal, M. F., Matson, W. R., and Bogdanov, M. B. (2005). Increased oxidative damage to DNA in an animal model of amyotrophic lateral sclerosis. Free Radic. Res. 39, 383–388. doi: 10.1080/10715760400027979
Ahn, S., Ginty, D. D., and Linden, D. J. (1999). A late phase of cerebellar long-term depression requires activation of CaMKIV and CREB. Neuron 23, 559–568. doi: 10.1016/s0896-6273(00)80808-9
Ahnesorg, P., Smith, P., and Jackson, S. P. (2006). XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 124, 301–313. doi: 10.1016/j.cell.2005.12.031
Alam, Z. I., Jenner, A., Daniel, S. E., Lees, A. J., Cairns, N., Marsden, C. D., et al. (1997). Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 69, 1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x
Bae, J. R., and Kim, S. H. (2016). Impairment of SO D1–G93 A motility is linked to mitochondrial movement in axons of hippocampal neurons. Arch. Pharm. Res. 39, 1144–1150. doi: 10.1007/s12272-016-0798-5
Bae, J. S., Simon, N. G., Menon, P., Vucic, S., and Kiernan, M. C. (2013). The puzzling case of hyperexcitability in amyotrophic lateral sclerosis. J. Clin. Neurol. 9, 65–74. doi: 10.3988/jcn.2013.9.2.65
Baez, M. V., Cercato, M. C., and Jerusalinsky, D. A. (2018). NMDA receptor subunits change after synaptic plasticity induction and learning and memory acquisition. Neural Plast. 2018:5093048. doi: 10.1155/2018/5093048
Bezprozvanny, I., and Hayden, M. R. (2004). Deranged neuronal calcium signaling and Huntington disease. Biochem. Biophys. Res. Commun. 322, 1310–1317. doi: 10.1016/j.bbrc.2004.08.035
Browne, S. E., Bowling, A. C., MacGarvey, U., Baik, M. J., Berger, S. C., Muqit, M. M., et al. (1997). Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann. Neurol. 41, 646–653. doi: 10.1002/ana.410410514
Brunet, A., Stuart-Lopez, G., Burg, T., Scekic-Zahirovic, J., and Rouaux, C. (2020). Cortical circuit dysfunction as a potential driver of amyotrophic lateral sclerosis. Front. Neurosci. 14:363. doi: 10.3389/fnins.2020.00363
Buck, D., Malivert, L., de Chasseval, R., Barraud, A., Fondaneche, M. C., Sanal, O., et al. (2006). Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 124, 287–299. doi: 10.1016/j.cell.2005.12.030
Buskila, Y., Kekesi, O., Bellot-Saez, A., Seah, W., Berg, T., Trpceski, M., et al. (2019). Dynamic interplay between H-current and M-current controls motoneuron hyperexcitability in amyotrophic lateral sclerosis. Cell Death Dis. 10:310. doi: 10.1038/s41419-019-1538-9
Cabelof, D. C., Raffoul, J. J., Yanamadala, S., Guo, Z., and Heydari, A. R. (2002). Induction of DNA polymerase beta-dependent base excision repair in response to oxidative stress in vivo. Carcinogenesis 23, 1419–1425. doi: 10.1093/carcin/23.9.1419
Canugovi, C., Shamanna, R. A., Croteau, D. L., and Bohr, V. A. (2014). Base excision DNA repair levels in mitochondrial lysates of Alzheimer’s disease. Neurobiol. Aging 35, 1293–1300. doi: 10.1016/j.neurobiolaging.2014.01.004
Canugovi, C., Yoon, J. S., Feldman, N. H., Croteau, D. L., Mattson, M. P., Bohr, V. A., et al. (2012). Endonuclease VIII-like 1 (NEIL1) promotes short-term spatial memory retention and protects from ischemic stroke-induced brain dysfunction and death in mice. Proc. Natl. Acad. Sci. U S A 109, 14948–14953. doi: 10.1073/pnas.1204156109
Cardinale, A., Racaniello, M., Saladini, S., De Chiara, G., Mollinari, C., de Stefano, M. C., et al. (2012). Sublethal doses of beta-amyloid peptide abrogate DNA-dependent protein kinase activity. J. Biol. Chem. 287, 2618–2631. doi: 10.1074/jbc.M111.276550
Castro-Perez, E., Soto-Soto, E., Perez-Carambot, M., Dionisio-Santos, D., Saied-Santiago, K., Ortiz-Zuazaga, H. G., et al. (2016). Identification and characterization of the V(D)J recombination activating gene 1 in long-term memory of context fear conditioning. Neural Plast. 2016:1752176. doi: 10.1155/2016/1752176
Chatterjee, N., and Walker, G. C. (2017). Mechanisms of DNA damage, repair and mutagenesis. Environ. Mol. Mutagen. 58, 235–263. doi: 10.1002/em.22087
Chen, K. H., Yakes, F. M., Srivastava, D. K., Singhal, R. K., Sobol, R. W., Horton, J. K., et al. (1998). Up-regulation of base excision repair correlates with enhanced protection against a DNA damaging agent in mouse cell lines. Nucleic Acids Res. 26, 2001–2007. doi: 10.1093/nar/26.8.2001
Choi, S., Klingauf, J., and Tsien, R. W. (2000). Postfusional regulation of cleft glutamate concentration during LTP at “silent synapses”. Nat. Neurosci. 3, 330–336. doi: 10.1038/73895
Ciccia, A., and Elledge, S. J. (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. doi: 10.1016/j.molcel.2010.09.019
Corona, J. C., Tovar-y-Romo, L. B., and Tapia, R. (2007). Glutamate excitotoxicity and therapeutic targets for amyotrophic lateral sclerosis. Expert Opin. Ther. Targets 11, 1415–1428. doi: 10.1517/14728222.11.11.1415
Crowe, S. L., Movsesyan, V. A., Jorgensen, T. J., and Kondratyev, A. (2006). Rapid phosphorylation of histone H2A.X following ionotropic glutamate receptor activation. Eur. J. Neurosci. 23, 2351–2361. doi: 10.1111/j.1460-9568.2006.04768.x
Deak, F., Shin, O. H., Tang, J., Hanson, P., Ubach, J., Jahn, R., et al. (2006). Rabphilin regulates SNARE-dependent re-priming of synaptic vesicles for fusion. EMBO J. 25, 2856–2866. doi: 10.1038/sj.emboj.7601165
DNA damage repair (Internet). (2021). Available online at: https://www.nature.com/collections/hwnqqcstyj/.
Dunah, A. W., and Standaert, D. G. (2001). Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J. Neurosci. 21, 5546–5558. doi: 10.1523/JNEUROSCI.21-15-05546.2001
Enokido, Y., Tamura, T., Ito, H., Arumughan, A., Komuro, A., Shiwaku, H., et al. (2010). Mutant huntingtin impairs Ku70-mediated DNA repair. J. Cell Biol. 189, 425–443. doi: 10.1083/jcb.200905138
Farg, M. A., Konopka, A., Soo, K. Y., Ito, D., and Atkin, J. D. (2017). The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 26, 2882–2896. doi: 10.1093/hmg/ddx170
Fishel, M. L., Vasko, M. R., and Kelley, M. R. (2007). DNA repair in neurons: so if they don’t divide what’s to repair? Mutat. Res. 614, 24–36. doi: 10.1016/j.mrfmmm.2006.06.007
Foran, E., and Trotti, D. (2009). Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid. Redox Signal. 11, 1587–1602. doi: 10.1089/ars.2009.2444
Goldberg, M. S., Pisani, A., Haburcak, M., Vortherms, T. A., Kitada, T., Costa, C., et al. (2005). Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 45, 489–496. doi: 10.1016/j.neuron.2005.01.041
Gonzalez-Hunt, C. P., and Sanders, L. H. (2021). DNA damage and repair in Parkinson’s disease: recent advances and new opportunities. J. Neurosci. Res. 99, 180–189. doi: 10.1002/jnr.24592
Healy, D. G., Abou-Sleiman, P. M., Valente, E. M., Gilks, W. P., Bhatia, K., Quinn, N., et al. (2004). DJ-1 mutations in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 75, 144–145.
Hegde, M. L., Gupta, V. B., Anitha, M., Harikrishna, T., Shankar, S. K., Muthane, U., et al. (2006). Studies on genomic DNA topology and stability in brain regions of Parkinson’s disease. Arch. Biochem. Biophys. 449, 143–156. doi: 10.1016/j.abb.2006.02.018
Hogan, M. K., Hamilton, G. F., and Horner, P. J. (2020). Neural stimulation and molecular mechanisms of plasticity and regeneration: a review. Front. Cell. Neurosci. 14:271. doi: 10.3389/fncel.2020.00271
Horowitz, M. P., Milanese, C., Di Maio, R., Hu, X., Montero, L. M., Sanders, L. H., et al. (2011). Single-cell redox imaging demonstrates a distinctive response of dopaminergic neurons to oxidative insults. Antioxid. Redox Signal. 15, 855–871. doi: 10.1089/ars.2010.3629
Hou, Y., Lautrup, S., Cordonnier, S., Wang, Y., Croteau, D. L., Zavala, E., et al. (2018). NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. U S A 115, E1876–E1885. doi: 10.1073/pnas.1718819115
Hunt, D. L., and Castillo, P. E. (2012). Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr. Opin. Neurobiol. 22, 496–508. doi: 10.1016/j.conb.2012.01.007
Iyer, R. R., and Pluciennik, A. (2021). DNA mismatch repair and its role in Huntington’s disease. J. Huntingtons Dis. 10, 75–94. doi: 10.3233/JHD-200438
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi: 10.1038/nature08467
Jankovic, J. (2008). Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376. doi: 10.1136/jnnp.2007.131045
Jeon, G. S., Kim, K. Y., Hwang, Y. J., Jung, M. K., An, S., Ouchi, M., et al. (2012). Deregulation of BRCA1 leads to impaired spatiotemporal dynamics of gamma-H2AX and DNA damage responses in Huntington’s disease. Mol. Neurobiol. 45, 550–563. doi: 10.1007/s12035-012-8274-9
Jiang, X., Zhu, D., Okagaki, P., Lipsky, R., Wu, X., Banaudha, K., et al. (2003). N-methyl-D-aspartate and TrkB receptor activation in cerebellar granule cells: an in vitro model of preconditioning to stimulate intrinsic survival pathways in neurons. Ann. N Y Acad. Sci. 993, 134–145. doi: 10.1111/j.1749-6632.2003.tb07522.x
Ju, B. G., Lunyak, V. V., Perissi, V., Garcia-Bassets, I., Rose, D. W., Glass, C. K., et al. (2006). A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 312, 1798–1802. doi: 10.1126/science.1127196
Julian, T. H., Glascow, N., Barry, A. D. F., Moll, T., Harvey, C., Klimentidis, Y. C., et al. (2021). Physical exercise is a risk factor for amyotrophic lateral sclerosis: convergent evidence from mendelian randomisation, transcriptomics and risk genotypes. EBioMedicine 68:103397. doi: 10.1016/j.ebiom.2021.103397
Kandel, E. R. (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. doi: 10.1126/science.1067020
Kim, D. H., Ko, I. G., Kim, B. K., Kim, T. W., Kim, S. E., Shin, M. S., et al. (2010). Treadmill exercise inhibits traumatic brain injury-induced hippocampal apoptosis. Physiol. Behav. 101, 660–665. doi: 10.1016/j.physbeh.2010.09.021
Kim, Y. J., and Wilson, D. M. 3rd (2012). Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 5, 3–13. doi: 10.2174/1874467211205010003
Kirtay, M., Sell, J., Marx, C., Haselmann, H., Ceanga, M., Zhou, Z. W., et al. (2021). ATR regulates neuronal activity by modulating presynaptic firing. Nat. Commun. 12:4067. doi: 10.1038/s41467-021-24217-2
Kitada, T., Pisani, A., Karouani, M., Haburcak, M., Martella, G., Tscherter, A., et al. (2009). Impaired dopamine release and synaptic plasticity in the striatum of parkin−/− mice. J. Neurochem. 110, 613–621. doi: 10.1111/j.1471-4159.2009.06152.x
Kitada, T., Pisani, A., Porter, D. R., Yamaguchi, H., Tscherter, A., Martella, G., et al. (2007). Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. U S A 104, 11441–11446. doi: 10.1073/pnas.0702717104
Koltai, E., Szabo, Z., Atalay, M., Boldogh, I., Naito, H., Goto, S., et al. (2010). Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mech. Ageing Dev. 131, 21–28. doi: 10.1016/j.mad.2009.11.002
Konopka, A., Whelan, D. R., Jamali, M. S., Perri, E., Shahheydari, H., Toth, R. P., et al. (2020). Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Mol. Neurodegener. 15:51. doi: 10.1186/s13024-020-00386-4
Koza, P., Beroun, A., Konopka, A., Gorkiewicz, T., Bijoch, L., Torres, J. C., et al. (2019). Neuronal TDP-43 depletion affects activity-dependent plasticity. Neurobiol. Dis. 130:104499. doi: 10.1016/j.nbd.2019.104499
Krieger, C., and Duchen, M. R. (2002). Mitochondria, Ca2+ and neurodegenerative disease. Eur. J. Pharmacol. 447, 177–188. doi: 10.1016/s0014-2999(02)01842-3
Kruman, I. I., Pedersen, W. A., Springer, J. E., and Mattson, M. P. (1999). ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp. Neurol. 160, 28–39. doi: 10.1006/exnr.1999.7190
Lang, A. E., and Lozano, A. M. (1998). Parkinson’s disease. Second of two parts. N. Engl. J. Med. 339, 1130–1143. doi: 10.1056/NEJM199810153391607
Le, W. D., Xu, P., Jankovic, J., Jiang, H., Appel, S. H., Smith, R. G., et al. (2003). Mutations in NR4A2 associated with familial Parkinson disease. Nat. Genet. 33, 85–89. doi: 10.1038/ng1066
Lee, D. Y., Jeon, G. S., Shim, Y. M., Seong, S. Y., Lee, K. W., Sung, J. J., et al. (2015). Modulation of SOD1 subcellular localization by transfection with wild- or mutant-type SOD1 in primary neuron and astrocyte cultures from ALS mice. Exp. Neurobiol. 24, 226–234. doi: 10.5607/en.2015.24.3.226
Li, X., Marshall, P. R., Leighton, L. J., Zajaczkowski, E. L., Wang, Z., Madugalle, S. U., et al. (2019). The DNA repair-associated protein gadd45gamma regulates the temporal coding of immediate early gene expression within the prelimbic prefrontal cortex and is required for the consolidation of associative fear memory. J. Neurosci. 39, 970–983. doi: 10.1523/JNEUROSCI.2024-18.2018
Li, X., Patel, J. C., Wang, J., Avshalumov, M. V., Nicholson, C., Buxbaum, J. D., et al. (2010). Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J. Neurosci. 30, 1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010
Lieber, M. R. (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211. doi: 10.1146/annurev.biochem.052308.093131
Lin, X., Kapoor, A., Gu, Y., Chow, M. J., Peng, J., Zhao, K., et al. (2020). Contributions of DNA damage to Alzheimer’s disease. Int. J. Mol. Sci. 21:1666. doi: 10.3390/ijms21051666
Liu, J., Yeo, H. C., Overvik-Douki, E., Hagen, T., Doniger, S. J., Chyu, D. W., et al. (2000). Chronically and acutely exercised rats: biomarkers of oxidative stress and endogenous antioxidants. J. Appl. Physiol. (1985) 89, 21–28. doi: 10.1152/jappl.2000.89.1.21
Lovell, M. A., Xie, C., and Markesbery, W. R. (2000). Decreased base excision repair and increased helicase activity in Alzheimer’s disease brain. Brain Res. 855, 116–123. doi: 10.1016/s0006-8993(99)02335-5
Lu, C., Fu, W., and Mattson, M. P. (2001). Caspase-mediated suppression of glutamate (AMPA) receptor channel activity in hippocampal neurons in response to DNA damage promotes apoptosis and prevents necrosis: implications for neurological side effects of cancer therapy and neurodegenerative disorders. Neurobiol. Dis. 8, 194–206. doi: 10.1006/nbdi.2000.0377
Lu, T., Pan, Y., Kao, S. Y., Li, C., Kohane, I., Chan, J., et al. (2004). Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891. doi: 10.1038/nature02661
Ma, A., and Dai, X. (2018). The relationship between DNA single-stranded damage response and double-stranded damage response. Cell Cycle 17, 73–79. doi: 10.1080/15384101.2017.1403681
Machamer, J. B., Collins, S. E., and Lloyd, T. E. (2014). The ALS gene FUS regulates synaptic transmission at the Drosophila neuromuscular junction. Hum. Mol. Genet. 23, 3810–3822. doi: 10.1093/hmg/ddu094
Machiela, E., and Southwell, A. L. (2020). Biological aging and the cellular pathogenesis of Huntington’s disease. J. Huntingtons Dis. 9, 115–128. doi: 10.3233/JHD-200395
Madabhushi, R., and Kim, T. K. (2018). Emerging themes in neuronal activity-dependent gene expression. Mol. Cell Neurosci. 87, 27–34. doi: 10.1016/j.mcn.2017.11.009
Madabhushi, R., Gao, F., Pfenning, A. R., Pan, L., Yamakawa, S., Seo, J., et al. (2015). Activity-Induced DNA breaks govern the expression of neuronal early-response genes. Cell 161, 1592–1605. doi: 10.1016/j.cell.2015.05.032
Madabhushi, R., Pan, L., and Tsai, L. H. (2014). DNA damage and its links to neurodegeneration. Neuron 83, 266–282. doi: 10.1016/j.neuron.2014.06.034
Maraganore, D. M., Lesnick, T. G., Elbaz, A., Chartier-Harlin, M. C., Gasser, T., Kruger, R., et al. (2004). UCHL1 is a Parkinson’s disease susceptibility gene. Ann. Neurol. 55, 512–521. doi: 10.1002/ana.20017
Martin, J. H. (2005). The corticospinal system: from development to motor control. Neuroscientist 11, 161–173. doi: 10.1177/1073858404270843
Martinez-Silva, M. L., Imhoff-Manuel, R. D., Sharma, A., Heckman, C. J., Shneider, N. A., Roselli, F., et al. (2018). Hypoexcitability precedes denervation in the large fast-contracting motor units in two unrelated mouse models of ALS. eLife 7:e30955. doi: 10.7554/eLife.30955
Marx, F. P., Holzmann, C., Strauss, K. M., Li, L., Eberhardt, O., Gerhardt, E., et al. (2003). Identification and functional characterization of a novel R621C mutation in the synphilin-1 gene in Parkinson’s disease. Hum. Mol. Genet. 12, 1223–1231. doi: 10.1093/hmg/ddg134
Mateos-Aparicio, P., and Rodriguez-Moreno, A. (2019). The impact of studying brain plasticity. Front. Cell. Neurosci. 13:66. doi: 10.3389/fncel.2019.00066
Mattson, M. P., Dou, P., and Kater, S. B. (1988). Outgrowth-regulating actions of glutamate in isolated hippocampal pyramidal neurons. J. Neurosci. 8, 2087–2100. doi: 10.1523/JNEUROSCI.08-06-02087.1988
McKinnon, P. J. (2016). Topoisomerases and the regulation of neural function. Nat. Rev. Neurosci. 17, 673–679. doi: 10.1038/nrn.2016.101
Medina, D. X., Orr, M. E., and Oddo, S. (2014). Accumulation of C-terminal fragments of transactive response DNA-binding protein 43 leads to synaptic loss and cognitive deficits in human TDP-43 transgenic mice. Neurobiol. Aging 35, 79–87. doi: 10.1016/j.neurobiolaging.2013.07.006
Mele, M., Leal, G., and Duarte, C. B. (2016). Role of GABAA R trafficking in the plasticity of inhibitory synapses. J. Neurochem. 139, 997–1018. doi: 10.1111/jnc.13742
Menon, P., Higashihara, M., van den Bos, M., Geevasinga, N., Kiernan, M. C., Vucic, S., et al. (2020). Cortical hyperexcitability evolves with disease progression in ALS. Ann. Clin. Transl. Neurol. 7, 733–741. doi: 10.1002/acn3.51039
Merlo, D., Cuchillo-Ibanez, I., Parlato, R., and Rammes, G. (2016). DNA damage, neurodegeneration and synaptic plasticity. Neural Plast. 2016:1206840. doi: 10.1155/2016/1206840
Misiak, M., Vergara Greeno, R., Baptiste, B. A., Sykora, P., Liu, D., Cordonnier, S., et al. (2017). DNA polymerase beta decrement triggers death of olfactory bulb cells and impairs olfaction in a mouse model of Alzheimer’s disease. Aging Cell 16, 162–172. doi: 10.1111/acel.12541
Mitra, J., Guerrero, E. N., Hegde, P. M., Liachko, N. F., Wang, H., Vasquez, V., et al. (2019). Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. U S A 116, 4696–4705. doi: 10.1073/pnas.1818415116
Mizuno, Y., Hattori, N., Mori, H., Suzuki, T., and Tanaka, K. (2001). Parkin and Parkinson’s disease. Curr. Opin. Neurol. 14, 477–482. doi: 10.1097/00019052-200108000-00008
Murphy, D. D., Rueter, S. M., Trojanowski, J. Q., and Lee, V. M. (2000). Synucleins are developmentally expressed and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 20, 3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000
Nussbaum, R. L., and Polymeropoulos, M. H. (1997). Genetics of Parkinson’s disease. Hum. Mol. Genet. 6, 1687–1691. doi: 10.1093/hmg/6.10.1687
Parker, J. A., Metzler, M., Georgiou, J., Mage, M., Roder, J. C., Rose, A. M., et al. (2007). Huntingtin-interacting protein 1 influences worm and mouse presynaptic function and protects Caenorhabditis elegans neurons against mutant polyglutamine toxicity. J. Neurosci. 27, 11056–11064. doi: 10.1523/JNEUROSCI.1941-07.2007
Paulsen, J. S. (2011). Cognitive impairment in huntington disease: diagnosis and treatment. Curr. Neurol. Neurosci. Rep. 11, 474–483. doi: 10.1007/s11910-011-0215-x
Pecina-Slaus, N., Kafka, A., Salamon, I., and Bukovac, A. (2020). Mismatch repair pathway, genome stability and cancer. Front. Mol. Biosci. 7:122. doi: 10.3389/fmolb.2020.00122
Perry, S., Han, Y., Das, A., and Dickman, D. (2017). Homeostatic plasticity can be induced and expressed to restore synaptic strength at neuromuscular junctions undergoing ALS-related degeneration. Hum. Mol. Genet. 26, 4153–4167. doi: 10.1093/hmg/ddx304
Phukan, J., Pender, N. P., and Hardiman, O. (2007). Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 6, 994–1003. doi: 10.1016/S1474-4422(07)70265-X
Picconi, B., Piccoli, G., and Calabresi, P. (2012). Synaptic dysfunction in Parkinson’s disease. Adv. Exp. Med. Biol. 533–572. doi: 10.1007/978-3-7091-0932-8_24
Polidori, M. C., Mecocci, P., Browne, S. E., Senin, U., and Beal, M. F. (1999). Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci. Lett. 272, 53–56. doi: 10.1016/s0304-3940(99)00578-9
Radak, Z., Apor, P., Pucsok, J., Berkes, I., Ogonovszky, H., Pavlik, G., et al. (2003). Marathon running alters the DNA base excision repair in human skeletal muscle. Life Sci. 72, 1627–1633. doi: 10.1016/s0024-3205(02)02476-1
Radak, Z., Atalay, M., Jakus, J., Boldogh, I., Davies, K., Goto, S., et al. (2009). Exercise improves import of 8-oxoguanine DNA glycosylase into the mitochondrial matrix of skeletal muscle and enhances the relative activity. Free Radic. Biol. Med. 46, 238–243. doi: 10.1016/j.freeradbiomed.2008.10.022
Radak, Z., Naito, H., Kaneko, T., Tahara, S., Nakamoto, H., Takahashi, R., et al. (2002). Exercise training decreases DNA damage and increases DNA repair and resistance against oxidative stress of proteins in aged rat skeletal muscle. Pflugers Arch. 445, 273–278. doi: 10.1007/s00424-002-0918-6
Ramana, C. V., Boldogh, I., Izumi, T., and Mitra, S. (1998). Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc. Natl. Acad. Sci. U S A 95, 5061–5066. doi: 10.1073/pnas.95.9.5061
Rao-Mirotznik, R., Buchsbaum, G., and Sterling, P. (1998). Transmitter concentration at a three-dimensional synapse. J. Neurophysiol. 80, 3163–3172. doi: 10.1152/jn.1998.80.6.3163
Reus, G. Z., Abaleira, H. M., Michels, M., Tomaz, D. B., dos Santos, M. A., Carlessi, A. S., et al. (2015). Anxious phenotypes plus environmental stressors are related to brain DNA damage and changes in NMDA receptor subunits and glutamate uptake. Mutat. Res. 772, 30–37. doi: 10.1016/j.mrfmmm.2014.12.005
Rui, Q., Ni, H., Li, D., Gao, R., and Chen, G. (2018). The role of LRRK2 in neurodegeneration of Parkinson disease. Curr. Neuropharmacol. 16, 1348–1357. doi: 10.2174/1570159X16666180222165418
Ryu, J. H., Paik, I. Y., Woo, J. H., Shin, K. O., Cho, S. Y., Roh, H. T., et al. (2016). Impact of different running distances on muscle and lymphocyte DNA damage in amateur marathon runners. J. Phys. Ther. Sci. 28, 450–455. doi: 10.1589/jpts.28.450
Sanders, L. H., McCoy, J., Hu, X., Mastroberardino, P. G., Dickinson, B. C., Chang, C. J., et al. (2014). Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson’s disease. Neurobiol. Dis. 70, 214–223. doi: 10.1016/j.nbd.2014.06.014
Schaser, A. J., Osterberg, V. R., Dent, S. E., Stackhouse, T. L., Wakeham, C. M., Boutros, S. W., et al. (2019). Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 9:10919. doi: 10.1038/s41598-019-47227-z
Scott, T. L., Wicker, C. A., Suganya, R., Dhar, B., Pittman, T., Horbinski, C., et al. (2017). Polyubiquitination of apurinic/apyrimidinic endonuclease 1 by Parkin. Mol. Carcinogenesis 56, 325–336. doi: 10.1002/mc.22495
Sepe, S., Milanese, C., Gabriels, S., Derks, K. W., Payan-Gomez, C., van, I. W. F., et al. (2016). Inefficient DNA repair is an aging-related modifier of Parkinson’s disease. Cell Rep. 15, 1866–1875. doi: 10.1016/j.celrep.2016.04.071
Shahheydari, H., Ragagnin, A., Walker, A. K., Toth, R. P., Vidal, M., Jagaraj, C. J., et al. (2017). Protein quality control and the amyotrophic lateral sclerosis/frontotemporal dementia continuum. Front. Mol. Neurosci. 10:119. doi: 10.3389/fnmol.2017.00119
Shanbhag, N. M., Evans, M. D., Mao, W., Nana, A. L., Seeley, W. W., Adame, A., et al. (2019). Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 7:77. doi: 10.1186/s40478-019-0723-5
Shen, X., Chen, J., Li, J., Kofler, J., and Herrup, K. (2016). Neurons in vulnerable regions of the Alzheimer’s disease brain display reduced ATM signaling. eNeuro 3:ENEURO.0124-15.2016. doi: 10.1523/ENEURO.0124-15.2016
Sheng, J. G., Mrak, R. E., and Griffin, W. S. (1998). Progressive neuronal DNA damage associated with neurofibrillary tangle formation in Alzheimer disease. J. Neuropathol. Exp. Neurol. 57, 323–328. doi: 10.1097/00005072-199804000-00003
Shkreta, L., and Chabot, B. (2015). The RNA splicing response to DNA damage. Biomolecules 5, 2935–2977. doi: 10.3390/biom5042935
Silva, A. R., Santos, A. C., Farfel, J. M., Grinberg, L. T., Ferretti, R. E., Campos, A. H., et al. (2014). Repair of oxidative DNA damage, cell-cycle regulation and neuronal death may influence the clinical manifestation of Alzheimer’s disease. PLoS One 9:e99897. doi: 10.1371/journal.pone.0099897
Siu, P. M., Pei, X. M., Teng, B. T., Benzie, I. F., Ying, M., Wong, S. H., et al. (2011). Habitual exercise increases resistance of lymphocytes to oxidant-induced DNA damage by upregulating expression of antioxidant and DNA repairing enzymes. Exp. Physiol. 96, 889–906. doi: 10.1113/expphysiol.2011.058396
Stetler, R. A., Gao, Y., Zukin, R. S., Vosler, P. S., Zhang, L., Zhang, F., et al. (2010). Apurinic/apyrimidinic endonuclease APE1 is required for PACAP-induced neuroprotection against global cerebral ischemia. Proc. Natl. Acad. Sci. U S A 107, 3204–3209. doi: 10.1073/pnas.1000030107
Stott, R. T., Kritsky, O., and Tsai, L. H. (2021). Profiling DNA break sites and transcriptional changes in response to contextual fear learning. PLoS One 16:e0249691. doi: 10.1371/journal.pone.0249691
Su, J. H., Deng, G., and Cotman, C. W. (1997). Neuronal DNA damage precedes tangle formation and is associated with up-regulation of nitrotyrosine in Alzheimer’s disease brain. Brain Res. 774, 193–199. doi: 10.1016/s0006-8993(97)81703-9
Suberbielle, E., Sanchez, P. E., Kravitz, A. V., Wang, X., Ho, K., Eilertson, K., et al. (2013). Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat. Neurosci. 16, 613–621. doi: 10.1038/nn.3356
Sykora, P., Misiak, M., Wang, Y., Ghosh, S., Leandro, G. S., Liu, D., et al. (2015). DNA polymerase beta deficiency leads to neurodegeneration and exacerbates Alzheimer disease phenotypes. Nucleic Acids Res. 43, 943–959. doi: 10.1093/nar/gku1356
Thompson, L. H., and Limoli, C. L. (2000). “Origin, recognition, signaling and repair of DNA double-strand breaks in mammalian cells,” in Madame Curie Bioscience Database [Internet]. Austin, TX: Landes Bioscience.
Tiwari, V. K., Burger, L., Nikoletopoulou, V., Deogracias, R., Thakurela, S., Wirbelauer, C., et al. (2012). Target genes of topoisomerase IIbeta regulate neuronal survival and are defined by their chromatin state. Proc. Natl. Acad. Sci. U S A 109, E934–E943. doi: 10.1073/pnas.1119798109
Ulas, J., and Cotman, C. W. (1996). Dopaminergic denervation of striatum results in elevated expression of NR2A subunit. Neuroreport 7, 1789–1793. doi: 10.1097/00001756-199607290-00020
Uyeda, A., Onishi, K., Hirayama, T., Hattori, S., Miyakawa, T., Yagi, T., et al. (2020). Suppression of DNA double-strand break formation by DNA polymerase beta in active DNA demethylation is required for development of hippocampal pyramidal neurons. J. Neurosci. 40, 9012–9027. doi: 10.1523/JNEUROSCI.0319-20.2020
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Vasquez, V., Mitra, J., Hegde, P. M., Pandey, A., Sengupta, S., Mitra, S., et al. (2017). Chromatin-bound oxidized alpha-synuclein causes strand breaks in neuronal genomes in in vitro models of Parkinson’s disease. J. Alzheimers Dis. 60, S133–S150. doi: 10.3233/JAD-170342
Vilela, T. C., de Andrade, V. M., Radak, Z., and de Pinho, R. A. (2020). The role of exercise in brain DNA damage. Neural Regen. Res. 15, 1981–1985. doi: 10.4103/1673-5374.282237
Vilela, T. C., Muller, A. P., Damiani, A. P., Macan, T. P., da Silva, S., Canteiro, P. B., et al. (2017). Strength and aerobic exercises improve spatial memory in aging rats through stimulating distinct neuroplasticity mechanisms. Mol. Neurobiol. 54, 7928–7937. doi: 10.1007/s12035-016-0272-x
Vucic, S., and Kiernan, M. C. (2006). Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain 129, 2436–2446. doi: 10.1093/brain/awl172
Wang, J., Jacob, N. K., Ladner, K. J., Beg, A., Perko, J. D., Tanner, S. M., et al. (2009). RelA/p65 functions to maintain cellular senescence by regulating genomic stability and DNA repair. EMBO Rep. 10, 1272–1278. doi: 10.1038/embor.2009.197
Wang, W. Y., Pan, L., Su, S. C., Quinn, E. J., Sasaki, M., Jimenez, J. C., et al. (2013). Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 16, 1383–1391. doi: 10.1038/nn.3514
Wang, H., and Xu, X. (2017). Microhomology-mediated end joining: new players join the team. Cell Biosci. 7:6. doi: 10.1186/s13578-017-0136-8
Weissman, L., Jo, D. G., Sorensen, M. M., de Souza-Pinto, N. C., Markesbery, W. R., Mattson, M. P., et al. (2007). Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 35, 5545–5555. doi: 10.1093/nar/gkm605
Weller, J., and Budson, A. (2018). Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res. 27:F1000 Faculty Rev-1161. doi: 10.12688/f1000research.14506.1
Welty, S., Teng, Y., Liang, Z., Zhao, W., Sanders, L. H., Greenamyre, J. T., et al. (2018). RAD52 is required for RNA-templated recombination repair in post-mitotic neurons. J. Biol. Chem. 293, 1353–1362. doi: 10.1074/jbc.M117.808402
Wezyk, M., Szybinska, A., Wojsiat, J., Szczerba, M., Day, K., Ronnholm, H., et al. (2018). Overactive BRCA1 affects presenilin 1 in induced pluripotent stem cell-derived neurons in Alzheimer’s disease. J. Alzheimers Dis. 62, 175–202. doi: 10.3233/JAD-170830
Wu, W., Hill, S. E., Nathan, W. J., Paiano, J., Callen, E., Wang, D., et al. (2021). Neuronal enhancers are hotspots for DNA single-strand break repair. Nature 593, 440–444. doi: 10.1038/s41586-021-03468-5
Yang, J. L., Lin, Y. T., Chuang, P. C., Bohr, V. A., and Mattson, M. P. (2014). BDNF and exercise enhance neuronal DNA repair by stimulating CREB-mediated production of apurinic/apyrimidinic endonuclease 1. Neuromol. Med. 16, 161–174. doi: 10.1007/s12017-013-8270-x
Yang, J. L., Sykora, P., Wilson, D. M., Mattson, M. P. 3rd, and Bohr, V. A. (2011). The excitatory neurotransmitter glutamate stimulates DNA repair to increase neuronal resiliency. Mech. Ageing Dev. 132, 405–411. doi: 10.1016/j.mad.2011.06.005
Yang, J. L., Tadokoro, T., Keijzers, G., Mattson, M. P., and Bohr, V. A. (2010). Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated up-regulation of apurinic endonuclease 1. J. Biol. Chem. 285, 28191–28199. doi: 10.1074/jbc.M109.082883
Yang, J. L., Weissman, L., Bohr, V. A., and Mattson, M. P. (2008). Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair (Amst). 7, 1110–1120. doi: 10.1016/j.dnarep.2008.03.012
Yu, H., Harrison, F. E., and Xia, F. (2018). Altered DNA repair; an early pathogenic pathway in Alzheimer’s disease and obesity. Sci. Rep. 8:5600. doi: 10.1038/s41598-018-23644-4
Yu, H., Su, Y., Shin, J., Zhong, C., Guo, J. U., Weng, Y. L., et al. (2015). Tet3 regulates synaptic transmission and homeostatic plasticity via DNA oxidation and repair. Nat. Neurosci. 18, 836–843. doi: 10.1038/nn.4008
Zang, D. W., Lopes, E. C., and Cheema, S. S. (2005). Loss of synaptophysin-positive boutons on lumbar motor neurons innervating the medial gastrocnemius muscle of the SOD1G93A G1H transgenic mouse model of ALS. J. Neurosci. Res. 79, 694–699. doi: 10.1002/jnr.20379
Zhang, H., Li, Q., Graham, R. K., Slow, E., Hayden, M. R., Bezprozvanny, I., et al. (2008). Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington’s disease. Neurobiol. Dis. 31, 80–88. doi: 10.1016/j.nbd.2008.03.010
Zhang, J., Perry, G., Smith, M. A., Robertson, D., Olson, S. J., Graham, D. G., et al. (1999). Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am. J. Pathol. 154, 1423–1429. doi: 10.1016/S0002-9440(10)65396-5
Keywords: synaptic plasticity, neural plasticity, DNA damage, DNA repair, neurodegeneration
Citation: Konopka A and Atkin JD (2022) The Role of DNA Damage in Neural Plasticity in Physiology and Neurodegeneration. Front. Cell. Neurosci. 16:836885. doi: 10.3389/fncel.2022.836885
Received: 16 December 2021; Accepted: 09 May 2022;
Published: 23 June 2022.
Edited by:
Xuejun Li, University of Illinois at Chicago, United StatesReviewed by:
Rosanna Parlato, University of Heidelberg, GermanyCopyright © 2022 Konopka and Atkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Konopka, YW5uYS5rb25vcGthQG1xLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.