 Junhui Chen
Junhui Chen Mingchang Li2†
Mingchang Li2† Kun Xiong
Kun Xiong- 1Department of Human Anatomy and Neurobiology, School of Basic Medical Science, Central South University, Changsha, Hunan, China
- 2Department of Neurosurgery, Renmin Hospital of Wuhan University, Wuhan, China
- 3Department of Neurosurgery, 904th Hospital of Joint Logistic Support Force of PLA, Wuxi Clinical College of Anhui Medical University, Wuxi, China
Subarachnoid haemorrhage (SAH) is a common cerebrovascular disease with high disability and mortality rates worldwide. The pathophysiological mechanisms involved in an aneurysm rupture in SAH are complex and can be divided into early brain injury and delayed brain injury. The initial mechanical insult results in brain tissue and vascular disruption with hemorrhages and neuronal necrosis. Following this, the secondary injury results in diffused cerebral damage in the peri-core area. However, the molecular mechanisms of neuronal death following an aneurysmal SAH are complex and currently unclear. Furthermore, multiple cell death pathways are stimulated during the pathogenesis of brain damage. Notably, particular attention should be devoted to necrosis, apoptosis, autophagy, necroptosis, pyroptosis and ferroptosis. Thus, this review discussed the mechanism of neuronal death and its influence on brain injury after SAH.
Introduction
Subarachnoid haemorrhage (SAH) is a common cerebrovascular disease, accounting for approximately 5% of all strokes. According to the latest report by the European Stroke Association, the incidence of SAH is reported to be more than 9/100,000 (Feigin et al., 2009; GBD 2019 Stroke Collaborators, 2021). The incidences of SAH in Asia and Iceland rank the highest, with Japan accounting for approximately 22.7/100,000 cases. Moreover, more than 85% of all SAH cases are caused by intracranial aneurysms, leading to high mortality and disability rates (Steiner et al., 2013). A meta-analysis reported that the short-term mortality of SAH is as high as 8.3–66.7%, the mean median of direct death before admission was 8.3% and the overall favorable prognosis rate was 36–55% (Nieuwkamp et al., 2009). The majority of SAH survivors usually have long-term cognitive dysfunction, affective disorders, loss of hearing and smell and decreased quality of life after surgery (Rinkel and Algra, 2011). Additionally, >60% of SAH survivors have memory problems, >75% have language problems (Al-Khindi et al., 2010) and almost 95% of patients with SAH have at least one cognitive or affective disorder (Passier et al., 2010). The treatment for patients with SAH is lengthy and expensive, which places a serious burden on the national economy. Since the beginning of the 21st century, the average hospitalization cost of each patient with a SAH aneurysm in the first year in the United States exceeded $65,000 (Qureshi et al., 2007) while those in the United Kingdom paid £510 million a year (Rivero-Arias et al., 2010).
The pathophysiological processes and mechanisms involved in aneurysm rupture in SAH are complex. Nonetheless, a previous study divided the pathological process into two stages: early brain injury (EBI) and delayed brain injury (DBI) (Chen S. et al., 2014). Additionally, various animal and cell experimental studies on SAH report that a large number of red blood cells enter the subarachnoid space, brain parenchyma or ventricle and their subsequent rapid lysis produce a large amount of hemoglobin and corresponding decomposition products (Macdonald, 2014; Lawton and Vates, 2017). These factors exert various neurotoxic effects and can subsequently induce secondary brain injury after SAH (Bulters et al., 2018; Chen et al., 2019c, 2021). Therefore, EBI is considered the most important cause of acute brain damage and neurological dysfunction in patients with SAH. Currently, it is gaining increasing attention as a research hotspot. Neuronal death is considered a major cause of EBI (Macdonald and Schweizer, 2017). Hence, a reduction in the number of neuronal deaths could improve nerve dysfunction and eventually improve the prognosis of patients (Dong et al., 2016; Ding et al., 2022). Thus, further study on the mechanism of EBI and neuronal death is vitally important. In the present review, we discuss the mechanism of neuronal death after SAH and its influence on brain injury (Table 1).

Table 1. Neuronal death pathways and associated morphological and biochemical hallmark features.
Neuronal necrosis
Neuronal necrosis is the death of neurons induced by extreme, chemical or severe pathological factors and is a passive pathological process that does not consume energy (Kroemer et al., 2009). The morphology of necrotic neurons exhibits increased neuronal membrane permeability, neuronal organelle oedema and deformation, nuclear changes, DNA dissolution, intracellular calcium overload, sodium ion influx, potassium ion outflow, plasma concentration changes (Friedrich et al., 2012). Furthermore, degraded lysosomes release various cellular autolysins that eventually destroy the plasma membrane and cause cell death (Kroemer et al., 2009). This pathological process is also an acute passive neuronal death process that is observed after SAH. The specific mechanism and molecular process of neuronal necrosis after SAH are scarce, to the best of our knowledge, as it is a passive, unregulated and irreversible pathological process (Zhu et al., 2012). Friedrich et al. (2012) reported that the activation of endothelial and parenchymal cell apoptosis and neuronal necrosis occurred within 10 min after SAH and is associated with poor outcomes. Thus, the prevention of apoptosis and necrosis could potentially improve impaired functions of the circumventricular organs after SAH (Edebali et al., 2014). However, neuronal necrosis is the most common form of neuronal death in EBI after SAH and is also the main cause of early death, coma and severe neurological dysfunction in patients after SAH (Friedrich et al., 2012). Although neuronal necrosis is a passive and unregulated process, early clinical intervention against its inducing factors can significantly reduce its occurrence (Zhu et al., 2012). Therefore, identifying and inhibiting the inducers of neuronal necrosis is key to regulating this process, which could also be a new research direction for neuronal necrosis after SAH. Additionally, Figure 1 briefly summarizes the clinical causes of neuronal necrosis after SAH.
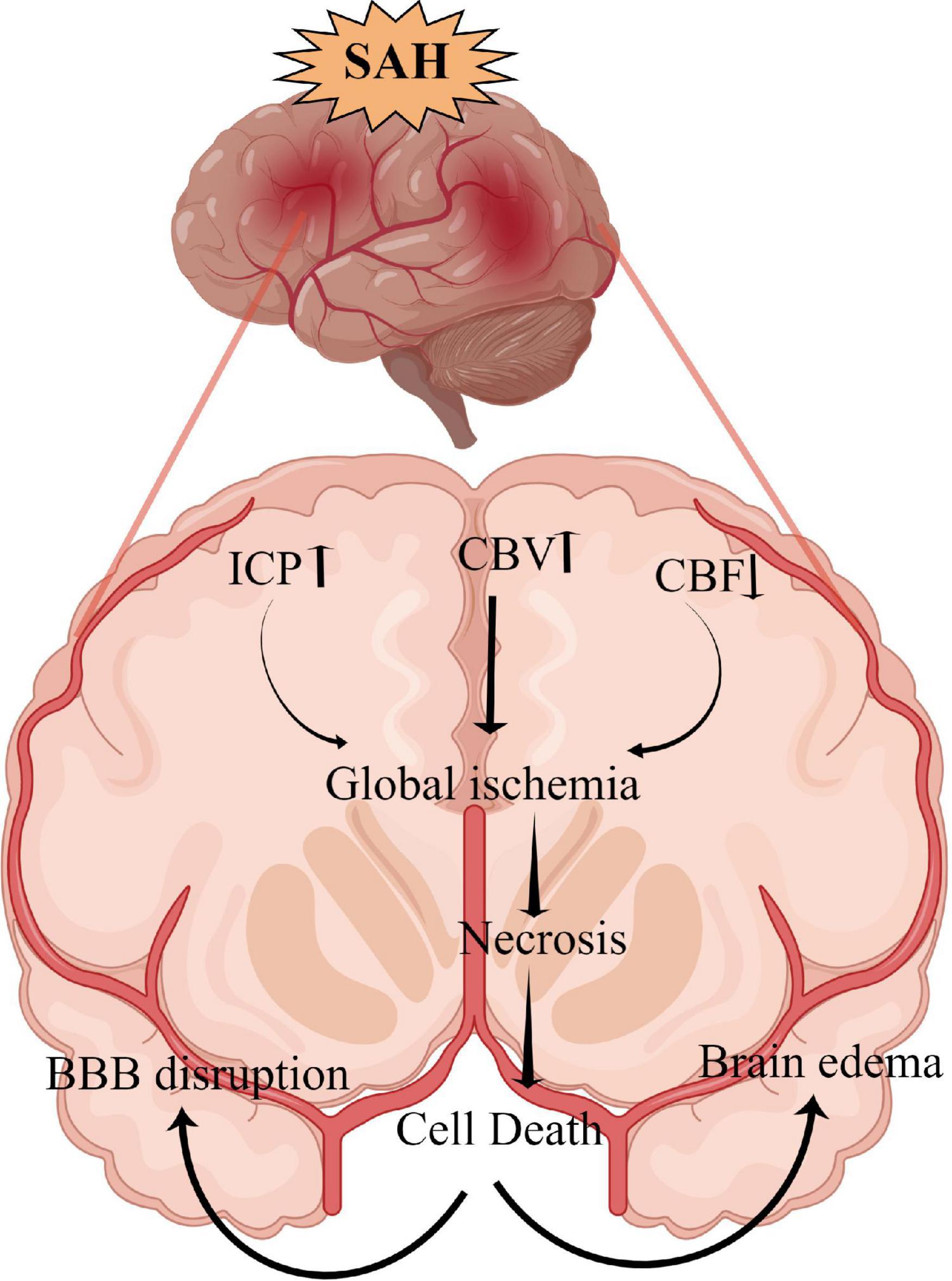
Figure 1. The overall scheme of neuronal necrosis from subarachnoid hemorrhage to early brain injury (EBI).
Intracranial hypertension
SAH can lead to extensive hemorrhage, including intraventricular hematoma and intracerebral hematoma, and the mass effect directly leads to increased intracranial pressure (ICP) (Macdonald and Schweizer, 2017). Heuer et al. (2004) reported that intracranial hypertension (>20 mmHg) occurs in more than 50% of patients with SAH, with rates as high as 60–70% in poor-grade SAH. Similarly, Zoerle et al. (2015) reported that >80% of patients with SAH had at least one episode of intracranial hypertension following SAH and 36% had an average ICP of >20 mmHg during hospitalization. Increased ICP and complications, such as cerebral ischemia and brain oedema, can directly induce neuronal necrosis, affecting the final prognosis of patients (Said et al., 2021; Narotam et al., 2022). Although the relationship between ICP and prognosis remains unclear, the definite presence of intracranial hypertension after SAH increases the mortality rates and leads to poor outcomes. Moreover, intracranial hypertension and its etiology (hydrocephalus, cerebral oedema, intracerebral hematoma and intraventricular hematoma.) can be used as an independent predictor of poor patient outcome (Claassen et al., 2002; Heuer et al., 2004; Zoerle et al., 2015; Wan et al., 2016).
Currently, there are no recommended guidelines for the treatment of intracranial hypertension after SAH (Svedung Wettervik et al., 2022). From our experience, early-time intraventricular ICP monitoring is a very important therapeutic strategy to relieve ICP in patients with SAH. It is also essential to monitor ICP and cerebral perfusion pressure to avoid severe cerebral ischemia, which can be avoided by maintaining appropriate perfusion pressure regardless of vasospasm (Chen et al., 2018a). Additionally, intraventricular ICP monitoring can aid in properly draining cerebrospinal fluid, reducing ICP, improving obstructive hydrocephalus, draining blood-cerebrospinal fluid to improve vasospasm and reducing toxic brain injury (He et al., 2015; Chen J. et al., 2016). However, not all patients with SAH require ventricular ICP monitoring but it can be considered for poor-grade patients with SAH as the invasive procedure could increase the risk of additional injury and intracranial infection (Chen et al., 2018a). Olson et al. (2013) reported that continuous cerebrospinal fluid drainage and intermittent ICP monitoring increased the incidence of intracranial infection, cerebrospinal fluid leakage and bleeding (clinicaltrials.gov identifier NCT01169454) in a randomized controlled study of 60 patients. Hence, it is necessary to adopt individualized precision therapy to control intracranial hypertension, thereby reducing neuronal necrosis and ameliorating EBI.
Cerebral ischemia
Cerebral ischemia is a common complication after SAH, with >30% of patients experiencing delayed cerebral ischemia (Stienen et al., 2014). Decreased cerebral hypoperfusion leads to cerebral ischemia and cerebral infarction, ultimately inducing severe neurological dysfunction, brain injury and even death (Güresir et al., 2022; Hostettler et al., 2022). Cerebral ischemia is caused by various factors, including cerebral vasospasm, micro thrombosis, blood-brain barrier destruction, inflammatory reactions and cerebral vascular self-regulation dysfunction (Macdonald, 2014). After SAH, a large amount of hemoglobin enters the subarachnoid space to directly stimulate intracranial blood vessels, thereby activating calcium channels in vascular smooth muscle cells and resulting in smooth muscle contraction and vascular spasm (Pluta, 2005). Endothelin-1 (ET-1) is produced in large quantities by astrocytes and white blood cells during inflammation or cerebral ischemia and has a strong vasoconstriction function (Chen et al., 2018b; Croci et al., 2019). Previous studies have demonstrated that many molecular signaling pathways, such as p38/MAPK (Huang et al., 2017), RhoA (González-Montelongo et al., 2018) and Nrf2-ARE (Zhang et al., 2010; Zhao et al., 2016), are involved in the development of cerebrovascular spasms (CVS) after SAH. Therefore, early diagnosis and treatment of CVS and cerebral ischemia could improve the prognosis of patients. Regrettably, a large number of drugs are effective in basic research but ineffective in clinical trials. For example, clazosentan has been considered for the targeted treatment of CVS; however, a Phase 3 clinical trial found that while it improved CVS, it did not improve the long-term outcomes for patients with SAH (Macdonald et al., 2011). Previous studies report that statins can protect against CVS and EBI after SAH (Chen J. H. et al., 2016; Chen et al., 2018b, 2020a, 2021); however, another study indicates that statins together with nimodipine can reduce CVS and cerebral infarction but have no benefits on 6 months of clinical outcome or 30-day all-cause mortality (Chen et al., 2020b). Additionally, magnesium is gaining widespread attention; however, it has also proven to be ineffective in Phase 2 randomized controlled trials (Mees et al., 2012). Recent clinical studies on cilostazol have demonstrated that it can significantly reduce the incidence of cerebral infarction, delay ischemia after SAH (Sugimoto et al., 2018), reduce the occurrence of secondary stroke and improve the prognosis of patients (Shinohara et al., 2010; Toyoda et al., 2019). However, its effect on ameliorating CVS and micro thrombosis after SAH and improving prognosis require further study using a large sample, multicentre, randomized controlled trial. Additionally, the relationship between CVS and neuronal necrosis after SAH remains unclear and requires further exploration.
Brain oedema
Brain oedema after SAH can be divided into three types: vasogenic cerebral oedema, cytotoxic cerebral oedema, and mixed cerebral oedema (Nag et al., 2009). Vasogenic cerebral oedema is the most common type of brain oedema in the clinical setting, whereas cytotoxic cerebral oedema is caused by increased permeability due to cell membrane disruption and swelling (Kaufmann and Cardoso, 1992). Notably, after SAH, different types of cerebral oedema mainly occur at different periods and stages (Nag et al., 2009). The expression of matrix metalloproteinase-9 is increased after SAH, which leads to the destruction of tight junction proteins between the vascular endothelium and subsequently results in the increased permeability of the blood-brain barrier and aggravated brain oedema (Lee et al., 2007; Sun X. G. et al., 2019). The activation of aquaporin-4 (AQP4) is also an important mechanism affecting brain oedema, wherein water molecules enter brain tissues in large quantities through the activated AQP4 and then aggravate brain oedema and neuronal necrosis in cytotoxic cerebral oedema (Huang et al., 2014; Chen J. H. et al., 2016). In vasogenic cerebral oedema, AQP4 plays an important role in improving oedema, which was confirmed by the significantly reduced rate of resolution of cerebral oedema in AQP4 knockout cells (Papadopoulos et al., 2004; Tourdias et al., 2011). Progressive cerebral oedema leads to increased ICP and malignant intracranial hypertension and subsequently to cerebral ischemia and neuronal necrosis (Bjerring et al., 2011; Halstead and Geocadin, 2019). Therefore, the early management of cerebral oedema after SAH is vitally important in improving the prognosis of patients with SAH, especially those with poor-grade aneurysmal SAH. Powell et al. (1976) also reported that the extent of swelling was significantly decreased with a dehydrating agent (mannitol) treatment and osmolality elevation decreased the extent of eventual myocardial necrosis. Additionally, dehydrating agents also improve brain tissue oxygenation by limiting astrocyte swelling and restoring capillary perfusion, thereby alleviating early brain injury (Halstead and Geocadin, 2019). Hence, exploring the treatment of neuronal necrosis with dehydrating drugs and AQP4-based targeted drugs has therapeutic potential.
Neuronal apoptosis
The mechanical compression of hematoma, toxic damage of hemoglobin and oxidative stress lead to the extensive apoptosis of cortical neurons and hippocampal neurons after SAH (Chen J. H. et al., 2016; Chen et al., 2018b, 2019c). The complex process of neuronal apoptosis involves exogenous death receptor pathways, P53-mediated apoptosis pathways, caspase-dependent and -non-dependent pathways and endogenous mitochondrial pathways (Gao et al., 2000; Sendoel et al., 2010). The BcL-2 gene family promotes the release of cytochrome C from the mitochondrial membrane and its binding with apoptosis protease activating factor 1 (Apaf-1) to regulate mitochondrial membrane permeability (Hasegawa et al., 2011; Jaafaru et al., 2019). The c/Apaf-1 complex further regulates the activation of the apoptosis factor Caspase-3 and induces neuronal apoptosis (Hasegawa et al., 2011). Recent study report that a novel membrane-bound bile acid receptor (TGR5) is involved in the process of oxidative stress and apoptosis, wherein TGR5 and its agonists inhibit neuronal apoptosis and improve neural function via the cAMP/PKCε/ALDH2 pathway in a rat SAH model (Zuo et al., 2019). Furthermore, protein-coupled receptor 30 (GPR30) and its agonist G1 are also involved in neuronal apoptosis after SAH, which can inhibit neuronal apoptosis through the src/EGFR/STAT3 signaling pathway (Peng et al., 2019). A previous study showed that statins can alleviate neuronal apoptosis and EBI by inhibiting caspase-3 after SAH (Chen J. H. et al., 2016). Additionally, Netrin-1 can activate PPARγ and Bcl-2 expressions, inhibit the proapoptotic factors Bax and NF-κB and ameliorate EBI after SAH (Chen et al., 2019c). Similarly, Gao et al. also reported that the early overexpression of PDK4 after SAH has the potential to attenuate neuronal apoptosis by reducing oxidative stress via the ROS/ASK1/P38 pathway (Gao X. et al., 2022). A recent study also demonstrated that early fluoxetine administration can protect against apoptosis by regulating the Notch1/ASK1/p38 MAPK signaling pathway, thereby ameliorating EBI after SAH (Liu M. et al., 2022). Neuronal apoptosis caused by SAH is widely and thoroughly studied, involving many signaling pathways and molecular mechanisms. Future studies need to assess and identify specific molecular mechanisms that can be regulated in the induction of apoptosis, thereby contributing to the development of clinically viable treatment options (Figure 2).
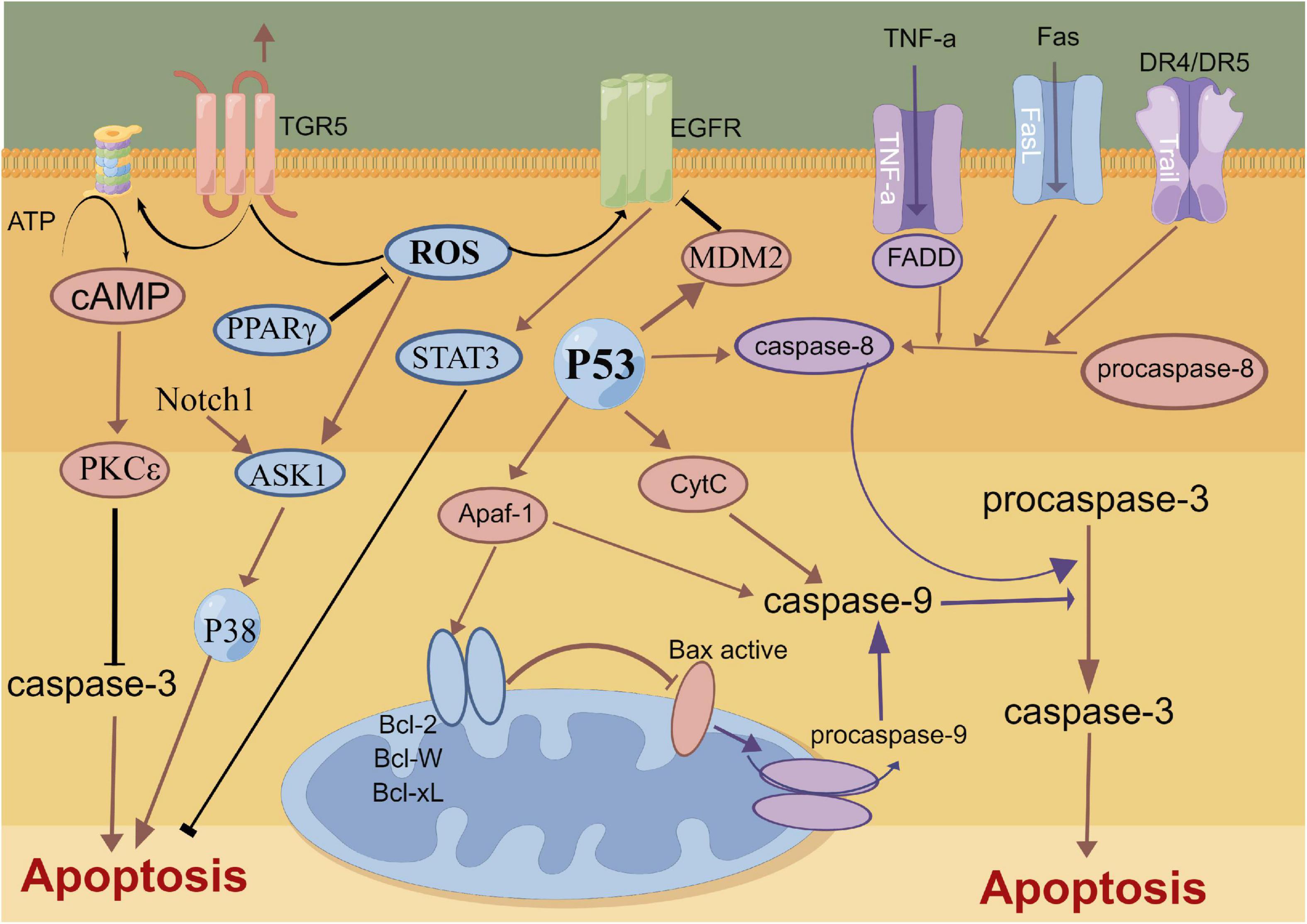
Figure 2. Molecular mechanisms of apoptosis from subarachnoid hemorrhage to early brain injury (EBI).
Neuronal pyroptosis
Pyroptosis, a newly reported cell death mechanism, is a form of programmed neuronal cell death initiated by Caspase-1 and an important cause of neurological damage (Bergsbaken et al., 2009). Pyroptosis, which was first proposed in 2001, differs from apoptosis with respect to its mechanism, which is associated with inflammation (Aglietti et al., 2016; Liu et al., 2016; Vande Walle and Lamkanfi, 2016). Pyroptosis is a programmed cell death mediated by GSDMD (gasdermin protein family, including Gasdermin A, Gasdermin B, Gasdermin C, DFNA5 and DFNB59), with the inflammasome playing an important role in the pyroptosis process (Kayagaki et al., 2015). Gasdermin, an important element of cell pyrosis, is also widely distributed in the central nervous system (Bergsbaken et al., 2009). Hence, neuronal pyrosis plays an important role in the development of central nervous system-related diseases and is an important mechanism of brain injury (Ito et al., 2015; Van Rossom et al., 2015; An et al., 2019; Kuwar et al., 2019; She et al., 2019; Sun Y. B. et al., 2019). In the classic Caspase-1-induced cell pyroptosis process, inflammasomes (NLRP1, NLRP3, NLRC4 and AIM2) sense the danger signal and induce the connector molecule ASC to activate Caspase-1, thus cleaving GSDMD (Swanson et al., 2019; Chen et al., 2022). Subsequently, cellular pores are formed on the cell membrane, which results in water and ion influx and ultimately cell swelling and death (Shi et al., 2015; Broz and Dixit, 2016). Non-classical pathway-induced cell pyroptosis is activated by the oligomerization of the non-classical inflammasome (in mice, a complex of Caspase-11 precursors and lipopolysaccharides), which binds the intracellular Caspase-11 precursors to lipopolysaccharides (Abu Khweek and Amer, 2020). Following this, GSDMD is cleaved by activation of the Caspase-4/5/11 pathway, which is independent of Caspase-1, to induce cell pyroptosis (Kayagaki et al., 2013; Diamond et al., 2015). Moreover, in animal ischemic stroke experiments, microglial TREM-1 induced the activation of spleen tyrosine kinase (SYK), which increased the level of the N-terminal fragment of GSDMD (Liang et al., 2020). Thereafter, pores were formed in the microglia to promote the release of intracellular inflammatory factors, leading to the pyroptosis of the microglia. Additionally, microglial TREM-1 receptors bind to SYK and induce neuroinflammatory damage after stroke (Xu et al., 2019). Kuwar et al. (2019) also reported that inflammasome inhibitors could significantly inhibit the pyroptosis of neurons and improve brain injury after traumatic brain injury. Additionally, statins have been reported to improve neurological outcomes and reduce neuronal death against neural pyroptosis and neuroinflammation in an animal SAH model (Chen et al., 2021). Gu et al. (2022) demonstrated that the activation of RKIP could attenuate neuronal pyroptosis and brain injury after ICH through the ASC/Caspase-1/GSDMD pathway. Similarly, the inhibition of the pyroptosis of astrocytes and microglia in the central nervous system can also indirectly inhibit the pyroptosis of neurons, thereby protecting neurons and alleviating brain injury (Chavez et al., 2021; Kerr et al., 2022). Collectively, these studies highlight the sophisticated interplay between pyroptosis-related molecules and inflammasome components that could be leveraged to prevent hyperinflammation after SAH (Figure 3). Thus, pyroptosis can be a new research direction in the exploration of the mechanism of SAH.
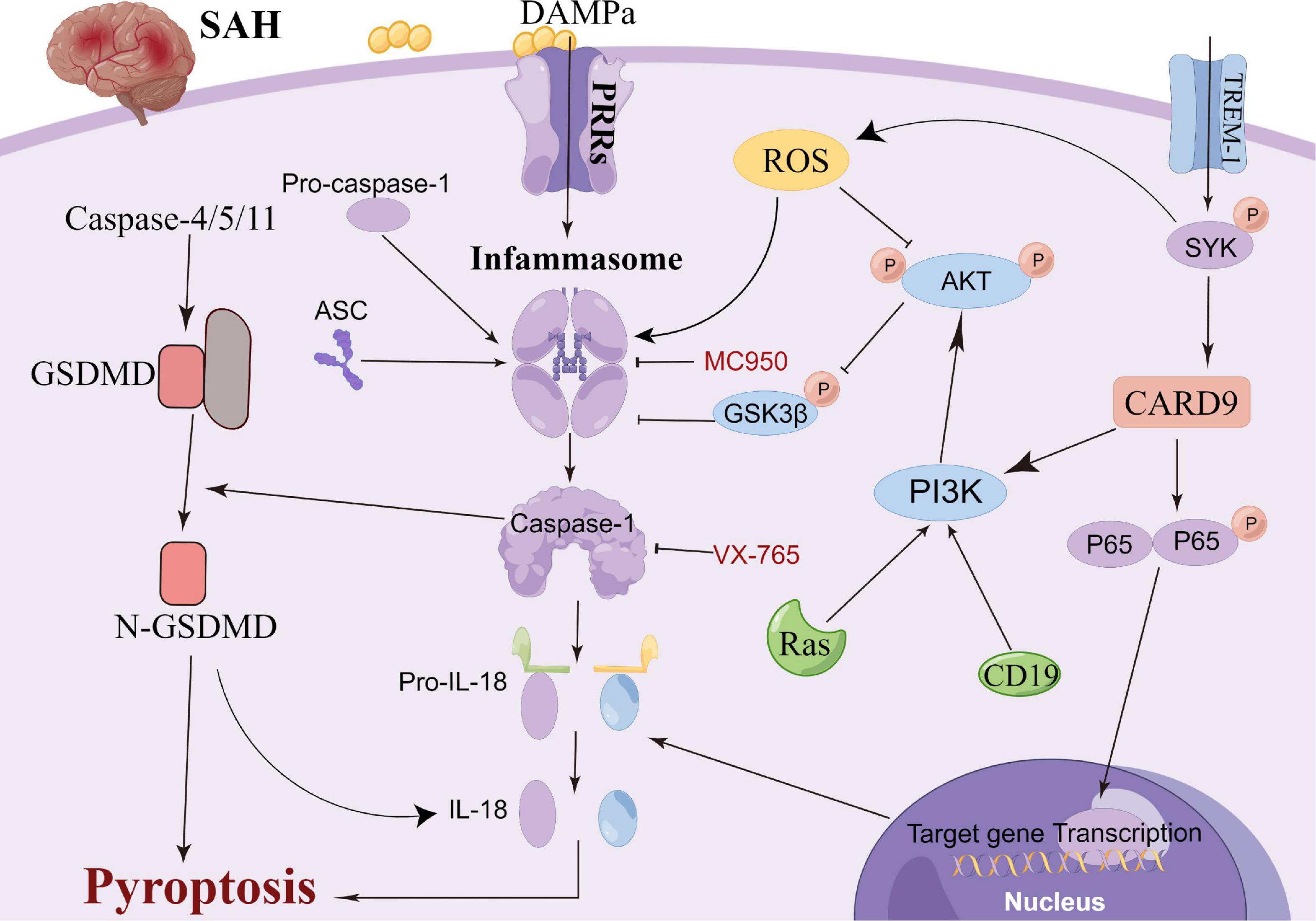
Figure 3. Molecular mechanisms of pyroptosis from subarachnoid hemorrhage to early brain injury (EBI).
Neuronal autophagy
Autophagy is a cellular repair process that stabilizes the intracellular environment via the lysosomal degradation of selected cytoplasmic components and programmed death of old, dysfunctional or unnecessary cytoplasmic entities (Ho et al., 2018). Autophagy is tightly regulated and involves multiple molecular mechanisms; autophagy plays an important role after SAH (Chen J. et al., 2014; Li et al., 2017; Ho et al., 2018; Sun C. et al., 2019; Sun C. M. et al., 2019). The main morphological changes have been observed in the autophagosomes and autophagolysosomes under an electron microscope (Harris and Rubinsztein, 2012; Chen J. et al., 2014; Li et al., 2017; Ho et al., 2018; Sun C. et al., 2019; Sun C. M. et al., 2019). Lee et al. (2009) reported that neuronal autophagy was significantly activated in the early injury stage of SAH, and different modes of autophagy activation in different parts of SAH indicated different injury mechanisms. Wang et al. (2012) reported that a small amount of autophagy exists in normal brain neurons; however, it is activated in large quantities after SAH. Autophagy can improve cerebral oedema, protect the blood-brain barrier, reduce cortical neuronal apoptosis and improve clinical prognosis after intervention with rapamycin (RAP) and 3-methyladenine (3-MA) (Hu X. et al., 2022; Pang et al., 2022). Lu et al. reported that melatonin enhances autophagy in brain vessel endothelial cells, then protect blood-brain barrier integrity. In the animal CVS model after SAH, the autophagy pathway was activated in the basilar artery wall after SAH, and CysC-induced autophagy played a beneficial role in preventing SAH-induced CVS (Liu et al., 2013). Additionally, many autophagy-related drugs have achieved good neuroprotective effects in basic research (Li et al., 2017; Chen et al., 2020a; Du et al., 2021; Chang et al., 2022). Sun C. et al. (2019) reported that osteopontin enhanced the autophagy response after SAH via the activation of the FAK-ERK signaling pathway, thereby improving long-term neurological function. Furthermore, Li and Han (2018) confirmed using in vivo and in vitro models that resveratrol increased the LC3-II/I ratio, AMPK phosphorylation and SIRT1 expression, thereby activating autophagy and reducing the release of inflammatory factors and neuronal apoptosis. Li et al. (2017) reported that fluoxetine significantly decreased the expression of NLRP3 and Caspase-1 and upregulated the expression of Beclin-1, which was reversed by the autophagy inhibitor 3-MA. Additionally, fluoxetine can enhance autophagy by inhibiting the activation of the NLRP3 inflammasome, thus playing a neuroprotective role, which could be attributed to the interaction between autophagy and pyroptosis (Li et al., 2017). Arachidonyl-2-chloroethylamide (ACEA), a highly selective CB1R agonist, was also reported to alleviate oxidative stress and neurological dysfunction by promoting mitophagy after SAH via the CB1R/Nrf1/PINK1 signaling pathway (Liu B. et al., 2022). Similarly, Zeng et al. (2021) demonstrated that the autophagy protein NRBF2 alleviated endoplasmic reticulum stress-associated neuroinflammation and oxidative stress by promoting autophagosome maturation, which occurs through interactions with Rab7 after SAH, and ultimately improving EBI. Hence, autophagy plays an important role in neuronal death, blood?brain barrier and CVS after SAH. Further studies focusing on elucidating the structural basis of the autophagy interaction for pyroptosis induction and developing autophagy-related drugs need to be explored in clinical trials (Figure 4).
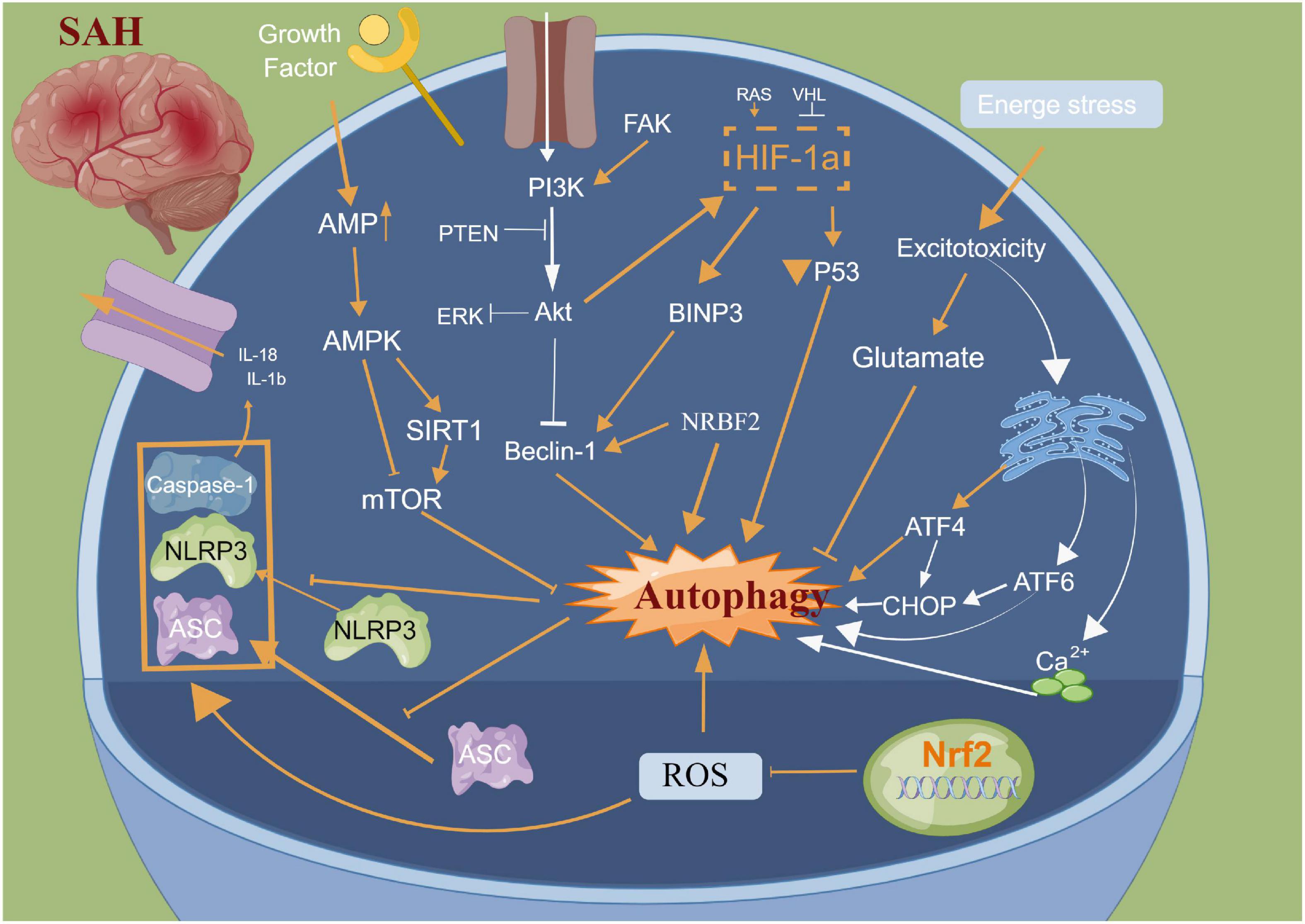
Figure 4. Molecular mechanisms of autophagy from subarachnoid hemorrhage to early brain injury (EBI).
Neuronal necroptosis
Necroptosis is a regulated form of cell death that relies heavily on receptor interactions of serine-threonine kinase 3 (RIPK3) and the mixed lineage kinase domain (MLKL) and usually presents with morphological characteristics of necrosis (Galluzzi et al., 2017). Unlike apoptosis, necroptosis is a caspase-independent mode of cell death. It is now known that serine/threonine kinases RIPK1, RIPK3 and MLKL are critical for necroptosis induced by the tumour necrosis factor (TNF) (Holler et al., 2000; He et al., 2009)superfamily, TLR3 or TLR4 and interferon receptors (Kaiser et al., 2013). TNF-α-induced necroptosis is the most characteristic signal transduction pathway. TNF-α homotrimer binds to TNF-1 to form a membrane signaling complex-1, which includes TRADD, RIPK1, TRAF2 and cIAP1/cIAP2, and subsequently activates NF-κB and MAP kinases (Chen and Goeddel, 2002). The polyubiquitination of RIPK1 by CIAP1/CIAP2 and LUBC is essential for the activation of the NF-κB signaling pathway (Gerlach et al., 2011). RIPK1 and RIPK3 in the necrosome are phosphorylated by their amino-terminal kinase domains, resulting in cell death through a signaling cascade (He et al., 2009). A previous review summarized that necroptosis is widely associated with various cardiovascular diseases, nerve regeneration diseases, kidney injury and other diseases (Galluzzi et al., 2017). Degterev et al. (2005) observed that necroptosis leads to delayed ischemic brain injury in mice through a mechanism that is different from apoptosis, thereby identifying a previously undescribed basic cell death pathway and providing a new therapeutic target for stroke. Chen et al. (2019a) reported that anti-TNF-α treatment can significantly improve endothelial cell necroptosis and blood-brain barrier damage and further improve nerve function after stroke. In a SAH experimental study, celastrol administration decreased the expression levels of necroptosis-related proteins RIPK3 and MLKL, which exhibited neuroprotective effects on EBI (Xu et al., 2021). Similarly, necrostatin-1, a small-molecule inhibitor of necroptosis, can potentially prevent SAH-induced necroptosis by suppressing the activity of the RIPK3/MLKL signaling pathway (Chen et al., 2019b). Therefore, the inhibition of necroptosis can significantly improve neural function, and its mechanism could be related to the phosphorylation of RIPK1 and RIPK3 (Shen et al., 2017; Yuan et al., 2019). Yang et al. (2018) reported that the specific and potent inhibitor of necroptosis Necrostatin-1 (Nec-1) mitigated SAH-induced synaptic impairments in the hippocampus by inhibiting necroptosis via the CREB-BDNF pathway. Additionally, Nec-1 has been reported to prevent CVS in animals models (Sahin et al., 2021). However, the clinical efficacy of drugs targeting the mechanism of necroptosis remains unclear owing to a lack of relevant clinical studies. Further studies on anti-necroptosis-related drugs that can be used in clinical settings against brain injury after SAH are essential. Additionally, clinical trials should be performed to evaluate the safety and clinical efficacy of anti-necroptosis-related drugs, such as Nec-1 (Figure 5).
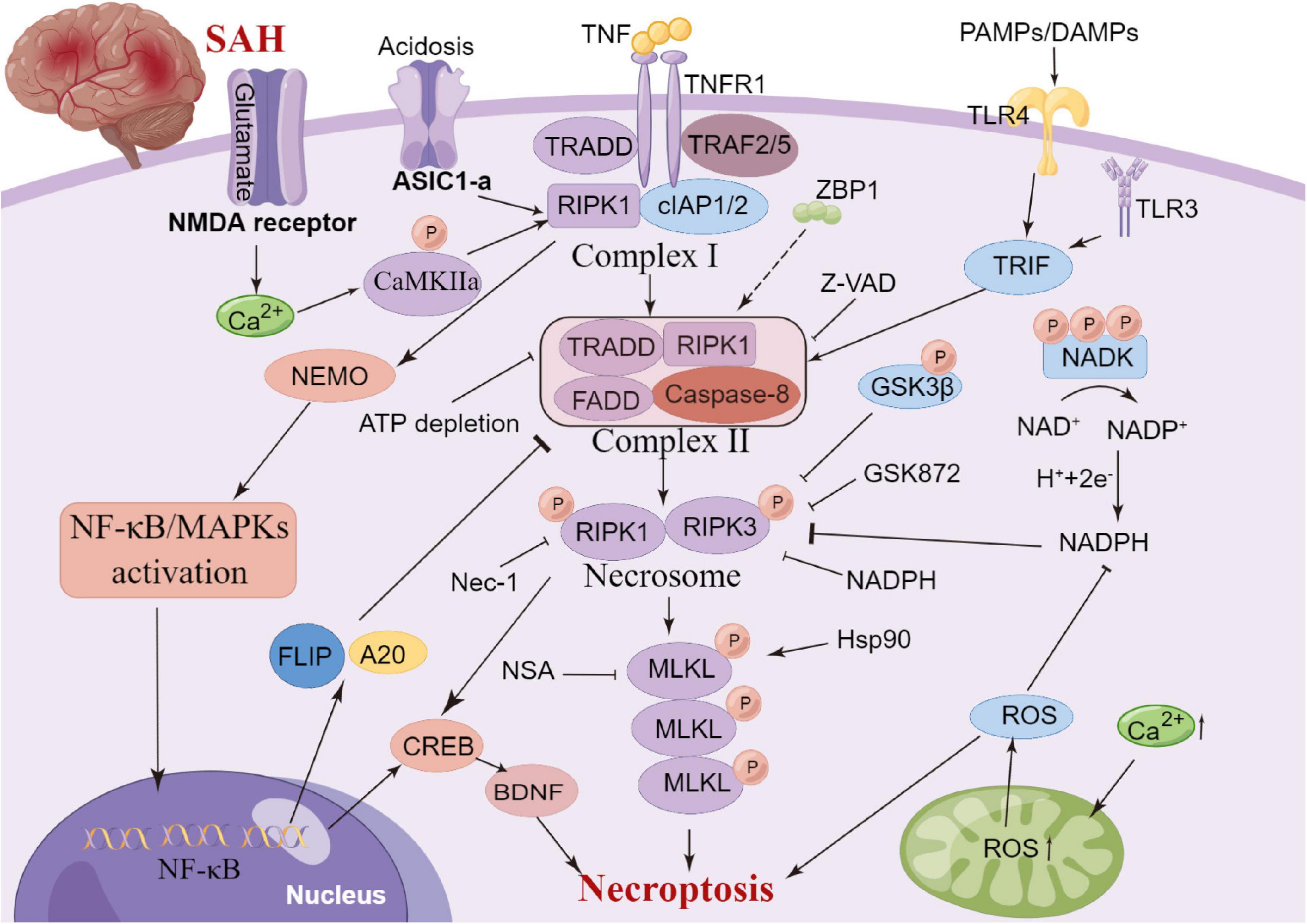
Figure 5. Molecular mechanisms of necroptosis from subarachnoid hemorrhage to early brain injury (EBI).
Neuronal ferroptosis
Recently, a newly identified cell death mechanism called ferroptosis has been widely explored. It is a non-apoptotic form of iron-dependent programmed cell death and differs from traditional cell death processes, such as apoptosis and autophagy (Dixon et al., 2012; Chen et al., 2020c). Moreover, ferroptosis cannot be completely inhibited by necroptosis/apoptosis/autophagy-related inhibitors and intracellular calcium overload (Zille et al., 2017). The classical morphological characteristics of ferroptosis are the disappearance of the mitochondrial cristae, significantly narrowed mitochondria, thickening of the lipid bilayer membrane and reduced cell connections that lead to cell separation (Chen et al., 2020c). Furthermore, the biological characteristics mainly include the metabolic dysfunction of iron ions, depletion of glutathione (GSH), accumulation of iron-dependent lipid reactive oxygen species (ROS) and decreased levels or inhibition of glutathione peroxidase 4 (GPX4) (Chen et al., 2020c). The mechanisms of ferroptosis are complex and involve various cell signaling pathways (Chen et al., 2020c, 2021). Recent studies have also confirmed that ferroptosis is widely observed in diseases of the central nervous system, including cerebral ischemia/reperfusion (Tuo et al., 2022), SAH (Gao S. et al., 2022), ICH (Dharmalingam et al., 2020) and traumatic brain injury (Rui et al., 2021). The GPX4–GSH–cysteine system, FSP1–CoQ10–NAD (P)H system and GCH1-BH4-DHFR system are the primary anti-ferroptosis systems. A better understanding of three primary anti-ferroptosis systems aids in developing better therapeutic strategies for patients with brain injury after SAH (Figure 6).
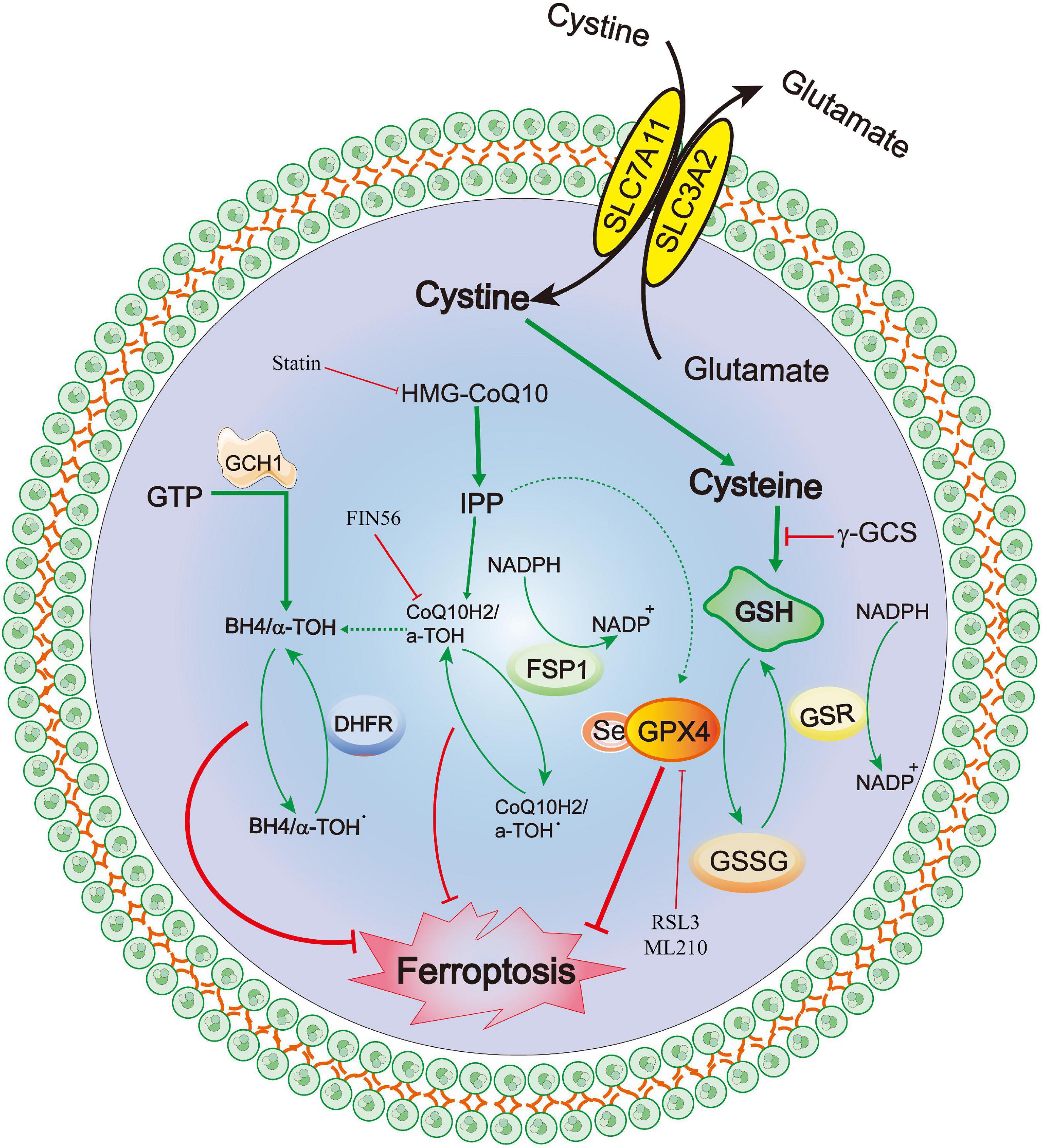
Figure 6. Molecular mechanisms of ferroptosis from subarachnoid hemorrhage to early brain injury (EBI).
GPX4–GSH–cysteine system
Ferroptosis was first reported to induce iron-dependent cell death via the small molecules erastin and 1S,3R-RSL3 (RSL3), a classical feature of ferroptosis is that it is independent of Caspase activation after erastin and RSL3 treatment (Dixon et al., 2012; Wang et al., 2016). Erastin targets the Xc- system (comprising SLC3A2 and SLC7A11), which transports cysteine into cells and glutamate out of cells, Cysteine is required for the synthesis of GSH, an important antioxidant that protects cells from oxidative damage (Dixon et al., 2012). Furthermore, erastin inhibits intracellular cysteine, leading to a significant decrease in intracellular GSH levels, ROS levels and oxidative damage (Dixon et al., 2012). GPX4 is the core of ferroptosis and requires GSH as a substrate for lipid repair (Yang et al., 2014). Reduced cysteine levels lead to the depletion of GSH levels and reduction of GPX4 levels and activity, resulting in the accumulation of unrepaired lipid peroxides and ferrous ions (Friedmann Angeli et al., 2014; Stockwell et al., 2017). GSH is a very important component of Fe2+ in unstable iron pools, it binds to Fe2+ to prevent lipid peroxidation, directly inhibiting GSH biosynthesis and triggering ferroptosis (Hider and Kong, 2011). Additionally, GPX4 can reduce the level of lipid hydrogen peroxide via GSH and convert it to ethanol or free hydrogen peroxide (Stockwell et al., 2017; Gaschler et al., 2018; Forcina and Dixon, 2019), which can be inhibited by RSL-3 (Dixon et al., 2012; Yang et al., 2014), and FINO2 (Gaschler et al., 2018). Conversely, selenium is a key regulator of GPX4 activity (Ingold et al., 2015, 2018; Friedmann Angeli and Conrad, 2018; Alim et al., 2019) and its supplementation after stroke can effectively inhibit ferroptosis and cell death after haemorrhagic or ischemic stroke (Alim et al., 2019).
FSP1–CoQ10–NAD(P)H
Recently, a novel apoptosis-inducing factor mitochondria-associated 2 (AIFM2) gene, also known as AIF homologous mitochondria-related death inducer, has been reported as an anti-ferroptosis gene (Bersuker et al., 2019; Doll et al., 2019). Doll et al. (2019) renamed AIFM2 as FSP1 (ferroptosis suppressor protein 1, FSP1). It has been reported to supplement the anti-iron death effect in the absence of GPX4 by using NAD(P)H to catalyze CoQ10 regeneration and then alleviate lipid peroxidation. Shimada et al. also confirmed that NADPH levels could be used as an important marker of the sensitivity of many cancers cell lines to ferroptosis (Shimada et al., 2016). The FSP1-CoQ10-NAD(P)H pathway is a parallel anti-ferroptosis system independent of the cystine-GSH-GPX4 system that inhibits lipid peroxidation and anti-ferroptosis effects, FSP1 has a dual regulatory role: inducing apoptosis and preventing ferroptosis (Wu et al., 2002; Bersuker et al., 2019; Dar et al., 2021; Tonnus et al., 2021). Furthemore, the C-terminal fragment of FSP1 located in the outer membrane of the mitochondria induced apoptosis while the two N-terminal fragments located in the nucleus cannot induce apoptosis (Wu et al., 2002). Kaku et al. (2015) reported that the translocation of endogenous FSP1 into the nucleus is a necessary step in the apoptosis mechanism. Additionally, FSP1 is an oxidoreductase of CoQ10 (CoQ10), and the nutmeg acylation of the N-terminal of FSP1 is a lipid modification that promotes FSP1 translocation to the plasma membrane, thereby mediating NADH-dependent CoQ reduction on the plasma membrane to inhibit CoQ10 activity (Wu et al., 2002; Bersuker et al., 2019; Dar et al., 2021; Drijvers et al., 2021; Song Z. et al., 2021; Tonnus et al., 2021). Notably, FSP1 still plays an anti-ferroptosis role even after GPX4 knockdown. Therefore, FSP1-CoQ10-NAD(P)H is an independent pathway that cooperates with the cystine-GSH-GPX4 system to inhibit ferroptosis. In a Parkinson’s disease model, Song L. M. et al. (2021) demonstrated that inhibiting the upregulation of long-chain acyl-CoA synthase 4 (ACSL4) and downregulation of ferroptosis inhibitor 1 (FSP1) can effectively prevent MPTP-induced ferroptosis, thereby identifying a promising therapeutic target for Parkinson’s disease.
GCH1-BH4-DHFR
Exogenous dopamine or melatonin has been shown to inhibit ferroptosis induced by heme (Song L. M. et al., 2021). Tetrahydrobiopterin (BH4) is a natural nutrient that is involved in the biosynthesis of neurotransmitters, for example, 5-hydroxytryptamine, dopamine, adrenaline and melatonin can be used as cofactors of various enzymes, such as tryptophan hydroxylase, nitric oxide (NO) synthase and glycerol ether monooxygenase (Werner et al., 2011). BH4 plays a redox role in the formation of NO, which is catalyzed by L-arginine and NADPH (Hu Q. et al., 2022). The oxidation of BH4 to BH2 leads to the uncoupling of NOS to form O2⋅−, which reacts rapidly with NO to form peroxynitrite and further decouple NOS (Song L. M. et al., 2021). Deng et al. (2020) also confirmed that the activation of the NO pathway was associated with tissue damage related to ferroptosis. Additionally, Soula et al. (2020) indicated that BH4 synthesis is a necessary molecule required to protect cells from death. Moreover, the expression of GTP cyclohydrolase 1 (GCH1) determines the effectiveness of BH4 and its dependence on GPX4 inhibition (Kraft et al., 2020; Cronin et al., 2022). BH4 prevents the accumulation of lipid peroxides independently of its cofactor and is an effective free radical trapping agent and antioxidant in the lipid membrane, Dihydrofolate reductase catalyzes BH4 regeneration, methotrexate and GPX4 have been reported to synergistically inhibit ferroptosis (Hu Q. et al., 2022). Additionally, GCH1-mediated BH4 production prevents ferroptosis by inhibiting lipid peroxidation (Kraft et al., 2020; Hu Q. et al., 2022). In the hypertension model reported by Wang et al. (2021), the antihypertensive effect of L-phenylalanine could be mediated by enhancing BH4 biosynthesis and reducing superoxide levels produced by NO synthase, thereby reducing ROS and improving NO levels and protecting blood vessels and renal function. Hence, the GCH1-BH4 signaling pathway, as an endogenous antioxidant pathway, inhibits iron death through a mechanism independent of the GPX4/GSH system (Kuang et al., 2020; Wei et al., 2020).
Conclusions and prospects
The pathological processes and mechanisms of SAH rupture are extremely complex. Regardless of the main mechanism, blood entering the subarachnoid space or brain tissue is considered the inducer. Furthermore, the subsequent red blood cell lysis products release various toxic substances and lead to the death of neurons, ultimately leading to secondary brain injury (Bulters et al., 2018). Increasing basic studies have also demonstrated that ferroptosis-specific inhibitors, including liproxstatin-1 and ferrostatin-1, can decrease lipid peroxidation and neuronal ferroptosis after SAH in vivo and in vitro by regulating GPX4 and ACSL4 (Cao et al., 2021; Li et al., 2021; Qu et al., 2021). The present review expounds on the roles and related mechanisms of neuron necrosis, apoptosis, pyroptosis, necroptosis, autophagy and ferroptosis in brain injury after SAH. These mechanisms are interwoven and interact to jointly regulate brain injury after SAH. Furthermore, we explored the interrelated molecular mechanisms and targets to provide new ideas for drug synthesis. This review provides novel research directions that can aid in improving the prognosis of patients with SAH.
Author contributions
JC, KX, and YW designed the study. JC and ML drafted the manuscript. All authors discussed the results and revised the manuscript and read and approved the final manuscript.
Funding
This work was supported by the Jiangsu Provincial Natural Science Foundation (Grant No: BK20201140), Top talent support program for young and middle-aged people of Wuxi Health Committee (HB2020119) and Wuxi Science and Technology Development Foundation (Grant No: N20201008).
Acknowledgments
We thank all the participants in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abu Khweek, A., and Amer, A. O. (2020). Pyroptotic and non-pyroptotic effector functions of caspase-11. Immunol. Rev. 297, 39–52. doi: 10.1111/imr.12910
Aglietti, R. A., Estevez, A., Gupta, A., Ramirez, M. G., Liu, P. S., Kayagaki, N., et al. (2016). GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. U.S.A. 113, 7858–7863. doi: 10.1073/pnas.1607769113
Alim, I., Caulfield, J. T., Chen, Y., Swarup, V., Geschwind, D. H., Ivanova, E., et al. (2019). Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177, 1262–1279.e25. doi: 10.1016/j.cell.2019.03.032
Al-Khindi, T., Macdonald, R. L., and Schweizer, T. A. (2010). Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke 41, e519–e536. doi: 10.1161/strokeaha.110.581975
An, P., Xie, J., Qiu, S., Wang, J., Xiu, X., Li, L., et al. (2019). Hispidulin exhibits neuroprotective activities against cerebral ischemia reperfusion injury through suppressing NLRP3-mediated pyroptosis. Life Sci. 232, 116599. doi: 10.1016/j.lfs.2019.116599
Bergsbaken, T., Fink, S., and Cookson, B. T. (2009). Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109. doi: 10.1038/nrmicro2070
Bersuker, K., Hendricks, J. M., Li, Z., Magtanong, L., Ford, B., Tang, P. H., et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. doi: 10.1038/s41586-019-1705-2
Bjerring, P., Eefsen, M., Larsen, F., Bernal, W., and Wendon, J. (2011). Hypermagnesemia does not prevent intracranial hypertension and aggravates cerebral hyperperfusion in a rat model of acute hyperammonemia. Hepatology 53, 1986–1994. doi: 10.1002/hep.24274
Broz, P., and Dixit, V. M. (2016). Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420. doi: 10.1038/nri.2016.58
Bulters, D., Gaastra, B., Zolnourian, A., Alexander, S., Ren, D., Blackburn, S. L., et al. (2018). Haemoglobin scavenging in intracranial bleeding: Biology and clinical implications. Nat. Rev. Neurol. 14, 416–432. doi: 10.1038/s41582-018-0020-0
Cao, Y., Li, Y., He, C., Yan, F., Li, J. R., Xu, H. Z., et al. (2021). Selective ferroptosis inhibitor liproxstatin-1 attenuates neurological deficits and neuroinflammation after subarachnoid hemorrhage. Neurosci. Bull. 37, 535–549. doi: 10.1007/s12264-020-00620-5
Chang, H., Lin, C., Li, Z., Shen, Y., Zhang, G., and Mao, L. (2022). T3 alleviates neuroinflammation and reduces early brain injury after subarachnoid haemorrhage by promoting mitophagy via PINK 1-parkin pathway. Exp. Neurol. 357:114175. doi: 10.1016/j.expneurol.2022.114175
Chavez, L., Meguro, J., Chen, S., de Paiva, V. N., Zambrano, R., Eterno, J. M., et al. (2021). Circulating extracellular vesicles activate the pyroptosis pathway in the brain following ventilation-induced lung injury. J. Neuroinflammation 18:310. doi: 10.1186/s12974-021-02364-z
Chen, J., Xuan, Y., Chen, Y., Wu, T., Chen, L., Guan, H., et al. (2019c). Netrin-1 alleviates subarachnoid haemorrhage-induced brain injury via the PPARγ/NF-KB signalling pathway. J. Cell. Mol. Med. 23, 2256–2262. doi: 10.1111/jcmm.14105
Chen, A. Q., Fang, Z., Chen, X. L., Yang, S., Zhou, Y. F., Mao, L., et al. (2019a). Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death Dis. 10:487. doi: 10.1038/s41419-019-1716-9
Chen, J., Jin, H., Xu, H., Peng, Y., Jie, L., Xu, D., et al. (2019b). The neuroprotective effects of necrostatin-1 on subarachnoid hemorrhage in rats are possibly mediated by preventing blood-brain barrier disruption and RIP3-mediated necroptosis. Cell Transplant. 28, 1358–1372. doi: 10.1177/0963689719867285
Chen, G., and Goeddel, D. V. (2002). TNF-R1 signaling: A beautiful pathway. Science 296, 1634–1635. doi: 10.1126/science.1071924
Chen, J. H., Li, M. C., Zhu, X., Yang, S., Zhang, C., Wu, T., et al. (2020a). Atorvastatin reduces cerebral vasospasm and infarction after aneurysmal subarachnoid hemorrhage in elderly Chinese adults. Aging 12, 2939–2951. doi: 10.18632/aging.102788
Chen, J. H., Wu, T., Xia, W. Y., Shi, Z. H., Zhang, C. L., Chen, L., et al. (2020b). An early neuroprotective effect of atorvastatin against subarachnoid hemorrhage. Neural Regen. Res. 15, 1947–1954. doi: 10.4103/1673-5374.280326
Chen, J., Wang, Y., Wu, J., Yang, J., Li, M., and Chen, Q. (2020c). The potential value of targeting ferroptosis in early brain injury after acute CNS disease. Front. Mol. Neurosci. 13:110. doi: 10.3389/fnmol.2020.00110
Chen, J., Zhu, J., He, J., Wang, Y., Chen, L., Zhang, C., et al. (2016). Ultra-early microsurgical treatment within 24 h of SAH improves prognosis of poor-grade aneurysm combined with intracerebral hematoma. Oncol. Lett. 11, 3173–3178. doi: 10.3892/ol.2016.4327
Chen, J. H., Yang, L. K., Chen, L., Wang, Y. H., Wu, Y., Jiang, B. J., et al. (2016). Atorvastatin ameliorates early brain injury after subarachnoid hemorrhage via inhibition of AQP4 expression in rabbits. Int. J. Mol. Med. 37, 1059–1066. doi: 10.3892/ijmm.2016.2506
Chen, S., Feng, H., Sherchan, P., Klebe, D., Zhao, G., Sun, X., et al. (2014). Controversies and evolving new mechanisms in subarachnoid hemorrhage. Prog. Neurobiol. 115, 64–91. doi: 10.1016/j.pneurobio.2013.09.002
Chen, J., Wang, L., Wu, C., Hu, Q., Gu, C., Yan, F., et al. (2014). Melatonin-enhanced autophagy protects against neural apoptosis via a mitochondrial pathway in early brain injury following a subarachnoid hemorrhage. J. Pineal Res. 56, 12–19. doi: 10.1111/jpi.12086
Chen, J., Zhang, C., Yan, T., Yang, L., Wang, Y., Shi, Z., et al. (2021). Atorvastatin ameliorates early brain injury after subarachnoid hemorrhage via inhibition of pyroptosis and neuroinflammation. J. Cell. Physiol. 236, 6920–6931. doi: 10.1002/jcp.30351
Chen, J.-H., Li, P.-P., Yang, K., Chen, L., Zhu, J., Hu, X., et al. (2018a). Value of ventricular intracranial pressure monitoring for traumatic bifrontal contusions. World Neurosurg. 113, E690–E701. doi: 10.1016/j.wneu.2018.02.122
Chen, J. H., Wu, T., Yang, L. K., Chen, L., Zhu, J., Li, P. P., et al. (2018b). Protective effects of atorvastatin on cerebral vessel autoregulation in an experimental rabbit model of subarachnoid hemorrhage. Mol. Med. Rep. 17, 1651–1659. doi: 10.3892/mmr.2017.8074
Chen, Y., Zhou, Y., Wang, Q., Chen, J., Chen, H., Xie, H. H., et al. (2022). κConciliatory anti-allergic decoction attenuates pyroptosis in RSV-infected asthmatic mice and lipopolysaccharide (LPS)-induced 16HBE cells by inhibiting TLR3/NLRP3/NF-B/IRF3 signaling pathway. J. Immunol. Res. 2022:1800401. doi: 10.1155/2022/1800401
Claassen, J., Carhuapoma, J. R., Kreiter, K. T., Du, E. Y., Connolly, E. S., and Mayer, S. A. (2002). Global cerebral edema after subarachnoid hemorrhage: Frequency, predictors, and impact on outcome. Stroke 33, 1225–1232. doi: 10.1161/01.str.0000015624.29071.1f
Croci, D., Nevzati, E., Danura, H., Schöpf, S., Fandino, J., Marbacher, S., et al. (2019). The relationship between IL-6, ET-1 and cerebral vasospasm, in experimental rabbit subarachnoid hemorrhage. J. Neurosurg. Sci. 63, 245–250. doi: 10.23736/s0390-5616.16.03876-5
Cronin, S., Rao, S., Tejada, M., Turnes, B. L., Licht-Mayer, S., Omura, T., et al. (2022). Phenotypic drug screen uncovers the metabolic GCH1/BH4 pathway as key regulator of EGFR/KRAS-mediated neuropathic pain and lung cancer. Sci. Transl. Med. 14:eabj1531. doi: 10.1126/scitranslmed.abj1531
Dar, H. H., Anthonymuthu, T. S., Ponomareva, L. A., Souryavong, A. B., Shurin, G. V., Kapralov, A. O., et al. (2021). A new thiol-independent mechanism of epithelial host defense against Pseudomonas aeruginosa: iNOS/NO(•) sabotage of theft-ferroptosis. Redox Biol. 45:102045. doi: 10.1016/j.redox.2021.102045
Degterev, A., Huang, Z., Boyce, M., Jagtap, P., Mizushima, N., Cuny, G. D., et al. (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119. doi: 10.1038/nchembio711
Deng, G., Li, Y., Ma, S., Gao, Z., Zeng, T., Chen, L., et al. (2020). Caveolin-1 dictates ferroptosis in the execution of acute immune-mediated hepatic damage by attenuating nitrogen stress. Free Radic. Biol. Med. 148, 151–161. doi: 10.1016/j.freeradbiomed.2019.12.026
Dharmalingam, P., Talakatta, G., Mitra, J., Wang, H., Derry, P. J., Nilewski, L. G., et al. (2020). Pervasive genomic damage in experimental intracerebral hemorrhage: Therapeutic potential of a mechanistic-based carbon nanoparticle. ACS Nano 14, 2827–2846. doi: 10.1021/acsnano.9b05821
Diamond, C. E., Khameneh, H. J., Brough, D., and Mortellaro, A. (2015). Novel perspectives on non-canonical inflammasome activation. Immunotargets Ther. 4, 131–141. doi: 10.2147/itt.S57976
Ding, P., Zhu, Q., Sheng, B., Yang, H., Xu, H. J., Tao, T., et al. (2022). Alpha-ketoglutarate alleviates neuronal apoptosis induced by central insulin resistance through inhibiting S6K1 phosphorylation after subarachnoid hemorrhage. Oxid. Med. Cell. Longev. 2022:9148257. doi: 10.1155/2022/9148257
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Doll, S., Freitas, F. P., Shah, R., Aldrovandi, M., da Silva, M. C., Ingold, I., et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. doi: 10.1038/s41586-019-1707-0
Dong, Y., Fan, C., Hu, W., Jiang, S., Ma, Z., Yan, X., et al. (2016). Melatonin attenuated early brain injury induced by subarachnoid hemorrhage via regulating NLRP3 inflammasome and apoptosis signaling. J. Pineal Res. 60, 253–262. doi: 10.1111/jpi.12300
Drijvers, J. M., Gillis, J. E., Muijlwijk, T., Nguyen, T. H., Gaudiano, E. F., Harris, I. S., et al. (2021). Pharmacologic Screening Identifies Metabolic Vulnerabilities of CD8(+) T Cells. Cancer Immunol. Res. 9, 184–199. doi: 10.1158/2326-6066.Cir-20-0384
Du, Y., Lu, Z., Yang, D., Wang, D., Jiang, L., Shen, Y., et al. (2021). MerTK inhibits the activation of the NLRP3 inflammasome after subarachnoid hemorrhage by inducing autophagy. Brain Res. 1766, 147525. doi: 10.1016/j.brainres.2021.147525
Edebali, N., Tekin, I., Açıkgöz, B., Açıkgöz, S., Barut, F., Sevinç, N., et al. (2014). Apoptosis and necrosis in the circumventricular organs after experimental subarachnoid hemorrhage as detected with annexin V and caspase 3 immunostaining. Neurol. Res. 36, 1114–1120. doi: 10.1179/1743132814y.0000000437
Feigin, V. L., Lawes, C. M., Bennett, D. A., Barker-Collo, S. L., and Parag, V. (2009). Worldwide stroke incidence and early case fatality reported in 56 population-based studies: A systematic review. Lancet Neurol. 8, 355–369. doi: 10.1016/s1474-4422(09)70025-0
Forcina, G. C., and Dixon, S. J. (2019). GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics 19, e1800311. doi: 10.1002/pmic.201800311
Friedmann Angeli, J. P., and Conrad, M. (2018). Selenium and GPX4, a vital symbiosis. Free Radic. Biol. Med. 127, 153–159. doi: 10.1016/j.freeradbiomed.2018.03.001
Friedmann Angeli, J. P., Schneider, M., Proneth, B., Tyurina, Y. Y., Tyurin, V. A., Hammond, V. J., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191. doi: 10.1038/ncb3064
Friedrich, V., Flores, R., and Sehba, F. A. (2012). Cell death starts early after subarachnoid hemorrhage. Neurosci. Lett. 512, 6–11. doi: 10.1016/j.neulet.2012.01.036
Galluzzi, L., Kepp, O., Chan, F. K., and Kroemer, G. (2017). Necroptosis: Mechanisms and relevance to disease. Annu. Rev. Pathol. 12, 103–130. doi: 10.1146/annurev-pathol-052016-100247
Gao, X., Gao, Y. Y., Yan, H. Y., Liu, G.-J., Zhou, Y., Tao, T., et al. (2022). PDK4 decrease neuronal apoptosis via inhibiting ROS-ASK1/P38 pathway in early brain injury after subarachnoid hemorrhage. Antioxid. Redox Signal. 36, 505–524. doi: 10.1089/ars.2021.0083
Gao, S., Zhou, L., Lu, J., Fang, Y., Wu, H., Xu, W., et al. (2022). Cepharanthine Attenuates early brain injury after subarachnoid hemorrhage in mice via inhibiting 15-lipoxygenase-1-mediated microglia and endothelial cell ferroptosis. Oxid. Med. Cell. Longev. 2022:4295208. doi: 10.1155/2022/4295208
Gao, Y., Ferguson, D., Xie, W., Manis, J. P., Sekiguchi, J., Frank, K. M., et al. (2000). Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 404, 897–900. doi: 10.1038/35009138
Gaschler, M. M., Andia, A. A., Liu, H., Csuka, J. M., Hurlocker, B., Vaiana, C. A., et al. (2018). FINO(2) initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 14, 507–515. doi: 10.1038/s41589-018-0031-6
GBD 2019 Stroke Collaborators (2021). Global, regional, and national burden of stroke and its risk factors, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 20, 795–820. doi: 10.1016/s1474-4422(21)00252-0
Gerlach, B., Cordier, S. M., Schmukle, A. C., Emmerich, C. H., Rieser, E., Haas, T. L., et al. (2011). Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471, 591–596. doi: 10.1038/nature09816
González-Montelongo, M. D. C., Egea-Guerrero, J. J., Murillo-Cabezas, F., González-Montelongo, R., Ruiz de Azúa-López, Z., Rodríguez-Rodríguez, A., et al. (2018). Relation of RhoA in peripheral blood mononuclear cells with severity of aneurysmal subarachnoid hemorrhage and vasospasm. Stroke 49, 1507–1510. doi: 10.1161/strokeaha.117.020311
Gu, L., Sun, M., Li, R., Tao, Y., Luo, X., Xu, J., et al. (2022). Activation of RKIP binding ASC attenuates neuronal pyroptosis and Brain Injury via Caspase-1/GSDMD signaling pathway after intracerebral hemorrhage in mice. Transl. Stroke Res. 13, 1037–1054. doi: 10.1007/s12975-022-01009-4
Güresir, E., Welchowski, T., Lampmann, T., Brandecker, S., Güresir, A., Wach, J., et al. (2022). Delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: The results of induced hypertension only after the IMCVS Trial-A prospective cohort study. J. Clin. Med. 11:5850. doi: 10.3390/jcm11195850
Halstead, M., and Geocadin, R. G. (2019). The medical management of cerebral edema: Past, present, and future therapies. Neurotherapeutics 16, 1133–1148. doi: 10.1007/s13311-019-00779-4
Harris, H., and Rubinsztein, D. C. (2012). Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol. 8, 108–117. doi: 10.1038/nrneurol.2011.200
Hasegawa, Y., Suzuki, H., Sozen, T., Altay, O., and Zhang, J. H. (2011). apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta Neurochir. Suppl. 110(Pt 1), 43–48.
He, J. Q., Chen, J. H., Zhu, J., Chen, L., Zhang, C. L., Yang, L. K., et al. (2015). Prognosis of ultra-early microsurgery combined with extraventricular drainage in patients with poor-grade aneurysms. Int. J. Clin. Exp. Med. 8, 9723–9729.
He, S., Wang, L., Miao, L., Wang, T., Du, F., Zhao, L., et al. (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111. doi: 10.1016/j.cell.2009.05.021
Heuer, G. G., Smith, M. J., Elliott, J. P., Winn, H. R., and LeRoux, P. D. (2004). Relationship between intracranial pressure and other clinical variables in patients with aneurysmal subarachnoid hemorrhage. J. Neurosurg. 101, 408–416. doi: 10.3171/jns.2004.101.3.0408
Hider, R. C., and Kong, X. L. (2011). Glutathione: A key component of the cytoplasmic labile iron pool. Biometals 24, 1179–1187. doi: 10.1007/s10534-011-9476-8
Ho, W. M., Akyol, O., Reis, H., Reis, C., McBride, D., Thome, C., et al. (2018). Autophagy after subarachnoid hemorrhage: Can cell death be good? Curr. Neuropharmacol. 16, 1314–1319. doi: 10.2174/1570159x15666171123200646
Holler, N., Zaru, R., Micheau, O., Thome, M., Attinger, A., Valitutti, S., et al. (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495. doi: 10.1038/82732
Hostettler, I., Kreiser, K., Lange, N., Schwendinger, N., Trost, D., Frangoulis, S., et al. (2022). Treatment during cerebral vasospasm phase-complication association and outcome in aneurysmal subarachnoid haemorrhage. J. Neurol. 269, 5553–5560. doi: 10.1007/s00415-022-11212-w
Hu, X., Zhang, H., Zhang, Q., Yao, X., Ni, W., and Zhou, K. (2022). Emerging role of STING signalling in CNS injury: Inflammation, autophagy, necroptosis, ferroptosis and pyroptosis. J. Neuroinflammation 19:242. doi: 10.1186/s12974-022-02602-y
Hu, Q., Wei, W., Wu, D., Huang, G., Li, M., Li, W., et al. (2022). Blockade of GCH1/BH4 axis activates ferritinophagy to mitigate the resistance of colorectal cancer to erastin-induced ferroptosis. Front. Cell. Dev. Biol. 10:810327. doi: 10.3389/fcell.2022.810327
Huang, J., Lu, W. T., Sun, S. Q., Yang, Z. B., Huang, S. Q., Gan, S. W., et al. (2014). Upregulation and lysosomal degradation of AQP4 in rat brains with bacterial meningitis. Neurosci. Lett. 566, 156–161. doi: 10.1016/j.neulet.2014.02.054
Huang, Q., Wang, G., Hu, Y. L., Liu, J. X., Yang, J., Wang, S., et al. (2017). Study on the expression and mechanism of inflammatory factors in the brain of rats with cerebral vasospasm. Eur. Rev. Med. Pharmacol. Sci. 21, 2887–2894.
Ingold, I., Aichler, M., Yefremova, E., Roveri, A., Buday, K., Doll, S., et al. (2015). Expression of a catalytically inactive mutant form of glutathione peroxidase 4 (Gpx4) confers a dominant-negative effect in male fertility. J. Biol. Chem. 290, 14668–14678. doi: 10.1074/jbc.M115.656363
Ingold, I., Berndt, C., Schmitt, S., Doll, S., Poschmann, G., Buday, K., et al. (2018). Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 172, 409–422.e421. doi: 10.1016/j.cell.2017.11.048
Ito, M., Shichita, T., Okada, M., Komine, R., Noguchi, Y., Yoshimura, A., et al. (2015). Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 6:7360. doi: 10.1038/ncomms8360
Jaafaru, M., Nordin, N., Rosli, R., Shaari, K., Bako, H. Y., Noor, N. M., et al. (2019). Prospective role of mitochondrial apoptotic pathway in mediating GMG-ITC to reduce cytotoxicity in HO-induced oxidative stress in differentiated SH-SY5Y cells. Biomed. Pharmacother. 119:109445. doi: 10.1016/j.biopha.2019.109445
Kaiser, W. J., Sridharan, H., Huang, C., Mandal, P., Upton, J. W., Gough, P. J., et al. (2013). Toll-like receptor 3-mediated necrosis via TRIF. RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279. doi: 10.1074/jbc.M113.462341
Kaku, Y., Tsuchiya, A., Kanno, T., and Nishizaki, T. (2015). HUHS1015 induces necroptosis and caspase-independent apoptosis of MKN28 human gastric cancer cells in association with AMID accumulation in the nucleus. Anticancer Agents Med. Chem. 15, 242–247. doi: 10.2174/1871520614666140922122700
Kaufmann, A., and Cardoso, E. J. J. (1992). Aggravation of vasogenic cerebral edema by multiple-dose mannitol. J. Neurosurg. 77, 584–589. doi: 10.3171/jns.1992.77.4.0584
Kayagaki, N., Stowe, I. B., Lee, B. L., O’Rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. doi: 10.1038/nature15541
Kayagaki, N., Wong, M. T., Stowe, I. B., Ramani, S. R., Gonzalez, L. C., Akashi-Takamura, S., et al. (2013). Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249. doi: 10.1126/science.1240248
Kerr, N. A., Sanchez, J., O’Connor, G., Watson, B. D., Daunert, S., Bramlett, H. M., et al. (2022). Inflammasome-regulated pyroptotic cell death in disruption of the gut-brain axis after stroke. Transl. Stroke Res. 13, 898–912. doi: 10.1007/s12975-022-01005-8
Kraft, V., Bezjian, C. T., Pfeiffer, S., Ringelstetter, L., Müller, C., Zandkarimi, F., et al. (2020). GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent. Sci. 6, 41–53. doi: 10.1021/acscentsci.9b01063
Kroemer, G., Galluzzi, L., Vandenabeele, P., Abrams, J., Alnemri, E. S., Baehrecke, E. H., et al. (2009). Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 16, 3–11. doi: 10.1038/cdd.2008.150
Kuang, F., Liu, J., Tang, D., and Kang, R. (2020). Oxidative damage and antioxidant defense in ferroptosis. Front. Cell. Dev. Biol. 8:586578. doi: 10.3389/fcell.2020.586578
Kuwar, R., Rolfe, A., Di, L., Xu, H., He, L., Jiang, Y., et al. (2019). A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J. Neuroinflammation 16:81. doi: 10.1186/s12974-019-1471-y
Lawton, M. T., and Vates, G. E. (2017). Subarachnoid Hemorrhage. N. Engl. J. Med. 377, 257–266. doi: 10.1056/NEJMcp1605827
Lee, C. Z., Xue, Z., Zhu, Y., Yang, G. Y., and Young, W. L. (2007). Matrix metalloproteinase-9 inhibition attenuates vascular endothelial growth factor-induced intracerebral hemorrhage. Stroke 38, 2563–2568. doi: 10.1161/strokeaha.106.481515
Lee, J.-Y., He, Y., Sagher, O., Keep, R., Hua, Y., and Xi, G. (2009). Activated autophagy pathway in experimental subarachnoid hemorrhage. Brain Res. 1287, 126–135. doi: 10.1016/j.brainres.2009.06.028
Li, J. R., Xu, H. Z., Nie, S., Peng, Y. C., Fan, L. F., Wang, Z. J., et al. (2017). Fluoxetine-enhanced autophagy ameliorates early brain injury via inhibition of NLRP3 inflammasome activation following subrachnoid hemorrhage in rats. J. Neuroinflammation 14:186. doi: 10.1186/s12974-017-0959-6
Li, Y., Liu, Y., Wu, P., Tian, Y., Liu, B., Wang, J., et al. (2021). Inhibition of ferroptosis alleviates early brain injury after subarachnoid hemorrhage in vitro and in vivo via reduction of lipid peroxidation. Cell. Mol. Neurobiol. 41, 263–278. doi: 10.1007/s10571-020-00850-1
Li, Z., and Han, X. (2018). Resveratrol alleviates early brain injury following subarachnoid hemorrhage: Possible involvement of the AMPK/SIRT1/autophagy signaling pathway. Biol. Chem. 399, 1339–1350. doi: 10.1515/hsz-2018-0269
Liang, Y., Song, P., Zhu, Y., Xu, J. M., Zhu, P. Z., Liu, R. R., et al. (2020). TREM-1-targeting LP17 attenuates cerebral ischemia-induced neuronal injury by inhibiting oxidative stress and pyroptosis. Biochem. Biophys. Res. Commun. 529, 554–561. doi: 10.1016/j.bbrc.2020.05.056
Liu, M., Zhong, W., Li, C., and Su, W. (2022). Fluoxetine attenuates apoptosis in early brain injury after subarachnoid hemorrhage through Notch1/ASK1/p38 MAPK signaling pathway. Bioengineered 13, 8396–8411. doi: 10.1080/21655979.2022.2037227
Liu, B., Tian, Y., Li, Y., Wu, P., Zhang, Y., Zheng, J., et al. (2022). ACEA attenuates oxidative stress by promoting mitophagy via CB1R/Nrf1/PINK1 pathway after subarachnoid hemorrhage in rats. Oxid. Med. Cell. Longev. 2022:1024279. doi: 10.1155/2022/1024279
Liu, X., Zhang, Z., Ruan, J., Pan, Y., Magupalli, V. G., Wu, H., et al. (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. doi: 10.1038/nature18629
Liu, Y., Cai, H., Wang, Z., Li, J., Wang, K., Yu, Z., et al. (2013). Induction of autophagy by cystatin C: A potential mechanism for prevention of cerebral vasospasm after experimental subarachnoid hemorrhage. Eur. J. Med. Res. 18:21. doi: 10.1186/2047-783x-18-21
Macdonald, R. L. (2014). Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 10, 44–58. doi: 10.1038/nrneurol.2013.246
Macdonald, R. L., and Schweizer, T. A. (2017). Spontaneous subarachnoid haemorrhage. Lancet 389, 655–666. doi: 10.1016/s0140-6736(16)30668-7
Macdonald, R. L., Higashida, R. T., Keller, E., Mayer, S. A., Molyneux, A., Raabe, A., et al. (2011). Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: A randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet Neurol. 10, 618–625. doi: 10.1016/s1474-4422(11)70108-9
Mees, S. M. D., Algra, A., Vandertop, W. P., van Kooten, F., Kuijsten, H. A., Boiten, J., et al. (2012). Magnesium for aneurysmal subarachnoid haemorrhage (MASH-2): A randomised placebo-controlled trial. The Lancet 380, 44–49. doi: 10.1016/s0140-6736(12)60724-7
Nag, S., Manias, J. L., and Stewart, D. J. (2009). Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol. 118, 197–217. doi: 10.1007/s00401-009-0541-0
Narotam, P. K., Garton, A., Morrison, J., Nathoo, N., and Narotam, N. (2022). Brain oxygen-directed management of aneurysmal subarachnoid hemorrhage. Temporal patterns of cerebral ischemia during acute brain attack, early brain injury, and territorial sonographic vasospasm. World Neurosurg. 166, e215–e236. doi: 10.1016/j.wneu.2022.06.149
Nieuwkamp, D. J., Setz, L. E., Algra, A., Linn, F. H., de Rooij, N. K., and Rinkel, G. J. (2009). Changes in case fatality of aneurysmal subarachnoid haemorrhage over time, according to age, sex, and region: A meta-analysis. Lancet Neurol. 8, 635–642. doi: 10.1016/s1474-4422(09)70126-7
Olson, D. M., Zomorodi, M., Britz, G. W., Zomorodi, A. R., Amato, A., and Graffagnino, C. (2013). Continuous cerebral spinal fluid drainage associated with complications in patients admitted with subarachnoid hemorrhage. J. Neurosurg. 119, 974–980. doi: 10.3171/2013.6.Jns122403
Pang, L., Liu, Z., Zhou, K., Chen, P., Pan, E., Che, Y., et al. (2022). ACE2 rescues impaired autophagic flux through the PI3K/AKT pathway after subarachnoid hemorrhage. Neurochem. Res. 47, 601–612. doi: 10.1007/s11064-021-03469-w
Papadopoulos, M. C., Manley, G. T., Krishna, S., and Verkman, A. S. (2004). Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 18, 1291–1293. doi: 10.1096/fj.04-1723fje
Passier, P. E., Visser-Meily, J. M., van Zandvoort, M. J., Post, M. W., Rinkel, G. J., and van Heugten, C. (2010). Prevalence and determinants of cognitive complaints after aneurysmal subarachnoid hemorrhage. Cerebrovasc. Dis. 29, 557–563. doi: 10.1159/000306642
Peng, J., Zuo, Y., Huang, L., Okada, T., Liu, S., Zuo, G., et al. (2019). Activation of GPR30 with G1 attenuates neuronal apoptosis via src/EGFR/stat3 signaling pathway after subarachnoid hemorrhage in male rats. Exp. Neurol. 320:113008. doi: 10.1016/j.expneurol.2019.113008
Pluta, R. M. (2005). Delayed cerebral vasospasm and nitric oxide: Review, new hypothesis, and proposed treatment. Pharmacol. Ther. 105, 23–56. doi: 10.1016/j.pharmthera.2004.10.002
Powell, W. J. Jr., DiBona, D. R., Flores, J., Frega, N., and Leaf, A. (1976). Effects of hyperosmotic mannitol in reducing ischemic cell swelling and minimizing myocardial necrosis. Circulation 53(3 Suppl.), I45–I49.
Qu, X. F., Liang, T. Y., Wu, D. G., Lai, N. S., Deng, R. M., Ma, C., et al. (2021). Acyl-CoA synthetase long chain family member 4 plays detrimental role in early brain injury after subarachnoid hemorrhage in rats by inducing ferroptosis. CNS Neurosci. Ther. 27, 449–463. doi: 10.1111/cns.13548
Qureshi, A. I., Suri, M. F., Nasar, A., Kirmani, J. F., Ezzeddine, M. A., Divani, A. A., et al. (2007). Changes in cost and outcome among US patients with stroke hospitalized in 1990 to 1991 and those hospitalized in 2000 to 2001. Stroke 38, 2180–2184. doi: 10.1161/strokeaha.106.467506
Rinkel, G. J., and Algra, A. (2011). Long-term outcomes of patients with aneurysmal subarachnoid haemorrhage. Lancet Neurol. 10, 349–356. doi: 10.1016/s1474-4422(11)70017-5
Rivero-Arias, O., Gray, A., and Wolstenholme, J. (2010). Burden of disease and costs of aneurysmal subarachnoid haemorrhage (aSAH) in the United Kingdom. Cost Eff. Resour. Alloc. 8:6. doi: 10.1186/1478-7547-8-6
Rui, T., Wang, H., Li, Q., Cheng, Y., Gao, Y., Fang, X., et al. (2021). Deletion of ferritin H in neurons counteracts the protective effect of melatonin against traumatic brain injury-induced ferroptosis. J. Pineal Res. 70:e12704. doi: 10.1111/jpi.12704
Sahin, M. H., Akyuz, E., and Kadioglu, H. H. (2021). The Effects of Necrostatin-1 on Cerebral Vasospasm- Induced Subarachnoid Hemorrhage. Turk. Neurosurg. [Epub ahead of print]. doi: 10.5137/1019-5149.Jtn.35167-21.4
Said, M., Gümüs, M., Herten, A., Dinger, T. F., Chihi, M., Darkwah Oppong, M., et al. (2021). Subarachnoid Hemorrhage Early Brain Edema Score (SEBES) as a radiographic marker of clinically relevant intracranial hypertension and unfavorable outcome after subarachnoid hemorrhage. Eur. J. Neurol. 28, 4051–4059. doi: 10.1111/ene.15033
Sendoel, A., Kohler, I., Fellmann, C., Lowe, S. W., and Hengartner, M. O. (2010). HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature 465, 577–583. doi: 10.1038/nature09141
She, Y., Shao, L., Zhang, Y., Hao, Y., Cai, Y., Cheng, Z., et al. (2019). Neuroprotective effect of glycosides in Buyang Huanwu Decoction on pyroptosis following cerebral ischemia-reperfusion injury in rats. J. Ethnopharmacol. 242:112051. doi: 10.1016/j.jep.2019.112051
Shen, H., Liu, C., Zhang, D., Yao, X., Zhang, K., Li, H., et al. (2017). Role for RIP1 in mediating necroptosis in experimental intracerebral hemorrhage model both in vivo and in vitro. Cell Death Dis. 8:e2641. doi: 10.1038/cddis.2017.58
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Shimada, K., Skouta, R., Kaplan, A., Yang, W. S., Hayano, M., Dixon, S. J., et al. (2016). Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503. doi: 10.1038/nchembio.2079
Shinohara, Y., Katayama, Y., Uchiyama, S., Yamaguchi, T., Handa, S., Matsuoka, K., et al. (2010). Cilostazol for prevention of secondary stroke (CSPS 2): An aspirin-controlled, double-blind, randomised non-inferiority trial. Lancet Neurol 9, 959–968. doi: 10.1016/s1474-4422(10)70198-8
Song, Z., Jia, G., Ma, P., and Cang, S. (2021). Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. 276:119399. doi: 10.1016/j.lfs.2021.119399
Song, L. M., Xiao, Z. X., Zhang, N., Zhang, N., Yu, X. Q., Cui, W., et al. (2021). Apoferritin improves motor deficits in MPTP-treated mice by regulating brain iron metabolism and ferroptosis. iScience 24:102431. doi: 10.1016/j.isci.2021.102431
Soula, M., Weber, R. A., Zilka, O., Alwaseem, H., La, K., Yen, F., et al. (2020). Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 16, 1351–1360. doi: 10.1038/s41589-020-0613-y
Steiner, T., Juvela, S., Unterberg, A., Jung, C., Forsting, M., Rinkel, G., et al. (2013). European stroke organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc. Dis. 35, 93–112. doi: 10.1159/000346087
Stienen, M. N., Smoll, N. R., Weisshaupt, R., Fandino, J., Hildebrandt, G., Studerus-Germann, A., et al. (2014). Delayed cerebral ischemia predicts neurocognitive impairment following aneurysmal subarachnoid hemorrhage. World Neurosurg. 82, e599–e605. doi: 10.1016/j.wneu.2014.05.011
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Sugimoto, K., Nomura, S., Shirao, S., Inoue, T., Ishihara, H., Kawano, R., et al. (2018). Cilostazol decreases duration of spreading depolarization and spreading ischemia after aneurysmal subarachnoid hemorrhage. Ann. Neurol. 84, 873–885. doi: 10.1002/ana.25361
Sun, X. G., Duan, H., Jing, G., Wang, G., Hou, Y., Zhang, M., et al. (2019). Inhibition of TREM-1 attenuates early brain injury after subarachnoid hemorrhage via downregulation of p38MAPK/MMP-9 and preservation of ZO-1. Neuroscience 406, 369–375. doi: 10.1016/j.neuroscience.2019.03.032
Sun, Y. B., Zhao, H., Mu, D. L., Zhang, W., Cui, J., Wu, L., et al. (2019). Dexmedetomidine inhibits astrocyte pyroptosis and subsequently protects the brain in in vitro and in vivo models of sepsis. Cell Death Dis. 10:167. doi: 10.1038/s41419-019-1416-5
Sun, C., Enkhjargal, B., Reis, C., Zhang, T., Zhu, Q., Zhou, K., et al. (2019). Osteopontin-enhanced autophagy attenuates early brain injury via FAK-ERK pathway and improves long-term outcome after subarachnoid hemorrhage in rats. Cells 8:980. doi: 10.3390/cells8090980
Sun, C. M., Enkhjargal, B., Reis, C., Zhou, K. R., Xie, Z. Y., Wu, L. Y., et al. (2019). Osteopontin attenuates early brain injury through regulating autophagy-apoptosis interaction after subarachnoid hemorrhage in rats. CNS Neurosci. Ther. 25, 1162–1172. doi: 10.1111/cns.13199
Svedung Wettervik, T., Hånell, A., Howells, T., Ronne Engström, E., Lewén, A., and Enblad, P. (2022). ICP, CPP, and PRx in traumatic brain injury and aneurysmal subarachnoid hemorrhage: Association of insult intensity and duration with clinical outcome. J. Neurosurg. [Epub ahead of print]. doi: 10.3171/2022.5.Jns22560
Swanson, K., Deng, M., and Ting, J. P. (2019). The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489. doi: 10.1038/s41577-019-0165-0
Tonnus, W., Meyer, C., Steinebach, C., Belavgeni, A., von Mässenhausen, A., Gonzalez, N. Z., et al. (2021). Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat. Commun. 12:4402. doi: 10.1038/s41467-021-24712-6
Tourdias, T., Mori, N., Dragonu, I., Cassagno, N., Boiziau, C., Aussudre, J., et al. (2011). Differential aquaporin 4 expression during edema build-up and resolution phases of brain inflammation. J. Neuroinflammation 8:143. doi: 10.1186/1742-2094-8-143
Toyoda, K., Uchiyama, S., Yamaguchi, T., Easton, J. D., Kimura, K., Hoshino, H., et al. (2019). Dual antiplatelet therapy using cilostazol for secondary prevention in patients with high-risk ischaemic stroke in Japan: A multicentre, open-label, randomised controlled trial. Lancet Neurol. 18, 539–548. doi: 10.1016/s1474-4422(19)30148-6
Tuo, Q. Z., Liu, Y., Xiang, Z., Yan, H. F., Zou, T., Shu, Y., et al. (2022). Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal Transduct. Target. Ther. 7:59. doi: 10.1038/s41392-022-00917-z
Van Rossom, S., Op de Beeck, K., Hristovska, V., Winderickx, J., and Van Camp, G. (2015). The deafness gene DFNA5 induces programmed cell death through mitochondria and MAPK-related pathways. Front. Cell. Neurosci. 9:231. doi: 10.3389/fncel.2015.00231
Vande Walle, L., and Lamkanfi, M. (2016). Pyroptosis. Curr. Biol. 26, R568–R572. doi: 10.1016/j.cub.2016.02.019
Wan, A., Jaja, B. N., Schweizer, T. A., Macdonald, R. L., and on behalf of the Sahit collaboration (2016). Clinical characteristics and outcome of aneurysmal subarachnoid hemorrhage with intracerebral hematoma. J. Neurosurg. 125, 1344–1351. doi: 10.3171/2015.10.Jns151036
Wang, Y., An, R., Umanah, G. K., Nambiar, K., Eacker, S. M., Kim, B., et al. (2016). A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 354:aad6872. doi: 10.1126/science.aad6872
Wang, Z., Cheng, C., Yang, X., and Zhang, C. (2021). L-phenylalanine attenuates high salt-induced hypertension in Dahl SS rats through activation of GCH1-BH4. PLoS One 16:e0250126. doi: 10.1371/journal.pone.0250126
Wang, Z., Shi, X.-Y., Yin, J., Zuo, G., Zhang, J., Chen, G., et al. (2012). Role of autophagy in early brain injury after experimental subarachnoid hemorrhage. J. Mol. Neurosci. 46, 192–202. doi: 10.1007/s12031-011-9575-6
Wei, X., Yi, X., Zhu, X. H., and Jiang, D. S. (2020). Posttranslational modifications in ferroptosis. Oxid. Med. Cell. Longev. 2020:8832043. doi: 10.1155/2020/8832043
Werner, E. R., Blau, N., and Thöny, B. (2011). Tetrahydrobiopterin: Biochemistry and pathophysiology. Biochem. J. 438, 397–414. doi: 10.1042/bj20110293
Wu, M., Xu, L. G., Li, X., Zhai, Z., and Shu, H. B. (2002). AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J. Biol. Chem. 277, 25617–25623. doi: 10.1074/jbc.M202285200
Xu, H., Cai, Y., Yu, M., Sun, J., Cai, J., Li, J., et al. (2021). Celastrol protects against early brain injury after subarachnoid hemorrhage in rats through alleviating blood-brain barrier disruption and blocking necroptosis. Aging 13, 16816–16833. doi: 10.18632/aging.203221
Xu, P., Zhang, X., Liu, Q., Xie, Y., Shi, X., Chen, J., et al. (2019). Microglial TREM-1 receptor mediates neuroinflammatory injury via interaction with SYK in experimental ischemic stroke. Cell Death Dis. 10:555. doi: 10.1038/s41419-019-1777-9
Yang, C., Li, T., Xue, H., Wang, L., Deng, L., Xie, Y., et al. (2018). Inhibition of necroptosis rescues SAH-Induced synaptic impairments in hippocampus via CREB-BDNF Pathway. Front. Neurosci. 12:990. doi: 10.3389/fnins.2018.00990
Yang, W. S., SriRamaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi: 10.1016/j.cell.2013.12.010
Yuan, S., Yu, Z., Zhang, Z., Zhang, J., Zhang, P., Li, X., et al. (2019). RIP3 participates in early brain injury after experimental subarachnoid hemorrhage in rats by inducing necroptosis. Neurobiol. Dis. 129, 144–158. doi: 10.1016/j.nbd.2019.05.004
Zeng, H., Chen, H., Li, M., Zhuang, J., Peng, Y., Zhou, H., et al. (2021). Autophagy protein NRBF2 attenuates endoplasmic reticulum stress-associated neuroinflammation and oxidative stress via promoting autophagosome maturation by interacting with Rab7 after SAH. J. Neuroinflammation 18:210. doi: 10.1186/s12974-021-02270-4
Zhang, J., Zhu, Y., Zhou, D., Wang, Z., and Chen, G. (2010). Recombinant human erythropoietin (rhEPO) alleviates early brain injury following subarachnoid hemorrhage in rats: Possible involvement of Nrf2-ARE pathway. Cytokine 52, 252–257. doi: 10.1016/j.cyto.2010.08.011
Zhao, X., Wen, L., Dong, M., Dong, M., and Lu, X. (2016). Sulforaphane activates the cerebral vascular Nrf2-ARE pathway and suppresses inflammation to attenuate cerebral vasospasm in rat with subarachnoid hemorrhage. Brain Res. 1653, 1–7. doi: 10.1016/j.brainres.2016.09.035
Zhu, X., Tao, L., Tejima-Mandeville, E., Qiu, J., Park, J., Garber, K., et al. (2012). Plasmalemma permeability and necrotic cell death phenotypes after intracerebral hemorrhage in mice. Stroke 43, 524–531. doi: 10.1161/strokeaha.111.635672
Zille, M., Karuppagounder, S. S., Chen, Y., Gough, P. J., Bertin, J., Finger, J., et al. (2017). Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke 48, 1033–1043. doi: 10.1161/strokeaha.116.015609
Zoerle, T., Lombardo, A., Colombo, A., Longhi, L., Zanier, E. R., Rampini, P., et al. (2015). Intracranial pressure after subarachnoid hemorrhage. Crit. Care Med. 43, 168–176. doi: 10.1097/ccm.0000000000000670
Zuo, G., Zhang, T., Huang, L., Araujo, C., Peng, J., Travis, Z., et al. (2019). Activation of TGR5 with INT-777 attenuates oxidative stress and neuronal apoptosis via cAMP/PKCε/ALDH2 pathway after subarachnoid hemorrhage in rats. Free Radic. Biol. Med. 143, 441–453. doi: 10.1016/j.freeradbiomed.2019.09.002
Keywords: cell death, SAH, necrosis, apoptosis, ferroptosis, autophagy, pyroptosis
Citation: Chen J, Li M, Liu Z, Wang Y and Xiong K (2022) Molecular mechanisms of neuronal death in brain injury after subarachnoid hemorrhage. Front. Cell. Neurosci. 16:1025708. doi: 10.3389/fncel.2022.1025708
Received: 30 August 2022; Accepted: 08 November 2022;
Published: 13 December 2022.
Edited by:
Anwen Shao, Zhejiang University, ChinaReviewed by:
Amit Kumar, Burke Neurological Institute (BNI), United StatesZhaohui He, First Affiliated Hospital of Chongqing Medical University, China
Copyright © 2022 Chen, Li, Liu, Wang and Xiong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuhai Wang, d2FuZ3l1aGFpMTUxNkAxNjMuY29t; Kun Xiong, eGlvbmdrdW4yMDAxQDE2My5jb20=
†These authors have contributed equally to this work