 Chih Hung Lo
Chih Hung Lo Mario Skarica
Mario Skarica Mohammad Mansoor
Mohammad Mansoor Shaan Bhandarkar
Shaan Bhandarkar Steven Toro
Steven Toro David Pitt
David Pitt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci. , 11 August 2021
Sec. Non-Neuronal Cells
Volume 15 - 2021 | https://doi.org/10.3389/fncel.2021.726479
This article is part of the Research Topic Non-neuronal Cell Heterogeneity in the Nervous System During Health and Disease View all 10 articles
The emergence of single cell technologies provides the opportunity to characterize complex immune/central nervous system cell assemblies in multiple sclerosis (MS) and to study their cell population structures, network activation and dynamics at unprecedented depths. In this review, we summarize the current knowledge of astrocyte subpopulations in MS tissue and discuss the challenges associated with resolving astrocyte heterogeneity with single-nucleus RNA-sequencing (snRNA-seq). We further discuss multiplexed imaging techniques as tools for defining population clusters within a spatial context. Finally, we will provide an outlook on how these technologies may aid in answering unresolved questions in MS, such as the glial phenotypes that drive MS progression and/or neuropathological differences between different clinical MS subtypes.
Due to the resounding success of current MS medications in treating relapsing-remitting MS (RRMS), research interest is increasingly focused on disease progression and neurorepair where current treatments are ineffective. In contrast to RRMS, progressive disease is thought be driven by central nervous system (CNS)-intrinsic processes, where infiltration with peripheral immune cells plays only a minor role. Thus, the attention in MS research is arguably shifting from the peripheral immune system to CNS cells. The CNS-intrinsic processes associated with progression include chronic, diffuse activation of glia cells at the rim of chronic active lesions and in the normal appearing white matter (NAWM) (Chen et al., 2014; Absinta et al., 2019; Sucksdorff et al., 2020), cortical demyelination and neuro-axonal degeneration (Dutta et al., 2006). Identifying the glial subpopulations that contribute to MS progression is likely to yield new targets for therapeutic intervention in progressive MS (Ponath et al., 2018b; Wilbanks et al., 2019; Guerrero and Sicotte, 2020).
In this review, we are focusing on astrocytes. Astrocytes are substantially more numerous than microglia cells, accounting for 20–40% of the total glial population, depending on the brain region (von Bartheld et al., 2016). In a homeostatic state, astrocytes play critical roles in brain function, including synapse formation and elimination (Lee et al., 2021), establishing and maintaining network circuitry, control of neurotransmitters release and uptake (Anderson and Swanson, 2000), modulation of blood-brain barrier (BBB) function (Horng et al., 2017) and blood flow, as well as maintenance of ion and water homeostasis among others (Sofroniew and Vinters, 2009; Ponath et al., 2018b).
CNS pathologies such as trauma, infection, neurodegeneration, and inflammatory demyelination lead to prominent astrocyte responses with morphological, molecular, and functional remodeling. Reactive astrocytes can promote tissue survival by forming a functional barrier around damaged tissue and contribute to tissue repair (Faulkner et al., 2004; Tyzack et al., 2014; Anderson et al., 2016). However, reactive astrocytes may also become dysfunctional and adopt disease-promoting phenotypes. In MS, this includes recruitment and activation of peripheral immune cells and resident microglia (Ponath et al., 2018a), and potentially direct neurotoxic effects on oligodendrocytes and neurons (Liddelow et al., 2017). In addition, reactive astrocytes have reduced capabilities for glutamate uptake (Pitt et al., 2000; Fang et al., 2012) and other homeostatic functions (Schirmer et al., 2019), which may result in indirect neurotoxicity.
Here, we review the progress made in identifying astrocyte populations in MS and its mouse model, experimental autoimmune encephalomyelitis (EAE). We will discuss the limitations of current snRNA-seq methods in identifying astrocyte subpopulations and the potential of highly multiplexed imaging in further elucidating astrocyte heterogeneity by adding spatial information.
In contrast to oligodendrocytes and microglia, astrocytes are morphologically heterogenous at a resting state. At least nine morphologically distinct astrocyte subpopulations have been identified, of which four subtypes are present within the human neocortex (protoplasmic, radial cells, marginal glia, and perivascular glia) (Matyash and Kettenmann, 2010). Additionally, astrocytes are highly diverse in their functional properties, including calcium dynamics, gap junction coupling, expression of transmitter receptors, membrane currents, and glutamate transporters (Matyash and Kettenmann, 2010).
One of the first efforts to identify astrocyte subpopulations was based on marker expression. Genetically green fluorescent protein (GFP)-labeled astrocytes were isolated and screened for 81 cell surface antigens with fluorescence-activated cell sorting (FACS) (John Lin et al., 2017). This identified five distinct astrocyte subpopulations present across cortex, brainstem, and olfactory bulb, which were subsequently demonstrated to emerge at different developmental stages and to have distinct gene-enrichment and functional properties.
Since their selection as the “Method of the Year 2013” by Nature Methods (2014), single-nucleus and single-cell RNA sequencing (scRNA-seq) have become routine technologies to identify cellular subpopulations based on transcriptome-wide gene expression at single-cell resolution. A study that employed this approach identified astrocyte subtypes in mouse cortex using scRNA-seq and single-cell spatial transcriptomics. By applying multiplexed single-molecule fluorescence in situ hybridization (smFISH), the study found that cortical astrocytes segregated into a superficial, mid and deep layer pattern, which did not match the neuronal layering (Bayraktar et al., 2020). However, the spatial clusters did not align with astrocyte clustering based on single-cell transcriptomics, indicating that single cell genomics and smFISH produce different patterns of astrocyte molecular diversity, potentially due to difference in parametric depth.
A separate scRNA-seq study that used Smart-seq2 followed by unsupervised clustering distinguished five distinct astrocyte subtypes in adult mouse cortex and hippocampus (Batiuk et al., 2020). The authors noted that the population structure of astrocytes differed from that of neurons, by lacking distinct cellular hierarchies, suggesting multiple axes of heterogeneity. The transcriptomic differences suggest astrocyte subtype specialization across major astrocyte functions, including synaptogenesis, phagocytosis, synapse function/plasticity, neurotransmission, and others. In addition, the subpopulations were shown to be differentially distributed across the cortex and hippocampus and exhibited distinct morphologies and differential Ca2+ signaling (Batiuk et al., 2020).
Together, the single-cell studies provide evidence for transcriptomic heterogeneity of astrocytes, albeit with less distinct cellular hierarchies than neuronal subtypes. The transcriptomic clusters correspond to distinct morphological features and spatial distribution and have implications for different physiological functions. These studies have focused on cortical astrocytes and less is known about astrocyte diversity in white matter (Köhler et al., 2021).
A hallmark of virtually all CNS pathologies is prominent astrocyte responses. The contribution of reactive astrocytes to a given CNS disease is complex and likely driven by multiple concurrent astrocyte phenotypes that vary with disease, disease stage, brain region, age and genetic predisposition (Ponath et al., 2018a; Escartin et al., 2021). Therefore, defining the population structures of reactive astrocytes in different CNS conditions will provide critical insight into their pathogenesis.
In EAE, earlier studies determined astrocytic gene expression in different neuroanatomic regions of mice using a Ribo-tagging approach in transgenic animals, which allows isolation of mRNA from astrocytes only (Itoh et al., 2018). This demonstrated regional differences in astrocytes transcriptomes, including differential expression of complement and cholesterol synthesis pathway genes in astrocytes isolated from spinal cord, optic nerve, cerebellum, hippocampus, and cerebral cortex, consistent with heterogeneity of astrocyte reactivity across different CNS regions.
In an early transcriptome study, astrocyte responses were stratified into neurotoxic and neuroprotective phenotypes termed A1 and A2 (Liddelow et al., 2017), in analogy with the now abandoned concept of M1/M2 (macrophages) and Th1/Th2 (lymphocytes) polarization (Ransohoff, 2016). The A1 phenotype exhibited functional deficits affecting phagocytosis and synapse formation, and was toxic to neurons and oligodendrocytes in vitro. A1 phenotypes were found to be present in neurodegenerative diseases and MS, based on the expression of complement component 3, whereas A2 astrocytes, which are characterized by upregulation of several neurotrophic factors, were prevalent in ischemia. This binary classification was initially received with considerable enthusiasm; however, in subsequent studies, only subsets or a mix of A1 and A2 signature genes were found to be upregulated in human diseases and mouse models of acute and chronic CNS injury (Al-Dalahmah et al., 2020; Das et al., 2020; Zhou et al., 2020). A recent consensus statement recommended to move beyond the A1–A2 labels, as they do not capture the functional diversity of reactive astrocytes, and to use multidimensional data to establish distinctive astrocyte phenotypes (Escartin et al., 2021).
A recent study by Quintana et al. that used scRNA-seq and epigenetic analyses in combination with in vivo CRISPR-Cas9-based genome editing, identified seven astrocyte subpopulations in EAE (Wheeler et al., 2020). The dominant cluster was characterized by activation of pro-inflammatory and neurotoxic pathways such as the unfolded protein response, activation of NF-κB and inducible nitric oxide synthase pathways. This cluster was driven by downregulation of genes targeted by transcription factor NRF2, which limit oxidative stress and inflammation (Wheeler et al., 2020). Moreover, this population was stabilized by epigenetic modifications driven by MAFG and MAT2α signaling, further supporting the notion that epigenetic changes to the CNS contribute to MS pathogenesis (Huynh et al., 2014). In MS lesion tissue, astrocytes were demonstrated to have increased expression of MAFG and decreased expression of NRF2. Recently, the same group identified an anti-inflammatory subset of astrocytes in EAE that was characterized by co-expression of the lysosomal protein, LAMP1, and the death receptor ligand, TRAIL (Sanmarco et al., 2021). This astrocyte population limits inflammation by inducing T cell apoptosis and is driven by IFNγ, produced by natural killer cells within the meninges. A similar population of astrocytes characterized by IFNγ and TRAIL signaling was identified in human brain and this population was reduced by over 80% in MS tissue (Sanmarco et al., 2021).
Astrocytes can be isolated intact from fresh murine CNS tissue, although enzymatic dissociation has been shown to induce early activation genes (Lacar et al., 2016). In adult human CNS, the method of choice for single-cell transcriptomics is snRNA-seq, because the high cellular interconnectivity makes it difficult to isolate astrocytes and neurons in intact form. snRNA-seq has the additional advantages that it avoids perturbation of gene expression by enzymatic dissociation during whole cell isolation, and that it can be applied to archival frozen material from biobanks (Ding et al., 2020).
Acceptance of snRNA-seq was initially limited, because of differences in RNA quantity and quality between nucleus and cytosol. It was subsequently established that nuclei can be confidently matched to their representing cells, that transcriptomes from single nuclei and whole cells correlate highly, albeit with an abundance of nascent transcripts in nuclei (Lake et al., 2017; Bakken et al., 2018). A recent comparison of snRNA-seq and scRNA-seq data from human microglia demonstrated that a high number of genes implicated in microglia activation in Alzheimer’s disease (AD) was substantially reduced in nuclei (Thrupp et al., 2020). This highlights that single-nucleus data are highly sparse and noisy, particularly in glial cells, requiring improved methodologies for recovery of biological signals (see below).
An additional problem of snRNA-seq studies is that the ratios between different CNS cell types differ to those determined by neuro-histological cell quantification (von Bartheld et al., 2016). The neuroanatomic literature suggests that glia cells are approximately 1.5–2 times more numerous than neurons in cortical gray matter, and 3–4 times more numerous in combined cortex and white matter. Oligodendrocytes are the most frequent glial cell type (45–75%), followed by astrocytes (19–40%), and microglia (10%), depending on the brain region (von Bartheld et al., 2016).
In snRNA-seq data, human brain astrocytes and microglia are consistently under-represented. In several landmark studies of human adult cortex, astrocytes constituted between 1.9 and 7.2%, and microglia between <1 and 2.1% of total nuclei (Lake et al., 2018; Zhu et al., 2018; Hodge et al., 2019). In the two snRNA-seq studies of MS white matter lesion tissue, the predominant nuclear populations were neurons, oligodendrocytes and oligodendrocyte precursor cells constituting 80% of total CNS nuclei, whereas astrocyte nuclei accounted for 7 and 11% and microglia nuclei 2.4 and 2.9%, respectively (Jäkel et al., 2019; Schirmer et al., 2019). A possible reason for this underrepresentation is that they are preferentially lost during the preparation steps presumably due to different mechanical properties of astrocyte/microglia nuclei. Moreover, neurons express up to 10 times more RNA than glial cells, which leads to overrepresentation of neuronal RNA in the final library composition (Zeisel et al., 2015). Collectively, this leads to low representation of astrocyte/microglia nuclei, reduced library complexity and consequently low resolution of population structure heterogeneity for glial cells.
In two recent studies, Quintana et al. performed scRNA-seq of astrocytes derived from fresh, rapid autopsy materials of MS patients using 10x Genomics v2.0 chemistry (Wheeler et al., 2020). This data set was integrated with previously published snRNA-seq results of astrocytes in MS and healthy brains. The authors used this approach to confirm the presence of a specific astrocyte population in MS, which was previously identified in EAE and characterized by increased MAFG activation, GM-CSF signaling and pro-inflammatory pathway activity. The authors demonstrated that this population was present in 12 out of 20 MS patients and was strongly expanded in MS compared to controls. The same approach was used to validate that an anti-inflammatory LAMP1+TRAIL+ astrocyte subpopulation that was reduced in EAE compared to naïve mice, was also downregulated in MS (Sanmarco et al., 2021). It is notable that the 48 human samples used in these studies yielded only a total of 9,700 astrocyte nuclei, averaging 200 astrocyte nuclei per sample. This further illustrates that capture of astrocytes/astrocyte nuclei from human brain is sparse and that the analysis is limited by the low number of astrocyte representation.
There is currently a limited understanding of how isolation and purification protocols may change the nuclear yield of various CNS cellular constituents (Box et al., 2020; Denisenko et al., 2020; Ding et al., 2020). New sample preparation protocols have to be designed to address the structural and metabolic vulnerabilities of different nuclei from different brain regions. In addition, standard FACS technology relies on increased pressures (10–70 psi) for hydrodynamic focusing, and application of high voltage charges to the sorted sample path, which inflicts substantial damage on nuclei and may introduce sampling bias by depleting sensitive nuclear populations. Microfluidic sorters have become a mature technology that operates on atmospheric pressure and uses mechanical valves (Utharala et al., 2018; Berlanda et al., 2021). The reduced for electromechanical manipulation greatly aids the preservation of nuclei and the selective enrichment of sensitive and rare nuclei.
Moreover, new genomic protocols and high-throughput strategies are constantly emerging. Since their inception, 3-prime-based scRNA-seq protocols (Ramsköld et al., 2012; Macosko et al., 2015) have improved their sensitivity from initially 5–10 to 20% of RNA molecules captured per cell. Similarly, Smart-seq protocols have doubled their sensitivity from 30–40 to 70% (Smart-seq3), allowing for collection of the majority of transcripts, including isoforms, which ultimately improves the ability to identify biologically meaningful cell clusters (Hagemann-Jensen et al., 2020).
Finally, multi-omics approaches that combine single-cell transcriptomics with epigenetic mapping, proteomics and lineage tracing (Stoeckius et al., 2018; Gaublomme et al., 2019), provide ever-increasing molecular details of cellular states (Ludwig et al., 2019). These details can be further integrated with spatial transcriptomics which provides additional information such as phenotype localization within a given microenvironment and cell-to-cell interactions (Giesen et al., 2014; Liu et al., 2020). Applied to MS, these methodological advances will eventually provide a more complete picture of the cellular constituents that drive MS pathology, including rare and underrepresented cell types.
An additional important aspect of defining cell populations is to determine their spatial organization within a tissue environment such as MS lesions, including their spatial interactions and the environmental cues that drive their specific expression profiles. Moreover, the spatial resolution of phenotypes that have been determined by single cell genomics, will confirm and/or further improve the phenotypic separation. As with single cell genomics, substantial progress has been made in recent years with multiplexed spatial profiling of RNA and proteins in tissue.
Technologies to quantify single-cell RNA levels in spatial context have been rapidly evolving. SmFISH and sequential FISH (seqFISH) approaches use fluorescence-labeled small oligonucleotides to probe single mRNA transcripts. SeqFISH uses multiple rounds of hybridization and has been shown to be scalable to the genome level in vitro (Eng et al., 2017). SmFISH has been used to define astrocyte subpopulations in different CNS regions (Batiuk et al., 2020; Bayraktar et al., 2020) and to map snRNA-seq-derived phenotypes onto MS lesion tissue (Jäkel et al., 2019; Schirmer et al., 2019). A further development that combines super-resolution microscopy with FISH, termed seqFISH+, achieves, in theory, multiplexing of >20,000 genes in single cells with high accuracy and sub-diffraction-limit resolution (Eng et al., 2019).
Additionally, a method termed deterministic barcoding in tissue for spatial omics sequencing (DBiT-seq) delivers DNA barcodes to tissue at a specific location via a microfluidics platform and sequences the spatially barcoded mRNA and proteins (Liu et al., 2020). Under ideal conditions, DBiT-seq is capable of spatially profiling thousands of mRNAs with next generation sequencing that can be co-mapped with proteins, albeit not at single-cell resolution. These technologies target the whole transcriptome but are currently not as robust as snRNA-seq with lower gene coverage and reading depth.
Highly multiplexed imaging has been driven by imaging mass cytometry (IMC), a technology that, like CyTOF, uses antibodies conjugated to lanthanide isotopes (Giesen et al., 2014; Baharlou et al., 2019). Tissue sections are simultaneously labeled with up to one hundred antibodies, laser-ablated at high resolution and followed by time-of-flight mass spectrometry. IMC has several draw-backs that include the need for comprehensive validation and optimization of antibodies, high cost of antibody conjugation and the ablation/mass spectrometry procedure, lack of an amplification step, which reduces sensitivity for low-abundance proteins, and a standard magnification of only 16×, which may provide insufficient resolution. A low tech approach to highly multiplexed imaging is serial immunofluorescence staining of tissue sections with repetitive cycles of immunolabeling, scanning and antibody removal, termed iterative indirect immunofluorescent imaging (4i) (Gut et al., 2018). Similar to classical immunofluorescence labeling, this approach uses off-the-shelf antibodies, and allows for amplifications steps with secondary antibodies, high magnification imaging and acquisition of large tissue areas with a scanning microscope. The number of cycles is limited by decreasing tissue integrity. In our experience, the multiplexed fluorescent signals are quantitatively reproducible over at least 20 cycles without detectable loss of antigen, increase in background or tissue distortion. Per round, typically 2–4 antibodies can be applied, which allows for staining with a total of 40+ markers. Currently, a microfluidics-assisted version of 4i is being developed that automates most of this method and presumably better preserves tissue integrity. Other multiplexed protein imaging methods, such as nanostring (Decalf et al., 2019; Merritt et al., 2020) and CODEX (Schürch et al., 2020), rely on barcoding of antibodies with unique oligonucleotide sequences. These technologies allow for a one-time antibody application, followed by multiple rounds of binding of unique oligonucleotide reporters, each with a spectrally distinct dye, to assay the corresponding antibody barcodes.
The different multiplexed imaging methods all require computational pipelines that include image registration (if multiple scans have been acquired), segmentation of cell types of interest with workflows such as ilastik pixel classification (Berg et al., 2019) and CellProfiler (Carpenter et al., 2006), and extraction of single cell information for cellular phenotype clustering that are visualized with dimensionality reduction methods such as principal component analysis (PCA) and t-distributed stochastic neighbor embedding (tSNE) (Amir et al., 2013), as with scRNA-seq or snRNA-seq data. The additional spatial data are analyzed with computational pipelines [e.g., histoCAT (Schapiro et al., 2017)] to localize cellular subpopulations within the tissue environment, to identify spatial interactions and to enrich phenotype transition trajectories for spatial context (Moon et al., 2019). Some of these tools are particularly relevant for tissues with complex cell morphologies such as CNS, where spatial analysis can focus separately on the cell body and peripheral processes. As highly multiplexed imaging methods evolve and are more broadly applied, new computational approaches for studying spatial patterns of cellular and molecular organization are emerging. This includes spatial variance component analysis (SVCA) (Arnol et al., 2019), which uses the marker profiles and spatial coordinates of each cell, to quantify the sources of variation for marker expression, such as cell-to-cell interactions, and intrinsic and environmental effects.
We have applied IMC to acute MS lesion tissue to investigate the landscape of myeloid and astrocyte phenotypes in acute and chronic active MS lesions (Park et al., 2019). In this study, we identified five astrocyte subtypes and six myeloid cell subpopulations based on expression patterns of 13 glial activation markers. We found that the different glial subpopulations localized to different lesional zones and exhibited subtype-specific spatial interactions. Moreover, we were able to elucidate astrocyte and microglia phenotypic transitions and quantify the effects of cell-intrinsic factors vs cell-to-cell interactions on marker expression in individual cells (Park et al., 2019). In a separate study of acute MS lesions, we were able to demonstrate the activation patterns of lymphocytes and microglia determined in previous MS studies. In particular, we were able to discriminate between the different source of demyelinating macrophages (microglia vs blood-derived macrophages) and to segregate different B and T cell phenotypes (Ramaglia et al., 2019). These studies demonstrate that highly multiplexed imaging, in conjunction with a computational analysis pipeline, provides a wealth of insight into the functional states and spatial organization of glial cells in MS lesions that are not accessible with standard histology.
Single cell genomics, spatial mRNA profiling and highly multiplexed (protein) imaging offer different advantages and disadvantages. While both single cell genomics and spatial mRNA profiling target the whole transcriptome; RNA-seq is superior in isolated cells/nuclei compared to tissue with regard to gene coverage and sequencing depth. In contrast, highly multiplexed protein imaging has a parametric depth of only dozens to perhaps hundreds of markers, but allows for precise outlining of cells with complex, irregular morphologies such as astrocytes, which will improve the spatial analysis of individual cells. Although it is implicitly assumed that mRNA expression correlates with protein expression, this correlation varies substantially with different gene classes (Koussounadis et al., 2015), making it desirable to confirm RNA expression at a protein level, at least for key markers (Figure 1). Finally, single-cell gene expression matrix typically contains excessive zero entries (Yuan et al., 2017), resulting in sparse data sets, particularly in underrepresented cell types such as astrocytes. We observed that several key markers of astrocytes and microglia activation that were readily detectable with multiplexed imaging, were absent in single-cell transcriptomic datasets from MS lesions. Of note, spatial RNA transcriptomics and multiplexed protein profiling are not believed to lead to underrepresentation of specific cell types such as astrocytes and microglia. The technology is moving rapidly toward development of spatial multi-omics that incorporate mRNA profiling with protein profiling and epigenomics, as it is now possible with DBiT-seq (Liu et al., 2020).
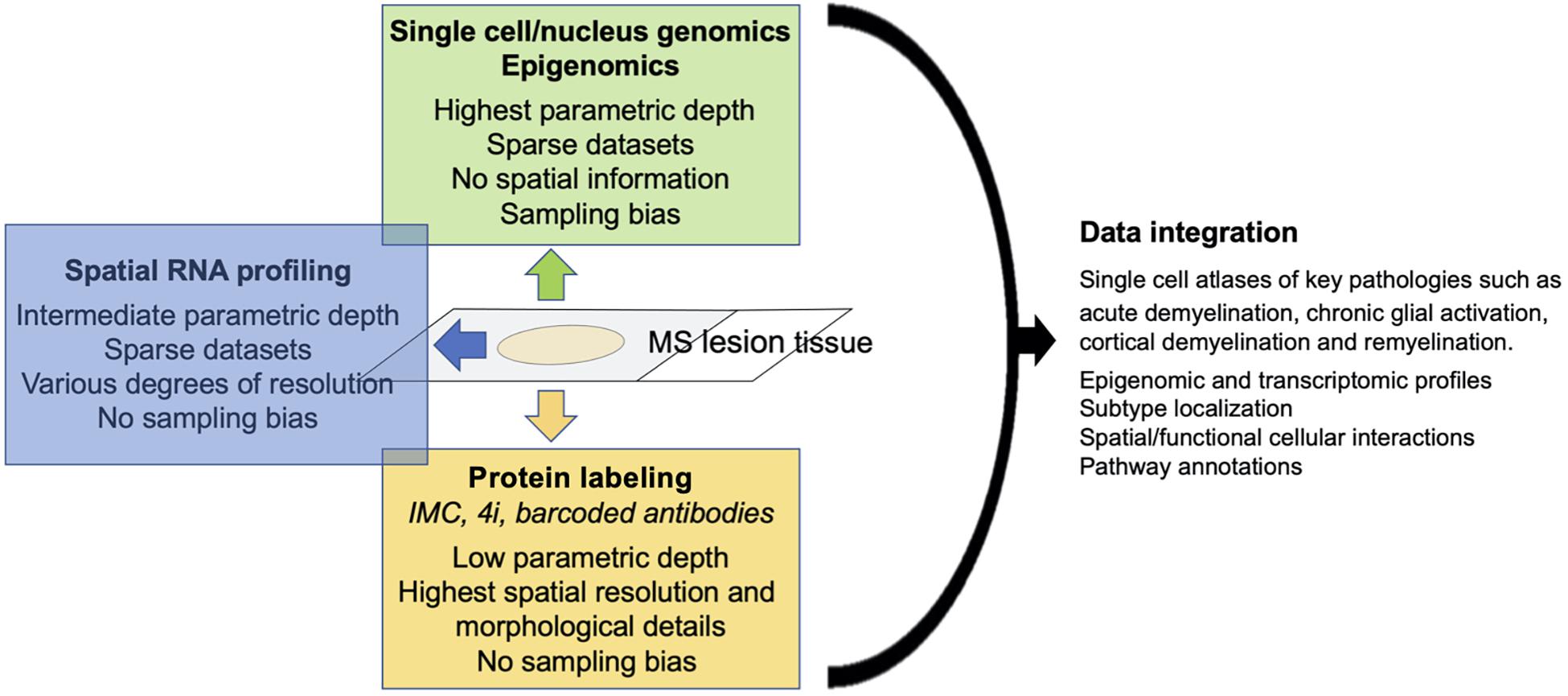
Figure 1. Schematic of strengths/weaknesses of single cell technologies and data output.
With optimized protocols to capture nuclei from astrocytes and other under-represented CNS cell types such as microglia, improvements in single cell genomics and multi-omics integration will allow generation of complete single-cell atlases of healthy CNS and neurological diseases. These atlases will include epigenomic and transcriptomic profiles, subpopulation localization, spatial and functional (receptor-ligand) cellular interactions and pathway annotations. Applied to carefully characterized MS tissue, this will identify the constituent subpopulations and cellular interactomes that are specific to key pathological processes such as acute demyelination, chronic glial activation in progressive MS, and remyelination. This comprehensive approach will identify therapeutic novel targets to ameliorate disease progression and to promote neurorepair.
CL, MS, MM, SB, ST, and DP wrote the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
DP is supported by the National Institute of Health Grants R01 NS112907, R01 NS118886, R01 NS102267, and R21 AI149731.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Absinta, M., Sati, P., Masuzzo, F., Nair, G., Sethi, V., Kolb, H., et al. (2019). Association of Chronic Active Multiple Sclerosis Lesions With Disability In Vivo. JAMA Neurol. 76, 1474–1483. doi: 10.1001/jamaneurol.2019.2399
Al-Dalahmah, O., Sosunov, A. A., Shaik, A., Ofori, K., Liu, Y., Vonsattel, J. P., et al. (2020). Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun. 8:19.
Amir, E.-A. D., Davis, K. L., Tadmor, M. D., Simonds, E. F., Levine, J. H., Bendall, S. C., et al. (2013). viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 31, 545–552. doi: 10.1038/nbt.2594
Anderson, C. M., and Swanson, R. A. (2000). Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32, 1–14. doi: 10.1002/1098-1136(200010)32:1<1::aid-glia10>3.0.co;2-w
Anderson, M. A., Burda, J. E., Ren, Y., Ao, Y., O’shea, T. M., Kawaguchi, R., et al. (2016). Astrocyte scar formation aids central nervous system axon regeneration. Nat. Publ. Group 532, 195–200. doi: 10.1038/nature17623
Arnol, D., Schapiro, D., Bodenmiller, B., Saez-Rodriguez, J., and Stegle, O. (2019). Modeling Cell-Cell Interactions from Spatial Molecular Data with Spatial Variance Component Analysis. Cell Rep. 29, 202–211.e206.
Baharlou, H., Canete, N. P., Cunningham, A. L., Harman, A. N., and Patrick, E. (2019). Mass Cytometry Imaging for the Study of Human Diseases—Applications and Data Analysis Strategies. Front. Immunol. 10:2657. doi: 10.3389/fimmu.2019.02657
Bakken, T. E., Hodge, R. D., Miller, J. A., Yao, Z., Nguyen, T. N., Aevermann, B., et al. (2018). Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One 13:e0209648. doi: 10.1371/journal.pone.0209648
Batiuk, M. Y., Martirosyan, A., Wahis, J., De Vin, F., Marneffe, C., Kusserow, C., et al. (2020). Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 11:1220.
Bayraktar, O. A., Bartels, T., Holmqvist, S., Kleshchevnikov, V., Martirosyan, A., Polioudakis, D., et al. (2020). Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat. Neurosci. 23, 500–509. doi: 10.1038/s41593-020-0602-1
Berg, S., Kutra, D., Kroeger, T., Straehle, C. N., Kausler, B. X., Haubold, C., et al. (2019). ilastik: interactive machine learning for (bio)image analysis. Nat. Methods 16, 1226–1232. doi: 10.1038/s41592-019-0582-9
Berlanda, S. F., Breitfeld, M., Dietsche, C. L., and Dittrich, P. S. (2021). Recent Advances in Microfluidic Technology for Bioanalysis and Diagnostics. Anal. Chem. 93, 311–331. doi: 10.1021/acs.analchem.0c04366
Box, A., Delay, M., Tighe, S., Chittur, S. V., Bergeron, A., Cochran, M., et al. (2020). Evaluating the Effects of Cell Sorting on Gene Expression. J. Biomol. Tech. 31, 100–111. doi: 10.7171/jbt.2020-3103-004
Carpenter, A. E., Jones, T. R., Lamprecht, M. R., Clarke, C., Kang, I. H., Friman, O., et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7:R100.
Chen, W., Gauthier, S. A., Gupta, A., Comunale, J., Liu, T., Wang, S., et al. (2014). Quantitative susceptibility mapping of multiple sclerosis lesions at various ages. Radiology 271, 183–192. doi: 10.1148/radiol.13130353
Das, S., Li, Z., Noori, A., Hyman, B. T., and Serrano-Pozo, A. (2020). Meta-analysis of mouse transcriptomic studies supports a context-dependent astrocyte reaction in acute CNS injury versus neurodegeneration. J. Neuroinflamm. 17:227.
Decalf, J., Albert, M. L., and Ziai, J. (2019). New tools for pathology: a user’s review of a highly multiplexed method for in situ analysis of protein and RNA expression in tissue. J. Pathol. 247, 650–661. doi: 10.1002/path.5223
Denisenko, E., Guo, B. B., Jones, M., Hou, R., De Kock, L., Lassmann, T., et al. (2020). Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 21:130.
Ding, J., Adiconis, X., Simmons, S. K., Kowalczyk, M. S., Hession, C. C., Marjanovic, N. D., et al. (2020). Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 38, 737–746.
Dutta, R., Mcdonough, J., Yin, X., Peterson, J., Chang, A., Torres, T., et al. (2006). Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Anna. Neurol. 59, 478–489. doi: 10.1002/ana.20736
Eng, C.-H. L., Shah, S., Thomassie, J., and Cai, L. (2017). Profiling the transcriptome with RNA SPOTs. Nat. Methods 14, 1153–1155. doi: 10.1038/nmeth.4500
Eng, C. L., Lawson, M., Zhu, Q., Dries, R., Koulena, N., Takei, Y., et al. (2019). Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239. doi: 10.1038/s41586-019-1049-y
Escartin, C., Galea, E., Lakatos, A., O’callaghan, J. P., Petzold, G. C., Serrano-Pozo, A., et al. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 24, 312–325.
Fang, J., Han, D., Hong, J., Tan, Q., and Tian, Y. (2012). The chemokine, macrophage inflammatory protein-2γ, reduces the expression of glutamate transporter-1 on astrocytes and increases neuronal sensitivity to glutamate excitotoxicity. J. Neuroinflamm. 9, 5–1.
Faulkner, J. R., Herrmann, J. E., Woo, M. J., Tansey, K. E., Doan, N. B., and Sofroniew, M. V. (2004). Reactive astrocytes protect tissue and preserve function after spinal cord injury. J. Neurosci. 24, 2143–2155. doi: 10.1523/jneurosci.3547-03.2004
Gaublomme, J. T., Li, B., Mccabe, C., Knecht, A., Yang, Y., Drokhlyansky, E., et al. (2019). Nuclei multiplexing with barcoded antibodies for single-nucleus genomics. Nat. Commun. 10:2907.
Giesen, C., Wang, H. A. O., Schapiro, D., Zivanovic, N., Jacobs, A., Hattendorf, B., et al. (2014). Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422. doi: 10.1038/nmeth.2869
Guerrero, B. L., and Sicotte, N. L. (2020). Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 11:374. doi: 10.3389/fimmu.2020.00374
Gut, G., Herrmann, M. D., and Pelkmans, L. (2018). Multiplexed protein maps link subcellular organization to cellular states. Science 3:361.
Hagemann-Jensen, M., Ziegenhain, C., Chen, P., Ramsköld, D., Hendriks, G. J., Larsson, A. J. M., et al. (2020). Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 38, 708–714. doi: 10.1038/s41587-020-0497-0
Hodge, R. D., Bakken, T. E., Miller, J. A., Smith, K. A., Barkan, E. R., Graybuck, L. T., et al. (2019). Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68.
Horng, S., Therattil, A., Moyon, S., Gordon, A., Kim, K., Argaw, A. T., et al. (2017). Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J. Clin. Investig. 127, 3136–3151. doi: 10.1172/jci91301
Huynh, J. L., Garg, P., Thin, T. H., Yoo, S., Dutta, R., Trapp, B. D., et al. (2014). Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat. Neurosci. 17, 121–130. doi: 10.1038/nn.3588
Itoh, N., Itoh, Y., Tassoni, A., Ren, E., Kaito, M., Ohno, A., et al. (2018). Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc. Natl. Acad. Sci. 115, E302–E309.
Jäkel, S., Agirre, E., Mendanha Falcão, A., Van Bruggen, D., Lee, K. W., Knuesel, I., et al. (2019). Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566, 543–547. doi: 10.1038/s41586-019-0903-2
John Lin, C.-C., Yu, K., Hatcher, A., Huang, T.-W., Lee, H. K., Carlson, J., et al. (2017). Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 20, 396–405. doi: 10.1038/nn.4493
Köhler, S., Winkler, U., and Hirrlinger, J. (2021). Heterogeneity of Astrocytes in Grey and White Matter. Neurochem. Res. 46, 3–14. doi: 10.1007/s11064-019-02926-x
Koussounadis, A., Langdon, S. P., Um, I. H., Harrison, D. J., and Smith, V. A. (2015). Relationship between differentially expressed mRNA and mRNA-protein correlations in a xenograft model system. Sci. Rep. 5:10775.
Lacar, B., Linker, S. B., Jaeger, B. N., Krishnaswami, S. R., Barron, J. J., Kelder, M. J. E., et al. (2016). Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun. 7:11022.
Lake, B. B., Chen, S., Sos, B. C., Fan, J., Kaeser, G. E., Yung, Y. C., et al. (2018). Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 36, 70–80. doi: 10.1038/nbt.4038
Lake, B. B., Codeluppi, S., Yung, Y. C., Gao, D., Chun, J., Kharchenko, P. V., et al. (2017). A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Scient. Rep. 7:1202.
Lee, J.-H., Kim, J.-Y., Noh, S., Lee, H., Lee, S. Y., Mun, J. Y., et al. (2021). Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nat. Publ. Group 590, 612–617. doi: 10.1038/s41586-020-03060-3
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487.
Liu, Y., Yang, M., Deng, Y., Su, G., Enninful, A., Guo, C. C., et al. (2020). High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 183, 1665–1681.e1618.
Ludwig, L. S., Lareau, C. A., Ulirsch, J. C., Christian, E., Muus, C., Li, L. H., et al. (2019). Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell 176.1325–1339.e1322.
Macosko, E. Z., Basu, A., Satija, R., Nemesh, J., Shekhar, K., Goldman, M., et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. doi: 10.1016/j.cell.2015.05.002
Matyash, V., and Kettenmann, H. (2010). Heterogeneity in astrocyte morphology and physiology. Brain Res. Rev. 63, 2–10. doi: 10.1016/j.brainresrev.2009.12.001
Merritt, C. R., Ong, G. T., Church, S. E., Barker, K., Danaher, P., Geiss, G., et al. (2020). Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 38, 586–599. doi: 10.1038/s41587-020-0472-9
Moon, K. R., Van Dijk, D., Wang, Z., Gigante, S., Burkhardt, D. B., Chen, W. S., et al. (2019). Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 37, 1482–1492. doi: 10.1038/s41587-019-0336-3
Park, C., Ponath, G., Levine-Ritterman, M., Bull, E., Swanson, E. C., De Jager, P. L., et al. (2019). The landscape of myeloid and astrocyte phenotypes in acute multiple sclerosis lesions. Acta Neuropathol. Commun. 7:130.
Pitt, D., Werner, P., and Raine, C. S. (2000). Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 6, 67–70. doi: 10.1038/71555
Ponath, G., Lincoln, M. R., Levine-Ritterman, M., Park, C., Dahlawi, S., Mubarak, M., et al. (2018a). Enhanced astrocyte responses are driven by a genetic risk allele associated with multiple sclerosis. Nat. Commun. 9:5337. doi: 10.1038/s41467-018-07785-8
Ponath, G., Park, C., and Pitt, D. (2018b). The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 9:217–217.
Ramaglia, V., Sheikh-Mohamed, S., Legg, K., Park, C., Rojas, O. L., Zandee, S., et al. (2019). Multiplexed imaging of immune cells in staged multiple sclerosis lesions by mass cytometry. eLife 8:e48051.
Ramsköld, D., Luo, S., Wang, Y.-C., Li, R., Deng, Q., Faridani, O. R., et al. (2012). Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 30, 777–782. doi: 10.1038/nbt.2282
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Sanmarco, L. M., Wheeler, M. A., Gutiérrez-Vázquez, C., Polonio, C. M., Linnerbauer, M., Pinho-Ribeiro, F. A., et al. (2021). Gut-licensed IFNγ+ NK cells drive LAMP1+TRAIL+ anti-inflammatory astrocytes. Nature 590, 473–479. doi: 10.1038/s41586-020-03116-4
Schapiro, D., Jackson, H. W., Raghuraman, S., Fischer, J. R., Zanotelli, V. R. T., Schulz, D., et al. (2017). histoCAT: analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat. Methods 14, 873–876. doi: 10.1038/nmeth.4391
Schirmer, L., Velmeshev, D., Holmqvist, S., Kaufmann, M., Werneburg, S., Jung, D., et al. (2019). Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 573, 75–82. doi: 10.1038/s41586-019-1404-z
Schürch, C. M., Bhate, S. S., Barlow, G. L., Phillips, D. J., Noti, L., Zlobec, I., et al. (2020). Coordinated Cellular Neighborhoods Orchestrate Antitumoral Immunity at the Colorectal Cancer Invasive. Front. Cell 182, 1341–1359.e19. doi: 10.1016/j.cell.2020.07.005
Sofroniew, M. V., and Vinters, H. V. (2009). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Stoeckius, M., Zheng, S., Houck-Loomis, B., Hao, S., Yeung, B. Z., Mauck, W. M., et al. (2018). Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19:224.
Sucksdorff, M., Matilainen, M., Tuisku, J., Polvinen, E., Vuorimaa, A., Rokka, J., et al. (2020). Brain TSPO-PET predicts later disease progression independent of relapses in multiple sclerosis. Brain 76:1474.
Thrupp, N., Sala Frigerio, C., Wolfs, L., Skene, N. G., Fattorelli, N., Poovathingal, S., et al. (2020). Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep. 32:108189. doi: 10.1016/j.celrep.2020.108189
Tyzack, G. E., Sitnikov, S., Barson, D., Adams-Carr, K. L., Lau, N. K., Kwok, J. C., et al. (2014). Astrocyte response to motor neuron injury promotes structural synaptic plasticity via STAT3-regulated TSP-1 expression. Nat. Commun. 5:4294.
Utharala, R., Tseng, Q., Furlong, E. E. M., and Merten, C. A. (2018). A Versatile, Low-Cost, Multiway Microfluidic Sorter for Droplets, Cells, and Embryos. Anal. Chem. 90, 5982–5988. doi: 10.1021/acs.analchem.7b04689
von Bartheld, C. S., Bahney, J., and Herculano-Houzel, S. (2016). The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J. Comp. Neurol. 524, 3865–3895. doi: 10.1002/cne.24040
Wheeler, M. A., Clark, I. C., Tjon, E. C., Li, Z., Zandee, S. E. J., Couturier, C. P., et al. (2020). MAFG-driven astrocytes promote CNS inflammation. Nature 578, 593–599. doi: 10.1038/s41586-020-1999-0
Wilbanks, B., Maher, L. J. III, and Rodriguez, M. (2019). Glial cells as therapeutic targets in progressive multiple sclerosis. Expert Rev. Neurother. 19, 481–494. doi: 10.1080/14737175.2019.1614443
Yuan, G.-C., Cai, L., Elowitz, M., Enver, T., Fan, G., Guo, G., et al. (2017). Challenges and emerging directions in single-cell analysis. Genome Biol. 18:84.
Zeisel, A., Muñoz-Manchado, A. B., Codeluppi, S., Lönnerberg, P., La Manno, G., Juréus, A., et al. (2015). Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142. doi: 10.1126/science.aaa1934
Zhou, Y., Song, W. M., Andhey, P. S., Swain, A., Levy, T., Miller, K. R., et al. (2020). Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 26, 131–142. doi: 10.1038/s41591-019-0695-9
Keywords: astrocytes, multiple sclerosis, single nucleus sequencing, multiplexed imaging, experimental autoimmune encephalomyelitis
Citation: Lo CH, Skarica M, Mansoor M, Bhandarkar S, Toro S and Pitt D (2021) Astrocyte Heterogeneity in Multiple Sclerosis: Current Understanding and Technical Challenges. Front. Cell. Neurosci. 15:726479. doi: 10.3389/fncel.2021.726479
Received: 16 June 2021; Accepted: 15 July 2021;
Published: 11 August 2021.
Edited by:
Jason R. Plemel, University of Alberta, CanadaReviewed by:
Luke Michael Healy, McGill University, CanadaCopyright © 2021 Lo, Skarica, Mansoor, Bhandarkar, Toro and Pitt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Pitt, ZGF2aWQucGl0dEB5YWxlLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.