 Junyun He
Junyun He Rodney M. Ritzel
Rodney M. Ritzel Junfang Wu
Junfang Wu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 09 April 2021
Sec. Cellular Neuropathology
Volume 15 - 2021 | https://doi.org/10.3389/fncel.2021.662971
This article is part of the Research Topic Advances in CNS Repair, Regeneration, and Neuroplasticity: From Basic Mechanisms to Therapeutic Strategies View all 32 articles
The voltage-gated proton channel Hv1 is a newly discovered ion channel that is highly conserved among species. It is known that Hv1 is not only expressed in peripheral immune cells but also one of the major ion channels expressed in tissue-resident microglia of the central nervous systems (CNS). One key role for Hv1 is its interaction with NADPH oxidase 2 (NOX2) to regulate reactive oxygen species (ROS) and cytosolic pH. Emerging data suggest that excessive ROS production increases and requires proton currents through Hv1 in the injured CNS, and manipulations that ablate Hv1 expression or induce loss of function may provide neuroprotection in CNS injury models including stroke, traumatic brain injury, and spinal cord injury. Recent data demonstrating microglial Hv1-mediated signaling in the pathophysiology of the CNS injury further supports the idea that Hv1 channel may function as a key mechanism in posttraumatic neuroinflammation and neurodegeneration. In this review, we summarize the main findings of Hv1, including its expression pattern, cellular mechanism, role in aging, and animal models of CNS injury and disease pathology. We also discuss the potential of Hv1 as a therapeutic target for CNS injury.
The central nervous system (CNS) injuries or diseases such as stroke, traumatic brain injury (TBI), spinal cord injury (SCI), and multiple sclerosis (MS), are the leading causes of death and disability in the United States and around the world. To date, no neuroprotective or neurorestorative therapy has clearly improved long-term recovery. This may reflect, in part, an incomplete understanding of the complex pathobiological mechanisms involved. Numerous experimental observations including ours (Faden et al., 2016; Skovira et al., 2016; Li Y. et al., 2020; Ritzel et al., 2020) indicate that persistent neuroinflammation contributes to long-term neurological functional deficits. However, no preferred current treatments specifically target neuroinflammatory mechanisms for CNS damage. Thus, identifying new targets to effectively reduce CNS injury-induced neuroinflammation remains an important research direction.
As the major cellular component of the innate immune system in the CNS, microglia play a critical role in neuroinflammation following CNS trauma (Loane and Byrnes, 2010). In response to injury, microglia can produce neuroprotective factors, clear cellular debris and orchestrate neurorestorative processes that are beneficial for neurological recovery. However, dysregulated microglia can also produce high levels of pro-inflammatory and cytotoxic mediators that can injure cells and hinder CNS repair (Loane and Kumar, 2016). Theoretically, resident microglia should work in concert to fine-tune inflammatory responses, scavenge debris, and promote remodeling and repair after injury, thereby contributing to successful wound healing. However, microglia often exhibit a sustained neurotoxic phenotype continuing for months to years after CNS trauma, associated with progressive neurodegeneration and related neurological dysfunction (Faden et al., 2016).
Proton (H+)-sensitive channel and exchanger in microglia include the hydrogen voltage gated channel 1 (HVCN1) and Na+/H+ exchanger (NHE). The HVCN1 gene encodes a voltage-gated protein channel protein (Hv1/VSOP) that is primarily expressed in phagocytes in the immune system (Ramsey et al., 2006; Sasaki et al., 2006) and is one of the major ion channels expressed in CNS-resident microglia (Wu et al., 2012). It is reported that Hv1 is not expressed by neurons or astrocytes in the mouse brain (Wu et al., 2012). In contrast, NHE1 is ubiquitously expressed in all cell types in the CNS. While NHE1 is important for maintaining the basal intracellular pH (pHi) in microglia, cell migration, and calcium triggering, Hv1 mediates pH regulation and membrane hyperpolarization in microglia (Luo et al., 2021). Although the voltage-dependent proton current was first reported forty years ago (Thomas and Meech, 1982), its gene and molecular structure were not identified until 2006 (Ramsey et al., 2006; Sasaki et al., 2006). The crystal structure is a chimeric construct composed of the parts of 3 different proteins and forms a trimer, in contrast to the dimer-forming Hv1 channel (Takeshita et al., 2014). To date, one key physiological function of Hv1 is the regulation of NADPH oxidase (NOX) activity in phagocytes (Ramsey et al., 2009). Under pathological conditions, microglial Hv1 is required for NOX-dependent generation of reactive oxygen species (ROS) by providing charge compensation for exported electrons via proton extrusion, thereby relieving the intracellular acidosis that occurs following respiratory burst. Excessive NOX/ROS expression and tissue acidosis can damage neurons and glia, and participates in the pathophysiology of neuroinflammatory disorders (von Leden et al., 2017). However, Hv1 also functions independently of the NOX/ROS pathway, for example in snail neurons, basophils, osteoclasts, and cancer cells (Seredenina et al., 2015). Early work from experimental stroke models using transgenic knockout (KO) mice provides a strong rationale for Hv1 as a potential therapeutic target for the treatment of ischemic brain injury (Wu et al., 2012).
Generally, ion channels including Hv1 are potent drug targets (Seredenina et al., 2015). To facilitate the development of clinically useful Hv1-active drugs, this review summarizes the current research on the function and mechanisms of the Hv1 channel in CNS injury models including various ischemic and traumatic brain injuries, neurodegenerative disease, and spinal cord injury. We also discuss recent studies on disrupted Hv1 in age-related pathologies, highlighting its involvement in chronic CNS injury and neurodegenerative diseases. The temporal and spatial expression of Hv1 channel are reviewed in CNS resident cells as well as on injury-responsive immune cells. Potential cellular mechanism studies addressing the role of Hv1 in CNS injury-mediated pathobiology are highlighted, including microglial Hv1-triggered oxidative stress and neuroinflammation. Lastly, we review recent novel mechanistic discoveries on Hv1-driven intracellular and extracellular acidosis in injured brain and spinal cord tissues, and provide therapeutic options, with an emphasis on the depletion or inhibition of the Hv1 channel for the prevention of acidotoxicity, oxidative stress damage, and inflammatory neurodegeneration after CNS trauma.
The voltage-dependent proton current was first reported in neurons of the land snail Helix aspersa (Thomas and Meech, 1982). Decades later in 2006 the Hv1 gene was identified simultaneously by two groups in both Mus musculus (Sasaki et al., 2006) and Homo Sapiens (Ramsey et al., 2006). The human Hv1 (hHv1) gene is located on the chromosome 12 at q24.11 and has seven exons. The hHv1 protein is predicted to be 273 amino acids in length (31.7 kD, pI = 6.62) and forms four transmembrane (TM) segments (s1–s4) (see Figure 1). The TM structure and placement of charged residues in hydrophobic domains is conserved among the vertebrate Hv1 orthologs. The N-terminal of Hv1 contains a voltage-sensitive domain (VSD) but is not conserved in sequence. The VSD of Hv1 is unique due to the lack of pore domain which is totally different from the other voltage-gated ion channels, and therefore Sasaki et al. (2006) first named it voltage sensor domain only protein (VSOP). The VSD has dual roles of voltage-sensing and proton permeation. Its gating is sensitive to pH and Zn2+ (Ramsey et al., 2006). H168 is crucial for ΔpH-dependent gating of the hHv1 (Cherny et al., 2018). The crystal structure of mouse Hv1 was presented in the resting state at 3.45Å resolution, providing a platform for understanding the general principles of voltage-sensing and proton permeation (Takeshita et al., 2014).
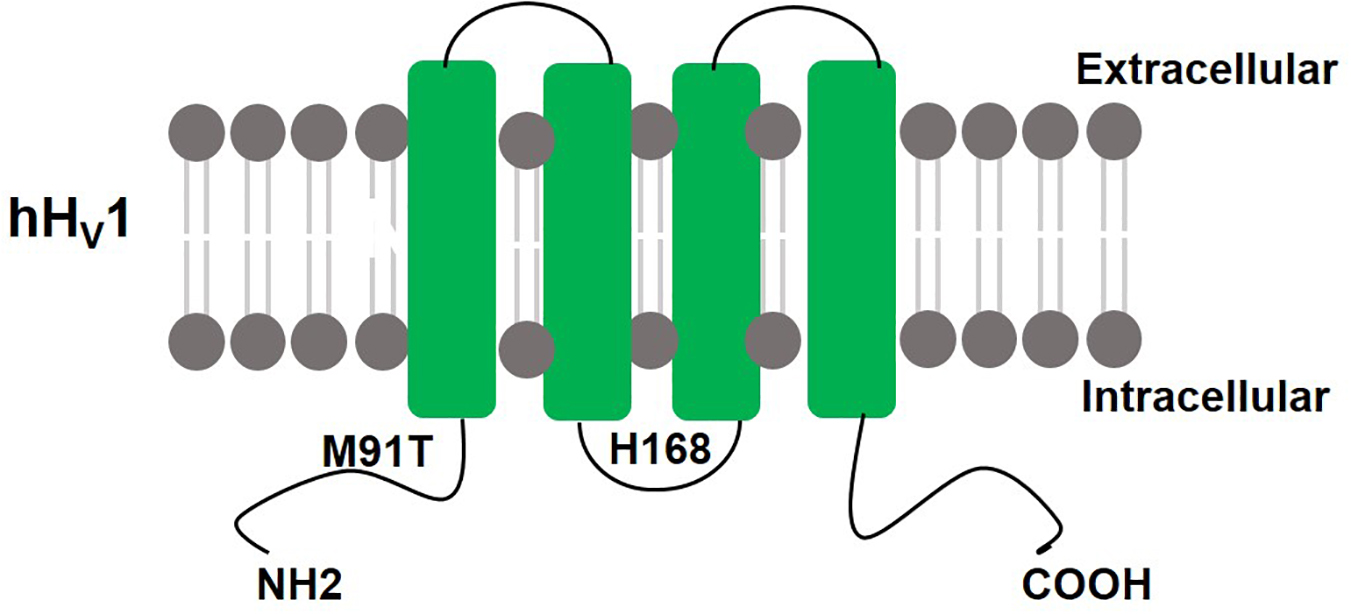
Figure 1. Schematic structure of hHv1: hHv1 has four transmembrane domains with N terminus and C terminus lie inside the cells. H168 is crucial for ΔpH-dependent gating, M91T mutant requires approximately 0.5 pH units more alkaline pH values than wild-type Hv1 to activate proton extrusion.
There are two isoforms of Hv1, long and truncated. HVCN1S is a short isoform lacking the first 20 amino acids via alternative mRNA splicing (Hondares et al., 2014). It is specifically expressed at higher levels in chronic lymphocytic leukemia (CLL) cells and other B cell lines than is the longer isoform, but only weakly expressed in normal B cells. HVCN1S mediates stronger currents upon PKC phosphorylation and increases BCR signaling, proliferation, and chemokine-dependent migration, which together confer a growth advantage to malignant B cells and contribute to the disease pathogenesis of CLL (Hondares et al., 2014).
In human sperm, Hv1 is post-translationally cleaved to a N-terminus truncated form that lacks the first 68 amino acids, generating Hv1Sper which has a molecular weight of 25 kDa (Berger et al., 2017). The pH-control of Hv1Sper and Hv1 is distinctively different. Voltage gating of Hv1 is intimately coupled to ΔpH across the cell membrane regardless of the changes in the intracellular and extracellular pH. While in Hv1Sper, gating is sensitive not only to ΔpH, but also to the changes in intracellular and extracellular pH. As a result, Hv1 can only export protons and alkalize sperm, whereas Hv1Sper can either alkalize or acidify sperm, depending on the pH of the environment (Berger et al., 2017).
A missense mutation of Hv1 at the 91 site from methionine to threonine (M91T) in epithelial cells requires approximately 0.5 pH units more alkaline mucosal pH values than wild-type Hv1 to activate proton extrusion, providing both functional and molecular indication that Hv1 mediates pH-regulated acid secretion by the airway epithelium (Iovannisci et al., 2010).
The Hv1 channel is a membrane protein that is highly expressed in cells of the immune system including eosinophils (DeCoursey et al., 2001), neutrophils (DeCoursey et al., 2000), basophils (Musset et al., 2008), B cells (Capasso et al., 2010), T cells (Schilling et al., 2002), monocytes (Musset et al., 2012a), macrophages (Kapus et al., 1993), dendritic cells (Szteyn et al., 2012), and mast cells (Kuno et al., 1997), suggesting that Hv1-driven signaling may be involved in a variety of immunological processes. In the CNS, Hv1 mRNA expression patterns in rat primary cultured neurons, microglia, and astrocytes revealed that its expression is predominantly limited to microglia (Li et al., 2021). The basal level of Hv1 in cultured astrocytes and neurons was extremely low or undetectable (Wu et al., 2012; Li et al., 2021). In the healthy mouse brain, Hv1 was found to be exclusively expressed in microglia, but not in neurons or astrocytes (Wu et al., 2012). The expression level of Hv1 in the mouse CNS is developmentally increased, i.e., non-detectable in the neonatal stage, detectable in adulthood by western blot, and 2–3-fold higher in the aged brain compared with that in the adult brain (Wu, 2014; Zhang et al., 2020). This likely reflects the increased activation state of microglia that occurs with normal aging (Harry, 2013; Norden and Godbout, 2013).
In response to CNS insults, increased Hv1 protein expression was observed in whole brain lysates at 24 h after permanent middle cerebral artery occlusion (MCAO) (Wu et al., 2012). In a mouse moderate/severe brain injury using a controlled cortical impact (CCI) model of experimental TBI, Hv1 mRNA and protein expression were remarkably increased at 24 h post-injury and remained elevated for up to 4 weeks in the ipsilateral cortex and hippocampus (Ritzel et al., 2021). qPCR analysis revealed a virtual absence of Hv1 mRNA expression in the microglia-depleted brain (Ritzel et al., 2021), further confirming that expression of this channel is largely restricted to microglia within the CNS. Dysregulation of Hv1 expression in the injured spinal cord after moderate/severe contusion in mice was also recently reported (Li et al., 2021). qPCR analysis showed a rapid up-regulation (40-fold) of Hv1 mRNA, starting at 3 days post-injury and continuing for up to 28 days in both male and female mice. This was confirmed in CD11b+ microglia/macrophages isolated from the adult spinal cord at 3 days post-injury. In situ, cells immunoreactive for Hv1 coexpress ionized calcium-binding adaptor molecule 1 (Iba1), a known marker for microglia/macrophages (Li et al., 2020b). Furthermore, a nearly four-fold increase in Hv1 protein expression was observed in spinal cord tissue at 3 days post-injury (Li et al., 2021). These results demonstrate that the expression level of the Hv1 channel is dysregulated following CNS injury.
The best-described function of Hv1 is the regulation of NOX2 activity in phagocytes. During respiratory burst, electrons from NADPH are transferred outside of the cell, leading to membrane depolarization; at the same time, protons are accumulated in the cytoplasm, leading to acidification of the cytosol (El Chemaly et al., 2010). NOX activity, depolarization, and intracellular acidification are key stimuli to activate Hv1. Both membrane depolarization and cytosol acidification inhibit the activity of NOX2, which can be reversed by Hv1-mediated extrusion of protons from the cytoplasm, thereby maintaining physiological membrane potential and re-establishing normal pH. Inhibition of Hv1 activity would thus portend a decrease in proton extrusion that could lead to intracellular acidosis. For example, in Hv1-deficient neutrophils, the cytosolic pH is more acidic than in wild-type (WT) cells following stimulation with phorbol myristate acetate (PMA) (El Chemaly et al., 2010). In addition to superoxide production, Hv1 also sustains calcium entry and migratory capacity (El Chemaly et al., 2010).
Hv1-deficient eosinophils have also been shown to produce significantly less ROS upon PMA stimulation compared with WT eosinophils (Zhu et al., 2013). However, Hv1-depleted eosinophils did not show an impairment in calcium mobilization or migration ability as demonstrated in neutrophils. Interestingly, Hv1-deficient eosinophils underwent significantly greater cell death after PMA stimulation, suggesting that intracellular acidosis during respiratory burst may be more severe in this cell type (Zhu et al., 2013).
In adaptive immune cells, Hv1 modulates B cell antigen receptor (BCR) signal strength in B cells (Capasso et al., 2010). Generation of ROS was lower in Hv1-deficient B cells, resulting in attenuated BCR signaling via impaired BCR-dependent oxidation of Src homology region 2 domain-containing phosphatase-1 (SHP-1). Hv1 KO mice showed reduced activation of the spleen tyrosine kinase (SYK) and protein kinase B (PKB, also known as Akt) signaling pathways, impaired mitochondrial respiration and glycolysis, and diminished antibody responses (Capasso et al., 2010). While little is known regarding the role of Hv1 in T cells, there is evidence that ablating Hv1 in Jurkat T cells induces acidification which promotes apoptotic cell death (Asuaje et al., 2017). It remains to be seen whether Hv1 plays a similar role in modulating T cell receptor (TCR) signaling pathways and downstream effector functions.
In microglia, Hv1 channels were reported to conduct protons to the extracellular space as charge compensation and to help sustain ROS production by NOX2 (Wu et al., 2012). However, Kawai et al. (2017, 2018) observed that Hv1-deficiency enhanced the extracellular ROS signal in primary cultured microglia, as well as in Kupffer cells (Kawai et al., 2020). This may be apparently paradoxical considering numerous evidence showing that Hv1 supports ROS generation, but still sheds light on the complex regulatory mechanism of Hv1 in context of diverse physiological states of microglia.
Within the medullary thick ascending limb, Hv1 is found to localize in the mitochondria membrane where it modulates the formation of ROS by complex I on the mitochondrial membrane rather than by NOX-mediated ROS production. It points to a novel pathway of Hv1 independent of its traditional role in maintaining cell membrane potential and intracellular pH (Patel et al., 2019).
It is noteworthy that in many instances Hv1 functions independently of NOX. As a proton channel, Hv1 is responsible for conducting protons and thus regulating the intracellular or extracellular pH. For example, in human spermatozoa, Hv1 is highly expressed in the flagellum. Activated Hv1 allows outward conduction of protons, resulting in intracellular alkalization and activation of spermatozoa (Lishko et al., 2010). Hv1 activity has been suggested to play a role in a number of functions in sperm, including motility, capacitation, sperm-zona pellucida interaction, acrosome reaction, and sperm-oocyte fusion (Lishko et al., 2010; Musset et al., 2012b; Zhao et al., 2018; Yeste et al., 2020).
In osteoclast cells, Hv1 helps release H+ ions to acidify the extracellular milieu and dissolve mineralized bone, promoting bone resorption (Nordstrom et al., 1995). In airway epithelia, Hv1 is involved in the maintenance of the physiological pH of the airway and lung environment (Fischer et al., 2002; Cho et al., 2009; Iovannisci et al., 2010; Fischer, 2012). Hv1 activation by PMA or anti-IgE in basophils may cause histamine release, which is coupled with the ability of Hv1 to maintain cytosolic pH but not linked with NOX2 due to its lack of expression in basophils (Musset et al., 2008). This same mechanism of regulating histamine release is also found in mast cells (Marone et al., 1986).
In addition to regulating the intracellular pH, Hv1 is also responsible for regulating the pH of phagocytic vacuole of neutrophils. In human cells, the vacuolar pH is around 9. By contrast, in Hv1 KO mouse neutrophils, the vacuolar pH rose above 11, and the cytosol acidified excessively, demonstrating that Hv1 plays an important role in charge compensation (Levine et al., 2015). Furthermore, Hv1 is required for proper endosomal acidification and subsequent toll-like receptor 9 (TLR9) signaling in plasmacytoid dendritic cells (Montes-Cobos et al., 2020). Hv1 deficiency delayed endosomal acidification, consequently limiting protease activity and the secretion of type I interferons (IFN-I). Whether Hv1 impacts antigen presentation by dendritic cells in the context of microbial infection remains to be seen.
Aging is a major risk factor for many human pathologies, including metabolic, inflammatory, and neurodegenerative conditions. The role of ROS and Ca2+ in aging and age-related disease has been extensively reviewed (Madreiter-Sokolowski et al., 2020). Each have been strongly implicated in the development and progression of age-related pathologies. By ensuring Ca2+ entry through charge compensation, the Hv1 proton channel helps to sustain NOX2-induced ROS production and other phagocyte effector functions critical for innate immunity (El Chemaly et al., 2010). Age-associated defects in neutrophil migration, adhesion, and oxidative burst activity have been documented in young Hv1-deficient mice (Ramsey et al., 2009; El Chemaly et al., 2010; Capasso et al., 2011; Okochi et al., 2020). These observations have led many to examine the role of Hv1 in the context of aging. Given its role in rebalancing pH and charges across the plasma membrane, dysregulated Hv1 activity could provide both a mechanism of age-related disease and a therapeutic target. Although few studies have examined the relationship between Hv1 and innate immune function in naturally aging mice, emerging data suggest that Hv1 activity may play a key role in age-dependent responses to injury.
Studies investigating Hv1 expression across the lifespan have identified age- and region-dependent increases in the brain. The highest expression level was seen in the striatum of WT mice and was associated with increased expression of markers of microglia (e.g., CD11b, CX3CR1, and CD68) (Kawai et al., 2021). These findings suggest that Hv1 may be involved in the transition of microglia from a ramified phenotype to an ameboid morphology indicative of the heightened activation state seen with advanced age (Kawai et al., 2021). Interestingly, despite showing preferential expression in the striatum, no functional significance could be detected in this, or any other, region of the brain in young KO mice. However, the authors found that morphological features of microglia activation and differential gene expression in the cortex were significantly increased in aged KO mice. While this study highlights a role for Hv1 in age-related microglia activation and chronic neuroinflammation, it is important to point out that the KO mice at all ages showed no severe neurological impairment. It remains to be seen whether these age-related changes are adaptive, if Hv1 activity itself is increased in the same region-dependent manner, and which environmental factors are responsible for promoting its expression.
Inflammation subsequent to microglia activation has been demonstrated to be a key driver of age-related cognitive deficits (Udeochu et al., 2016). Due to its high expression in microglia, it is not unreasonable to assume Hv1 could play a detrimental role in neurological decline. However, presently there is no data to support this notion. A study assessing neuroinflammation after postoperative surgery found that Hv1 protein expression was significantly increased in the hippocampus of 18 months old WT mice, but the functional importance of this change with relation to cognition remains to be seen (Zhang et al., 2020). One possible consequence of increased Hv1 activity includes higher production of ROS and oxidative stress damage. Recent work has demonstrated a concomitant increase in NOX2 and ROS levels within the aging hippocampus (Fan et al., 2019). Indeed, recent evidence suggests that Hv1 may promote transcription of oxidative stress genes (e.g., Nrf2, Sod2, and Gpx1) in an age-dependent manner (Kawai et al., 2017). Utilizing global KO mice of various ages, the authors demonstrated that functional Hv1 is increasingly more important during early middle age (i.e., 6 months), when its loss-of-function appears to coincide with higher expression of oxidative stress genes. That deletion of a gene partially required for NOX2-mediated ROS production would result in increased oxidative stress seems paradoxical unless one is to consider the possibility that it removes a brake on, or is overcompensated by increased expression of, Hv1-independent drivers of ROS production. In the same study it was also reported that neuroprotection to experimental stroke was seen in old, but not young mice. While these results are surprising, it supports an adaptive role for Hv1 in age-related responses to brain injury. Consistent with notion, microarray profiling revealed higher expression of Hv1 and other oxidative stress genes in the ischemic hemisphere of old mice compared to young mice (Sieber et al., 2014). The immunological response to stroke is dramatically altered with age, with older mice showing significantly greater neutrophil infiltration and higher rates of hemorrhagic transformation (Ritzel et al., 2018). It is possible that the age-dependent protection seen in older KO mice is due to decreased transmigration and reactivity of neutrophils in the ischemic brain, rather than microglia-driven. Nevertheless, neuroprotection in young KO mice has been documented in models of traumatic CNS injury, hypoperfusion-induced white matter injury, and multiple sclerosis (Li et al., 2021; Ritzel et al., 2021). Moreover, our group (Ritzel et al., 2021) showed that protection persisted for weeks and months following TBI. These findings indicate there could be important interactions between age, Hv1, and the onset, duration, and type of injury.
It is worth reiterating that Hv1 expression is not exclusive to phagocytes of myeloid lineage, as some groups have reported expression in B and T lymphocytes (Fernandez et al., 2016). In B cells, Hv1 has been shown to regulate the strength of BCR signaling (Capasso et al., 2010). Despite the well characterized effects of aging on B cells and autoimmunity, few studies have examined the age-dependency of Hv1 function in adaptive immunity (Ma et al., 2019). One report indicates Hv1 KO mice develop an autoimmune disorder phenotype with advanced age (Sasaki et al., 2013). This was characterized by splenomegaly at 6 months of age, higher CD44 expression on T cells, and increased production and deposition of autoantibodies in the kidney coincident with nephritis. If confirmed, these findings suggest that Hv1 may play a more important role in the adaptive arm of the immune system than previously appreciated. However, these results should be interpreted with caution, as it has been suggested that these age-associated phenotypic features may be dependent on external and intrinsic factors such as housing conditions and C57Bl/6 substrains, respectively (Capasso, 2014).
One major caveat to these studies is the over interpretation of results generated using germline KO mice, which may have hidden developmental aberrations that affect the context of Hv1 activity, including the potential for other genes to compensate for this activity. For this reason, the advent of cell type-specific conditional gene deletion and temporal-controlled inducible gene deletion systems will prove indispensable for understanding how the aging process effects Hv1 activity, and by extension, the pathogenesis of age-related pathology. Furthermore, future studies using pharmacologic inhibitors to selectively target Hv1 should include aged animals to determine whether there is an enhanced therapeutic response. The established roles of ROS, ionic dysregulation, inflammation, and metabolism in aging and disease continue to provide sound rationale for establishing the underlying role of Hv1 in this process.
A detrimental role for Hv1 in stroke was first reported in 2012 (Wu et al., 2012). By utilizing Hv1 KO mice and both transient and permanent MCAO ischemic stroke models, Wu and colleagues showed that mice constitutively lacking Hv1 had reduced infarct volumes and more favorable neurological deficit scores at 24 h after stroke independent of reperfusion. This protection was due to a reduction in ROS attributable to the interaction between microglial Hv1 and NOX2. The authors demonstrated this by generating bone marrow chimeric mice that lacked either microglial or bone marrow-derived Hv1. These data supported the notion that brain resident microglial Hv1 are the primary mediators for NOX/ROS-induced brain damage 1d after MCAO. However, during the first week of injury, large numbers of the blood-borne leukocytes entered the brain which highly express Hv1 (Schilling et al., 2003; Gelderblom et al., 2009). Thus, the contribution of peripheral Hv1 cannot be excluded in the post-acute stages of stroke.
In a photothrombotic stroke model, Hv1 KO mice displayed smaller brain infarction and fewer motor coordination deficits compared to the WT group during the first three days after injury (Tian et al., 2016), in part by reducing the pro-inflammatory (M1)- and increasing the anti-inflammatory (M2)-type polarization of microglia and macrophages (Tian et al., 2016). Furthermore, Kawai et al. (2017) reported the neuroprotective effects of Hv1 deficiency on infarct volume in experimental stoke occur in an age-dependent manner. ROS production was noted to be slightly decreased in Hv1 KO mice at younger stages of development (1 day, 5 days, 3 weeks old), but drastically increased in older KO mice (6 months old) compared with age-matched WT mice. Neuroprotection was seen in older Hv1 KO mice, whereas no protection was evident in the younger KO mice (9 weeks old) (Kawai et al., 2017). These findings were confirmed in Hv1 mutant Dahl salt-sensitive rats in both transient and permanent MCAO models. Although proton currents were large in mouse microglia and virtually absent in rat microglia (Wu et al., 2012), Hv1 KO rats exhibited less edema and bleeding in the brain, indicative of attenuated cerebrovascular injury (Li et al., 2019). Taken together, these results suggest that ablation of Hv1 is a promising therapeutic target for the treatment of ischemia stroke. The therapeutic effects of Hv1 deficiency in various animal models is summarized in Table 1. Limitations of these studies reflect the fact that transgenic mice do not relate directly to a therapeutic approach.
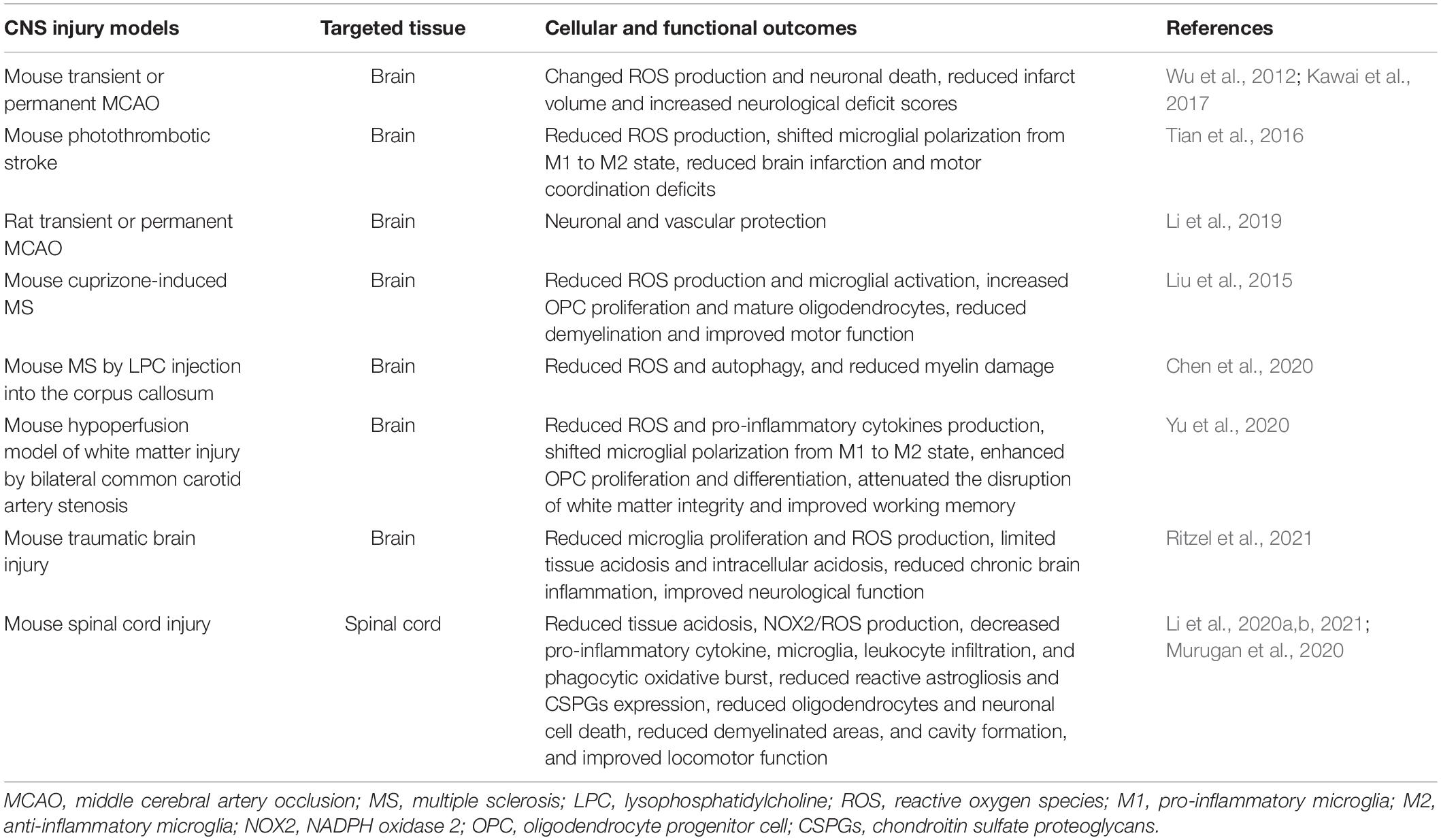
Table 1. Therapeutic effect of Hv1 deficiency (Hv1 knockout) in various animal models.
Multiple sclerosis (MS) is a chronic CNS disease characterized by autoimmune attack of the myelin sheath by T lymphocytes, resulting in inflammation and demyelination. In a cuprizone-induced mouse model of MS, Liu et al. (2015) showed that mice lacking Hv1 are partially protected from demyelination and motor deficits compared to WT mice. This effect was associated with reduced ROS production, attenuated microglial activation, increased oligodendrocyte progenitor cell proliferation, and increased numbers of mature oligodendrocytes, reaffirming the role of the Hv1 proton channel in controlling NOX-dependent ROS production in the pathogenesis of MS (Liu et al., 2015). Using an established two-point model of demyelination by injecting lysophosphatidylcholine (LPC) into the corpus callosum, a recent study identified that LPC-mediated myelin damage was reduced in Hv1 KO mice, evidenced by reduced ROS generation and autophagic activation in microglia (Chen et al., 2020). The link between Hv1 and autophagic function in microglia requires further investigation.
Oligodendrocyte progenitor cells (OPC) are believed to be the most fragile cells in hypoxia-ischemia-induced demyelination (Yu et al., 2018). To investigate the contribution of Hv1 to OPC damage, Yu et al. (2018) employed an in vitro model of co-culture with microglia. The authors found that, following oxygen-glucose deprivation, OPCs co-cultured with Hv1 KO microglia had attenuated apoptosis and greater proliferation and differentiation than those co-cultured with WT microglia. This protection was associated with decreased phosphorylation and activation of extracellular signal-regulated kinase (ERK) 1/2 and p38 mitogen-activated protein kinase (MAPK), and attenuated production of ROS and pro-inflammatory cytokines in microglia (Yu et al., 2018).
In a hypoperfusion model of white matter injury created by bilateral common carotid artery stenosis, genetic ablation of Hv1 attenuated the disruption of white matter integrity and improved working memory by enhancing OPC proliferation and differentiation into mature oligodendrocytes (Yu et al., 2020). Microglia-OPC co-cultures suggested that PI3K/Akt signaling was involved in Hv1 deficiency-induced anti-inflammatory-type microglial polarization and concomitant OPC differentiation. Thus, these results suggest that Hv1-NOX-ROS signaling contributes to the pathological changes in CNS demyelination.
Although it is well known that Hv1 regulates intracellular pH, and aids in compensation for NOX-dependent generation of ROS, a direct connection between acidosis, ROS, and inflammation has not been established until recently. Work from our group has demonstrated that Hv1 is a key driver of tissue acidosis, oxidative stress, and neuroinflammation in models of traumatic brain injury and spinal cord injury (Li et al., 2021; Ritzel et al., 2021; see Figure 2).
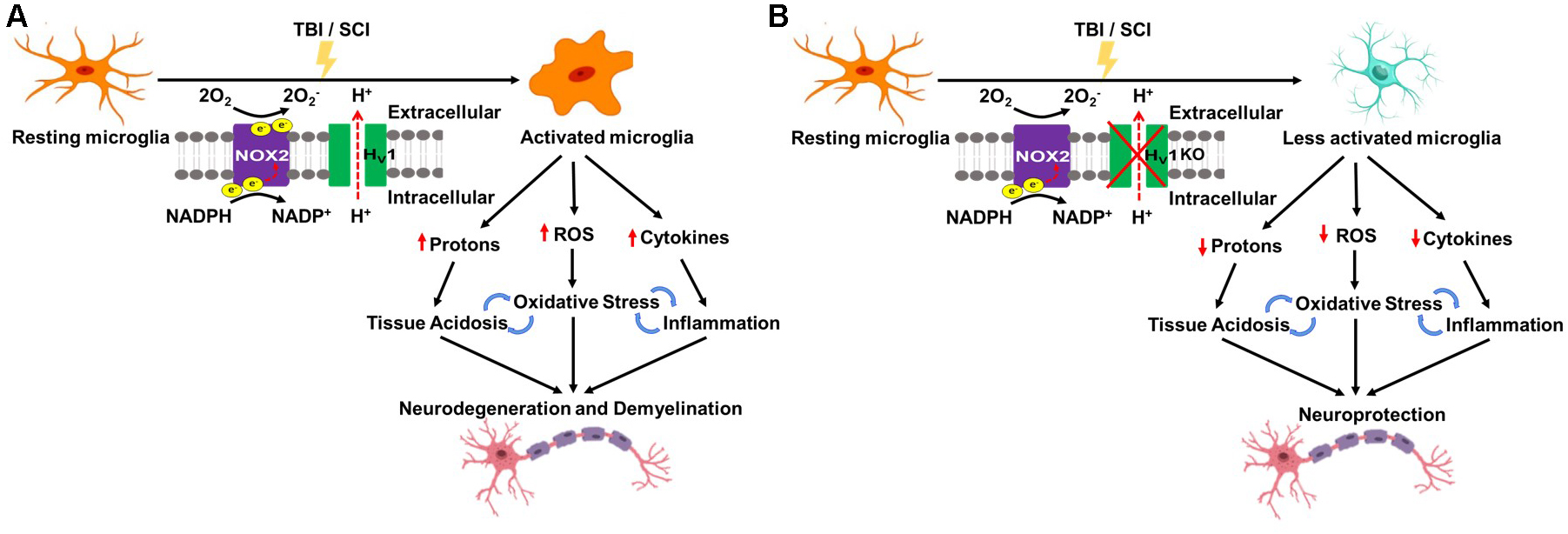
Figure 2. Schematic of Hv1 as a key driver of tissue acidosis, oxidative stress, and neuroinflammation in traumatic central nervous system (CNS) injury. (A) Traumatic brain injury (TBI) and spinal cord injury (SCI) lead to Hv1-driven microglia activation which causes tissue acidosis, oxidative stress, and an increase in proinflammatory cytokine levels. Consequently, there is an increase in neuronal loss, functional deficits, and poor long-term recovery. (B) Hv1 deficiency (KO) limits microglia activation and thereby rectifies trauma-induced reductions in environmental pH, abolishes ROS production, ameliorates neuroinflammation, and as a result, slows the progressive worsening of neuronal injury and functional deficits following SCI/TBI.
Tissue acidosis in the CNS is a common occurrence in ischemic stroke and the brain/spinal cord trauma and a strong predictor of acute clinical function and long-term outcome (Marmarou, 1992; Marmarou et al., 1993; Clausen et al., 2005). The underlying mechanisms of pathological acidosis in CNS injury are not well understood, however, growing evidence suggests a positive association with inflammation. In a well-characterized CCI mouse model of experimental TBI, we investigated the cellular and molecular mechanisms of brain acidosis (Ritzel et al., 2021). We demonstrated that TBI causes intracellular and extracellular acidosis that persist for weeks after injury. Microglia proliferation and production of ROS were increased during the first week. When microglia were depleted by the colony stimulating factor 1 receptor (CSF1R) inhibitor, PLX5622, TBI-induced extracellular acidosis, oxidative stress, and inflammation were markedly decreased during the acute stages of injury. These data suggest that TBI-activated microglia participate in the regulation of pathological brain acidosis through a proton extrusion mechanism, indicating a direct connection between acidosis and inflammation. Moreover, we believe this may be a chronic process that contributes to chronic brain inflammation and progressive neurodegeneration. Using Hv1 KO mice, we demonstrated attenuated ROS production and increased intracellular acidosis in both microglia and brain-infiltrating myeloid cells during the acute period after TBI. Importantly, Hv1 KO mice exhibited lasting neuroprotection and improved functional recovery months after injury, suggesting therapeutic strategies to alleviate acidosis could have possible benefits even in the chronic stages of injury. Our data provide new cellular and molecular insights into the relationship between inflammation, acidosis, and head injury (Ritzel et al., 2021).
Using a contusion model of SCI in adult female mice, we demonstrated that depletion of Hv1 significantly attenuated tissue acidosis, NOX2 expression, ROS production, proinflammatory cytokine production, microglia proliferation, leukocyte infiltration, and phagocytic oxidative burst at 3 days post-injury (Li et al., 2021). Tissue pH levels were markedly lower in the first week after SCI. Tissue acidosis was most evident at the injury site, but also extended into proximal regions of the cervical and lumbar cord. Tissue ROS levels and expression of Hv1 were also significantly increased during the first week of injury. In the long term, Hv1 KO mice exhibited significantly improved locomotor function and reduced histopathology. Meanwhile, other groups have reported similar results as well as Hv1’s role in attenuating reactive astrogliosis, and reducing oligodendrocyte apoptosis in SCI models (Li et al., 2020a; Murugan et al., 2020). Overall, these data suggest an important role for Hv1 in regulating extracellular acidosis, NOX2-mediated ROS production, and functional outcome following SCI (Li et al., 2021). Thus, the Hv1 proton channel represents a potential target that may lead to novel therapeutic strategies for TBI and SCI.
However, injured brain and spinal cord are invaded by blood-borne leukocytes expressing high levels of Hv1 and NOX. To further investigate whether peripheral Hv1 contributes to tissue damage after CNS trauma, bone marrow chimeric mice will be needed to examine respective contribution of Hv1 in microglia and peripheral immune cells to neuroinflammation as well as to functional outcomes.
Since Hv1 is involved in the regulation of NOX2, it offers a potential advantage as a drug target over NOX2, as NOX2 deficiency completely abolishes ROS production that is beneficial and necessary for supporting the function and viability of cells under non-injury conditions. In contrast, it is postulated that Hv1 deficiency may keep at least 30% of ROS, enough to supply vital cellular functions and maintenance (Seredenina et al., 2015). As an acid extruder that is independent of NOX2, Hv1 is indicated in the treatment of not only CNS injury but also cancer, obesity, diabetes, and allergy (Bare et al., 2020; Kawai et al., 2020; Pang et al., 2020). In addition to the development of Hv1 inhibitors, Hv1 activators might also be of interest for certain types of pathologies such as male infertility and possibly autoimmune disease (Seredenina et al., 2015).
Zn2+ and other polyvalent cations have been used for many years as proton current inhibitors. These compete with H+ for binding to the external surface, which changes the membrane potential perceived by the channel. However, this inhibition is non-specific, as Zn2+ ions are implicated in many other physiological processes. Thus, the usefulness of Zn2+ as an Hv1 channel blocker is limited. Hanatoxin, a toxin from the venom of the tarantula Grammostola spatulate, is another non-specific blocker of Hv1. This molecule binds to the paddle motif, which is highly conserved among different voltage-gated ion channels (Seredenina et al., 2015).
Some guanidine derivatives have been shown to have the ability to inhibit Hv1 activity. One of these compounds, 2-guanidinobenzimidazole (2GBI) binds the channel’s voltage-sensitive domain only in the open conformation. The binding site is within the proton permeation pathway and faces the cytoplasm (Hong et al., 2013). However, 2GBI is too polar to permeate the cytoplasmic membrane, and thus prevents it from being considered a potential Hv1 inhibitor. A modified version of 2GBI (CIGBI) that makes it more permeable to the cell membrane was later identified to bind Hv1 more efficiently, making a step forward to the development of pharmacological treatments for diseases caused by Hv1 hyperactivity (Hong et al., 2014). Nonetheless, the relative low potency and uncertainty about specificity against Hv1 remains a problem (Pupo and Gonzalez Leon, 2014).
Several proton current inhibitors, such as 4-aminopyridine, amantadine, amiloride, D600, nicardipine, imipramine, DM, chlorpromazine, clozapine, haloperidol, and rimantadine have potentially indirect effects on Hv1 function, presumably by increasing intracellular pH (Shin et al., 2015). In particular, epigallocatechin-3-gallate might interfere with Hv1 channel activity by modifying the lipid bilayer structure (Seredenina et al., 2015; Fernandez et al., 2016). Additionally, a toxic peptide C6 was identified as a high potential Hv1 blocker with specificity and high affinity (Kd = 1 nM) (Tang et al., 2020). Two C6 peptides bind to each dimeric channel on the S3–S4 loop of the VSD, shifting human Hv1 activation to more positive voltages, slowing opening and speeding closure, which consequently diminishes the membrane depolarization (Zhao et al., 2018). The scorpion toxin anti-tumor analgesic peptide (AGAP/W38F) is another novel selective Hv1 channel antagonist. Its binding pocket on the Hv1 channel partly overlaps that of Zn2+ with even less pH dependence. Therefore, AGAP/W38F could be a useful probe for exploring the structure–function relationship of the Hv1 channel and may have strong therapeutic potential (Tang et al., 2020).
Currently, no drug-like molecules are known to activate Hv1. Unsaturated long chain fatty acids such as arachidonic acid (AA) are known to modulate the activities of various ion channels, including Hv1 (Kawanabe and Okamura, 2016). Application of AA, for example, rapidly induced a robust increase in the amplitude of the proton current through Hv1. The application of phospholipase A2 (PLA2), which generates AA from cell membrane phospholipids, stimulated Hv1 activity to a similar extent as the direct application of 20 μM AA, suggesting that endogenous AA may regulate Hv1 channel activity (Kawanabe and Okamura, 2016).
Ever since the identification of the gene in 2006, there has been growing interest in the Hv1 proton channel and its role in immunity, CNS injury, metabolic disease and cancer. Yet many questions remain unanswered. Though it is widely accepted that Hv1 interacts with NOX2 to regulate the production of ROS, it is not clear which one is upstream. In acute CNS injury, Hv1 deficiency diminishes ROS production, reduces expression of inflammatory cytokines, and ameliorates extracellular acidosis. However, whether this is primarily caused by the residential microglia or infiltrating myeloid cells is still not clear. In the future, the development of cell-specific Hv1 KO mice will be necessary to address the respective contributions of each phagocyte population to the pathogenesis of acidotic and oxidative stress damage following CNS injury.
Structural exploration could not only help with the understanding of the molecular mechanisms of channel activation and proton permeation (Chamberlin et al., 2015; Li et al., 2015; De La Rosa et al., 2018), but also facilitate the discovery of drugs targeting Hv1 (Lee et al., 2018; Jardin et al., 2020). These drug candidates, in turn, will help further our understanding of Hv1 function in health and disease.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The work was supported by the National Institutes of Health Grants R01 NS110825, R01 NS094527, RF1 NS110637, R01 NS110635 to JW, and K99 NS116032 to RR.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Asuaje, A., Smaldini, P., Martin, P., Enrique, N., Orlowski, A., Aiello, E. A., et al. (2017). The inhibition of voltage-gated H(+) channel (HVCN1) induces acidification of leukemic Jurkat T cells promoting cell death by apoptosis. Pflugers Arch. 469, 251–261. doi: 10.1007/s00424-016-1928-0
Bare, D. J., Cherny, V. V., DeCoursey, T. E., Abukhdeir, A. M., and Morgan, D. (2020). Expression and function of voltage gated proton channels (Hv1) in MDA-MB-231 cells. PLoS One 15:e0227522. doi: 10.1371/journal.pone.0227522
Berger, T. K., Fussholler, D. M., Goodwin, N., Bonigk, W., Muller, A., Dokani Khesroshahi, N., et al. (2017). Post-translational cleavage of Hv1 in human sperm tunes pH- and voltage-dependent gating. J. Physiol. 595, 1533–1546. doi: 10.1113/jp273189
Capasso, M. (2014). Regulation of immune responses by proton channels. Immunology 143, 131–137. doi: 10.1111/imm.12326
Capasso, M., Bhamrah, M. K., Henley, T., Boyd, R. S., Langlais, C., Cain, K., et al. (2010). HVCN1 modulates BCR signal strength via regulation of BCR-dependent generation of reactive oxygen species. Nat. Immunol. 11, 265–272. doi: 10.1038/ni.1843
Capasso, M., DeCoursey, T. E., and Dyer, M. J. (2011). pH regulation and beyond: unanticipated functions for the voltage-gated proton channel, HVCN1. Trends Cell Biol. 21, 20–28. doi: 10.1016/j.tcb.2010.09.006
Chamberlin, A., Qiu, F., Wang, Y., Noskov, S. Y., and Larsson, H. P. (2015). Mapping the gating and permeation pathways in the voltage-gated proton channel Hv1. J. Mol. Biol. 427, 131–145. doi: 10.1016/j.jmb.2014.11.018
Chen, M., Yang, L. L., Hu, Z. W., Qin, C., Zhou, L. Q., Duan, Y. L., et al. (2020). Deficiency of microglial Hv1 channel is associated with activation of autophagic pathway and ROS production in LPC-induced demyelination mouse model. J. Neuroinflamm. 17:333. doi: 10.1186/s12974-020-02020-y
Cherny, V. V., Morgan, D., Thomas, S., Smith, S. M. E., and DeCoursey, T. E. (2018). Histidine(168) is crucial for DeltapH-dependent gating of the human voltage-gated proton channel, hHV1. J. Gen. Physiol. 150, 851–862. doi: 10.1085/jgp.201711968
Cho, D. Y., Hajighasemi, M., Hwang, P. H., Illek, B., and Fischer, H. (2009). Proton secretion in freshly excised sinonasal mucosa from asthma and sinusitis patients. Am. J. Rhinol. Allergy 23, e10–e13. doi: 10.2500/ajra.2009.23.3389
Clausen, T., Khaldi, A., Zauner, A., Reinert, M., Doppenberg, E., Menzel, M., et al. (2005). Cerebral acid-base homeostasis after severe traumatic brain injury. J. Neurosurg. 103, 597–607. doi: 10.3171/jns.2005.103.4.0597
De La Rosa, V., Bennett, A. L., and Ramsey, I. S. (2018). Coupling between an electrostatic network and the Zn(2+) binding site modulates Hv1 activation. J. Gen. Physiol. 150, 863–881. doi: 10.1085/jgp.201711822
DeCoursey, T. E., Cherny, V. V., DeCoursey, A. G., Xu, W., and Thomas, L. L. (2001). Interactions between NADPH oxidase-related proton and electron currents in human eosinophils. J. Physiol. 535(Pt 3), 767–781. doi: 10.1111/j.1469-7793.2001.00767.x
DeCoursey, T. E., Cherny, V. V., Zhou, W., and Thomas, L. L. (2000). Simultaneous activation of NADPH oxidase-related proton and electron currents in human neutrophils. Proc. Natl. Acad. Sci. U.S.A. 97, 6885–6889. doi: 10.1073/pnas.100047297
El Chemaly, A., Okochi, Y., Sasaki, M., Arnaudeau, S., Okamura, Y., and Demaurex, N. (2010). VSOP/Hv1 proton channels sustain calcium entry, neutrophil migration, and superoxide production by limiting cell depolarization and acidification. J. Exp. Med. 207, 129–139. doi: 10.1084/jem.20091837
Faden, A. I., Wu, J., Stoica, B. A., and Loane, D. J. (2016). Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br. J. Pharmacol. 173, 681–691. doi: 10.1111/bph.13179
Fan, L. M., Geng, L., Cahill-Smith, S., Liu, F., Douglas, G., McKenzie, C. A., et al. (2019). Nox2 contributes to age-related oxidative damage to neurons and the cerebral vasculature. J. Clin. Invest. 129, 3374–3386. doi: 10.1172/JCI125173
Fernandez, A., Pupo, A., Mena-Ulecia, K., and Gonzalez, C. (2016). Pharmacological modulation of proton channel Hv1 in cancer therapy: future perspectives. Mol. Pharmacol. 90, 385–402. doi: 10.1124/mol.116.103804
Fischer, H. (2012). Function of proton channels in lung epithelia. Wiley Interdiscip. Rev. Membr. Transp. Signal 1, 247–258. doi: 10.1002/wmts.17
Fischer, H., Widdicombe, J. H., and Illek, B. (2002). Acid secretion and proton conductance in human airway epithelium. Am. J. Physiol. Cell Physiol. 282, C736–C743. doi: 10.1152/ajpcell.00369.2001
Gelderblom, M., Leypoldt, F., Steinbach, K., Behrens, D., Choe, C. U., Siler, D. A., et al. (2009). Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 40, 1849–1857. doi: 10.1161/STROKEAHA.108.534503
Harry, G. J. (2013). Microglia during development and aging. Pharmacol. Ther. 139, 313–326. doi: 10.1016/j.pharmthera.2013.04.013
Hondares, E., Brown, M. A., Musset, B., Morgan, D., Cherny, V. V., Taubert, C., et al. (2014). Enhanced activation of an amino-terminally truncated isoform of the voltage-gated proton channel HVCN1 enriched in malignant B cells. Proc. Natl. Acad. Sci. U.S.A. 111, 18078–18083. doi: 10.1073/pnas.1411390111
Hong, L., Kim, I. H., and Tombola, F. (2014). Molecular determinants of Hv1 proton channel inhibition by guanidine derivatives. Proc. Natl. Acad. Sci. U.S.A. 111, 9971–9976. doi: 10.1073/pnas.1324012111
Hong, L., Pathak, M. M., Kim, I. H., Ta, D., and Tombola, F. (2013). Voltage-sensing domain of voltage-gated proton channel Hv1 shares mechanism of block with pore domains. Neuron 77, 274–287. doi: 10.1016/j.neuron.2012.11.013
Iovannisci, D., Illek, B., and Fischer, H. (2010). Function of the HVCN1 proton channel in airway epithelia and a naturally occurring mutation, M91T. J. Gen. Physiol. 136, 35–46. doi: 10.1085/jgp.200910379
Jardin, C., Chaves, G., and Musset, B. (2020). Assessing structural determinants of Zn(2+) binding to human HV1 via multiple MD simulations. Biophys. J. 118, 1221–1233. doi: 10.1016/j.bpj.2019.12.035
Kapus, A., Romanek, R., Qu, A. Y., Rotstein, O. D., and Grinstein, S. (1993). A pH-sensitive and voltage-dependent proton conductance in the plasma membrane of macrophages. J. Gen. Physiol. 102, 729–760. doi: 10.1085/jgp.102.4.729
Kawai, T., Kayama, K., Tatsumi, S., Akter, S., Miyawaki, N., Okochi, Y., et al. (2020). Regulation of hepatic oxidative stress by voltage-gated proton channels (Hv1/VSOP) in Kupffer cells and its potential relationship with glucose metabolism. FASEB J. 34, 15805–15821. doi: 10.1096/fj.202001056RRR
Kawai, T., Okochi, Y., Ozaki, T., Imura, Y., Koizumi, S., Yamazaki, M., et al. (2017). Unconventional role of voltage-gated proton channels (VSOP/Hv1) in regulation of microglial ROS production. J. Neurochem. 142, 686–699. doi: 10.1111/jnc.14106
Kawai, T., Takao, K., Akter, S., Abe, M., Sakimura, K., Miyakawa, T., et al. (2021). Heterogeneity of microglial proton channel in different brain regions and its relationship with aging. J. Neurochem. doi: 10.1111/jnc.15292 [Epub ahead of print].
Kawai, T., Tatsumi, S., Kihara, S., Sakimura, K., and Okamura, Y. (2018). Mechanistic insight into the suppression of microglial ROS production by voltage-gated proton channels (VSOP/Hv1). Channels (Austin) 12, 1–8. doi: 10.1080/19336950.2017.1385684
Kawanabe, A., and Okamura, Y. (2016). Effects of unsaturated fatty acids on the kinetics of voltage-gated proton channels heterologously expressed in cultured cells. J. Physiol. 594, 595–610. doi: 10.1113/jp271274
Kuno, M., Kawawaki, J., and Nakamura, F. (1997). A highly temperature-sensitive proton current in mouse bone marrow-derived mast cells. J. Gen. Physiol. 109, 731–740. doi: 10.1085/jgp.109.6.731
Lee, M., Bai, C., Feliks, M., Alhadeff, R., and Warshel, A. (2018). On the control of the proton current in the voltage-gated proton channel Hv1. Proc. Natl. Acad. Sci. U.S.A. 115, 10321–10326. doi: 10.1073/pnas.1809766115
Levine, A. P., Duchen, M. R., de Villiers, S., Rich, P. R., and Segal, A. W. (2015). Alkalinity of neutrophil phagocytic vacuoles is modulated by HVCN1 and has consequences for myeloperoxidase activity. PLoS One 10:e0125906. doi: 10.1371/journal.pone.0125906
Li, Q., Shen, R., Treger, J. S., Wanderling, S. S., Milewski, W., Siwowska, K., et al. (2015). Resting state of the human proton channel dimer in a lipid bilayer. Proc. Natl. Acad. Sci. U.S.A. 112, E5926–E5935. doi: 10.1073/pnas.1515043112
Li, W., Ward, R., Dong, G., Ergul, A., and O’Connor, P. (2019). Neurovascular protection in voltage-gated proton channel Hv1 knock-out rats after ischemic stroke: interaction with Na(+) /H(+) exchanger-1 antagonism. Physiol. Rep. 7:e14142. doi: 10.14814/phy2.14142
Li, X., Liu, R., Yu, Z., He, D., Zong, W., Wang, M., et al. (2020a). Microglial Hv1 exacerbates secondary damage after spinal cord injury in mice. Biochem. Biophys. Res. Commun. 525, 208–215. doi: 10.1016/j.bbrc.2020.02.012
Li, X., Yu, Z., Zong, W., Chen, P., Li, J., Wang, M., et al. (2020b). Deficiency of the microglial Hv1 proton channel attenuates neuronal pyroptosis and inhibits inflammatory reaction after spinal cord injury. J. Neuroinflamm. 17:263. doi: 10.1186/s12974-020-01942-x
Li, Y., Ritzel, R. M., He, J., Cao, T., Sabirzhanov, B., Li, H., et al. (2021). The voltage-gated proton channel Hv1 plays a detrimental role in contusion spinal cord injury via extracellular acidosis-mediated neuroinflammation. Brain Behav. Immun. 91, 267–283. doi: 10.1016/j.bbi.2020.10.005
Li, Y., Ritzel, R. M., Khan, N., Cao, T., He, J., Lei, Z., et al. (2020). Delayed microglial depletion after spinal cord injury reduces chronic inflammation and neurodegeneration in the brain and improves neurological recovery in male mice. Theranostics 10, 11376–11403. doi: 10.7150/thno.49199
Lishko, P. V., Botchkina, I. L., Fedorenko, A., and Kirichok, Y. (2010). Acid extrusion from human spermatozoa is mediated by flagellar voltage-gated proton channel. Cell 140, 327–337. doi: 10.1016/j.cell.2009.12.053
Liu, J., Tian, D., Murugan, M., Eyo, U. B., Dreyfus, C. F., Wang, W., et al. (2015). Microglial Hv1 proton channel promotes cuprizone-induced demyelination through oxidative damage. J. Neurochem. 135, 347–356. doi: 10.1111/jnc.13242
Loane, D. J., and Byrnes, K. R. (2010). Role of microglia in neurotrauma. Neurotherapeutics 7, 366–377. doi: 10.1016/j.nurt.2010.07.002
Loane, D. J., and Kumar, A. (2016). Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 275(Pt 3), 316–327. doi: 10.1016/j.expneurol.2015.08.018
Luo, L., Song, S., Ezenwukwa, C. C., Jalali, S., Sun, B., and Sun, D. (2021). Ion channels and transporters in microglial function in physiology and brain diseases. Neurochem. Int. 142:104925. doi: 10.1016/j.neuint.2020.104925
Ma, S., Wang, C., Mao, X., and Hao, Y. (2019). B cell dysfunction associated with aging and autoimmune diseases. Front. Immunol. 10:318. doi: 10.3389/fimmu.2019.00318
Madreiter-Sokolowski, C. T., Thomas, C., and Ristow, M. (2020). Interrelation between ROS and Ca(2+) in aging and age-related diseases. Redox Biol. 36:101678. doi: 10.1016/j.redox.2020.101678
Marmarou, A. (1992). Intracellular acidosis in human and experimental brain injury. J. Neurotrauma 9(Suppl. 2), S551–S562.
Marmarou, A., Holdaway, R., Ward, J. D., Yoshida, K., Choi, S. C., Muizelaar, J. P., et al. (1993). Traumatic brain tissue acidosis: experimental and clinical studies. Acta Neurochir. Suppl. (Wien) 57, 160–164. doi: 10.1007/978-3-7091-9266-5_23
Marone, G., Columbo, M., de Paulis, A., Cirillo, R., Giugliano, R., and Condorelli, M. (1986). Physiological concentrations of zinc inhibit the release of histamine from human basophils and lung mast cells. Agents Actions 18, 103–106. doi: 10.1007/BF01987995
Montes-Cobos, E., Huscher, B., Engler, J. B., Woo, M. S., Binkle, L., Bauer, S., et al. (2020). Voltage-gated proton channel Hv1 controls TLR9 activation in plasmacytoid dendritic cells. J. Immunol. 205, 3001–3010. doi: 10.4049/jimmunol.2000404
Murugan, M., Zheng, J., Wu, G., Mogilevsky, R., Zheng, X., Hu, P., et al. (2020). The voltage-gated proton channel Hv1 contributes to neuronal injury and motor deficits in a mouse model of spinal cord injury. Mol. Brain 13:143. doi: 10.1186/s13041-020-00682-6
Musset, B., Cherny, V. V., and DeCoursey, T. E. (2012a). Strong glucose dependence of electron current in human monocytes. Am. J. Physiol. Cell Physiol. 302, C286–C295. doi: 10.1152/ajpcell.00335.2011
Musset, B., Clark, R. A., DeCoursey, T. E., Petheo, G. L., Geiszt, M., Chen, Y., et al. (2012b). NOX5 in human spermatozoa: expression, function, and regulation. J. Biol. Chem. 287, 9376–9388. doi: 10.1074/jbc.M111.314955
Musset, B., Morgan, D., Cherny, V. V., MacGlashan, D. W. Jr., Thomas, L. L., Rios, E., et al. (2008). A pH-stabilizing role of voltage-gated proton channels in IgE-mediated activation of human basophils. Proc. Natl. Acad. Sci. U.S.A. 105, 11020–11025. doi: 10.1073/pnas.0800886105
Norden, D. M., and Godbout, J. P. (2013). Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 39, 19–34. doi: 10.1111/j.1365-2990.2012.01306.x
Nordstrom, T., Rotstein, O. D., Romanek, R., Asotra, S., Heersche, J. N., Manolson, M. F., et al. (1995). Regulation of cytoplasmic pH in osteoclasts. Contribution of proton pumps and a proton-selective conductance. J. Biol. Chem. 270, 2203–2212. doi: 10.1074/jbc.270.5.2203
Okochi, Y., Umemoto, E., and Okamura, Y. (2020). Hv1/VSOP regulates neutrophil directional migration and ERK activity by tuning ROS production. J. Leukoc. Biol. 107, 819–831. doi: 10.1002/JLB.2A0320-110RR
Pang, H., Li, J., Du, H., Gao, Y., Lv, J., Liu, Y., et al. (2020). Loss of voltage-gated proton channel Hv1 leads to diet-induced obesity in mice. BMJ Open Diabetes Res. Care 8:e000951. doi: 10.1136/bmjdrc-2019-000951
Patel, B., Zheleznova, N. N., Ray, S. C., Sun, J., Cowley, A. W. Jr., and O’Connor, P. M. (2019). Voltage gated proton channels modulate mitochondrial reactive oxygen species production by complex I in renal medullary thick ascending limb. Redox Biol. 27:101191. doi: 10.1016/j.redox.2019.101191
Pupo, A., and Gonzalez Leon, C. (2014). In pursuit of an inhibitory drug for the proton channel. Proc. Natl. Acad. Sci. U.S.A. 111, 9673–9674. doi: 10.1073/pnas.1408808111
Ramsey, I. S., Moran, M. M., Chong, J. A., and Clapham, D. E. (2006). A voltage-gated proton-selective channel lacking the pore domain. Nature 440, 1213–1216. doi: 10.1038/nature04700
Ramsey, I. S., Ruchti, E., Kaczmarek, J. S., and Clapham, D. E. (2009). Hv1 proton channels are required for high-level NADPH oxidase-dependent superoxide production during the phagocyte respiratory burst. Proc. Natl. Acad. Sci. U.S.A. 106, 7642–7647. doi: 10.1073/pnas.0902761106
Ritzel, R. M., He, J., Li, Y., Cao, T., Khan, N., Shim, B., et al. (2021). Proton extrusion during oxidative burst in microglia exacerbates pathological acidosis following traumatic brain injury. Glia 69, 746–764. doi: 10.1002/glia.23926
Ritzel, R. M., Lai, Y. J., Crapser, J. D., Patel, A. R., Schrecengost, A., Grenier, J. M., et al. (2018). Aging alters the immunological response to ischemic stroke. Acta Neuropathol. 136, 89–110. doi: 10.1007/s00401-018-1859-2
Ritzel, R. M., Li, Y., He, J., Khan, N., Doran, S. J., Faden, A. I., et al. (2020). Sustained neuronal and microglial alterations are associated with diverse neurobehavioral dysfunction long after experimental brain injury. Neurobiol. Dis. 136:104713. doi: 10.1016/j.nbd.2019.104713
Sasaki, M., Takagi, M., and Okamura, Y. (2006). A voltage sensor-domain protein is a voltage-gated proton channel. Science 312, 589–592. doi: 10.1126/science.1122352
Sasaki, M., Tojo, A., Okochi, Y., Miyawaki, N., Kamimura, D., Yamaguchi, A., et al. (2013). Autoimmune disorder phenotypes in Hvcn1-deficient mice. Biochem. J. 450, 295–301. doi: 10.1042/bj20121188
Schilling, M., Besselmann, M., Leonhard, C., Mueller, M., Ringelstein, E. B., and Kiefer, R. (2003). Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp. Neurol. 183, 25–33. doi: 10.1016/s0014-4886(03)00082-7
Schilling, T., Gratopp, A., DeCoursey, T. E., and Eder, C. (2002). Voltage-activated proton currents in human lymphocytes. J. Physiol. 545, 93–105. doi: 10.1113/jphysiol.2002.028878
Seredenina, T., Demaurex, N., and Krause, K. H. (2015). Voltage-gated proton channels as novel drug targets: from NADPH oxidase regulation to sperm biology. Antioxid. Redox Signal. 23, 490–513. doi: 10.1089/ars.2013.5806
Shin, H., Kim, J., and Song, J. H. (2015). Clozapine and olanzapine inhibit proton currents in BV2 microglial cells. Eur. J. Pharmacol. 755, 74–79. doi: 10.1016/j.ejphar.2015.03.003
Sieber, M. W., Guenther, M., Jaenisch, N., Albrecht-Eckardt, D., Kohl, M., Witte, O. W., et al. (2014). Age-specific transcriptional response to stroke. Neurobiol. Aging 35, 1744–1754. doi: 10.1016/j.neurobiolaging.2014.01.012
Skovira, J. W., Wu, J., Matyas, J. J., Kumar, A., Hanscom, M., Kabadi, S. V., et al. (2016). Cell cycle inhibition reduces inflammatory responses, neuronal loss, and cognitive deficits induced by hypobaria exposure following traumatic brain injury. J. Neuroinflamm. 13:299. doi: 10.1186/s12974-016-0769-2
Szteyn, K., Yang, W., Schmid, E., Lang, F., and Shumilina, E. (2012). Lipopolysaccharide-sensitive H+ current in dendritic cells. Am. J. Physiol. Cell Physiol. 303, C204–C212. doi: 10.1152/ajpcell.00059.2012
Takeshita, K., Sakata, S., Yamashita, E., Fujiwara, Y., Kawanabe, A., Kurokawa, T., et al. (2014). X-ray crystal structure of voltage-gated proton channel. Nat. Struct. Mol. Biol. 21, 352–357. doi: 10.1038/nsmb.2783
Tang, D., Yang, Y., Xiao, Z., Xu, J., Yang, Q., Dai, H., et al. (2020). Scorpion toxin inhibits the voltage-gated proton channel using a Zn(2+) -like long-range conformational coupling mechanism. Br. J. Pharmacol. 177, 2351–2364. doi: 10.1111/bph.14984
Thomas, R. C., and Meech, R. W. (1982). Hydrogen ion currents and intracellular pH in depolarized voltage-clamped snail neurones. Nature 299, 826–828. doi: 10.1038/299826a0
Tian, D. S., Li, C. Y., Qin, C., Murugan, M., Wu, L. J., and Liu, J. L. (2016). Deficiency in the voltage-gated proton channel Hv1 increases M2 polarization of microglia and attenuates brain damage from photothrombotic ischemic stroke. J. Neurochem. 139, 96–105. doi: 10.1111/jnc.13751
Udeochu, J. C., Shea, J. M., and Villeda, S. A. (2016). Microglia communication: parallels between aging and Alzheimer’s disease. Clin. Exp. Neuroimmunol. 7, 114–125. doi: 10.1111/cen3.12307
von Leden, R. E., Yauger, Y. J., Khayrullina, G., and Byrnes, K. R. (2017). Central nervous system injury and nicotinamide adenine dinucleotide phosphate oxidase: oxidative stress and therapeutic targets. J. Neurotrauma 34, 755–764. doi: 10.1089/neu.2016.4486
Wu, L. J. (2014). Voltage-gated proton channel HV1 in microglia. Neuroscientist 20, 599–609. doi: 10.1177/1073858413519864
Wu, L. J., Wu, G., Akhavan Sharif, M. R., Baker, A., Jia, Y., Fahey, F. H., et al. (2012). The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat. Neurosci. 15, 565–573. doi: 10.1038/nn.3059
Yeste, M., Llavanera, M., Mateo-Otero, Y., Catalan, J., Bonet, S., and Pinart, E. (2020). HVCN1 channels are relevant for the maintenance of sperm motility during in vitro capacitation of pig spermatozoa. Int. J. Mol. Sci. 21:3255. doi: 10.3390/ijms21093255
Yu, Y., Luo, X., Li, C., Ding, F., Wang, M., Xie, M., et al. (2020). Microglial Hv1 proton channels promote white matter injuries after chronic hypoperfusion in mice. J. Neurochem. 152, 350–367. doi: 10.1111/jnc.14925
Yu, Y., Yu, Z., Xie, M., Wang, W., and Luo, X. (2018). Hv1 proton channel facilitates production of ROS and pro-inflammatory cytokines in microglia and enhances oligodendrocyte progenitor cells damage from oxygen-glucose deprivation in vitro. Biochem. Biophys. Res. Commun. 498, 1–8. doi: 10.1016/j.bbrc.2017.06.197
Zhang, Z. J., Zheng, X. X., Zhang, X. Y., Zhang, Y., Huang, B. Y., and Luo, T. (2020). Aging alters Hv1-mediated microglial polarization and enhances neuroinflammation after peripheral surgery. CNS Neurosci. Ther. 26, 374–384. doi: 10.1111/cns.13271
Zhao, R., Kennedy, K., De Blas, G. A., Orta, G., Pavarotti, M. A., Arias, R. J., et al. (2018). Role of human Hv1 channels in sperm capacitation and white blood cell respiratory burst established by a designed peptide inhibitor. Proc. Natl. Acad. Sci. U.S.A. 115, E11847–E11856. doi: 10.1073/pnas.1816189115
Keywords: voltage-gated proton channel Hv1, NOX2, acidification, ROS, microglia, traumatic brain injury, spinal cord injury, stroke
Citation: He J, Ritzel RM and Wu J (2021) Functions and Mechanisms of the Voltage-Gated Proton Channel Hv1 in Brain and Spinal Cord Injury. Front. Cell. Neurosci. 15:662971. doi: 10.3389/fncel.2021.662971
Received: 02 February 2021; Accepted: 18 March 2021;
Published: 09 April 2021.
Edited by:
Anwen Shao, Zhejiang University, ChinaReviewed by:
Kimberly R. Byrnes, Uniformed Services University of the Health Sciences, United StatesCopyright © 2021 He, Ritzel and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junfang Wu, anVuZmFuZy53dUBzb20udW1hcnlsYW5kLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.