 Agnes L. Nishimura
Agnes L. Nishimura Natalia Arias
Natalia Arias- 1Department of Basic and Clinical Neuroscience, UK Dementia Research Institute, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology and Neuroscience, King’s College London, London, United Kingdom
- 2INEUROPA, Instituto de Neurociencias del Principado de Asturias, Oviedo, Spain
Amyotrophic Lateral Sclerosis (ALS) is a complex neurodegenerative disease caused by degeneration of motor neurons (MNs). ALS pathogenic features include accumulation of misfolded proteins, glutamate excitotoxicity, mitochondrial dysfunction at distal axon terminals, and neuronal cytoskeleton changes. Synergies between loss of C9orf72 functions and gain of function by toxic effects of repeat expansions also contribute to C9orf72-mediated pathogenesis. However, the impact of haploinsufficiency of C9orf72 on neurons and in synaptic functions requires further examination. As the motor neurons degenerate, the disease symptoms will lead to neurotransmission deficiencies in the brain, spinal cord, and neuromuscular junction. Altered neuronal excitability, synaptic morphological changes, and C9orf72 protein and DPR localization at the synapses, suggest a potential involvement of C9orf72 at synapses. In this review article, we provide a conceptual framework for assessing the putative involvement of C9orf72 as a synaptopathy, and we explore the underlying and common disease mechanisms with other neurodegenerative diseases. Finally, we reflect on the major challenges of understanding C9orf72-ALS as a synaptopathy focusing on integrating mitochondrial and neuronal cytoskeleton degeneration as biomarkers and potential targets to treat ALS neurodegeneration.
Understanding Amyotrophic Lateral Sclerosis as A Synaptopathy: The Role of C9orf72
ALS as a Synaptopathy
Amyotrophic lateral sclerosis (ALS; also known as motor neuron disease) has a remarkably variable clinical presentation and progression, with ~75% of patients presenting upper and lower limb weakness (Vucic et al., 2014). The remaining 25% of patients present bulbar signs and symptoms, including impairment of speech and swallowing difficulties (Vucic et al., 2014). Approximately 20–50% of ALS patients also present frontotemporal dementia (FTD) symptoms, which obscure diagnosis and prognosis (Vucic et al., 2014; Genc et al., 2017). Despite variable clinical presentations, cortical post-mortem pathological features are highly conserved across sporadic and familial forms of ALS (Genc et al., 2017). These include dendritic and synaptic degeneration in the cortex and corticospinal motor neurons (CSMNs; Fogarty, 2019).
Cortical synaptic degeneration landmark has inspired Braak and colleagues to classify ALS as a disease of large axons neurons with discrete stages of trans-synaptic spread (Braak et al., 2013). This classification suggests that the disease starts in CSMNs, descends to MNs, and progresses to extramotor areas (Braak et al., 2013; Brettschneider et al., 2013). This pathological insight is consistent with cortical hyperexcitability as an early clinical feature of ALS (Vucic et al., 2014). Moreover, the trans-synaptic spread hypothesis suggests a mechanism of spreading misfolded protein aggregates to distant populations of neurons through a prion-like transmission mechanism (Braak et al., 2013). This is consistent with the hypothesis of ALS being a synaptopathy (Fogarty, 2019). Synaptopathy is a broad definition of diseases with synaptic dysfunction, regardless of the disease mechanisms (Lepeta et al., 2016). In the broadest definition, synaptopathy encompasses a wide range of features, which in due course, will lead to synaptic dysfunction. These features include changes in Ca2+ levels at synapses, glutamate excitotoxicity, structural changes in pre- and postsynaptic anchoring proteins, altered synaptic structure and function, which is often associated with dendritic spine loss, dysfunctional neurotransmitter release (quantal content and frequency), impaired maintenance, and regeneration of axons by Schwann cells, and cognitive deficit (Lepeta et al., 2016; Fogarty, 2019). Furthermore, synaptopathies lead to neuronal loss, mitochondrial dysfunction, accumulation of misfolded proteins associated with defective proteostasis, and defective neuromuscular junctions (NMJ; Figure 1; Wishart et al., 2006; Lepeta et al., 2016; Fogarty, 2019).
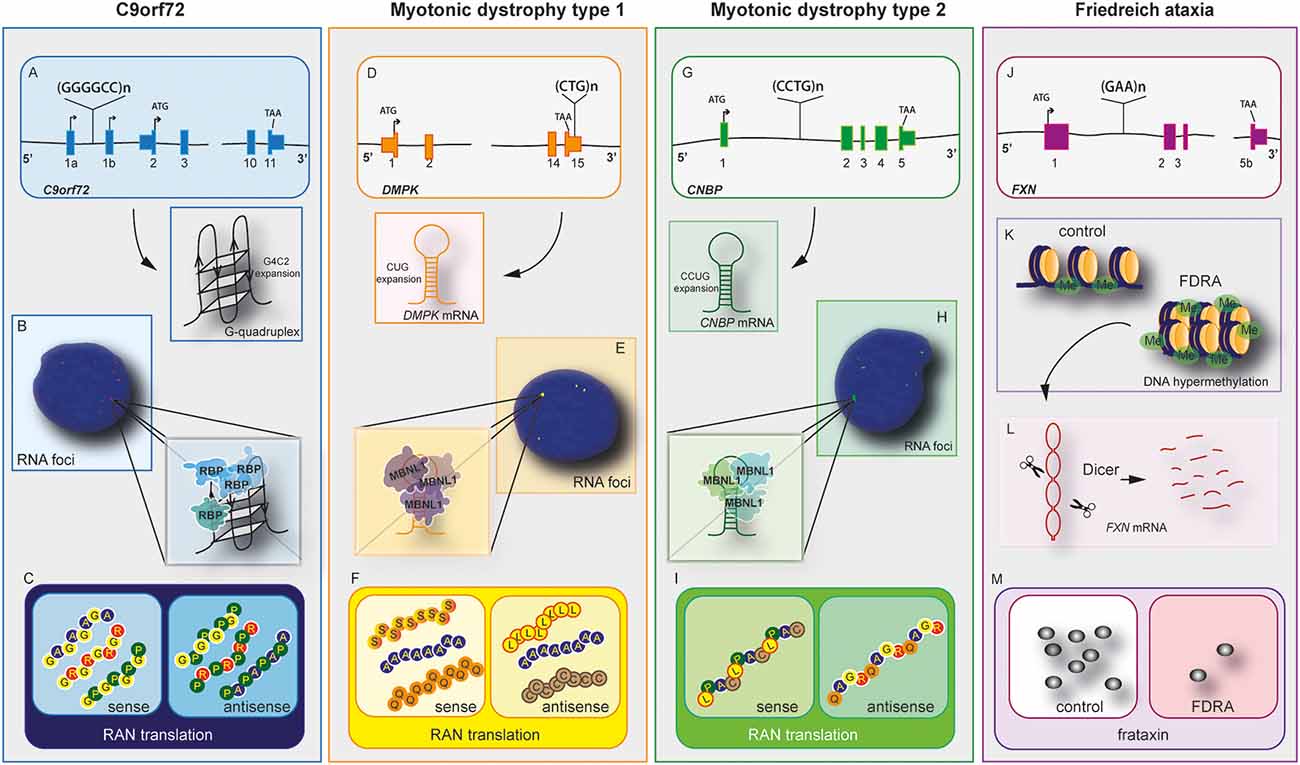
Figure 1. C9orf72 and other repeat expansion disease mechanisms. Nucleotide repeat expansions located at intron or untranslated regions (UTRs) cause three main disease-related features, haploinsufficiency in C9orf72 (A), myotonic Dystrophy type 1 (DM1; D), myotonic dystrophy type 2 (G), and Friedreich’s ataxia (FRDA) (J). mRNAs containing repeat expansions form G-quadruplex (C9orf72) and RNA hairpin structures (myotonic dystrophy type 1 and 2). These RNA hairpins will accumulate and form RNA foci (B,E,H), which will sequester RNA binding proteins (RBPs) including hnRNP-H in C9orf72 and Muscleblind like Splicing Regulator 1 (MBNL1) in myotonic dystrophy type 1 and 2. Repeat-associated non-AUG (RAN) translation produces peptides from the sense and antisense strands in C9orf72 (C), myotonic dystrophy type 1 (F), and myotonic dystrophy type 2 (I). The exception lies in FRDA disease mechanisms. In this disease, the repeat expansion causes DNA hypermethylation and gene expression dysregulation of neighboring genes (K). The mRNA forms hairpins, which are degraded by Dicer (L), leading to downregulation of frataxin (M). In this disease, RNA foci and RAN translation were not observed.
In this line, several studies have demonstrated abnormal morphology and function of synapses in ALS, with the nature of changes dependent on the disease timeline (Genc et al., 2017; Fogarty, 2018). Moreover, notable changes observed in ALS post-mortem samples are neuronal loss, change in the dendritic spine density, and morphology in excitatory neurotransmission sites, particularly in pyramidal cells such as CSMNs (Genc et al., 2017; Fogarty, 2018). This feature is also observed in different rodent models of ALS (Jara et al., 2012; Fogarty et al., 2017). In this review, we will discuss how changes in different pathways will lead to synaptic dysfunction in C9orf72-ALS pathology.
C9orf72 Role in Synaptic Dysfunction
The identification of an expansion of the hexanucleotide repeat GGGGCC (or G4C2) in intron 1 of the C9orf72 gene has opened a new field of investigation on long non-coding RNAs and neurodegenerative diseases. The G4C2 expansion was first identified in 2011 in patients with ALS and FTD (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Despite C9orf72 being initially involved in these two diseases, G4C2 expansions have also been observed in other neurodegenerative conditions including Creutzfeldt–Jakob disease, progressive supranuclear palsy, ataxia, Huntington disease-like syndrome, corticobasal syndrome, Parkinson’s disease, and Alzheimer’s disease (Woollacott and Mead, 2014).
Three plausible pathogenic mechanisms have been linked to the G4C2 expansion and they include: (a) loss of function mechanism due to G4C2 repeat expansion leading to downregulation of C9orf72 protein expression; (b) toxic gain of function by recruitment of other RNA-binding proteins into G4C2 RNA foci; and (c) aberrant dipeptide repeat proteins (DPR)s produced by repeat-associated non-AUG (RAN) translation. Six DPRs are produced from both strands generating poly-glycine-alanine [poly (GA)], poly-glycine-arginine [poly (GR)], poly-glycine-proline [poly (GP)] from the sense strand and poly-proline arginine [poly (PR)], poly-proline-alanine [poly (PA)], and poly-proline-glycine [poly (PG) or poly (GP)] from the antisense strand. These DPRs display different toxic properties in distinct animal and cellular models (Ash et al., 2013; Mori et al., 2013; Zu et al., 2013; Mizielinska et al., 2014; Haeusler et al., 2016; Lee et al., 2020) and their roles in synaptic dysfunction will be further discussed.
Little was known about the role of C9orf72 protein when it was first associated with ALS and FTD. Levine et al. (2013) reported that C9orf72 protein sequence and structure analyses share distant homology to Differentially Expressed in Normal and Neoplasia (DENN; Levine et al., 2013). The DENN protein is a GDP/GTP exchange factor (GEF) that activates Rab-GTPases. Thus, the homology to DENN protein suggests that C9orf72 may regulate membrane traffic and promote Rab-GTPase switches. Rab-GTPases are distributed in the soma and neurites and specific subtypes of Rab-GTPases, such as RAB3 and RAB8, are enriched at synapses in flies (Kiral et al., 2018). Atkinson et al. (2015) reported that C9orf72 protein is localized in different compartments in the cells during mouse development (Atkinson et al., 2015). At the embryonic stage and P1, C9orf72 forms small puncta throughout the neuropil, whereas at P7 and P56, C9orf72 is diffused in the nucleus and soma. Furthermore, C9orf72 protein is found in the synaptosome fraction, and the C9orf72 isoform 1 is enriched in the synaptic fraction in the adult mouse brain. Corroborating these data, Frick et al. (2018) showed similar findings using novel C9orf72 antibodies in wild-type mouse brain tissues (Frick et al., 2018). These authors observed that C9orf72 is mainly located in pre-synaptic compartments in the mouse brain and interacts with the RAB3 protein family (Frick et al., 2018). C9orf72 forms complex with SMCR8 and WDR41 (Sellier et al., 2016; Sullivan et al., 2016; Yang et al., 2016) with a function as GDP/GTP exchange factor for RAB3, RAB8a, RAB39b, and ULK1, suggesting a potential involvement in synaptic transmission and autophagy (Sellier et al., 2016; Yang et al., 2016; Frick et al., 2018; Xiao et al., 2019). Furthermore, synaptic fractionation of C9orf72 knockout mouse brain revealed that C9orf72, SMCR8, and RAB39b proteins were significantly depleted, with concomitant upregulation of GluR1 (Xiao et al., 2019). In this line, it has been shown that downregulation of C9orf72 partly impairs autophagy leading to accumulation of TDP-43 and p62, evidence of misfolded proteins accumulation (Sellier et al., 2016). Moreover, this role of C9orf72 protein in synaptic dysfunction has been supported by the localization of this protein in pre- and post-synaptic compartments (Xiao et al., 2019).
Studies using induced pluripotent stem cells (iPSCs) derived from patients with the C9orf72 expansion revealed that mutant lines displayed altered synaptic activity. Sareen et al. (2013) reported that neurons derived from C9orf72 patients were less excitable than control neurons, whereas Devlin et al. (2015) demonstrated that mutant C9orf72 lines presented hyperexcitability followed by a progressive loss of synaptic activity. Furthermore, two independent reports showed C9orf72 fly models presented dysfunctional neuromuscular junction transmission, with fewer active zones (Freibaum et al., 2015; Zhang et al., 2015). Herranz-Martin et al. (2017) described mice lines overexpressing 102×G4C2 repeats mediated by adeno-associated virus (AAV) and observed abnormalities in the NMJ. Lower acetylcholine receptors were observed in the C9orf72 mice compared to controls.
Interestingly, dipeptide repeats may also play a role in synaptic dysfunction. Xu and Xu (2018) induced the expression of different DPRs in Drosophila models. They observed that poly (GR) and poly (PR) overexpressing flies presented altered synaptic buttons at NMJs and degeneration of glutamatergic neurons due to excitotoxicity mechanisms. In contrast, Jensen et al. (2020) observed that poly(GA) peptides are located in neurites and are less mobile in overexpressing poly(GA) mouse models.
Finally, it is important to point out that despite C9orf72-ALS presenting features compatible with synaptopathy (glutamate excitotoxicity, accumulation of misfolded proteins, and mitochondrial dysfunction at distal axons) a few disease features deviate from the synaptopathy hypothesis. Such an example is the contribution of neuroinflammation in the ALS disease process (Fogarty, 2019). This inflammatory reaction is a result of a complex cascade of events characterized by infiltration of T-lymphocytes and macrophages and activation of microglia and reactive astrocytes (Philips and Robberecht, 2011). It is believed that neuroinflammation is activated to combat the mutant misfolded protein, however, in a bizarre turn of events, it becomes a hazardous process that contributes to neuronal damage instead (Philips and Robberecht, 2011). Furthermore, the limitations of these collective studies lie on their descriptive nature, represented by a snapshot of temporal and spatial changes. This is partially due to the experimental challenges and limitations in using cellular models to investigate synaptic dysfunction in vitro. The combination of electrophysiology and neuroimaging studies in patients and in animal models may provide more insights into synaptic dysfunction in C9orf72-ALS. Whether synaptic dysfunction is causal or a consequence of disease progression, more research into this field is needed to understand how synaptic dysfunction contributes to C9orf72 pathology in ALS.
The Role of Synaptic Neuroplasticity and Mitochondria Dysfunction in C9orf72 Neurodegeneration
Neuroplasticity or neuronal plasticity involves structural and functional adaptations of neuronal circuits to changes due to learning and memory, environmental influences, and brain damage (Malenka and Nicoll, 1999; Cheng et al., 2010). Structural plasticity processes in the mature nervous system are known as synaptic plasticity and include dendritic spine growth and synaptogenesis (Mattson, 2007). Long–term potentiation (LTP) and long–term depression (LTD) are relevant examples of neuroplasticity (Malenka and Bear, 2004). During LTP and synaptic activation, several studies have reported changes in mitochondria (Mattson and Liu, 2003), including energy production, calcium buffering and pump activity, and mitochondrial gene expression (Wieraszko, 1982; Stanton and Schanne, 1986; Williams et al., 1998). In this line, Ho et al. (2020) have shown that haploinsufficiency of C9orf72 impairs LTP in the dentate gyrus (DG-LTP) and LTD in the hippocampal CA1 area (CA1-LTD) as well as adult neurogenesis in the hippocampus.
Mitochondria are organelles found in all cells and they are highly abundant at axonal terminals and dendrites of neurons (Mattson, 2007). Mitochondria regulate calcium homeostasis by removing calcium from the cytoplasm in response to calcium influx into the cell, and they release calcium in response to certain stimuli, such as synaptic transmission (Simpson, 2000; Mattson, 2007). This is accompanied by energy production (ATP) and it also plays a role in redox signaling. This makes mitochondria pivotal in the regulation of synaptic transmission and maintenance of neuronal integrity and function (Mattson et al., 2008; Cheng et al., 2010; Amaral and Pozzo-Miller, 2012; Ivannikov et al., 2013; Manczak et al., 2013; Brot et al., 2014; Su et al., 2014). In dendrites, mitochondria are located mainly in the dendritic shafts and are also found to be associated with spines (Cameron et al., 1991; Popov et al., 2005; Valenti et al., 2014). In response to synaptic stimulation, mitochondria are redistributed toward dendritic protrusions to enhance their activity. Li et al. (2004) reported that decreasing the dendritic mitochondrial content led to a loss of synapses and spines, whereas the number of spines and synapses significantly increased by the accumulation of mitochondria in the dendrites. Furthermore, new spine and synapse formation are enhanced by the aggregation of mitochondria in dendrites. Thus, there are mechanisms for mutual regulation of synaptic plasticity and mitochondrial distribution and activity.
In C9orf72-ALS, mitochondrial dysfunction has been observed in different models and systems. Dafinca et al. (2016) reported that MNs derived from C9orf72 patients show swollen mitochondria and elevated levels of cytochrome c. In addition, a significant reduction in the mitochondrial membrane potential was observed. This indicates a reduced capacity to regulate cytosolic calcium (Dafinca et al., 2016). A few years later, the same group observed that C9orf72 MNs present a low capacity to uptake mitochondrial Ca2+ compared with control and corrected MNs. This contributed to glutamate excitotoxicity (Dafinca et al., 2020). Glutamate toxicity is an important feature in neurodegeneration and glutamate regulates synaptic plasticity and modifies cellular energy metabolism (Mattson et al., 1995). Glutamate plays a critical role in synaptic plasticity by activating receptors coupled to calcium influx (Mattson, 2007). In addition, it regulates downstream signaling-pathways via activation of kinases (such as PKA, PKC, and ERKs) and transcription factors that are important for long-term alterations of plasticity (Roberson et al., 1999). The protein activation processes are often ATP-dependent, and glutamate also stimulates an increase in mitochondrial oxygen consumption and thereby ATP production (Schuchmann et al., 2005; Mattson, 2007). These features are altered in C9orf72-ALS pathology.
Another study has suggested that mitochondrial ATP production is dysregulated in C9orf72 disease (Mehta et al., 2021). This source of energy is necessary for membrane potential generation, synaptic vesicle recruitment and release, as well as protein phosphorylation reactions (Stefani et al., 1997; Attwell and Laughlin, 2001; Valenti et al., 2014). All of these processes are critical for neuroplasticity and can be modified by changes in ATP production and release (Mattson and Liu, 2003). Mehta et al. (2021) have shown that C9orf72 MNs have shorter axons caused by altered mitochondrial bioenergetic function (Mehta et al., 2021). The authors found reduced gene expression of mitochondria encoded electron transport chain transcripts in MNs-iPSCs from C9orf72 patients. These findings were confirmed in ventral horn spinal MNs of C9orf72-ALS post-mortem tissue. Moreover, the authors demonstrated that the fast axonal transport of mitochondrial cargo was impaired, which is dependent on mitochondrial ATP (Zala et al., 2013). This study showed that axonal phenotypes in C9orf72-ALS were associated with concomitant metabolic dysfunction, owing to defective mitochondrial respiration.
In this line, Choi et al. (2019) established a mouse model of C9orf72, in which poly (GR) was expressed in the brain in a spatially and temporally controlled manner. These authors found that poly (GR) accumulates in the soma and dendrites of neurons in an age-dependent manner. Poly (GR) expression induced ALS/FTD-associated synaptic dysfunction and social behavioral deficits as well as neuronal cell loss, microgliosis, and DNA damage. The increased DNA damage is probably a consequence of compromised mitochondrial morphology and function, which may result in increased oxidative stress that in turn causes DNA damage. This was also observed in C9orf72 iPSC-derived motor neurons (Lopez-Gonzalez et al., 2016). More importantly, poly (GR) preferentially bound to ATP synthase F1 subunit alpha (Atp5a1), which is a subunit of mitochondrial complex V. It has been observed that this protein expression was decreased in poly (GR)-expressing primary cortical neurons cultured from CamKII: (GR) 80 mice, and its expression was reduced in the cortex of 6-months old CamKII: (GR) 80 mice and in the frontal cortex of patients with C9orf72-related ALS/FTD. Other mitochondrial proteins’ expression remained unchanged (Choi et al., 2019). Furthermore, recent in vitro studies showed that the reduction of dendritic mitochondrial content through increased mitophagy leads to inhibition of dendrite growth during neuronal polarization (Brot et al., 2014) and to dendrite shortening in mature neuronal cultures (Cherra et al., 2013). Taken together, sufficient dendritic mitochondrial content is required for proper development and maintenance of dendrites, as well as synapse and spine formation. However, the functional role of mitochondria in dendritic protrusions remains to be determined. As in axonal growth cones (Morris and Hollenbeck, 1993), it is possible that changes in ATP demand and required calcium buffering capacity underlie the number of mitochondria in growing spines.
Finally, we want to highlight that dendritic spines rarely contain mitochondria, thus the spine pathology observed in C9orf72 disease may also involve alterations in other organelles near the spines. These include smooth endoplasmic reticulum (SER) or endosomal multivesicular bodies (Spacek and Harris, 1997). Although the contribution of these organelles to neurodegeneration is unclear, they have been studied to a much lesser extent than mitochondria dysfunction and changes in spine distribution. Spine organelles alterations, postsynaptic cell damage, and excitotoxic caused by excessive presynaptic glutamate release are features observed in C9orf72-ALS pathology.
Involvement of Neuronal Cytoskeleton in C9orf72 Plasticity
Like any other eukaryotic cells, neurons are dependent on their cytoskeleton to maintain their shape, promote cell motility, and to organize their intracellular components (Pollard, 2003). The main components of cytoskeleton include actin, neurofilaments (NFs), and microtubules (MTs). Cytoskeleton dynamics are crucial to promote intracellular transport and to provide structural scaffolding for specialized structures. These include the axonal initial segment, axon and, presynaptic boutons. Dendrites are supported by microtubules, whereas filopodia and simple spines rely on actin remodeling (Neukirchen and Bradke, 2011).
Recent studies have shown that mutations causing neurodegenerative disorders are associated with dysfunction of cytoskeletal components (McMurray, 2000). These mutations often cause the formation of protein aggregates such as tau in AD (Matsuo et al., 1994; Castellani, 2020), alpha-synuclein in PD (Spillantini et al., 1997), Huntingtin (Arrasate and Finkbeiner, 2012), and TDP-43 in ALS (Neumann et al., 2006) and may influence vesicular biogenesis, trafficking and affect synaptic transmission (Table 1). Moreover, these mutations can initiate a cascade of events that include mitochondrial dysfunction, oxidative stress, activate DNA damage response and cause neuronal death. Thus, the dysfunction of the cytoskeleton is likely a common feature observed in several neurodegenerative diseases (McMurray, 2000; Table 1). The dysregulation of cytoskeleton components in C9orf72 pathology is discussed as follows.
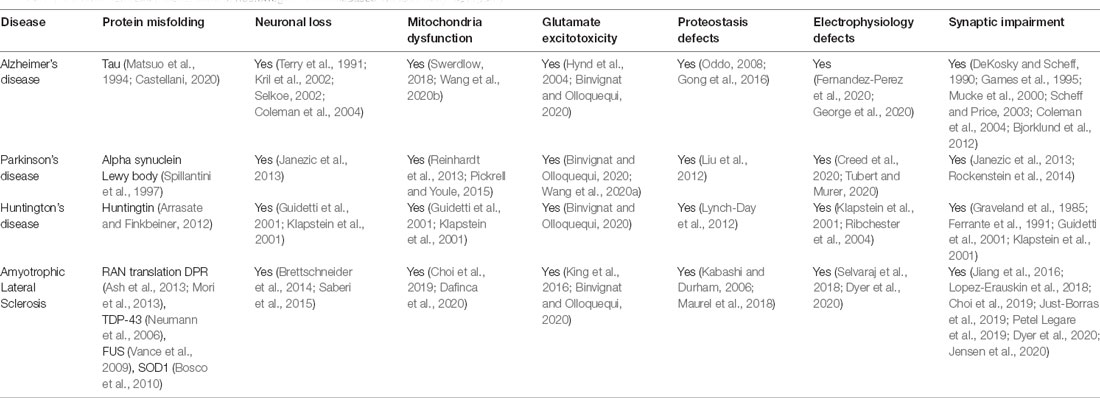
Table 1. Common disease mechanisms of neurodegenerative diseases will lead to synaptopathy.
The actin dynamics in neurons regulate extension and direction of axon growth, whereas in the dendritic spines, it plays a fundamental role in assembly and disassembly of synapses (Coles and Bradke, 2015).
Dysregulation of actin remodeling is a process thought to be implicated in ALS neurodegeneration (Hensel and Claus, 2018). In C9orf72 pathology, actin remodeling is also impaired. Giampetruzzi et al. (2019) have shown that actin homeostasis is disrupted in C9orf72-ALS. In this study, the authors transfected primary motor neurons with a synthetic construct expressing 80× GGGGCC repeats (G4C2-80). No differences in F-actin levels at the growth cone were found between conditions, however, the authors observed promoting actin filament assembly in fibroblasts from ALS patients can alleviate defects in nuclear-cytoplasmic transport defects (Giampetruzzi et al., 2019).
In order to understand the key regulators of actin dynamics in C9orf72-ALS, Sivadasan et al. (2016) over-expressed influenza hemagglutinin (HA)-tagged human C9orf72 protein and immunoprecipitated interacting proteins from mouse neuroblastoma NSC-34 cells. These authors found that cofilin, Arp2/3, and coronin were among statistically significant interaction partners of C9orf72. Special attention needs to be given to cofilin which promotes actin assembly or disassembly depending on the concentration of the former. This concentration is relative to actin, but also to other actin-binding proteins (Bravo-Cordero et al., 2013). The activity of cofilin is dependent on its phosphorylation at Ser3 residue, which inactivates its function in F-actin assembly (Moriyama et al., 1996). The authors observed that the overexpression of C9orf72-HA protein reduced phospho-cofilin (Ser3), while knockdown increased cofilin phosphorylation at Ser3 without changing total cofilin levels. These results were replicated in human cells with C9orf72 intronic expansion and in post-mortem cerebellar brain tissue from C9orf72-ALS patients (Sivadasan et al., 2016). Finally, the study shed some light on the influence of C9orf72 on actin dynamics. In this regard, Sivadasan et al. (2016) found that the number of newly generated actin filaments was reduced in C9orf72-depleted motor neurons which was accompanied by a significant reduction in the velocity of actin movement in axonal growth cones in motor neurons.
In the same line, Radwan et al. (2020) have demonstrated that Arginine-rich DPRs (PR and GR) impede the assembly of the actin cytoskeleton and this is correlated to a significant reduction in F-actin levels. Thus, modulation of the actin dynamics could represent potential therapeutic strategies for C9orf72-ALS pathology.
NFs are also known to contribute to the growth and stability of axons in both central and peripheral nerves, as well as to maintain mitochondrial stability (Gentil et al., 2015) and MT content (Bocquet et al., 2009). Additionally, NFs are also integral components of synapses. This is based on Cochard and Paulin (1984) study on mouse embryonic neuronal axons where NFs were present, pointing out their role in early development. Other studies have also shown that synapses contain a unique pool of NFs (Yuan et al., 2015a, b). These NFs isolated from brain synaptosomes were distinguishable both morphologically and biochemically from other NFs from different parts of the neuron (Yuan et al., 2003).
More interestingly, it has been shown that changes in phosphorylation of synaptic neurofilament light chain (NfL) are associated with calcium/calmodulin-dependent protein kinase II activation during modulation of LTP (Hashimoto et al., 2000), which it is altered in C9orf72-ALS (Ho et al., 2020). These results suggest that disturbed LTP may be caused by alterations in synaptic neurofilament proteins in C9orf72-ALS. In addition, it is important to highlight the role of NFs isoforms in regulating glutamatergic and dopaminergic synaptic neurotransmission (Schwartz et al., 1995). In this respect, several studies have demonstrated that NfL are expected to stabilize NMDA receptors within the neuronal plasma membrane (Ehlers et al., 1998; Ratnam and Teichberg, 2005; Gafson et al., 2020). These results are supported by observations that the cellular distribution of the NMDA GluN1 receptors could be linked to the phosphorylation states of NFs subunits (Ehlers et al., 1998; Terry-Lorenzo et al., 2000).
Taken together, any alteration in NFs subunits could be disrupting the synaptic scaffolding, leading to changes in LTP and LTD, and neurotransmission. Ultimately, these changes can lead to neuronal and cognitive changes as observed in C9orf72-ALS neurodegeneration. In this line, NFs have been used as a sensitive biomarker for neurodegeneration in different neurological disorders, including in C9orf72-ALS (Gendron et al., 2017; Khalil et al., 2018). During axonal degeneration, NFs are released into cerebrospinal fluid (CSF) and blood (Zucchi et al., 2020). In C9orf72 patients, phosphorylated neurofilament heavy (pNfH) proteins are found in the CSF and predict disease progression and survival. Patients expressing higher levels of pNFH exhibit faster disease progression and shorter survival (Gendron et al., 2017). Benatar et al. (2019) also investigated the phosphorylation status of NfH, as well as NfL in C9orf72 patients (Benatar et al., 2019). The authors observed an increase in the absolute levels of NfL proteins, however, pNfH levels remained unchanged in serum. No differences in both neurofilaments were found in CSF or blood. Specifically, in C9orf72 patients, NfL and pNfH were elevated ~3.5 years prior to phenoconversion, highlighting the importance of tracking neurofilament proteins as biomarkers of C9orf72-ALS (Benatar et al., 2019). In addition, Lu et al. (2015) have shown an increase of the NfL isoform and a modest upregulation of heavy and medium NF chain subunits in the blood of ALS patients. Thus, the levels of NFs could help to understand the relationship between genotype, the duration of the pre-symptomatic phase and predict survival of C9orf72-ALS.
At last, during axonal degeneration, NF accumulates in bundles and forms spheroid structures known as axonal swellings or blebs. These abnormal structures recruit hyperphosphorylated NfH and NfM in axons and gradually disrupt axonal transport, causing axonal fragmentation (Zucchi et al., 2020).
Axonal Transport Degeneration in C9orf72
Axonal transport is a key cellular process by which RNA, organelles, synaptic vesicles, proteins and, lipids are trafficked in bidirectional ends (Sleigh et al., 2019). Both anterograde and retrograde transports are MT-dependent and key for MN survival, maintenance, and functionality (Chevalier-Larsen and Holzbaur, 2006). The length of the MNs axons is highly dependent on cytoskeletal architecture and axonal transport stability. In C9orf72-ALS pathology the cytoskeleton integrity is compromised, leading to a disruption in the axonal transport necessary to maintain synapse integrity. Indeed, distal axonopathy has been observed in many neurodegenerative diseases, including ALS (Sleigh et al., 2019). This leads to defective axonal transport, a key initiating contributor to the selective vulnerability of motor nerves (Baldwin et al., 2016). The expression of the different pathogenic ALS mutations in SOD1 (Zhang et al., 1997; Williamson and Cleveland, 1999), TARDBP (Wang et al., 2013; Magrane et al., 2014), FUS, and C9orf72 (Baldwin et al., 2016) contributes to axonal transport defects, which are early pathology events that precedes neuronal loss and clinical symptoms.
As we have previously described, actin dynamics and neurofilaments alterations have been observed in C9orf72-ALS pathology. These observations could explain the disruption of cytoskeleton integrity and/or MT-dependent transport mechanisms which lead to the inability of MNs to supply their synapses with essential components and/or to convey information back to the cell body, potentially triggering the degeneration processes associated with C9orf72-ALS.
In this line, it has been recently proposed that mutations in NFL and NFH genes might affect the axonal integrity of MNs by altering the transport of other essential components in ALS (Zucchi et al., 2020). Two experiments were key to demonstrate their contribution to the disease. Lee et al. (1994) overexpressed NfL in mice and they observed degeneration of MNs, perikaryal and axonal swellings with the presence of intermediate filament spheroids, and NMJ denervation presenting as muscle atrophy. In the same hypothesis line, Julien et al. (1995) overexpressed NfH in mice observing swellings of proximal axons in the spinal cord, progressive axonopathy, and atrophy of muscle fibers. More interestingly, in this study not only the axonal transport of NF proteins was altered, but also actin, tubulin, and mitochondria.
In addition, other proteins have been involved in the axonal transport defects in C9orf72 models. Liang et al. (2019) observed that transgenic C9orf72 BAC mice presented a loss of function of C9orf72 protein as well as Smcr8. Both proteins form a complex (C9orf72-Smcr8) which associates with dynein causing the disruption in the axonal transport in MNs and impairment of retrograde transport resulted in stalling of autophagosomes. Furthermore, abnormal axon swellings formation was found in the spinal cord and NMJ in these mutant mice.
Moreover, in the 2016’s study, Baldwin and colleagues observed that Drosophila G4C2 overexpressing lines presented defects in axonal transport which increased the stationary mitochondria (Baldwin et al., 2016). They observed that the presence of DPRs, especially the PR-36, was causing a severe disruption of vesicle transport leading to mitochondria stalling. All these results together question whether mutations in NFs lead to a secondary effect or contribute to worsening the disease phenotype in synergy with C9orf72 haploinsufficiency and/or DPRs accumulation.
Recently, Fumagalli et al. (2021) demonstrated the existence of inhibitory interactions of arginine-rich DPRs with axonal transport machinery in C9orf72-associated ALS/FTD. In this study, the authors observed impaired microtubule-based transport in iPSC-derived MNs from C9orf72-ALS/FTD patients, including elevated stalled cargoes. This is in line with Abo-Rady et al. (2020) who reported that lysosome trafficking is also impaired in neurons with the C9orf72 repeat expansion. Moreover, Fumagalli et al. (2021) also showed that when control iPSC-derived motor neurons and Drosophila neurons were exposed to poly-PR and poly-GR, comparable effects in microtubule transport deficits were found. Protein interaction studies performed in MNs and post-mortem patient tissues revealed an association of arginine-rich DPRs with kinesin-1, dynein, and, microtubules. More importantly, the single-molecule imaging of purified components demonstrated that the arginine-rich DPRs directly impede motility by binding both the unstructured tubulin tails of microtubules and dynein and kinesin-1 motor complexes. However, while the DPRs increase the binding to microtubules and the duration of its processive runs to the active form of human kinesin-1 isoform (KIF5B), DPRs showed opposite results with dynein in a dose-dependent manner. These results support a direct inhibitory effect of arginine-rich DPRs on axonal transport and possibly different processes may contribute to trafficking defects observed in C9of72-ALS/FTD neurons. Nonetheless, these data highlight the relevance of targeting the cytoskeleton integrity as a therapy for C9orf72-ALS pathology.
C9orf72 Common Disease Mechanisms
Repeat Expansion Diseases
C9orf72 mechanism of disease is unique within the ALS spectrum as the three plausible disease mechanisms have been observed in different systems including cellular and animal models. Furthermore, downregulation of C9orf72 protein, the presence of RNA foci and DPRs have been observed in patient’s post-mortem samples and iPSCs derived neurons (Balendra and Isaacs, 2018; Shi et al., 2018; Westergard et al., 2019; Dafinca et al., 2020). Despite the unusual pathomechanism of disease, C9orf72 holds similarities with other expansion diseases (Rodriguez and Todd, 2019), such as Myotonic Dystrophy type 1 (DM1), Myotonic Dystrophy type 2 (DM2; Figure 2), and Friedreich ataxia (FA; Nguyen et al., 2019; Table 2).
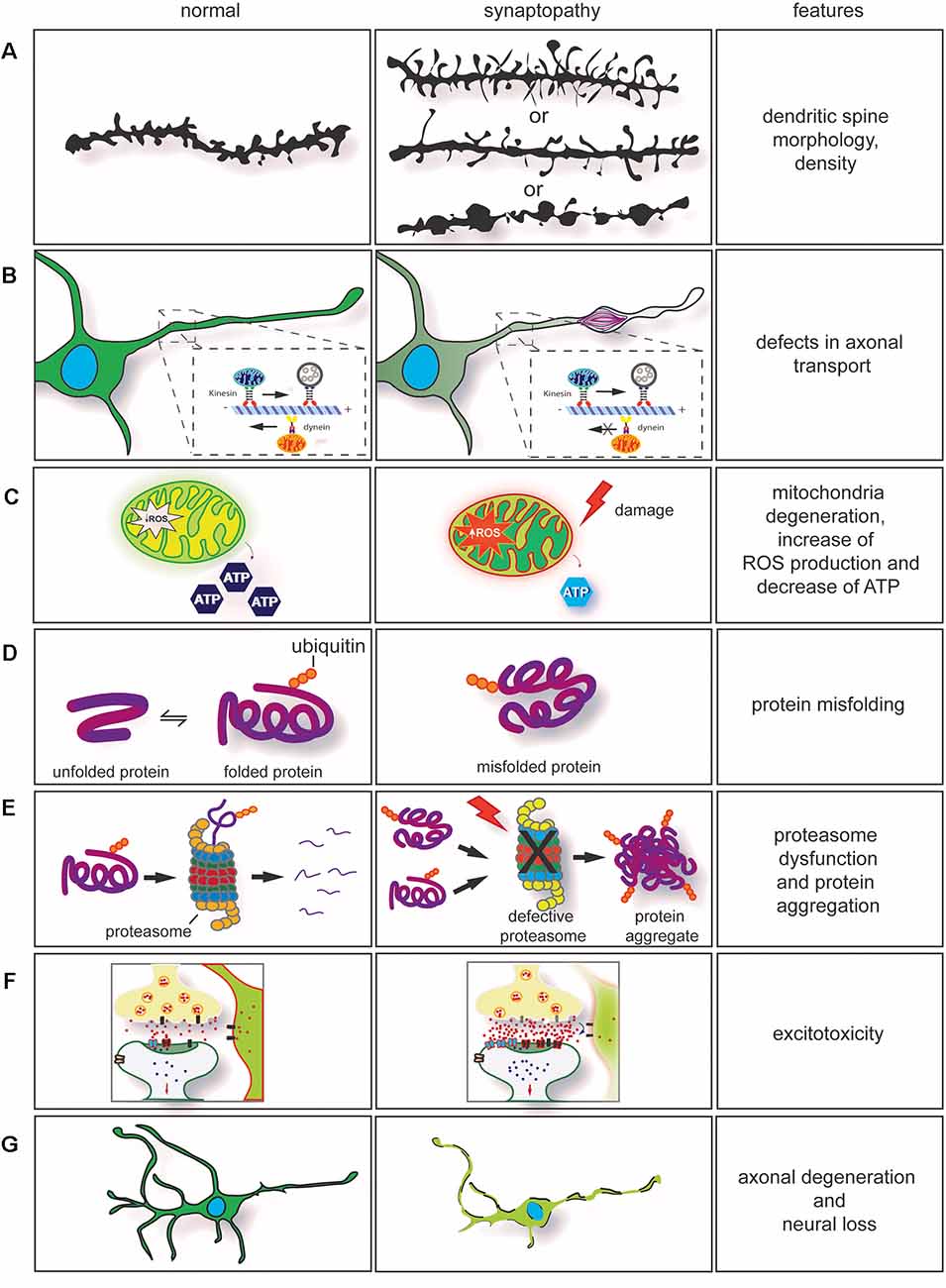
Figure 2. Dysregulation of key cellular mechanisms leads to synaptopathy. (A) Drawing of dendritic spines in controls (left) and in neurological diseases (right). The morphology and density of dendritic spines are altered in several neurodegenerative conditions. In synaptopathies (A, middle section), immature spines are normally long and thin, and density can be increased or decreased. On few occasions, neurite swellings with long and thin spines are observed. (B) Axonal transport impairment in neurodegenerative conditions leads to synaptopathy. Retrograde and anterograde transport disruption leads to stalling of mitochondria transport. Furthermore, rearrangement of neurofilaments (NFs) and damage caused by the neurodegeneration process will lead to formation of axonal swelling impairing axonal transport. (C) Mitochondria dysfunction [ATP production, change in homeostasis, and increase of reactive oxygen species (ROS)] are observed in neurodegenerative conditions. These contribute to synaptic dysfunction. (D) Mutations in genes involved in neurodegenerative conditions often cause protein misfolding and accumulation in neurons. (E) In association with proteasome dysfunction, protein misfolding will form insoluble protein accumulation, enhancing the neurodegeneration process. This contributes to axonal degeneration and axonal transport impairment. (F) Impairment in axonal transport, alteration in calcium buffering, mitochondria degeneration, formation of ROS associated with dysregulation of synaptic proteins can enhance glutamate excitotoxicity. (G) Neuronal degeneration and neuronal loss are also observed in neurodegenerative conditions. They are a result of the neurodegeneration process.
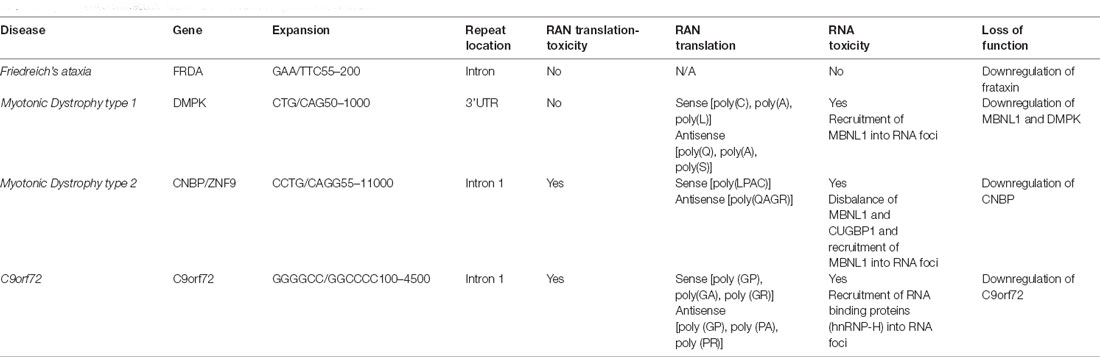
Table 2. Common molecular features observed in expansion diseases.
DM1 is a genetic disease caused by myotonia, cardiopathy, muscular dystrophy, and cataracts (Meola and Cardani, 2015). It is caused by the expansion of CTG in the 3’ untranslated region (UTR) of myotonic dystrophy protein kinase (DMPK; Brook et al., 1992). RNA toxicity is the main pathological feature observed in DM1, which causes the formation of RNA foci and recruitment of an RNA-binding protein known as muscle blind-like 1 (MBNL1) protein, a protein involved in alternative splicing (Miller et al., 2000). The sequestration of MBNL in RNA foci leads to upregulation of CUG-binding protein (CUGBP1; Lin et al., 2006). This imbalance of MBNL and CUGBP1 causes splicing defects on several mRNAs (Philips et al., 1998). DM1 seems to produce RAN translation products, including poly (C), poly (A), and poly (L) from the sense strand, and poly (Q), poly (A), and poly (S) from the antisense strand. However, it is unclear whether these DPR products are pathogenic (Zu et al., 2017). Downregulation of DMPK leads to mild myopathy in knockout mice, suggesting it plays a minor role in the disease (Jansen et al., 1996).
DM2 is less frequent than DM1 and the clinical presentation varies (Meola and Cardani, 2015). DM2 is caused by an expansion of CCTG repeats in the intron of cellular nucleic acid binding protein (CNBP), also known as Zinc finger factor 9 (ZNF9). The disease mechanism of DM2 is similar to DM1 and is caused by RNA toxicity with the formation of RNA foci with recruitment of MBNL1 protein and disbalance between MBNL1 and CUGBP1 (Jones et al., 2011). Furthermore, RAN translation produces the peptides poly (LPAC; sense) and poly (QAGR; antisense). Contrary to DM1, DM2 RAN translated peptides are present in autopsy tissues from patients with DM2 and seem to be toxic in cells independent of RNA gain of function (Zu et al., 2017). Haploinsufficiency of CNBP causes abnormal feature reflecting DM2 phenotype in murine models (Chen et al., 2007).
Friedreich’s ataxia (FRDA) is another repeat expansion disease caused by GAA repeats in intron 1 of frataxin (FXN) gene, which encodes for a mitochondrial protein. FRDA affects the peripheral and central nervous system (Lin et al., 2017). The expansion causes haploinsufficiency of FXN leading to epigenetic modifications on the gene, reducing transcription of frataxin protein. In addition to common disease mechanisms, DM and FDRA present synaptic dysfunction (Hernandez-Hernandez et al., 2013; Lin et al., 2017). Transgenic models for myotonic DM1 revealed that the CTG expansion leads to upregulation of RAB3A protein, leading to dysfunction in neurotransmission and mouse behavior (Hernandez-Hernandez et al., 2013). In contrast, FA’s transgenic mice presented early VGLUT1 depletion and dysregulated synaptic input deficits and cerebellar circuit (Lin et al., 2017).
Neurodegenerative Diseases
As we have previously described, C9orf72 localization and function and DPR localization at synapses indicated C9orf72 disease is a synaptopathy within the neuromotor system. Moreover, other neurological diseases have also shown features of synaptopathy with altered synaptic structure and function. Among these neurodegenerative conditions, we include Parkinson’s disease (PD), Huntington’s (HD), prion pathologies, and Alzheimer’s disease (AD; Table 1). Early onset neurological diseases such as schizophrenia (SCZ), bipolar disorder (BP), and autism spectrum disorders (ASD) are also synaptopathies (Lepeta et al., 2016).
By comparing these neurodegenerative diseases’ pathology and progression, common features among synaptopathies diseases are observed (Table 1). This allows us to deepen our understanding of the neurodegeneration process and may help us to design therapies that target one pathophysiological pathway common to other synaptopathies.
In AD, a progressive loss of dendritic spines in hippocampal pyramidal neurons (Scheibel, 1979b; Ferrer and Gullotta, 1990), dentate granule cells (de Ruiter and Uylings, 1987; Gertz et al., 1987; Einstein et al., 1994), and neocortical pyramidal neurons (Scheibel, 1979a; Catala et al., 1988; Baloyannis et al., 1992) have been observed. Moreover, synapse loss in AD is accompanied by a compensatory increase in synaptic bouton’s size (Scheff et al., 1990; Scheff and Price, 1993). A similar neuronal loss and extensive dendritic spine loss in the cortex have also been found in FTD (Baloyannis et al., 2001), in neocortical neurons in Pick’s disease (Ferrer, 1999), and ALS (Ferrer et al., 1991). Moreover, a reduction in dendritic spines in dopaminergic projections of striatal neurons in PD (McNeill et al., 1988) and in locus ceruleus and substantia nigra in rodent models for PD (Shors et al., 2001) have been also observed. Furthermore, a decrease in spine density has been found in severe HD cases (Ferrante et al., 1991). This observation was confirmed in the striatal and cortical regions of HD transgenic mice (Guidetti et al., 2001). However, an increased spine density has also been observed in HD models (Ferrante et al., 1991).
Finally, we also want to highlight that neuronal loss, mitochondria dysfunction, glutamate excitotoxicity, proteostasis defects, and synaptic impairment are other features common to other synaptopathies (Table 1). Whether these events are the endpoint of signaling cascades or common mechanisms of neurodegenerative diseases, it remains to be elucidated.
Conclusion
In this review article, we addressed how the expansion in the C9orf72 gene leads to the formation of RNA foci, DPRs, and downregulation of C9orf72 transcripts. Furthermore, we discuss how C9orf72 may have a function at synapses and how the convergence of a wide range of cellular defects is observed in other neurodegenerative conditions. Dysregulation of key cellular mechanisms, such as dendritic spine morphology defects, defects in the axonal transport and axonal degeneration, mitochondria degeneration, protein misfolding, protein aggregation and proteasome dysfunction, excitotoxicity, and neuronal loss have always been addressed separately. However, these disease features commonalities are often presented together, and this evidence can no longer be overlooked. These converging mechanisms can provide insights into the pathogenic pathways leading to synaptopathy and will aid better identification of disease hallmarks of neurodegenerative diseases. Furthermore, it may offer potential targets for intervention to prevent synaptic degeneration in ALS caused by C9orf72 repeat expansion.
Author Contributions
AN: writing—original draft preparation, reviewing, and editing. NA: wrote the first draft of the manuscript, supervision, conceptualization, and reviewing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by UK Dementia Research Institute, the Ministry of Science, Innovation and Universities (Ministerio de Ciencia, Innovación y Universidades; PSI2017-83893-R), and the Ministry of Economy and Business (Ministerio de Economía, Industria y Competitividad, Gobierno de España; PSI2017-90806-REDT; Spain).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the Alzheimer’s Research UK, King’s College London Network Centre, UK Dementia Research Institute, and AINDACE foundation for their technical and human support.
References
Abo-Rady, M., Kalmbach, N., Pal, A., Schludi, C., Janosch, A., Richter, T., et al. (2020). Knocking out C9ORF72 exacerbates axonal trafficking defects associated with hexanucleotide repeat expansion and reduces levels of heat shock proteins. Stem Cell Rep. 14, 390–405. doi: 10.1016/j.stemcr.2020.01.010
Amaral, M. D., and Pozzo-Miller, L. (2012). Intracellular Ca2+ stores and Ca2+ influx are both required for BDNF to rapidly increase quantal vesicular transmitter release. Neural Plast. 2012:203536. doi: 10.1155/2012/203536
Arrasate, M., and Finkbeiner, S. (2012). Protein aggregates in Huntington’s disease. Exp. Neurol. 238, 1–11. doi: 10.1016/j.expneurol.2011.12.013
Ash, P. E., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., Dejesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Atkinson, R. A., Fernandez-Martos, C. M., Atkin, J. D., Vickers, J. C., and King, A. E. (2015). C9ORF72 expression and cellular localization over mouse development. Acta Neuropathol. Commun. 3:59. doi: 10.1186/s40478-015-0238-7
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Baldwin, K. R., Godena, V. K., Hewitt, V. L., and Whitworth, A. J. (2016). Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum. Mol. Genet. 25, 2378–2392. doi: 10.1093/hmg/ddw105
Balendra, R., and Isaacs, A. M. (2018). C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558. doi: 10.1038/s41582-018-0047-2
Baloyannis, S. J., Manolidis, S. L., and Manolidis, L. S. (1992). The acoustic cortex in Alzheimer’s disease. Acta Otolaryngol. Suppl. 494, 1–13. doi: 10.3109/00016489209137072
Baloyannis, S. J., Manolidis, S. L., and Manolidis, L. S. (2001). The acoustic cortex in frontal dementia. Acta Otolaryngol. 121, 289–292. doi: 10.1080/000164801300043884
Benatar, M., Wuu, J., Lombardi, V., Jeromin, A., Bowser, R., Andersen, P. M., et al. (2019). Neurofilaments in pre-symptomatic ALS and the impact of genotype. Amyotroph. Lateral Scler. Frontotemporal Degener. 20, 538–548. doi: 10.1080/21678421.2019.1646769
Binvignat, O., and Olloquequi, J. (2020). Excitotoxicity as a target against neurodegenerative processes. Curr. Pharm. Des. 26, 1251–1262. doi: 10.2174/1381612826666200113162641
Bjorklund, N. L., Reese, L. C., Sadagoparamanujam, V. M., Ghirardi, V., Woltjer, R. L., and Taglialatela, G. (2012). Absence of amyloid beta oligomers at the postsynapse and regulated synaptic Zn2+ in cognitively intact aged individuals with Alzheimer’s disease neuropathology. Mol. Neurodegener. 7:23. doi: 10.1186/1750-1326-7-23
Bocquet, A., Berges, R., Frank, R., Robert, P., Peterson, A. C., and Eyer, J. (2009). Neurofilaments bind tubulin and modulate its polymerization. J. Neurosci. 29, 11043–11054. doi: 10.1523/JNEUROSCI.1924-09.2009
Bosco, D. A., Morfini, G., Karabacak, N. M., Song, Y., Gros-Louis, F., Pasinelli, P., et al. (2010). Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403. doi: 10.1038/nn.2660
Braak, H., Brettschneider, J., Ludolph, A. C., Lee, V. M., Trojanowski, J. Q., and Del Tredici, K. (2013). Amyotrophic lateral sclerosis—a model of corticofugal axonal spread. Nat. Rev. Neurol. 9, 708–714. doi: 10.1038/nrneurol.2013.221
Bravo-Cordero, J. J., Magalhaes, M. A., Eddy, R. J., Hodgson, L., and Condeelis, J. (2013). Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell Biol. 14, 405–415. doi: 10.1038/nrm3609
Brettschneider, J., Arai, K., Del Tredici, K., Toledo, J. B., Robinson, J. L., Lee, E. B., et al. (2014). TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol. 128, 423–437. doi: 10.1007/s00401-014-1299-6
Brettschneider, J., Del Tredici, K., Toledo, J. B., Robinson, J. L., Irwin, D. J., Grossman, M., et al. (2013). Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 74, 20–38. doi: 10.1002/ana.23937
Brook, J. D., McCurrach, M. E., Harley, H. G., Buckler, A. J., Church, D., Aburatani, H., et al. (1992). Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG). repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68, 799–808. doi: 10.1016/0092-8674(92)90154-5
Brot, S., Auger, C., Bentata, R., Rogemond, V., Menigoz, S., Chounlamountri, N., et al. (2014). Collapsin response mediator protein 5 (CRMP5). induces mitophagy, thereby regulating mitochondrion numbers in dendrites. J. Biol. Chem. 289, 2261–2276. doi: 10.1074/jbc.M113.490862
Cameron, H. A., Kaliszewski, C. K., and Greer, C. A. (1991). Organization of mitochondria in olfactory bulb granule cell dendritic spines. Synapse 8, 107–118. doi: 10.1002/syn.890080205
Castellani, R. J. (2020). The significance of tau aggregates in the human. Brain Sci. 10:972. doi: 10.3390/brainsci10120972
Catala, I., Ferrer, I., Galofre, E., and Fabregues, I. (1988). Decreased numbers of dendritic spines on cortical pyramidal neurons in dementia. A quantitative Golgi study on biopsy samples. Hum. Neurobiol. 6, 255–259.
Cheng, A., Hou, Y., and Mattson, M. P. (2010). Mitochondria and neuroplasticity. ASN Neuro 2:e00045. doi: 10.1042/AN20100019
Chen, W., Wang, Y., Abe, Y., Cheney, L., Udd, B., and Li, Y. P. (2007). Haploinsuffciency for Znf9 in Znf9+/– mice is associated with multiorgan abnormalities resembling myotonic dystrophy. J. Mol. Biol. 368, 8–17. doi: 10.1016/j.jmb.2007.01.088
Cherra, S. J., 3rd, Steer, E., Gusdon, A. M., Kiselyov, K., and Chu, C. T. (2013). Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am. J. Pathol. 182, 474–484. doi: 10.1016/j.ajpath.2012.10.027
Chevalier-Larsen, E., and Holzbaur, E. L. (2006). Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta 1762, 1094–1108. doi: 10.1016/j.bbadis.2006.04.002
Choi, S. Y., Lopez-Gonzalez, R., Krishnan, G., Phillips, H. L., Li, A. N., Seeley, W. W., et al. (2019). C9ORF72-ALS/FTD-associated poly (GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci. 22, 851–862. doi: 10.1038/s41593-019-0397-0
Cochard, P., and Paulin, D. (1984). Initial expression of neurofilaments and vimentin in the central and peripheral nervous system of the mouse embryo in vivo. J. Neurosci. 4, 2080–2094. doi: 10.1523/JNEUROSCI.04-08-02080.1984
Coleman, P., Federoff, H., and Kurlan, R. (2004). A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology 63, 1155–1162. doi: 10.1212/01.wnl.0000140626.48118.0a
Coles, C. H., and Bradke, F. (2015). Coordinating neuronal actin-microtubule dynamics. Curr. Biol. 25, R677–R691. doi: 10.1016/j.cub.2015.06.020
Creed, R. B., Roberts, R. C., Farmer, C. B., McMahon, L. L., and Goldberg, M. S. (2020). Increased glutamate transmission onto dorsal striatum spiny projection neurons in Pink1 knockout rats. Neurobiol. Dis. 150:105246. doi: 10.1016/j.nbd.2020.105246
Dafinca, R., Barbagallo, P., Farrimond, L., Candalija, A., Scaber, J., Ababneh, N. A., et al. (2020). Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in iPS-derived motor neurons from patients with ALS/FTD. Stem Cell Reports 14, 892–908. doi: 10.1016/j.stemcr.2020.03.023
Dafinca, R., Scaber, J., Ababneh, N., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 34, 2063–2078. doi: 10.1002/stem.2388
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
DeKosky, S. T., and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464. doi: 10.1002/ana.410270502
Devlin, A. C., Burr, K., Borooah, S., Foster, J. D., Cleary, E. M., Geti, I., et al. (2015). Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 6:5999. doi: 10.1038/ncomms6999
de Ruiter, J. P., and Uylings, H. B. (1987). Morphometric and dendritic analysis of fascia dentata granule cells in human aging and senile dementia. Brain Res. 402, 217–229. doi: 10.1016/0006-8993(87)90028-x
Dyer, M. S., Reale, L. A., Lewis, K. E., Walker, A. K., Dickson, T. C., Woodhouse, A., et al. (2020). Mislocalisation of TDP-43 to the cytoplasm causes cortical hyperexcitability and reduced excitatory neurotransmission in the motor cortex. J. Neurochem. [Online ahead of print]. doi: 10.1111/jnc.15214
Ehlers, M. D., Fung, E. T., O’Brien, R. J., and Huganir, R. L. (1998). Splice variant-specific interaction of the NMDA receptor subunit NR1 with neuronal intermediate filaments. J. Neurosci. 18, 720–730. doi: 10.1523/JNEUROSCI.18-02-00720.1998
Einstein, G., Buranosky, R., and Crain, B. J. (1994). Dendritic pathology of granule cells in Alzheimer’s disease is unrelated to neuritic plaques. J. Neurosci. 14, 5077–5088. doi: 10.1523/JNEUROSCI.14-08-05077.1994
Fernandez-Perez, E. J., Gallegos, S., Armijo-Weingart, L., Araya, A., Riffo-Lepe, N. O., Cayuman, F., et al. (2020). Changes in neuronal excitability and synaptic transmission in nucleus accumbens in a transgenic Alzheimer’s disease mouse model. Sci. Rep. 10:19606. doi: 10.1038/s41598-020-76456-w
Ferrante, R. J., Kowall, N. W., and Richardson, E. P., Jr. (1991). Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J. Neurosci. 11, 3877–3887. doi: 10.1523/JNEUROSCI.11-12-03877.1991
Ferrer, I. (1999). Neurons and their dendrites in frontotemporal dementia. Dement. Geriatr. Cogn. Disord. 10, 55–60. doi: 10.1159/000051214
Ferrer, I., and Gullotta, F. (1990). Down’s syndrome and Alzheimer’s disease: dendritic spine counts in the hippocampus. Acta Neuropathol. 79, 680–685. doi: 10.1007/BF00294247
Ferrer, I., Roig, C., Espino, A., Peiro, G., and Matias Guiu, X. (1991). Dementia of frontal lobe type and motor neuron disease. A Golgi study of the frontal cortex. J. Neurol. Neurosurg. Psychiatry. 54, 932–934. doi: 10.1136/jnnp.54.10.932
Fogarty, M. J. (2018). Driven to decay: excitability and synaptic abnormalities in amyotrophic lateral sclerosis. Brain Res. Bull. 140, 318–333. doi: 10.1136/jnnp.54.10.932
Fogarty, M. J. (2019). Amyotrophic lateral sclerosis as a synaptopathy. Neural Regen. Res. 14, 189–192. doi: 10.4103/1673-5374.244782
Fogarty, M. J., Mu, E. W. H., Lavidis, N. A., Noakes, P. G., and Bellingham, M. C. (2017). Motor areas show altered dendritic structure in an amyotrophic lateral sclerosis mouse model. Front. Neurosci. 11:609. doi: 10.3389/fnins.2017.00609
Freibaum, B. D., Lu, Y., Lopez-Gonzalez, R., Kim, N. C., Almeida, S., Lee, K. H., et al. (2015). GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133. doi: 10.1038/nature14974
Frick, P., Sellier, C., Mackenzie, I. R. A., Cheng, C. Y., Tahraoui-Bories, J., Martinat, C., et al. (2018). Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol. Commun. 6:72. doi: 10.1186/s40478-018-0579-0
Fumagalli, L., Young, F. L., Boeynaems, S., De Decker, M., Mehta, A. R., Swijsen, A., et al. (2021). C9orf72-derived arginine-containing dipeptide repeats associate with axonal transport machinery and impede microtubule-based motility. Sci. Adv. 7:eabg3013. doi: 10.1126/sciadv.abg3013
Gafson, A. R., Barthelemy, N. R., Bomont, P., Carare, R. O., Durham, H. D., Julien, J. P., et al. (2020). Neurofilaments: neurobiological foundations for biomarker applications. Brain 143, 1975–1998. doi: 10.1093/brain/awaa098
Games, D., Adams, D., Alessandrini, R., Barbour, R., Berthelette, P., Blackwell, C., et al. (1995). Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373, 523–527. doi: 10.1038/373523a0
Genc, B., Jara, J. H., Lagrimas, A. K., Pytel, P., Roos, R. P., Mesulam, M. M., et al. (2017). Apical dendrite degeneration, a novel cellular pathology for Betz cells in ALS. Sci. Rep. 7:41765. doi: 10.1038/srep41765
Gendron, T. F., Group, C. O. N. S., Daughrity, L. M., Heckman, M. G., Diehl, N. N., Wuu, J., et al. (2017). Phosphorylated neurofilament heavy chain: a biomarker of survival for C9ORF72-associated amyotrophic lateral sclerosis. Ann. Neurol. 82, 139–146. doi: 10.1002/ana.24980
Gentil, B. J., Tibshirani, M., and Durham, H. D. (2015). Neurofilament dynamics and involvement in neurological disorders. Cell Tissue Res. 360, 609–620. doi: 10.1007/s00441-014-2082-7
George, A. A., Vieira, J. M., Xavier-Jackson, C., Gee, M. T., Cirrito, J. R., Bimonte-Nelson, H., et al. (2020). Implications of oligomeric amyloid-beta (oAβ42). signaling through alpha7beta2-nicotinic acetylcholine receptors (nAChR) on basal forebrain cholinergic neuronal intrinsic excitability and cognitive decline. J. Neurosci. 41, 555–575. doi: 10.1523/JNEUROSCI.0876-20.2020
Gertz, H. J., Cervos-Navarro, J., and Ewald, V. (1987). The septo-hippocampal pathway in patients suffering from senile dementia of Alzheimer’s type. Evidence for neuronal plasticity? Neurosci. Lett. 76, 228–232. doi: 10.1016/0304-3940(87)90720-8
Giampetruzzi, A., Danielson, E. W., Gumina, V., Jeon, M., Boopathy, S., Brown, R. H., et al. (2019). Modulation of actin polymerization affects nucleocytoplasmic transport in multiple forms of amyotrophic lateral sclerosis. Nat. Commun. 10:3827. doi: 10.1038/s41467-019-11837-y
Gong, B., Radulovic, M., Figueiredo-Pereira, M. E., and Cardozo, C. (2016). The ubiquitin-proteasome system: potential therapeutic targets for Alzheimer’s disease and spinal cord injury. Front. Mol. Neurosci. 9:4. doi: 10.3389/fnmol.2016.00004
Graveland, G. A., Williams, R. S., and DiFiglia, M. (1985). Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 227, 770–773. doi: 10.1126/science.3155875
Guidetti, P., Charles, V., Chen, E. Y., Reddy, P. H., Kordower, J. H., Whetsell, W. O., et al. (2001). Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp. Neurol. 169, 340–350. doi: 10.1006/exnr.2000.7626
Haeusler, A. R., Donnelly, C. J., and Rothstein, J. D. (2016). The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat. Rev. Neurosci. 17, 383–395. doi: 10.1038/nrn.2016.38
Hashimoto, R., Nakamura, Y., Komai, S., Kashiwagi, Y., Tamura, K., Goto, T., et al. (2000). Site-specific phosphorylation of neurofilament-L is mediated by calcium/calmodulin-dependent protein kinase II in the apical dendrites during long-term potentiation. J. Neurochem. 75, 373–382. doi: 10.1046/j.1471-4159.2000.0750373.x
Hensel, N., and Claus, P. (2018). The actin cytoskeleton in SMA and ALS: how does it contribute to motoneuron degeneration? Neuroscientist 24, 54–72. doi: 10.1177/1073858417705059
Hernandez-Hernandez, O., Guiraud-Dogan, C., Sicot, G., Huguet, A., Luilier, S., Steidl, E., et al. (2013). Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behaviour. Brain 136, 957–970. doi: 10.1093/brain/aws367
Herranz-Martin, S., Chandran, J., Lewis, K., Mulcahy, P., Higginbottom, A., Walker, C., et al. (2017). Viral delivery of C9orf72 hexanucleotide repeat expansions in mice leads to repeat-length-dependent neuropathology and behavioural deficits. Dis. Model. Mech. 10, 859–868. doi: 10.1242/dmm.029892
Ho, W. Y., Navakkode, S., Liu, F., Soong, T. W., and Ling, S. C. (2020). Deregulated expression of a longevity gene, Klotho, in the C9orf72 deletion mice with impaired synaptic plasticity and adult hippocampal neurogenesis. Acta Neuropathol. Commun. 8:155. doi: 10.1186/s40478-020-01030-4
Hynd, M. R., Scott, H. L., and Dodd, P. R. (2004). Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 45, 583–595. doi: 10.1016/j.neuint.2004.03.007
Ivannikov, M. V., Sugimori, M., and Llinas, R. R. (2013). Synaptic vesicle exocytosis in hippocampal synaptosomes correlates directly with total mitochondrial volume. J. Mol. Neurosci. 49, 223–230. doi: 10.1007/s12031-012-9848-8
Janezic, S., Threlfell, S., Dodson, P. D., Dowie, M. J., Taylor, T. N., Potgieter, D., et al. (2013). Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl. Acad. Sci. U S A 110, E4016–E4025. doi: 10.1073/pnas.1309143110
Jansen, G., Groenen, P. J., Bachner, D., Jap, P. H., Coerwinkel, M., Oerlemans, F., et al. (1996). Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat. Genet. 13, 316–324. doi: 10.1038/ng0796-316
Jara, J. H., Villa, S. R., Khan, N. A., Bohn, M. C., and Ozdinler, P. H. (2012). AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol. Dis. 47, 174–183. doi: 10.1016/j.nbd.2012.03.036
Jensen, B. K., Schuldi, M. H., McAvoy, K., Russell, K. A., Boehringer, A., Curran, B. M., et al. (2020). Synaptic dysfunction induced by glycine-alanine dipeptides in C9orf72-ALS/FTD is rescued by SV2 replenishment. EMBO Mol. Med. 12:e10722. doi: 10.15252/emmm.201910722
Jiang, J., Zhu, Q., Gendron, T. F., Saberi, S., McAlonis-Downes, M., Seelman, A., et al. (2016). Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90, 535–550. doi: 10.1016/j.neuron.2016.04.006
Jones, K., Jin, B., Iakova, P., Huichalaf, C., Sarkar, P., Schneider-Gold, C., et al. (2011). RNA Foci, CUGBP1 and ZNF9 are the primary targets of the mutant CUG and CCUG repeats expanded in myotonic dystrophies type 1 and type 2. Am. J. Pathol. 179, 2475–2489. doi: 10.1016/j.ajpath.2011.07.013
Julien, J. P., Côté, F., and Collard, J. F. (1995). Mice overexpressing the human neurofilament heavy gene as a model of ALS. Neurobiol. Aging. 16, 487–490. doi: 10.1016/0197-4580(94)00169-2
Just-Borras, L., Hurtado, E., Cilleros-Mane, V., Biondi, O., Charbonnier, F., Tomas, M., et al. (2019). Overview of impaired BDNF signaling, their coupled downstream serine-threonine kinases and SNARE/SM complex in the neuromuscular junction of the amyotrophic lateral sclerosis model SOD1–G93A mice. Mol. Neurobiol. 56, 6856–6872. doi: 10.1007/s12035-019-1550-1
Kabashi, E., and Durham, H. D. (2006). Failure of protein quality control in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 1038–1050. doi: 10.1016/j.bbadis.2006.06.006
Khalil, M., Teunissen, C. E., Otto, M., Piehl, F., Sormani, M. P., Gattringer, T., et al. (2018). Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 14, 577–589. doi: 10.1038/s41582-018-0058-z
King, A. E., Woodhouse, A., Kirkcaldie, M. T., and Vickers, J. C. (2016). Excitotoxicity in ALS: overstimulation, or overreaction? Exp. Neurol. 275, 162–171. doi: 10.1016/j.expneurol.2015.09.019
Kiral, F. R., Kohrs, F. E., Jin, E. J., and Hiesinger, P. R. (2018). Rab GTPases and membrane trafficking in neurodegeneration. Curr. Biol. 28, R471–R486. doi: 10.1016/j.cub.2018.02.010
Klapstein, G. J., Fisher, R. S., Zanjani, H., Cepeda, C., Jokel, E. S., Chesselet, M. F., et al. (2001). Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington’s disease transgenic mice. J. Neurophysiol. 86, 2667–2677. doi: 10.1152/jn.2001.86.6.2667
Kril, J. J., Patel, S., Harding, A. J., and Halliday, G. M. (2002). Neuron loss from the hippocampus of Alzheimer’s disease exceeds extracellular neurofibrillary tangle formation. Acta Neuropathol. 103, 370–376. doi: 10.1007/s00401-001-0477-5
Lee, Y. B., Baskaran, P., Gomez-Deza, J., Chen, H. J., Nishimura, A. L., Smith, B. N., et al. (2020). C9orf72 poly GA RAN-translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum. Mol. Genet. 26, 4765–4777. doi: 10.1093/hmg/ddx350
Lee, M. K., Marszalek, J. R., and Cleveland, D. W. (1994). A mutant neurofilament subunit causes massive, selective motor neuron death: implications for the pathogenesis of human motor neuron disease. Neuron 13, 975–988. doi: 10.1016/0896-6273(94)90263-1
Lepeta, K., Lourenco, M. V., Schweitzer, B. C., Martino Adami, P. V., Banerjee, P., Catuara-Solarz, S., et al. (2016). Synaptopathies: synaptic dysfunction in neurological disorders - a review from students to students. J. Neurochem. 138, 785–805. doi: 10.1111/jnc.13713
Levine, T. P., Daniels, R. D., Gatta, A. T., Wong, L. H., and Hayes, M. J. (2013). The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29, 499–503. doi: 10.1093/bioinformatics/bts725
Li, Z., Okamoto, K., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Liang, C., Shao, Q., Zhang, W., Yang, M., Chang, Q., Chen, R., et al. (2019). Smcr8 deficiency disrupts axonal transport-dependent lysosomal function and promotes axonal swellings and gain of toxicity in C9ALS/FTD mouse models. Hum. Mol. Genet. 28, 3940–3953. doi: 10.1093/hmg/ddz230
Lin, H., Magrane, J., Clark, E. M., Halawani, S. M., Warren, N., Rattelle, A., et al. (2017). Early VGLUT1-specific parallel fiber synaptic deficits and dysregulated cerebellar circuit in the KIKO mouse model of Friedreich ataxia. Dis. Model. Mech. 10, 1529–1538. doi: 10.1242/dmm.030049
Lin, X., Miller, J. W., Mankodi, A., Kanadia, R. N., Yuan, Y., Moxley, R. T., et al. (2006). Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 15, 2087–2097. doi: 10.1093/hmg/ddl132
Liu, G. H., Qu, J., Suzuki, K., Nivet, E., Li, M., Montserrat, N., et al. (2012). Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 491, 603–607. doi: 10.1038/nature11557
Lopez-Erauskin, J., Tadokoro, T., Baughn, M. W., Myers, B., McAlonis-Downes, M., Chillon-Marinas, C., et al. (2018). ALS/FTD-linked mutation in FUS suppresses intra-axonal protein synthesis and drives disease without nuclear loss-of-function of FUS. Neuron 100, 816.e7–830.e7. doi: 10.1016/j.neuron.2018.09.044
Lopez-Gonzalez, R., Lu, Y., Gendron, T. F., Karydas, A., Tran, H., Yang, D., et al. (2016). Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92, 383–391. doi: 10.1016/j.neuron.2016.09.015
Lu, C. H., Petzold, A., Topping, J., Allen, K., Macdonald-Wallis, C., Clarke, J., et al. (2015). Plasma neurofilament heavy chain levels and disease progression in amyotrophic lateral sclerosis: insights from a longitudinal study. J. Neurol. Neurosurg. Psychiatry 86, 565–573. doi: 10.1136/jnnp-2014-307672
Lynch-Day, M. A., Mao, K., Wang, K., Zhao, M., and Klionsky, D. J. (2012). The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a009357. doi: 10.1101/cshperspect.a009357
Magrane, J., Cortez, C., Gan, W. B., and Manfredi, G. (2014). Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 23, 1413–1424. doi: 10.1093/hmg/ddt528
Malenka, R. C., and Bear, M. F. (2004). LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. doi: 10.1016/j.neuron.2004.09.012
Malenka, R. C., and Nicoll, R. A. (1999). Long-term potentiation–a decade of progress? Science 285, 1870–1874. doi: 10.1126/science.285.5435.1870
Manczak, M., Sheiko, T., Craigen, W. J., and Reddy, P. H. (2013). Reduced VDAC1 protects against Alzheimer’s disease, mitochondria and synaptic deficiencies. J Alzheimers Dis. 37, 679–690. doi: 10.3233/JAD-130761
Matsuo, E. S., Shin, R. W., Billingsley, M. L., Van deVoorde, A., O’Connor, M., Trojanowski, J. Q., et al. (1994). Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 13, 989–1002. doi: 10.1016/0896-6273(94)90264-x
Mattson, M. P. (2007). Mitochondrial regulation of neuronal plasticity. Neurochem. Res. 32, 707–715. doi: 10.1007/s11064-006-9170-3
Mattson, M. P., Gleichmann, M., and Cheng, A. (2008). Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766. doi: 10.1016/j.neuron.2008.10.010
Mattson, M. P., and Liu, D. (2003). Mitochondrial potassium channels and uncoupling proteins in synaptic plasticity and neuronal cell death. Biochem. Biophys. Res. Commun. 304, 539–549. doi: 10.1016/s0006-291x(03)00627-2
Mattson, M. P., Lovell, M. A., Furukawa, K., and Markesbery, W. R. (1995). Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J. Neurochem. 65, 1740–1751. doi: 10.1046/j.1471-4159.1995.65041740.x
Maurel, C., Dangoumau, A., Marouillat, S., Brulard, C., Chami, A., Hergesheimer, R., et al. (2018). Causative genes in amyotrophic lateral sclerosis and protein degradation pathways: a link to neurodegeneration. Mol. Neurobiol. 55, 6480–6499. doi: 10.1007/s12035-017-0856-0
McMurray, C. T. (2000). Neurodegeneration: diseases of the cytoskeleton? Cell Death Differ. 7, 861–865. doi: 10.1038/sj.cdd.4400764
McNeill, T. H., Brown, S. A., Rafols, J. A., and Shoulson, I. (1988). Atrophy of medium spiny I striatal dendrites in advanced Parkinson’s disease. Brain Res. 455, 148–152. doi: 10.1016/0006-8993(88)90124-2
Mehta, A. R., Gregory, J. M., Dando, O., Carter, R. N., Burr, K., Nanda, J., et al. (2021). Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol. 141, 257–279. doi: 10.1007/s00401-020-02252-5
Meola, G., and Cardani, R. (2015). Myotonic dystrophies: an update on clinical aspects, genetic, pathology and molecular pathomechanisms. Biochim. Biophys. Acta 1852, 594–606. doi: 10.1016/j.bbadis.2014.05.019
Miller, J. W., Urbinati, C. R., Teng-Umnuay, P., Stenberg, M. G., Byrne, B. J., Thornton, C. A., et al. (2000). Recruitment of human muscleblind proteins to (CUG)(n). expansions associated with myotonic dystrophy. EMBO J. 19, 4439–4448. doi: 10.1093/emboj/19.17.4439
Mizielinska, S., Gronke, S., Niccoli, T., Ridler, C. E., Clayton, E. L., Devoy, A., et al. (2014). C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192–1194. doi: 10.1126/science.1256800
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Moriyama, K., Iida, K., and Yahara, I. (1996). Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells 1, 73–86. doi: 10.1046/j.1365-2443.1996.05005.x
Morris, R. L., and Hollenbeck, P. J. (1993). The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J. Cell Sci. 104, 917–927.
Mucke, L., Masliah, E., Yu, G. Q., Mallory, M., Rockenstein, E. M., Tatsuno, G., et al. (2000). High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000
Neukirchen, D., and Bradke, F. (2011). Neuronal polarization and the cytoskeleton. Semin. Cell Dev. Biol. 22, 825–833. doi: 10.1016/j.semcdb.2011.08.007
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nguyen, L., Cleary, J. D., and Ranum, L. P. W. (2019). Repeat-associated non-ATG translation: molecular mechanisms and contribution to neurological disease. Annu. Rev. Neurosci. 42, 227–247. doi: 10.1016/j.semcdb.2011.08.007
Oddo, S. (2008). The ubiquitin-proteasome system in Alzheimer’s disease. J. Cell Mol. Med. 12, 363–373. doi: 10.1111/j.1582-4934.2008.00276.x
Petel Legare, V., Harji, Z. A., Rampal, C. J., Allard-Chamard, X., Rodriguez, E. C., Armstrong, G. A. B. (2019). Augmentation of spinal cord glutamatergic synaptic currents in zebrafish primary motoneurons expressing mutant human TARDBP (TDP-43). Sci. Rep. 9:9122. doi: 10.1038/s41598-019-45530-3
Philips, A. V., Timchenko, L. T., and Cooper, T. A. (1998). Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 280, 737–741. doi: 10.1126/science.280.5364.737
Philips, T., and Robberecht, W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 10, 253–263. doi: 10.1016/S1474-4422(11)70015-1
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Pollard, T. D. (2003). The cytoskeleton, cellular motility and the reductionist agenda. Nature 422, 741–745. doi: 10.1038/nature01598
Popov, V., Medvedev, N. I., Davies, H. A., and Stewart, M. G. (2005). Mitochondria form a filamentous reticular network in hippocampal dendrites but are present as discrete bodies in axons: a three-dimensional ultrastructural study. J. Comp. Neurol. 492, 50–65. doi: 10.1002/cne.20682
Radwan, M., Ang, C. S., Ormsby, A. R., Cox, D., Daly, J. C., Reid, G. E., et al. (2020). Arginine in C9ORF72 dipolypeptides mediates promiscuous proteome binding and multiple modes of toxicity. Mol. Cell. Proteomics 19, 640–654. doi: 10.1074/mcp.RA119.001888
Ratnam, J., and Teichberg, V. I. (2005). Neurofilament-light increases the cell surface expression of the N-methyl-D-aspartate receptor and prevents its ubiquitination. J. Neurochem. 92, 878–885. doi: 10.1111/j.1471-4159.2004.02936.x
Reinhardt, P., Schmid, B., Burbulla, L. F., Schondorf, D. C., Wagner, L., Glatza, M., et al. (2013). Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12, 354–367. doi: 10.1016/j.stem.2013.01.008
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Ribchester, R. R., Thomson, D., Wood, N. I., Hinks, T., Gillingwater, T. H., Wishart, T. M., et al. (2004). Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington’s disease mutation. Eur. J. Neurosci. 20, 3092–3114. doi: 10.1111/j.1460-9568.2004.03783.x
Roberson, E. D., English, J. D., Adams, J. P., Selcher, J. C., Kondratick, C., and Sweatt, J. D. (1999). The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J. Neurosci. 19, 4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999
Rockenstein, E., Nuber, S., Overk, C. R., Ubhi, K., Mante, M., Patrick, C., et al. (2014). Accumulation of oligomer-prone alpha-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 137, 1496–1513. doi: 10.1093/brain/awu057
Rodriguez, C. M., and Todd, P. K. (2019). New pathologic mechanisms in nucleotide repeat expansion disorders. Neurobiol. Dis. 130:104515. doi: 10.1016/j.nbd.2019.104515
Saberi, S., Stauffer, J. E., Schulte, D. J., and Ravits, J. (2015). Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol. Clin. 33, 855–876. doi: 10.1016/j.ncl.2015.07.012
Sareen, D., O’Rourke, J. G., Meera, P., Muhammad, A. K., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5:208ra149. doi: 10.1126/scitranslmed.3007529
Scheff, S. W., DeKosky, S. T., and Price, D. A. (1990). Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol. Aging 11, 29–37. doi: 10.1016/0197-4580(90)90059-9
Scheff, S. W., and Price, D. A. (1993). Synapse loss in the temporal lobe in Alzheimer’s disease. Ann. Neurol. 33, 190–199. doi: 10.1002/ana.410330209
Scheff, S. W., and Price, D. A. (2003). Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiol. Aging 24, 1029–1046. doi: 10.1016/j.neurobiolaging.2003.08.002
Scheibel, A. B. (1979a). Dendritic changes in senile and presenile dementias. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 57, 107–124.
Scheibel, A. B. (1979b). The hippocampus: organizational patterns in health and senescence. Mech. Ageing Dev. 9, 89–102. doi: 10.1016/0047-6374(79)90123-4
Schuchmann, S., Buchheim, K., Heinemann, U., Hosten, N., and Buttgereit, F. (2005). Oxygen consumption and mitochondrial membrane potential indicate developmental adaptation in energy metabolism of rat cortical neurons. Eur. J. Neurosci. 21, 2721–2732. doi: 10.1111/j.1460-9568.2005.04109.x
Schwartz, M. L., Bruce, J., Shneidman, P. S., and Schlaepfer, W. W. (1995). Deletion of 3′-untranslated region alters the level of mRNA expression of a neurofilament light subunit transgene. J. Biol. Chem. 270, 26364–26369. doi: 10.1074/jbc.270.44.26364
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Sellier, C., Campanari, M. L., Julie Corbier, C., Gaucherot, A., Kolb-Cheynel, I., Oulad-Abdelghani, M., et al. (2016). Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–1297. doi: 10.15252/embj.201593350
Selvaraj, B. T., Livesey, M. R., Zhao, C., Gregory, J. M., James, O. T., Cleary, E. M., et al. (2018). C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca(2+)-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 9:347. doi: 10.1038/s41467-017-02729-0
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W. H., Hung, S. T., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325. doi: 10.1038/nm.4490
Shors, T. J., Chua, C., and Falduto, J. (2001). Sex differences and opposite effects of stress on dendritic spine density in the male versus female hippocampus. J. Neurosci. 21, 6292–6297. doi: 10.1523/JNEUROSCI.21-16-06292.2001
Simpson, P. B. (2000). The local control of cytosolic Ca2+ as a propagator of CNS communication—integration of mitochondrial transport mechanisms and cellular responses. J. Bioenerg. Biomembr. 32, 5–13. doi: 10.1023/a:1005552126516
Sivadasan, R., Hornburg, D., Drepper, C., Frank, N., Jablonka, S., Hansel, A., et al. (2016). C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat. Neurosci. 19, 1610–1618. doi: 10.1038/nn.4407
Sleigh, J. N., Rossor, A. M., Fellows, A. D., Tosolini, A. P., and Schiavo, G. (2019). Axonal transport and neurological disease. Nat. Rev. Neurol. 15, 691–703. doi: 10.1038/s41582-019-0257-2
Spacek, J., and Harris, K. M. (1997). Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J. Neurosci. 17, 190–203. doi: 10.1523/JNEUROSCI.17-01-00190.1997
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Stanton, P. K., and Schanne, F. A. (1986). Hippocampal long-term potentiation increases mitochondrial calcium pump activity in rat. Brain Res. 382, 185–188. doi: 10.1016/0006-8993(86)90130-7
Stefani, G., Onofri, F., Valtorta, F., Vaccaro, P., Greengard, P., and Benfenati, F. (1997). Kinetic analysis of the phosphorylation-dependent interactions of synapsin I with rat brain synaptic vesicles. J Physiol. 504, 501–515. doi: 10.1111/j.1469-7793.1997.501bd.x
Su, B., Ji, Y. S., Sun, X. L., Liu, X. H., and Chen, Z. Y. (2014). Brain-derived neurotrophic factor (BDNF)-induced mitochondrial motility arrest and presynaptic docking contribute to BDNF-enhanced synaptic transmission. J. Biol. Chem. 289, 1213–1226. doi: 10.1074/jbc.M113.526129
Sullivan, P. M., Zhou, X., Robins, A. M., Paushter, D. H., Kim, D., Smolka, M. B., et al. (2016). The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 4:51. doi: 10.1186/s40478-016-0324-5
Swerdlow, R. H. (2018). Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimers Dis. 62, 1403–1416. doi: 10.3233/JAD-170585
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. doi: 10.1002/ana.410300410
Terry-Lorenzo, R. T., Inoue, M., Connor, J. H., Haystead, T. A., Armbruster, B. N., Gupta, R. P., et al. (2000). Neurofilament-L is a protein phosphatase-1-binding protein associated with neuronal plasma membrane and post-synaptic density. J. Biol. Chem. 275, 2439–2446. doi: 10.1074/jbc.275.4.2439
Tubert, C., and Murer, M. G. (2020). What’s wrong with the striatal cholinergic interneurons in Parkinson’s disease? Focus on intrinsic excitability. Eur. J. Neurosci. 53, 2100–2116. doi: 10.1111/ejn.14742
Valenti, D., de Bari, L., De Filippis, B., Henrion-Caude, A., and Vacca, R. A. (2014). Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: an overview of down syndrome, autism, fragile X and rett syndrome. Neurosci. Biobehav. Rev. 46, 202–217. doi: 10.1016/j.neubiorev.2014.01.012
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K., Nishimura, A., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Vucic, S., Rothstein, J. D., and Kiernan, M. C. (2014). Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci. 37, 433–442. doi: 10.1016/j.tins.2014.05.006
Wang, W., Li, L., Lin, W. L., Dickson, D. W., Petrucelli, L., Zhang, T., et al. (2013). The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 22, 4706–4719. doi: 10.1093/hmg/ddt319
Wang, J., Wang, F., Mai, D., and Qu, S. (2020a). Molecular mechanisms of glutamate toxicity in Parkinson’s disease. Front. Neurosci. 14:585584. doi: 10.3389/fnins.2020.585584
Wang, W., Zhao, F., Ma, X., Perry, G., and Zhu, X. (2020b). Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener. 15:30. doi: 10.1186/s13024-020-00376-6
Westergard, T., McAvoy, K., Russell, K., Wen, X., Pang, Y., Morris, B., et al. (2019). Repeat-associated non-AUG translation in C9orf72-ALS/FTD is driven by neuronal excitation and stress. EMBO Mol. Med. 11:e9423. doi: 10.15252/emmm.201809423
Wieraszko, A. (1982). Changes in the hippocampal slices energy metabolism following stimulation and long-term potentiation of Schaffer collaterals-pyramidal cell synapses tested with the 2-deoxyglucose technique. Brain Res. 237, 449–457. doi: 10.1016/0006-8993(82)90456-5
Williamson, T. L., and Cleveland, D. W. (1999). Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat. Neurosci. 2, 50–56. doi: 10.1038/4553
Williams, J. M., Thompson, V. L., Mason-Parker, S. E., Abraham, W. C., and Tate, W. P. (1998). Synaptic activity-dependent modulation of mitochondrial gene expression in the rat hippocampus. Mol. Brain Res. 60, 50–56. doi: 10.1016/s0169-328x(98)00165-x
Wishart, T. M., Parson, S. H., and Gillingwater, T. H. (2006). Synaptic vulnerability in neurodegenerative disease. J. Neuropathol. Exp. Neurol. 65, 733–739. doi: 10.1097/01.jnen.0000228202.35163.c4
Woollacott, I. O., and Mead, S. (2014). The C9ORF72 expansion mutation: gene structure, phenotypic and diagnostic issues. Acta Neuropathol. 127, 319–332. doi: 10.1007/s00401-014-1253-7
Xiao, S., McKeever, P. M., Lau, A., and Robertson, J. (2019). Synaptic localization of C9orf72 regulates post-synaptic glutamate receptor 1 levels. Acta Neuropathol. Commun. 7:161. doi: 10.1186/s40478-019-0812-5
Xu, W., and Xu, J. (2018). C9orf72 dipeptide repeats cause selective neurodegeneration and cell-autonomous excitotoxicity in Drosophila glutamatergic neurons. J. Neurosci. 38, 7741–7752. doi: 10.1523/JNEUROSCI.0908-18.2018
Yang, M., Liang, C., Swaminathan, K., Herrlinger, S., Lai, F., Shiekhattar, R., et al. (2016). A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv. 2:e1601167. doi: 10.1126/sciadv.1601167
Yuan, A., Rao, M. V., Kumar, A., Julien, J. P., and Nixon, R. A. (2003). Neurofilament transport in vivo minimally requires hetero-oligomer formation. J. Neurosci. 23, 9452–9458. doi: 10.1523/JNEUROSCI.23-28-09452.2003
Yuan, A., Sershen, H., Veeranna, Basavarajappa, B. S., Kumar, A., Hashim, A., et al. (2015a). Functions of neurofilaments in synapses. Mol. Psychiatry 20:915. doi: 10.1038/mp.2015.99
Yuan, A., Sershen, H., Veeranna, Basavarajappa, B. S., Kumar, A., Hashim, A., et al. (2015b). Neurofilament subunits are integral components of synapses and modulate neurotransmission and behavior in vivo. Mol. Psychiatry 20, 986–994. doi: 10.1038/mp.2015.45
Zala, D., Hinckelmann, M. V., Yu, H., Lyra da Cunha, M. M., Liot, G., Cordelieres, F. P., et al. (2013). Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 152, 479–491. doi: 10.1016/j.cell.2012.12.029
Zhang, K., Donnelly, C. J., Haeusler, A. R., Grima, J. C., Machamer, J. B., Steinwald, P., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. doi: 10.1038/nature14973
Zhang, B., Tu, P., Abtahian, F., Trojanowski, J. Q., and Lee, V. M. (1997). Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J. Cell Biol. 139, 1307–1315. doi: 10.1083/jcb.139.5.1307
Zu, T., Cleary, J. D., Liu, Y., Banez-Coronel, M., Bubenik, J. L., Ayhan, F., et al. (2017). RAN translation regulated by muscleblind proteins in myotonic dystrophy type 2. Neuron 95, 1292.e5–1305.e5. doi: 10.1016/j.neuron.2017.08.039
Zu, T., Liu, Y., Banez-Coronel, M., Reid, T., Pletnikova, O., Lewis, J., et al. (2013). RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. U S A 110, E4968–E4977. doi: 10.1073/pnas.1315438110
Keywords: synaptic transmission, dendrites, dendritic spines, synaptopathies, neurodegeneration, amyotrophic lateral sclerosis
Citation: Nishimura AL and Arias N (2021) Synaptopathy Mechanisms in ALS Caused by C9orf72 Repeat Expansion. Front. Cell. Neurosci. 15:660693. doi: 10.3389/fncel.2021.660693
Received: 29 January 2021; Accepted: 03 May 2021;
Published: 01 June 2021.
Edited by:
Xin Qi, Case Western Reserve University, United StatesReviewed by:
Isabella Zanella, University of Brescia, ItalyJian-Fu Chen, University of Southern California, United States
Copyright © 2021 Nishimura and Arias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agnes L. Nishimura, YWduZXMubmlzaGltdXJhQGtjbC5hYy51aw==; Natalia Arias, bmF0YWxpYS5hcmlhc0BrY2wuYWMudWs=