 Riffat Mehboob
Riffat Mehboob Anna Marchenkova
Anna Marchenkova Arn M. J. M. van den Maagdenberg
Arn M. J. M. van den Maagdenberg Andrea Nistri
Andrea Nistri- 1Department of Neuroscience, International School for Advanced Studies (SISSA), Trieste, Italy
- 2Research Unit, Faculty of Allied Health Sciences, University of Lahore, Lahore, Pakistan
- 3Department of Neurology, Leiden University Medical Center, Leiden, Netherlands
- 4Department of Human Genetics, University Medical Center, Leiden, Netherlands
Trigeminal sensory neurons of transgenic knock-in (KI) mice expressing the R192Q missense mutation in the α1A subunit of neuronal voltage-gated CaV2.1 Ca2+ channels, which leads to familial hemiplegic migraine type 1 (FHM1) in patients, exhibit a hyperexcitability phenotype. Here, we show that the expression of NaV1.7 channels, linked to pain states, is upregulated in KI primary cultures of trigeminal ganglia (TG), as shown by increased expression of its α1 subunit. In the majority of TG neurons, NaV1.7 channels are co-expressed with ATP-gated P2X3 receptors (P2X3R), which are important nociceptive sensors. Reversing the trigeminal phenotype with selective CaV2.1 channel inhibitor ω-agatoxin IVA inhibited NaV1.7 overexpression. Functionally, KI neurons revealed a TTX-sensitive inward current of larger amplitude that was partially inhibited by selective NaV1.7 blocker Tp1a. Under current-clamp condition, Tp1a raised the spike threshold of both wild-type (WT) and KI neurons with decreased firing rate in KI cells. NaV1.7 activator OD1 accelerated firing in WT and KI neurons, a phenomenon blocked by Tp1a. Enhanced expression and function of NaV1.7 channels in KI TG neurons resulted in higher excitability and facilitated nociceptive signaling. Co-expression of NaV1.7 channels and P2X3Rs in TGs may explain how hypersensitivity to local stimuli can be relevant to migraine.
Introduction
Familial hemiplegic migraine type 1 (FHM1) is an autosomal dominant subtype of migraine with aura (IHS, 2013), with similar clinical features (Thomsen et al., 2002)–apart from the hemiparesis–and trigger factors (Hansen et al., 2011) as observed for the common forms of migraine. FHM1 is caused by gain-of-function missense mutations in the pore-forming α1 subunit of neuronal voltage-gated CaV2.1 Ca2+ channels (Ophoff et al., 1996; Tottene et al., 2002, 2009; Pietrobon, 2013). Introduction of the R192Q missense mutation, previously found in patients with hemiplegic migraine (Ophoff et al., 1996), in the orthologous mouse Cacna1a gene resulted in a transgenic FHM1 R192Q knock-in (“R192Q KI”) mouse model (van den Maagdenberg et al., 2004) that expresses key features of migraine pathophysiology and was instrumental in unraveling mechanisms of migraine pathophysiology (Ferrari et al., 2015; Pietrobon and Brennan, 2019). For instance, the mutation causes a gain of function of CaV2.1 channels leading to enhanced neurotransmission and cortical hyperexcitability. In addition, an enhanced susceptibility to experimentally induced cortical spreading depolarization (van den Maagdenberg et al., 2004; Eikermann-Haerter et al., 2009; Tottene et al., 2009) and signs of unilateral head pain were observed (Langford et al., 2010; Chanda et al., 2013).
The R192Q mutation also affects the trigeminovascular system, which provides innervation and supplies blood to the orofacial region, asd therefore, is regarded an important contributor and site for migraine pain transduction (Goadsby et al., 2017). For instance, trigeminal ganglia (TG) of R192Q KI mice show a neuroinflammatory profile (Franceschini et al., 2013; Hullugundi et al., 2013), and their TG neurons show upregulated activity of pain-sensing purinergic P2X3 receptors (P2X3Rs) (Nair et al., 2010; Hullugundi et al., 2013), and neuronal hyperexcitability (Fioretti et al., 2011; Hullugundi et al., 2014; Marchenkova et al., 2016a). The hyperexcitability state of KI TG neurons is shown by a lower firing threshold (Fioretti et al., 2011; Hullugundi et al., 2014; Marchenkova et al., 2016a) suggesting that differences in subthreshold conductances, which can facilitate the voltage trajectory to spike discharge, increase the probability to reach the action potential (AP) threshold (Dib-Hajj et al., 2013). Such function can be attributed to various subtypes of subthreshold voltage-gated NaV Na+ currents (Raman and Bean, 1997; Ogata et al., 2001), some of which are expressed by primary sensory neurons and have been associated with pathological pain conditions (Toledo-Aral et al., 1997; Wood et al., 2004; Dib-Hajj et al., 2010; Habib et al., 2015).
Gain-of-function mutations in α1 subunits of voltage-gated Na+ channels NaV1.7, 1.8 and 1.9 have been linked to pain in humans (Emery et al., 2016; Hoffmann et al., 2018; Shields et al., 2018). Hence there is a rationale to develop selective blockers of NaV1.7 channels as potential drug targets for the treatment of pain-related diseases (Payandeh and Hackos, 2018). Here we focused on the expression and function of NaV1.7 channels in wild-type (WT) and KI TG neurons. Both Na+ channel types are tightly linked to pain pathways and strongly expressed in sensory neurons (Wood et al., 2004; Dib-Hajj et al., 2010; Habib et al., 2015). NaV1.7 expression is upregulated in chronic inflammatory (Gould et al., 2000; Black et al., 2004; Strickland et al., 2008), neuropathic and acute pain (Cai et al., 2016).
Like all NaV1 channels, NaV1.7 consist of a pore-forming α1 subunit and an auxiliary β subunit (Isom et al., 1992; Ogata and Ohishi, 2002). Both Na+ channel types are TTX-sensitive with fast kinetics (Klugbauer et al., 1995; Toledo-Aral et al., 1997). NaV1.7 current contributes to subthreshold modulatory conductance. Because of their slow voltage-dependent inactivation in response to a small, slow depolarization, NaV1.7 channels can generate a substantial ramp current, thereby amplifying and facilitating membrane depolarization to reach the spike threshold (Cummins et al., 1998; Herzog et al., 2003). It was shown that rat dorsal root ganglion (DRG) neurons with a NaV1.7 channel gain-of-function mutation exhibit increased spontaneous and evoked firing (Yang et al., 2016).
We here characterized the expression and function of NaV1.7 channels in WT and R192Q KI TG neurons to explore their contribution to the enhanced neuronal excitability seen in KI TG neurons, which may have relevance to understanding migraine pathophysiology.
Materials and Methods
Mouse Trigeminal Ganglion Cultures
Experiments were performed on TG cultures from homozygous CaV2.1 FHM1 R192Q KI and WT mouse littermates. KI mice were obtained from Leiden University Medical Center (van den Maagdenberg et al., 2004) and bred and maintained locally in accordance with the Italian Animal Welfare Act. Genotyping was routinely performed by PCR, as previously reported (Nair et al., 2010). All experimental protocols were in accordance with the EU guidelines (2010/63/EU) and Italian legislation (D.L. 4/3/2014, no. 26), and were approved by the SISSA ethical committee.
For primary culture preparation, TG from P11–P13 mice of both sexes were dissected and enzymatically dissociated for 12 min at 37°C in a mixture containing 0.25 mg/mL trypsin, 1 mg/mL collagenase, and 0.2 mg/mL DNase (Sigma, Milan, Italy) in F-12 medium (Invitrogen, Milan, Italy). Cells were plated on poly-L-lysine-coated petri dishes in F12 medium with 10% fetal calf serum, and used after a 24-h incubation period in accordance with previously described protocols (Simonetti et al., 2006; Hullugundi et al., 2014). For each cell culture series to be used for each experimental protocol (i.e., testing WT, mutant, untreated, treated groups) we harvested six TGs from three mice to yield several culture dishes. For all experiments, KI and WT cultures were used in parallel, at the same time, to allow for direct comparison of experimental data.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 20 min at room temperature (RT) and washed three times with PBS. To reduce non-specific antibody binding, cells were kept in blocking solution (5% fetal bovine serum, 5% of albumin from bovine serum, 0.1% Triton-X 100 in PBS) for 1 h at RT. Cells were then incubated with primary antibodies against NaV1.7 α1 subunit (mouse anti-SCN9A/PN1, monoclonal, 1:500 in blocking buffer; Acris Antibodies GmbH Cat# AM12054PU-N, RRID:AB_10654661, Herford, Germany) and β-tubulin III (mouse T5076, 1:1,000; Sigma, Milan) was used to specifically mark neurons. Incubation with secondary antibodies (Alexa Fluor 594 goat anti-rabbit, 1:250 and Alexa Fluor 488 goat-anti-mouse 1:250; Invitrogen) was done for 1 h at RT.
For the immunofluorescent staining of NaV1.7-P2X3R co-expression, cells were incubated with both primary NaV1.7 α1 subunit antibody (mouse anti-SCN9A/PN1, monoclonal, 1:500 in blocking buffer; Acris) and P2X3R antibody (guinea pig anti-P2X3R, polyclonal, 1:100 in blocking buffer; Neuromics, Edina, MN, United States). Next, an incubation was performed with secondary antibodies (Alexa Fluor 594 goat-anti-mouse, 1:250 and Alexa Fluor 488 goat anti-guinea pig, 1:250; Invitrogen) for 1 h at RT.
Cell nuclei were counterstained with DAPI (1 μg/mL; Sigma) for 5 min. Coverslips were mounted on a microscope slide with Vectashield Mounting Medium (H-1000; Vector Laboratories, Peterborough, United Kingdom). Images from coverslips were acquired using Zeiss Axioscop fluorescence microscope (Zurich, Switzerland). For cell counting, 10 images of each section/experimental condition (100–600 cells) were taken from at least three experiments. Immunoreactive cells and co-expressions were counted using ImageJ manual cell counter plugin1. For a number of experiments, CaV2.1-specific blocker ω-agatoxin IVA (200 nM; Sigma) was applied overnight (37°C) to cultures (Nair et al., 2010), which were subsequently processed as indicated above.
Western Blotting
Primary TG cell cultures were washed in PBS, scraped and placed in ice-cold lysis buffer (10 mM Tris pH 7.4, 150 mM NaCl, 2 mM EDTA, 2% n-octyl-beta-D-glucopyranoside, 100 mM NaF) containing a cocktail of protease inhibitors (Roche Applied Science, Basel, Switzerland). Cells were disrupted mechanically using a syringe and incubated on ice for 20 min. The lysate was then centrifuged (13,000 rpm) at 4°C for 20 min. The supernatant was transferred to a new Eppendorf tube and the same buffer volume was added. For each experiment a minimum of 3 WT (six ganglia and several culture dishes) and 3 KI (six ganglia and several culture dishes) animals were used. A total of 12 ganglia were used for one experiment. One well had a sample from one TG of one animal. Samples were separated on 8% SDS gel, transferred to a PVDF membrane that was subsequently incubated with anti-NaV1.7 α1 subunit antibody (mouse anti-SCN9A/PN1, monoclonal, 1:1,000; Acris) for 1 h at RT. Mouse anti-rabbit IgG–HRP-conjugated antibody (Jackson ImmunoResearch, Suffolk, United Kingdom) was used as secondary antibody that was incubated for 1 h at RT. Signals were detected with the enhanced chemiluminescence light system ECL (Amersham Biosciences, Piscataway, NJ, United States) and recorded with the digital imaging system Alliance 4.7 (Uvitec, Cambridge, United Kingdom). Quantification of the optical density of the bands was performed with the ImageJ software plug-in. Expression of anti-β-actin antibody (A5441; Sigma) was used as normalization control of gel loading as indicated in the figure legends.
Electrophysiology
Whole-cell patch-clamp experiments were performed on small- to medium-sized TG neurons thought to be nociceptors (capacitance below 25 pF) after 24 h in culture (Nair et al., 2010; Hullugundi et al., 2013) using a Patch Clamp PC-501A amplifier (Warner Instrument Corporation, Hamden, CT, United States). Cells were continuously superfused at a rate of 2–3 mL/min with physiological solution containing (in mM): 152 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES (pH adjusted to 7.4 with NaOH) as previously described (Nair et al., 2010; Hullugundi et al., 2013). Electrophysiological responses were filtered at 2 KHz and acquired by means of a DigiData 1,200 interface and pClamp8.2 software (Molecular Devices, Sunnyvale, CA, United States); series resistance was compensated by 75–90%.
For current-clamp experiments glass pipettes (4–5 MΩ resistance) were filled with the following solution (in mM):125 K-gluconate, 5 KCl, 2 MgCl2, 2 Mg2ATP3, 10 HEPES, and 10 EGTA (pH adjusted to 7.2 with KOH). The liquid junction potential was 14.5 mV, and data were corrected accordingly. Cells were routinely held at −70 mV, apart from the experiments on spontaneous firing when neurons were recorded also at −60 and −50 mV. Depolarizing current pulses (300 ms, 45 pA) were used to elicit cell-specific firing activity from TG neurons, in line with previously used protocols (Hullugundi et al., 2014; Marchenkova et al., 2016a, b). The electrophysiological properties of WT and KI neurons obtained in our lab were published in detail (Hullugundi et al., 2014). It is important to note that neurons can be classified into different categories of spike firing patterns and that this observation is best achieved by using a standard current pulse (300 ms, 45 pA) that elicits a comparable depolarization level. Hence, we used this approach for the experiments.
For voltage-clamp experiments glass pipettes (3–4 MΩ resistance) were filled with a solution containing (in mM): 130 CsCl, 20 HEPES, 1 MgCl2, 3 ATP-Mg, 5 EGTA, pH 7.2 adjusted with CsOH. The liquid junction potential was 5 mV, and data were corrected accordingly. Cells were held at −75 mV (after correction for liquid junction potential). Subthreshold inward currents were evoked using a 100-ms square current pulse of 30 mV amplitude (from −75 to −45 mV). External TEA (Sigma; 10 mM) was used to minimize K+ currents (Elliott and Elliott, 1993; Yoshimura and de Groat, 1996). TTX (1 μM; Sigma) was used to eliminate the TTX-sensitive part of the Na+ inward current (Elliott and Elliott, 1993; Klugbauer et al., 1995; Yoshimura and de Groat, 1996; Tan et al., 2014).
The spider venom toxin ProTx-III [m-TRTX–Tp1a] (Tp1a; Smartox Biotechnology, Saint-Egrève d’Hères, France) (Cardoso et al., 2015) was used to inhibit NaV1.7 channels selectively. Cells were incubated with 7 nM Tp1a for 30 min to ensure strong block of NaV1.7 channels (IC50 2.1 nM) (Cardoso et al., 2015), and to minimize block of NaV1.3 current (IC50 11.5 nM (Cardoso et al., 2015). Data were collected from cells in culture dishes (same culture series), averaged and processed in parallel for comparison. The α-scorpion toxin OD1 (Smartox Biotechnology) was used to activate NaV1.7 channels (EC50 4.5 nM) (Jalali et al., 2005; Maertens et al., 2006; Motin et al., 2016) OD1 (10 nM) was applied either directly to the recorded neuron using a fast superfusion system (Rapid Solution Changer RSC-200; BioLogic Science Instruments, Claix, France) or via the bathing solution. In the latter tests we used a pair design for each cell with comparison of effects before and after drug application.
Data Analysis
Data are shown as mean ± standard error of the mean (SEM), with “n” indicating the number of analyzed cells (electrophysiology) or experiments (molecular biology). Thus, for immunofluorescence and molecular biology data, each experiment involved three WT and three KI mice that yielded six ganglia per genotype from which culture dishes were prepared and used for testing. Statistical relevance of the sample size was verified with the online sample size analysis software biomath.info2 and cG93ZXJhbmRzYW1wbGVzaXplLmNvbQ==3 (power = 0.8, type I error rate = 0.05). For statistical analysis the Student’s t-test or the Mann–Whitney rank sum test was used after the software-directed choice of parametric or non-parametric data, respectively (Matlab4 ; Sigma Plot and Sigma Stat, version 14, Chicago, IL, United States5); chi-square test was used to compare proportions. A p-value less than 0.05 was accepted as indicative of a statistically significant difference.
When analyzing firing threshold, an algorithm was used that allowed automated threshold detection based on first and second discrete time derivatives of the voltage time-series (Hullugundi et al., 2014; Marchenkova et al., 2016a). The parameters for threshold calculation were determined empirically and kept constant for all analyzed recordings.
Results
Expression of NaV1.7 α1 Subunit in TG Primary Cultures
Figure 1A shows immunofluorescent signals of NaV1.7 α1 subunits. Systematic cell counting indicated that the majority of β-tubulin III-positive cells in WT and KI cultures also expressed NaV1.7 channels (76 ± 0.1%, n = 7 WT cultures, and 84 ± 0.0%, n = 7 KI cultures; p = 0.016, Mann–Whitney test; see also Figure 1B). Western blot analysis of NaV1.7 α1 protein showed that the antibody recognized a single band of the expected size (226 kDa) in mouse TG homogenates (Figure 1B). There was, however, a significant difference in the amount of NaV1.7 protein expressed by WT and KI culture homogenates (Figure 1B; p = 0.048, Mann–Whitney test; n = 8 experiments). Our data shows that the highest percent of neurons expressing NaV1.7 were in the 13- to 15- and 16- to 18-μm-diameter groups with similar distribution for WT and KI neurons (Figure 1C).
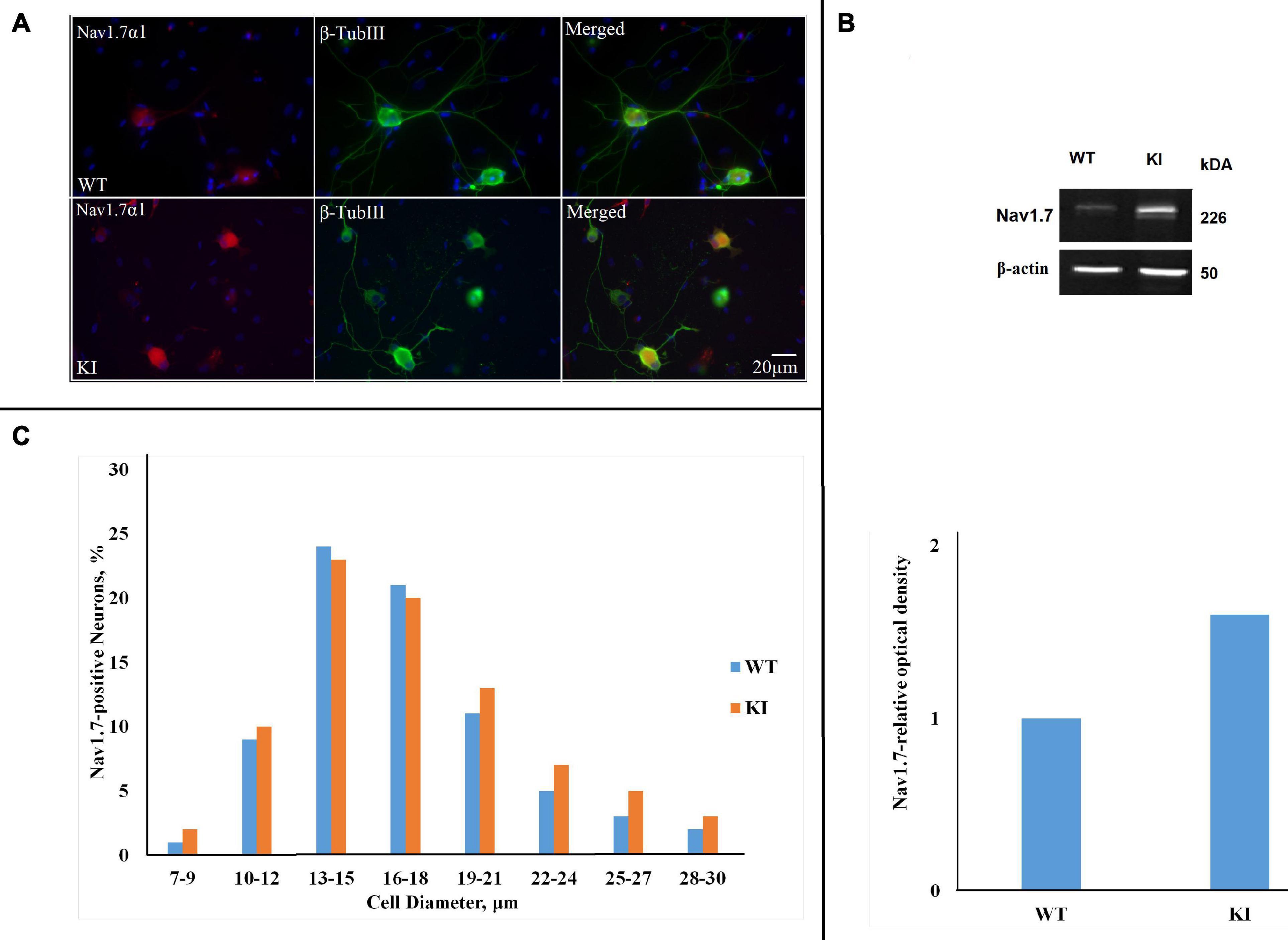
Figure 1. NaV1.7 α1 subunit expression in wild-type (WT) and R192Q KI TG cultures. (A) Representative immunofluorescent examples of NaV1.7 α1 expression in WT and R192Q KI mouse trigeminal ganglia (TG) cultures. Nuclei are visualized with DAPI (blue); scale bar = 20 μm. Note extensive co-staining of NaV1.7 α1 (red) and β-tubulin III (green) protein in both WT and KI samples. (B) Western blot example showing protein expression of NaV1.7 α1 in TG from P12 mice. β-actin was used as a loading control. Histograms representing NaV1.7 α1 relative optical density values for each group (*p = 0.46, Mann-Whitney test; n = 8 experiments). Note the significant difference between WT and KI groups. (C) The histograms quantify the cell diameter distribution of NaV1.7 α1 immunofluorescence in different subgroups of WT and KI cultures (n = 3 experiments). The percentages of positive neurons for NaV1.7 were calculated by considering the total number of neurons labeled by β-Tubulin III as standard.
Previous studies have demonstrated a lower CaV2.1 activation threshold and increased CaV2.1 current in several types of KI neurons (van den Maagdenberg et al., 2004; Tottene et al., 2009; Fioretti et al., 2011), and in our TG neuron preparations upregulated P2X3R function (Nair et al., 2010) and a neuroinflammatory ganglion profile (Franceschini et al., 2013). This KI TG phenotype could be reversed by pretreatment with ω-agatoxin IV (200 nM), a selective blocker of CaV2.1 channels (Nair et al., 2010; Marchenkova et al., 2016b). Hence, we adopted the same approach to investigate whether CaV2.1 inhibition could reverse overexpression of NaV1.7 channels in KI TG cultures. After overnight treatment with the blocker, NaV1.7 expression remained unchanged in WT cells (75 ± 0.8%; p = 0.732, Mann–Whitney test; n = 3 experiments), whereas it was down to 59 ± 0.8% in KI cultures (p = 0.003; n = 3 experiments) (Figure 2).
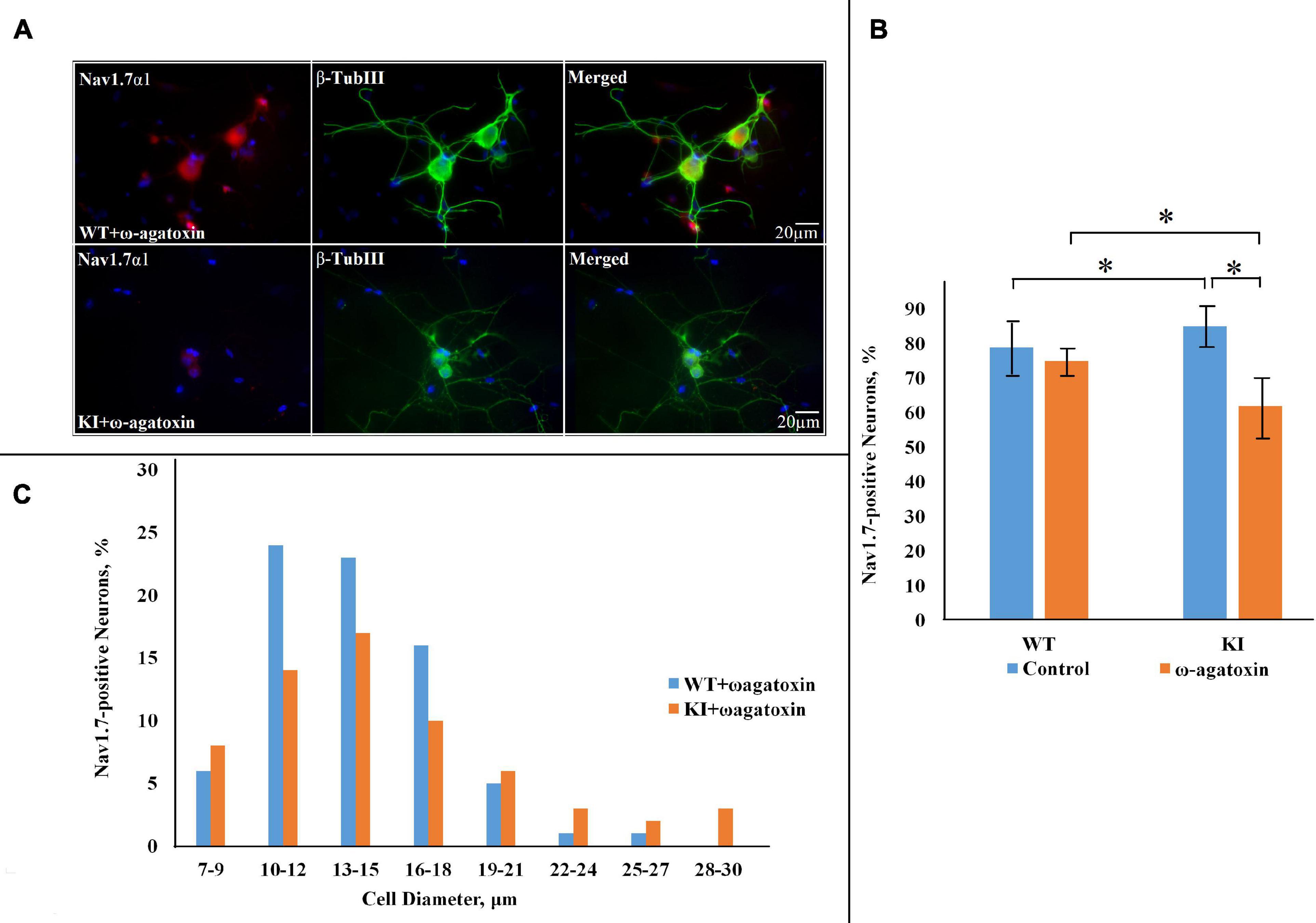
Figure 2. NaV1.7 α1 subunit expression in WT and R192Q KI TG cultures after pretreatment with ω-agatoxin IVA. (A) Representative immunofluorescent examples of NaV1.7 α1 expression in WT and R192Q KI mouse TG cultures. ω-agatoxin IVA (200 nM, overnight: in this and subsequent Figures the toxin is abbreviated as ω-Agatoxin) was used to specifically block CaV2.1 channels (that are mutated in the KI model). Nuclei are visualized with DAPI (blue); scale bar = 20 μm. Note extensive co-staining of NaV1.7 α1 (red) and β-tubulin III (green) protein in both WT and KI samples. (B) Histograms represent the percentage of NaV1.7-positive cells in WT and KI cultures. Note significant difference between WT and KI control groups (*p = 0.015, Mann–Whitney test; n = 7 experiments), and the decrease in NaV1.7 positive neurons in the KI after application of ω-agatoxin IVA (p = 0.035; n = 3 experiments). (C) The histograms quantify the cell diameter distribution of NaV1.7 α1 immunofluorescence in different subgroups in WT and KI ω-agatoxin IVA-treated cultures.
Since P2X3R-expressing neurons are important contributors to trigeminal pain (Wirkner et al., 2005; Burnstock, 2006), we also investigated whether P2X3R immunoreactivity was co-expressed with NaV1.7 α1 immunopositivity. Figure 3A shows examples of neurons in which both proteins were co-expressed: it is noteworthy that NaV1.7 α1 immunoreactivity was mainly observed in soma, whereas P2X3R expression was more diffuse in neurons (see Figure 3A for merged images). Histograms show that the majority of NaV1.7 α1-positive neurons were also positive for P2X3R in both WT and KI cultures (Figure 3B; p = 0.95, Mann–Whitney test; n = 3 experiments), and the neuronal diameter of the double-positive neurons is in the same range as that of individually stained neurons (Figure 3C).
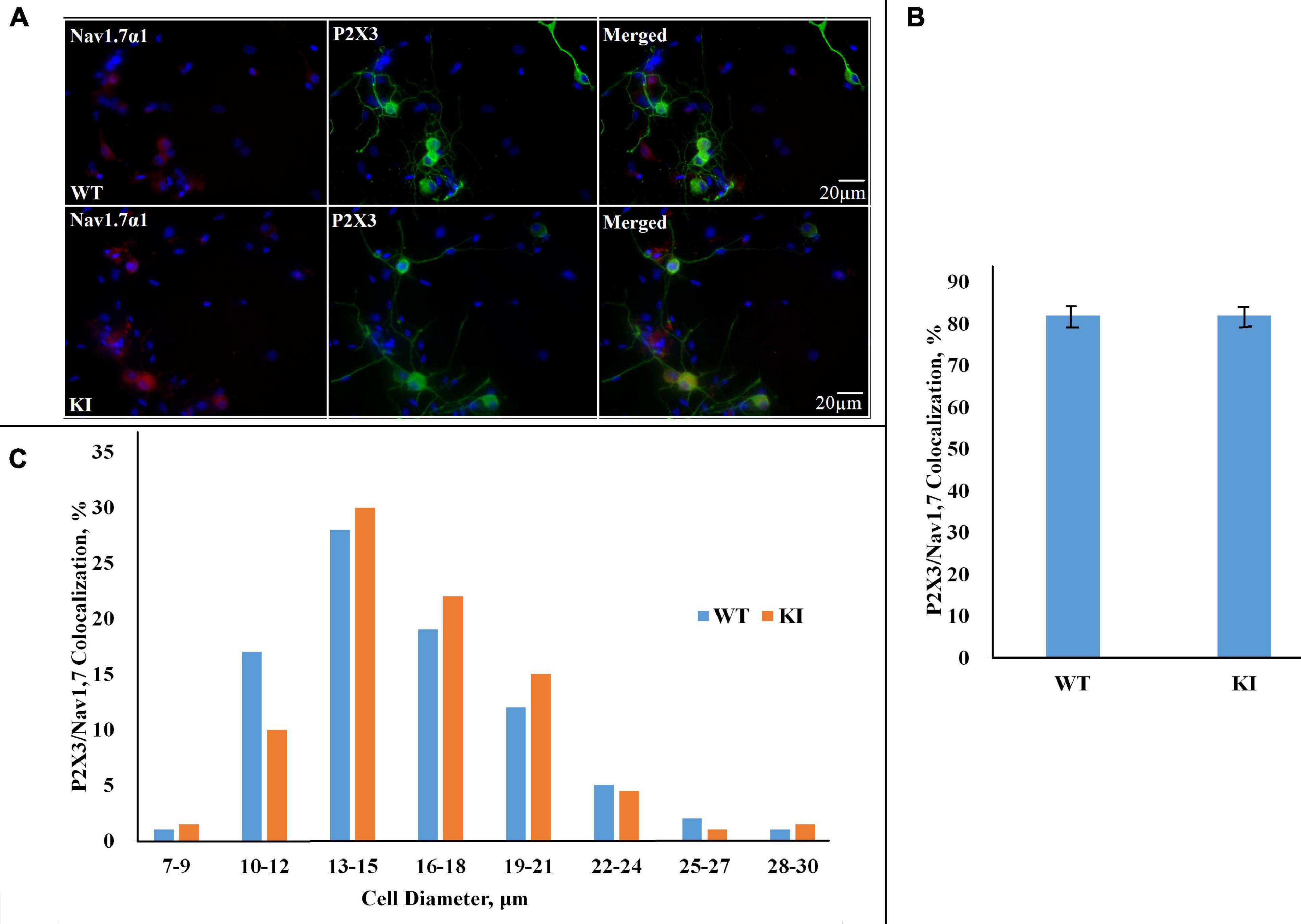
Figure 3. Co-expression of NaV1.7 α1 subunit and P2X3R in WT and R192Q KI cultures. (A) Representative examples of NaV1.7 α1–P2X3 immunostaining in WT and R192Q KI mouse TG cultures. Nuclei are visualized with DAPI (blue); scale bar = 20 μm. Note the extensive co-staining of NaV1.7 α1 (red) and P2X3 (green) protein in both WT and KI samples. (B) Histograms represent percentage co-expression of NaV1.7 α1–P2X3 proteins in WT and KI cultures (p = 0.95, Mann–Whitney test; n = 3 experiments). (C) The histograms quantify the cell diameter distribution of NaV1.7 α1–P2X3R immunofluorescence in different subgroups in WT and KI cultures.
Observations that a gain-of-function mutation of NaV1.7 channels led to increased neuronal excitability in rat DRGs (Yang et al., 2016) prompted us to investigate whether the observed increase in NaV1.7 channel expression in KI TG cultures contributes to the development of the hyperexcitability phenotype typical for a subpopulation of KI TG neurons (Fioretti et al., 2011; Hullugundi et al., 2014; Marchenkova et al., 2016b).
NaV1.7 Current in TG Neurons
To study subthreshold NaV1.7 currents, voltage-clamp experiments were performed with Cs+-filled pipettes and extracellular TEA to minimize outward K+ currents. A step-change in voltage from holding potential of −75 to −45 mV elicited a transient inward current whose peak was significantly larger in KI compared to WT neurons (Figure 4A, control; p = 0.039, two-sample Student’s t-test). Application of 1 μM TTX to each neuron significantly reduced WT and KI currents (p = 0.2 × 10–4 and 0.6 × 10–5, respectively, two-sample Student’s t-test) (Figure 4A). Residual TTX-resistant current was very similar in WT and KI neurons (p = 0.97, two-sample Student’s t-test) (Figure 4A). Figure 4B shows an example of control and residual currents recorded from WT TG neurons (similar traces were obtained for KI TG neurons). By subtracting the TTX-resistant current from the total control current, the TTX-sensitive current was obtained that was clearly larger in KI neurons (Figure 4C). On a different batch of cultures we observed that pretreatment of TG neurons with selective NaV1.7 blocker toxin Tp1a (7 nM, 30 min) (Cardoso et al., 2015) reduced the amplitude of the total inward current in both WT and KI neurons (p = 0.026 and 0.028, respectively, two-sample Student’s t-test) to the same level (p = 0.61, two-sample Student’s t-test), as shown in Figure 4D. Subsequent subtraction of Tp1a-resistant current from control current resulted in a Tp1a-sensitive current that was larger in KI neurons (Figure 4E). The total inward current as well as the TTX-sensitive current declined during sustained voltage command to baseline with an analogous time-constant of 19.3 ± 1.2 ms for WT (n = 14) and 19.0 ± 1.5 ms for KI (n = 11) neurons (p = 0.86, two-sample Student’s t-test). The similar values between genotypes suggest that there was no systematic change in kinetic properties, but rather a dissimilar expression as indicated by the histochemical observations.
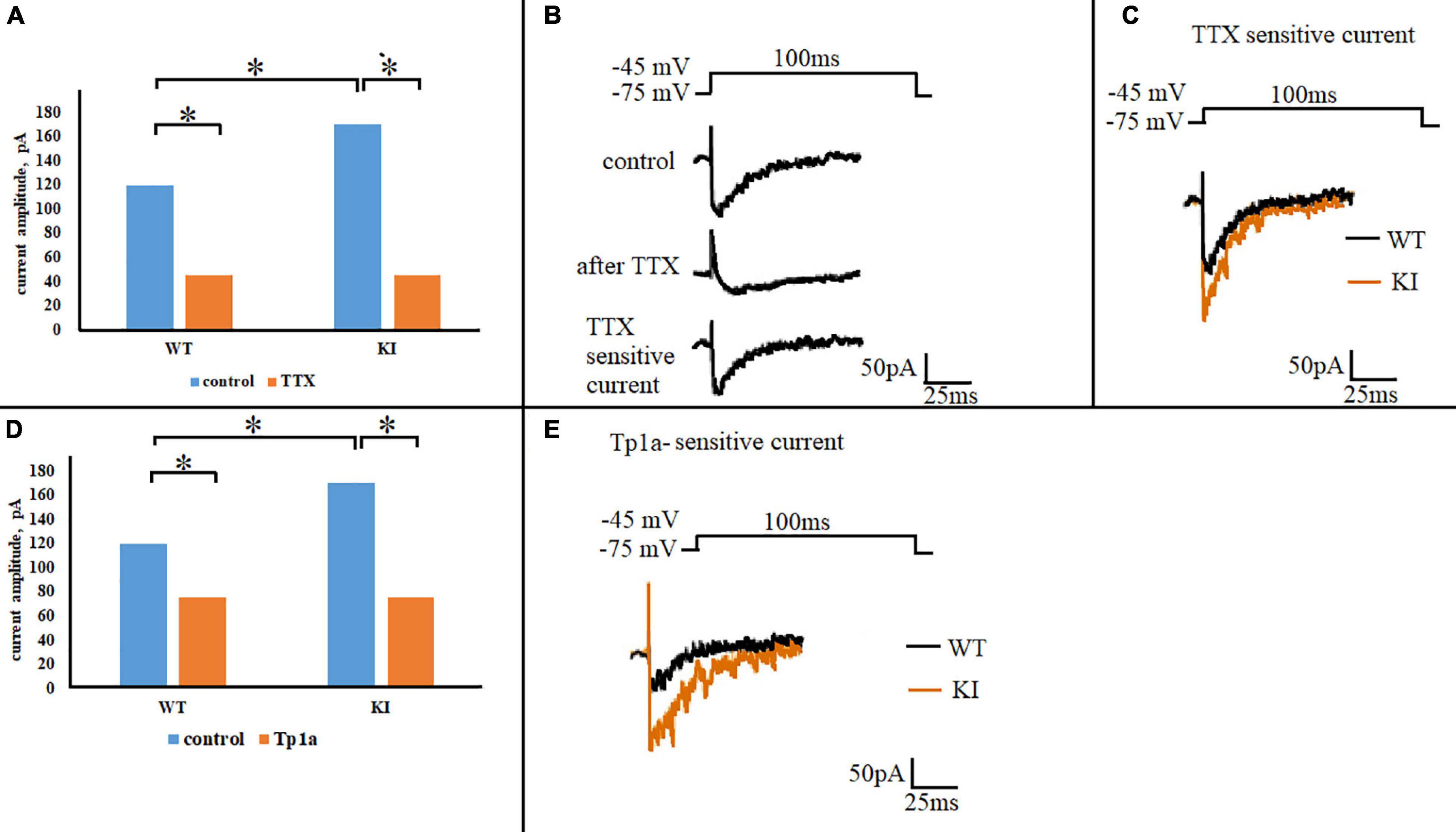
Figure 4. NaV1.7 current in WT and R192Q KI TG neurons. (A) Mean amplitudes of the currents evoked by a square pulse 100 ms depolarizing step from –75 to –45 mV, in WT and R192Q KI TG neurons under control conditions and after application of 1 μM TTX. Note larger control current in KI (*p = 0.039, two-sample Student’s t-test). Number of cells in each group: n = 29 (WT, control), n = 20 (WT, TTX), n = 43 (KI, control), i = 24 (KI, TTX). (B) Representative traces of inward currents recorded from one WT neuron in response to the same stimulation in control and after 1 μM TTX; the trace of TTX-sensitive current was obtained by subtraction of the TTX-resistant current from the control. (C) Superimposed representative traces of WT and KI TTX-sensitive currents obtained by subtraction; note larger KI current. (D) Histograms represent the calculated mean amplitudes of the currents evoked by the same stimulus (100-ms step from –75 to –45 mV) in control and after Tp1a (7 nM, 30 min). *Indicates statistically significant change (two-sample Student’s t-test, p-values are given in the text body). Number of cells in each group: n = 29 (WT, control), n = 28 (WT, Tp1a), n = 43 (KI, control), n = 38 (KI, Tp1a). (E) Superimposed representative traces of WT and KI Tp1a-sensitive (NaV1.7) currents obtained by subtraction of the TTX-resistant current from the control; note larger NaV1.7 current in KI.
Blocking NaV1.7 Channels Reduces Evoked Firing of TG Neurons
One objective of the study was to investigate whether manipulations aimed at changing the NaV1.7 conductance could differentially affect the excitability of WT or KI neurons. Hence, we first compared control firing of these two groups of cells by employing the same firing protocol to standardize the spike-generating characteristics of the heterogeneous class of TG neurons. To this end, current-clamp experiments were performed and compared for responses of WT and KI neurons to a depolarizing square current pulse (45 pA, 300 ms) in control solution and after 30 min of 7 nM Tp1a application. This current pulse elicited cell-specific responses from WT and KI neurons, including not-spiking (NS), single-spike (SS), and multiple-firing (MF) cells (see examples in Figure 5A), similar to those previously reported (Hullugundi et al., 2014; Marchenkova et al., 2016a). Blocking NaV1.7 channels with Tp1a did not significantly affect the distribution of firing patterns in WT and KI cultures (chi-square test for proportions) (Figure 5B). However, significant differences in the number of elicited spikes and firing threshold were detected after Tp1a application (Figures 5C–E). In fact, in KI neurons Tp1a decreased spike numbers/pulse to the level of WT (p = 0.008, Mann–Whitney test) (Figure 5C), and, likewise, shifted the threshold value for AP generation (p = 5.9 × 10–9, two-sample Student’s t-test) (Figures 5D,E). Where KI TG neurons have a more negative AP threshold (Figures 5D,E) (see also Marchenkova et al., 2016a), Tp1a application brought WT and KI threshold values to a less negative (i.e., depolarized) level. The effect of Tp1a on WT neurons was similar, although less pronounced with a decreased number of generated spikes (p = 0.055, Mann–Whitney test) (Figure 5C) and depolarized firing threshold (p = 0.019, two-sample Student’s t-test) (Figure 5D).
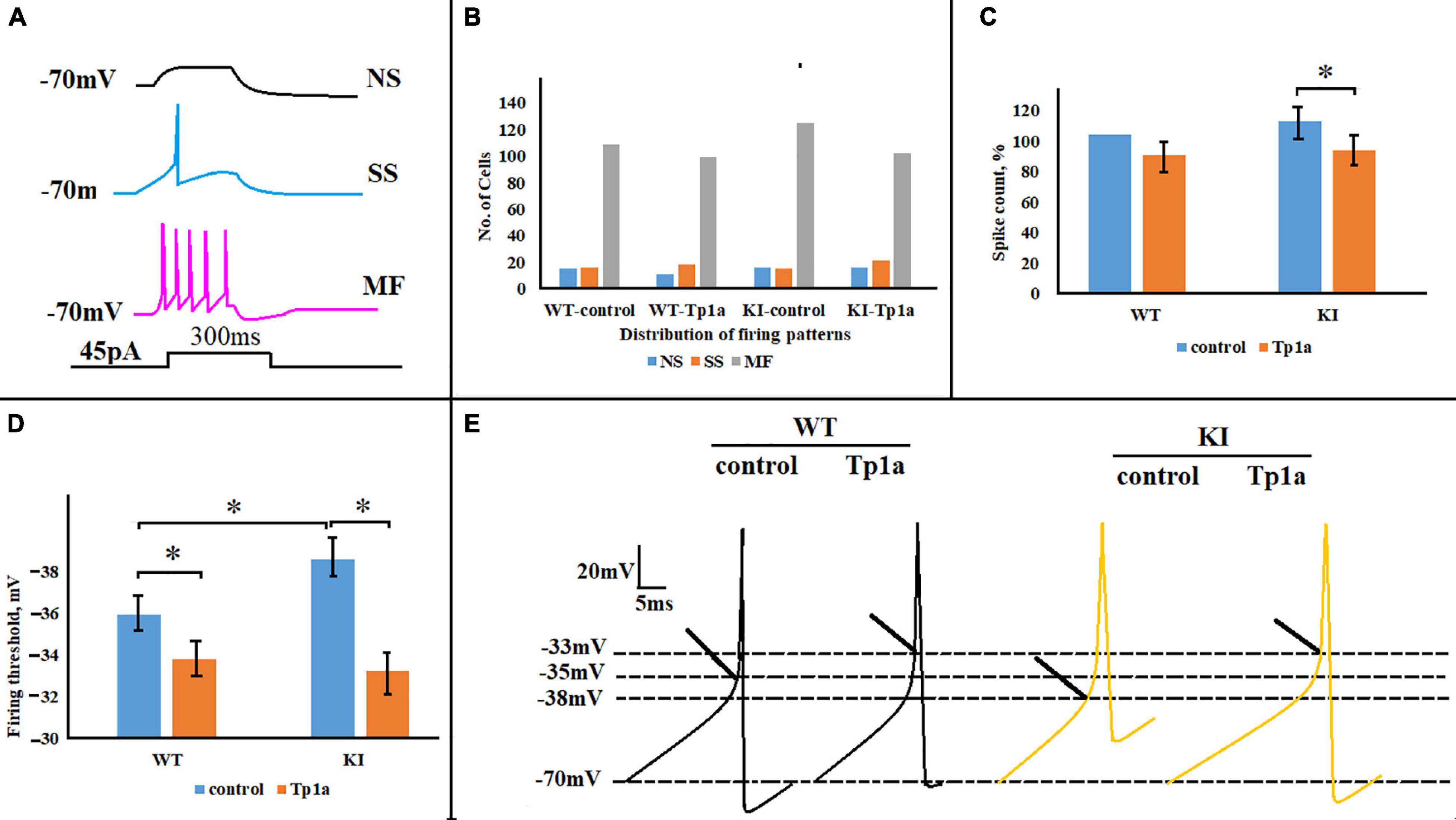
Figure 5. Effects of Tp1a on firing of TG neurons evoked by a 45-pA 300-ms square current pulse. (A) Cell-specific firing patterns recorded from three different WT TG neurons in response to the same stimulation. NS, no-spiking cell; SS, single-spike cell; MF, multiple-firing cell. (B) Distribution of firing patterns in WT and R192Q KI TG cultures is very similar in control and after application of Tp1a (7 nM, 30 min); chi-square test for proportions. Number of cells in each group for NS, SS, and MF cells, respectively: n (WT, control) = 15, 16, 109; n (WT, Tp1a) = 11, 18, 99; n (KI, control) = 16, 15, 125; n (KI, Tp1a) = 16, 21, 102. (C) Histograms represent the percent change of the number of spikes, generated by WT and KI neurons in control and after 30 min application of 7 nM Tp1a. *Indicates statistically significant change (Mann–Whitney test, p-values are given in the text body). Number of cells in each group: n (WT, control) = 109, n (WT, Tp1a) = 99, n (KI, control) = 133, n (KI, Tp1a) = 102. (D) Averaged firing threshold of WT and KI neurons in control and after Tp1a (7 nM, 30 min). *Indicates statistically significant change (two-sample Student’s t-test, p-values are given in the text body). Number of cells is as in (C). Note more negative KI threshold value in control and depolarized WT and KI thresholds after Tp1a application. (E) Representative traces of the first generated AP by WT and KI control and Tp1a-treated (7 nM, 30 min) neurons. Arrows indicate AP threshold.
In addition to firing properties, Tp1a also changed the size of the rebound effect, observed as a transient depolarization (“off response”) after a 300-ms hyperpolarizing pulse. Figure 6A shows representative recordings of the off response in WT and KI TG neurons, whereas Figure 6B summarizes the averaged values for all cells. The off response in control Krebs solution was statistically larger in KI neurons (p = 0.003, two-sample Students t-test), but decreased to the WT level (p = 0.009, two-sample Student’s t-test) after 30 min application of 7 nM Tp1a (Figures 6A,B). Conversely, Tp1a did not affect the off response in WT neurons (p = 0.3, two-sample Student’s t-test) (Figures 6A,B). These data support the notion that NaV1.7 channel activation is particularly important for subthreshold behavior of KI neurons.
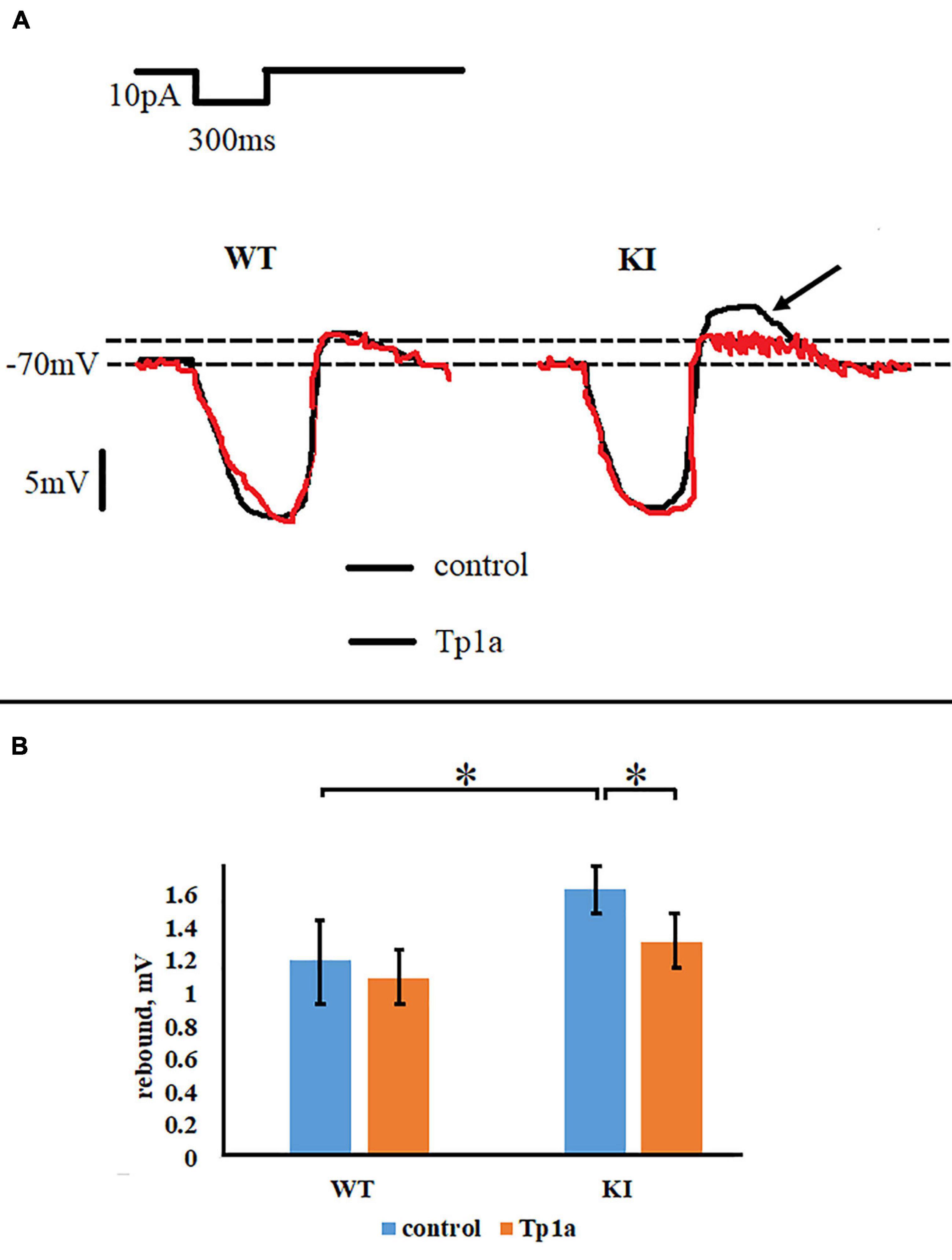
Figure 6. Tp1a reduces rebound effect after a 300-ms hyperpolarizing pulse of 10 pA in R192Q KI TG neurons. (A) Representative responses of WT and R192Q KI neurons, recorded in control and after application of Tp1a (7 nM, 30 min). Arrow shows higher rebound in KI which is reduced by Tp1a. (B) Histograms represent rebound effect in WT and KI neurons under control conditions and after 30 min application of 7 nM Tp1a. *Indicates statistically significant change (two-sample Student’s t-test, p-values are given in the text body). Number of cells in each group: n = 90 (WT, control), n = 86 (WT, Tp1a), n = 109 (KI, control), n = 96 (KI, Tp1a). Note small but statistically significant difference between WT and KI neurons, and a reduction of the rebound effect in KI after Tp1a application.
Activation of NaV1.7 Channels Stimulates TG Neuron Firing
Next, we examined the effect of the NaV1.7 channel activator OD1 (Jalali et al., 2005; Maertens et al., 2006; Motin et al., 2016) on the firing properties of TG neurons. Figure 7A shows representative traces of firing activity, recorded from WT and KI neurons, in response to 45-pA, 300-ms square current pulses in control condition, after application of 10 nM OD1, and when OD1 was applied to cells pretreated with Tp1a (7 nM, 30 min). Figure 7B indicates the average changes in spike threshold and spike count values when OD1 was either directly applied to the control cells (middle bars) or to the cells pretreated with 7 nM Tp1a (right bars). Application of OD1 (10 nM, 10 min) to the recorded cells brought AP threshold to more negative potentials both in WT and KI neurons (Figure 7B shows the difference in%; p = 0.005 for WT and p = 0.001 for KI, Mann–Whitney test). The effect of OD1 was absent following 30 min pretreatment with 7 nM Tp1a. Application of OD1 also significantly increased the number of generated spikes in response to 45 pA current stimulation (Figure 7B shows the difference in%; p = 0.021 for WT and 0.046 for KI, Mann–Whitney test), whereas pretreatment with Tp1a prevented full manifestation of this effect.
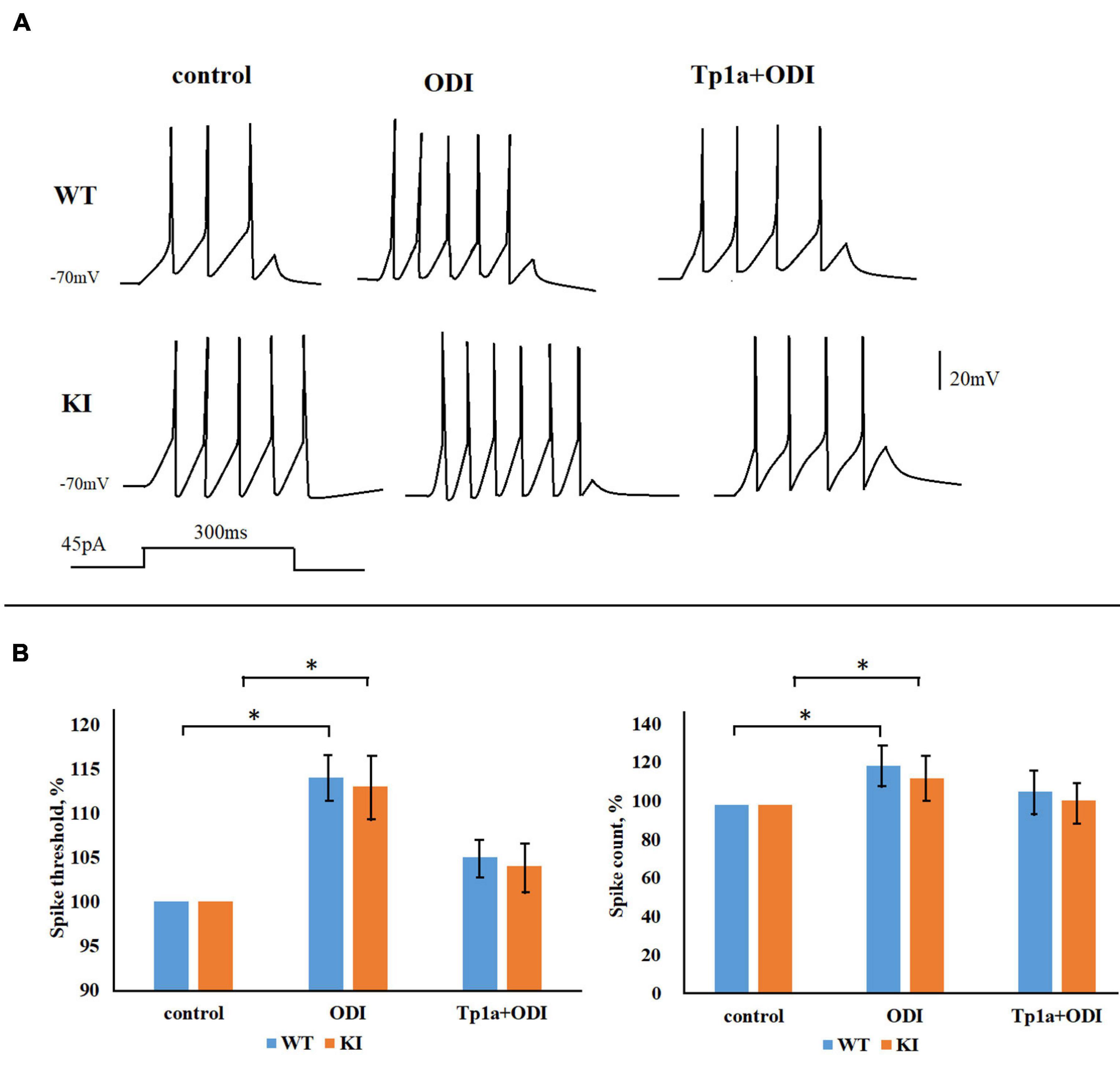
Figure 7. OD1 elevates firing of TG neurons in response to a 45-pA 300-ms square current pulse. (A) Representative traces obtained from WT and R192Q KI TG neurons in control and after application of 10 nM OD1 (paired data, recordings from the same cell) or after application of 10 nM OD1 on cells pretreated with Tp1a (7 nM, 30 min; recordings from different cells). (B) OD1 application makes the AP threshold more negative and increases spike count both in WT and KI neurons (paired data); *Indicates statistically significant change (Mann-Whitney test, p-values are given in the text body). Number of cells in each group: n = 37 (WT, control), n = 36 (WT, OD1), i = 28 (KI, control), n = 25 (KI, OD1). The effect of OD1 is decreased when cells were pretreated with Tp1a (7 nM, 30 min); *Indicates statistically significant change (Mann–Whitney test, p-values are given in the text body).
Larger NaV1.7 Current Underlies Higher Spontaneous Firing Observed in KI Neurons
The observation that NaV1.7 activity was acting within the subthreshold voltage range led us to investigate whether this current might influence not only electrically-evoked firing but also spontaneous spike discharges that occur when the neuronal membrane potential fluctuates around (and above) the resting membrane potential. Consequently, we studied the spontaneous generation of APs by WT and KI TG neurons held at −70, −60, and −50 mV. This phenomenon was investigated in each neuron by changing the holding membrane potential within the −70 to −50 mV range and observing the sustained spike discharge (see examples in Figure 8D depicting higher spike discharge of a KI neuron than a WT one). In control solution WT and KI cultures showed the same percent of no active cells at −70 mV and the majority of neurons generating APs at −50 mV (Figure 8A). Figure 8A also shows that while few WT cells could spontaneously fire at −60 mV, more KI neurons did so with or without OD1 application. These results are consistent with the constitutively higher excitability of KI neurons with propensity to fire with smaller depolarizations (Hullugundi et al., 2014). Blocking NaV1.7 channels with Tp1a (7 nM, 30 min) did not affect the number of firing neurons at −50 mV, but significantly reduced the percent of cells that were active at −60 mV in both WT and KI cultures (p = 0.048 for WT and 0.016 for KI, Fisher test) (Figure 8A). It is noteworthy that large differences were detected in spontaneous firing frequency between WT and KI cells as the latter generated, on average, twice as many APs than WT cells at −50 mV (p = 0.007, two-sample Student’s t-test) (Figure 8B). This difference was abolished by Tp1a, which decreased the firing frequency of KI neurons (p = 0.006) to that of WT, without affecting WT neurons (Figure 8B).
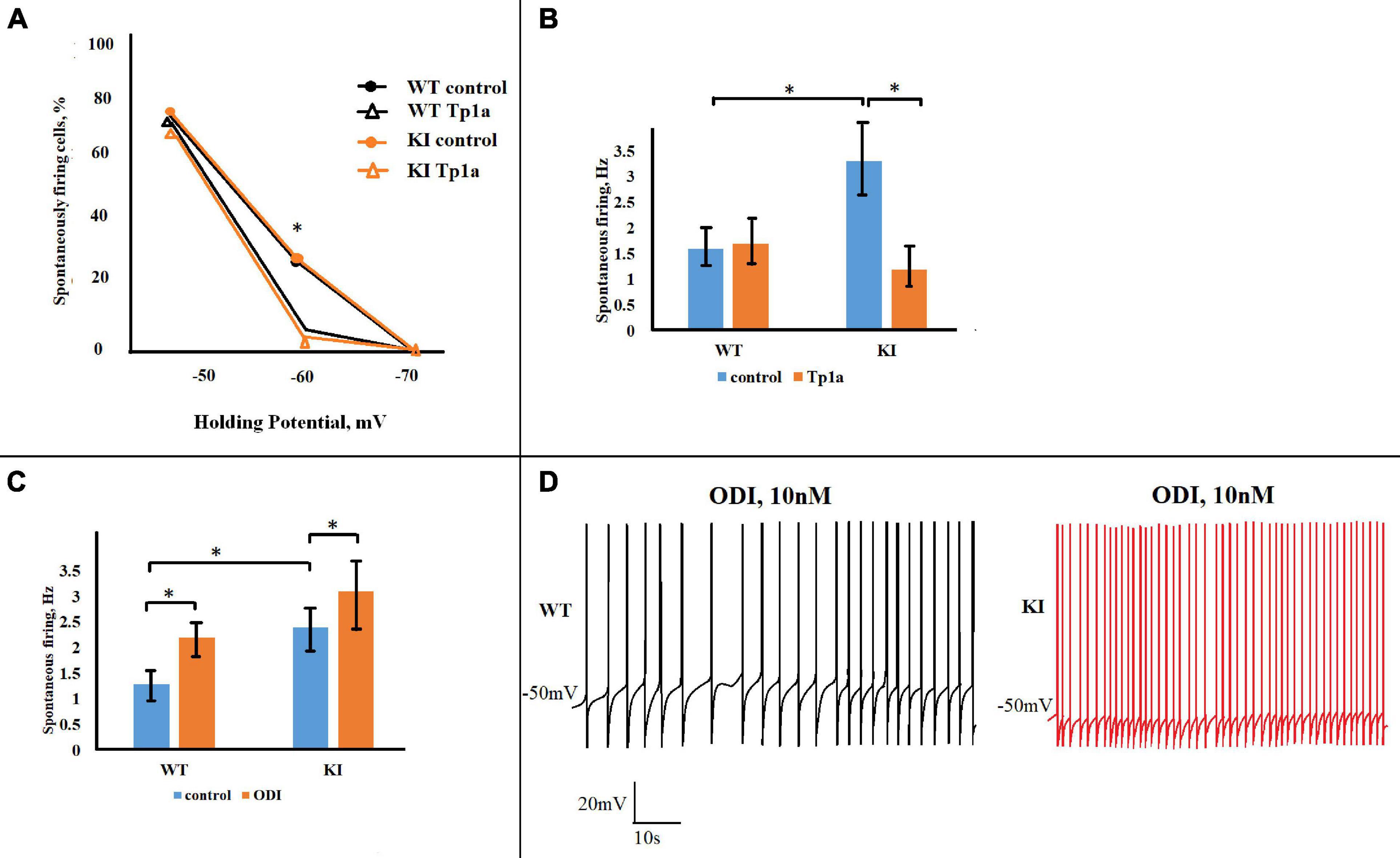
Figure 8. Opposite effects of Tp1a and OD1 on spontaneous firing in TG neurons. (A) Diagram shows percentage of firing WT and R192Q KI TG neurons at different holding potentials in control and after Tp1a (7 nM, 30 min). *Indicates statistically significant change (Fisher test for proportions, p-values are given in the text body). Number of analyzed neurons at –70, –60, and –50 mV, respectively, are: n (WT, control) = 40, 40, 40; n (WT, Tp1a) = 32, 32, 31; n (KI, control) = 42, 42, 37; n (KI, Tp1a) = 33, 33, 33. (B) Bar charts represent average firing frequencies of WT and KI neurons in control and after Tp1a (7 nM, 30 min). *Indicates statistically significant change (paired Student’s t-test, p-values are given in the text body). Number of cells in each group: n = 31 (WT, control), n = 30 (WT, Tp1a), n = 23 (KI, control), n = 23 (KI, Tp1a). KI neurons fire with significantly higher frequency, which is reduced to the WT level after Tp1a application. (C) Application of 10 nM OD1 (paired data, recordings from the same cell) increases firing frequency of both WT and KI neurons at –50 mV. *Indicates statistically significant change (paired Student’s t-test, p-values are given in the text). Number of cells in each group: n = 17 (WT), n = 10 (KI), data are paired. (D) Examples of WT and KI neurons, firing at –50 mV before and after application of 10 nM OD1.
Finally, we examined to what extent facilitating the activation of NaV1.7 channels with OD1 influenced spontaneous firing of WT and KI TG neurons. Consistent with its effect on pulse-evoked responses, OD1 significantly increased spontaneous firing frequency of both WT and KI neurons at −50 mV (p = 0.002 for WT and p = 0.02 for KI, two-sample Student’s t-test) (Figure 8C). It is worth noting that the OD1 effect was proportionally more pronounced in WT neurons (as exemplified in Figure 8D), with their firing frequency being elevated to the level of that in KI neurons.
Discussion
In this study we investigated the differential expression and function of TTX-sensitive NaV1.7 channels in trigeminal sensory neurons of WT mice and mice that express the R192Q missense mutation in the α1 subunit of CaV2.1 calcium channels that causes FHM1 in patients (Ferrari et al., 2015; Pietrobon and Brennan, 2019). The main finding of our study was a higher expression of NaV1.7 channels with larger NaV1.7 current that lowers the threshold for generating APs in KI TG neurons, at least in those of very young mice. We propose that this phenomenon is underlying the hyperexcitability phenotype we observed for KI neurons (Marchenkova et al., 2016a). This phenomenon may contribute to the activation of pain mechanisms, as shown to certain extent in KI mice by Chanda et al. (2013), and perhaps in FHM1 patients. We wish to mention that the hyperexcitability phenotype in KI neurons is not straightforward to interpret, when taking into account data from other laboratories. For instance, Fioretti et al. (2011) have reported a gain of function of CaV2.1 channel activity in a subpopulation of adult TG neurons not innervating meninges. Noteworthy, Fioretti et al. employed a different cell culture preparation method that yielded acutely dissociated neurons with minimal processes. Conversely, our procedure produced neurons with long processes (see Figures 1, 2) that may have a distinct chemosensitivity and electroresponsiveness. Our former work based on single cell Ca2+ imaging has indicated a wider distribution of CaV2.1 activity. In particular, our previous studies have shown that there is similar sensitivity of WT and KI neurons to capsaicin (Hullugundi et al., 2013), and that there are minimal differences in their rheobase and input resistance (Hullugundi et al., 2014). Indeed, KI cells fired more spikes with P2X3 or TRPV1 activation, suggesting a special chemosensitivity phenotype to KI TG neurons.
Subthreshold Currents in TG Neurons
Neurons, including trigeminal sensory neurons, communicate through APs generated in response to a stimulus (Sunada et al., 1990; Chudler et al., 1991; Coste et al., 2008). The timing and frequency of spikes are essential for encoding information (Eggermont, 1998; Masuda and Aihara, 2007) and, therefore, should be correctly modulated in response to changes in the extracellular milieu. Various conductances are known to play a role in regulating neuronal AP threshold and firing frequency, including currents through various K+ (Takeda et al., 2011), low-threshold Ca2+ (Kim et al., 2001; Llinas and Steriade, 2006) subthreshold Na+ (Waxman et al., 1999; Dib-Hajj et al., 2010; Yang et al., 2016) and hyperpolarization-activated cyclic nucleotide-gated (HCN) (Biel et al., 2009; Cho et al., 2011; He et al., 2014) channels. In the present study, we show that in the FHM1 R192Q KI mouse model TG neuron hyperexcitability phenotype is conferred by increased expression and function of NaV1.7 Na+ channels. One cannot exclude though the potential role of other subthreshold conductances. Afterall, TG neurons also exhibit low-threshold T-type Ca2+ (Yoshida and Oka, 1998; Borgland et al., 2001; Ikeda and Matsumoto, 2003; Ross et al., 2009) and Ih (Wells et al., 2007; Cho et al., 2009, 2011) currents, which positively regulate neuronal firing (Kim et al., 2001; Llinas and Steriade, 2006; Cho et al., 2011) and play a role in pathological pain conditions (Emery et al., 2011; Seo et al., 2013). Although at −70 mV a minor fraction of HCN channels might be open as the Ih activation threshold is cell-dependent (Biel et al., 2009; He et al., 2014), rat TG neurons show very little Ih current at this membrane potential (Cho et al., 2011). In addition, Ih current has much slower kinetics than the NaV1.7 current (Vijayaragavan et al., 2001; Biel et al., 2009; Cho et al., 2011; He et al., 2014) implying their distinct modulation of neuronal firing properties. While the low-threshold T-type Ca2+ current has kinetics similar to NaV1.7 current, this conductance is mostly inactivated at the resting membrane potential (−60 to −70 mV), and requires significant hyperpolarization to remove voltage-dependent channel inactivation (Lovinger and White, 1989; Yoshida and Oka, 1998; Seo et al., 2013) making it an unlikely contributor to firing modulation in our mouse model. Our results accord with previous studies (Shields et al., 2018; Wu et al., 2019; Neff et al., 2020; Siebenga et al., 2020) reporting pharmacological evidence that Nav1.7 inhibition by its blockers may be involved in relieving pain.
Higher Expression of NaV1.7 Channels Contributes to the KI TG Neuronal Hyperexcitability Phenotype
Hyperexcitability of TG neurons in FHM1 R192Q KI mice was previously shown in response to a short current pulse or chemical stimulus (Hullugundi et al., 2014; Marchenkova et al., 2016a). Here we examined genotypic differences between WT and KI TG neurons with respect to neuronal excitability by assessing, under current-clamp condition, spike threshold and firing frequency at different holding potentials. Although no neurons fired at −70 mV, holding them at −60 mV was sufficient to evoke spontaneous firing in a subpopulation of cells. Because NaV1.7 channel conductance becomes activated at this membrane potential (Yang et al., 2016), its inhibition by Tp1a validated the role of NaV1.7 in subthreshold properties facilitating the onset of firing. Whereas the percentage of WT and KI neurons generating APs at each test voltage did not differ, KI neurons fired with significantly higher frequency, a difference again abolished by Tp1a. Notably, Tp1a did not affect WT firing frequency, implying that the NaV1.7 channel current did not play a significant role in regulating repetitive firing of WT neurons under control conditions per se. Its role, however, appeared prominent when NaV1.7 channel expression and function are upregulated in mutated TG neurons. This is consistent with results from a study of DRG neurons that express a gain-of-function NaV1.7 mutation associated with inherited erythromelalgia and persistent pain states (Yang et al., 2016).
Upregulation of NaV1.7 channel function in KI TG neurons most likely originated from a higher expression of the channel. Indeed, in KI cultures, the number of NaV1.7-positive neurons was increased with an apparently higher expression of NaV1.7 α1 protein in KI than in WT neurons, although immunohistochemistry is at best semi-quantitative. Consistent with these findings, reversing the KI phenotype with CaV2.1 blocker ω-agatoxin IVA (Nair et al., 2010; Gnanasekaran et al., 2013) significantly reduced the number of NaV1.7-positive neurons in KI cultures. There was no detectable difference in NaV1.7 current kinetics, suggesting no constitutive alteration of KI NaV1.7 channel properties. The activation of NaV1.7 channels by OD1 toxin largely increased spike activity in all neurons, albeit more strongly in WT cells. The lower magnitude of the OD1 effect in KI neurons could be attributed to the partially up-regulated properties of the NaV1.7 channels under basal conditions. The turnover of NaV1.7 channels is incompletely understood and is likely to be influenced by intracellular modulators including the concentration of free Ca2+. We might speculate that the experimentally-observed long-lasting pharmacological inhibition by ω-agatoxin IVA of functionally upregulated CaV2.1 channels of KI TG neurons was translated into a downregulation of NaV1.7 channels. Nonetheless, it is also possible that toxin-dependent partial deprivation of other endogenous substances, such as CGRP and TNFα [upregulated in KI cultures following facilitation of CaV2.1 activity (Franceschini et al., 2013; Hullugundi et al., 2013)], regulated NaV1.7 expression. The mechanisms of NaV1.7 modulation will require future studies.
Co-expression of NaV1.7 Channels and P2X3Rs
ATP-sensitive P2X3Rs are predominantly expressed in sensory ganglion neurons, including TG neurons (Vulchanova et al., 1997; Llewellyn-Smith and Burnstock, 1998) where they control transduction of nociceptive stimuli (Wirkner et al., 2005; Burnstock, 2006). Strong upregulation of P2X3R function and P2X3R-mediated excitability of TG neurons was shown to be associated with the hyperexcitability phenotype of the FHM1 R192Q KI model (Nair et al., 2010; Hullugundi et al., 2013; Marchenkova et al., 2016a,b) further supporting their proposed relevance for triggering migraine pain (Fabbretti, 2013; Yan and Dussor, 2014).
We found extensive co-expression of NaV1.7 and P2X3R in both WT and KI TG cultures with important implications for TG neuron function. The amount of synthesis/release of natural P2X3R agonist ATP that is known to be enhanced in pathological conditions including migraine (Burnstock et al., 2011) is considered one mechanism leading to the development of neuronal sensitization (Hamilton and McMahon, 2000). In KI TG neurons, the observed co-expression of NaV1.7 and P2X3R along with increased NaV1.7 activity can well contribute to the observed P2X3-mediated hyperexcitability, typical for KI TG neurons (Hullugundi et al., 2014; Marchenkova et al., 2016a,b). Taking into account their slow voltage-dependent inactivation (Cummins et al., 1998; Herzog et al., 2003) we propose that NaV1.7 channels are well-suited to amplify and facilitate depolarization produced by activated P2X3 receptors, leading to higher frequency of spike generation and sensitization of TG sensory neurons.
Thus, neuronal co-expression of NaV1.7 and P2X3R by small-size neurons within a basal neuroinflammatory milieu comprising of higher ATP levels in the TG of KI mice (Franceschini et al., 2013; Hullugundi et al., 2013) may turn out to be an interesting background for facilitating hypersensitivity to environmental stimuli.
Possible Relevance to Migraine
The observed hyperexcitability phenotype, namely higher expression of NaV1.7 channels with larger NaV1.7 current that lowers the threshold for generating APs in KI TG, was only studied in young mice (P11–P13), and findings should thus not be immediately extrapolated to older mice. In fact, Fioretti et al. (2011), who used mice of the same KI strain studied consequences of the R192Q mutation in 4- to 6-weeks-old animals and showed that the mutation produced a gain-of-function effect (i.e., an increase of Ca2+ influx through CaV2.1 channels) only in TG neurons that do not innervate the meninges. In TG neurons that innervate the meninges and release CGRP CaV2.1 channels seem unaffected by the mutation, suggesting that the R192Q mutation does not alter CGRP release from meninges. Whereas a study by Chan et al. (2019) that investigated the same mutant mice but of 13–14 weeks did not show increased basal nor KCl-stimulated CGRP release in TG either, an increased CGRP release was observed in young mutant mice by Ceruti et al. (2011). Future studies are warranted as they might detect a developmentally-regulated shift in neuronal chemosensitivity. In particular, future in vivo studies are needed to translate the in vitro observations to in vivo application.
At least under our culturing conditions, which involve mild dissociation and mild proteolytic treatment, we found no indication for a change in neuronal CaV2.1 expression in dissociated TG neurons versus intact ganglia (Nair et al., 2010).
With respect to the relevance for migraine pathophysiology, the vast majority of small-size TG neurons are highly sensitive to ATP, a very powerful algogenic mediator (Nair et al., 2010). It therefore follows that the large majority of cultured neurons are functional nociceptors. It is helpful to mention that “pure” nociceptors, i.e., responding solely to painful stimuli, are rare because most sensory neurons are polymodal nociceptors, i.e., activated by various sensory stimuli as well as painful ones over a broad receptive field. It should be noted that migraine pain does not originate exclusively from meninges as important trigger areas are, among others, also scalp and neck muscles.
Conclusion
NaV1.7 sodium channel antagonism as shown here in vitro may be an interesting approach to treat pain-related disorders of the trigeminal system, including migraine. This possibility deserves further exploration in in vivo pain models among those that compare effects of antagonists of NaV1.7 channels.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by SISSA Ethical Committee, Trieste, Italy.
Author Contributions
RM performed the immunofluorescence and Western blotting experiments. AM performed the electrophysiological experiments. AMJMvdM developed the genetic mouse model of hemiplegic migraine. AN supervised the study. All the authors have contributed in the writing and final approval of the manuscript. AMJMvdM and AN also critically reviewed the manuscript.
Funding
This work was supported by the Grant FP7-EUROHEADPAIN (No. 602633 to AMJMvdM and AN). The authors declare no competing financial interests.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
- ^ http://imagej.nih.gov/ij/
- ^ http://www.biomath.info/
- ^ http://powerandsamplesize.com
- ^ https://www.mathworks.com/products/matlab.html
- ^ https://systatsoftware.com/products/sigmaplot/
References
Biel, M., Wahl-Schott, C., Michalakis, S., and Zong, X. (2009). Hyperpolarization-activated cation channels: from genes to function. Physiol. Rev. 89, 847–885. doi: 10.1152/physrev.00029.2008
Black, J. A., Liu, S., Tanaka, M., Cummins, T. R., and Waxman, S. G. (2004). Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108, 237–247. doi: 10.1016/j.pain.2003.12.035
Borgland, S. L., Connor, M., and Christie, M. J. (2001). Nociceptin inhibits calcium channel currents in a subpopulation of small nociceptive trigeminal ganglion neurons in mouse. J. Physiol. 536, 35–47. doi: 10.1111/j.1469-7793.2001.t01-1-00035.x
Burnstock, G. (2006). Purinergic P2 receptors as targets for novel analgesics. Pharmacol. Ther. 110, 433–454. doi: 10.1016/j.pharmthera.2005.08.013
Burnstock, G., Krugel, U., Abbracchio, M. P., and Illes, P. (2011). Purinergic signalling: from normal behaviour to pathological brain function. Prog. Neurobiol. 95, 229–274. doi: 10.1016/j.pneurobio.2011.08.006
Cai, W., Cao, J., Ren, X., Qiao, L., Chen, X., Li, M., et al. (2016). ShRNA mediated knockdown of Nav1.7 in rat dorsal root ganglion attenuates pain following burn injury. BMC Anesthesiol. 16:59. doi: 10.1186/s12871-016-0215-0
Cardoso, F. C., Dekan, Z., Rosengren, K. J., Erickson, A., Vetter, I., Deuis, J. R., et al. (2015). Identification and characterization of ProTx-III [mu-TRTX-Tp1a], a new voltage-gated sodium channel inhibitor from venom of the Tarantula Thrixopelma pruriens. Mol. Pharmacol. 88, 291–303. doi: 10.1124/mol.115.098178
Ceruti, S., Villa, G., Fumagalli, M., Colombo, L., Magni, G., Zanardelli, M., et al. (2011). Calcitonin gene-related peptide-mediated enhancement of purinergic neuron/glia communication by the algogenic factor bradykinin in mouse trigeminal ganglia from wild-type and R192Q Cav2.1 Knock-in mice: implications for basic mechanisms of migraine pain. J. Neurosci. 31, 3638–3649. doi: 10.1523/JNEUROSCI.6440-10.2011
Chan, K. Y., Labastida-Ramírez, A., Ramírez-Rosas, M. B., Labruijere, S., Garrelds, I. M., Danser, A. H., et al. (2019). Trigeminovascular calcitonin gene-related peptide function in Cacna1a R192Q-mutated knock-in mice. J. Cereb. Blood Flow Metab. 39, 718–729. doi: 10.1177/0271678X17725673
Chanda, M. L., Tuttle, A. H., Baran, I., Atlin, C., Guindi, D., Hathaway, G., et al. (2013). Behavioral evidence for photophobia and stress-related ipsilateral head pain in transgenic Cacna1a mutant mice. Pain 154, 1254–1262. doi: 10.1016/j.pain.2013.03.038
Cho, H. J., Furness, J. B., and Jennings, E. A. (2011). Postnatal maturation of the hyperpolarization-activated cation current, I(h), in trigeminal sensory neurons. J. Neurophysiol. 106, 2045–2056. doi: 10.1152/jn.00798.2010
Cho, H. J., Staikopoulos, V., Ivanusic, J. J., and Jennings, E. A. (2009). Hyperpolarization-activated cyclic-nucleotide gated 4 (HCN4) protein is expressed in a subset of rat dorsal root and trigeminal ganglion neurons. Cell Tissue Res. 338, 171–177. doi: 10.1007/s00441-009-0869-8
Chudler, E. H., Foote, W. E., and Poletti, C. E. (1991). Responses of cat C1 spinal cord dorsal and ventral horn neurons to noxious and non-noxious stimulation of the head and face. Brain Res. 555, 181–192. doi: 10.1016/0006-8993(91)90341-r
Coste, J., Voisin, D. L., Luccarini, P., and Dallel, R. (2008). A role for wind-up in trigeminal sensory processing: intensity coding of nociceptive stimuli in the rat. Cephalalgia 28, 631–639. doi: 10.1111/j.1468-2982.2008.01568.x
Cummins, T. R., Howe, J. R., and Waxman, S. G. (1998). Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J. Neurosci. 18, 9607–9619. doi: 10.1523/JNEUROSCI.18-23-09607.1998
Dib-Hajj, S. D., Cummins, T. R., Black, J. A., and Waxman, S. G. (2010). Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 33, 325–347. doi: 10.1146/annurev-neuro-060909-153234
Dib-Hajj, S. D., Yang, Y., Black, J. A., and Waxman, S. G. (2013). The Na(V)1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 14, 49–62. doi: 10.1038/NRN3404
Eggermont, J. J. (1998). Is there a neural code? Neurosci. Biobehav. Rev. 22, 355–370. doi: 10.1016/S0149-7634(97)00021-3
Eikermann-Haerter, K., Dileköz, E., Kudo, C., Savitz, S. I., Waeber, C., Baum, M. J., et al. (2009). Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J. Clin. Invest. 119, 99–109. doi: 10.1016/j.jphysparis.2011.10.004
Elliott, A. A., and Elliott, J. R. (1993). Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J. Physiol. 463, 39–56. doi: 10.1113/jphysiol.1993.sp019583
Emery, E. C., Luiz, A. P., and Wood, J. N. (2016). Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin. Ther. Targets 20, 975–983. doi: 10.1517/14728222.2016.1162295
Emery, E. C., Young, G. T., Berrocoso, E. M., Chen, L., and McNaughton, P. (2011). HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science 333, 1462–1466. doi: 10.1126/science.1206243
Fabbretti, E. (2013). ATP P2X3 receptors and neuronal sensitization. Front. Cell Neurosci. 7:236. doi: 10.3389/fncel.2013.00236
Ferrari, M. D., Klever, R. R., Terwindt, G. M., Ayata, C., and van den Maagdenberg, A. M. J. M. (2015). Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol. 14, 65–80. doi: 10.1016/S1474-4422(14)70220-0
Fioretti, B., Catacuzzeno, L., Sforna, L., Gerke-Duncan, M. B., van den Maagdenberg, A. M. J. M., Franciolini, F., et al. (2011). Trigeminal ganglion neuron subtype-specific alterations of Ca(V)2.1 calcium current and excitability in a Cacna1a mouse model of migraine. J. Physiol. 589, 5879–5895. doi: 10.1113/jphysiol.2011.220533
Franceschini, A., Vilotti, S., Ferrari, M. D., van den Maagdenberg, A. M. J. M., Nistri, A., and Fabbretti, E. (2013). TNFalpha levels and macrophages expression reflect an inflammatory potential of trigeminal ganglia in a mouse model of familial hemiplegic migraine. PLoS One 8:e52394. doi: 10.1371/journal.pone.0052394
Gnanasekaran, A., Bele, T., Hullugundi, S., Simonetti, M., Ferrari, M. D., van den Maagdenberg, A. M. J. M., et al. (2013). Mutated CaV2.1 channels dysregulate CASK/P2X3 signaling in mouse trigeminal sensory neurons of R192Q Cacna1a knock-in mice. Mol. Pain 9:62. doi: 10.1186/1744-8069-9-62
Goadsby, P. J., Holland, P. R., Martins-Oliveira, M., Hoffmann, J., Schankin, C. and Akerman, S. (2017). Pathophysiology of migraine: a disorder of sensory processing. Physiol. Rev. 97, 553–622. doi: 10.1152/physrev.00034.2015
Gould, H. J., Gould, T. N., England, J. D., Paul, D., Liu, Z. P., and Levinson, S. R. (2000). A possible role for nerve growth factor in the augmentation of sodium channels in models of chronic pain. Brain Res. 854, 19–29. doi: 10.1016/s0006-8993(99)02216-7
Habib, A. M., Wood, J. N., and Cox, J. J. (2015). Sodium channels and pain. Handb. Exp. Pharmacol. 227, 39–56. doi: 10.1007/978-3-662-46450-2_3
Hamilton, S. G., and McMahon, S. B. (2000). ATP as a peripheral mediator of pain. J. Auton. Nerv. Syst. 81, 187–194. doi: 10.1016/s0165-1838(00)00137-5
Hansen, J. M., Hauge, A. W., Ashina, M., and Olesen, J. (2011). Trigger factors for familial hemiplegic migraine. Cephalalgia 31, 1274–1281. doi: 10.1177/0333102411415878
He, C., Chen, F., Li, B., and Hu, Z. (2014). Neurophysiology of HCN channels: from cellular functions to multiple regulations. Prog. Neurobiol. 112, 1–23. doi: 10.1016/j.pneurobio.2013.10.001
Herzog, R. I., Cummins, T. R., Ghassemi, F., Dib-Hajj, S. D., and Waxman, S. G. (2003). Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J. Physiol. 551, 741–750. doi: 10.1113/jphysiol.2003.047357
Hoffmann, T., Sharon, O., Wittmann, J., Carr, R. W., Vyshnevska, A., Col, R. D., et al. (2018). NaV1.7 and pain: contribution of peripheral nerves. Pain 159, 496–506. doi: 10.1097/j.pain.0000000000001119
Hullugundi, S. K., Ansuini, A., Ferrari, M. D., van den Maagdenberg, A. M. J. M., and Nistri, A. (2014). A hyperexcitability phenotype in mouse trigeminal sensory neurons expressing the R192Q Cacna1a missense mutation of familial hemiplegic migraine type-1. Neuroscience 266, 244–254. doi: 10.1016/j.neuroscience.2014.02.020
Hullugundi, S. K., Ferrari, M. D., van den Maagdenberg, A. M. J. M., and Nistri, A. (2013). The mechanism of functional up-regulation of P2X3 receptors of trigeminal sensory neurons in a genetic mouse model of familial hemiplegic migraine type 1 (FHM-1). PLoS One 8:e60677. doi: 10.1371/journal.pone.0060677
IHS (2013). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 33, 629–808. doi: 10.1177/0333102413485658
Ikeda, M., and Matsumoto, S. (2003). Classification of voltage-dependent Ca2+ channels in trigeminal ganglion neurons from neonatal rats. Life Sci. 73, 1175–1187. doi: 10.1016/s0024-3205(03)00414-4
Isom, L. L., De Jongh, K. S., Patton, D. E., Reber, B. F., Offord, J., Charbonneau, H., et al. (1992). Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256, 839–842. doi: 10.1126/science.256.5058.839
Jalali, A., Bosmans, F., Amininasab, M., Clynen, E., Cuypers, E., Zaremirakabadi, A., et al. (2005). OD1, the first toxin isolated from the venom of the scorpion Odonthobuthus doriae active on voltage-gated Na+ channels. FEBS Lett. 579, 4181–4186. doi: 10.1016/j.febslet.2005.06.052
Kim, D., Song, I., Keum, S., Lee, T., Jeong, M. J., Kim, S. S., et al. (2001). Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca(2+) channels. Neuron 31, 35–45. doi: 10.1016/s0896-6273(01)00343-9
Klugbauer, N., Lacinova, L., Flockerzi, V., and Hofmann, F. (1995). Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 14, 1084–1090.
Langford, D. J., Bailey, A. L., Chanda, M. L., Clarke, S. E., Drummond, T. E., Echols, S., et al. (2010). Coding of facial expressions of pain in the laboratory mouse. Nat. Methods 7, 447–449. doi: 10.1038/nmeth.1455
Llewellyn-Smith, I. J., and Burnstock, G. (1998). Ultrastructural localization of P2X3 receptors in rat sensory neurons. Neuroreport 9, 2545–2550. doi: 10.1097/00001756-199808030-00022
Llinas, R. R., and Steriade, M. (2006). Bursting of thalamic neurons and states of vigilance. J. Neurophysiol. 95, 3297–3308. doi: 10.1152/jn.00166.2006
Lovinger, D. M., and White, G. (1989). Post-natal development of burst firing behavior and the low-threshold transient calcium current examined using freshly isolated neurons from rat dorsal root ganglia. Neurosci. Lett. 102, 50–57. doi: 10.1016/0304-3940(89)90306-6
Maertens, C., Cuypers, E., Amininasab, M., Jalali, A., Vatanpour, H., and Tytgat, J. (2006). Potent modulation of the voltage-gated sodium channel Nav1.7 by OD1, a toxin from the scorpion Odonthobuthus doriae. Mol. Pharmacol. 70, 405–414. doi: 10.1124/mol.106.022970
Marchenkova, A., van den Maagdenberg, A. M. J. M., and Nistri, A. (2016a). Loss of inhibition by brain natriuretic peptide over P2X3 receptors contributes to enhanced spike firing of trigeminal ganglion neurons in a mouse model of familial hemiplegic migraine type-1. Neuroscience 331, 197–205. doi: 10.1016/j.neuroscience.2016.06.034
Marchenkova, A., Vilotti, S., Ntamati, N., van den Maagdenberg, A. M. J. M., and Nistri, A. (2016b). Inefficient constitutive inhibition of P2X3 receptors by brain natriuretic peptide system contributes to sensitization of trigeminal sensory neurons in a genetic mouse model of familial hemiplegic migraine. Mol. Pain 12:1744806916646110. doi: 10.1177/1744806916646110
Masuda, N., and Aihara, K. (2007). Dual coding hypotheses for neural information representation. Math. Biosci. 207, 312–321. doi: 10.1016/j.mbs.2006.09.009
Motin, L., Durek, T., and Adams, D. (2016). Modulation of human Nav1.7 channel gating by synthetic alpha-scorpion toxin OD1 and its analogs. Channels 10, 139–147. doi: 10.1080/19336950.2015.1120392
Nair, A., Simonetti, M., Birsa, N., Ferrari, M. D., van den Maagdenberg, A. M., Giniatullin, R., et al. (2010). Familial hemiplegic migraine Ca(v)2.1 channel mutation R192Q enhances ATP-gated P2X3 receptor activity of mouse sensory ganglion neurons mediating trigeminal pain. Mol. Pain 6:48. doi: 10.1186/1744-8069-6-48
Neff, R. A., Flinspach, M., Gibbs, A., Shih, A. Y., Minassian, N. A., Liu, Y., et al. (2020). Comprehensive engineering of the tarantula venom peptide huwentoxin-IV to inhibit the human voltage-gated sodium channel hNav1.7. J. Biol. Chem. 295, 1315–1327. doi: 10.1074/jbc.RA119.011318
Ogata, N., and Ohishi, Y. (2002). Molecular diversity of structure and function of the voltage-gated Na+ channels. Jpn. J. Pharmacol. 88, 365–377. doi: 10.1254/jjp.88.365
Ogata, N., Yamamoto, M., and Maruyama, H. (2001). The role of tetrodotoxin-resistant sodium channels in pain sensation studied on sns-knockout mice. Nihon Rinsho 59, 1688–1697.
Ophoff, R. A., Terwindt, G. M., Vergouwe, M. N., van Eijk, R., Oefner, P. J., Hoffman, J. E., et al. (1996). Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87, 543–552. doi: 10.1016/s0092-8674(00)81373-2
Payandeh, J., and Hackos, D. H. (2018). Selective Ligands and drug discovery targeting the voltage-gated sodium channel Nav1.7. Handb. Exp. Pharmacol. 246, 271–306. doi: 10.1007/164_2018_97
Pietrobon, D. (2013). Calcium channels and migraine. Biochim. Biophys. Acta 1828, 1655–1665. doi: 10.1016/j.bbamem.2012.11.012
Pietrobon, D., and Brennan, K. C. (2019). Genetic mouse models of migraine. J. Headache Pain 20:79. doi: 10.1186/s10194-019-1029-5
Raman, I. M., and Bean, B. P. (1997). Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J. Neurosci. 17, 4517–4526. doi: 10.1523/jneurosci.17-12-04517.1997
Ross, H. R., Gilmore, A. J., and Connor, M. (2009). Inhibition of human recombinant T-type calcium channels by the endocannabinoid N-arachidonoyl dopamine. Br. J. Pharmacol. 156, 740–750. doi: 10.1111/j.1476-5381.2008.00072.x
Seo, S. H., Li, H. Y., Perez-Reyes, E., and Lee, J. H. (2013). Effects of eugenol on T-type Ca2+ channel isoforms. J. Pharmacol. Exp. Ther. 347, 310–317. doi: 10.1124/jpet.113.207936
Shields, S. D., Deng, L., Reese, R. M., Dourado, M., Tao, J., Foreman, O., et al. (2018). Insensitivity to pain upon adult-onset deletion of Nav1.7 or its blockade with selective inhibitors. J. Neurosci. 38, 10180–10201. doi: 10.1523/jneurosci.1049-18.2018
Siebenga, P., van Amerongen, G., Hay, J. L., McDonnell, A., Gorman, D., Butt, R., et al. (2020). Lack of detection of the analgesic properties of PF-05089771, a Selective Nav 1.7 inhibitor, using a battery of pain models in healthy subjects. Clin. Transl. Sci. 13, 318–324. doi: 10.1111/cts.12712
Simonetti, M., Fabbro, A., D’Arco, M., Zweyer, M., Nistri, A., Giniatullin, R. and Fabbretti, E., (2006). Comparison of P2X and TRPV1 receptors in ganglia or primary culture of trigeminal neurons and their modulation by NGF or serotonin. Mol. Pain 2:11. doi: 10.1186/1744-8069-2-11
Strickland, I. T., Martindale, J. C., Woodhams, P. L., Reeve, A. J., Chessell, I. P., and McQueen, D. S. (2008). Changes in the expression of NaV1.7, NaV1.8 and NaV1.9 in a distinct population of dorsal rootganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur. J. Pain 12, 564–572. doi: 10.1016/j.ejpain.2007.09.001
Sunada, T., Kurasawa, K., Hirose, Y., and Nakamura, Y. (1990). Intracellular response properties of neurons in the spinal trigeminal nucleus to peripheral and cortical stimulation in the cat. Brain Res. 514, 189–197. doi: 10.1016/0006-8993(90)91415-D
Takeda, M., Tsuboi, Y., Kitagawa, J., Nakagawa, K., Iwata, K., and Matsumoto, S. (2011). Potassium channels as a potential therapeutic target for trigeminal neuropathic and inflammatory pain. Mol. Pain 7:5. doi: 10.1186/1744-8069-7-5
Tan, Z. Y., Piekarz, A. D., Priest, B. T., Knopp, K. L., Krajewski, J. L., McDermott, J. S., et al. (2014). Tetrodotoxin-resistant sodium channels in sensory neurons generate slow resurgent currents that are enhanced by inflammatory mediators. J. Neurosci. 34, 7190–7197. doi: 10.1523/jneurosci.5011-13.2014
Thomsen, L. L., Eriksen, M. K., Roemer, S. F., Andersen, I., Olesen, J., and Russel, M. B. (2002). A population-based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain 125, 1379–1391. doi: 10.1093/brain/awf132
Toledo-Aral, J. J., Moss, B. L., He, Z. J., Koszowski, A. G., Whisenand, T., Levinson, S. R., et al. (1997). Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc. Natl. Acad. Sci. U.S.A. 94, 1527–1532. doi: 10.1073/pnas.94.4.1527
Tottene, A., Conti, R., Fabbro, A., Vecchia, D., Shapovalova, M., Santello, M., et al. (2009). Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron 61, 762–773. doi: 10.1016/j.neuron.2009.01.027
Tottene, A., Fellin, T., Pagnutti, S., Luvisetto, S., Striessnig, J., Fletcher, C., et al. (2002). Familial hemiplegic migraine mutations increase Ca(2+) influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc. Natl. Acad. Sci. U.S.A. 99, 13284–13289. doi: 10.1073/pnas.192242399
van den Maagdenberg, A. M. J. M., Pietrobon, D., Pizzorusso, T., Kaja, S., Broos, L. A. M., Cesetti, T., et al. (2004). A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 41, 701–710. doi: 10.1016/s0896-6273(04)00085-6
Vijayaragavan, K., O’Leary, M. E., and Chahine, M. (2001). Gating properties of Na(v)1.7 and Na(v)1.8 peripheral nerve sodium channels. J. Neurosci. 21, 7909–7918. doi: 10.1523/jneurosci.21-20-07909.2001
Vulchanova, L., Riedl, M. S., Shuster, S. J., Buell, G., Surprenant, R. A., and North, R. A. (1997). Immunohistochemical study of the P2X2 and P2X3 receptor subunits in rat and monkey sensory neurons and their central terminals. Neuropharmacology 36, 1229–1242. doi: 10.1016/s0028-3908(97)00126-3
Waxman, S. G., Cummins, T. R., Dib-Hajj, S. D., Fjell, J., and Black, J. A. (1999). Sodium channels, excitability of primary sensory neurons, and the molecular basis of pain. Muscle Nerve 22, 1177–1187.
Wells, J. E., Rowland, K. C., and Proctor, E. K. (2007). Hyperpolarization-activated channels in trigeminal ganglia innervating healthy and pulp-exposed teeth. Int. Endod. J. 40, 715–721. doi: 10.1111/j.1365-2591.2007.01297.x
Wirkner, K., Stanchev, D., Koles, L., Klebingat, M., Dihazi, H., Flehmig, G., et al. (2005). Regulation of human recombinant P2X3 receptors by ecto-protein kinase C. J. Neurosci. 25, 7734–7742. doi: 10.1523/jneurosci.2028-05.2005
Wood, J. N., Boorman, J. P., and Baker, K. O. D. (2004). Voltage-gated sodium channels and pain pathways. J. Neurobiol. 61, 55–71. doi: 10.1002/neu.20094
Wu, X., Wang, Z., Chen, Y., Xu, D., Zhang, P., and Wang, X. (2019). Newly discovered action of HpTx3 from venom of Heteropoda venatoria on Nav1.7 and its pharmacological implications in Analgesia. Toxins 11:680. doi: 10.3390/toxins11120680
Yan, J., and Dussor, G. (2014). Ion channels and migraine. Headache 54, 619–639. doi: 10.1111/head.12323
Yang, Y., Huang, J., Malgorzata, A., Estacion, M., Macala, L., Shah, P., et al. (2016). Nav1.7-A1632G mutation from a family with inherited erythromelalgia: enhanced firing of dorsal root ganglia neurons evoked by thermal stimuli. J. Neurosci. 36, 7511–7522. doi: 10.1523/jneuroscI.0462-16.2016
Yoshida, S., and Oka, H. (1998). Membrane properties of dissociated trigeminal mesencephalic neurons of the adult rat. Neurosci. Res. 30, 227–234. doi: 10.1016/s0168-0102(98)00003-0
Keywords: sodium channel, calcium channel, nociception, migraine, purinergic receptors, transgenic mice
Citation: Mehboob R, Marchenkova A, van den Maagdenberg AMJM and Nistri A (2021) Overexpressed NaV1.7 Channels Confer Hyperexcitability to in vitro Trigeminal Sensory Neurons of CaV2.1 Mutant Hemiplegic Migraine Mice. Front. Cell. Neurosci. 15:640709. doi: 10.3389/fncel.2021.640709
Received: 11 December 2020; Accepted: 09 April 2021;
Published: 25 May 2021.
Edited by:
Maria Cristina D’Adamo, University of Malta, MaltaReviewed by:
Theodore R. Cummins, Indiana University, Purdue University Indianapolis, United StatesJudith Ann Strong, University of Cincinnati, United States
Copyright © 2021 Mehboob, Marchenkova, van den Maagdenberg and Nistri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Riffat Mehboob, bWVoYm9vYi5yaWZmYXRAZ21haWwuY29t
†These authors have contributed equally to this work