 Guendalina Bergonzoni
Guendalina Bergonzoni Jessica Döring
Jessica Döring Marta Biagioli
Marta Biagioli
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci. , 10 February 2021
Sec. Cellular Neurophysiology
Volume 15 - 2021 | https://doi.org/10.3389/fncel.2021.628010
This article is part of the Research Topic Cell Type Diversity in the Nervous System: From Genes to Function View all 19 articles
Huntington’s disease (HD) is a devastating neurodegenerative disorder caused by an aberrant expansion of the CAG tract within the exon 1 of the HD gene, HTT. HD progressively impairs motor and cognitive capabilities, leading to a total loss of autonomy and ultimate death. Currently, no cure or effective treatment is available to halt the disease. Although the HTT gene is ubiquitously expressed, the striatum appears to be the most susceptible district to the HD mutation with Medium-sized Spiny Neurons (MSNs) (D1R and D2R) representing 95% of the striatal neuronal population. Why are striatal MSNs so vulnerable to the HD mutation? Particularly, why do D1R- and D2R-MSNs display different susceptibility to HD? Here, we highlight significant differences between D1R- and D2R-MSNs subpopulations, such as morphology, electrophysiology, transcriptomic, functionality, and localization in the striatum. We discuss possible reasons for their selective degeneration in the context of HD. Our review suggests that a better understanding of cell type-specific gene expression dysregulation within the striatum might reveal new paths to therapeutic intervention or prevention to ameliorate HD patients’ life expectancy.
Huntington’s disease (HD) is a rare, progressive, neurodegenerative disorder characterized by devastating motor, cognitive, and psychiatric symptoms. The monogenic, autosomal dominant disease is caused by a CAG repeat expansion in exon 1 of the HD gene (HTT), encoding for the huntingtin protein (MacDonald et al., 1993). The worldwide prevalence of HD is estimated to be 2.71 per 100,000 individuals (Pringsheim et al., 2012) and the average age of onset is between 30 and 50 years (Roos, 2010). So far, no treatments are available to block or slow-down the HD pathologic process, albeit mutant huntingtin lowering strategies are currently tested in clinical trials as promising therapeutic (Hoffmann-La Roche, 2020; Wave Life Sciences Ltd, 2020).
Although mutant huntingtin protein is ubiquitously expressed in all human districts, the brain, wherein the striatum, is the primary deteriorating region in HD (Saudou and Humbert, 2016; Ghosh and Tabrizi, 2018). Most striatal functions are mediated by inhibitory Medium-sized Spiny Neurons (MSNs), which comprise 95% of neurons in this area with the remaining being interneurons. There are two subtypes of MSNs differentiable by the expression of the D1 and D2 dopamine families’ receptors, constituting the direct and indirect pathways, respectively (Lanciego et al., 2012). The dorsal striatum (neostriatum) is the input module to the cortico-basal ganglia-thalamo-cortical loop (CBGTC), a neuronal circuit necessary for voluntary movement control. In the direct pathway, glutamatergic cortical terminals activate dopamine receptor 1 (D1R)-expressing MSNs, which exert their inhibitory effect on the globus pallidus internal segment (GPi) (Entopeduncular nucleus, in rodents). Inhibitory neurons in this area, project to the ventral anterior/lateral motor thalamus. Thus, the stimulation of D1R-MSNs has a net excitatory effect on the motor thalamus, allowing the final switch of the motor cortex and the stimulation of skeletal muscles. On the other hand, in the indirect pathway, dopamine receptor 2 (D2R)-expressing, inhibitory MSNs are also stimulated by glutamate release of cortical terminals. D2R neurons connect to the GP through an indirect loop, such that, they first project to and inhibit the globus pallidus external segment (GPe). These neurons firstly connect to the subthalamic nucleus exciting the area through glutamate release. Finally, the excitation of inhibitory GPi neurons produces motor thalamus repression (Alexander et al., 1986; Bolam et al., 2000). Dysfunction and death of striatal MSNs are the main causes for the motor disorders associated with HD (Ghosh and Tabrizi, 2018). In this review, we provide an overview of key pathological pathways leading to striatal degeneration. Furthermore, we describe general characteristics and physiological differences between D1R- and D2R-MSNs and highlight distinct morphological and functional alterations of MSNs during the disease. Our review emphasizes the importance of understanding cell-type specific physiological differences contributing to striatal vulnerability which may provide insights toward new avenues of therapeutic intervention.
Because of unavailability of pre-symptomatic HD brain tissues, the reasons behind selective striatal vulnerability in HD were mostly investigated using animal models. In fact, the basal ganglia and, particularly, the cortico-striatal motor circuitry, appears to be conserved in mouse, minipig, and primates (Vodicka et al., 2005; Stephenson-Jones et al., 2011; Balsters et al., 2020). Thus, genetically engineered models, bearing normal or pathological CAG repeat lengths, have revealed important pathogenic mechanisms of the HD mutation (Menalled, 2005; Lerner et al., 2012; Peng et al., 2016; Table 1 and Figures 1A,B). Nevertheless, several salient features of human HD pathology–such as overt striatal atrophy, cortical degeneration, and onset of choreic movements–failed to fully replicate in animal models of the disease (Rubinsztein, 2002).
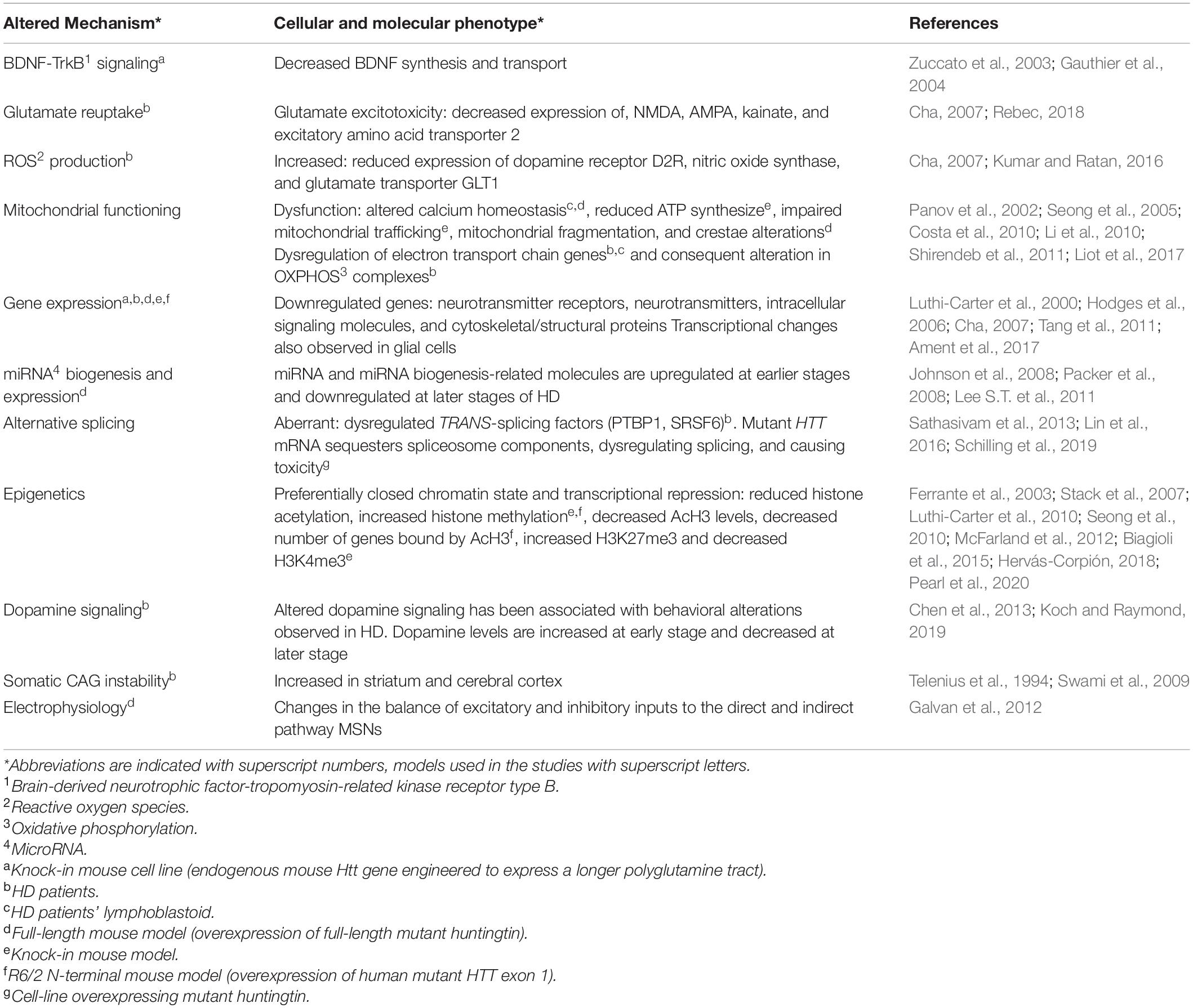
Table 1. Pathogenic mechanisms correlated with striatal degeneration in Huntington’s Disease.

Figure 1. HD profoundly alters the striatum and MSNs in both patients and mouse models, upper panel. (A) Schematic representation of HD patient brain. At the early stage of the disease (grade 1–2), patients already manifest mild brain atrophy and astrogliosis (Ross and Tabrizi, 2011; Rüb et al., 2016). MSNs undergo proliferative expansion and show increased number and size of spines (Ferrante et al., 1991). At the molecular level, a reduction of synaptic proteins and alterations in gene transcription are detected (DiProspero et al., 2004; Hodges et al., 2006). These changes continue during advanced stages (grade 3–4), when brain atrophy becomes prominent (Rüb et al., 2016). MSNs undergo degenerative changes and spine loss, and mutant huntingtin aggregates can be detected within nuclei (Ferrante et al., 1991; DiFiglia et al., 1997; Rüb et al., 2016). Altered energy metabolism and somatic instability of the CAG tract are detected (Gines et al., 2003; Swami et al., 2009; Liot et al., 2017). (B) Knock-in HD mouse models (CAG > 80) faithfully recapitulate the human HD mutation and mimic several aspects of the human condition. At early age (<4 months), mice show mild brain atrophy (Peng et al., 2016) and diffuse accumulation of mutant huntingtin within MSNs nuclei (Carty et al., 2015). At the molecular level, energy metabolism and transcription are altered (Gines et al., 2003; Mochel et al., 2012; Ament et al., 2017). Somatic instability is already detectable at this stage (Pinto et al., 2013). At later stages (>6 months), brain atrophy becomes more prominent and astrogliosis could be detected (Menalled, 2005; Lerner et al., 2012; Peng et al., 2016). MSNs show reduced dendritic complexity and spine loss, and nuclear mutant huntingtin aggregates inclusions (Lerner et al., 2012; Carty et al., 2015). The molecular alterations proceed and somatic instability becomes particularly evident (Lee J.M. et al., 2011). Specific pathways and phenotypes developed by D1R- and D2R-MSNs upon expression of mutant huntingtin, lower panel. (C,C′) Striatopallidal neurons (D2R-MSNs) are affected earlier than striatonigral ones (D1R-MSNs) by HD mutation (Sapp et al., 1995). (D,D′) Gene expression profiling on R6/2 mice revealed that neurotrophin pathway is specifically upregulated in D2R-MSNs, while mismatch repair (MMR) and synaptic functioning pathways seem to be altered in both MSNs subpopulations (Lee et al., 2020). (E) In both R6/2 and zQ175 mice, oxidative phosphorylation (OXPHOS) downregulation observed in D2R cells contribute to mitochondrial dysfunction (Lee et al., 2020), while (E′) OXPHOS genes are upregulated in D1R cells, possibly suggesting a homeostatic response (Lee et al., 2020). (F) In mice and rat striatal cell cultures overexpressing mutant huntingtin, D2R stimulation enhances mutant huntingtin aggregation and mitochondrial dysfunction (Charvin et al., 2005; Benchoua et al., 2008). (F′) In HdhQ111 models, dopamine and glutamate synergistically enhance MSNs sensitivity to mutant huntingtin toxicity through D1R activation (Paoletti et al., 2008). (G,G′) In 12 months old zQ175 KI mice, D1R neurons show proliferative expansion of the dendritic arborization and a significant reduction in the density of thin spines, while D2R neurons do not show significant differences (Goodliffe et al., 2018). (H,H′) In the same model, only D1R neurons exhibit reduced rheobase and action potential amplitude (arrows). Figure created with BioRender.com.
Huntington’s disease post-mortem brains revealed that MSNs exhibit altered morphology, with proliferative changes–recurving and branching of dendrites and increased number and size of spines–since early stages of the disease. Degenerative alterations–truncation of the dendritic arborization and loss of spines–are characteristics of severe grades (Ferrante et al., 1991; Figure 1A). MSNs of 3 months old R6/2 N-terminal transgenic line–which overexpress human mutant HTT exon 1 (Mangiarini et al., 1996)- and of 20–26 months old HdhQ140 knock-in mice–with the endogenous mouse Htt gene engineered to express a longer polyglutamine tract (Menalled et al., 2003)- do present similar decreased spine density and size of dendritic arborization (Klapstein et al., 2001; Lerner et al., 2012; Figure 1B).
Other studies in post-mortem brains also highlighted mutant huntingtin aggregates within neuronal MSNs nuclei (DiFiglia et al., 1997; Rüb et al., 2016; Figure 1A). Similarly, mutant huntingtin diffuse nuclear localization could be visualized at earlier ages (3 months) in MSNs of zQ175 knock-in mouse models (Menalled et al., 2012), while clear nuclear inclusions can be spotted only at later stages (8–12 months old) (Carty et al., 2015; Figure 1B).
Most observations point toward toxic gain-of-function for the pathogenic mechanisms. However, some data suggest that a loss-of-function mechanism should not be completely ruled out (Borrell-Pagès et al., 2006). Specifically, mutant huntingtin impairs the brain-derived neurotrophic factor-tropomyosin-related kinase receptor type B (BDNF-TrkB) signaling in striatal neurons (Table 1). This deficiency plays a pivotal role in dysfunction and death of MSNs and may represent a therapeutic target for HD treatment. Accordingly, several studies examined whether increasing levels of BDNF may be a viable strategy (Baydyuk and Xu, 2014). R6/2 mice, perfused with BDNF at 4 and 13 weeks of age showed less severe neurological dysfunction (Giampà et al., 2013), and significantly reduced motor coordination impairment (Giralt et al., 2011). However, the administration of citalopram, an antidepressant believed to increase BDNF levels, failed to improve motor and psychiatric symptoms in HD patients (Beglinger et al., 2014).
Recently, a significant increase in reactive oxygen species (ROS) production was described in the striatum of HD patients (Kumar and Ratan, 2016). ROS, produced by excitotoxicity or mitochondrial dysfunction, are important mediators of cell death (Gu et al., 1996; Browne et al., 1997). Coherently, mutant huntingtin seems to interfere with mitochondrial functioning (Table 1). Lymphoblasts from HD patients present mitochondrial fragmentation and cristae alterations (Costa et al., 2010), while cortical specimens from grade 3–4 HD patients display downregulation of complexes II, III, and IV of the oxidative phosphorylation (OXPHOS) pathway (Tabrizi et al., 2000; Shirendeb et al., 2011; Liot et al., 2017; Figure 1A). Energy metabolism alterations were also detected in R6/2 transgenic mice (Tabrizi et al., 2000), in HdhQ111 knock-in models with decreased cAMP levels in the striatum at 10 weeks of age (Gines et al., 2003; Mochel et al., 2012) and, finally, precursor cells from striatal primordia of knock-in mice (Trettel et al., 2000) show significantly reduced respiration and ATP production (Figure 1B). Thus, considering that striatal neurons require higher amounts of ATP to maintain their hyperpolarized resting membrane potential (Hammond, 2015), it is conceivable that they might result more sensitive to mitochondrial dysfunction.
Initial studies on HD mouse models and later on HD post-mortem striatum revealed that mutant huntingtin causes transcriptional dysregulation of signaling pathways, neuronal, gliosis, and neuroinflammatory genes. Moreover, studies on HdhQ111 knock-in models also demonstrated that transcriptional alterations can already be detected at 9 weeks of age (Cha, 2000; Luthi-Carter et al., 2000; Hodges et al., 2006; Ament et al., 2017; Table 1). Interestingly, striatal transcriptional changes are among the earliest detectable phenotypes in HD mouse models (Langfelder et al., 2016; Ament et al., 2017, 2018), which conform with HD patients (Seredenina and Luthi-Carter, 2012; Labadorf et al., 2015; Figures 1A,B).
Transcriptional dysregulation of synaptic proteins, such as complexin 2, dynamin, and PACSIN 1, correlates with neuronal morphological changes and reduction in the number of axonal fibers in early-stage HD patients (DiProspero et al., 2004; Han et al., 2010; Figure 1A). Furthermore, altered microRNA biogenesis and expression was reported in HD post-mortem tissues and in YAC128 murine models of full-length mutant huntingtin overexpression (Johnson et al., 2008; Packer et al., 2008; Lee S.T. et al., 2011; Table 1). Notably, mutant huntingtin can directly or indirectly compromise the epigenetic status of brain cells (Table 1), at least in part explaining the observed transcriptional dysregulation (Stack et al., 2007; Seong et al., 2010; McFarland et al., 2012; Biagioli et al., 2015; Hervás-Corpión, 2018; Pearl et al., 2020).
Recent RNAseq analysis of HD patients’ motor cortex revealed that mutant huntingtin interferes with RNA processing and induces aberrant alternative splicing (Table 1), affecting the expression levels of TRANS-splicing factors and/or trapping specific RNA binding proteins (Sathasivam et al., 2013; Lin et al., 2016; Schilling et al., 2019).
Dopaminergic inputs from the substantia nigra are crucial for proper signaling of striatal MSNs in the basal ganglia circuit. Indeed, substantia nigra pars compacta (SNc) modulates the direct and indirect pathways by releasing dopamine, which has an excitatory effect on D1R and an inhibitory one on D2R. Consequently, dopamine excites the direct pathways and inhibits the indirect pathway, producing an overall stimulation of the motor activity (Leisman et al., 2013).
Studies on HD patients suggest that early stages of the disease are characterized by an increase in dopamine levels, contributing to choreiform symptoms. This might be due to the inhibitory effect of MSNs projecting to the SNc, which, in early stages, may produce hyperactivation of this pathway. Conversely, as disease progresses, dopamine levels decrease–possibly because of dopaminergic nigrostriatal terminals loss–accounting for the late akinetic stage (Chen et al., 2013; Koch and Raymond, 2019; Table 1). Accordingly, studies on both patients and mouse models confirmed an increase in dopamine release and tyrosine hydroxylase levels in early HD, followed by a reduction of the same parameters in advanced disease conditions (Koch and Raymond, 2019).
Within striatal MSNs, a modulatory mechanism between dopamine and glutamate was observed. On one hand, dopamine binding to D1R stimulates surface expression of NMDA and AMPA receptors, resulting in an increased responsiveness of D1R-MSNs to glutamate release. On the other hand, dopamine binding to D2R decreases surface AMPA receptors, reducing their glutamate excitability (Surmeier et al., 2007). Interestingly, both in patients and murine models, glutamate signaling follows the same pattern of dopamine alterations, being increased during HD early stages and decreased at advanced stages (Chen et al., 2013), thus suggesting a cross-talk between these two neurotransmitters.
The expanded CAG repeat in the mutant huntingtin gene is unstable, undergoing progressive length increases over time and resulting in somatic mosaicism in selective human body districts (Table 1). Specifically, it is possible that high level of somatic CAG instability in the striatum and cerebral cortex (Telenius et al., 1994; Swami et al., 2009; Lee J.M. et al., 2011) contributes to HD pathology (Figures 1A,B). Knock-out of DNA mismatch repair (MMR) proteins in HdhQ111 knock-in mice showed that Msh2/3/6, Mlh1, and Mlh3 are modifiers of somatic CAG instability (Wheeler, 2003; Dragileva et al., 2009; Pinto et al., 2013). Importantly, genome-wide association analysis of a cohort of 9,000 HD patients confirmed MMR genes and specifically MLH1 as crucial HD genetic modifiers (Lee et al., 2015, 2019).
Medium-sized spiny neurons are characterized by a small to medium cellular body size (10–15 μm in diameter) and a radially oriented large dendritic tree covered by spines. Upon dopamine binding, D1R activates adenylyl cyclase (AC) signaling, leading to an excitatory effect, whereas D2R represses AC through Gi-protein signaling, resulting in inhibition (Lanciego et al., 2012). Striatopallidal (D2R) and striatonigral (D1R) neurons exhibit a random distribution in the murine rostral, dorsal striatum. However, a regionalization is observed in the caudal part, near the GPe, which comprises almost exclusively D1R-MSNs (Gangarossa et al., 2013). It is well accepted that D2R-MSNs are affected earlier than D1R-MSNs (Sapp et al., 1995; Figures 1C,C′) and, accordingly, GPe-targeting MSNs show substantial loss in patients at early stages of the disease (Albin et al., 1992). The lack of inhibition of the GPe by D2R-MSNs results in an excessive activation of the pallidal neurons, leading to choreiform movements observed in HD (Hedreen and Folstein, 1995). Nevertheless, in the latest stages, GPi-targeting MSNs of the direct pathway undergo marked decline, resulting in akinetic movements and rigidity (Deng et al., 2004; Lanciego et al., 2012).
Morphologically, striatonigral neurons show more primary dendrites and a more extended arborization than striatopallidal ones. Experimental simulation suggested that different dendritic areas may contribute to the divergent electrophysiological properties. Indeed, experiments performed on brain slices from D1R and D2R-EGFP BAC transgenic mice demonstrated that D1R neurons display a more hyperpolarized resting membrane potential and a greater rheobase (Gertler et al., 2008).
Accordingly, recent experiments using Drd1a-td Tomato mice revealed an increased intrinsic excitability for D2R-MSNs compared to D1R-MSNs. This might be due to the different rheobase, which is decreased in D2R neurons (Willett et al., 2019). Previous studies, however, pointed to a differential role of M1 muscarinic receptors activation, which downregulates Kir channel currents in striatopallidal MSNs, but not in striatonigral ones (Shen et al., 2007).
Considering that increased release of glutamate might contribute to MSNs degeneration (DiFiglia, 1990; Cepeda et al., 2007), it is noteworthy that D2R-MSNs receive more cortical inputs, mainly from pyramidal neurons (Francelle et al., 2014). Moreover, cortical axons making synapses with D2R-MSNs are larger in size, compared with the ones from D1R neurons (Lei, 2004). Altogether, these characteristics expose them to higher excitotoxicity, possibly reflecting on their greater susceptibility to cell death (Table 1). D2R-, but not D1R-, MSNs can form the protein complex with β-arrestin2, Akt, and protein phosphatase 2A (PP2A), which, in turn, reduces the phosphorylation of glycogen synthase kinase-3 (GSK3) (Harrison et al., 2013). GSK3 plays crucial roles in neuronal function, synapse formation, and neurite outgrowth (Beaulieu et al., 2004, 2005). Since both Rhes and Akt have been demonstrated to interact with and modulate mutant huntingtin toxicity, the Akt/β-Arrestin 2PP2A/GSK3 pathway may represent an additional mediator of D2R specific selective vulnerability (Colin et al., 2005; Lee et al., 2014).
Furthermore, TrkB is unequally expressed in striatal MSNs, with higher level in D2R-MSNs (Baydyuk and Xu, 2014). Thus, the aberrant BDNF-TrkB signaling caused by mutant huntingtin might have stronger effects in these cells (Table 1). Analysis of mouse striatum using single cell RNA sequencing (scRNA-seq) unveiled additional transcriptional differences between D1R- and D2R-MSNs. Further complexity emerged following the discovery of region-specific molecular markers for dorsal D2R neurons (Puighermanal et al., 2020), the identification of discrete subgroups of D1R and D2R neurons (Gokce et al., 2016) and of a possible third subtype of MSNs, which may have unique characteristics (Gokce et al., 2016). The existence of a third subpopulation of MSNs was also reported by Saunders et al. (2018), who observed a cluster of neurons in the striatum of C57BL6/N, co-expressing Drd1 and Adora2a, named as “eccentric” MSNs. It is still premature to point to a clear connection between these physiological differences and the unequal cellular vulnerability to HD of the two MSNs subpopulations. Nevertheless, evaluation and integration of these single-cell analyses with other molecular aspects, such as alternative splicing, somatic mosaicism, and epigenetics differences between MSNs subtypes (Table 1), will be instrumental to understand the molecular mechanisms impinging on different vulnerability of D1R- and D2R-MSNs.
Recent studies on HD patients described rostro-caudal and dorso-ventral degenerative gradients. Specifically, the caudal striatum displayed greater neuronal death compared to the rostral part, while the dorsal-medial area seemed to degenerate faster compared to the ventral-lateral striatum (Morigaki and Goto, 2017). Although the contribution of MSNs’ physiological regionalization to selective vulnerability in HD is not fully dissected, some unequal distribution of the subclasses (Gangarossa et al., 2013) and subgroups (Gokce et al., 2016; Puighermanal et al., 2020) of MSNs might play a role.
To dissect why D1R- and D2R-MSNs are differentially affected by HD, Lee et al. (2020) highlighted thousands dysregulated protein-coding genes implicated in OXPHOS, synaptic functioning and circadian entrainment by using translating ribosome affinity purification and snRNA-seq of D1R and D2R neurons of HD patients and mouse models (R6/2 and zQ175DN, a knock-in zQ175 line without neomycin cassette) (Franich et al., 2019). Strikingly, downregulation of OXPHOS and upregulation of neurotrophin pathway genes in D2R neurons indicated a cell-type specific response to the disease (Figure 1D). Notably, Lee et al. (2020) demonstrated that OXPHOS genes downregulation causes mitochondrial dysfunction (Figure 1E) and mitochondrial RNA release in the cytosol, which, in turn, activates protein kinase R and cellular toxicity through the interferon pathways. Coherently, it was shown previously that D2R contributes to mutant huntingtin aggregation and mitochondrial impairment (Charvin et al., 2005, 2008; Benchoua et al., 2008; Figure 1F). Moreover, the upregulation of MMR genes, implicated in somatic instability of the CAG tract (Table 1), in both D1R- and D2R-MSNs supported a possible predisposing feature for selective degeneration (Figures 1D,D′). However, additional studies will be needed to correlate these findings with HD progression and MSNs vulnerability (Lee et al., 2020).
On the other hand, analysis on YAC128 and BACHD mouse models demonstrated that glutamate transmission was increased in D1R neurons at early disease and decreased in both D1R and D2R cells at advanced stages (André et al., 2011). Since a modulatory mechanism between dopamine and glutamate was observed within healthy striatal MSNs (Surmeier et al., 2007), dopamine and glutamate might synergistically enhance sensitivity to mutant huntingtin toxicity through D1R but not D2R activation (Paoletti et al., 2008; Figure 1F′). Similarly, in a different study using 12 months old zQ175 knock-in models, striatonigral neurons showed more prominent morphological and electrophysiological changes than striatopallidal ones (Goodliffe et al., 2018; Figures 1G′,H′). While this view is in contrast with the well-established hypothesis that D2R neurons are selectively damaged in early stages of HD, nonetheless, these findings might highlight the presence of a compensatory mechanism in D1R neurons. It is interesting to note that, in the knock-in mouse model zQ175DN, Lee et al. (2020) reported an increased expression for OXPHOS genes in D1R neurons (Figure 1D′), which, indeed, may support the activation of a transcriptional protective response in this subclass of MSNs.
In conclusion, our review provides a general overview into key pathological pathways leading to neuronal cell death of striatal MSNs in HD. We specifically focus on differences between D1R- and D2R-MSNs, underpinning sensitizing or protective features that might determine diverse responses to the same mutation. From initial studies, a combination of cell-type specific and non-specific reactions seem to be activated in HD, sensitizing D2R-MSNs to cell death. However, the application of single cell techniques, such as, but not limited to, scRNA-seq, is nowadays pioneering a new field of discussion, addressing the contribution of each single cell type (neuronal or glial) to HD striatal vulnerability. Specifically, other cell clusters in the striatum, such as striatal interneurons and astroglia, seem to respond to the HD mutation with some altered genes and pathways as in D1R and D2R, while microglia, oligodendrocytes, and oligodendrocytes precursors seem to be less responsive (Lee et al., 2020). The role of astrocytes in HD has been previously proposed, since mutant huntingtin downregulates the expression of the glial glutamine transporter GLT-1, exacerbating neuronal excitotoxicity. Similarly, specific mutant huntingtin expression in astrocytes prompts motor function deficits, weight loss, and age-dependent neurological phenotypes in transgenic mouse models (Bradford et al., 2009, 2010). Previous studies have provided evidence that activated microglia and reactive astrocytes might contribute to human HD pathology, perpetuating inflammation (Palpagama et al., 2019). However, still debatable is the attribution of beneficial vs. detrimental effects to activated microglia and astrocytes. Moreover, the highly heterogeneous class of striatal interneurons, generally thought to be spared in HD (Cicchetti et al., 2000), still displays selective degeneration in presence of the HD mutation, with documented loss of only parvalbumin-positive interneurons (Cicchetti et al., 2000; Reiner and Deng, 2018). Therefore, a better understanding of the neuroinflammatory environment, but also a detailed clarification of the interneurons population in the HD brain is needed. Moreover, analysis of chromatin, genome-architecture, and spatial distribution will assist in the elucidation of single cell characteristics. This will offer a new angle of interpretation to selective vulnerability to HD and will possibly pave the way to new avenues of therapeutic intervention.
GB made substantial contributions to conception, design of the work, and writing of the manuscript. JD assisted with drafting and critical reading of the text. MB conceived, supervised the project, and wrote the paper. All authors contributed to the article and approved the submitted version.
This work was supported by the University of Trento, the HDSA Human Biology Project and the EHDN 1041 to MB. MB was a recipient of a Marie SkłodowskaŰCurie reintegration fellowship (The European Union’s Horizon 2020 Research and Innovation Program) under the grant agreement No. 706567.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Albin, R. L., Reiner, A., Anderson, K. D., Dure, L. S., Handelin, B., Balfour, R., et al. (1992). Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington’s disease. Ann. Neurol. 31, 425–430. doi: 10.1002/ana.410310412
Alexander, G. E., DeLong, M. R., and Strick, P. L. (1986). Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu. Rev. Neurosci. 9, 357–381. doi: 10.1146/annurev.ne.09.030186.002041
Ament, S. A., Pearl, J. R., Cantle, J. P., Bragg, R. M., Skene, P. J., Coffey, S. R., et al. (2018). Transcriptional regulatory networks underlying gene expression changes in Huntington’s disease. Mol. Syst. Biol. 14:e7435. doi: 10.15252/msb.20167435
Ament, S. A., Pearl, J. R., Grindeland, A., St Claire, J., Earls, J. C., Kovalenko, M., et al. (2017). High resolution time-course mapping of early transcriptomic, molecular and cellular phenotypes in Huntington’s disease CAG knock-in mice across multiple genetic backgrounds. Hum. Mol. Genet. 26, 913–922. doi: 10.1093/hmg/ddx006
André, V. M., Cepeda, C., Fisher, Y. E., Huynh, M., Bardakjian, N., Singh, S., et al. (2011). Differential electrophysiological changes in striatal output neurons in Huntington’s disease. J. Neurosci. 31, 1170–1182. doi: 10.1523/JNEUROSCI.3539-10.2011
Balsters, J. H., Zerbi, V., Sallet, J., Wenderoth, N., and Mars, R. B. (2020). Primate homologs of mouse cortico- striatal circuits. elife 9:e53680.
Baydyuk, M., and Xu, B. (2014). BDNF signaling and survival of striatal neurons. Front. Cell. Neurosci. 8:254. doi: 10.3389/fncel.2014.00254
Beaulieu, J.-M., Sotnikova, T. D., Marion, S., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. (2005). An Akt/β-Arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273. doi: 10.1016/j.cell.2005.05.012
Beaulieu, J.-M., Sotnikova, T. D., Yao, W. D., Kockeritz, L., Woodgett, J. R., Gainetdinov, R. R., et al. (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci.U.S.A. 101, 5099–5104. doi: 10.1073/pnas.0307921101
Beglinger, L. J., Adams, W. H., Langbehn, D., Fiedorowicz, J. G., Jorge, R., Biglan, K., et al. (2014). Results of the citalopram to enhance cognition in Huntington disease trial: citalopram in HD. Mov. Dis. 29, 401–405. doi: 10.1002/mds.25750
Benchoua, A., Trioulier, Y., Diguet, E., Malgorn, C., Gaillard, M. C., Dufour, N., et al. (2008). Dopamine determines the vulnerability of striatal neurons to the N-terminal fragment of mutant huntingtin through the regulation of mitochondrial complex II. Hum. Mol. Genet. 17, 1446–1456. doi: 10.1093/hmg/ddn033
Biagioli, M., Ferrari, F., Mendenhall, E. M., Zhang, Y., Erdin, S., Vijayvargia, R., et al. (2015). Htt CAG repeat expansion confers pleiotropic gains of mutant huntingtin function in chromatin regulation. Hum. Mol. Genet. 24, 2442–2457. doi: 10.1093/hmg/ddv006
Bolam, J. P., Hanley, J. J., Booth, P. A., and Bevan, M. D. (2000). Synaptic organisation of the basal ganglia. J. Anat. 196 (Pt 4), 527–542. doi: 10.1046/j.1469-7580.2000.19640527.x
Borrell-Pagès, M., Zala, D., Humbert, S., and Saudou, F. (2006). Huntington’s disease: from huntingtin function and dysfunction to therapeutic strategies. Cell. Mol. Life Sci. 63, 2642–2660. doi: 10.1007/s00018-006-6242-0
Bradford, J., Shin, J. Y., Roberts, M., Wang, C. E., Li, X. J., and Li, S. (2009). Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl. Acad. Sci.U.S.A. 106, 22480–22485. doi: 10.1073/pnas.0911503106
Bradford, J., Shin, J. Y., Roberts, M., Wang, C. E., Sheng, G., Li, S., et al. (2010). Mutant huntingtin in glial cells exacerbates neurological symptoms of huntington disease mice. J. Biol. Chem. 285, 10653–10661. doi: 10.1074/jbc.M109.083287
Browne, S. E., Bowling, A. C., MacGarvey, U., Baik, M. J., Berger, S. C., Muqit, M. M., et al. (1997). Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann. Neurol. 41, 646–653. doi: 10.1002/ana.410410514
Carty, N., Berson, N., Tillack, K., Thiede, C., Scholz, D., Kottig, K., et al. (2015). Characterization of HTT inclusion size, location, and timing in the zQ175 mouse model of Huntington’s disease: an in vivo high-content imaging study. PLoS One 10:e0123527. doi: 10.1371/journal.pone.0123527
Cepeda, C., Wu, N., André, V. M., Cummings, D. M., and Levine, M. S. (2007). The corticostriatal pathway in Huntington’s disease. Prog. Neurobiol. 81, 253–271. doi: 10.1016/j.pneurobio.2006.11.001
Cha, J.-H. J. (2000). Transcriptional dysregulation in Huntington’s disease. Trends Neurosci. 23, 387–392.
Cha, J.-H. J. (2007). Transcriptional signatures in Huntington’s disease. Prog. Neurobiol. 83, 228–248.
Charvin, D., Roze, E., Perrin, V., Deyts, C., Betuing, S., Pagès, C., et al. (2008). Haloperidol protects striatal neurons from dysfunction induced by mutated huntingtin in vivo. Neurobiol. Dis. 29, 22–29. doi: 10.1016/j.nbd.2007.07.028
Charvin, D., Vanhoutte, P., Pagès, C., Borrelli, E., and Caboche, J. (2005). Unraveling a role for dopamine in Huntington’s disease: the dual role of reactive oxygen species and D2 receptor stimulation. Proc. Natl. Acad. Sci.U.S.A. 102, 12218–12223. doi: 10.1073/pnas.0502698102
Chen, J. Y., Wang, E. A., Cepeda, C., and Levine, M. S. (2013). Dopamine imbalance in Huntington’s disease: a mechanism for the lack of behavioral flexibility. Front. Neurosci. 7:114. doi: 10.3389/fnins.2013.00114
Cicchetti, F., Prensa, L., Wu, Y., and Parent, A. (2000). Chemical anatomy of striatal interneurons in normal individuals and in patients with Huntington’s disease. Brain Res. Brain Res. Rev. 34, 80–101. doi: 10.1016/s0165-0173(00)00039-4
Colin, E., Régulier, E., Perrin, V., Dürr, A., Brice, A., Aebischer, P., et al. (2005). Akt is altered in an animal model of Huntington’s disease and in patients. Eur. J. Neurosci. 21, 1478–1488. doi: 10.1111/j.1460-9568.2005.03985.x
Costa, V., Giacomello, M., Hudec, R., Lopreiato, R., Ermak, G., Lim, D., et al. (2010). Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol. Med. 2, 490–503. doi: 10.1002/emmm.201000102
Deng, Y. P., Albin, R. L., Penney, J. B., Young, A. B., Anderson, K. D., and Reiner, A. (2004). Differential loss of striatal projection systems in Huntington’s disease: a quantitative immunohistochemical study. J. Chem. Neuroanatomy 27, 143–164. doi: 10.1016/j.jchemneu.2004.02.005
DiFiglia, M. (1990). Excitotoxic injury of the neostriatum: a model for Huntington’s disease. Trends Neurosci. 13, 286–289. doi: 10.1016/0166-2236(90)90111-M
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science (New York N.Y.) 277, 1990–1993. doi: 10.1126/science.277.5334.1990
DiProspero, N. A., Chen, E. Y., Charles, V., Plomann, M., Kordower, J. H., and Tagle, D. A. (2004). Early changes in Huntington’s disease patient brains involve alterations in cytoskeletal and synaptic elements. J. Neurocytol. 33, 517–533. doi: 10.1007/s11068-004-0514-8
Dragileva, E., Hendricks, A., Teed, A., Gillis, T., Lopez, E. T., Friedberg, E. C., et al. (2009). Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol. Dis. 33, 37–47. doi: 10.1016/j.nbd.2008.09.014
Ferrante, R. J., Kowall, N. W., and Richardson, E. P. (1991). Proliferative and degenerative changes in striatal spiny neurons in huntington’s disease: a combined study using the section-golgi method and calbindin D28k lmmunocytochemistry. J. Neurosci. 11, 3877–3887.
Ferrante, R. J., Kubilus, J. K., Lee, J., Ryu, H., Beesen, A., Zucker, B., et al. (2003). Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 23, 9418–9427.
Francelle, L., Galvan, L., and Brouillet, E. (2014). Possible involvement of self-defense mechanisms in the preferential vulnerability of the striatum in Huntington’s disease. Front. Cell. Neurosci. 8:295. doi: 10.3389/fncel.2014.00295
Franich, N. R., Hickey, M. A., Zhu, C., Osborne, G. F., Ali, N., Chu, T., et al. (2019). Phenotype onset in Huntington’s disease knock-in mice is correlated with the incomplete splicing of the mutant huntingtin gene. J. Neurosci. Res. 97, 1590–1605. doi: 10.1002/jnr.24493
Galvan, L., André, V. M., Wang, E. A., Cepeda, C., and Levine, M. S. (2012). Functional differences between direct and indirect striatal output pathways in huntington’s Disease. J. Huntingtons Dis. 1, 17–25. doi: 10.3233/JHD-2012-120009
Gangarossa, G., Espallergues, J., Mailly, P., De Bundel, D., de Kerchove d’Exaerde, A., Hervé, D., et al. (2013). Spatial distribution of D1R- and D2R-expressing medium-sized spiny neurons differs along the rostro-caudal axis of the mouse dorsal striatum. Front. Neural Circ. 7:124. doi: 10.3389/fncir.2013.00124
Gauthier, L. R., Charrin, B. C., Borrell-Pagès, M., Dompierre, J. P., Rangone, H., Cordelières, F. P., et al. (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118, 127–138. doi: 10.1016/j.cell.2004.06.018
Gertler, T. S., Chan, C. S., and Surmeier, D. J. (2008). Dichotomous anatomical properties of adult striatal medium spiny neurons. J. Neurosci. 28, 10814–10824. doi: 10.1523/JNEUROSCI.2660-08.2008
Ghosh, R., and Tabrizi, S. J. (2018). “Huntington disease,” in Handbook of Clinical Neurology, eds D. H. Geschwind, H. L. Paulson, and C. Klein (Amsterdam: Elsevier), 255–278. doi: 10.1016/B978-0-444-63233-3.00017-8
Giampà, C., Montagna, E., Dato, C., Melone, M. A., Bernardi, G., and Fusco, F. R. (2013). Systemic delivery of recombinant brain derived neurotrophic factor (BDNF) in the R6/2 mouse model of Huntington’s disease. PLoS One 8:e64037. doi: 10.1371/journal.pone.0064037
Gines, S., Seong, I. S., Fossale, E., Ivanova, E., Trettel, F., Gusella, J. F., et al. (2003). Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum. Mol. Genet. 12, 497–508. doi: 10.1093/hmg/ddg046
Giralt, A., Carretón, O., Lao-Peregrin, C., Martín, E. D., and Alberch, J. (2011). Conditional BDNF release under pathological conditions improves Huntington’s disease pathology by delaying neuronal dysfunction. Mol. Neurodegen. 6:71. doi: 10.1186/1750-1326-6-71
Gokce, O., Stanley, G. M., Treutlein, B., Neff, N. F., Camp, J. G., Malenka, R. C., et al. (2016). Cellular taxonomy of the mouse striatum as revealed by single-cell RNA-Seq. Cell Rep. 16, 1126–1137. doi: 10.1016/j.celrep.2016.06.059
Goodliffe, J. W., Song, H., Rubakovic, A., Chang, W., Medalla, M., Weaver, C. M., et al. (2018). Differential changes to D1 and D2 medium spiny neurons in the 12-month-old Q175+/- mouse model of Huntington’s Disease. PLoS One 13:e0200626. doi: 10.1371/journal.pone.0200626
Gu, M., Gash, M. T., Mann, V. M., Javoy-Agid, F., Cooper, J. M., and Schapira, A. H. (1996). Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 39, 385–389. doi: 10.1002/ana.410390317
Han, I., You, Y., Kordower, J. H., Brady, S. T., and Morfini, G. A. (2010). Differential vulnerability of neurons in Huntington’s disease: the role of cell type-specific features. J. Neurochem. 113, 1073–1091. doi: 10.1111/j.1471-4159.2010.06672.x
Harrison, L. M., Muller, S. H., and Spano, D. (2013). Effects of the ras homolog rhes on Akt/Protein kinase B and glycogen synthase kinase 3 phosphorylation in striatum. Neuroscience 236C, 21–30. doi: 10.1016/j.neuroscience.2012.12.062
Hedreen, J. C., and Folstein, S. E. (1995). Early loss of neostriatal striosome neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol. 54, 105–120. doi: 10.1097/00005072-199501000-00013
Hervás-Corpión, I. (2018). Early alteration of epigenetic-related transcription in Huntington’s disease mouse models. Sci. Rep. 8:9925.
Hodges, A., Strand, A. D., Aragaki, A. K., Kuhn, A., Sengstag, T., Hughes, G., et al. (2006). Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 15, 965–977. doi: 10.1093/hmg/ddl013
Hoffmann-La Roche (2020). A Randomized, Multicenter, Double-Blind, Placebo-Controlled, Phase III Clinical Study to Evaluate the Efficacy and Safety of Intrathecally Administered RO7234292 (RG6042) in Patients With Manifest Huntington’s Disease. Clinical trial registration NCT03761849. clinicaltrials.gov. Available online at: https://clinicaltrials.gov/ct2/show/NCT03761849 (accessed October 12, 2020).
Johnson, R., Zuccato, C., Belyaev, N. D., Guest, D. J., Cattaneo, E., Buckley, N. J., et al. (2008). A microRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol. Dis. 29, 438–445. doi: 10.1016/j.nbd.2007.11.001
Klapstein, G. J., Fisher, R. S., Zanjani, H., Cepeda, C., Jokel, E. S., Chesselet, M. F., et al. (2001). Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington’s disease transgenic mice. J. Neurophysiol. 86, 2667–2677. doi: 10.1152/jn.2001.86.6.2667
Koch, E. T., and Raymond, L. A. (2019). Dysfunctional striatal dopamine signaling in Huntington’s disease. J. Neurosci. Res. 97, 1636–1654. doi: 10.1002/jnr.24495
Kumar, A., and Ratan, R. R. (2016). Oxidative stress and Huntington’s disease: the good, the bad, and the ugly. J. Huntingtons Dis. 5, 217–237. doi: 10.3233/JHD-160205
Labadorf, A., Hoss, A. G., Lagomarsino, V., Latourelle, J. C., Hadzi, T. C., Bregu, J., et al. (2015). RNA sequence analysis of human huntington disease brain reveals an extensive increase in inflammatory and developmental gene expression. PloS One 10:e0143563. doi: 10.1371/journal.pone.0143563
Lanciego, J. L., Luquin, N., and Obeso, J. A. (2012). Functional neuroanatomy of the basal ganglia. Cold Spring Harb. Perspect. Med. 2:a009621. doi: 10.1101/cshperspect.a009621
Langfelder, P., Cantle, J. P., Chatzopoulou, D., Wang, N., Gao, F., Al-Ramahi, I., et al. (2016). Integrated genomics and proteomics define huntingtin CAG length–dependent networks in mice. Nat. Neurosci. 19, 623–633. doi: 10.1038/nn.4256
Lee, H., Fenster, R. J., Pineda, S. S., Gibbs, W. S., Mohammadi, S., Davila-Velderrain, J., et al. (2020). Cell type-specific transcriptomics reveals that mutant huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron 107, 891–908.e8. doi: 10.1016/j.neuron.2020.06.021
Lee, J. H., Sowada, M. J., Boudreau, R. L., Aerts, A. M., Thedens, D. R., Nopoulos, P., et al. (2014). Rhes suppression enhances disease phenotypes in Huntington’s disease mice. J. Huntingtons Dis. 3, 65–71. doi: 10.3233/JHD-140094
Lee, J.-M., Correia, K., Loupe, J., Kim, K.-H., Barker, D., Hong, E. P., et al. (2019). CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell 178, 887–900.e14. doi: 10.1016/j.cell.2019.06.036
Lee, J.-M., Pinto, R. M., Gillis, T., St Claire, J. C., and Wheeler, V. C. (2011). Quantification of age-dependent somatic CAG repeat instability in Hdh CAG knock-in mice reveals different expansion dynamics in striatum and liver. PLoS One 6:e23647. doi: 10.1371/journal.pone.0023647
Lee, J.-M., Wheeler, V. C., Chao, M. J., Vonsattel, J. P. G., Pinto, R. M., Lucente, D., et al. (2015). Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162, 516–526. doi: 10.1016/j.cell.2015.07.003
Lee, S.-T., Chu, K., Im, W. S., Yoon, H. J., Im, J. Y., Park, J. E., et al. (2011). Altered microRNA regulation in Huntington’s disease models. Exp. Neurol. 227, 172–179. doi: 10.1016/j.expneurol.2010.10.012
Lei, W. (2004). Evidence for differential cortical input to direct pathway versus indirect pathway striatal projection neurons in rats. J. Neurosci. 24, 8289–8299. doi: 10.1523/JNEUROSCI.1990-04.2004
Leisman, G., Melillo, R., and Carrick, F. R. (2013). “Clinical motor and cognitive neurobehavioral relationships in the basal ganglia,” in Basal Ganglia – An Integrative View, ed. E. Franz (Rijeka: InTech), 1–30. doi: 10.5772/55227
Lerner, R. P., Trejo Martinez Ldel, C., Zhu, C., Chesselet, M. F., and Hickey, M. A. (2012). Striatal atrophy and dendritic alterations in a knock-in mouse model of Huntington’s disease. Brain Res. Bull. 87, 571–578. doi: 10.1016/j.brainresbull.2012.01.012
Li, X.-J., Orr, A. L., and Li, S. (2010). Impaired mitochondrial trafficking in Huntington’s disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 1802, 62–65. doi: 10.1016/j.bbadis.2009.06.008
Lin, L., Park, J. W., Ramachandran, S., Zhang, Y., Tseng, Y. T., Shen, S., et al. (2016). Transcriptome sequencing reveals aberrant alternative splicing in Huntington’s disease. Hum. Mol. Genet. 25, 3454–3466. doi: 10.1093/hmg/ddw187
Liot, G., Valette, J., Pépin, J., Flament, J., and Brouillet, E. (2017). Energy defects in Huntington’s disease: why “in vivo” evidence matters’. Biochem. Biophys. Res. Commun. 483, 1084–1095. doi: 10.1016/j.bbrc.2016.09.065
Luthi-Carter, R., Strand, A., Peters, N. L., Solano, S. M., Hollingsworth, Z. R., Menon, A. S., et al. (2000). Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum. Mol. Genet. 9, 1259–1271. doi: 10.1093/hmg/9.9.1259
Luthi-Carter, R., Taylor, D. M., Pallos, J., Lambert, E., Amore, A., Parker, A., et al. (2010). SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci.U.S.A. 107, 7927–7932. doi: 10.1073/pnas.1002924107
MacDonald, M. E., Ambrose, C. M., Duyao, M. P., Myers, R. H., Lin, C., Srinidhi, L., et al. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-E
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., et al. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506. doi: 10.1016/S0092-8674(00)81369-0
McFarland, K. N., Das, S., Sun, T. T., Leyfer, D., Xia, E., Sangrey, G. R., et al. (2012). Genome-wide histone acetylation is altered in a transgenic mouse model of Huntington’s disease. PLoS One 7:e41423. doi: 10.1371/journal.pone.0041423
Menalled, L. B. (2005). Knock-in mouse models of Huntington’s disease. NeuroRX 2, 465–470. doi: 10.1602/neurorx.2.3.465
Menalled, L. B., Kudwa, A. E., Miller, S., Fitzpatrick, J., Watson-Johnson, J., Keating, N., et al. (2012). Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175’. PLoS One 7:e49838. doi: 10.1371/journal.pone.0049838
Menalled, L. B., Sison, J. D., Dragatsis, I., Zeitlin, S., and Chesselet, M. F. (2003). Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J. Comp. Neurol. 465, 11–26. doi: 10.1002/cne.10776
Mochel, F., Durant, B., Meng, X., O’Callaghan, J., Yu, H., Brouillet, E., et al. (2012). Early alterations of brain cellular energy homeostasis in huntington disease models. J. Biol. Chem. 287, 1361–1370. doi: 10.1074/jbc.M111.309849
Morigaki, R., and Goto, S. (2017). Striatal vulnerability in huntington’s disease: neuroprotection versus neurotoxicity. Brain Sci. 7:63. doi: 10.3390/brainsci7060063
Packer, A. N., Xing, Y., Harper, S. Q., Jones, L., and Davidson, B. L. (2008). The bifunctional microRNA miR-9/miR-9∗ regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 28, 14341–14346. doi: 10.1523/JNEUROSCI.2390-08.2008
Palpagama, T. H., Waldvogel, H. J., Faull, R. L. M., and Kwakowsky, A. (2019). The role of microglia and astrocytes in huntington’s disease. Front. Mol. Neurosci. 12:258. doi: 10.3389/fnmol.2019.00258
Panov, A. V., Gutekunst, C. A., Leavitt, B. R., Hayden, M. R., Burke, J. R., Strittmatter, W. J., et al. (2002). Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 5, 731–736. doi: 10.1038/nn884
Paoletti, P., Vila, I., Rifé, M., Lizcano, J. M., Alberch, J., and Ginés, S. (2008). Dopaminergic and glutamatergic signaling crosstalk in huntington’s disease neurodegeneration: the role of p25/Cyclin-dependent kinase 5. J. Neurosci. 28, 10090–10101. doi: 10.1523/JNEUROSCI.3237-08.2008
Pearl, J. R., Shetty, A. C., Cantle, J. P., Bergey, D. E., Bragg, R. M., Coffey, S. R., et al. (2020). Altered huntingtin-chromatin interactions predict transcriptional and epigenetic changes in Huntington’s disease. bioRixv [preprint] doi: 10.1101/2020.06.04.132571
Peng, Q., Wu, B., Jiang, M., Jin, J., Hou, Z., Zheng, J., et al. (2016). Characterization of behavioral, neuropathological, brain metabolic and key molecular changes in zQ175 knock-in mouse model of huntington’s disease. PLoS One 11:e0148839. doi: 10.1371/journal.pone.0148839
Pinto, R. M., Dragileva, E., Kirby, A., Lloret, A., Lopez, E., St Claire, J., et al. (2013). Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in huntington’s disease mice: genome-wide and candidate approaches. PLoS Genet. 9:e1003930. doi: 10.1371/journal.pgen.1003930
Pringsheim, T., Wiltshire, K., Day, L., Dykeman, J., Steeves, T., and Jette, N. (2012). The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov. Disord. 27, 1083–1091. doi: 10.1002/mds.25075
Puighermanal, E., Castell, L., Esteve-Codina, A., Melser, S., Kaganovsky, K., and Zussy, C. (2020). Functional and molecular heterogeneity of D2R neurons along dorsal ventral axis in the striatum. Nat. Commun. 11:1957. doi: 10.1038/s41467-020-15716-9
Rebec, G. V. (2018). Corticostriatal network dysfunction in Huntington’s disease: deficits in neural processing, glutamate transport, and ascorbate release. CNS Neurosci. Ther. 24, 281–291. doi: 10.1111/cns.12828
Reiner, A., and Deng, Y.-P. (2018). Disrupted striatal neuron inputs and outputs in Huntington’s disease. CNS Neurosci. Ther. 24, 250–280. doi: 10.1111/cns.12844
Roos, R. A. C. (2010). Huntington’s disease: a clinical review. Orphanet J. Rare Dis. 5:40. doi: 10.1186/1750-1172-5-40
Ross, C. A., and Tabrizi, S. J. (2011). Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98. doi: 10.1016/S1474-4422(10)70245-3
Rüb, U., Seidel, K., Heinsen, H., Vonsattel, J. P., den Dunnen, W. F., and Korf, H. W. (2016). Huntington’s disease (HD): the neuropathology of a multisystem neurodegenerative disorder of the human brain. Brain Pathol. (Zurich Switzerland) 26, 726–740. doi: 10.1111/bpa.12426
Rubinsztein, D. C. (2002). Lessons from animal models of Huntington’s disease. Trends Genet. 18, 202–209. doi: 10.1016/S0168-9525(01)02625-7
Sapp, E., Ge, P., Aizawa, H., Bird, E., Penney, J., Young, A. B., et al. (1995). Evidence for a preferential loss of enkephalin immunoreactivity in the external globus pallidus in low grade Huntington’s disease using high resolution image analysis. Neuroscience 64, 397–404. doi: 10.1016/0306-4522(94)00427-7
Sathasivam, K., Neueder, A., Gipson, T. A., Landles, C., Benjamin, A. C., Bondulich, M. K., et al. (2013). Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease’. Proc. Natl. Acad. Sci.U.S.A. 110, 2366–2370. doi: 10.1073/pnas.1221891110
Saudou, F., and Humbert, S. (2016). The biology of huntingtin. Neuron 89, 910–926. doi: 10.1016/j.neuron.2016.02.003
Saunders, A., Macosko, E. Z., Wysoker, A., Goldman, M., Krienen, F. M., de Rivera, H., et al. (2018). Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16. doi: 10.1016/j.cell.2018.07.028
Schilling, J., Broemer, M., Atanassov, I., Duernberger, Y., Vorberg, I., Dieterich, C., et al. (2019). Deregulated splicing is a major mechanism of RNA-Induced toxicity in Huntington’s disease. J. Mol. Biol. 431, 1869–1877. doi: 10.1016/j.jmb.2019.01.034
Seong, I. S., Ivanova, E., Lee, J. M., Choo, Y. S., Fossale, E., Anderson, M., et al. (2005). HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum. Mol. Genet. 14, 2871–2880. doi: 10.1093/hmg/ddi319
Seong, I. S., Woda, J. M., Song, J. J., Lloret, A., Abeyrathne, P. D., Woo, C. J., et al. (2010). Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 19, 573–583. doi: 10.1093/hmg/ddp524
Seredenina, T., and Luthi-Carter, R. (2012). What have we learned from gene expression profiles in Huntington’s disease? Neurobiol. Dis. 45, 83–98. doi: 10.1016/j.nbd.2011.07.001
Shen, W., Tian, X., Day, M., Ulrich, S., Tkatch, T., Nathanson, N. M., et al. (2007). Cholinergic modulation of Kir2 channels selectively elevates dendritic excitability in striatopallidal neurons. Nat. Neurosci. 10, 1458–1466. doi: 10.1038/nn1972
Shirendeb, U., Reddy, A. P., Manczak, M., Calkins, M. J., Mao, P., Tagle, D. A., et al. (2011). Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum. Mol. Genet. 20, 1438–1455. doi: 10.1093/hmg/ddr024
Stack, E. C., Del Signore, S. J., Luthi-Carter, R., Soh, B. Y., Goldstein, D. R., Matson, S., et al. (2007). Modulation of nucleosome dynamics in Huntington’s disease. Hum. Mol. Genet. 16, 1164–1175. doi: 10.1093/hmg/ddm064
Stephenson-Jones, M., Samuelsson, E., Ericsson, J., Robertson, B., and Grillner, S. (2011). Evolutionary conservation of the basal ganglia as a common vertebrate mechanism for action selection. Curr. Biol. 21, 1081–1091. doi: 10.1016/j.cub.2011.05.001
Surmeier, D. J., Ding, J., Day, M., Wang, Z., and Shen, W. (2007). D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 30, 228–235. doi: 10.1016/j.tins.2007.03.008
Swami, M., Hendricks, A. E., Gillis, T., Massood, T., Mysore, J., and Myers, R. H. (2009). Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum. Mol. Genet. 18, 3039–3047. doi: 10.1093/hmg/ddp242
Tabrizi, S. J., Workman, J., Hart, P. E., Mangiarini, L., Mahal, A., Bates, G., et al. (2000). Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann. Neurol. 47, 80–86.
Tang, B., Seredenina, T., Coppola, G., Kuhn, A., Geschwind, D. H., Luthi-Carter, R., et al. (2011). Gene expression profiling of R6/2 transgenic mice with different CAG repeat lengths reveals genes associated with disease onset and progression in Huntington’s disease. Neurobiol. Dis. 42, 459–467. doi: 10.1016/j.nbd.2011.02.008
Telenius, H., Kremer, B., Goldberg, Y. P., Theilmann, J., Andrew, S. E., Zeisler, J., et al. (1994). Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat. Genet. 6, 409–414. doi: 10.1038/ng0494-409
Trettel, F., Rigamonti, D., Hilditch-Maguire, P., Wheeler, V. C., Sharp, A. H., Persichetti, F., et al. (2000). Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 9, 2799–2809. doi: 10.1093/hmg/9.19.2799
Vodicka, P., Smetana, K. Jr., Dvoránková, B., Emerick, T., Xu, Y. Z., Ourednik, J., et al. (2005). The miniature pig as an animal model in biomedical research. Ann. N. Y. Acad. Sci. 1049, 161–171. doi: 10.1196/annals.1334.015
Wave Life Sciences Ltd (2020). A Multicenter, Randomized, Double-blind, Placebo-controlled, Phase 1b/2a Study of WVE-120101 Administered Intrathecally in Patients With Huntington’s Disease. Clinical trial registration NCT03225833. clinicaltrials.gov. Available online at: https://clinicaltrials.gov/ct2/show/NCT03225833 (accessed October 12, 2020).
Wheeler, V. C. (2003). Mismatch repair gene Msh2 modifies the timing of early disease in HdhQ111 striatum. Hum. Mol. Genet. 12, 273–281. doi: 10.1093/hmg/ddg056
Willett, J. A., Cao, J., Dorris, D. M., Johnson, A. G., Ginnari, L. A., and Meitzen, J. (2019). Electrophysiological properties of medium spiny neuron subtypes in the caudate-putamen of prepubertal male and female Drd1a -tdTomato line 6 BAC transgenic mice. Eneuro 6, ENEURO.0016-19.2019.
Keywords: Huntington’s disease, neurodegeneration, striatum, medium-sized spiny neurons, selective vulnerability, D1R, D2R
Citation: Bergonzoni G, Döring J and Biagioli M (2021) D1R- and D2R-Medium-Sized Spiny Neurons Diversity: Insights Into Striatal Vulnerability to Huntington’s Disease Mutation. Front. Cell. Neurosci. 15:628010. doi: 10.3389/fncel.2021.628010
Received: 10 November 2020; Accepted: 20 January 2021;
Published: 10 February 2021.
Edited by:
Giordano Lippi, The Scripps Research Institute, United StatesReviewed by:
Emmanuel Valjent, Centre National de la Recherche Scientifique (CNRS), FranceCopyright © 2021 Bergonzoni, Döring and Biagioli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Biagioli, bWFydGEuYmlhZ2lvbGlAdW5pdG4uaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.