 Kimya Nourbakhsh
Kimya Nourbakhsh Smita Yadav
Smita Yadav
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 10 February 2021
Sec. Cellular Neurophysiology
Volume 15 - 2021 | https://doi.org/10.3389/fncel.2021.624648
This article is part of the Research TopicConnecting Form and Function: Recent Advances in Understanding Dendrite Morphogenesis and PlasticityView all 14 articles
Dendrites undergo extensive growth and remodeling during their lifetime. Specification of neurites into dendrites is followed by their arborization, maturation, and functional integration into synaptic networks. Each of these distinct developmental processes is spatially and temporally controlled in an exquisite fashion. Protein kinases through their highly specific substrate phosphorylation regulate dendritic growth and plasticity. Perturbation of kinase function results in aberrant dendritic growth and synaptic function. Not surprisingly, kinase dysfunction is strongly associated with neurodevelopmental and psychiatric disorders. Herein, we review, (a) key kinase pathways that regulate dendrite structure, function and plasticity, (b) how aberrant kinase signaling contributes to dendritic dysfunction in neurological disorders and (c) emergent technologies that can be applied to dissect the role of protein kinases in dendritic structure and function.
Dendrites are specialized neuronal processes that receive and integrate synaptic or sensory input. While dendrites are extremely heterogeneous morphologically, dendrites of a certain neuron type generally exhibit stereotyped morphology (Jan and Jan, 2001). Dendrites are sculpted by intrinsic genetic programs, external cues, and patterns of neuronal activity during development (Cline, 2001; Jan and Jan, 2010). These factors together confer dendrites their type specific morphology, and ensure fidelity in forming synaptic connections. Owing to the critical role dendrites play in establishing neuronal connectivity, their dysfunction is strongly associated with several neurological disorders (Forrest et al., 2018). Over the last four decades, multiple molecular signaling pathways that mediate structural and functional development of dendrites have been identified. Protein kinases play a pivotal role in almost all aspects of dendritic development and function, while their dysregulation contributes extensively to disease states.
Kinases catalyze the transfer of a phosphate group from ATP molecule to hydroxyl group containing amino acids primarily serine, threonine and tyrosine on substrate proteins, in a process termed as phosphorylation (Cohen, 2002; Fabbro et al., 2015). Several characteristics make kinase signaling uniquely powerful and versatile: (1) Phosphorylation occurs in a highly specific yet reversible fashion. (2) Phosphorylation can affect substrates in distinct ways: such as induce a gain or loss of substrate activity, change substrate localization or interactome. (3) Kinases often act in cascades and are capable of remarkable signal integration and amplification that can be tuned to achieve a variety of responses (Cobb, 1999). (4) Kinases are highly druggable making them promising therapeutic targets (Cohen, 2002). Several protein kinases are genetically linked to neurological disorders. These include neurodevelopmental disorders such as autism spectrum disorder as well as neurodegenerative diseases such as Parkinson's disease, yet little is known about how dysfunction in kinase signaling leads to pathological states (Baltussen et al., 2017; Krahn et al., 2020). Phosphorylation mediated by kinases is countered by phosphatases, a large family of enzymes that catalytically remove phosphate groups from their substrates (Barford et al., 1998; Peng and Maller, 2010). Phosphatases are divided into subfamilies of serine/threonine and tyrosine phosphatases, and dysfunction of these enzymes is associated with several diseases (Tonks, 2006). Activity of phosphatases is tightly regulated by diverse mechanisms that include binding with inhibitory proteins, direct oxidation, and kinase mediated phosphorylation (den Hertog, 2003). Thus, the true phosphorylation status of a protein in time and space is determined by the opposing action of protein kinases and phosphatases.
Herein, we describe how kinase signaling exquisitely orchestrates each step of dendrite development, beginning at neurogenesis to its maturation into synaptic networks. Evidence for critical and causative role of kinase dysfunction in neurodevelopmental and degenerative disorders is presented. Finally, we detail the emergent technologies that will be instrumental in delineating kinase function in dendritic development and how kinase dysfunction leads to pathologies associated with neurological disorders.
Before the inception of neurogenesis, the developing brain is comprised of polarized neuroepithelial cells that line the neural tube (Martin-Belmonte and Mostov, 2008; Taverna et al., 2014). Apical-basal polarity is established early in the developing brain, and is achieved through junctional complexes and polarized protein trafficking. These are regulated by kinase signaling through the conserved Par complex. The Par complex is comprised of scaffolding proteins Par3 and Par6, and the kinase atypical protein kinase C (aPKC) that together establish the apical-basal polarity (McCaffrey and Macara, 2012). The Rho GTPase Cdc42 targets and activates the Par complex at the apical membrane, which is separated from the basolateral membrane domain by adherens junctions. In the mammalian neocortex, intrinsic neuroepithelial polarity set up by the Par complex asymmetrically orients the mitotic spindle, that remarkably, during cell cycle can give rise to asymmetric division (Rodríguez-Fraticelli et al., 2011; Lancaster and Knoblich, 2012). Radial glial cells arising from the asymmetrical division of polarized neuroepithelial cells can further differentiate into neural progenitor cells (NPCs) or neurons through asymmetrical division (Götz and Huttner, 2005). The arising NPCs further self-renew or undergo a terminal differentiation into neurons (Florio and Huttner, 2014). Radial glial cells retain the epithelial polarity set up by the Par complexes. Knockout of one of the isoforms of aPKC, aPKC-λ, in neuroepithelial cells and radial glial cells results in the loss of apical processes that cause disordered layering of the cortex, highlighting the role of aPKC in apical-basal polarity (Imai et al., 2006). Apically located Par complex promotes self-renewing of progenitors at the expense of neurogenic differentiation in the developing cerebral cortex (Costa et al., 2008; Sottocornola et al., 2010). While the Par complex proteins set up intrinsic polarity, extrinsic cues greatly affect neuronal polarity, migration and layer formation in brain development. Secreted factors such as reelin, semaphorins, and neurotrophic factors play important roles in instructing neuronal polarity and migration during early development, and are executed by distinct kinase pathways. We direct readers to in depth reviews detailing how these extrinsic cues and growth factors regulate early brain development (Huang and Reichardt, 2001; Yazdani and Terman, 2006; Jossin, 2020). Here we will focus primarily on the kinase signaling pathways important for dendritic growth, structure and functional maturation.
Most newly generated mammalian neurons migrate from the site of neurogenesis to their final destination where they are integrated into neural networks. It is during this migration process that they acquire axon-dendrite polarity (Polleux and Snider, 2010). Some neurons, such as retinal ganglion cells, acquire the polarity of the progenitors from which they arise. Others, such as cortical pyramidal neurons, consolidate multiple extended neurites into one leading and one lagging neurite that gives rise to axo-dendritic polarity (Barnes and Polleux, 2009; Polleux and Snider, 2010). On reaching their final destination, neurons extend their dendrites through growth, dendrites scale in size with organism development, and mature in an exquisitely controlled fashion. Control of dendritic development in response to neuronal activity and neurotrophic factors is mediated by kinases, which play an instrumental role in regulating dendritic size and maturation to form synaptic contacts (Figures 1A–D).
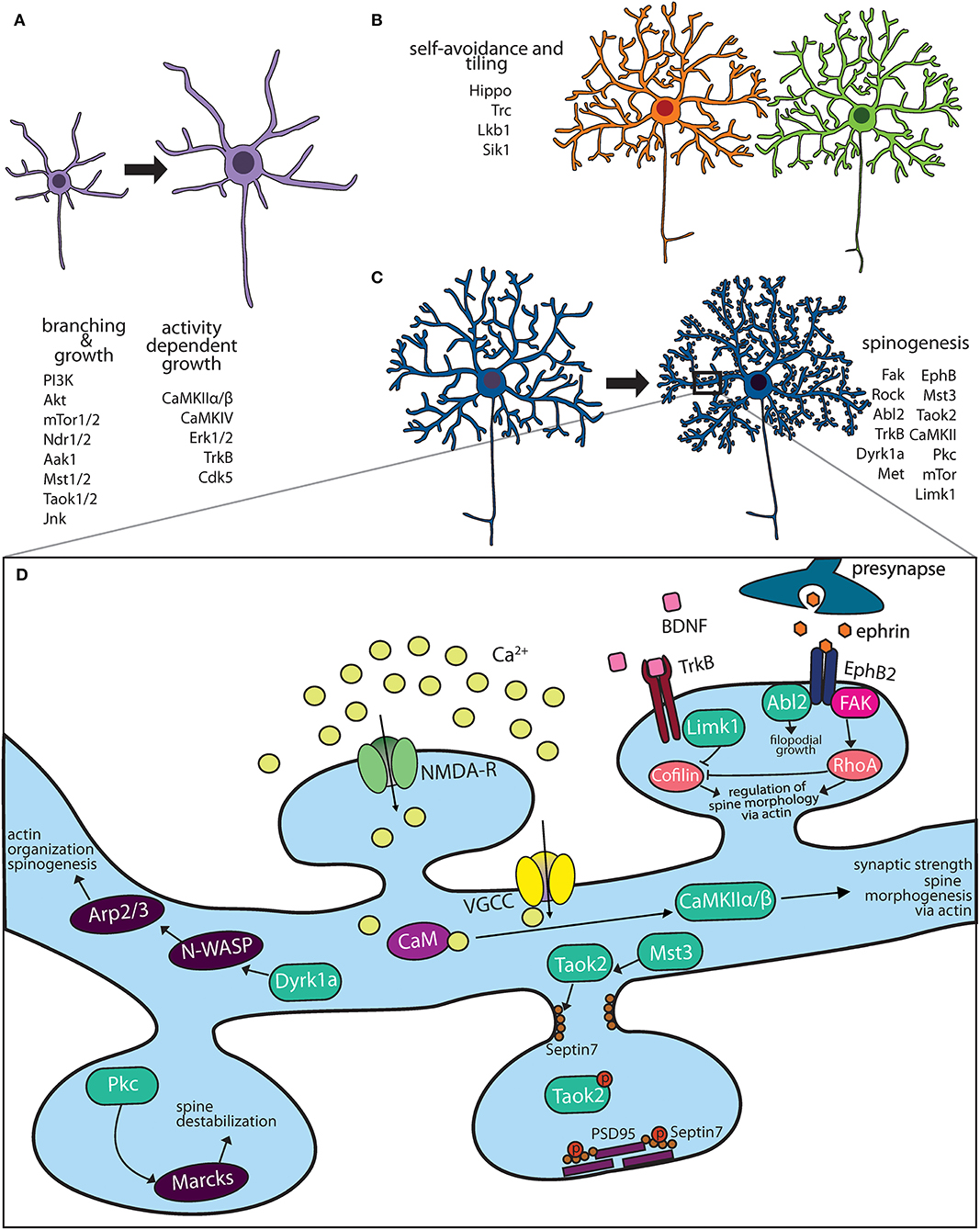
Figure 1. Kinase pathways mediate dendritic morphogenesis and maturation. (A) Immature dendritic neurites undergo expansive growth and branching during early development. The PI3K-Akt-mTor kinase and Hippo pathway are critical for growth. Activity dependent growth is primarily mediated by Ca+2 influx and downstream signaling by CaMK family members. (B) During arborization, dendrites of the same neuron avoid overlap through a principle known as self-avoidance, while neighboring neurons of the same type grow in well-defined territories. Kinases that mediate self-avoidance and dendritic tiling have been identified through screens in Drosophila peripheral sensory da neurons. (C) Most excitatory neurons on maturation form actin rich protrusions called dendritic spines that serve as sites of synapse formation. Several kinases that regulate the synaptic cytoskeleton are important regulators of spine formation in hippocampal and cortical neurons. (D) Calcium influx through N-methyl D-aspartate (NMDA) receptors and voltage gated calcium channels (VGCC) activate CaMK kinases, which mediate spine morphogenesis and plasticity. Mutations in several kinases important for dendritic spine formation are associated with neuropsychiatric diseases.
Neuronal activity during brain development profoundly impacts dendritic growth and retraction (Cline, 2001). Experiments where neuronal activity is manipulated such as in sensory deprivation (Wiesel and Hubel, 1963), pharmacological block of activity in Xenopus tadpoles (Rajan and Cline, 1998), or enhanced environmental enrichment in rodents (Volkmar and Greenough, 1972), induces dramatic alteration in both dendritic development and its structural complexity. Calcium influx in response to neuronal activity leads to kinase activation, providing the biochemical signal that mediates activity dependent dendritic growth dynamics (Ghosh and Greenberg, 1995). Calcium enters through glutamate receptor NMDA or voltage gated calcium channels (VGCC) and is sequestered by the calcium binding protein, calmodulin. In the Ca+2 bound state, calmodulin binds calcium/calmodulin-dependent protein kinases (CaMK), which then undergo autophosphorylation mediated activation. Several CaMKs have been implicated in dendritic development, and these act either via local effects or though transcriptional changes (Redmond and Ghosh, 2005). CaMKII has been extensively studied, most famously in relation to dendrite development and synaptic long-term potentiation. Two isoforms of CaMKII are expressed in the brain, CaMKIIα and CaMKIIβ, which each mediate distinct roles in dendrite development in hippocampal neurons, primarily on account of their differential ability to associate with actin (Shen et al., 1998). CaMKIIα expression in tectal neurons in tadpoles was found to stabilize dendritic arbors, as premature expression of CaMKIIα causes dendrites to slow their growth rate to that of more mature neurons. Conversely, blocking endogenous CaMKII maintains neurons in their rapid growth phase, such that dendritic arbors grow larger than normal (Wu et al., 1999; Zou and Cline, 1999). CaMKIIβ, on the other hand promotes actin polymerization, thereby increasing filopodial extension and growth of fine dendrites in rat hippocampal neurons (Fink et al., 2003). Another member of the CaM kinase family CaMKIV, is also crucial for activity dependent dendritic growth. Blocking CaMKIV signaling reduces calcium-induced dendritic growth, while expression of an activated form of CaMKIV in rat cortical neurons mimics the dendrite growth induced by calcium influx (Redmond et al., 2002). CaMKIV is enriched in the nucleus and its effect on dendrite growth is primarily mediated through transcriptional changes. Interfering with activity of transcriptional targets of CaMKIV blocks the ability of active CaMKIV to induce dendrite growth (Redmond and Ghosh, 2005). Importantly, activity induced changes in dendritic growth mediated by CaMKs are developmentally regulated. While the peak expression of CaMKIIβ and CaMKIV coincides with the period of maximal dendritic growth of cortical neurons in rodents, highest expression of CamKIIα occurs later during dendritic maturation when the arbor is elaborated (Cline, 2001; Wong and Ghosh, 2002; Redmond and Ghosh, 2005). Another kinase pathway activated in response of neuronal activity is Ras-MAPK signaling (Konur and Ghosh, 2005). In rat hippocampal pyramidal neurons, persistent dual phosphorylation of ERK1/2 (MAPKs) in response to calcium influx and Ras signaling is important for local cytoskeletal effects and transcriptional changes that lead to dendritic remodeling (Wu et al., 2001). One key protein important for dendritic growth is MAP2 (microtubule associated protein 2), which is phosphorylated by MAPK kinases in response to neuronal activity (Vaillant et al., 2002). In rat cortical neurons, both CaM kinases and Ras/MAPK signaling can regulate gene transcription in response to neuronal activity. This is mediated by phosphorylation of transcription factors, cAMP response element binding protein (CREB), calcium-responsive transactivator (CREST), LMO4 and NeuroD2 (Redmond et al., 2002; Aizawa et al., 2004; Redmond and Ghosh, 2005; Kashani et al., 2006). Both CREB and CREST bind the transcriptional coactivator, CREB binding protein (CBP) on phosphorylation, although the mechanisms by which transcription regulates dendritic growth are not well-understood (Aizawa et al., 2004). Many cytoplasmic and secreted proteins whose expression increases after activity have been identified. Some of these have shown clear and essential function in modulating dendrite and spine morphology such as candidate plasticity gene-15 (cpg15) (Nedivi et al., 1998), Arc (Peebles et al., 2010), Homer (Sala et al., 2001), and brain-derived neurotrophic factor (BDNF) (Lom and Cohen-Cory, 1999). Other than kinases belonging to the CaM kinase and MAP kinase families, cyclin dependent kinase 5 (CDK5) kinase is translocated to the nucleus in an activity dependent manner in hippocampal neurons where it regulates the transcription of BDNF by phosphorylating the transcriptional repressor MeCP2 (Cheung et al., 2007). It is likely that a quantitative and comprehensive analysis of activity induced kinases might reveal yet unidentified pathways that regulate activity dependent dendritic development.
Neurotrophins are secreted growth-promoting proteins that are essential for dendritic development both in peripheral and central nervous system (CNS). The four neutotrophins, nerve growth factor (NGF), brain derived growth factor (BDNF) and neurotrophins 3 and 4 (NT-3, NT-4) signal by binding and activating their receptors, which are members of the tropomyosin receptor kinases (Trk) and the structurally unrelated p75 neurotrophin receptor (Huang and Reichardt, 2001). Trk proteins are receptor tyrosine kinases that dimerize and transactivate on binding their respective neurotrophin ligand. In the mouse CNS, neurotrophins regulate the dendritic growth of pyramidal neurons in the developing neocortex (McAllister et al., 1995). Each of the four neurotrophins rapidly increases the length and complexity of dendrites of cortical pyramidal neurons when applied exogenously (McAllister et al., 1995). Experiments involving Trk “receptor bodies,” which are fusion proteins of the ligand-binding domains of each Trk receptor and the Fc domain of human IgG, clearly demonstrate that removing endogenous neurotrophins has dramatic consequences for the dendritic growth of pyramidal neurons in developing cortex (Shelton et al., 1995; McAllister et al., 1997). Consistent with the results of adding exogenous factors, endogenous BDNF is required for the growth and maintenance of dendritic arbors of layer 4 neurons whereas endogenous NT-3 is required for the growth and maintenance of dendritic arbors of layer 6 neurons. The distribution of receptors and secreted neurotrophin is exquisitely regulated. Not only do neurons in each cortical layer respond to subsets of neurotrophins, but also within a single cortical layer, each neurotrophin elicits a unique pattern of dendritic changes. Further, there are distinct changes in apical vs. basal dendrites evident at the individual neuron level (Horch and Katz, 2002). This precise level of neuronal and dendritic specificity suggests that neurotrophins do not simply enhance neuronal growth but, rather, act instructively to modulate particular patterns of dendritic arborization (McAllister, 2000). Another level of kinase regulation is added by Cdk5, which phosphorylates TrkB at the intracellular juxtamembrane region. In hippocampal cultures, reducing Cdk5 activity or expression of a TrkB mutant lacking the Cdk5 phosphorylation site abolishes BDNF triggered dendritic growth (Cheung et al., 2007). Thus, kinases act as both a signal detector (neurotrophin factors or calcium influx) and a biochemical switch (phosphorylation state) that turn on in response to stimuli and play critical roles in dendritic growth and remodeling (Figure 1A).
The phosphoinositide-3 kinase (PI3K)–Akt–mammalian target of rapamycin (mTOR) signaling pathway promotes dendritic growth and branching through upregulation of protein and lipid synthesis. The lipid kinase PI3K signals through AKT and is inhibited by phosphatase, Phosphatase and Tensin homolog deleted on chromosome Ten (PTEN). In neurons, mTOR senses and integrates nutrient and growth factor availability through extracellular signals such as BDNF, insulin, insulin-like growth factor 1 (IGF1), vascular endothelial growth factor (VEGF), ciliary neurotrophic factor (CNTF) and glutamate, to mediate neuronal proliferation, dendritic growth and synaptic function (Lipton and Sahin, 2014). Constitutively active Ras, PI3K, and Akt that activate the mTOR kinase induce growth and elaboration of dendrites in cultured hippocampal neurons. In these neurons, this effect can be potently blocked by mTOR inhibitor rapamycin or activation of PTEN that counteracts the action of lipid kinase PI3K (Jaworski et al., 2005). Loss of upstream regulators of mTOR either TSC1/2 or lipid phosphatase PTEN, result in structural neuronal defects. PTEN null mutant mice exhibit dendritic hypertrophy and macrocephaly (Kwon et al., 2003; Lugo et al., 2014). Loss of TSC1/2 increased size of both neuronal somata and dendritic arbors (Tavazoie et al., 2005). Conversely, dendritic complexity is reduced by inhibition of PI3K, knockdown of mTOR, or its effector p70 ribosomal S6 kinase that upregulates protein synthesis. MTOR is composed of two complexes mTORC1 and mTORC2. These share the mTOR kinase but comprise other distinct proteins such as Raptor and Rictor, respectively, that make the two complexes functionally distinct (Hoeffer and Klann, 2010). Both mTORC1 and mTORC2 are integral to the proper formation of dendrites and their arbor. Knockdown of their essential components Raptor and Rictor in rat hippocampal and cortical neurons inhibits dendritic growth (Urbanska et al., 2012).
The conserved Hippo kinase signaling pathway plays an important role in dendritic growth, tiling and function (Jan and Jan, 2010). Hippo is a conserved serine/threonine kinase of the Ste family, that is important for dendritic growth in Drosophila peripheral sensory neurons (Emoto et al., 2006). Hippo kinase mediates dendritic arbor growth and maintenance through the action of downstream kinase Wts. The mammalian homolog of hippo are MST1/2 kinases, which phosphorylates and activates kinases involved in cytoskeletal modulation including Ndr1/2 (nuclear Dbf2-related) kinases and Lats1/2 (large tumor suppressor 1/2) kinases (Emoto, 2011). In Drosophila and C. elegans sensory neurons, Ndr kinase is required for dendrite branching where it likely promotes neurite outgrowth and branching through Rho family GTPase (Emoto, 2011). Mammalian Ndr1/2 kinases analogous to the roles of their fly homolog Trc, limit dendrite branching and length in hippocampal primary cultures and in vivo (Ultanir et al., 2012). The ability of Ndr kinases to limit dendritic branching is mediated by its direct substrate Aak1 kinase which is a downstream substrate implicated in regulation of intracellular trafficking (Ultanir et al., 2012). In hippocampal neuron cultures, Ndr2 kinase was shown to regulate dendritic growth through phosphorylation of integrins, its subsequent translocation to the neurite tips where it facilitate neurite extension (Rehberg et al., 2014). The Tao family of Ste kinases are regulators of Hippo kinases signaling (Poon et al., 2011; Huang et al., 2014). The mammalian Tao kinase, Taok2, is a serine-threonine kinase that specifically regulates the development of basal, but not apical, dendrites in cortical pyramidal neurons (de Anda et al., 2012). The secreted guidance molecule Semaphorin3a activates its receptor Neuropilin, which in turn binds Taok2 and regulates Jnk kinase signaling development of basal dendrites (de Anda et al., 2012). Taok2 associates with microtubules (Mitsopoulos et al., 2003), as well as mediates septin phosphorylation (Yadav et al., 2017). Mechanistically, how interaction of Taok2 with various cytoskeletal elements contributes to its role in dendritic development is not well-understood.
Elaboration of the dendritic arbor through growth and branching is fine-tuned by mechanisms of self-avoidance and tiling, which prevent redundancy and increase efficient use of receptive field (Figure 1B). Dendrites avoid overlapping with other dendrites of the same neuron in a process known as self-avoidance, well-studied in Drosophila (Grueber et al., 2003; Jan and Jan, 2010). Dendrite self-avoidance is important in order to prevent self-crossing of dendrites, clumping of dendrites as well as to maximize the receptive field. In Purkinje cells, the liver kinase B1 (Lkb1) is developmentally expressed in the dendrites. Depletion of Lkb1 in these neurons results in increased dendritic crossing (Kuwako and Okano, 2018). Exogenous expression of salt inducible kinase 1 (Sik1), a downstream target of Lkb1 kinase is able to rescue self-crossing defects through regulation of the guidance cue receptor Robo2 (Kuwako and Okano, 2018), these data suggest that the Lkb1-Sik1 kinase pathways is required for dendritic self-avoidance in cerebellar Purkinje neurons. Dendritic tiling ensures that dendrites of different neurons of the same type avoid each other (Grueber and Sagasti, 2010; Jan and Jan, 2010). Dendritic tiling has been best studied in the Drosophila peripheral sensory da neurons and in retinal ganglion cells. In flies, loss of either the serine/threonine kinase, Tricornered (Trc), or Furry (Fry), a protein required for Trc kinase activity, leads to sensory neurons that produce excessive numbers of dendritic branches, which fail to tile normally (Emoto et al., 2004).
Dendritic spines are actin-rich protrusions on the dendritic membrane of most excitatory neurons. Early in development, thin actin filopodia extend from dendritic branches, which then mature into stable mushroom shaped spines likely on contact with the axonal membrane (Matus, 2000; Yuste and Bonhoeffer, 2004). However, spines retain remarkable structural plasticity even after development, and on exposure to the right stimuli can extensively retract, morph or become larger (Hering and Sheng, 2001). Spines are enriched in F-actin that contributes greatly to their structure and plasticity (Hotulainen and Hoogenraad, 2010). Repetitive firing of synapses, such as that which occurs during high-frequency synaptic stimulation of hippocampal neurons (long term potentiation), leads to an increase in F-actin which causes the spine to enlarge (Okamoto et al., 2004). Conversely, in long-term depression, decrease in F-actin/G-actin ratio causes dendritic spine shrinkage (Zhou et al., 2004). In addition to actin, other cytoskeletal elements such as septin and microtubules regulate dendritic spines structure. In cortical and hippocampal neurons, septin proteins mark the site of dendritic filopodia extension, and are important for dendritic spine stability (Xie et al., 2007; Ewers et al., 2014; Yadav et al., 2017). Interestingly, transient entry of microtubules into dendritic spines of hippocampal neurons regulates actin dynamics and spine morphology (Jaworski et al., 2009). It is therefore, not surprising, that several kinases (Figure 1C) and GTPases that regulate actin, septin and microtubule dynamics are implicated in spine formation and its plasticity (Hotulainen and Hoogenraad, 2010; Lin and Koleske, 2010).
Several kinases modulate spinogenesis through regulation of the actin cytoskeleton (Figure 1D). In addition to their role in dendrite development (described above) the calcium/calmodulin-dependent protein kinase II (CaMKII) is important for dendritic spine formation. In hippocampal neurons, the two main neuronal CaMKII isoforms have distinct roles, while CaMKIIα regulates synaptic strength, the CaMKIIβ isoform controls dendritic spine morphology and synapse number via its ability to bundle actin filaments (Fink et al., 2003; Okamoto et al., 2007). Knock-in mice expressing kinase dead CaMKIIα, show impaired learning and memory as well as a loss of long-term potentiation (Yamagata et al., 2009). Neuronal activity or NMDA receptor activation regulates spine morphogenesis by mediating Ca2+ influx into postsynaptic neurons, which modulate the activity of many actin binding proteins, including CaMKIIβ (Lisman et al., 2002). The dual specificity kinase Dyrk1a, negatively regulates filopodia and spine formation through phosphorylation of N-WASP, an actin filament assembly protein (Park et al., 2012). In cultured hippocampal neurons, N-WASP activates the actin branching protein Arp2/3, which is required for spine formation (Wegner et al., 2008). LIM-kinase1 (LIMK1) inhibits the activity of actin depolymerizing protein cofilin by phosphorylation (Yang et al., 1998) and hence affects dendritic spine morphology and synaptic function (Meng et al., 2002). LIM-Kinase1 (Limk1) functions as an actin destabilizer. Hippocampal neurons in Limk1 knockout mice exhibit aberrant spines and enhanced LTP (Meng et al., 2002). Another actin destabilizing kinase is PKC, several isoforms of which are enriched at the synapse. PKC phosphorylates myristoylated, alanine-rich C-kinase substrate (MARCKS) inhibiting its ability to cross link the actin cytoskeleton to membrane thereby destabilizing dendritic spines in hippocampal neurons (Calabrese and Halpain, 2005).
Receptor tyrosine kinases, notably Trk kinase and Eph/ephrin family members are important for dendritic spine formation. TrkB acts as a receptor for the neurotrophic factor BDNF and is crucial for neuronal plasticity such as structural remodeling associated with LTP (Huang et al., 2013). In rat hippocampal slices, BDNF stimulates activity of serine/threonine kinase p21 activated kinase (Pak), which inactivates cofilin through phosphorylation causing increase in spine size and stability. TrkB also activates Ras GTPase inducing spine enlargement and stability (Yasuda et al., 2006). Eph receptors expressed on dendrites are activated by ephrins on opposing membranes such as axonal/glial. Signaling through these receptors regulate dendritic spine and synapse formation or activity-induced LTP in hippocampal cultured neurons (Klein, 2009). Activation of Eph receptors leads to tyrosine phosphorylation of target molecules, such as proteoglycan syndecan-2, which clusters them at the postsynapse promoting spine maturation (Ethell et al., 2001). Activation of EphB on binding EphrinB leads to the receptor interaction with NMDA receptors and subsequent synaptic targeting of NMDA receptors (Dalva et al., 2000; Nolt et al., 2011). Further, Eph receptors also mediate structural changes in dendritic spines. Optogenetic local activation of expressed OptoEphb2 in dendrites led to rapid actin polymerization causing filopodial growth. While inhibition of Rac1 and Cdc42 did not abolish OptoEphB2-induced actin polymerization, Abelson tyrosine-protein kinase 2 (Abl2/Arg) were found to be downstream effector of filopodia growth in dendrites (Locke et al., 2018). Hippocampal neurons derived from EphB1/B2/B3 receptor triple knockout mice are unable to form mature dendritic spines (Henkemeyer et al., 2003), consistent with the essential role of Eph tyrosine kinases in spinogenesis. Another pathway that contributes to EphB2-mediated dendritic spine stabilization is FAK kinase that activates RhoA-ROCK-LIMK-1 pathway to suppress cofilin activity and remodel dendritic spines (Shi et al., 2009; Koleske, 2013).
In addition to actin, the septin cytoskeleton plays essential roles in dendritic spine morphogenesis (Figure 1D). Septin7 localizes to the base of dendritic spines at the spine neck and is required for spine formation (Tada et al., 2007; Xie et al., 2007) and spine stability (Ewers et al., 2014) in both hippocampal and cortical neurons. The serine/threonine kinase TAOK2 directly phosphorylates Septin7 at an evolutionarily conserved residue. In cultured hippocampal neurons, phosphorylation at its C-terminal tail promotes septin7 translocation from the base of the dendritic spine to spine head, where it associates and stabilizes the synaptic scaffold protein PSD95 (Yadav et al., 2017). TAOK2 depletion or expression of phospho-dead Septin7 leads to exuberant filopodial extension and inhibits spine maturation (Yadav et al., 2017).
Structural and functional dendritic defects are strongly associated with several neurodevelopmental and psychiatric disorders including autism spectrum disorder, schizophrenia, and Down syndrome (Raymond et al., 1996; Kulkarni and Firestein, 2012). Homeostatic balance between dendritic stability and instability is perturbed during neurodegenerative diseases or injury insults such as stroke. Most neurodegenerative diseases lead to dystrophy of dendrites and synapses, with some evidence suggesting that synaptic dysfunction precedes axonal degeneration (Gan et al., 2018). The evidence implicating kinase pathways in neurological disorders, and the potential mechanisms through which mutations in these kinase pathways contribute to disease are outlined below (Table 1).
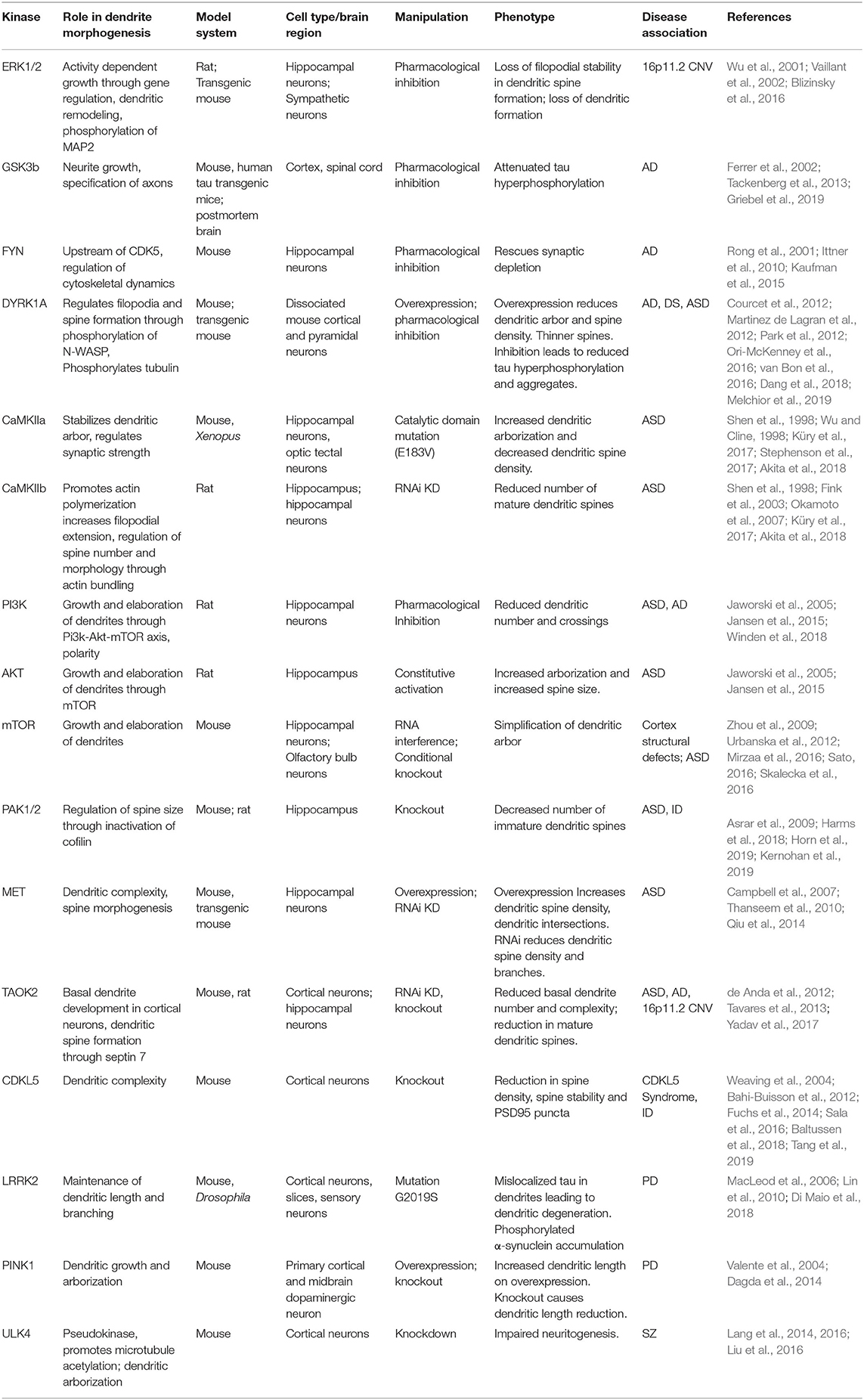
Table 1. Kinase signaling pathways implicated in neurological diseases and their role in dendrite development.
Autism spectrum disorder (ASD) is a neurodevelopmental disorder with a strong genetic basis, and is defined by deficits in social communication, language development as well as repeated behaviors (Geschwind and Levitt, 2007). Several kinases have been genetically associated with autism spectrum disorders. DYRK1A has one of strongest genetic association with ASD (De Rubeis et al., 2014; Iossifov et al., 2014). The encoded protein DYRK1a is a dual-specificity tyrosine phosphorylation-regulated serine/threonine kinase. Several ASD associated variants of DYRK1A affect its kinase function causing either loss or gain of kinase activity. Mouse hippocampal neurons transfected with these variants show defects in neuronal development including in dendritic outgrowth and dendritic spine density (Dang et al., 2018). Further overexpression of these pathogenic variants in developing mice embryos perturb neuronal migration in vivo (Dang et al., 2018). DYRK1A mutations lead to syndromic form of autism and intellectual disability with many shared features. DYRK1A is affected in 21q22 microdeletion in human, and is associated with growth retardation, primary microcephaly, facial dysmorphism, seizures, ataxic gait, absent speech and intellectual disability (Møller et al., 2008; Courcet et al., 2012; Ji et al., 2015; van Bon et al., 2016).
Aberrant signaling through the PI3K/AKT/mTOR pathway is associated with ASD (Winden et al., 2018). A range of structural brain abnormalities are associated with mutations in the mTOR pathway. Activating mutations in PI3K/AKT/mTOR pathway result in megalencephalies and hemimegalencephalies associated with ASD (Jansen et al., 2015). Mutations in PTEN are linked to macrocephaly and ASD (Butler et al., 2005). Pten knockout mice exhibit enlarged dendritic arbors and neuronal soma and exhibit autism-like behavior (Lugo et al., 2014). Mutations in genes involved in the mTOR signaling pathway have been identified in some cases of syndromic ASD (Mirzaa et al., 2016). While mutations that inhibit mTOR are associated with microcephaly, hyperactive mTOR signaling is associated with monogenic ASD. Inhibition of mTOR signaling is a potential pharmacotherapy for ASD (Sato, 2016).
TAOK2 is an autism susceptibility gene encoding a serine/threonine kinase. De novo mutations in TAOK2 have been found in ASD patients (Richter et al., 2018). TAOK2 is one of the two kinases within the 16p11.2 gene locus, a region prone to copy number variations (CNV) associated with ASD and schizophrenia (Kumar et al., 2008; Weiss et al., 2008; McCarthy et al., 2009). ASD patients with 16p11.2 CNV exhibit a strong correlation between TAOK2 expression and head circumference (Luo et al., 2012). Further, Taok2 knockout mice exhibit increased total brain volume compared to wildtype (Richter et al., 2018), suggesting that perturbation in TAOK2 gene dosage might contribute to reciprocal brain size difference associated with the 16p11.2 CNV. However, mechanisms of whether and how TAOK2 might affect brain size have not been elucidated. TAOK2 kinase regulates the microtubule cytoskeleton (Moore et al., 2000; Mitsopoulos et al., 2003), septin cytoskeleton (Yadav et al., 2017) and RhoA signaling (de Anda et al., 2012). It is likely that many of these signaling pathways converge to mediate structural changes in the brain. In cultured hippocampal neurons, depletion or expression of kinase dead TAOK2 results in a loss of mature mushroom-shaped spines (Ultanir et al., 2014; Yadav et al., 2017; Richter et al., 2018), increased shaft synapses (Yadav et al., 2017). Further in cortical neurons, TAOK2 is required for proper basal dendrite development (de Anda et al., 2012). Interestingly, de novo mutations in another member of the TAO family, TAOK1, are associated with neurodevelopmental delay (Dulovic-Mahlow et al., 2019).
Additional kinases associated with ASD are MET, CaMKII and the PAK kinases. MET is associated with ASD and encodes a receptor tyrosine kinase MET (Campbell et al., 2007; Thanseem et al., 2010). Loss or gain of function of Met leads to opposing changes in dendritic complexity, spine morphogenesis, and timing of glutamatergic synapse maturation in CA1 hippocampal neurons (Qiu et al., 2014). De novo mutations in CAMK2A and CAMK2B are associated with autism, intellectual disability and neurodevelopmental disorders (Küry et al., 2017; Akita et al., 2018; Chiocchetti et al., 2018). De novo mutation in the CaMKIIα catalytic domain (E183V) was identified in a ASD proband (Stephenson et al., 2017). In cultured hippocampal neurons, the E183V mutation reduces CaMKIIα targeting to dendritic spines, increases dendritic arborization and decreases dendritic spine density. Mice with a knock-in CaMKIIα-E183V mutation display aberrant behavioral phenotypes, including hyperactivity, social interaction deficits, and increased repetitive behavior (Stephenson et al., 2017). Characterization of 19 rare de novo CAMK2A or CAMK2B variants identified in individuals with intellectual disability revealed that mutations that decreased or increased CAMKII autophosphorylation at Thr286/Thr287 also affected neuronal migration (Küry et al., 2017). PAK1 and PAK2 kinases are associated with autism and neurodevelopmental delay (Harms et al., 2018; Horn et al., 2019; Kernohan et al., 2019). PAK1 mutations in patients with intellectual disability are located within or proximal to the autoinhibitory switch domain, suggesting a gain of function mechanism of disease (Harms et al., 2018). The Pak1/2 serine/threonine protein kinases regulate cell motility, cell cycle progression, apoptosis or proliferation through downstream GTPases Cdc42 and Rac1. PAK2 is encoded in the 3q29 genomic region, deletion of which can result in numerous neurodevelopmental defects including ASD (Quintero-Rivera et al., 2010). Haploinsufficiency of PAK2 in mice results in decreased spine density and synapse number in the hippocampus (Wang et al., 2018b). In addition to PAK1/2, mutations in PAK3 gene are associated with X-linked intellectual disability (Allen et al., 1998).
CDKL5 Syndrome is a rare X-linked genetic disorder that results in severe neurodevelopmental impairment, infantile seizures and intellectual disability (Weaving et al., 2004). Mutations in CDKL5 gene encoding a serine/threonine kinase is causative of the syndrome. The severity of the disease seems to depend on the site of mutation, where those in the kinase domain are more pathogenic (Bahi-Buisson et al., 2012). CDKL5 knockout mouse exhibit dendritic hypotrophy in granule cells of the hippocampus (Fuchs et al., 2014). Enhanced NMDAR signaling and circuit hyperexcitability were shown to underlie autistic-like features in mouse models of CDKL5 Syndrome (Tang et al., 2019). Recently, direct target substrates of CDKL5 in the brain were identified, which included microtubule regulators Microtubule Associated Protein 1S (MAP1S), Microtubule End Binding Protein 2 (EB2) and RhoGTPase activator ARHGEF2 (Baltussen et al., 2018). Importantly, hypophosphorylation of these targets were confirmed in vivo as well as in induced pluripotent stem cell (iPSC) derived neurons differentiated from CDKL5 patients, suggesting that these are physiologically relevant kinase and substrates. Further, this important study also identified the consensus sequence of CDKL5 phosphorylation as RPXpS motif (Baltussen et al., 2018), which will be useful for identification of other downstream substrates.
ULK4 is a rare susceptibility gene for schizophrenia (Lang et al., 2014; Tassano et al., 2018), a devastating neuropsychiatric disease with high heritability but few monogenetic associations. The ULK (UNC51-like) kinase family member ULK4 is classified as a pseudokinase, as it is catalytically inactive. While ULK can bind ATP molecules it does not have phosphotransfer activity (Khamrui et al., 2020). The expression of ULK4 is neuron-specific and developmentally regulated, and its depletion in mice leads to defects in neural proliferation, migration as well as reduced dendritic arborization of cortical neurons (Lang et al., 2016). Knockdown of Ulk4 disrupts the composition of microtubules by reducing tubulin acetylation. Targeted disruption of the Ulk4 in the cortex decreases the neural stem cell pool at birth, which significantly reduced cerebral cortex size in postnatal mice (Liu et al., 2016). The implications of these changes in pathogenesis of schizophrenia are unknown.
Copy number variation (CNV) in the 16p11.2 genomic locus are strongly associated with neurodevelopmental disorders including ASD, schizophrenia, and structural brain changes (Kumar et al., 2008; Weiss et al., 2008; McCarthy et al., 2009; Qureshi et al., 2014; Owen et al., 2018). This genomic locus spans 29 annotated genes, two of which encode kinases: TAOK2 and MAPK3. Transcriptomic analysis of 16p11.2 CNV patients suggests there is a strong correlation between TAOK2 gene expression and head circumference (Luo et al., 2012). Further, TAOK2 knockout mice exhibit increased total brain volume compared to wildtype (Richter et al., 2018). Dissociated primary cortical neurons from 16p11.2 microduplication mice model show increased dendritic elaboration, which was rescued with ERK1 inhibitors (Blizinsky et al., 2016), suggesting increase in MAPK3 dosage might contribute to changes in brain structure and function. Paradoxically, ERK1 inhibition in 16p11.2 deletion also seems to rescue cortical defects in mice models. It is important to note that mouse models of 16p11.2 CNVs do not faithfully recapitulate the human condition in terms of structural brain changes (Portmann et al., 2014; Deshpande et al., 2017), and human relevant models of this CNV are needed to understand the mechanisms of many defects associated with these CNVs. Interestingly, TAOK2 and MAPK3, are the only two genes in this locus that have been independently associated with autism (Pinto et al., 2014; Richter et al., 2018).
Down Syndrome (DS) is a severe neurodevelopmental disorder caused by presence of an extra copy (or parts) of chromosome 21 (Dierssen, 2012). Postmortem analysis of neuronal morphology in DS patients showed increased complexity of dendritic arbors in early postnatal period followed by much reduced dendritic length and arborization in older children (Becker et al., 1986). Among the DS Critical genes, increased dosage of DYRK1A has been identified to play a crucial role in the disease pathology. Dyrk1a overexpression in mouse neocortex inhibits neural stem cell proliferation and leads to premature neuronal differentiation (Yabut et al., 2010). Overexpression of Dyrk1A leads to reduced dendritic arbor complexity and synaptogenesis in layer II/III pyramidal cells, indicating this kinase is a major contributor to the dendritic phenotypes in DS (Martinez de Lagran et al., 2012). Both the mammalian Dyrk1a and its Drosophila ortholog minibrain regulate dendritic morphogenesis through direct phosphorylation of β-tubulin which inhibits microtubule polymerization (Ori-McKenney et al., 2016).
Alzheimer's disease (AD) is a progressive neurodegenerative disease that affects wide areas of the cerebral cortex and hippocampus. Human neuropathology data from AD patients suggests that dendritic abnormalities in AD are widespread and often are present in the early stages of disease. Dendritic abnormalities associated with AD include dystrophic dendrites, reduction in dendrite complexity, and loss of dendritic spines (Cochran et al., 2014). Hallmark features of AD include accumulation of extracellular insoluble forms of amyloid-β (Aβ) and intracellular aggregation of hyperphosphorylated microtubule associated protein tau in neurofibrillary tangles (Giacobini and Gold, 2013; Congdon and Sigurdsson, 2018). Aβ refers to peptides that are 38–43 amino acids in length, derived by the proteolytic cleavage of amyloid precursor protein (APP). Aβ is released into the extracellular matrix in oligomeric form where it is normally cleared by macrophages and microglia. Defects in efficient clearing of Aβ and certain oligomeric Aβ conformations lead to formation of fibrils that ultimately form amyloid plaques (Ittner and Ittner, 2018). Plaques induce neurotoxicity through numerous pathways, including through recruitment of the microtubule binding proteins tau (Masters et al., 2015). Experiments in hippocampal neurons show that Aβ oligomers accumulate at synapses, inducing clustering and dysfunction of metabotropic glutamate receptors (Renner et al., 2010). Exposure to Aβ oligomers disrupts polarized trafficking causing mis-sorting of axonal proteins including tau into somatodendritic compartment. Defects in axonal trafficking of tau promotes axonal degeneration and corresponding decrease in synaptic inputs (Zempel et al., 2010). Further, missorted tau in dendrites induces tubulin polyglutamylation which recruits the microtubule severing protein spastin, ultimately leading to dendritic dystrophy (Zempel et al., 2013). Of note, disease modifying treatments targeting Aβ, have so far failed in clinical trials. Recent evidence suggests that amyloid deposition is not strongly correlated with cognition in multivariate analyses. Hyperphosphorylated tau, as well as synaptic and neuronal loss are, however, associated with memory deficits (Giacobini and Gold, 2013). AD is associated with high amount of hyperphosphorylated tau. Tau contains 77 potential serine/threonine and 4 tyrosine phosphorylation sites clustered in the proline-rich region and the tail domain adjacent to the microtubule targeting domains (Noble et al., 2013) Tau hyperphosphorylation decreases its binding to microtubules. As tau becomes progressively hyperphosphorylated, deficits in molecular chaperones and degradation contribute to tau oligomerization and paired helical filament formation, ultimately forming neurofibrillary tangles (Iqbal et al., 2016; Ittner and Ittner, 2018). Emergent evidence suggests that AD is a synaptopathy (Li et al., 2018). Synaptic dysfunction due to pathogenic Aβ oligomers and tau pathology is one of the earliest signs of disease, preceding synaptic loss and neurodegeneration (Selkoe, 2002; Hoover et al., 2010; DeVos et al., 2018). In addition to its axonal role, tau plays a dendritic function important for postsynaptic targeting of Fyn kinase, a modulator of NMDA receptor activity. Tau KO mice exhibit disrupted postsynaptic targeting of Fyn, which reverses the excitotoxicity caused by NMDA receptor dysfunction due to Aβ toxicity (Ittner et al., 2010). In addition to Fyn kinase, dysfunction in several kinase pathways have been implicated in AD. Overactivation of Gsk3β (Lauretti et al., 2020) and Dyrk1a (Coutadeur et al., 2015) has been independently shown to increase tau phosphorylation as well as Aβ production. Overactivated TAOK2 kinase was found in the neurofibrillary tangles in AD postmortem brain (Tavares et al., 2013), and its inhibition reduces tau phosphorylation in cellular models (Giacomini et al., 2018). Another important kinase pathway that has emerged in AD is the Cdk5. Deregulation of Cdk5 by overexpression of its activator p25 triggers progressive neurodegeneration and neurofibrillary tangle formation in mice (Cruz and Tsai, 2004). In addition to its effect on microtubule stability and synaptic function, hyperphosphorylated tau promotes Aβ toxicity mediated neuropathology (Ittner et al., 2010; Mairet-Coello et al., 2013), hence targeting tau hyperphosphorylation might prove to be a viable therapeutic strategy for AD. Several small molecule inhibitors targeting kinases that phosphorylate tau are currently in clinical trial for AD (Table 2) (Tell and Hilgeroth, 2013; Krahn et al., 2020).
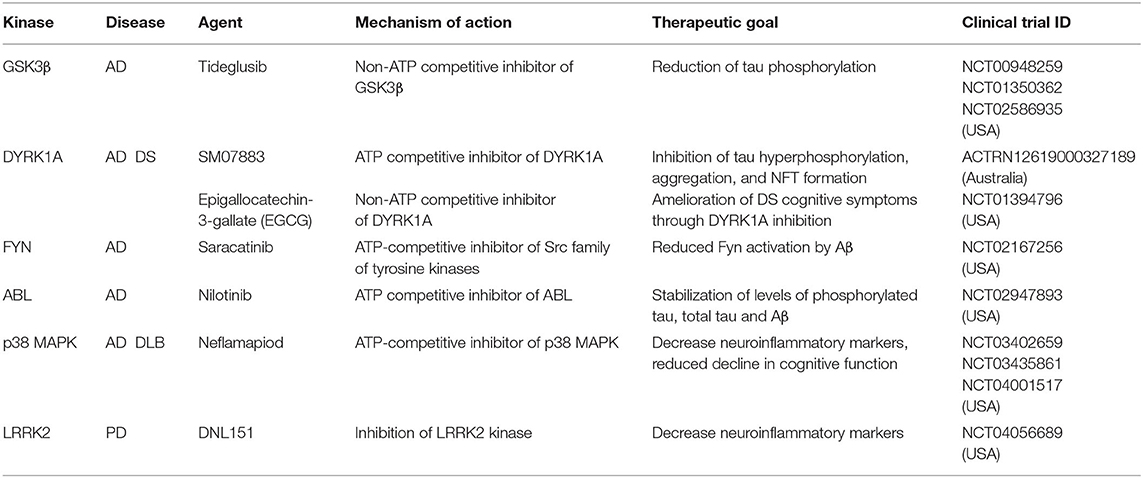
Table 2. Kinase inhibitors in clinical trials for the treatment of neurological disorders.
Parkinson's disease (PD) is pathologically defined by the neurodegeneration of dopaminergic neurons of the substantia nigra, and is characterized by the presence of cytoplasmic inclusions composed of α-synuclein protein aggregates called Lewy bodies (Poewe et al., 2017). Some of the strongest disease associated genes in Parkinson's disease encode for kinases; LRRK2 and PINK1. Mutations in leucine-rich repeat kinase 2 (LRRK2) underlie an autosomal-dominant, inherited form of PD. The PD-associated LRRK2 mutations display disinhibited kinase activity and induce a progressive reduction in dendrite length and branching in primary cortical cultures and in vivo mouse models (MacLeod et al., 2006). Increased LRRK2 kinase activity was observed in idiopathic PD, and in neurons exposed to mitochondrial toxins, suggesting that LRRK2 kinase activity might have a broader role in PD pathogenesis (Di Maio et al., 2018). Small-molecule LRRK2 kinase inhibitors have shown promise in preclinical models of PD, and has brought LRRK2 to the forefront of disease modifying efforts in PD (Tolosa et al., 2020). Homozygous loss of function mutations in PTEN-induced kinase 1 (PINK1) are associated with early onset PD (Valente et al., 2004). PINK1 encodes a serine/threonine kinase that acts as a sensor for mitochondrial health. In healthy mitochondria, PINK1 is targeted to mitochondria where it is rapidly degraded. In unhealthy membrane potential (ΔΨm)-deficient mitochondria, however, PINK1 accumulates and recruits an E3-ubiquitin ligase Parkin, which in turn initiates mitophagy (Okatsu et al., 2012). Loss of function PD associated PINK1 mutations perturb normal neuronal mitophagy, and accumulated damaged mitochondria in neurons lead to disease pathology. In mouse primary cortical and midbrain dopaminergic neurons, PINK1 kinase activity was found to promote dendritic arborization and its depletion resulted in shortened dendritic length (Dagda et al., 2014). While mechanisms through which PINK1 might regulate dendritic length are not well elucidated, there is some indication that control of mitochondrial motility and trafficking within dendrites by PINK1 could contribute to its dendritic role (Banerjee et al., 2017).
Kinase signaling occurs in a spatiotemporally precise fashion, however, traditional biochemical tools do not provide information on when and where kinase activation occurs during neuronal development. A major challenge in understanding kinase function is the identification of direct substrates of the kinase of interest (KOI).
A key method for studying temporal and spatial kinetics of kinase signaling is the use of kinase sensors (Figure 2A). Fundamentally, kinase sensors are comprised of two parts, a sensing unit, which is sensitive to a phosphorylation event on substrate and a reporting unit to indicate the phosphorylation state (Oldach and Zhang, 2014). Fluorescent kinase sensors work in three ways (Turk, 2005). FRET kinase sensors rely on Förster resonance energy transfer (FRET) between a fluorophore donor attached to a peptide designed to harbor the kinase specific phosphorylation site, and an acceptor fluorophore bound phospho-peptide binding motif (Sato et al., 2002). The phosphopeptide sensing motif interacts with the phosphorylated peptide, bringing donor and acceptor fluorophores together achieving resonant fluorescence that can be visualized through microscopy. Environmentally sensitive kinase sensors utilize a phosphorylation sequence peptide conjugated to a fluorophore that shifts wavelengths or intensity when in close proximity to phosphate (Yeh et al., 2002; Sharma et al., 2008). Chelation sensitive sensors use fluorophores sensitive to Mg2+ concentrations found in the ATP binding site of the kinase, paired with a phosphopeptide (Shults and Imperiali, 2003). Activation of ERK, PKA, and CaMKIIα in single dendritic spines during LTP has been studied using FRET based kinase sensors (Tang and Yasuda, 2017). Optogenetic regulation of kinases enables fast, reversible, and non-invasive manipulation of protein kinase activities providing exquisite control on regulation by bypassing upstream factors like growth factors, protein kinase inhibitors or chemical crosslinker that induce changes in kinase activity (Leopold et al., 2018). Design of opto-kinases is based on the plant photoreceptor domain light, oxygen, or voltage (LOV) or photoswitchable caging using Dronpa fluorophore. The light, oxygen, or voltage (LOV) domains are the sites for initial photochemistry in blue light photoreceptors in plant flavoprotein kinases, which have inspired creation of light activated kinases (Crosson and Moffat, 2002). LOV domains can be used to drive light mediated homodimerization of engineered tyrosine kinase domain thereby leading to their activation. The dimeric protein, pdDronpa, dissociates in cyan light and reassembles in violet light (Zhou et al., 2017). Switchable kinases have been designed by attaching two pdDronpa domains in the kinase domain thereby caging the kinase. When Dronpa dimers dissociate in cyan light it allows the kinase domains to come together to get activated, which can be rapidly shut down with violet light. Dronpa based photo-switchable (ps) psRaf1, psMEK1, psMEK2, and psCDK5 kinases were recently designed to uncover a direct and rapid inhibitory feedback loop from ERK to MEK1. Dronpa-kinases were also shown to work in vivo where they could acutely regulate synaptic vesicle transport (Zhou et al., 2017). Challenges such as low dynamic range, low signal to noise ratio, applicability to in vivo studies and control of expression level are some of the difficulties in design of useful kinase sensors (Oldach and Zhang, 2014). Iterative sensor optimization combined with improvements in imaging capabilities will further expand the scope of kinase sensors in understanding dendritic development and function.
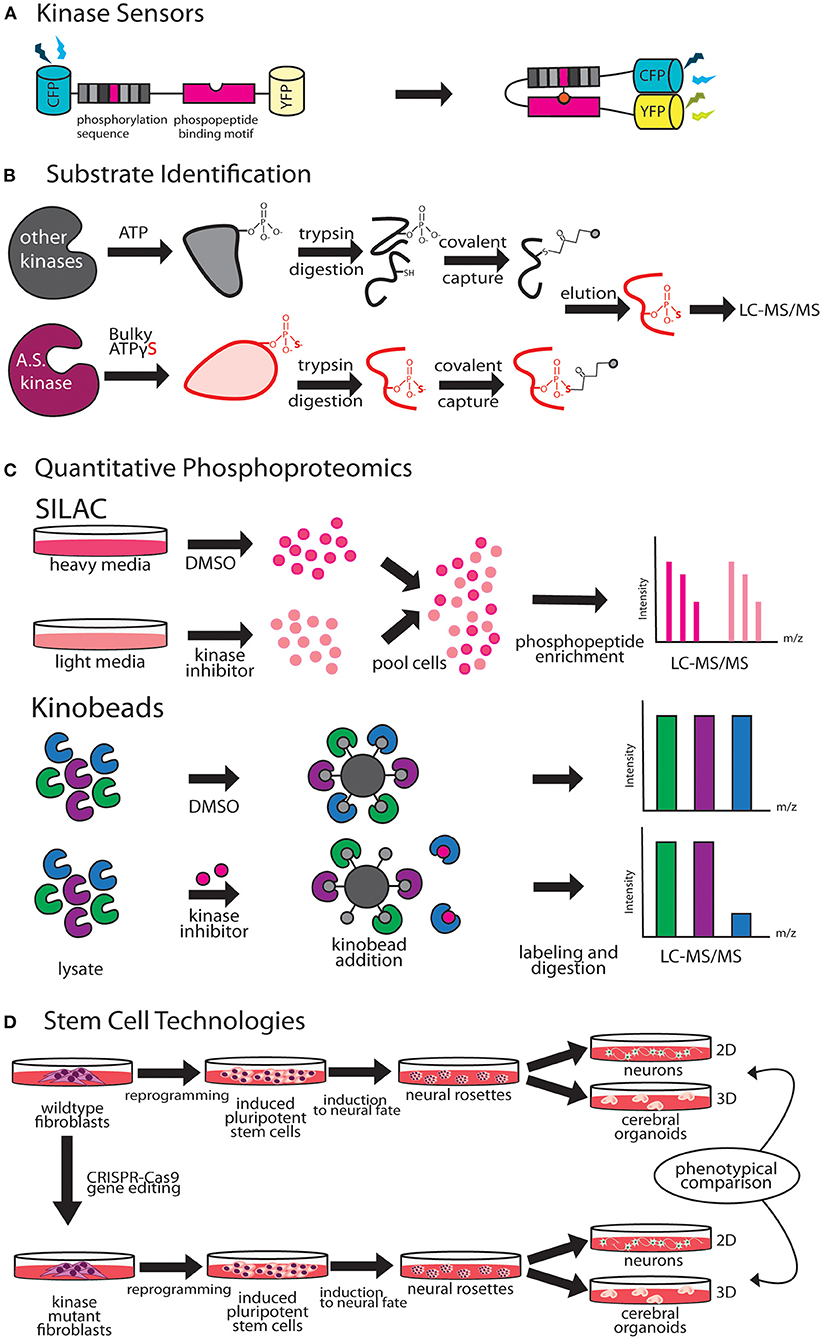
Figure 2. Emergent technologies to investigate kinase pathways. (A) FRET-based kinase sensors are comprised of kinase specific phosphopeptide sequence and a phosphopeptide binding motif. (B) Analog-sensitive kinases are an important tool for direct substrate identification through covalent capture of phosphopeptides and mass spectrometry. (C) Stable isotope labeling of amino acids in culture (SILAC), is a versatile metabolic labeling proteomic technique that can be applied to study the effect of kinase inhibitors on neuronal growth by quantifying changes in proteome. Kino-beads can be employed for profiling the neuronal kinome in healthy and disease states. (D) Human iPSCs have been used to model neurological diseases associated with kinase genes.
Cascades of protein phosphorylation downstream of kinase activation make precise identification of direct substrates difficult. A breakthrough in this field was the development of a technique that enables covalent capture of analog-sensitive kinase substrates (Figure 2B). This technique involves genetic engineering of the KOI to allow for utilization of bulkier ATPγS analogs. Thiophosphorylated proteins that represent direct substrates of KOI are covalently captured by thiol-reactive iodoacetyl agarose beads, and identified by mass spectrometry (Blethrow et al., 2008). As this method also yields the site of phosphorylation on the identified substrate, validation of substrate identity can be easily performed by point mutation of these sites in downstream biochemical assays. This method has been used for identification of direct targets of several kinases that play key role in dendrite morphogenesis including Cdkl5 (Baltussen et al., 2018), Taok2 (Yadav et al., 2017), and Hippo kinase members (Ultanir et al., 2012, 2014). Biological function of KOI and validated substrates can then be inferred through independent methods such as purified in vitro kinase assays and rescue of biological phenotypes with phosphomimetic substrates. An innovative application of this method was the finding that certain kinases such as PINK1, can utilize artificial ATP analogs (kinetin triphosphate), more efficiently than ATP, enhancing kinase activity of wild type as well as rescuing effects of low activity PD associated PINK1 mutant (Hertz et al., 2013). A limitation of this technique is that since phosphorylation reaction is performed in vitro in a cell or tissue lysate, the physiological context of kinase and substrate localization is lost. This can lead to false positive substrates, and therefore further validation of candidate substrates identified through this technique is essential.
Stable isotope labeling by amino acids in cell culture (SILAC) is a metabolic labeling technique that can be used to incorporate amino acids carrying specific heavy and light isotopes of C and N, allowing for simultaneous identification and quantitation of complex protein mixtures (Ong et al., 2002). SILAC can be employed in neuronal cultures or in vivo animal models to detect changes in the proteomic landscape upon different genetic or pharmacological perturbations (Krüger et al., 2008; Spellman et al., 2008). Therefore, SILAC can be a powerful method to study kinase pathways during dendrite development or disease (Figure 2C). SILAC was utilized to identify novel interactors of Tao kinases on phosphorylation by upstream kinase Mst3, to reveal unique interaction with cytoskeletal motor protein MyosinVa (Ultanir et al., 2014). Fragile X Syndrome is caused by transcriptional silencing of Fmr1, which encodes a protein that regulates mRNA translation in neuronal dendrites. SILAC labeling revealed profound up- or down- regulation of proteins related to synaptic structure and morphogenesis, dendritic mRNA transport, and synaptic transmission in Fmr1 knockout mouse cortical synapses compared to wild type (Liao et al., 2008). Improvements in SILAC technologies include use of “spike in” SILAC that can be utilized to compare more than three distinct comparisons (Geiger et al., 2010). However, optimization of proper spike-in-standard can be time consuming and variable based on the sample type (Wang et al., 2018a).
Quantitative phosphoproteomics has emerged as a powerful tool to perform unbiased and quantitative measurement of changes in signaling pathways in normal and diseased states (Hosp and Mann, 2017). Chemical labeling methods like tandem mass tag (TMT) or iTRAQ allow for multiplexing and are powerful when used with human tissue samples as labeling is performed in vitro after obtaining the tissue lysate (Glibert et al., 2015; Navarrete-Perea et al., 2018). Dendritic differentiation and maturation occur in distinct stages, and proteomic techniques can be applied to systematically delineate kinase pathways that mediate each of these processes. The developmental proteomic profile of cultured rat hippocampal neurons at different stages was recently mapped. Here, a combination of stable isotope labeling and high-resolution liquid chromatography-tandem mass spectrometry (LC-MS/MS) was utilized to detect extensive remodeling of the neuronal proteome, where one third of 4,500 proteins quantified were found to undergo 2-fold change in expression during neuronal differentiation (Frese et al., 2017). Phosphoproteomic techniques are especially powerful in identification of signaling aberrations in disease states. Haploinsufficiency of the gene SHANK3 causes Phelan-McDermid syndrome (PMDS) that is associated with a high risk of autism (Mitz et al., 2018). Unbiased, quantitative proteomics revealed profound changes in the phosphoproteome of Shank3-deficient neurons, including downregulation of Akt-mTORC1 signaling due to increased steady-state levels of upstream kinase, Cdc2-like kinase 2 (Clk2) (Bidinosti et al., 2016). Pharmacological and genetic activation of Akt or inhibition of Clk2 relieved synaptic and behavioral deficits in PMDS patient-derived neurons and mouse models, thereby highlighting the value of using unbiased proteomic approaches in discovery of novel drug targets. Another powerful example is the use of phosphoproteomics screening analysis of Parkinson disease associated kinase LRRK2, in combination with different pharmacological inhibitors that uncovered Rab GTPases as key LRRK2 substrates, and pointed toward a new disease mechanism in PD (Steger et al., 2016).
Immobilized broad-spectrum kinase inhibitors can be used in affinity pulldown to probe full-length kinases from whole neuronal or brain tissue proteomes (Bantscheff et al., 2007). This chemical proteomics method uses as multiplexed kinase inhibitor beads or kinobead-profiling enabling simultaneous profiling of over 200 kinases in a single experiment. Combining kinobead pulldown with SILAC or isobaric chemical labeling can increase analytical throughput dramatically, as well as allow comparison of proteomes from normal and disease states (Golkowski et al., 2017). In addition, kinobeads can be used to map drug-induced changes in the phosphorylation state of the kinome, enabling analysis of signaling downstream of target kinases (Golkowski et al., 2020). A limitation of kinobeads is that their applicability in providing spatial and temporal information on the kinome has not been proven. Most applications of kinobeads to date have focused on whole cell or tissue samples to query the kinome. ATP probes can be used in a similar fashion for profiling the entire kinome, kinases and other ATP utilizing proteins such as ATPases. This complementary chemical proteomic method trademarked as KiNativ, utilizes highly reactive biotinylated acyl phosphate derivatives of ATP as an affinity tag to profile the cellular kinome (Patricelli et al., 2007). In addition, the KiNativ platform can be used to determine the proteomic response to specific kinase inhibitors, which allows characterization of inhibitor interactions with endogenous kinases in native conditions (Patricelli et al., 2011). Application of these advanced kinome profiling techniques to better understand and delineate kinase signaling in dendritic development and disease states would likely reveal novel mechanistic insight (Figure 2C).
Human induced pluripotent stem cell (hiPSC) derived neurons can be a powerful tool to model dendritic development and for studying how kinase signaling contributes to development and dendritic dysfunction. Stem cells can be reprogrammed from affected patient fibroblasts or from health controls, and differentiated into different neuronal and non-neuronal cell types to model neurological diseases in vitro (Dolmetsch and Geschwind, 2011; Paşca et al., 2014). There are three primary applications of this technology in studying dendrite development. First, iPSCs from healthy individuals can be gene edited using CRISPR-Cas9 to introduce truncations and single nucleotide polymorphisms (SNPs) to model the effect of kinase disease variants (Vermilyea et al., 2020). Secondly, iPSCs derived from patients with known neurological disorder can be differentiated to study neuronal development (Figure 2D). Forebrain cortical neurons differentiated from iPSCs derived from fibroblasts of 16p11.2 deletion and duplication carriers, exhibit opposing changes in dendritic arbor size (Deshpande et al., 2017), suggesting that reciprocal changes in neuronal size could contribute to brain size defects associated with 16p11.2 CNVs. Neurons derived from iPS cells are an excellent human cellular model for investigating neuronal development in an isogenic background. For example, female iPSCs harboring CDKL5-mutations were shown to maintain X-chromosome inactivation, with clones expressing either mutant CDKL5 allele or the wild-type allele (Amenduni et al., 2011). Neurons differentiated from patient iPSCs with CDKL5 mutations develop increased number of dendritic protrusions (Ricciardi et al., 2012). Another study utilized iPSC derived neurons from patients with distinct mutations to show that loss of CDKL5 led to a decrease in phosphorylation of CDKL5 substrate EB2. A microtubule regulator, EB2 phosphorylation status was shown to be critical for microtubule dynamics within dendrites (Baltussen et al., 2018). Finally, human iPSC derived 2D neuronal and 3D organoid models can be used in high throughput screens, for identification of novel modulators of dendrite development. A recent high-throughput screening of human iPSC-derived neurons focused on neurite growth employed a collection of 4,421 bioactive compounds, and identified 108 hit compounds, including 37 approved drugs, that regulate neurite growth (Sherman and Bang, 2018). Human iPSC derived neurons can be used as a preclinical model for drug toxicity tests relevant to human physiology, an issue that has hampered progress in generation of new therapeutics (Inoue et al., 2014). Continued innovation in the field of stem cell technologies have greatly improved reproducibility and reliability of iPS derived neurons. The prolonged developmental timeline of human neuronal development compared to rodents (Dolmetsch and Geschwind, 2011), however, necessitates further improvement that enable longer in vitro 2D and 3D culturing capacities, and allow for co-culturing of various brain cell types to recapitulate in vivo dendritic development, maturation and synaptic pruning (Kelava and Lancaster, 2016).
Kinase signaling pathways act in a concerted fashion to mediate almost all aspects of dendritic development. These complex signaling pathways are elegantly intertwined, with key kinase signaling elements recurring throughout neuronal development. The human genome encodes a total of 518 kinases (Manning et al., 2002). While genetic models and GWAS analyses have identified several kinase-encoding genes implicated in neurological disease (Krahn et al., 2020), there remains much to discover about kinase function in dendrite development and how their dysregulation contributes to neuronal disease. Unbiased mapping of kinase signaling that instruct distinct stages of dendritic growth may reveal novel pathways that can be further genetically dissected. Comparative phosphoproteomic analyses of normal and disease states will serve as a powerful tool for future identification of novel therapeutic kinase targets. Advances in neurobiological and proteomic techniques will greatly facilitate the exploration of kinase pathways that impact dendrite structure and dysfunction in disease states.
SY and KN wrote the article and approved the submitted version.
We are thankful for the funding provided by National Institutes of Health R00MH108648 and R01MH121674 to SY. KN was supported by the NIH predoctoral Pharmacological Sciences Training Program T32GM007750.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Shao-En Ong (UW), Sujin Byeon, and Reilly Falter for their suggestions.
Aizawa, H., Hu, S.-C., Bobb, K., Balakrishnan, K., Ince, G., Gurevich, I., et al. (2004). Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science 303, 197–202. doi: 10.1126/science.1089845
Akita, T., Aoto, K., Kato, M., Shiina, M., Mutoh, H., Nakashima, M., et al. (2018). De novo variants in CAMK2A and CAMK2B cause neurodevelopmental disorders. Ann. Clin. Transl. Neurol. 5, 280–296. doi: 10.1002/acn3.528
Allen, K. M., Gleeson, J. G., Bagrodia, S., Partington, M. W., MacMillan, J. C., Cerione, R. A., et al. (1998). PAK3 mutation in nonsyndromic X-linked mental retardation. Nat. Genet. 20, 25–30. doi: 10.1038/1675
Amenduni, M., De Filippis, R., Cheung, A. Y. L., Disciglio, V., Epistolato, M. C., Ariani, F., et al. (2011). iPS cells to model CDKL5-related disorders. Eur. J. Hum. Genet. 19, 1246–1255. doi: 10.1038/ejhg.2011.131
Asrar, S., Meng, Y., Zhou, Z., Todorovski, Z., Huang, W. W., and Jia, Z. (2009). Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1). Neuropharmacology 56, 73–80. doi: 10.1016/j.neuropharm.2008.06.055
Bahi-Buisson, N., Villeneuve, N., Caietta, E., Jacquette, A., Maurey, H., Matthijs, G., et al. (2012). Recurrent mutations in the CDKL5 gene: genotype-phenotype relationships. Am. J. Med. Genet. A 158A, 1612–1619. doi: 10.1002/ajmg.a.35401
Baltussen, L. L., Negraes, P. D., Silvestre, M., Claxton, S., Moeskops, M., Christodoulou, E., et al. (2018). Chemical genetic identification of CDKL5 substrates reveals its role in neuronal microtubule dynamics. EMBO J. 37:137. doi: 10.15252/embj.201899763
Baltussen, L. L., Rosianu, F., and Ultanir, S. K. (2017). Kinases in synaptic development and neurological diseases. Prog. Neuropsychopharmacol. Biol. Psychiatry. 84(Pt B), 343–352. doi: 10.1016/j.pnpbp.2017.12.006
Banerjee, T. D., Dagda, R. Y., Dagda, M., Chu, C. T., Rice, M., Vazquez-Mayorga, E., et al. (2017). PINK1 regulates mitochondrial trafficking in dendrites of cortical neurons through mitochondrial PKA. J. Neurochem. 142, 545–559. doi: 10.1111/jnc.14083
Bantscheff, M., Eberhard, D., Abraham, Y., Bastuck, S., Boesche, M., Hobson, S., et al. (2007). Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 25, 1035–1044. doi: 10.1038/nbt1328
Barford, D., Das, A. K., and Egloff, M. P. (1998). The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct. 27, 133–164. doi: 10.1146/annurev.biophys.27.1.133
Barnes, A. P., and Polleux, F. (2009). Establishment of axon-dendrite polarity in developing neurons. Annu. Rev. Neurosci. 32, 347–381. doi: 10.1146/annurev.neuro.31.060407.125536
Becker, L. E., Armstrong, D. L., and Chan, F. (1986). Dendritic atrophy in children with Down's syndrome. Ann. Neurol. 20, 520–526. doi: 10.1002/ana.410200413
Bidinosti, M., Botta, P., Krüttner, S., Proenca, C. C., Stoehr, N., Bernhard, M., et al. (2016). CLK2 inhibition ameliorates autistic features associated with SHANK3 deficiency. Science 351, 1199–1203. doi: 10.1126/science.aad5487
Blethrow, J. D., Glavy, J. S., Morgan, D. O., and Shokat, K. M. (2008). Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc. Natl. Acad. Sci. U.S.A. 105, 1442–1447. doi: 10.1073/pnas.0708966105
Blizinsky, K. D., Diaz-Castro, B., Forrest, M. P., Schürmann, B., Bach, A. P., Martin-de-Saavedra, M. D., et al. (2016). Reversal of dendritic phenotypes in 16p11.2 microduplication mouse model neurons by pharmacological targeting of a network hub. Proc. Natl. Acad. Sci. U.S.A. 113, 8520–8525. doi: 10.1073/pnas.1607014113
Butler, M. G., Dasouki, M. J., Zhou, X.-P., Talebizadeh, Z., Brown, M., Takahashi, T. N., et al. (2005). Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 42, 318–321. doi: 10.1136/jmg.2004.024646
Calabrese, B., and Halpain, S. (2005). Essential role for the PKC target MARCKS in maintaining dendritic spine morphology. Neuron 48, 77–90. doi: 10.1016/j.neuron.2005.08.027
Campbell, D. B., D'Oronzio, R., Garbett, K., Ebert, P. J., Mirnics, K., Levitt, P., et al. (2007). Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann. Neurol. 62, 243–250. doi: 10.1002/ana.21180
Cheung, Z. H., Chin, W. H., Chen, Y., Ng, Y. P., and Ip, N. Y. (2007). Cdk5 is involved in BDNF-stimulated dendritic growth in hippocampal neurons. PLoS Biol. 5:e63. doi: 10.1371/journal.pbio.0050063
Chiocchetti, A. G., Yousaf, A., Bour, H. S., Haslinger, D., Waltes, R., Duketis, E., et al. (2018). Common functional variants of the glutamatergic system in Autism spectrum disorder with high and low intellectual abilities. J Neural Transm. 125, 259–271. doi: 10.1007/s00702-017-1813-9
Cline, H. T. (2001). Dendritic arbor development and synaptogenesis. Curr. Opin. Neurobiol. 11, 118–126. doi: 10.1016/S0959-4388(00)00182-3
Cobb, M. H. (1999). MAP kinase pathways. Prog. Biophys. Mol. Biol. 71, 479–500. doi: 10.1016/S0079-6107(98)00056-X
Cochran, J. N., Hall, A. M., and Roberson, E. D. (2014). The dendritic hypothesis for Alzheimer's disease pathophysiology. Brain Res. Bull. 103, 18–28. doi: 10.1016/j.brainresbull.2013.12.004
Cohen, P. (2002). Protein kinases–the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 1, 309–315. doi: 10.1038/nrd773
Congdon, E. E., and Sigurdsson, E. M. (2018). Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 14, 399–415. doi: 10.1038/s41582-018-0013-z
Costa, M. R., Wen, G., Lepier, A., Schroeder, T., and Götz, M. (2008). Par-complex proteins promote proliferative progenitor divisions in the developing mouse cerebral cortex. Development 135, 11–22. doi: 10.1242/dev.009951
Courcet, J.-B., Faivre, L., Malzac, P., Masurel-Paulet, A., Lopez, E., Callier, P., et al. (2012). The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J. Med. Genet. 49, 731–736. doi: 10.1136/jmedgenet-2012-101251
Coutadeur, S., Benyamine, H., Delalonde, L., de Oliveira, C., Leblond, B., Foucourt, A., et al. (2015). A novel DYRK1A (dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer's disease: effect on Tau and amyloid pathologies in vitro. J. Neurochem. 133, 440–451. doi: 10.1111/jnc.13018
Crosson, S., and Moffat, K. (2002). Photoexcited structure of a plant photoreceptor domain reveals a light-driven molecular switch. Plant Cell 14, 1067–1075. doi: 10.1105/tpc.010475
Cruz, J. C., and Tsai, L.-H. (2004). Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends Mol. Med. 10, 452–458. doi: 10.1016/j.molmed.2004.07.001
Dagda, R. K., Pien, I., Wang, R., Zhu, J., Wang, K. Z. Q., Callio, J., et al. (2014). Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J. Neurochem. 128, 864–877. doi: 10.1111/jnc.12494
Dalva, M. B., Takasu, M. A., Lin, M. Z., Shamah, S. M., Hu, L., Gale, N. W., et al. (2000). EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell 103, 945–956. doi: 10.1016/S0092-8674(00)00197-5
Dang, T., Duan, W. Y., Yu, B., Tong, D. L., Cheng, C., Zhang, Y. F., et al. (2018). Autism-associated Dyrk1a truncation mutants impair neuronal dendritic and spine growth and interfere with postnatal cortical development. Mol. Psychiatry 23, 747–758. doi: 10.1038/mp.2016.253
de Anda, F. C., Rosario, A. L., Durak, O., Tran, T., Gräff, J., Meletis, K., et al. (2012). Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat. Neurosci. 15, 1022–1031. doi: 10.1038/nn.3141
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
den Hertog, J. (2003). Regulation of protein phosphatases in disease and behaviour. EMBO Rep. 4, 1027–1032. doi: 10.1038/sj.embor.7400009
Deshpande, A., Yadav, S., Dao, D. Q., Wu, Z.-Y., Hokanson, K. C., Cahill, M. K., et al. (2017). Cellular phenotypes in human iPSC-derived neurons from a genetic model of autism spectrum disorder. Cell Rep. 21, 2678–2687. doi: 10.1016/j.celrep.2017.11.037
DeVos, S. L., Corjuc, B. T., Oakley, D. H., Nobuhara, C. K., Bannon, R. N., Chase, A., et al. (2018). Synaptic Tau seeding precedes tau pathology in human Alzheimer's disease brain. Front Neurosci 12:267. doi: 10.3389/fnins.2018.00267
Di Maio, R., Hoffman, E. K., Rocha, E. M., Keeney, M. T., Sanders, L. H., De Miranda, B. R., et al. (2018). LRRK2 activation in idiopathic Parkinson's disease. Sci. Transl. Med. 10:eaar5429. doi: 10.1126/scitranslmed.aar5429
Dierssen, M. (2012). Down syndrome: the brain in trisomic mode. Nat. Rev. Neurosci. 13, 844–858. doi: 10.1038/nrn3314
Dolmetsch, R., and Geschwind, D. H. (2011). The human brain in a dish: the promise of iPSC-derived neurons. Cell 145, 831–834. doi: 10.1016/j.cell.2011.05.034
Dulovic-Mahlow, M., Trinh, J., Kandaswamy, K. K., Braathen, G. J., Di Donato, N., Rahikkala, E., et al. (2019). De Novo variants in TAOK1 cause neurodevelopmental disorders. Am. J. Hum. Genet. 105, 213–220. doi: 10.1016/j.ajhg.2019.05.005
Emoto, K. (2011). The growing role of the Hippo–NDR kinase signalling in neuronal development and disease. J. Biochem. 150, 133–141. doi: 10.1093/jb/mvr080
Emoto, K., He, Y., Ye, B., Grueber, W. B., Adler, P. N., Jan, L. Y., et al. (2004). Control of dendritic branching and tiling by the Tricornered-kinase/Furry signaling pathway in Drosophila sensory neurons. Cell 119, 245–256. doi: 10.1016/j.cell.2004.09.036
Emoto, K., Parrish, J. Z., Jan, L. Y., and Jan, Y.-N. (2006). The tumour suppressor Hippo acts with the NDR kinases in dendritic tiling and maintenance. Nature 443, 210–213. doi: 10.1038/nature05090
Ethell, I. M., Irie, F., Kalo, M. S., Couchman, J. R., Pasquale, E. B., and Yamaguchi, Y. (2001). EphB/syndecan-2 signaling in dendritic spine morphogenesis. Neuron 31, 1001–1013. doi: 10.1016/S0896-6273(01)00440-8
Ewers, H., Tada, T., Petersen, J. D., Racz, B., Sheng, M., and Choquet, D. (2014). A septin-dependent diffusion barrier at dendritic spine necks. PLoS ONE 9:e113916. doi: 10.1371/journal.pone.0113916
Fabbro, D., Cowan-Jacob, S. W., and Moebitz, H. (2015). Ten things you should know about protein kinases: IUPHAR review 14. Br. J. Pharmacol. 172, 2675–2700. doi: 10.1111/bph.13096
Ferrer, I., Barrachina, M., and Puig, B. (2002). Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer's disease, Pick's disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 104, 583–591. doi: 10.1007/s00401-002-0587-8
Fink, C. C., Bayer, K.-U., Myers, J. W., Ferrell, J. E., Schulman, H., and Meyer, T. (2003). Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron 39, 283–297. doi: 10.1016/S0896-6273(03)00428-8
Florio, M., and Huttner, W. B. (2014). Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141, 2182–2194. doi: 10.1242/dev.090571
Forrest, M. P., Parnell, E., and Penzes, P. (2018). Dendritic structural plasticity and neuropsychiatric disease. Nat. Rev. Neurosci. 19, 215–234. doi: 10.1038/nrn.2018.16
Frese, C. K., Mikhaylova, M., Stucchi, R., Gautier, V., Liu, Q., Mohammed, S., et al. (2017). Quantitative map of proteome dynamics during neuronal differentiation. Cell Rep. 18, 1527–1542. doi: 10.1016/j.celrep.2017.01.025
Fuchs, C., Trazzi, S., Torricella, R., Viggiano, R., De Franceschi, M., Amendola, E., et al. (2014). Loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3β signaling. Neurobiol. Dis. 70, 53–68. doi: 10.1016/j.nbd.2014.06.006
Gan, L., Cookson, M. R., Petrucelli, L., and La Spada, A. R. (2018). Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 21, 1300–1309. doi: 10.1038/s41593-018-0237-7
Geiger, T., Cox, J., Ostasiewicz, P., Wisniewski, J. R., and Mann, M. (2010). Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods 7, 383–385. doi: 10.1038/nmeth.1446
Geschwind, D. H., and Levitt, P. (2007). Autism spectrum disorders: developmental disconnection syndromes. Curr. Opin. Neurobiol. 17, 103–111. doi: 10.1016/j.conb.2007.01.009
Ghosh, A., and Greenberg, M. E. (1995). Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 268, 239–247. doi: 10.1126/science.7716515
Giacobini, E., and Gold, G. (2013). Alzheimer disease therapy–moving from amyloid-β to tau. Nat. Rev. Neurol. 9, 677–686. doi: 10.1038/nrneurol.2013.223
Giacomini, C., Koo, C.-Y., Yankova, N., Tavares, I. A., Wray, S., Noble, W., et al. (2018). A new TAO kinase inhibitor reduces tau phosphorylation at sites associated with neurodegeneration in human tauopathies. Acta Neuropathol. Commun. 6, 37–16. doi: 10.1186/s40478-018-0539-8
Glibert, P., Meert, P., Van Steendam, K., Van Nieuwerburgh, F., De Coninck, D., Martens, L., et al. (2015). Phospho-iTRAQ: assessing isobaric labels for the large-scale study of phosphopeptide stoichiometry. J. Proteome Res. 14, 839–849. doi: 10.1021/pr500889v
Golkowski, M., Lau, H.-T., Chan, M., Kenerson, H., Vidadala, V. N., Shoemaker, A., et al. (2020). Pharmacoproteomics identifies kinase pathways that drive the epithelial-mesenchymal transition and drug resistance in hepatocellular carcinoma. Cell Syst. 11, 196–207.e197. doi: 10.1016/j.cels.2020.07.006
Golkowski, M., Vidadala, R. S. R., Lombard, C. K., Suh, H. W., Maly, D. J., and Ong, S.-E. (2017). Kinobead and single-shot LC-MS Profiling identifies selective PKD inhibitors. J. Proteome Res. 16, 1216–1227. doi: 10.1021/acs.jproteome.6b00817
Götz, M., and Huttner, W. B. (2005). The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 6, 777–788. doi: 10.1038/nrm1739
Griebel, G., Stemmelin, J., Lopez-Grancha, M., Boulay, D., Boquet, G., Slowinski, F., et al. (2019). The selective GSK3 inhibitor, SAR502250, displays neuroprotective activity and attenuates behavioral impairments in models of neuropsychiatric symptoms of Alzheimer's disease in rodents. Sci. Rep. 9:18045. doi: 10.1038/s41598-019-54557-5
Grueber, W. B., Jan, L. Y., and Jan, Y. N. (2003). Different levels of the homeodomain protein cut regulate distinct dendrite branching patterns of drosophila multidendritic neurons. Cell. 112, 805–818. doi: 10.1016/s0092-8674(03)00160-0
Grueber, W. B., and Sagasti, A. (2010). Self-avoidance and tiling: mechanisms of dendrite and axon spacing. Cold Spring Harb. Perspect. Biol. 2, a001750–a001750. doi: 10.1101/cshperspect.a001750
Harms, F. L., Kloth, K., Bley, A., Denecke, J., Santer, R., Lessel, D., et al. (2018). Activating mutations in PAK1, encoding p21-activated kinase 1, cause a neurodevelopmental disorder. Am. J. Hum. Genet. 103, 579–591. doi: 10.1016/j.ajhg.2018.09.005
Henkemeyer, M., Itkis, O. S., Ngo, M., Hickmott, P. W., and Ethell, I. M. (2003). Multiple EphB receptor tyrosine kinases shape dendritic spines in the hippocampus. J. Cell Biol. 163, 1313–1326. doi: 10.1083/jcb.200306033
Hering, H., and Sheng, M. (2001). Dendritic spines: structure, dynamics and regulation. Nat. Rev. Neurosci. 2, 880–888. doi: 10.1038/35104061
Hertz, N. T., Berthet, A., Sos, M. L., Thorn, K. S., Burlingame, A. L., Nakamura, K., et al. (2013). A neo-substrate that amplifies catalytic activity of parkinson's-disease-related kinase PINK1. Cell. 154, 737–747. doi: 10.1016/j.cell.2013.07.030
Hoeffer, C. A., and Klann, E. (2010). mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75. doi: 10.1016/j.tins.2009.11.003
Hoover, B. R., Reed, M. N., Su, J., Penrod, R. D., Kotilinek, L. A., Grant, M. K., et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. doi: 10.1016/j.neuron.2010.11.030
Horch, H. W., and Katz, L. C. (2002). BDNF release from single cells elicits local dendritic growth in nearby neurons. Nat. Neurosci. 5, 1177–1184. doi: 10.1038/nn927
Horn, S., Au, M., Basel-Salmon, L., Bayrak-Toydemir, P., Chapin, A., Cohen, L., et al. (2019). De novo variants in PAK1 lead to intellectual disability with macrocephaly and seizures. Brain 142, 3351–3359. doi: 10.1093/brain/awz264
Hosp, F., and Mann, M. (2017). A primer on concepts and applications of proteomics in neuroscience. Neuron 96, 558–571. doi: 10.1016/j.neuron.2017.09.025
Hotulainen, P., and Hoogenraad, C. C. (2010). Actin in dendritic spines: connecting dynamics to function. J. Cell Biol. 189, 619–629. doi: 10.1083/jcb.201003008
Huang, E. J., and Reichardt, L. F. (2001). Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736. doi: 10.1146/annurev.neuro.24.1.677
Huang, S. H., Wang, J., Sui, W. H., Chen, B., Zhang, X. Y., Yan, J., et al. (2013). BDNF-dependent recycling facilitates TrkB translocation to postsynaptic density during LTP via a Rab11-dependent pathway. J. Neurosci. 33, 9214–9230. doi: 10.1523/JNEUROSCI.3256-12.2013
Huang, X., Shi, L., Cao, J., He, F., Li, R., Zhang, Y., et al. (2014). The sterile 20-like kinase tao controls tissue homeostasis by regulating the hippo pathway in Drosophila adult midgut. J. Genet. Genomics 41, 429–438. doi: 10.1016/j.jgg.2014.05.007
Imai, F., Hirai, S.-I., Akimoto, K., Koyama, H., Miyata, T., Ogawa, M., et al. (2006). Inactivation of aPKClambda results in the loss of adherens junctions in neuroepithelial cells without affecting neurogenesis in mouse neocortex. Development 133, 1735–1744. doi: 10.1242/dev.02330
Inoue, H., Nagata, N., Kurokawa, H., and Yamanaka, S. (2014). iPS cells: a game changer for future medicine. Embo J. 33, 409–417. doi: 10.1002/embj.201387098
Iossifov, I., O'Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Iqbal, K., Liu, F., and Gong, C.-X. (2016). Tau and neurodegenerative disease: the story so far. Nat. Rev. Neurol. 12, 15–27. doi: 10.1038/nrneurol.2015.225
Ittner, A., and Ittner, L. M. (2018). Dendritic Tau in Alzheimer's disease. Neuron 99, 13–27. doi: 10.1016/j.neuron.2018.06.003
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Jan, Y.-N., and Jan, L. Y. (2010). Branching out: mechanisms of dendritic arborization. Nat. Rev. Neurosci. 11, 316–328. doi: 10.1038/nrn2836
Jansen, L. A., Mirzaa, G. M., Ishak, G. E., O'Roak, B. J., Hiatt, J. B., Roden, W. H., et al. (2015). PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 138, 1613–1628. doi: 10.1093/brain/awv045
Jaworski, J., Kapitein, L. C., Gouveia, S. M., Dortland, B. R., Wulf, P. S., Grigoriev, I., et al. (2009). Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron 61, 85–100. doi: 10.1016/j.neuron.2008.11.013
Jaworski, J., Spangler, S., Seeburg, D. P., Hoogenraad, C. C., and Sheng, M. (2005). Control of dendritic arborization by the phosphoinositide-3'-kinase-Akt-mammalian target of rapamycin pathway. J. Neurosci. 25, 11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005
Ji, J., Lee, H., Argiropoulos, B., Dorrani, N., Mann, J., Martinez-Agosto, J. A., et al. (2015). DYRK1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur. J. Hum. Genet. 23, 1473–1481. doi: 10.1038/ejhg.2015.71
Jossin, Y. (2020). Reelin functions, mechanisms of action and signaling pathways during brain development and maturation. Biomolecules 10:964. doi: 10.3390/biom10060964
Kashani, A. H., Qiu, Z., Jurata, L., Lee, S.-K., Pfaff, S., Goebbels, S., et al. (2006). Calcium activation of the LMO4 transcription complex and its role in the patterning of thalamocortical connections. J. Neurosci. 26, 8398–8408. doi: 10.1523/JNEUROSCI.0618-06.2006
Kaufman, A. C., Salazar, S. V., Haas, L. T., Yang, J., Kostylev, M. A., Jeng, A. T., et al. (2015). Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann. Neurol. 77, 953–971. doi: 10.1002/ana.24394
Kelava, I., and Lancaster, M. A. (2016). Dishing out mini-brains: current progress and future prospects in brain organoid research. Dev. Biol. 420, 199–209. doi: 10.1016/j.ydbio.2016.06.037
Kernohan, K. D., McBride, A., Hartley, T., Rojas, S. K., Care4Rare Canada Consortium, Dyment, D. A., et al. (2019). p21 protein-activated kinase 1 is associated with severe regressive autism, and epilepsy. Clin. Genet. 96, 449–455. doi: 10.1111/cge.13618
Khamrui, S., Ung, P. M. U., Secor, C., Schlessinger, A., and Lazarus, M. B. (2020). High-resolution structure and inhibition of the schizophrenia-linked pseudokinase ULK4. J. Am. Chem. Soc. 142, 33–37. doi: 10.1021/jacs.9b10458
Klein, R. (2009). Bidirectional modulation of synaptic functions by Eph/ephrin signaling. Nat. Neurosci. 12, 15–20. doi: 10.1038/nn.2231
Koleske, A. J. (2013). Molecular mechanisms of dendrite stability. Nat. Rev. Neurosci. 14, 536–550. doi: 10.1038/nrn3486
Konur, S., and Ghosh, A. (2005). Calcium signaling and the control of dendritic development. Neuron 46, 401–405. doi: 10.1016/j.neuron.2005.04.022
Krahn, A. I., Wells, C., Drewry, D. H., Beitel, L. K., Durcan, T. M., and Axtman, A. D. (2020). Defining the neural kinome: strategies and opportunities for small molecule drug discovery to target neurodegenerative diseases. ACS Chem. Neurosci. 11, 1871–1886. doi: 10.1021/acschemneuro.0c00176
Krüger, M., Moser, M., Ussar, S., Thievessen, I., Luber, C. A., Forner, F., et al. (2008). SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell 134, 353–364. doi: 10.1016/j.cell.2008.05.033
Kulkarni, V. A., and Firestein, B. L. (2012). The dendritic tree and brain disorders. Mol. Cell. Neurosci. 50, 10–20. doi: 10.1016/j.mcn.2012.03.005
Kumar, R. A., Karamohamed, S., Sudi, J., Conrad, D. F., Brune, C., Badner, J. A., et al. (2008). Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 17, 628–638. doi: 10.1093/hmg/ddm376
Küry, S., van Woerden, G. M., Besnard, T., Proietti Onori, M., Latypova, X., Towne, M. C., et al. (2017). de novo mutations in protein kinase genes CAMK2A and CAMK2B cause intellectual disability. Am. J. Hum. Genet. 101, 768–788. doi: 10.1016/j.ajhg.2017.10.003
Kuwako, K.-I., and Okano, H. (2018). The LKB1-SIK pathway controls dendrite self-avoidance in purkinje cells. Cell. Rep. 24, 2808–2818.e4. doi: 10.1016/j.celrep.2018.08.029
Kwon, C.-H., Zhu, X., Zhang, J., and Baker, S. J. (2003). mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 12923–12928. doi: 10.1073/pnas.2132711100
Lancaster, M. A., and Knoblich, J. A. (2012). Spindle orientation in mammalian cerebral cortical development. Curr. Opin. Neurobiol. 22, 737–746. doi: 10.1016/j.conb.2012.04.003
Lang, B., Pu, J., Hunter, I., Liu, M., Martin-Granados, C., Reilly, T. J., et al. (2014). Recurrent deletions of ULK4 in schizophrenia: a gene crucial for neuritogenesis and neuronal motility. J. Cell. Sci. 127, 630–640. doi: 10.1242/jcs.137604
Lang, B., Zhang, L., Jiang, G., Hu, L., Lan, W., Zhao, L., et al. (2016). Control of cortex development by ULK4, a rare risk gene for mental disorders including schizophrenia. Sci. Rep. 6, 31126–31114. doi: 10.1038/srep31126
Lauretti, E., Dincer, O., and Praticò, D. (2020). Glycogen synthase kinase-3 signaling in Alzheimer's disease. Biochim. Biophys. Acta Mol. Cell. Res. 1867:118664. doi: 10.1016/j.bbamcr.2020.118664
Leopold, A. V., Chernov, K. G., and Verkhusha, V. V. (2018). Optogenetically controlled protein kinases for regulation of cellular signaling. Chem. Soc. Rev. 47, 2454–2484. doi: 10.1039/C7CS00404D
Li, K., Wei, Q., Liu, F.-F., Hu, F., Xie, A.-J., Zhu, L.-Q., et al. (2018). Synaptic dysfunction in Alzheimer's disease: Aβ, Tau, and epigenetic alterations. Mol. Neurobiol. 55, 3021–3032. doi: 10.1007/s12035-017-0533-3
Liao, L., Park, S. K., Xu, T., Vanderklish, P., and Yates, J. R. (2008). Quantitative proteomic analysis of primary neurons reveals diverse changes in synaptic protein content in fmr1 knockout mice. Proc. Natl. Acad. Sci. U.S.A. 105, 15281–15286. doi: 10.1073/pnas.0804678105
Lin, C. H., Tsai, P. I., Wu, R. M., and Chien, C. T. (2010). LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3β. J. Neurosci. 30, 13138–13149. doi: 10.1523/JNEUROSCI.1737-10.2010
Lin, Y.-C., and Koleske, A. J. (2010). Mechanisms of synapse and dendrite maintenance and their disruption in psychiatric and neurodegenerative disorders. Annu. Rev. Neurosci. 33, 349–378. doi: 10.1146/annurev-neuro-060909-153204
Lipton, J. O., and Sahin, M. (2014). The neurology of mTOR. Neuron 84, 275–291. doi: 10.1016/j.neuron.2014.09.034
Lisman, J., Schulman, H., and Cline, H. (2002). The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 3, 175–190. doi: 10.1038/nrn753
Liu, M., Guan, Z., Shen, Q., Flinter, F., Domínguez, L., Ahn, J. W., et al. (2016). Ulk4 regulates neural stem cell pool. Stem Cells 34, 2318–2331. doi: 10.1002/stem.2423
Locke, C., Machida, K., Tucker, C. L., Wu, Y., and Yu, J. (2018). Correction: optogenetic activation of EphB2 receptor in dendrites induced actin polymerization by activating Arg kinase. Biol. Open 7:bio034694. doi: 10.1242/bio.034694
Lom, B., and Cohen-Cory, S. (1999). Brain-derived neurotrophic factor differentially regulates retinal ganglion cell dendritic and axonal arborization in vivo. J. Neurosci. 19, 9928–9938. doi: 10.1523/JNEUROSCI.19-22-09928.1999
Lugo, J. N., Smith, G. D., Arbuckle, E. P., White, J., Holley, A. J., Floruta, C. M., et al. (2014). Deletion of PTEN produces autism-like behavioral deficits and alterations in synaptic proteins. Front. Mol. Neurosci. 7:27. doi: 10.3389/fnmol.2014.00027
Luo, R., Sanders, S. J., Tian, Y., Voineagu, I., Huang, N., Chu, S. H., et al. (2012). Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Am. J. Hum. Genet. 91, 38–55. doi: 10.1016/j.ajhg.2012.05.011
MacLeod, D., Dowman, J., Hammond, R., Leete, T., Inoue, K., and Abeliovich, A. (2006). The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52, 587–593. doi: 10.1016/j.neuron.2006.10.008
Mairet-Coello, G., Courchet, J., Pieraut, S., Courchet, V., Maximov, A., and Polleux, F. (2013). The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Aβ oligomers through Tau phosphorylation. Neuron 78, 94–108. doi: 10.1016/j.neuron.2013.02.003
Manning, G., Whyte, D. B., Martinez, R., Hunter, T., and Sudarsanam, S. (2002). The protein kinase complement of the human genome. Science 298, 1912–1934. doi: 10.1126/science.1075762
Martin-Belmonte, F., and Mostov, K. (2008). Regulation of cell polarity during epithelial morphogenesis. Curr. Opin. Cell Biol. 20, 227–234. doi: 10.1016/j.ceb.2008.01.001
Martinez de Lagran, M., Benavides-Piccione, R., Ballesteros-Yañez, I., Calvo, M., Morales, M., Fillat, C., et al. (2012). Dyrk1A influences neuronal morphogenesis through regulation of cytoskeletal dynamics in mammalian cortical neurons. Cereb. Cortex 22, 2867–2877. doi: 10.1093/cercor/bhr362
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., and Cummings, J. L. (2015). Alzheimer's disease. Nat. Rev. Dis. Primers 1, 15056–15018. doi: 10.1038/nrdp.2015.56
Matus, A. (2000). Actin-based plasticity in dendritic spines. Science 290, 754–758. doi: 10.1126/science.290.5492.754
McAllister, A. K. (2000). Cellular and molecular mechanisms of dendrite growth. Cereb. Cortex 10, 963–973. doi: 10.1093/cercor/10.10.963
McAllister, A. K., Katz, L. C., and Lo, D. C. (1997). Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron 18, 767–778. doi: 10.1016/S0896-6273(00)80316-5
McAllister, A. K., Lo, D. C., and Katz, L. C. (1995). Neurotrophins regulate dendritic growth in developing visual cortex. Neuron 15, 791–803. doi: 10.1016/0896-6273(95)90171-X
McCaffrey, L. M., and Macara, I. G. (2012). Signaling pathways in cell polarity. Cold Spring Harb. Perspect. Biol. 4, a009654–a009654. doi: 10.1101/cshperspect.a009654
McCarthy, S. E., Makarov, V., Kirov, G., Addington, A. M., McClellan, J., Yoon, S., et al. (2009). Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 41, 1223–1227. doi: 10.1038/ng.474
Melchior, B., Mittapalli, G. K., Lai, C., Duong-Polk, K., Stewart, J., Güner, B., et al. (2019). Tau pathology reduction with SM07883, a novel, potent, and selective oral DYRK1A inhibitor: a potential therapeutic for Alzheimer's disease. Aging Cell 18:e13000. doi: 10.1111/acel.13000
Meng, Y., Zhang, Y., Tregoubov, V., Janus, C., Cruz, L., Jackson, M., et al. (2002). Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35, 121–133. doi: 10.1016/S0896-6273(02)00758-4
Mirzaa, G. M., Campbell, C. D., Solovieff, N., Goold, C., Jansen, L. A., Menon, S., et al. (2016). Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol. 73, 836–845. doi: 10.1001/jamaneurol.2016.0363
Mitsopoulos, C., Zihni, C., Garg, R., Ridley, A. J., and Morris, J. D. H. (2003). The prostate-derived sterile 20-like kinase (PSK) regulates microtubule organization and stability. J. Biol. Chem. 278, 18085–18091. doi: 10.1074/jbc.M213064200
Mitz, A. R., Philyaw, T. J., Boccuto, L., Shcheglovitov, A., Sarasua, S. M., Kaufmann, W. E., et al. (2018). Identification of 22q13 genes most likely to contribute to Phelan McDermid syndrome. Eur. J. Hum. Genet. 26, 293–302. doi: 10.1038/s41431-017-0042-x
Møller, R. S., Kübart, S., Hoeltzenbein, M., Heye, B., Vogel, I., Hansen, C. P., et al. (2008). Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am. J. Hum. Genet. 82, 1165–1170. doi: 10.1016/j.ajhg.2008.03.001
Moore, T. M., Garg, R., Johnson, C., Coptcoat, M. J., Ridley, A. J., and Morris, J. D. (2000). PSK, a novel STE20-like kinase derived from prostatic carcinoma that activates the c-Jun N-terminal kinase mitogen-activated protein kinase pathway and regulates actin cytoskeletal organization. J. Biol. Chem. 275, 4311–4322. doi: 10.1074/jbc.275.6.4311
Navarrete-Perea, J., Yu, Q., Gygi, S. P., and Paulo, J. A. (2018). Streamlined tandem mass tag (SL-TMT) protocol: an efficient strategy for quantitative (phospho)proteome profiling using tandem mass tag-synchronous precursor selection-MS3. J. Proteome Res. 17, 2226–2236. doi: 10.1021/acs.jproteome.8b00217
Nedivi, E., Wu, G. Y., and Cline, H. T. (1998). Promotion of dendritic growth by CPG15, an activity-induced signaling molecule. Science 281, 1863–1866. doi: 10.1126/science.281.5384.1863
Noble, W., Hanger, D. P., Miller, C. C. J., and Lovestone, S. (2013). The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 4:83. doi: 10.3389/fneur.2013.00083
Nolt, M. J., Lin, Y., Hruska, M., Murphy, J., Sheffler-Colins, S. I., Kayser, M. S., et al. (2011). EphB controls NMDA receptor function and synaptic targeting in a subunit-specific manner. J. Neurosci. 31, 5353–5364. doi: 10.1523/JNEUROSCI.0282-11.2011
Okamoto, K.-I., Nagai, T., Miyawaki, A., and Hayashi, Y. (2004). Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat. Neurosci. 7, 1104–1112. doi: 10.1038/nn1311
Okamoto, K.-I., Narayanan, R., Lee, S. H., Murata, K., and Hayashi, Y. (2007). The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc. Natl. Acad. Sci. U.S.A. 104, 6418–6423. doi: 10.1073/pnas.0701656104
Okatsu, K., Oka, T., Iguchi, M., Imamura, K., Kosako, H., Tani, N., et al. (2012). PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 3, 1016–1010. doi: 10.1038/ncomms2016
Oldach, L., and Zhang, J. (2014). Genetically encoded fluorescent biosensors for live-cell visualization of protein phosphorylation. Chem. Biol. 21, 186–197. doi: 10.1016/j.chembiol.2013.12.012
Ong, S.-E., Blagoev, B., Kratchmarova, I., Kristensen, D. B., Steen, H., Pandey, A., et al. (2002). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics 1, 376–386. doi: 10.1074/mcp.M200025-MCP200
Ori-McKenney, K. M., McKenney, R. J., Huang, H. H., Li, T., Meltzer, S., Jan, L. Y., et al. (2016). Phosphorylation of β-tubulin by the down syndrome kinase, minibrain/DYRK1a, regulates microtubule dynamics and dendrite morphogenesis. Neuron 90, 551–563. doi: 10.1016/j.neuron.2016.03.027
Owen, J. P., Bukshpun, P., Pojman, N., Thieu, T., Chen, Q., Lee, J., et al. (2018). Brain MR imaging findings and associated outcomes in carriers of the reciprocal copy number variation at 16p11.2. Radiology 286, 217–226. doi: 10.1148/radiol.2017162934
Park, J., Sung, J. Y., Park, J., Song, W.-J., Chang, S., and Chung, K. C. (2012). Dyrk1A negatively regulates the actin cytoskeleton through threonine phosphorylation of N-WASP. J. Cell. Sci. 125, 67–80. doi: 10.1242/jcs.086124
Paşca, S. P., Panagiotakos, G., and Dolmetsch, R. E. (2014). Generating human neurons in vitro and using them to understand neuropsychiatric disease. Annu. Rev. Neurosci. 37, 479–501. doi: 10.1146/annurev-neuro-062012-170328
Patricelli, M. P., Nomanbhoy, T. K., Wu, J., Brown, H., Zhou, D., Zhang, J., et al. (2011). In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol. 18, 699–710. doi: 10.1016/j.chembiol.2011.04.011
Patricelli, M. P., Szardenings, A. K., Liyanage, M., Nomanbhoy, T. K., Wu, M., Weissig, H., et al. (2007). Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 46, 350–358. doi: 10.1021/bi062142x
Peebles, C. L., Yoo, J., Thwin, M. T., Palop, J. J., Noebels, J. L., and Finkbeiner, S. (2010). Arc regulates spine morphology and maintains network stability in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 18173–18178. doi: 10.1073/pnas.1006546107
Peng, A., and Maller, J. L. (2010). Serine/threonine phosphatases in the DNA damage response and cancer. Oncogene 29, 5977–5988. doi: 10.1038/onc.2010.371
Pinto, D., Delaby, E., Merico, D., Barbosa, M., Merikangas, A., Klei, L., et al. (2014). Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 94, 677–694. doi: 10.1016/j.ajhg.2014.03.018
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primers 3, 17013–17021. doi: 10.1038/nrdp.2017.13
Polleux, F., and Snider, W. (2010). Initiating and growing an axon. Cold Spring Harb. Perspect. Biol. 2, a001925–a001925. doi: 10.1101/cshperspect.a001925
Poon, C. L. C., Lin, J. I., Zhang, X., and Harvey, K. F. (2011). The sterile 20-like kinase Tao-1 controls tissue growth by regulating the Salvador-Warts-Hippo pathway. Dev. Cell 21, 896–906. doi: 10.1016/j.devcel.2011.09.012
Portmann, T., Yang, M., Mao, R., Panagiotakos, G., Ellegood, J., Dolen, G., et al. (2014). Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell. Rep. 7, 1077–1092. doi: 10.1016/j.celrep.2014.03.036
Qiu, S., Lu, Z., and Levitt, P. (2014). MET receptor tyrosine kinase controls dendritic complexity, spine morphogenesis, and glutamatergic synapse maturation in the hippocampus. J. Neurosci. 34, 16166–16179. doi: 10.1523/JNEUROSCI.2580-14.2014
Quintero-Rivera, F., Sharifi-Hannauer, P., and Martinez-Agosto, J. A. (2010). Autistic and psychiatric findings associated with the 3q29 microdeletion syndrome: case report and review. Am. J. Med. Genet. A 152A, 2459–2467. doi: 10.1002/ajmg.a.33573
Qureshi, A. Y., Mueller, S., Snyder, A. Z., Mukherjee, P., Berman, J. I., Roberts, T. P. L., et al. (2014). Opposing brain differences in 16p11.2 deletion and duplication carriers. J. Neurosci. 34, 11199–11211. doi: 10.1523/JNEUROSCI.1366-14.2014
Rajan, I., and Cline, H. T. (1998). Glutamate receptor activity is required for normal development of tectal cell dendrites in vivo. J. Neurosci. 18, 7836–7846. doi: 10.1523/JNEUROSCI.18-19-07836.1998
Raymond, G. V., Bauman, M. L., and Kemper, T. L. (1996). Hippocampus in autism: a Golgi analysis. Acta Neuropathol. 91, 117–119. doi: 10.1007/s004010050401
Redmond, L., and Ghosh, A. (2005). Regulation of dendritic development by calcium signaling. Cell Calcium 37, 411–416. doi: 10.1016/j.ceca.2005.01.009
Redmond, L., Kashani, A. H., and Ghosh, A. (2002). Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 34, 999–1010. doi: 10.1016/S0896-6273(02)00737-7
Rehberg, K., Kliche, S., Madencioglu, D. A., Thiere, M., Müller, B., Meineke, B. M., et al. (2014). The serine/threonine kinase Ndr2 controls integrin trafficking and integrin-dependent neurite growth. J. Neurosci. 34, 5342–5354. doi: 10.1523/JNEUROSCI.2728-13.2014
Renner, M., Lacor, P. N., Velasco, P. T., Xu, J., Contractor, A., Klein, W. L., et al. (2010). Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 66, 739–754. doi: 10.1016/j.neuron.2010.04.029
Ricciardi, S., Ungaro, F., Hambrock, M., Rademacher, N., Stefanelli, G., Brambilla, D., et al. (2012). CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat. Cell Biol. 14, 911–923. doi: 10.1038/ncb2566
Richter, M., Murtaza, N., Scharrenberg, R., White, S. H., Johanns, O., Walker, S., et al. (2018). Altered TAOK2 activity causes autism-related neurodevelopmental and cognitive abnormalities through RhoA signaling. Mol. Psychiatry 20:1237. doi: 10.1038/s41380-018-0025-5
Rodríguez-Fraticelli, A. E., Gálvez-Santisteban, M., and Martin-Belmonte, F. (2011). Divide and polarize: recent advances in the molecular mechanism regulating epithelial tubulogenesis. Curr. Opin. Cell. Biol. 23, 638–646. doi: 10.1016/j.ceb.2011.07.002
Rong, Y., Lu, X., Bernard, A., Khrestchatisky, M., and Baudry, M. (2001). Tyrosine phosphorylation of ionotropic glutamate receptors by Fyn or Src differentially modulates their susceptibility to calpain and enhances their binding to spectrin and PSD-95. J. Neurochem. 79, 382–390. doi: 10.1046/j.1471-4159.2001.00565.x
Sala, C., Piëch, V., Wilson, N. R., Passafaro, M., Liu, G., and Sheng, M. (2001). Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31, 115–130. doi: 10.1016/S0896-6273(01)00339-7
Sala Della, G., Putignano, E., Chelini, G., Melani, R., Calcagno, E., Michele Ratto, G., et al. (2016). Dendritic spine instability in a mouse model of CDKL5 disorder is rescued by insulin-like growth factor 1. Biol. Psychiatry 80, 302–311. doi: 10.1016/j.biopsych.2015.08.028
Sato, A. (2016). mTOR, a potential target to treat autism spectrum disorder. CNS Neurol. Disord. Drug Targets 15, 533–543. doi: 10.2174/1871527315666160413120638
Sato, M., Ozawa, T., Inukai, K., Asano, T., and Umezawa, Y. (2002). Fluorescent indicators for imaging protein phosphorylation in single living cells. Nat. Biotechnol. 20, 287–294. doi: 10.1038/nbt0302-287
Selkoe, D. J. (2002). Alzheimer's disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Sharma, V., Wang, Q., and Lawrence, D. S. (2008). Peptide-based fluorescent sensors of protein kinase activity: design and applications. Biochim. Biophys. Acta 1784, 94–99. doi: 10.1016/j.bbapap.2007.07.016
Shelton, D. L., Sutherland, J., Gripp, J., Camerato, T., Armanini, M. P., Phillips, H. S., et al. (1995). Human trks: molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesins. J. Neurosci. 15, 477–491. doi: 10.1523/JNEUROSCI.15-01-00477.1995
Shen, K., Teruel, M. N., Subramanian, K., and Meyer, T. (1998). CaMKIIbeta functions as an F-actin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron 21, 593–606. doi: 10.1016/S0896-6273(00)80569-3
Sherman, S. P., and Bang, A. G. (2018). High-throughput screen for compounds that modulate neurite growth of human induced pluripotent stem cell-derived neurons. Dis. Model. Mech. 11:dmm031906. doi: 10.1242/dmm.031906
Shi, Y., Pontrello, C. G., DeFea, K. A., Reichardt, L. F., and Ethell, I. M. (2009). Focal adhesion kinase acts downstream of EphB receptors to maintain mature dendritic spines by regulating cofilin activity. J. Neurosci. 29, 8129–8142. doi: 10.1523/JNEUROSCI.4681-08.2009
Shults, M. D., and Imperiali, B. (2003). Versatile fluorescence probes of protein kinase activity. J. Am. Chem. Soc. 125, 14248–14249. doi: 10.1021/ja0380502
Skalecka, A., Liszewska, E., Bilinski, R., Gkogkas, C., Khoutorsky, A., Malik, A. R., et al. (2016). mTOR kinase is needed for the development and stabilization of dendritic arbors in newly born olfactory bulb neurons. Dev. Neurobiol. 76, 1308–1327. doi: 10.1002/dneu.22392
Sottocornola, R., Royer, C., Vives, V., Tordella, L., Zhong, S., Wang, Y., et al. (2010). ASPP2 binds Par-3 and controls the polarity and proliferation of neural progenitors during CNS development. Dev. Cell 19, 126–137. doi: 10.1016/j.devcel.2010.06.003
Spellman, D. S., Deinhardt, K., Darie, C. C., Chao, M. V., and Neubert, T. A. (2008). Stable isotopic labeling by amino acids in cultured primary neurons: application to brain-derived neurotrophic factor-dependent phosphotyrosine-associated signaling. Mol. Cell Proteomics 7, 1067–1076. doi: 10.1074/mcp.M700387-MCP200
Steger, M., Tonelli, F., Ito, G., Davies, P., Trost, M., Vetter, M., et al. (2016). Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5:809. doi: 10.7554/eLife.12813.023
Stephenson, J. R., Wang, X., Perfitt, T. L., Parrish, W. P., Shonesy, B. C., Marks, C. R., et al. (2017). A novel human CAMK2A mutation disrupts dendritic morphology and synaptic transmission, and causes ASD-related behaviors. J. Neurosci. 37, 2216–2233. doi: 10.1523/JNEUROSCI.2068-16.2017
Tackenberg, C., Grinschgl, S., Trutzel, A., Santuccione, A. C., Frey, M. C., Konietzko, U., et al. (2013). NMDA receptor subunit composition determines beta-amyloid-induced neurodegeneration and synaptic loss. Cell Death Dis. 4:e608. doi: 10.1038/cddis.2013.129
Tada, T., Simonetta, A., Batterton, M., Kinoshita, M., Edbauer, D., and Sheng, M. (2007). Role of septin cytoskeleton in spine morphogenesis and dendrite development in neurons. Curr. Biol. 17, 1752–1758. doi: 10.1016/j.cub.2007.09.039
Tang, S., Terzic, B., Wang, I.-T. J., Sarmiento, N., Sizov, K., Cui, Y., et al. (2019). Altered NMDAR signaling underlies autistic-like features in mouse models of CDKL5 deficiency disorder. Nat. Commun. 10, 2655–2614. doi: 10.1038/s41467-019-10689-w
Tang, S., and Yasuda, R. (2017). Imaging ERK and pka activation in single dendritic spines during structural plasticity. Neuron 93, 1315–1324.e3. doi: 10.1016/j.neuron.2017.02.032
Tassano, E., Uccella, S., Giacomini, T., Striano, P., Severino, M., Porta, S., et al. (2018). Intragenic microdeletion of ULK4 and partial microduplication of BRWD3 in siblings with neuropsychiatric features and obesity. Cytogenet. Genome Res. 156, 14–21. doi: 10.1159/000491871
Tavares, I. A., Touma, D., Lynham, S., Troakes, C., Schober, M., Causevic, M., et al. (2013). Prostate-derived sterile 20-like kinases (PSKs/TAOKs) phosphorylate tau protein and are activated in tangle-bearing neurons in Alzheimer disease. J. Biol. Chem. 288, 15418–15429. doi: 10.1074/jbc.M112.448183
Tavazoie, S. F., Alvarez, V. A., Ridenour, D. A., Kwiatkowski, D. J., and Sabatini, B. L. (2005). Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 8, 1727–1734. doi: 10.1038/nn1566
Taverna, E., Götz, M., and Huttner, W. B. (2014). The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu. Rev. Cell. Dev. Biol. 30, 465–502. doi: 10.1146/annurev-cellbio-101011-155801
Tell, V., and Hilgeroth, A. (2013). Recent developments of protein kinase inhibitors as potential AD therapeutics. Front. Cell Neurosci. 7:189. doi: 10.3389/fncel.2013.00189
Thanseem, I., Nakamura, K., Miyachi, T., Toyota, T., Yamada, S., Tsujii, M., et al. (2010). Further evidence for the role of MET in autism susceptibility. Neurosci. Res. 68, 137–141. doi: 10.1016/j.neures.2010.06.014
Tolosa, E., Vila, M., Klein, C., and Rascol, O. (2020). LRRK2 in Parkinson disease: challenges of clinical trials. Nat. Rev. Neurol. 16, 97–107. doi: 10.1038/s41582-019-0301-2
Tonks, N. K. (2006). Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846. doi: 10.1038/nrm2039
Turk, B. E. (2005). Measuring kinase activity: finding needles in a haystack. Nat. Methods 2, 251–252. doi: 10.1038/nmeth0405-251
Ultanir, S. K., Hertz, N. T., Li, G., Ge, W.-P., Burlingame, A. L., Pleasure, S. J., et al. (2012). Chemical genetic identification of NDR1/2 kinase substrates AAK1 and Rabin8 Uncovers their roles in dendrite arborization and spine development. Neuron 73, 1127–1142. doi: 10.1016/j.neuron.2012.01.019
Ultanir, S. K., Yadav, S., Hertz, N. T., Oses-Prieto, J. A., Claxton, S., Burlingame, A. L., et al. (2014). MST3 kinase phosphorylates TAO1/2 to enable myosin Va function in promoting spine synapse development. Neuron 84, 968–982. doi: 10.1016/j.neuron.2014.10.025
Urbanska, M., Gozdz, A., Swiech, L. J., and Jaworski, J. (2012). Mammalian target of rapamycin complex 1 (mTORC1) and 2 (mTORC2) control the dendritic arbor morphology of hippocampal neurons. J. Biol. Chem. 287, 30240–30256. doi: 10.1074/jbc.M112.374405
Vaillant, A. R., Zanassi, P., Walsh, G. S., Aumont, A., Alonso, A., and Miller, F. D. (2002). Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron 34, 985–998. doi: 10.1016/S0896-6273(02)00717-1
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M. K., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
van Bon, B. W. M., Coe, B. P., Bernier, R., Green, C., Gerdts, J., Witherspoon, K., et al. (2016). Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol. Psychiatry 21, 126–132. doi: 10.1038/mp.2015.5
Vermilyea, S. C., Babinski, A., Tran, N., To, S., Guthrie, S., Kluss, J. H., et al. (2020). In vitro CRISPR/Cas9-directed gene editing to model LRRK2 G2019S Parkinson's Disease in common marmosets. Sci. Rep. 10:3447. doi: 10.1038/s41598-020-60273-2
Volkmar, F. R., and Greenough, W. T. (1972). Rearing complexity affects branching of dendrites in the visual cortex of the rat. Science 176, 1445–1447. doi: 10.1126/science.176.4042.1445
Wang, X., He, Y., Ye, Y., Zhao, X., Deng, S., He, G., et al. (2018a). SILAC-based quantitative MS approach for real-time recording protein-mediated cell-cell interactions. Sci. Rep. 8, 8441–8449. doi: 10.1038/s41598-018-26262-2
Wang, Y., Zeng, C., Li, J., Zhou, Z., Ju, X., Xia, S., et al. (2018b). PAK2 haploinsufficiency results in synaptic cytoskeleton impairment and autism-related behavior. Cell. Rep. 24, 2029–2041. doi: 10.1016/j.celrep.2018.07.061
Weaving, L. S., Christodoulou, J., Williamson, S. L., Friend, K. L., McKenzie, O. L. D., Archer, H., et al. (2004). Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 75, 1079–1093. doi: 10.1086/426462
Wegner, A. M., Nebhan, C. A., Hu, L., Majumdar, D., Meier, K. M., Weaver, A. M., et al. (2008). N-wasp and the arp2/3 complex are critical regulators of actin in the development of dendritic spines and synapses. J. Biol. Chem. 283, 15912–15920. doi: 10.1074/jbc.M801555200
Weiss, L. A., Shen, Y., Korn, J. M., Arking, D. E., Miller, D. T., Fossdal, R., et al. (2008). Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 358, 667–675. doi: 10.1056/NEJMoa075974
Wiesel, T. N., and Hubel, D. H. (1963). Single-cell responses in striate cortex of kittens deprived of vision in one eye. J. Neurophysiol. 26, 1003–1017. doi: 10.1152/jn.1963.26.6.1003
Winden, K. D., Ebrahimi-Fakhari, D., and Sahin, M. (2018). Abnormal mTOR activation in autism. Annu. Rev. Neurosci. 41, 1–23. doi: 10.1146/annurev-neuro-080317-061747
Wong, R. O. L., and Ghosh, A. (2002). Activity-dependent regulation of dendritic growth and patterning. Nat. Rev. Neurosci. 3, 803–812. doi: 10.1038/nrn941
Wu, G. Y., and Cline, H. T. (1998). Stabilization of dendritic arbor structure in vivo by CaMKII. Science 279, 222–226. doi: 10.1126/science.279.5348.222
Wu, G. Y., Deisseroth, K., and Tsien, R. W. (2001). Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat. Neurosci. 4, 151–158. doi: 10.1038/83976
Wu, G. Y., Zou, D. J., Rajan, I., and Cline, H. (1999). Dendritic dynamics in vivo change during neuronal maturation. J. Neurosci. 19, 4472–4483. doi: 10.1523/JNEUROSCI.19-11-04472.1999
Xie, Y., Vessey, J. P., Konecna, A., Dahm, R., Macchi, P., and Kiebler, M. A. (2007). The GTP-binding protein Septin 7 is critical for dendrite branching and dendritic-spine morphology. Curr. Biol. 17, 1746–1751. doi: 10.1016/j.cub.2007.08.042
Yabut, O., Domogauer, J., and D'Arcangelo, G. (2010). Dyrk1A overexpression inhibits proliferation and induces premature neuronal differentiation of neural progenitor cells. J. Neurosci. 30, 4004–4014. doi: 10.1523/JNEUROSCI.4711-09.2010
Yadav, S., Oses-Prieto, J. A., Peters, C. J., Zhou, J., Pleasure, S. J., Burlingame, A. L., et al. (2017). TAOK2 kinase mediates PSD95 stability and dendritic spine maturation through septin7 phosphorylation. Neuron 93, 379–393. doi: 10.1016/j.neuron.2016.12.006
Yamagata, Y., Kobayashi, S., Umeda, T., Inoue, A., Sakagami, H., Fukaya, M., et al. (2009). Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIalpha in dendritic spine enlargement, long-term potentiation, and learning. J. Neurosci. 29, 7607–7618. doi: 10.1523/JNEUROSCI.0707-09.2009
Yang, N., Higuchi, O., Ohashi, K., Nagata, K., Wada, A., Kangawa, K., et al. (1998). Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393, 809–812. doi: 10.1038/31735
Yasuda, R., Harvey, C. D., Zhong, H., Sobczyk, A., van Aelst, L., and Svoboda, K. (2006). Supersensitive Ras activation in dendrites and spines revealed by two-photon fluorescence lifetime imaging. Nat. Neurosci. 9, 283–291. doi: 10.1038/nn1635
Yazdani, U., and Terman, J. R. (2006). The semaphorins. Genome Biol. 7, 211–214. doi: 10.1186/gb-2006-7-3-211
Yeh, R.-H., Yan, X., Cammer, M., Bresnick, A. R., and Lawrence, D. S. (2002). Real time visualization of protein kinase activity in living cells. J. Biol. Chem. 277, 11527–11532. doi: 10.1074/jbc.M111300200
Yuste, R., and Bonhoeffer, T. (2004). Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat. Rev. Neurosci. 5, 24–34. doi: 10.1038/nrn1300
Zempel, H., Luedtke, J., Kumar, Y., Biernat, J., Dawson, H., Mandelkow, E., et al. (2013). Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. Embo J. 32, 2920–2937. doi: 10.1038/emboj.2013.207
Zempel, H., Thies, E., Mandelkow, E., and Mandelkow, E.-M. (2010). A beta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 30, 11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010
Zhou, J., Blundell, J., Ogawa, S., Kwon, C. H., Zhang, W., Sinton, C., et al. (2009). Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J. Neurosci. 29, 1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009
Zhou, Q., Homma, K. J., and Poo, M.-M. (2004). Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44, 749–757. doi: 10.1016/j.neuron.2004.11.011
Zhou, X. X., Fan, L. Z., Li, P., Shen, K., and Lin, M. Z. (2017). Optical control of cell signaling by single-chain photoswitchable kinases. Science 355, 836–842. doi: 10.1126/science.aah3605
Keywords: dendrites, kinases, neurodevelopmental diseases, neurological disorder, kinome
Citation: Nourbakhsh K and Yadav S (2021) Kinase Signaling in Dendritic Development and Disease. Front. Cell. Neurosci. 15:624648. doi: 10.3389/fncel.2021.624648
Received: 31 October 2020; Accepted: 06 January 2021;
Published: 10 February 2021.
Edited by:
Peter Soba, University Medical Center Hamburg-Eppendorf, GermanyReviewed by:
Hee Jung Chung, University of Illinois at Urbana-Champaign, United StatesCopyright © 2021 Nourbakhsh and Yadav. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Smita Yadav, c21pdGF5QHV3LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.