
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 27 October 2020
Sec. Cellular Neuropathology
Volume 14 - 2020 | https://doi.org/10.3389/fncel.2020.599717
This article is part of the Research Topic “Inside-out” vs “Outside-in” Paradigms in Multiple Sclerosis Etiopathogenesis View all 10 articles
 Haley E. Titus1†
Haley E. Titus1† Yanan Chen2†
Yanan Chen2† Joseph R. Podojil1,3
Joseph R. Podojil1,3 Andrew P. Robinson1
Andrew P. Robinson1 Roumen Balabanov2
Roumen Balabanov2 Brian Popko2
Brian Popko2 Stephen D. Miller1,3,4*
Stephen D. Miller1,3,4*Multiple Sclerosis (MS) is an immune-mediated neurological disorder, characterized by central nervous system (CNS) inflammation, oligodendrocyte loss, demyelination, and axonal degeneration. Although autoimmunity, inflammatory demyelination and neurodegeneration underlie MS, the initiating event has yet to be clarified. Effective disease modifying therapies need to both regulate the immune system and promote restoration of neuronal function, including remyelination. The challenge in developing an effective long-lived therapy for MS requires that three disease-associated targets be addressed: (1) self-tolerance must be re-established to specifically inhibit the underlying myelin-directed autoimmune pathogenic mechanisms; (2) neurons must be protected from inflammatory injury and degeneration; (3) myelin repair must be engendered by stimulating oligodendrocyte progenitors to remyelinate CNS neuronal axons. The combined use of chronic and relapsing remitting experimental autoimmune encephalomyelitis (C-EAE, R-EAE) (“outside-in”) as well as progressive diphtheria toxin A chain (DTA) and cuprizone autoimmune encephalitis (CAE) (“inside-out”) mouse models allow for the investigation and specific targeting of all three of these MS-associated disease parameters. The “outside-in” EAE models initiated by myelin-specific autoreactive CD4+ T cells allow for the evaluation of both myelin-specific tolerance in the absence or presence of neuroprotective and/or remyelinating agents. The “inside-out” mouse models of secondary inflammatory demyelination are triggered by toxin-induced oligodendrocyte loss or subtle myelin damage, which allows evaluation of novel therapeutics that could promote remyelination and neuroprotection in the CNS. Overall, utilizing these complementary pre-clinical MS models will open new avenues for developing therapeutic interventions, tackling MS from the “outside-in” and/or “inside-out”.
Multiple Sclerosis presents most often in young adulthood and is chronic as most patients live with the disease for decades. Recent studies on prevalence uncovered that nearly a million people live with MS within the United States (Culpepper et al., 2019; Nelson et al., 2019; Wallin et al., 2019). Approximately 85% of patients present with the relapsing-remitting form of MS (RRMS) that involves episodes of neurological deficits followed by phases of recovery (Steinman, 2009). The disease often slowly converts to a secondary-progressive form of MS (SPMS) that shows significant and irreversible neurological impairment (Stadelmann, 2011). The primary-progressive form of MS (PPMS) appears in the remaining patients and results in rapid progressive neurological decline (Miller and Leary, 2007).
With advances in technology, in addition to the traditional family history portion of a patient’s medical record, further characterization of a patient’s predisposition to disease can be evaluated by genomic sequencing. For example, recent studies have identified over 200 genomic and proteomic anomalies prevalent in the MS patient population, all directly or indirectly linked to the immune system (International Multiple Sclerosis Genetics Consortium et al., 2013; Manconi et al., 2018; International Multiple Sclerosis Genetics Consortium, 2019; Kotelnikova et al., 2019). Clinical presentation of the disease as well as chronic neuropathology highlight the destructive nature of the interaction between the immune system and the CNS.
The “outside-in” hypothesis constitutes a primary pathogenesis of autoimmune inflammation followed by a secondary pathogenesis of myelin degradation (Table 1). The “inside-out” hypothesis is a primary pathogenesis of oligodendrocyte (OL) injury/myelin destabilization and a secondary pathogenesis due to activation of a reactive inflammatory response (Table 1). Experimental murine models can recapitulate patient clinical presentations and pathological changes including inflammatory demyelination, axonal pathology, and immune cell infiltration utilizing both “outside-in” immune mediated demyelination and the “inside-out” CNS demyelination/neurodegeneration driven models (Table 2).

Table 1. “Inside-out” and “Outside-in” hypotheses of MS pathophysiology.
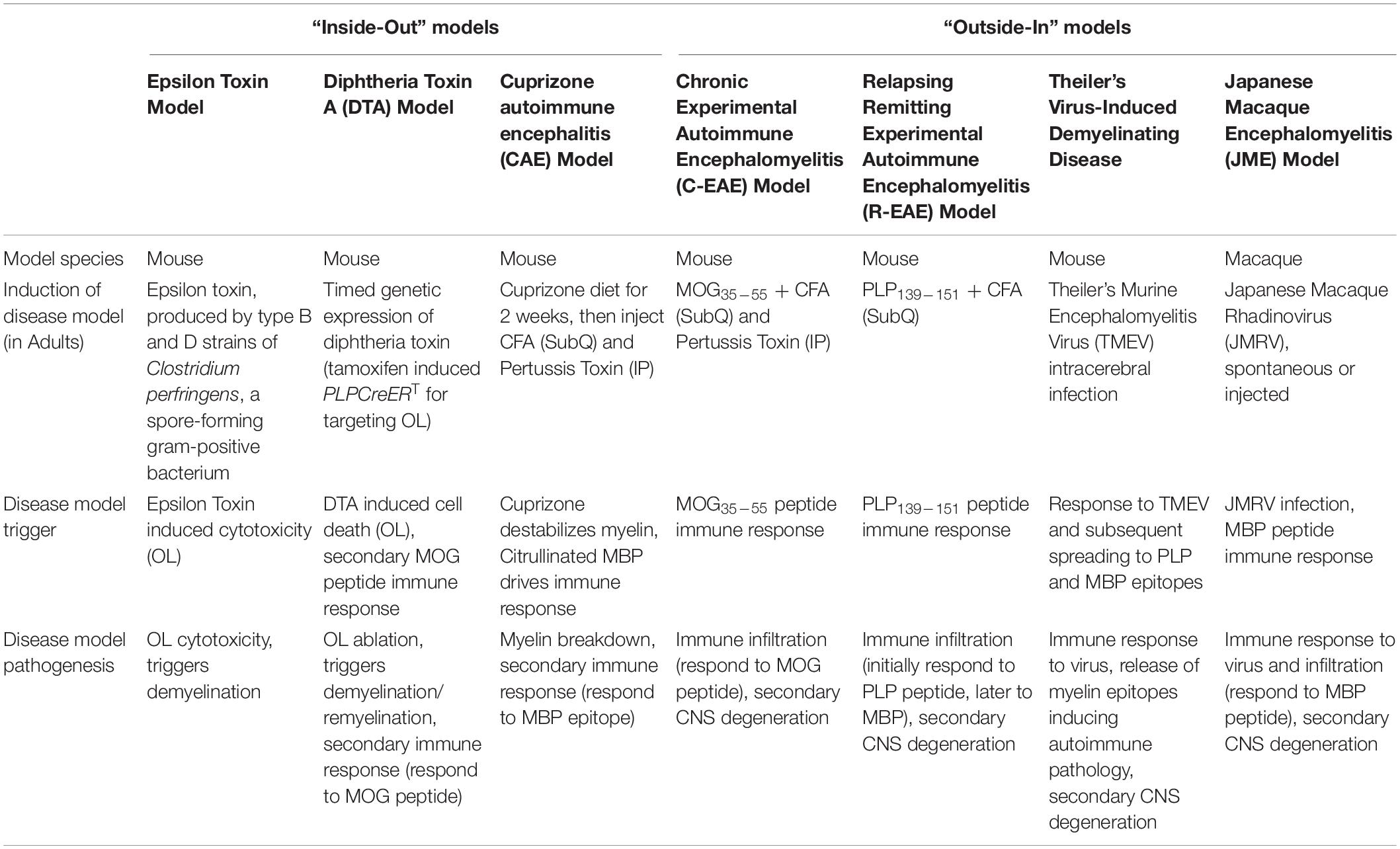
Table 2. “Inside-out” and “Outside-in” disease model systems highlighted in this review.
Presently, there is no cure for MS and the long-term treatment of MS patients is based on disease-modifying therapies that either suppress or modulate the immune system, and symptomatic management. Ideally, the mechanism(s) of action for an efficacious therapy would function to specifically target the root cause of the immune and CNS dysfunction. First, the underlying autoimmunity must be mitigated through re-establishing self-tolerance (McCarthy et al., 2014; Luo et al., 2016; Pearson et al., 2017, 2019). Second, neurodegeneration must be mitigated to protect the remaining function of CNS neurons. Third, tissue repair within the CNS must restore oligodendrocyte insulation and myelin sheath formation around damaged axons (Rodgers et al., 2013). As the etiology of the disease is unknown, utilizing both “outside-in” and “inside-out” models for single selective immune regulatory and myelin repair therapy as well as combination therapy in pre-clinical trials are imperative for success in developing effective therapeutics in patient clinical trials. This review will focus on the “outside-in” models, “inside-out” models of MS, and the multi-directional feedback between the immune system and the CNS.
The process of drug discovery, approval, and future patient use requires initial testing in experimental models that recapitulate hallmarks of the human disease state. Initial inflammatory demyelination in Multiple Sclerosis and subsequent neurodegeneration is a result of multi-directional feedback involving CNS resident cells (i.e., oligodendrocytes, neurons, and microglia) and infiltrating immune cells (i.e., autoreactive T cells and B cells, inflammatory monocytes, and macrophages) (McFarland and Martin, 2007; Bhat and Steinman, 2009; Weiner, 2009). While the primary etiology of MS remains unknown and is likely multi-determinant, the disease involves the activation of the peripheral adaptive immune system against CNS myelin epitopes. However, the triggering event that initiates this autoimmune response is not understood. Antigen presenting cells (APCs) (i.e., dendritic cells, monocytes, macrophages, microglia, and B cells) activate naïve CD4+ T cells and promote differentiation of CD4+ Th17 and Th1 cells through inflammatory cytokines (IL-1β, IL-6, and IL-23; and IL-12, respectively).
Activated microglia are rapidly recruited to sites of CNS damage (Duan et al., 2009). These APCs upregulate the expression of MHCII and other costimulatory molecules, such as CD40, CD80, and CD86 (Windhagen et al., 1995; Gerritse et al., 1996; Zrzavy et al., 2017). These observations suggest that activated APCs within the CNS possess the capacity to present antigens to infiltrating T cells. However, in vitro data show that microglia have a limited capacity to activate CD4+ T cells (Mack et al., 2003). In contrast to the in vitro findings, a dynamic alteration in the presence and phenotype of microglia within the CNS has been reported with regard to the absence or presence of lesions within the local microenvironment (Esiri and Reading, 1987; Ferguson et al., 1997; Prineas et al., 2001; Zrzavy et al., 2017). For example, microglia activation was more pronounced and increased with the length of disease even in the normal-appearing white matter sections from MS patients as compared to control tissue samples (Zrzavy et al., 2017). Additionally, active lesions from MS patients contained microglia and macrophages expressing a pro-inflammatory phenotype (Zrzavy et al., 2017), suggesting the ability of these cells to activate CD4+ T cells.
Besides microglia and macrophages, recent evidence suggests that B cells may serve as an important APC population in RRMS disease pathogenesis. To test this hypothesis, the ability of memory B cells from RRMS patients to activate CD4+ T cell in response to MBP and MOG was compared to naïve B cells from health donors. The data show that the naïve B cells from healthy donors did not activate the CD4+ T cells in the presence of MBP and MOG, while the memory B cells from RRMS patients did activate the CD4+ T cells (Harp et al., 2010). In the context of anti-CD20 therapy, which deletes peripheral B cells, the aforementioned findings suggest that the depletion of B cells following anti-CD20 treatment may be due in-part to the loss of B cells as an APC population. This possibility is supported by studies utilizing whole MOG protein-induced EAE in C57BL/6 mice, in which MOG-specific B cells are activated (Hausler et al., 2018).
In the “inside-out” model of MS (Figure 1), amplified inflammation, driven by peripherally derived autoreactive CD4+ Th17 and Th1 cells, directly and indirectly leads to further myelin destruction (Glass et al., 2010; Prinz and Kalinke, 2010). Based on the presence of T cell-mediated inflammation within the CNS of MS patients, the field historically utilizes “outside-in” experimental models of disease (Figure 2). These experimental models include relapsing-remitting experimental autoimmune encephalomyelitis (R-EAE) and Theiler’s murine encephalomyelitis virus (TMEV) infection in SJL/J mice, chronic experimental autoimmune encephalomyelitis (C-EAE) in C57BL/6 mice, and more recently a non-human primate model of virus-induced Japanese macaque encephalomyelitis (JME) (Table 2).
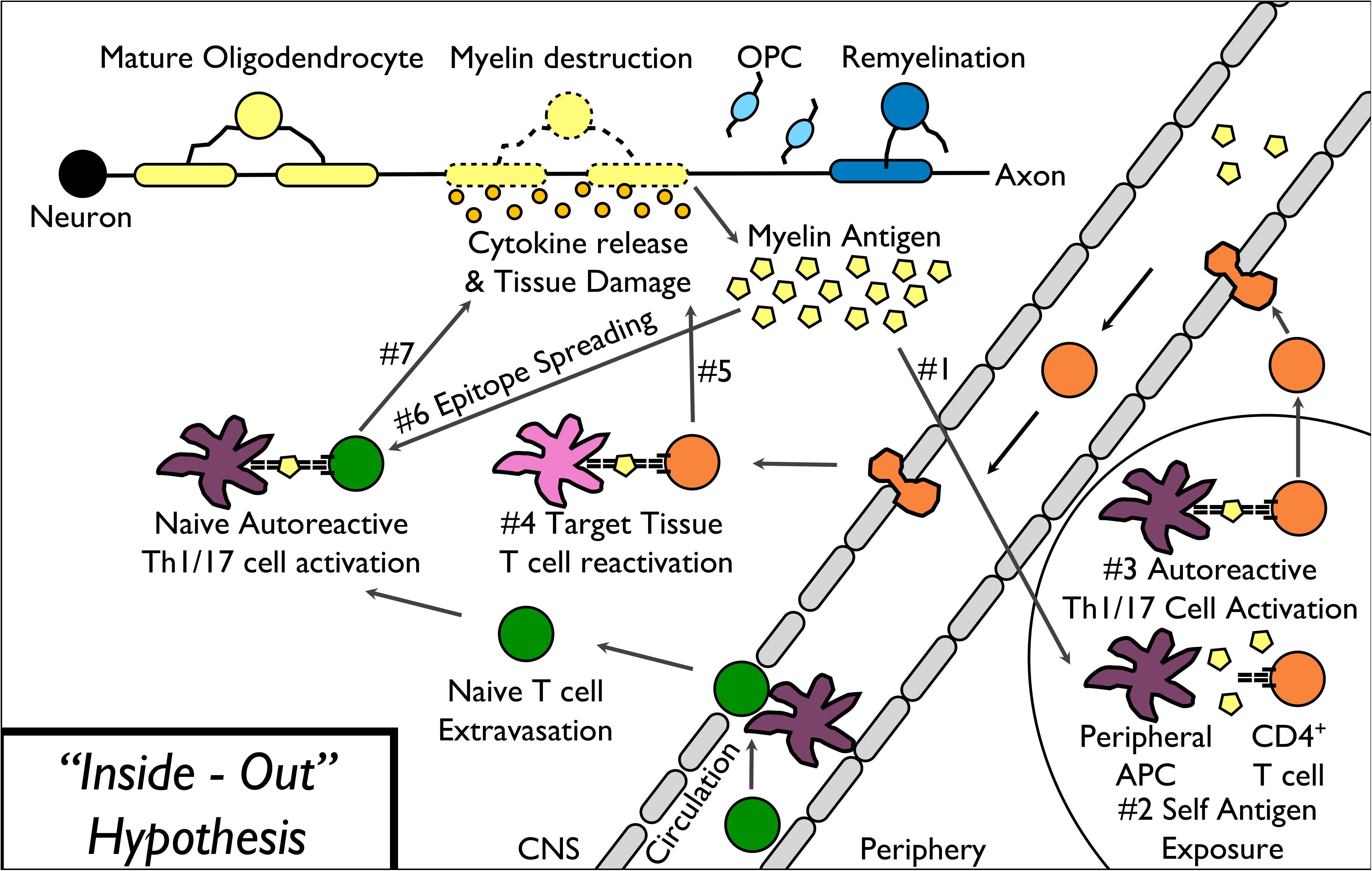
Figure 1. “Inside-Out” model of MS pathophysiology. The “inside-out” model of MS pathogenesis begins with the release of myelin antigens from injured or destabilized myelin to the periphery (1) followed by the presentation of myelin epitopes to (2) and activation of autoreactive T cells (3). Activated autoreactive T cells then migrate into the CNS, are reactivated by CNS-resident APCs (4), and release cytokines leading to direct as well as indirect damage to myelin (5). Additional myelin epitopes released by the primary T cell response induce epitope spreading (6) leading to additional myelin destruction (7).
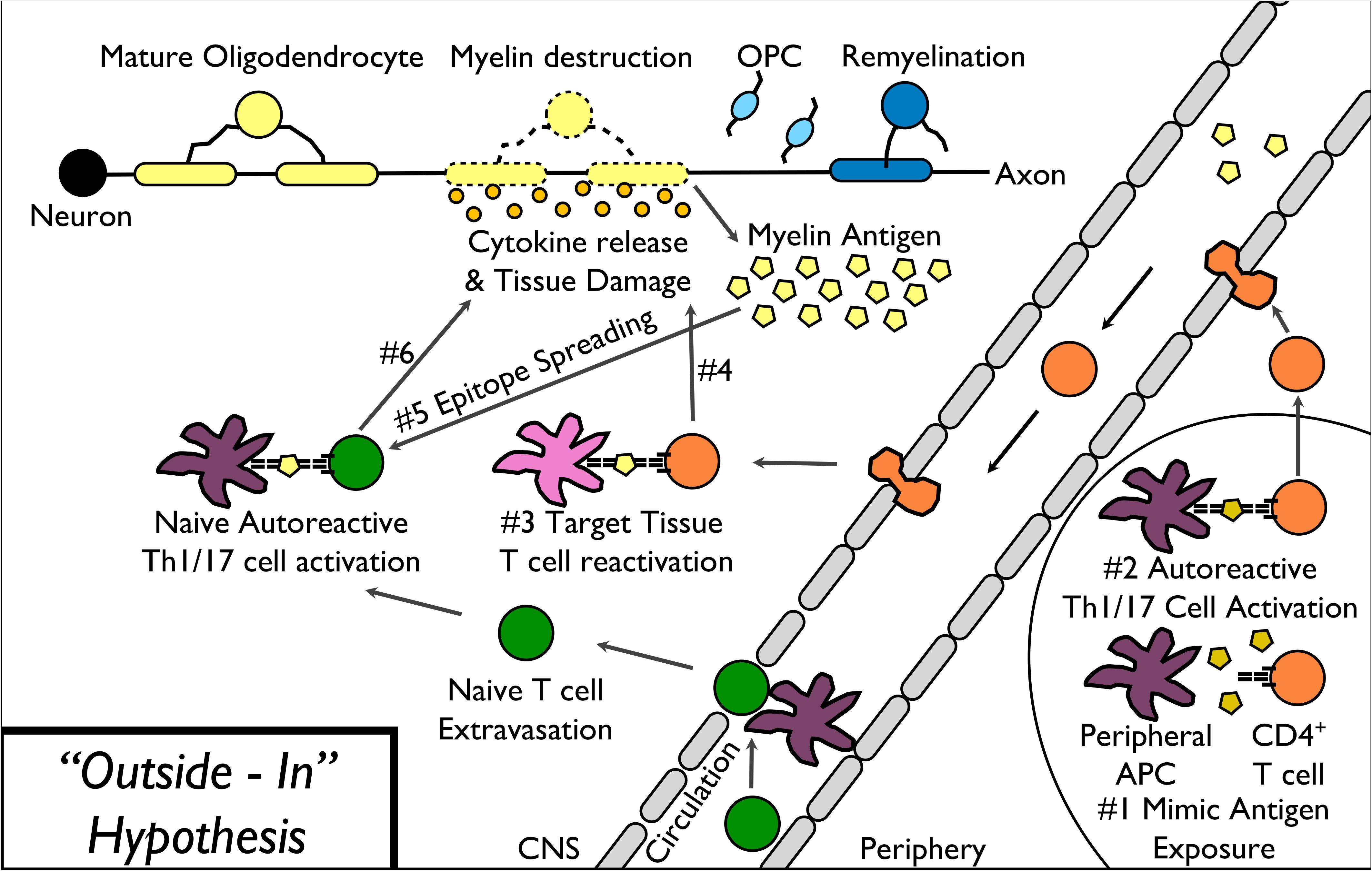
Figure 2. “Outside-In” model of MS pathophysiology. The “outside-in” model of MS pathogenesis begins with activation of myelin-specific T cells in response to a myelin peptide mimic epitope expressed on a pathogenic virus or other microbe exposure (1–2). Activated autoreactive T cells then migrate into the CNS, are reactivated by CNS-resident APCs (3), and release cytokines leading to direct as well as indirect damage to myelin (4). Additional myelin epitopes released by the primary T cell response induce epitope spreading (5) leading to additional myelin destruction (6).
In mice, the MS disease processes, including myelination defects, axonal pathology, and immune cell infiltration can be experimentally recapitulated. Classically, the experimental autoimmune encephalomyelitis (EAE) mouse model has been used to mimic autoimmune demyelination in response to a peripheral immune-priming event serving as an “outside-in” approach. Both relapsing-remitting MS (RR-MS) and primary-progressive MS (PP-MS) can be arguably modeled by EAE induced via subcutaneous priming of different mouse strains with specific myelin peptides in complete Freund’s adjuvant (CFA). CD4+ Th1/17 cells primed in the peripheral lymph nodes, traffic to the CNS, and are re-stimulated with endogenous myelin antigens leading to effector responses and clinical disease. Priming SJL/J mice with proteolipid protein (PLP)139–151/CFA results in multiple clinical relapses (R-EAE) and priming C57BL/6 mice with myelin oligodendrocyte glycoprotein (MOG)35–55/CFA and pertussis toxin or infection of SJL/mice with TMEV induces an acute phase of disease followed by chronic progression (C-EAE) as measured by clinical scoring (Theiler and Gard, 1940; Veillette et al., 1989; Ben-Nun et al., 1991; Burns et al., 1991; Sun et al., 1991; Trotter et al., 1991; Soderstrom et al., 1993; Zhang et al., 1993; Steinman et al., 2002; Sospedra and Martin, 2005; Robinson et al., 2014; Terry et al., 2016). The use of the R-EAE and C-EAE mouse models allow assessment of motor function via clinical scoring, immune cell function, promotion of oligodendrocyte proliferation/maturation, and formation of new myelin.
Ideally, re-establishing self-tolerance would be induced by antigen-specific immune therapy (i.e. immune tolerance) in which the remaining immune system functions remain intact. The utilization of both R-EAE and C-EAE models of disease have been used to identify the underlying epitope-spreading mechanism within autoimmune disease. Epitope spreading in R-EAE has been clearly defined during the various phases of disease (Miller et al., 1995). During the immune response to a foreign or self-protein, the initial CD4+ T cell response focuses on one or two antigenic peptide epitopes within the immunogenic protein. These initial immunogenic epitope(s) are termed the dominant epitope(s). As the immune response progresses, the process of epitope spreading occurs, which is defined as the activation of additional antigen-specific CD4+ T cells that express T cell receptors specific for additional antigens that are not the dominant epitope(s) (Lehmann et al., 1992, 1993; Vanderlugt and Miller, 2002). For example, in an SJL/J mouse primed with PLP139–151/CFA, PLP139–151-specific CD4+ T cell reactivity is induced within 3 days of priming in the draining lymph nodes for the site of PLP139–151/CFA injection, and this dominant epitope-specific CD4+ T cell response is maintained throughout the disease course. Immediately before, and continuing during, the primary relapse phase of disease, PLP178–191 reactivity (termed intramolecular epitope spreading, i.e., spreading from one peptide epitope to another peptide epitope contained within the same protein) is detected by T cell proliferation and delayed-type hyper-sensitivity (DTH) assays. During the secondary relapse phase of disease, MBP84–104 responses (termed intermolecular epitope spreading, i.e., spreading from one peptide epitope to another peptide epitope contained within a different protein) are detectible. Conversely, if SJL/J mice are primed with PLP178–191/CFA, the acute phase of disease is mediated by CD4+ T cell responses to the initiating PLP178–191 epitope. Subsequently, PLP139–151 CD4+ T cells are detectible within the spleen and cervical lymph nodes during the primary disease relapse, and MBP84–104 specific CD4+ T cells during the secondary disease relapse. Published data show that while the detection of the spread epitope-specific CD4+ T cells (PLP178–191 or PLP139–151 specific CD4+ T cells depending on the peptide used to induce disease) does not occur until the primary disease relapse, these spread epitope-specific CD4+ T cells are initially activated during the acute phase of disease via antigen presenting cells presenting spread epitope peptides within the CNS (McMahon et al., 2005). Similarly, infection of SJL/J mice with TMEV, results in the bystander immune-mediated CNS damage leading to initial epitope spreading to PLP139–151 followed by responses to additional myelin epitopes. The development of these responses correlates with the extent of myelin destruction during the acute disease phase (McRae et al., 1995). The hierarchy of dominant epitopes is due to a combination of differential protein processing and presentation by various APCs, and the precursor frequency of the antigen-specific CD4+ T cells (Lehmann et al., 1998; Moon et al., 2007).
The epitope spreading phenomena during autoimmune disease has been confirmed by the use of antigen-specific tolerance therapies. For example, immune tolerance is readily induced by coupling of peptides to donor splenocytes (SP) using the chemical crosslinker 1-ethyl-3-(3- dimethylaminopropyl) carbodiimide (ECDI) (Wetzig et al., 1979). Antigen coupled to splenocytes (Ag-SP) delivers the antigen to APCs that present the cargo antigen in a tolerogenic manner. The non-specific crosslinking of antigen to the cell surface while inducing apoptosis allows the donor cells to be perceived by the host in a non-inflammatory (non-immunogenic) manner. Ag-SP have been employed to prevent and treat the relapsing EAE model of MS (Podojil and Miller, 2009), and type 1 diabetes (T1D) in the non-obese diabetic (NOD) mouse (Prasad et al., 2012). A recent publication summarized the results of a phase I trial in MS patients using apoptotic ECDI-fixed peripheral blood mononuclear cells (PBMCs) pulsed with a cocktail of myelin peptides, illustrating the safety and efficacy of this procedure in human autoimmune disease (Lutterotti et al., 2013). Importantly, the mechanistic aspects of this study provided an important proof-of-principle that induced peripheral tolerance can be successfully employed to induce unresponsiveness in human autoreactive T cells.
More recently, biodegradable carboxylated nanoparticles composed of poly(lactic-co-glycolic) acid (PLGA) were shown to induce antigen-specific tolerance for prevention and treatment of EAE when encephalitogenic peptides were EDCI fixed to the surface of the particles or encapsulated within the particles (Getts et al., 2012; Hunter et al., 2014). Administration of Ag-bearing PLGA nanoparticles results in significantly reduced CNS infiltration of encephalitogenic Th1 (IFN-γ) and Th17 (IL-17a and GM-CSF) cells as well as inflammatory monocytes/MΦs. Tolerance was most effectively induced by intravenous infusion of Ag-PLG (Getts et al., 2012), though intraperitoneal delivery was also able to attenuate disease scores. The intravenous route likely has greater efficacy due to direct trafficking and uptake of the nanoparticles by APCs in the liver and spleen via the macrophage receptor of collagenous structure (MARCO) scavenger receptor (Getts et al., 2011).
As outlined above, we have previously extensively studied and reviewed (Croxford et al., 2002; Munz et al., 2009) the immunopathogenesis of TMEV-induced demyelinating disease (TMEV-IDD) “outside-in” model of MS. Briefly, TMEV is a picornavirus which naturally enters the CNS via a fecal-oral transmission route and enters the CNS via a retrograde transport mechanism. In experimental TMEV-IDD, disease is induced by intracerebral injection of TMEV which then induces a persist infection of microglia which, in susceptible mouse strains, stimulates inflammatory anti-viral immune responses (Th1, Th17, and CD8) which cause bystander damage to oligodendrocytes and myelin in the CNS. Released myelin antigens then activate myelin epitope-specific autoimmune responses via the process of epitope spreading (Miller et al., 1997) leading to a chronic demyelinating and a spastic course of paralysis. We also showed that a strain of TMEV engineered to express molecular mimic of the myelin PLP139–151 epitope sharing only 3 of the 13 amino acid residues (critically including the primary MHC binding and the primary and secondary T cell receptor binding residues), could induce demyelinating disease via the process of molecular mimicry (Olson et al., 2001). Collectively these studies indicate that myelin-specific autoimmune pathology can be induced by infection both via bystander damage induced release of self-antigens (epitope spreading) and molecular mimicry.
Japanese macaque encephalomyelitis (JME) is an inflammatory demyelinating disease that occurs spontaneously in a colony of Japanese macaques (JM) at the Oregon National Primate Research Center (Axthelm et al., 2011; Blair et al., 2016). The disease only occurs in specific lineages within the colony, and is triggered by a novel gamma-herpes virus, Japanese macaque rhadinovirus (JMRV), that occurs spontaneously in 1–3% of the JM colony and with targeted breeding 2–5% (Axthelm et al., 2011). If needed, based on population and time constraints, disease can be induced by intracranial injections of JMRV into animals from affected lineages (Estep et al., 2013) with the advantage of a known consistent location for histology and MRI/DTI. Animals with JME display clinical signs resembling Multiple Sclerosis, such as; ataxia, paresis, and magnetic resonance imaging reveals multiple T2-weighted hyperintensities and gadolinium-enhancing lesions in the central nervous system (i.e., brainstem, cerebellum, and cervical spinal cord). The prevalent myelin epitope is myelin basic protein (MBP). Comparable to disease manifestation in MS patients, the CNS of animals with JME present with active lesions that contain CD4+ Th1 and Th17 cells, CD8+ T cells, and oligoclonal bands are present within the CSF (Blair et al., 2016).
In addition to testing immune modulatory therapies, therapies that potentially promote myelin repair by stimulating oligodendrocyte progenitor cell expansion, homing and/or differentiation can be assessed in the EAE and TMEV-IDD mouse and JME primate models of MS. Researchers can examine clinical disease progression in the form of sensory and motor function, CNS immune and inflammatory responses, flow cytometry-based enumeration of cells of the oligodendrocyte lineage, and changes in myelin. The use of the MS-like animal disease models provides a robust platform for assessing combined immune regulation and myelin repair therapies in an “outside-in” model of CNS immune-induced demyelinating disease. These in vivo platforms for testing new myelin repair drugs will hopefully lead to the translation of a novel drug, either alone or in combination with immune regulatory drugs, for the treatment of MS.
The “inside-out” model proposed by Stys et al. (2012) argues that primary degeneration of oligodendrocytes and myelin is the initial event of MS, and might occur in the earliest years before the onset of symptoms. Primary oligodendrocyte death and/or subtle myelinopathy can precede and subsequently drive a secondary autoimmune attack, resulting in inflammatory demyelination in MS (Figure 1). Therefore, there has been a search for agents that could trigger these CNS events, resulting in the onset of an immune response to myelin.
In the earliest stage of MS, the histologic description of formation of nascent lesions without inflammatory infiltration argues for the possibility of an “inside-out” mechanism (Barnett and Prineas, 2004; Prineas and Parratt, 2012). Observation of oligodendrocyte apoptosis along with blood brain barrier disruption in the nascent lesions indicates that MS might arise from an environmental insult targeting oligodendrocytes, such as a toxin or virus. Epsilon toxin is produced by the type B and the type D strains of Clostridium perfringens, a spore-forming gram-positive bacterium mostly found in the intestines of ruminant animals (Blackwell et al., 1983; Uzal et al., 2004; Uzal and Songer, 2008). Epsilon toxin is converted into an active form, crosses the intestinal mucosa and disseminates via the bloodstream, massively accumulating in the brain and kidneys (Finnie, 1984, 2003; Tamai et al., 2003). The toxin has the capability to cross the blood brain barrier and infiltrate the brain parenchyma, which results in MS-like symptoms (Dorca-Arevalo et al., 2008; Popoff, 2011). Over three decades ago, Murrell et al. (1986) first hypothesized that epsilon toxin is the potential toxin that triggers MS, even though humans are not natural hosts for C. perfringens types B and D. More recently, C. perfringens type B was isolated from the stool of a female remitting-relapsing MS patient with an onset 3 months previously (Rumah et al., 2013). Furthermore, epsilon toxin specific antibodies were found in serum and/or CSF of 10% of MS patients and 1% of healthy controls from the banked samples in the United States (Rumah et al., 2013). Similarly, immunoreactivity toward epsilon toxin in serum is higher in MS patients than in controls in the United Kingdom (Wagley et al., 2019). In light of these observations and clinical evidence, it has been proposed that epsilon toxin exposure may play a role in initiating MS lesion formation by binding to oligodendrocytes, myelin and white matter (Lonchamp et al., 2010; Wioland et al., 2015). Although the mechanism underlying the effect of epsilon toxin on oligodendrocytes and subsequent demyelination is not yet clear, several lines of evidence in vitro indicate that epsilon toxin selectively attacks mature oligodendrocytes and triggers demyelination (Linden et al., 2015; Wioland et al., 2015; Bossu et al., 2020). It has been shown that myelin and lymphocyte protein (MAL) could be a candidate epsilon toxin receptor on oligodendrocytes (Rumah et al., 2015). Once bound to oligodendrocytes, epsilon toxin could lead to the rise of glutamate and subsequent activation of mGluR, which activates intracellular Ca2+signaling and eventually triggers demyelination (Lonchamp et al., 2010; Kostic et al., 2013; Wioland et al., 2015). In line with this evidence, it is highly possible that an agent cytotoxic to oligodendrocytes may trigger MS.
One of the proposed factors in the alternative “inside-out” theory for initiating MS is the primary cytotoxic death of oligodendrocytes. A toxin-induced ablation of oligodendrocytes is useful for testing whether such oligodendrocyte death could trigger anti-CNS autoimmunity (Jakel and Dimou, 2017). Plp1-CreERT; ROSA26-eGFP-DTA (DTA) is a mouse model of oligodendrocyte ablation accomplished via an oligodendrocyte specific activation of toxin expression in adult mice. The A subunit of diphtheria toxin (DT-A) induces cell death by catalyzing the inactivation of elongation factor 2, thereby halting global protein synthesis (Collier, 2001). The Plp1-CreERT mouse line drives expression of the tamoxifen-regulated Cre recombinase under control of the oligodendrocyte-specific myelin proteolipid protein (PLP) transcriptional control region (Doerflinger et al., 2003). The DT-A expression in oligodendrocytes is the result of tamoxifen-induced Cre-mediated recombination of ROSA26-eGFP-DTA locus via Plp1-CreERT (Traka et al., 2010). The expression of the DT-A subunit specifically in CNS myelinating oligodendrocytes results in widespread oligodendrocyte ablation, CNS demyelination, and the subsequent development of severe neurological symptoms. There is no breakdown of the blood brain barrier or detectible increase in CD4+ T cell into the CNS despite local inflammation, and there is no apparent loss of CNS axons during the initial demyelination event. However, the specific phenotype(s) of the T cells within the CNS during this initial demyelination phase of disease have not been fully characterized. The clinical symptoms of the acute phase of the disease are ameliorated during a recovery phase that correlates with the repopulation of mature oligodendrocytes and robust remyelination in the following 6–7 weeks (Traka et al., 2010). Interestingly, as the recovered mice age, they develop a secondary lethal progressive demyelinating disease starting around 40 weeks after tamoxifen injection that is mediated by MOG35–55-specific CD4+ T cell infiltration into the CNS during the late stages of disease (Traka et al., 2016). While the MOG35–55-specific CD4+ T cell responses are detectable in the peripheral lymphoid organs at 40 weeks post induction and are not present at 10 weeks, there is a significant increased number of CD4+ T cells in the CNS at 10 weeks (Traka et al., 2016). This increase in CNS CD4+ T cells may correlate with the expansion of myelin-specific T cells, similar to the initial activation and expansion of spread-epitope-specific CD4+ T cells within the CNS in EAE (McMahon et al., 2005; Bailey et al., 2007). The late-stage pathology is due to the induction of CD4+ T cell-mediated autoimmune responses secondary to oligodendrocyte ablation via a non-immune-mediated event (i.e. DT-mediated toxicitiy). This is supported by the major findings that adoptive transfer of the myelin-specific CD4+ T cells derived from DTA mice into naïve mice consistently results in the induction of mild neurological symptoms and inflammatory CNS lesions in the recipients, and induction of immune tolerance using the MOG35–55-coupled PLG nanoparticles significantly inhibits the progression of late-onset disease symptoms in DTA mice protecting animals from eventual fatal demyelinating disease (Traka et al., 2016).
In addition to the DTA mouse model, other genetic mouse models were later developed to achieve a faster oligodendrocyte ablation via expressing diphtheria-toxin receptor (DTR) under the MOG-promoter accompanied by direct administration of diphtheria toxin (Ghosh et al., 2011; Locatelli et al., 2012; Oluich et al., 2012; Gritsch et al., 2014). However, some studies reported that a secondary anti-CNS immunity did not develop in these mice, that is most likely due to premature death of these mice (Locatelli et al., 2012; Gritsch et al., 2014). The development of CNS immunity after oligodendrocyte death appears to be a slow process, taking several months in the DTA mouse model described above.
The DTA mouse model supports the “inside-out” theory, recapitulating pathological evidence showing that the loss of oligodendrocytes and subsequent demyelination may result in the induction of autoreactivity against myelin antigens as well as secondarily lead to inflammation and demyelination in the CNS. The unique DTA mouse model system allows fundamental unanswered questions concerning the molecular and cellular mechanisms associated with the induction of the autoimmune response to contribute to the understanding of MS disease pathogenesis and to the development and testing of remyelination therapies.
In addition to diphtheria-toxin, cuprizone is a demyelinating neurotoxin that has been used in testing “inside-out” hypothesis. Long-term of cuprizone feeding in mice lead to oligodendrocyte death, demyelination and gliosis (Matsushima and Morell, 2001; Sen et al., 2019b). Unlike DTA model, cuprizone feeding did not evoke a peripheral immune response in the CNS (Caprariello et al., 2018; Sen et al., 2019a). Some studies reported that the failure of cuprizone feeding to trigger in triggering CNS immune response is due to the atrophy of immune organs like the spleen and thymus (Solti et al., 2015; Partridge et al., 2016; Sen et al., 2019a). A more recent study reported that cuprizone induced demyelination can trigger an “inside-out” immune response when the BBB is disrupted by pertussis toxin (Almuslehi et al., 2020).
Accumulating clinical evidence suggests that primary myelin destabilization by citrullination releases immunogenic myelin debris and subsequently drives a secondary autoimmune attack (Moscarello et al., 1994; Cao et al., 1999; Stys et al., 2012). Excessive citrullination of myelin basic protein (MBP) had been found in normal appearing white matter from postmortem MS brain tissues and the extent of modified myelin is related to the severity of MS (Moscarello et al., 1994; Wood et al., 1996; Bradford et al., 2014). Citrullination is a post-translational modification mediated by peptidylarginine deiminase (PAD). Citrullination occurs when a positively charged arginine residue is deiminated to a neutrally charged citrulline (Vossenaar et al., 2003; Moscarello et al., 2007). Due to the changed charge in the protein, citrullinated MBP is partially unfolded and cannot stabilize a compact myelin sheath (Beniac et al., 2000; Bakhti et al., 2013). Studies have shown that deiminated MBP with citrulline is more susceptible to proteolytic digestion and that the digestion rate is remarkably correlated with the amount of citrulline present in MBP peptides (Cao et al., 1999; D’Souza and Moscarello, 2006; Musse et al., 2006). Increased breakdown of citrullinated MBP results in generating immunodominant epitopes (Musse et al., 2006); potentially triggering autoimmunity and eliciting destructive inflammatory demyelination (Raijmakers et al., 2005).
A newly developed mouse model of cuprizone autoimmune encephalitis (CAE) provides direct evidence to support the causative relationship between primary abnormalities of myelin and inflammation (Caprariello et al., 2018), whereby biochemical destabilizing myelin triggers a secondary inflammatory demyelination comparable to active MS lesions. The CAE is initiated with a 2-week exposure of neurotoxicant cuprizone to perturb myelin without causing overt demyelination, followed by an immune boost of complete Freund’s adjuvant (without exogenous antigen) and pertussis toxin. After 2 weeks, these mice develop inflammatory demyelination which resembles pathology found in MS patients and the EAE mouse model (Caprariello et al., 2018). The histopathological changes of the CAE model are characterized by periventricular and white matter tract gadolinium enhancement of MRI of the brain as well as overt demyelination and cellular infiltration within the corpus callosum. Gadolinium enhancement indicates breakdown of the BBB as a result of active inflammation. However, removal of the immune boost abrogates these responses, implying the importance of an immune-permissive environment. Most importantly, suppression of the destructive immune response by administration of peptidyl arginine deiminase (PAD) inhibitors to the CAE mice suggests that citrullinated proteins altered by abbreviated cuprizone exposure possibly drive the inflammatory demyelinated lesions in CAE.
Although genetic mutations of myelin proteins as well as traumatic brain injury-associated dysmyelination have been linked to later development of MS, it is not yet clear what initially triggers citrullination of myelin proteins or subtle dysmyelination (Warshawsky et al., 2005; Donovan et al., 2014; Sidaway, 2017; Cloake et al., 2018).
Adrenoleukodystrophy (ALD) is an X-linked neurometabolic disorder due to mutations in a proximal transporter, adenosine triphosphate (ATP)-binding cassette, subfamily, member 1 gene (ABCD1) (Moser et al., 2007; Kemp et al., 2016). The clinical presentation of ALD is complex; involving adrenal insufficiency and myelopathy (de Beer et al., 2014; Kemp et al., 2016). Approximately 60% of male patients develop rapidly progressive inflammatory cerebral demyelination (Moser et al., 1992), which clinically coincides with a progressive neurological decline similar to MS (Ferrer et al., 2010; Brandao de Paiva et al., 2018). However, the complex mechanisms on how this metabolic disease is transitioned to a fatal neuroinflammatory disease remains elusive. ABCD1 mutation may prevent transport of very long-chain fatty acids (VLCFAs) into peroxisomes for oxidation and degradation (Moser et al., 2007; Kemp et al., 2016). Some studies suggest that the accumulation of VLCFAs in myelin could mediate myelin instability and initial demyelination, which are believed to contribute to initiation of the inflammatory disease (Ho et al., 1995; Ito et al., 2001; Singh et al., 2009). The findings of CD1 (antigen presenting molecule) -mediated lipid antigen presentation in cerebral ALD lesions supported the hypothesis that VLCFA-containing proteolipid protein in myelin may be a potential lipid antigen for triggering autoimmunity after myelin breakdown (Ito et al., 2001). The lesions progress rapidly accompanied by the opening of blood brain barrier and invasions of inflammatory cells (Powers et al., 1992; Ito et al., 2001). Further evidence for the involvement of different components of the immune system in the pathogenesis of demyelinating ALD was reviewed in Hudspeth and Raymond (2007). Interestingly, the demyelinating progress arrested in a small percentage of patients with initial cerebral demyelination, in which the disruption of the blood brain barrier does not occur (Korenke et al., 1996). The importance of blood brain barrier in ALD was emphasized by several pieces of evidence that suggested potential environmental factors, such as head trauma (Weller et al., 1992; Raymond et al., 2010), possibly increase permeability of the blood brain barrier, which either trigger or precipitate the demyelination.
Traumatic brain injury (TBI) is most commonly caused by an external head impact that injures the brain. Demyelination and irreversible axon damage, particular in the corpus callosum, represent major pathological features frequently observed in TBI patients (Rutgers et al., 2008; Armstrong et al., 2016a; Chung et al., 2018; O’Phelan et al., 2018). Nevertheless, the progression of white matter injury is poorly understood in TBI. Oligodendrocytes are known to be vulnerable to oxidative stress and excitotoxicity following traumatic injury (Lotocki et al., 2011; Giacci and Fitzgerald, 2018). The loss of oligodendrocytes could significantly contribute to underlying demyelination after injury (Dent et al., 2015; Armstrong et al., 2016b) and activate neuroinflammation (Mierzwa et al., 2015). Furthermore, the observation that a persistent adaptive immune response in the CNS developed in the mice weeks after TBI (Daglas et al., 2019) fits within the “inside-out” theory. Interestingly, neuroinflammation, which persists for years after TBI, has recently been shown to largely contribute to neurodegeneration and long-term neurological dysfunction (Bazarian et al., 2009; Amor et al., 2010; Daglas et al., 2019). In particular, genetic depletion of CD8+ T cells in TBI mouse model improves neurological outcomes (Daglas et al., 2019), which indicates the importance of neuroinflammation in the progression of TBI. Encouragingly, damping neuroinflammation with immunomodulatory nanoparticles results in reduced neuropathology and neurophysiological abnormalities following TBI, suggesting a potential therapeutic strategy (Sharma et al., 2020).
Pre-clinical and clinical findings in ALD as well as TBI could shed light on potential MS therapeutics and vice versa. Current MS treatments are mainly directed to immune suppression, but the CAE model provides evidence for a potential “inside-out” mechanism of initiation of chronic demyelination and could serve as a compelling preclinical model of MS translational studies for the development of myelin-protective strategies in early stages of the disease.
As we have highlighted the “outside-in” (Figure 2) peripheral immune driven models and “inside-out” (Figure 1) neurodegenerative models (Table 2), the next section will focus on the relevance for the clinic. The “outside-in” pathogenesis (Figure 2) begins with activation of myelin-specific T cells in response to a myelin peptide mimic epitope expressed on a pathogenic virus or other microbe exposure. Activated autoreactive T cells then migrate into the CNS, are reactivated by CNS-resident APCs, and release cytokines leading to direct as well as indirect damage to myelin. Additional myelin epitopes released by the primary T cell response induce epitope spreading, leading to additional myelin destruction. During repair, a mild inflammatory reaction can stimulate oligodendrocyte precursor cells and “protective autoimmunity” utilizing T regulatory cells (Schwartz and Raposo, 2014). Unfortunately, the eventual failure of myelin repair during RRMS leads to chronically demyelinated axons, which degenerate over time and contribute to disease progression (Franklin and Ffrench-Constant, 2008; Trapp and Nave, 2008). Neuronal injury occurs, in part, as a result of inflammation mediated by myelin-specific CD4+ T cells. Direct and indirect effects of neuroantigen-specific Th1/17 cells can lead to demyelination and subsequent neuronal dysfunction by mechanisms that include activation of microglia and infiltrating inflammatory monocytes/macrophages by pro-inflammatory cytokines (IFN-γ, IL-17, and GM-CSF), which then produce proteases and additional pro-inflammatory cytokines, nitric oxide (NO) and reactive oxygen species (ROS), which induce myelin and axonal damage (Glass et al., 2010). Neuronal loss is thought to be a consequence of the demyelination, which causes dramatic ionic and energy imbalances in axons resulting from the loss of the structural and trophic support provided by oligodendrocytes (Trapp and Nave, 2008; Nave, 2010). This subsequent “inside-out” pathogenesis (Figure 1) can then spread and lead to further loss of axonal integrity. This perpetuates the release of myelin antigens to the periphery followed by the presentation of myelin epitopes to and activation of autoreactive T cells; ultimately leading to the progressive diffuse atrophy of the brain.
Disease modifying therapies for MS include immunomodulatory and immunosuppressive medications that suppress or modulate the self-reactive immune responses. While most of the medications exert their effects in the peripheral immune organs or blood stream, some also have the capacity to modulate the local immune responses and oligodendrocytes in the CNS.
In patients, Dimethyl fumarate (Tecfidera) decreased B cell CD40 expression (disrupted B-cell activation), decreased memory T cells, and decreased T cell proliferation and activation; resulting in lymphopenia (Linker and Gold, 2013). The mechanism of action of dimethyl fumarate within the CNS involves both Nrf-2-dependent as well as independent pathways for neuroprotection involving diminished neuroinflammation (Mills et al., 2018; Yadav et al., 2019). The Nrf2-dependent pathway promotes neuroprotection, oligodendrocyte survival, and decreases astrocyte activation (Linker et al., 2011; Kalinin et al., 2013; Wang et al., 2015; Zarrouk et al., 2017). The Nrf2-independent pathway also increases neuroprotection and decreases astrocyte activation, specifically reactive oxygen species production (i.e., Nitric Oxide) (Lin et al., 2011). Additionally, dimethyl fumarate targets innate immunity, in the form of microglia, resulting in diminished activation by the Nrf2 independent pathway (Parodi et al., 2015).
In patients, Fingolimod (Gilenya) suppresses migration of peripheral lymphocytes (Brinkmann et al., 2010; Francis et al., 2014). The mechanism of action of Fingolimod is the modulation of S1P receptor expression, most notably S1P1 receptor associated with lymphocytes, diminishing the number of T cells infiltrating into the CNS by retaining T cells in the lymph nodes (Brinkmann et al., 2002; Mandala et al., 2002; Fujino et al., 2003; Pham et al., 2008). Additionally, Fingolimod is neuroprotective, functioning within the CNS on neurons (Balatoni et al., 2007; Lee et al., 2010), oligodendrocyte lineage cells (Zhang et al., 2015), and decreasing the hyperactivity of reactive astrocytes (Choi et al., 2011). Of note, cumulatively Fingolimod has been shown to improve white matter integrity in relapsing remitting MS patients (Gurevich et al., 2018). Fingolimod can have “off target” effects as it can interact with multiple S1PR subtypes (S1PR1, S1PR3, S1PR4, and S1PR5) in a variety of tissues, including the heart (Chaudhry et al., 2017). The field has shifted to developing new therapies to mitigate these side effects, that selectively target subtype 1 of S1PR, yet this may diminish the neuroprotective capacity and immune suppression as S1PR5 is associated with oligodendrocyte function and natural killer cells (Chaudhry et al., 2017).
Natalizumab (Tysabri) is a monoclonal antibody against very late activating antigen (VLA)-α4 integrin and can bind to a majority of leukocytes, impeding cross over through the blood brain barrier into the CNS, thereby diminishing the aberrant heightened immune surveillance and inflammation (Stuve et al., 2006). However, use of Natalizumab in patients infected with John Cunningham virus (JCV) can result in progressive multifocal leukoencephalopathy (PML) in a subset of patients (Clerico et al., 2017; Ho et al., 2017; Fragoso et al., 2019; Ryerson et al., 2019). JCV is an opportunistic virus causing oligodendrocyte destruction, demyelination, and eventually a detrimental inflammatory reaction. Natalizumab associated PML, leading to subsequent CNS inflammation and worsening of MS, underscores the interplay between the generation of free antigen (viral and myelin), T cell immune surveillance, and the rebalancing mechanisms of neuroinflammation. Overall, despite vast strides in disease modifying therapy (DMT) options for MS patients over the last few decades, substantial risk for adverse side effects remain with the existing therapies.
As Multiple Sclerosis is a syndrome with multiple clinical presentations and not a single disease entity, it is likely that both immune (outside-in) and neurodegenerative (inside-out) driven molecular pathways can initiate the etiopathogenesis of MS in different patients (Table 1). Additionally, it is important to consider that these two mechanisms are not mutually exclusive as, regardless of the initiating event, both Immune-mediated and neurodegenerative processes are important components of both types of models with the difference being the timing of the two processes. Each highlighted model has benefits yet limitations and not all pre-clinical models of MS were covered (Lassmann and Bradl, 2017). We highlighted both conventional as well as new experimental models for testing novel MS therapeutics, while exploring the underlying role of the adaptive and innate immune systems (Table 2). The three therapeutic targets for balancing immune dysfunction and preventing neurodegeneration necessary for effective amelioration of MS progression include: re-establishing self-tolerance, neuroprotection, and promotion of remyelination.
Chronic immunosuppression and immunomodulation are the most commonly used therapeutic strategies for MS, outside of symptom management. In addition to traditional disease-modifying therapies, immune reconstitution therapy (IRT) has emerged as a novel treatment paradigm (AlSharoqi et al., 2020; Derfuss et al., 2020). The latter is based on partial or full ablation of the immune system aiming to destroy self-reactive clones and restore normal function. Though attractive in principle, immune reconstitution at present is an uncontrolled process whose long-term efficacy and side effects remain to be established. Furthermore, IRT is a costly therapy that is available only in certain medical centers and typically reserved for patients with highly active disease. Immunotherapy based on re-establishing of self tolerance is likely to be more advantageous to patients in terms of disease control and avoidance of immunosuppressive side effects. Such strategy also may set the basis for personalized therapies of MS, where patient specific autoimmune responses are targeted by tolerizing agents. The Miller lab has recently demonstrated an effective means of ameliorating ongoing disease in EAE mouse models of MS by inducing tolerance in autoreactive CD4+ T cells using intravenous (i.v.) infusion of 500 nM poly(lactic-co-glycolic acid) nanoparticles coupled with or encapsulating myelin peptides (Ag-PLG) that effectively reduces disease burden in relapsing-remitting (R-EAE) and in chronic-progressive (C-EAE) mouse models of experimental autoimmune encephalomyelitis (EAE) by reducing inflammatory cell activation and pro-inflammatory Th1/17 cytokine production (Getts et al., 2012, 2013; Hunter et al., 2014; McCarthy et al., 2017). Using myelin peptide-coupled autologous apoptotic leukocytes, we had previously demonstrated successful tolerance induction in MS patients (Lutterotti et al., 2013). Clinical testing of the Ag-PLG tolerance platform will be initiated in MS patients within the next year. We have recently shown, in a phase 1/2a trial in human celiac disease, safety and efficacy of PLG nanoparticles encapsulating gliadin (Kelly et al., 2019).
Interestingly, a recent single cell transcriptome study of oligodendrocyte lineage cells from the spinal cord of EAE mice indicated that oligodendrocytes and OPCs may not be passive targets of the immune attack, but rather involved in antigen-processing and presentation during the development of MS (Falcao et al., 2018). This possibility is supported by a previous study demonstrating that IFN-γ stimulated the production of chemokines from oligodendrocytes, while transgenic mice that suppresses oligodendrocyte responsiveness to IFN-γ developed an accelerated EAE onset (Balabanov et al., 2007).
The Popko lab has worked to enhance the protection of oligodendrocytes and myelin by augmenting the integrated stress response (ISR), a mechanism that protects endangered cells from inflammatory insults. Using a variety of mouse models of inflammatory demyelination, they have shown that genetic manipulations that compromise the ISR increase the susceptibility of oligodendrocytes in response to CNS inflammation (Lin et al., 2005, 2007) and that the genetic enhancement of the ISR, in contrast, provides increased protection to oligodendrocytes (Lin et al., 2008, 2013). Encouragingly, it has been shown that the ISR modulators, guanabenz and Sephin1, are able to protect oligodendrocytes against inflammatory stress through enhancing the ISR in MS mouse models (Way et al., 2015; Chen et al., 2019). Based on the “inside-out” theory, oligodendrocyte protection diminishes demyelination and reduces the generation of myelin debris, which likely decreases the exposure of myelin fragments and limits the autoimmune response. The success of these studies attests to the potential of oligodendrocyte protective therapeutics in MS.
At present, there are not any FDA approved therapies approved for myelin repair in MS despite successful pre-clinical trials. Utilizing both “outside-in” and “inside-out” models allows a comprehensive study of the multi-directional feedback between the CNS and periphery. As both immune dysregulation as well as inflammatory demyelination and neurodegeneration lead to disease progression, the field will need both “outside-in” and “inside-out” models to test single and combination therapies. Our collective goal as a field, of clinicians and scientists, is to improve patient outcomes and quality of life for those living with Multiple Sclerosis. In summary, the availability and utilization of these diverse models allows the MS field a robust platform for developing novel therapeutics targeting the autoimmune response, neuronal stress and promoting myelin repair.
HET, YC, JP, RB, BP, and SDM conceived and outlined the manuscript and edited the manuscript. HET and YC wrote the manuscript. AR, HET, and SDM contributed to figures preparation. All authors contributed to the article and approved the submitted version.
Relevant work in the authors’ laboratories has been supported by grants from the NIH [NS099334 and AI142059 (SDM), NS034939, NS109372, and NS067550 (BP)], the National Multiple Sclerosis Society [RG 4952-A-5 (SDM and BP)], the Myelin Repair Foundation (SDM and BP), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (BP), the Rampy MS Research Foundation (BP), the Johnnie Walker’s MS Foundation (SDM), the David and Amy Fulton Foundation (SDM), the Cramer Family Foundation (SDM), and a National Multiple Sclerosis Society Postdoctoral Fellowship FG 20125-A-1 (HET).
SDM is an academic co-founder, scientific advisory board member, paid consultant, and grantee of Cour Pharmaceutical Development Company, Inc; scientific advisory board member and grantee of NextCure, Inc., scientific advisory board member of Takeda Pharmaceutical Company and Myeloid Therapeutics. RB has received honorariums and research support from Biogen, Sanofi Genzyme, Genentech, and Alexion Pharmaceuticals, Inc. JP was employed by company Cour Pharmaceutical Development Company, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Almuslehi, M. S. M., Sen, M. K., Shortland, P. J., Mahns, D. A., and Coorssen, J. R. (2020). CD8 T-cell recruitment into the central nervous system of cuprizone-fed mice: relevance to modeling the etiology of multiple sclerosis. Front. Cell Neurosci. 14:43. doi: 10.3389/fncel.2020.00043
AlSharoqi, I. A., Aljumah, M., Bohlega, S., Boz, C., Daif, A., El-Koussa, S., et al. (2020). Immune reconstitution therapy or continuous immunosuppression for the management of active relapsing-remitting multiple sclerosis patients? A Narrative Review. Neurol. Ther. 9, 55–66. doi: 10.1007/s40120-020-00187-3
Amor, S., Puentes, F., Baker, D., and Van Der Valk, P. (2010). Inflammation in neurodegenerative diseases. Immunology 129, 154–169.
Armstrong, R. C., Mierzwa, A. J., Marion, C. M., and Sullivan, G. M. (2016a). White matter involvement after TBI: clues to axon and myelin repair capacity. Exp. Neurol. 275(Pt 3), 328–333. doi: 10.1016/j.expneurol.2015.02.011
Armstrong, R. C., Mierzwa, A. J., Sullivan, G. M., and Sanchez, M. A. (2016b). Myelin and oligodendrocyte lineage cells in white matter pathology and plasticity after traumatic brain injury. Neuropharmacology 110, 654–659. doi: 10.1016/j.neuropharm.2015.04.029
Axthelm, M. K., Bourdette, D. N., Marracci, G. H., Su, W., Mullaney, E. T., Manoharan, M., et al. (2011). Japanese macaque encephalomyelitis: a spontaneous multiple sclerosis-like disease in a nonhuman primate. Ann. Neurol. 70, 362–373. doi: 10.1002/ana.22449
Bailey, S. L., Schreiner, B., Mcmahon, E. J., and Miller, S. D. (2007). CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ T(H)-17 cells in relapsing EAE. Nat. Immunol. 8, 172–180. doi: 10.1038/ni1430
Bakhti, M., Snaidero, N., Schneider, D., Aggarwal, S., Mobius, W., Janshoff, A., et al. (2013). Loss of electrostatic cell-surface repulsion mediates myelin membrane adhesion and compaction in the central nervous system. Proc. Natl. Acad. Sci. U.S.A. 110, 3143–3148. doi: 10.1073/pnas.1220104110
Balabanov, R., Strand, K., Goswami, R., Mcmahon, E., Begolka, W., Miller, S. D., et al. (2007). Interferon-gamma-oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J. Neurosci. 27, 2013–2024. doi: 10.1523/jneurosci.4689-06.2007
Balatoni, B., Storch, M. K., Swoboda, E. M., Schonborn, V., Koziel, A., Lambrou, G. N., et al. (2007). FTY720 sustains and restores neuronal function in the DA rat model of MOG-induced experimental autoimmune encephalomyelitis. Brain Res. Bull. 74, 307–316. doi: 10.1016/j.brainresbull.2007.06.023
Barnett, M. H., and Prineas, J. W. (2004). Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann. Neurol. 55, 458–468. doi: 10.1002/ana.20016
Bazarian, J. J., Cernak, I., Noble-Haeusslein, L., Potolicchio, S., and Temkin, N. (2009). Long-term neurologic outcomes after traumatic brain injury. J. Head Trauma Rehabil. 24, 439–451. doi: 10.1097/htr.0b013e3181c15600
Beniac, D. R., Wood, D. D., Palaniyar, N., Ottensmeyer, F. P., Moscarello, M. A., and Harauz, G. (2000). Cryoelectron microscopy of protein-lipid complexes of human myelin basic protein charge isomers differing in degree of citrullination. J. Struct. Biol. 129, 80–95. doi: 10.1006/jsbi.1999.4200
Ben-Nun, A., Liblau, R. S., Cohen, L., Lehmann, D., Tournier-Lasserve, E., Rosenzweig, A., et al. (1991). Restricted T-cell receptor V beta gene usage by myelin basic protein-specific T-cell clones in multiple sclerosis: predominant genes vary in individuals. Proc. Natl. Acad. Sci. U.S.A. 88, 2466–2470. doi: 10.1073/pnas.88.6.2466
Bhat, R., and Steinman, L. (2009). Innate and adaptive autoimmunity directed to the central nervous system. Neuron 64, 123–132. doi: 10.1016/j.neuron.2009.09.015
Blackwell, T. E., Butler, D. G., and Bell, J. A. (1983). Enterotoxemia in the goat: the humoral response and local tissue reaction following vaccination with two different bacterin-toxoids. Can. J. Comp. Med. 47, 127–132.
Blair, T. C., Manoharan, M., Rawlings-Rhea, S. D., Tagge, I., Kohama, S. G., Hollister-Smith, J., et al. (2016). Immunopathology of Japanese macaque encephalomyelitis is similar to multiple sclerosis. J. Neuroimmunol. 291, 1–10. doi: 10.1016/j.jneuroim.2015.11.026
Bossu, J. L., Wioland, L., Doussau, F., Isope, P., Popoff, M. R., and Poulain, B. (2020). Epsilon toxin from Clostridium perfringens causes inhibition of potassium inward rectifier (Kir) channels in oligodendrocytes. Toxins 12:36. doi: 10.3390/toxins12010036
Bradford, C. M., Ramos, I., Cross, A. K., Haddock, G., Mcquaid, S., Nicholas, A. P., et al. (2014). Localisation of citrullinated proteins in normal appearing white matter and lesions in the central nervous system in multiple sclerosis. J. Neuroimmunol. 273, 85–95. doi: 10.1016/j.jneuroim.2014.05.007
Brandao de Paiva, A. R., Pucci Filho, C. R., Porto, A. M., Feltrin, F. S., Kok, F., and Camargo, C. H. F. (2018). When multiple sclerosis and X-linked adrenoleukodystrophy are tangled: a challenging case. Neurol. Clin. Pract. 8, 156–158. doi: 10.1212/cpj.0000000000000431
Brinkmann, V., Billich, A., Baumruker, T., Heining, P., Schmouder, R., Francis, G., et al. (2010). Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 9, 883–897. doi: 10.1038/nrd3248
Brinkmann, V., Davis, M. D., Heise, C. E., Albert, R., Cottens, S., Hof, R., et al. (2002). The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 277, 21453–21457. doi: 10.1074/jbc.c200176200
Burns, J., Littlefield, K., Gill, J., and Trotter, J. (1991). Autoantigen-induced self lysis of human myelin basic protein-specific T lymphocytes. J. Neuroimmunol. 35, 227–236. doi: 10.1016/0165-5728(91)90177-9
Cao, L., Goodin, R., Wood, D., Moscarello, M. A., and Whitaker, J. N. (1999). Rapid release and unusual stability of immunodominant peptide 45-89 from citrullinated myelin basic protein. Biochemistry 38, 6157–6163. doi: 10.1021/bi982960s
Caprariello, A. V., Rogers, J. A., Morgan, M. L., Hoghooghi, V., Plemel, J. R., Koebel, A., et al. (2018). Biochemically altered myelin triggers autoimmune demyelination. Proc. Natl. Acad. Sci. U.S.A. 115, 5528–5533. doi: 10.1073/pnas.1721115115
Chaudhry, B. Z., Cohen, J. A., and Conway, D. S. (2017). Sphingosine 1-Phosphate receptor modulators for the treatment of multiple sclerosis. Neurotherapeutics 14, 859–873. doi: 10.1007/s13311-017-0565-4
Chen, Y., Podojil, J. R., Kunjamma, R. B., Jones, J., Weiner, M., Lin, W., et al. (2019). Sephin1, which prolongs the integrated stress response, is a promising therapeutic for multiple sclerosis. Brain 142, 344–361. doi: 10.1093/brain/awy322
Choi, J. W., Gardell, S. E., Herr, D. R., Rivera, R., Lee, C. W., Noguchi, K., et al. (2011). FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. U.S.A. 108, 751–756. doi: 10.1073/pnas.1014154108
Chung, S., Fieremans, E., Wang, X., Kucukboyaci, N. E., Morton, C. J., Babb, J., et al. (2018). White matter tract integrity: an indicator of axonal pathology after mild traumatic brain injury. J. Neurotrauma 35, 1015–1020. doi: 10.1089/neu.2017.5320
Clerico, M., Artusi, C. A., Di Liberto, A., Rolla, S., Bardina, V., Barbero, P., et al. (2017). Long-term safety evaluation of natalizumab for the treatment of multiple sclerosis. Expert Opin. Drug Saf. 16, 963–972. doi: 10.1080/14740338.2017.1346082
Cloake, N. C., Yan, J., Aminian, A., Pender, M. P., and Greer, J. M. (2018). L1 mutations in patients with multiple sclerosis: identification of a new mutation and potential pathogenicity of the mutations. J. Clin. Med. 7:342. doi: 10.3390/jcm7100342
Collier, R. J. (2001). Understanding the mode of action of diphtheria toxin: a perspective on progress during the 20th century. Toxicon 39, 1793–1803. doi: 10.1016/s0041-0101(01)00165-9
Croxford, J. L., Olson, J. K., and Miller, S. D. (2002). Epitope spreading and molecular mimicry as triggers of autoimmunity in the Theiler’s virus-induced demyelinating disease model of multiple sclerosis. Autoimmun. Rev. 1, 251–260. doi: 10.1016/s1568-9972(02)00080-0
Culpepper, W. J., Marrie, R. A., Langer-Gould, A., Wallin, M. T., Campbell, J. D., Nelson, L. M., et al. (2019). Validation of an algorithm for identifying MS cases in administrative health claims datasets. Neurology 92, e1016–e1028.
Daglas, M., Draxler, D. F., Ho, H., Mccutcheon, F., Galle, A., Au, A. E., et al. (2019). Activated CD8(+) T cells cause long-term neurological impairment after traumatic brain injury in mice. Cell Rep. 29, 1178.e6–1191.e6.
de Beer, M., Engelen, M., and Van Geel, B. M. (2014). Frequent occurrence of cerebral demyelination in adrenomyeloneuropathy. Neurology 83, 2227–2231. doi: 10.1212/wnl.0000000000001074
Dent, K. A., Christie, K. J., Bye, N., Basrai, H. S., Turbic, A., Habgood, M., et al. (2015). Oligodendrocyte birth and death following traumatic brain injury in adult mice. PLoS One 10:e0121541. doi: 10.1371/journal.pone.0121541
Derfuss, T., Mehling, M., Papadopoulou, A., Bar-Or, A., Cohen, J. A., and Kappos, L. (2020). Advances in oral immunomodulating therapies in relapsing multiple sclerosis. Lancet Neurol. 19, 336–347. doi: 10.1016/s1474-4422(19)30391-6
Doerflinger, N. H., Macklin, W. B., and Popko, B. (2003). Inducible site-specific recombination in myelinating cells. Genesis 35, 63–72. doi: 10.1002/gene.10154
Donovan, V., Kim, C., Anugerah, A. K., Coats, J. S., Oyoyo, U., Pardo, A. C., et al. (2014). Repeated mild traumatic brain injury results in long-term white-matter disruption. J. Cereb. Blood Flow Metab. 34, 715–723. doi: 10.1038/jcbfm.2014.6
Dorca-Arevalo, J., Soler-Jover, A., Gibert, M., Popoff, M. R., Martin-Satue, M., and Blasi, J. (2008). Binding of epsilon-toxin from Clostridium perfringens in the nervous system. Vet. Microbiol. 131, 14–25. doi: 10.1016/j.vetmic.2008.02.015
D’Souza, C. A., and Moscarello, M. A. (2006). Differences in susceptibility of MBP charge isomers to digestion by stromelysin-1 (MMP-3) and release of an immunodominant epitope. Neurochem. Res. 31, 1045–1054. doi: 10.1007/s11064-006-9116-9
Duan, Y., Sahley, C. L., and Muller, K. J. (2009). ATP and NO dually control migration of microglia to nerve lesions. Dev. Neurobiol. 69, 60–72. doi: 10.1002/dneu.20689
Esiri, M. M., and Reading, M. C. (1987). Macrophage populations associated with multiple sclerosis plaques. Neuropathol. Appl. Neurobiol. 13, 451–465. doi: 10.1111/j.1365-2990.1987.tb00074.x
Estep, R. D., Hansen, S. G., Rogers, K. S., Axthelm, M. K., and Wong, S. W. (2013). Genomic characterization of Japanese macaque rhadinovirus, a novel herpesvirus isolated from a nonhuman primate with a spontaneous inflammatory demyelinating disease. J. Virol. 87, 512–523. doi: 10.1128/jvi.02194-12
Falcao, A. M., Van Bruggen, D., Marques, S., Meijer, M., Jakel, S., Agirre, E., et al. (2018). Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 24, 1837–1844. doi: 10.1038/s41591-018-0236-y
Ferguson, B., Matyszak, M. K., Esiri, M. M., and Perry, V. H. (1997). Axonal damage in acute multiple sclerosis lesions. Brain 120(Pt 3), 393–399. doi: 10.1093/brain/120.3.393
Ferrer, I., Aubourg, P., and Pujol, A. (2010). General aspects and neuropathology of X-linked adrenoleukodystrophy. Brain Pathol. 20, 817–830. doi: 10.1111/j.1750-3639.2010.00390.x
Finnie, J. W. (1984). Ultrastructural changes in the brain of mice given Clostridium perfringens type D epsilon toxin. J. Comp. Pathol. 94, 445–452. doi: 10.1016/0021-9975(84)90031-8
Finnie, J. W. (2003). Pathogenesis of brain damage produced in sheep by Clostridium perfringens type D epsilon toxin: a review. Aust. Vet. J. 81, 219–221. doi: 10.1111/j.1751-0813.2003.tb11474.x
Fragoso, Y. D., Brooks, J. B. B., Eboni, A. C. B., Fezer, L., Da Gama, P. D., Gomes, S., et al. (2019). Seroconversion of JCV antibodies is strongly associated to natalizumab therapy. J. Clin. Neurosci. 61, 112–113. doi: 10.1016/j.jocn.2018.10.128
Francis, G., Kappos, L., O’connor, P., Collins, W., Tang, D., Mercier, F., et al. (2014). Temporal profile of lymphocyte counts and relationship with infections with fingolimod therapy. Mult. Scler. 20, 471–480. doi: 10.1177/1352458513500551
Franklin, R. J., and Ffrench-Constant, C. (2008). Remyelination in the CNS: from biology to therapy. Nat. Rev. Neurosci. 9, 839–855. doi: 10.1038/nrn2480
Fujino, M., Funeshima, N., Kitazawa, Y., Kimura, H., Amemiya, H., Suzuki, S., et al. (2003). Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J. Pharmacol. Exp. Ther. 305, 70–77. doi: 10.1124/jpet.102.045658
Gerritse, K., Laman, J. D., Noelle, R. J., Aruffo, A., Ledbetter, J. A., Boersma, W. J., et al. (1996). CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 93, 2499–2504. doi: 10.1073/pnas.93.6.2499
Getts, D. R., Martin, A. J., Mccarthy, D. P., Terry, R. L., Hunter, Z. N., Yap, W. T., et al. (2012). Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat. Biotechnol. 30, 1217–1224. doi: 10.1038/nbt.2434
Getts, D. R., Mccarthy, D. P., and Miller, S. D. (2013). Exploiting apoptosis for therapeutic tolerance induction. J. Immunol. 191, 5341–5346. doi: 10.4049/jimmunol.1302070
Getts, D. R., Turley, D. M., Smith, C. E., Harp, C. T., Mccarthy, D., Feeney, E. M., et al. (2011). Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells. J. Immunol. 187, 2405–2417. doi: 10.4049/jimmunol.1004175
Ghosh, A., Manrique-Hoyos, N., Voigt, A., Schulz, J. B., Kreutzfeldt, M., Merkler, D., et al. (2011). Targeted ablation of oligodendrocytes triggers axonal damage. PLoS One 6:e22735. doi: 10.1371/journal.pone.0022735
Giacci, M., and Fitzgerald, M. (2018). Oligodendroglia are particularly vulnerable to oxidative damage after neurotrauma in vivo. J. Exp. Neurosci. 12:1179069518810004.
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Gritsch, S., Lu, J., Thilemann, S., Wortge, S., Mobius, W., Bruttger, J., et al. (2014). Oligodendrocyte ablation triggers central pain independently of innate or adaptive immune responses in mice. Nat.Commun. 5:5472.
Gurevich, M., Waknin, R., Stone, E., and Achiron, A. (2018). Fingolimod-improved axonal and myelin integrity of white matter tracts associated with multiple sclerosis-related functional impairments. CNS Neurosci. Ther. 24, 412–419. doi: 10.1111/cns.12796
Harp, C. T., Ireland, S., Davis, L. S., Remington, G., Cassidy, B., Cravens, P. D., et al. (2010). Memory B cells from a subset of treatment-naive relapsing-remitting multiple sclerosis patients elicit CD4(+) T-cell proliferation and IFN-gamma production in response to myelin basic protein and myelin oligodendrocyte glycoprotein. Eur. J. Immunol. 40, 2942–2956. doi: 10.1002/eji.201040516
Hausler, D., Hausser-Kinzel, S., Feldmann, L., Torke, S., Lepennetier, G., Bernard, C. C. A., et al. (2018). Functional characterization of reappearing B cells after anti-CD20 treatment of CNS autoimmune disease. Proc. Natl. Acad. Sci. U.S.A. 115, 9773–9778. doi: 10.1073/pnas.1810470115
Ho, J. K., Moser, H., Kishimoto, Y., and Hamilton, J. A. (1995). Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J. Clin. Invest. 96, 1455–1463. doi: 10.1172/jci118182
Ho, P. R., Koendgen, H., Campbell, N., Haddock, B., Richman, S., and Chang, I. (2017). Risk of natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: a retrospective analysis of data from four clinical studies. Lancet Neurol. 16, 925–933. doi: 10.1016/s1474-4422(17)30282-x
Hudspeth, M. P., and Raymond, G. V. (2007). Immunopathogenesis of adrenoleukodystrophy: current understanding. J. Neuroimmunol. 182, 5–12. doi: 10.1016/j.jneuroim.2006.10.009
Hunter, Z., Mccarthy, D. P., Yap, W. T., Harp, C. T., Getts, D. R., Shea, L. D., et al. (2014). A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS Nano 8, 2148–2160. doi: 10.1021/nn405033r
International Multiple Sclerosis Genetics Consortium, A. H., Patsopoulos, N. A., Xifara, D. K., Davis, M. F., Kemppinen, A., et al. (2013). Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 45, 1353–1360. doi: 10.1038/ng.2770
International Multiple Sclerosis Genetics Consortium (2019). Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365:eaav7188. doi: 10.1126/science.aav7188
Ito, M., Blumberg, B. M., Mock, D. J., Goodman, A. D., Moser, A. B., Moser, H. W., et al. (2001). Potential environmental and host participants in the early white matter lesion of adreno-leukodystrophy: morphologic evidence for CD8 cytotoxic T cells, cytolysis of oligodendrocytes, and CD1-mediated lipid antigen presentation. J. Neuropathol. Exp. Neurol. 60, 1004–1019. doi: 10.1093/jnen/60.10.1004
Jakel, S., and Dimou, L. (2017). Glial cells and their function in the adult brain: a journey through the history of their ablation. Front. Cell Neurosci. 11:24. doi: 10.3389/fncel.2017.00024
Kalinin, S., Polak, P. E., Lin, S. X., Braun, D., Guizzetti, M., Zhang, X., et al. (2013). Dimethyl fumarate regulates histone deacetylase expression in astrocytes. J. Neuroimmunol. 263, 13–19. doi: 10.1016/j.jneuroim.2013.07.007
Kelly, C., Murray, J., Leffler, D., Bledsoe, A., Smithson, G., Podojil, J., et al. (2019). CNP-101 prevents gluten challenge induced immune activation in adults with celiac disease. Proc. United Eur. Gastroenterol. 7. Available online at: https://ueg.eu/library/cnp-101-prevents-gluten-challenge-induced-immune-activation-in-adults-with-celiac-disease/208600
Kemp, S., Huffnagel, I. C., Linthorst, G. E., Wanders, R. J., and Engelen, M. (2016). Adrenoleukodystrophy - neuroendocrine pathogenesis and redefinition of natural history. Nat. Rev. Endocrinol. 12, 606–615. doi: 10.1038/nrendo.2016.90
Korenke, G. C., Pouwels, P. J., Frahm, J., Hunneman, D. H., Stoeckler, S., Krasemann, E., et al. (1996). Arrested cerebral adrenoleukodystrophy: a clinical and proton magnetic resonance spectroscopy study in three patients. Pediatr. Neurol. 15, 103–107. doi: 10.1016/0887-8994(95)00156-5
Kostic, M., Zivkovic, N., and Stojanovic, I. (2013). Multiple sclerosis and glutamate excitotoxicity. Rev. Neurosci. 24, 71–88.
Kotelnikova, E., Kiani, N. A., Messinis, D., Pertsovskaya, I., Pliaka, V., Bernardo-Faura, M., et al. (2019). MAPK pathway and B cells overactivation in multiple sclerosis revealed by phosphoproteomics and genomic analysis. Proc. Natl. Acad. Sci. U.S.A. 116, 9671–9676. doi: 10.1073/pnas.1818347116
Lassmann, H., and Bradl, M. (2017). Multiple sclerosis: experimental models and reality. Acta Neuropathol. 133, 223–244. doi: 10.1007/s00401-016-1631-4
Lee, C. W., Choi, J. W., and Chun, J. (2010). Neurological S1P signaling as an emerging mechanism of action of oral FTY720 (fingolimod) in multiple sclerosis. Arch. Pharm. Res. 33, 1567–1574. doi: 10.1007/s12272-010-1008-5
Lehmann, P. V., Forsthuber, T., Miller, A., and Sercarz, E. E. (1992). Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature 358, 155–157. doi: 10.1038/358155a0
Lehmann, P. V., Sercarz, E. E., Forsthuber, T., Dayan, C. M., and Gammon, G. (1993). Determinant spreading and the dynamics of the autoimmune T-cell repertoire. Immunol. Today 14, 203–208. doi: 10.1016/0167-5699(93)90163-f
Lehmann, P. V., Targoni, O. S., and Forsthuber, T. G. (1998). Shifting T-cell activation thresholds in autoimmunity and determinant spreading. Immunol. Rev. 164, 53–61. doi: 10.1111/j.1600-065x.1998.tb01207.x
Lin, S. X., Lisi, L., Dello Russo, C., Polak, P. E., Sharp, A., Weinberg, G., et al. (2011). The anti-inflammatory effects of dimethyl fumarate in astrocytes involve glutathione and haem oxygenase-1. ASN Neuro 3:e00055.
Lin, W., Bailey, S. L., Ho, H., Harding, H. P., Ron, D., Miller, S. D., et al. (2007). The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J. Clin. Invest. 117, 448–456. doi: 10.1172/jci29571
Lin, W., Harding, H. P., Ron, D., and Popko, B. (2005). Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J. Cell Biol. 169, 603–612. doi: 10.1083/jcb.200502086
Lin, W., Kunkler, P. E., Harding, H. P., Ron, D., Kraig, R. P., and Popko, B. (2008). Enhanced integrated stress response promotes myelinating oligodendrocyte survival in response to interferon-gamma. Am. J. Pathol. 173, 1508–1517. doi: 10.2353/ajpath.2008.080449
Lin, W., Lin, Y., Li, J., Fenstermaker, A. G., Way, S. W., Clayton, B., et al. (2013). Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J. Neurosci. 33, 5980–5991. doi: 10.1523/jneurosci.1636-12.2013
Linden, J. R., Ma, Y., Zhao, B., Harris, J. M., Rumah, K. R., Schaeren-Wiemers, N., et al. (2015). Clostridium perfringens epsilon toxin causes selective death of mature oligodendrocytes and central nervous system demyelination. mBio 6:e02513.
Linker, R. A., and Gold, R. (2013). Dimethyl fumarate for treatment of multiple sclerosis: mechanism of action, effectiveness, and side effects. Curr. Neurol. Neurosci. Rep. 13:394.
Linker, R. A., Lee, D. H., Ryan, S., Van Dam, A. M., Conrad, R., Bista, P., et al. (2011). Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 134, 678–692. doi: 10.1093/brain/awq386
Locatelli, G., Wortge, S., Buch, T., Ingold, B., Frommer, F., Sobottka, B., et al. (2012). Primary oligodendrocyte death does not elicit anti-CNS immunity. Nat. Neurosci. 15, 543–550. doi: 10.1038/nn.3062
Lonchamp, E., Dupont, J. L., Wioland, L., Courjaret, R., Mbebi-Liegeois, C., Jover, E., et al. (2010). Clostridium perfringens epsilon toxin targets granule cells in the mouse cerebellum and stimulates glutamate release. PLoS One 5:e13046. doi: 10.1371/journal.pone.0013046
Lotocki, G., De Rivero Vaccari, J. P., Alonso, O., Molano, J. S., Nixon, R., Safavi, P., et al. (2011). Oligodendrocyte vulnerability following traumatic brain injury in rats. Neurosci. Lett. 499, 143–148. doi: 10.1016/j.neulet.2011.05.056
Luo, X., Miller, S. D., and Shea, L. D. (2016). Immune tolerance for autoimmune disease and cell transplantation. Annu. Rev. Biomed. Eng. 18, 181–205. doi: 10.1146/annurev-bioeng-110315-020137
Lutterotti, A., Yousef, S., Sputtek, A., Sturner, K. H., Stellmann, J. P., Breiden, P., et al. (2013). Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci. Transl. Med. 5:188ra175.
Mack, C. L., Vanderlugt-Castaneda, C. L., Neville, K. L., and Miller, S. D. (2003). Microglia are activated to become competent antigen presenting and effector cells in the inflammatory environment of the Theiler’s virus model of multiple sclerosis. J. Neuroimmunol. 144, 68–79. doi: 10.1016/j.jneuroim.2003.08.032
Manconi, B., Liori, B., Cabras, T., Vincenzoni, F., Iavarone, F., Lorefice, L., et al. (2018). Top-down proteomic profiling of human saliva in multiple sclerosis patients. J. Proteomics 187, 212–222. doi: 10.1016/j.jprot.2018.07.019
Mandala, S., Hajdu, R., Bergstrom, J., Quackenbush, E., Xie, J., Milligan, J., et al. (2002). Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296, 346–349. doi: 10.1126/science.1070238
Matsushima, G. K., and Morell, P. (2001). The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 11, 107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x
McCarthy, D. P., Hunter, Z. N., Chackerian, B., Shea, L. D., and Miller, S. D. (2014). Targeted immunomodulation using antigen-conjugated nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 6, 298–315. doi: 10.1002/wnan.1263
McCarthy, D. P., Yap, J. W., Harp, C. T., Song, W. K., Chen, J., Pearson, R. M., et al. (2017). An antigen-encapsulating nanoparticle platform for TH1/17 immune tolerance therapy. Nanomedicine 13, 191–200. doi: 10.1016/j.nano.2016.09.007
McFarland, H. F., and Martin, R. (2007). Multiple sclerosis: a complicated picture of autoimmunity. Nat. Immunol. 8, 913–919. doi: 10.1038/ni1507
McMahon, E. J., Bailey, S. L., Castenada, C. V., Waldner, H., and Miller, S. D. (2005). Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat. Med. 11, 335–339. doi: 10.1038/nm1202
McRae, B. L., Vanderlugt, C. L., Dal Canto, M. C., and Miller, S. D. (1995). Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J. Exp. Med. 182, 75–85. doi: 10.1084/jem.182.1.75
Mierzwa, A. J., Marion, C. M., Sullivan, G. M., Mcdaniel, D. P., and Armstrong, R. C. (2015). Components of myelin damage and repair in the progression of white matter pathology after mild traumatic brain injury. J. Neuropathol. Exp. Neurol. 74, 218–232. doi: 10.1097/nen.0000000000000165
Miller, D. H., and Leary, S. M. (2007). Primary-progressive multiple sclerosis. Lancet Neurol. 6, 903–912.
Miller, S. D., Mcrae, B. L., Vanderlugt, C. L., Nikcevich, K. M., Pope, J. G., Pope, L., et al. (1995). Evolution of the T-cell repertoire during the course of experimental immune-mediated demyelinating diseases. Immunol. Rev. 144, 225–244. doi: 10.1111/j.1600-065x.1995.tb00071.x
Miller, S. D., Vanderlugt, C. L., Begolka, W. S., Pao, W., Yauch, R. L., Neville, K. L., et al. (1997). Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 3, 1133–1136. doi: 10.1038/nm1097-1133
Mills, E. A., Ogrodnik, M. A., Plave, A., and Mao-Draayer, Y. (2018). Emerging understanding of the mechanism of action for dimethyl fumarate in the treatment of multiple sclerosis. Front. Neurol. 9:5. doi: 10.3389/fneur.2018.00005
Moon, J. J., Chu, H. H., Pepper, M., Mcsorley, S. J., Jameson, S. C., Kedl, R. M., et al. (2007). Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27, 203–213. doi: 10.1016/j.immuni.2007.07.007
Moscarello, M. A., Mastronardi, F. G., and Wood, D. D. (2007). The role of citrullinated proteins suggests a novel mechanism in the pathogenesis of multiple sclerosis. Neurochem. Res. 32, 251–256. doi: 10.1007/s11064-006-9144-5
Moscarello, M. A., Wood, D. D., Ackerley, C., and Boulias, C. (1994). Myelin in multiple sclerosis is developmentally immature. J. Clin. Invest. 94, 146–154. doi: 10.1172/jci117300
Moser, H. W., Mahmood, A., and Raymond, G. V. (2007). X-linked adrenoleukodystrophy. Nat. Clin. Pract. Neurol. 3, 140–151.
Moser, H. W., Moser, A. B., Smith, K. D., Bergin, A., Borel, J., Shankroff, J., et al. (1992). Adrenoleukodystrophy: phenotypic variability and implications for therapy. J. Inherit. Metab. Dis. 15, 645–664. doi: 10.1007/bf01799621
Munz, C., Lunemann, J. D., Getts, M. T., and Miller, S. D. (2009). Antiviral immune responses: triggers of or triggered by autoimmunity? Nat. Rev. Immunol. 9, 246–258. doi: 10.1038/nri2527
Murrell, T. G., O’donoghue, P. J., and Ellis, T. (1986). A review of the sheep-multiple sclerosis connection. Med. Hypotheses 19, 27–39. doi: 10.1016/0306-9877(86)90134-9
Musse, A. A., Boggs, J. M., and Harauz, G. (2006). Deimination of membrane-bound myelin basic protein in multiple sclerosis exposes an immunodominant epitope. Proc. Natl. Acad. Sci. U.S.A. 103, 4422–4427. doi: 10.1073/pnas.0509158103
Nave, K. A. (2010). Myelination and support of axonal integrity by glia. Nature 468, 244–252. doi: 10.1038/nature09614
Nelson, L. M., Wallin, M. T., Marrie, R. A., Culpepper, W. J., Langer-Gould, A., Campbell, J., et al. (2019). A new way to estimate neurologic disease prevalence in the United States: Illustrated with MS. Neurology 92, 469–480. doi: 10.1212/wnl.0000000000007044
Olson, J. K., Croxford, J. L., Calenoff, M. A., Dal Canto, M. C., and Miller, S. D. (2001). A virus-induced molecular mimicry model of multiple sclerosis. J. Clin. Invest. 108, 311–318. doi: 10.1172/jci200113032
Oluich, L. J., Stratton, J. A., Xing, Y. L., Ng, S. W., Cate, H. S., Sah, P., et al. (2012). Targeted ablation of oligodendrocytes induces axonal pathology independent of overt demyelination. J. Neurosci. 32, 8317–8330. doi: 10.1523/jneurosci.1053-12.2012
O’Phelan, K. H., Otoshi, C. K., Ernst, T., and Chang, L. (2018). Common patterns of regional brain injury detectable by diffusion tensor imaging in otherwise normal-appearing white matter in patients with early moderate to severe traumatic brain injury. J. Neurotrauma 35, 739–749. doi: 10.1089/neu.2016.4944
Parodi, B., Rossi, S., Morando, S., Cordano, C., Bragoni, A., Motta, C., et al. (2015). Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol. 130, 279–295. doi: 10.1007/s00401-015-1422-3
Partridge, M. A., Gopinath, S., Myers, S. J., and Coorssen, J. R. (2016). An initial top-down proteomic analysis of the standard cuprizone mouse model of multiple sclerosis. J. Chem. Biol. 9, 9–18. doi: 10.1007/s12154-015-0138-0
Pearson, R. M., Casey, L. M., Hughes, K. R., Miller, S. D., and Shea, L. D. (2017). In vivo reprogramming of immune cells: technologies for induction of antigen-specific tolerance. Adv. Drug Deliv. Rev. 114, 240–255. doi: 10.1016/j.addr.2017.04.005
Pearson, R. M., Podojil, J. R., Shea, L. D., King, N. J. C., Miller, S. D., and Getts, D. R. (2019). Overcoming challenges in treating autoimmuntity: development of tolerogenic immune-modifying nanoparticles. Nanomedicine 18, 282–291. doi: 10.1016/j.nano.2018.10.001
Pham, T. H., Okada, T., Matloubian, M., Lo, C. G., and Cyster, J. G. (2008). S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity 28, 122–133. doi: 10.1016/j.immuni.2007.11.017
Podojil, J. R., and Miller, S. D. (2009). Molecular mechanisms of T-cell receptor and costimulatory molecule ligation/blockade in autoimmune disease therapy. Immunol. Rev. 229, 337–355. doi: 10.1111/j.1600-065x.2009.00773.x
Popoff, M. R. (2011). Epsilon toxin: a fascinating pore-forming toxin. FEBS J. 278, 4602–4615. doi: 10.1111/j.1742-4658.2011.08145.x
Powers, J. M., Liu, Y., Moser, A. B., and Moser, H. W. (1992). The inflammatory myelinopathy of adreno-leukodystrophy: cells, effector molecules, and pathogenetic implications. J. Neuropathol. Exp. Neurol. 51, 630–643. doi: 10.1097/00005072-199211000-00007
Prasad, S., Xu, D., and Miller, S. D. (2012). Tolerance strategies employing antigen-coupled apoptotic cells and carboxylated PLG nanoparticles for the treatment of type 1 diabetes. Rev. Diabet Stud. 9, 319–327. doi: 10.1900/rds.2012.9.319
Prineas, J. W., Kwon, E. E., Cho, E. S., Sharer, L. R., Barnett, M. H., Oleszak, E. L., et al. (2001). Immunopathology of secondary-progressive multiple sclerosis. Ann. Neurol. 50, 646–657.
Prineas, J. W., and Parratt, J. D. (2012). Oligodendrocytes and the early multiple sclerosis lesion. Ann. Neurol. 72, 18–31. doi: 10.1002/ana.23634
Prinz, M., and Kalinke, U. (2010). New lessons about old molecules: how type I interferons shape Th1/Th17-mediated autoimmunity in the CNS. Trends Mol. Med. 16, 379–386. doi: 10.1016/j.molmed.2010.06.001
Raijmakers, R., Vogelzangs, J., Croxford, J. L., Wesseling, P., Van Venrooij, W. J., and Pruijn, G. J. (2005). Citrullination of central nervous system proteins during the development of experimental autoimmune encephalomyelitis. J. Comp. Neurol. 486, 243–253. doi: 10.1002/cne.20529
Raymond, G. V., Seidman, R., Monteith, T. S., Kolodny, E., Sathe, S., Mahmood, A., et al. (2010). Head trauma can initiate the onset of adreno-leukodystrophy. J. Neurol. Sci. 290, 70–74. doi: 10.1016/j.jns.2009.11.005
Robinson, A. P., Harp, C. T., Noronha, A., and Miller, S. D. (2014). The experimental autoimmune encephalomyelitis (EAE) model of MS: utility for understanding disease pathophysiology and treatment. Handb. Clin. Neurol. 122, 173–189. doi: 10.1016/b978-0-444-52001-2.00008-x
Rodgers, J. M., Robinson, A. P., and Miller, S. D. (2013). Strategies for protecting oligodendrocytes and enhancing remyelination in multiple sclerosis. Discov. Med. 16, 53–63.
Rumah, K. R., Linden, J., Fischetti, V. A., and Vartanian, T. (2013). Isolation of Clostridium perfringens type B in an individual at first clinical presentation of multiple sclerosis provides clues for environmental triggers of the disease. PLoS One 8:e76359. doi: 10.1371/journal.pone.0076359
Rumah, K. R., Ma, Y., Linden, J. R., Oo, M. L., Anrather, J., Schaeren-Wiemers, N., et al. (2015). The myelin and lymphocyte protein MAL is required for binding and activity of Clostridium perfringens epsilon-toxin. PLoS Pathog. 11:e1004896. doi: 10.1371/journal.ppat.1004896
Rutgers, D. R., Fillard, P., Paradot, G., Tadie, M., Lasjaunias, P., and Ducreux, D. (2008). Diffusion tensor imaging characteristics of the corpus callosum in mild, moderate, and severe traumatic brain injury. AJNR Am. J. Neuroradiol. 29, 1730–1735. doi: 10.3174/ajnr.a1213
Ryerson, L. Z., Foley, J., Chang, I., Kister, I., Cutter, G., Metzger, R. R., et al. (2019). Risk of natalizumab-associated PML in patients with MS is reduced with extended interval dosing. Neurology 93, e1452–e1462.
Schwartz, M., and Raposo, C. (2014). Protective autoimmunity: a unifying model for the immune network involved in CNS repair. Neuroscientist 20, 343–358. doi: 10.1177/1073858413516799
Sen, M. K., Almuslehi, M. S. M., Gyengesi, E., Myers, S. J., Shortland, P. J., Mahns, D. A., et al. (2019a). Suppression of the peripheral immune system limits the central immune response following cuprizone-feeding: relevance to modelling multiple sclerosis. Cells 8:1314. doi: 10.3390/cells8111314
Sen, M. K., Mahns, D. A., Coorssen, J. R., and Shortland, P. J. (2019b). Behavioural phenotypes in the cuprizone model of central nervous system demyelination. Neurosci. Biobehav. Rev. 107, 23–46. doi: 10.1016/j.neubiorev.2019.08.008
Sharma, S., Ifergan, I., Kurz, J. E., Linsenmeier, R. A., Xu, D., Cooper, J. G., et al. (2020). Intravenous immunomodulatory nanoparticle treatment for traumatic brain injury. Ann. Neurol. 87, 442–455. doi: 10.1002/ana.25675
Sidaway, P. (2017). Multiple sclerosis: concussion during adolescence linked to increased risk of MS. Nat. Rev. Neurol. 13:640. doi: 10.1038/nrneurol.2017.135
Singh, J., Khan, M., and Singh, I. (2009). Silencing of Abcd1 and Abcd2 genes sensitizes astrocytes for inflammation: implication for X-adrenoleukodystrophy. J. Lipid Res. 50, 135–147. doi: 10.1194/jlr.m800321-jlr200
Soderstrom, M., Link, H., Sun, J. B., Fredrikson, S., Kostulas, V., Hojeberg, B., et al. (1993). T cells recognizing multiple peptides of myelin basic protein are found in blood and enriched in cerebrospinal fluid in optic neuritis and multiple sclerosis. Scand. J. Immunol. 37, 355–368. doi: 10.1111/j.1365-3083.1993.tb02565.x
Solti, I., Kvell, K., Talaber, G., Veto, S., Acs, P., Gallyas, F., et al. (2015). Thymic atrophy and apoptosis of CD4+CD8+ thymocytes in the cuprizone model of multiple sclerosis. PLoS One 10:e0129217. doi: 10.1371/journal.pone.0129217
Sospedra, M., and Martin, R. (2005). Immunology of multiple sclerosis. Annu. Rev. Immunol. 23, 683–747.
Stadelmann, C. (2011). Multiple sclerosis as a neurodegenerative disease: pathology, mechanisms and therapeutic implications. Curr. Opin. Neurol. 24, 224–229. doi: 10.1097/wco.0b013e328346056f
Steinman, L. (2009). A molecular trio in relapse and remission in multiple sclerosis. Nat. Rev. Immunol. 9, 440–447. doi: 10.1038/nri2548
Steinman, L., Martin, R., Bernard, C., Conlon, P., and Oksenberg, J. R. (2002). Multiple sclerosis: deeper understanding of its pathogenesis reveals new targets for therapy. Annu. Rev. Neurosci. 25, 491–505. doi: 10.1146/annurev.neuro.25.112701.142913
Stuve, O., Marra, C. M., Jerome, K. R., Cook, L., Cravens, P. D., Cepok, S., et al. (2006). Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann. Neurol. 59, 743–747.
Stys, P. K., Zamponi, G. W., Van Minnen, J., and Geurts, J. J. (2012). Will the real multiple sclerosis please stand up? Nat. Rev. Neurosci. 13, 507–514. doi: 10.1038/nrn3275
Sun, J., Link, H., Olsson, T., Xiao, B. G., Andersson, G., Ekre, H. P., et al. (1991). T and B cell responses to myelin-oligodendrocyte glycoprotein in multiple sclerosis. J. Immunol. 146, 1490–1495.
Tamai, E., Ishida, T., Miyata, S., Matsushita, O., Suda, H., Kobayashi, S., et al. (2003). Accumulation of Clostridium perfringens epsilon-toxin in the mouse kidney and its possible biological significance. Infect. Immun. 71, 5371–5375. doi: 10.1128/iai.71.9.5371-5375.2003
Terry, R. L., Ifergan, I., and Miller, S. D. (2016). Experimental autoimmune encephalomyelitis in mice. Methods Mol. Biol. 1304, 145–160.
Theiler, M., and Gard, S. (1940). Encephalomyelitis of Mice : iii. Epidemiology. J. Exp. Med. 72, 79–90.
Traka, M., Arasi, K., Avila, R. L., Podojil, J. R., Christakos, A., Miller, S. D., et al. (2010). A genetic mouse model of adult-onset, pervasive central nervous system demyelination with robust remyelination. Brain 133, 3017–3029. doi: 10.1093/brain/awq247
Traka, M., Podojil, J. R., Mccarthy, D. P., Miller, S. D., and Popko, B. (2016). Oligodendrocyte death results in immune-mediated CNS demyelination. Nat. Neurosci. 19, 65–74. doi: 10.1038/nn.4193
Trapp, B. D., and Nave, K. A. (2008). Multiple sclerosis: an immune or neurodegenerative disorder? Annu. Rev. Neurosci. 31, 247–269. doi: 10.1146/annurev.neuro.30.051606.094313
Trotter, J. L., Hickey, W. F., Van Der Veen, R. C., and Sulze, L. (1991). Peripheral blood mononuclear cells from multiple sclerosis patients recognize myelin proteolipid protein and selected peptides. J. Neuroimmunol. 33, 55–62. doi: 10.1016/0165-5728(91)90034-5
Uzal, F. A., Kelly, W. R., Morris, W. E., Bermudez, J., and Baison, M. (2004). The pathology of peracute experimental Clostridium perfringens type D enterotoxemia in sheep. J. Vet. Diagn. Invest. 16, 403–411. doi: 10.1177/104063870401600506
Uzal, F. A., and Songer, J. G. (2008). Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. J. Vet. Diagn. Invest. 20, 253–265.
Vanderlugt, C. L., and Miller, S. D. (2002). Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat. Rev. Immunol. 2, 85–95. doi: 10.1038/nri724
Veillette, A., Zuniga-Pflucker, J. C., Bolen, J. B., and Kruisbeek, A. M. (1989). Engagement of CD4 and CD8 expressed on immature thymocytes induces activation of intracellular tyrosine phosphorylation pathways. J. Exp. Med. 170, 1671–1680. doi: 10.1084/jem.170.5.1671
Vossenaar, E. R., Zendman, A. J., Van Venrooij, W. J., and Pruijn, G. J. (2003). PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays 25, 1106–1118. doi: 10.1002/bies.10357
Wagley, S., Bokori-Brown, M., Morcrette, H., Malaspina, A., D’arcy, C., Gnanapavan, S., et al. (2019). Evidence of Clostridium perfringens epsilon toxin associated with multiple sclerosis. Mult. Scler 25, 653–660. doi: 10.1177/1352458518767327
Wallin, M. T., Culpepper, W. J., Campbell, J. D., Nelson, L. M., Langer-Gould, A., Marrie, R. A., et al. (2019). The prevalence of MS in the United States: A population-based estimate using health claims data. Neurology 92, e1029–e1040.
Wang, Q., Chuikov, S., Taitano, S., Wu, Q., Rastogi, A., Tuck, S. J., et al. (2015). Dimethyl fumarate protects neural stem/progenitor cells and neurons from oxidative damage through Nrf2-ERK1/2 MAPK Pathway. Int. J. Mol. Sci. 16, 13885–13907. doi: 10.3390/ijms160613885
Warshawsky, I., Rudick, R. A., Staugaitis, S. M., and Natowicz, M. R. (2005). Primary progressive multiple sclerosis as a phenotype of a PLP1 gene mutation. Ann. Neurol. 58, 470–473. doi: 10.1002/ana.20601
Way, S. W., Podojil, J. R., Clayton, B. L., Zaremba, A., Collins, T. L., Kunjamma, R. B., et al. (2015). Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat. Commun. 6:6532.
Weiner, H. L. (2009). The challenge of multiple sclerosis: how do we cure a chronic heterogeneous disease? Ann. Neurol. 65, 239–248. doi: 10.1002/ana.21640
Weller, M., Liedtke, W., Petersen, D., Opitz, H., and Poremba, M. (1992). Very-late-onset adrenoleukodystrophy: possible precipitation of demyelination by cerebral contusion. Neurology 42, 367–370. doi: 10.1212/wnl.42.2.367
Wetzig, R., Hanson, D. G., Miller, S. D., and Claman, H. N. (1979). Binding of ovalbumin to mouse spleen cells with and without carbodiimide. J. Immunol. Methods 28, 361–368. doi: 10.1016/0022-1759(79)90201-1
Windhagen, A., Newcombe, J., Dangond, F., Strand, C., Woodroofe, M. N., Cuzner, M. L., et al. (1995). Expression of costimulatory molecules B7-1 (CD80). B7-2 (CD86), and interleukin 12 cytokine in multiple sclerosis lesions. J. Exp. Med. 182, 1985–1996. doi: 10.1084/jem.182.6.1985
Wioland, L., Dupont, J. L., Doussau, F., Gaillard, S., Heid, F., Isope, P., et al. (2015). Epsilon toxin from Clostridium perfringens acts on oligodendrocytes without forming pores, and causes demyelination. Cell Microbiol. 17, 369–388. doi: 10.1111/cmi.12373
Wood, D. D., Bilbao, J. M., O’connors, P., and Moscarello, M. A. (1996). Acute multiple sclerosis (Marburg type) is associated with developmentally immature myelin basic protein. Ann. Neurol. 40, 18–24. doi: 10.1002/ana.410400106
Yadav, S. K., Soin, D., Ito, K., and Dhib-Jalbut, S. (2019). Insight into the mechanism of action of dimethyl fumarate in multiple sclerosis. J. Mol. Med. 97, 463–472. doi: 10.1007/s00109-019-01761-5
Zarrouk, A., Nury, T., Karym, E. M., Vejux, A., Sghaier, R., Gondcaille, C., et al. (2017). Attenuation of 7-ketocholesterol-induced overproduction of reactive oxygen species, apoptosis, and autophagy by dimethyl fumarate on 158N murine oligodendrocytes. J. Steroid. Biochem. Mol. Biol. 169, 29–38. doi: 10.1016/j.jsbmb.2016.02.024
Zhang, J., Zhang, Z. G., Li, Y., Ding, X., Shang, X., Lu, M., et al. (2015). Fingolimod treatment promotes proliferation and differentiation of oligodendrocyte progenitor cells in mice with experimental autoimmune encephalomyelitis. Neurobiol. Dis. 76, 57–66. doi: 10.1016/j.nbd.2015.01.006
Zhang, Y., Burger, D., Saruhan, G., Jeannet, M., and Steck, A. J. (1993). The T-lymphocyte response against myelin-associated glycoprotein and myelin basic protein in patients with multiple sclerosis. Neurology 43, 403–407. doi: 10.1212/wnl.43.2.403
Keywords: multiple sclerosis, etiopathogenesis, demyelination, autoimmunity, animal models
Citation: Titus HE, Chen Y, Podojil JR, Robinson AP, Balabanov R, Popko B and Miller SD (2020) Pre-clinical and Clinical Implications of “Inside-Out” vs. “Outside-In” Paradigms in Multiple Sclerosis Etiopathogenesis. Front. Cell. Neurosci. 14:599717. doi: 10.3389/fncel.2020.599717
Received: 27 August 2020; Accepted: 06 October 2020;
Published: 27 October 2020.
Edited by:
Antonio Luchicchi, VU University Medical Center, NetherlandsReviewed by:
Jason R. Plemel, University of Alberta, CanadaCopyright © 2020 Titus, Chen, Podojil, Robinson, Balabanov, Popko and Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen D. Miller, cy1kLW1pbGxlckBub3J0aHdlc3Rlcm4uZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.