 Mar Puigdellívol
Mar Puigdellívol David H. Allendorf
David H. Allendorf Guy C. Brown
Guy C. Brown- Department of Biochemistry, University of Cambridge, Cambridge, United Kingdom
Microglia are brain macrophages that mediate neuroinflammation and contribute to and protect against neurodegeneration. The terminal sugar residue of all glycoproteins and glycolipids on the surface of mammalian cells is normally sialic acid, and addition of this negatively charged residue is known as “sialylation,” whereas removal by sialidases is known as “desialylation.” High sialylation of the neuronal cell surface inhibits microglial phagocytosis of such neurons, via: (i) activating sialic acid receptors (Siglecs) on microglia that inhibit phagocytosis and (ii) inhibiting binding of opsonins C1q, C3, and galectin-3. Microglial sialylation inhibits inflammatory activation of microglia via: (i) activating Siglec receptors CD22 and CD33 on microglia that inhibit phagocytosis and (ii) inhibiting Toll-like receptor 4 (TLR4), complement receptor 3 (CR3), and other microglial receptors. When activated, microglia release a sialidase activity that desialylates both microglia and neurons, activating the microglia and rendering the neurons susceptible to phagocytosis. Activated microglia also release galectin-3 (Gal-3), which: (i) further activates microglia via binding to TLR4 and TREM2, (ii) binds to desialylated neurons opsonizing them for phagocytosis via Mer tyrosine kinase, and (iii) promotes Aβ aggregation and toxicity in vivo. Gal-3 and desialylation may increase in a variety of brain pathologies. Thus, Gal-3 and sialidases are potential treatment targets to prevent neuroinflammation and neurodegeneration.
Introduction
Microglia are brain macrophages that mediate neuroinflammation and can protect against neurodegeneration, for example, by removing protein aggregates, phagocytosing debris, and aiding repair. However, microglia can in some circumstances also contribute to neurodegeneration, for example, by mediating chronic neuroinflammation, or by excessive phagocytosis of synapses or neurons. This article reviews the roles of sialylation and galectin-3 (Gal-3) in microglia-mediated neuroinflammation and neurodegeneration. That means we will be reviewing the effects of changes in (a) sialylation of brain cells and (b) extracellular Gal-3 on microglial activation and neurodegeneration. In general, Gal-3 is upregulated and released by microglia during neuroinflammation and promotes neuroinflammation and phagocytosis (Shin, 2013; Chen et al., 2014). Conversely, sialylation of microglia and neurons inhibits neuroinflammation and phagocytosis, in part by blocking Gal-3 binding. Moreover, neuroinflammation can promote desialylation.
Microglia constantly survey the brain with their long and rapidly moving processes, looking for signs of pathogens, damage, or protein aggregates. If they detect such signs they become “activated,” i.e., they retract their processes, express inflammatory genes, produce reactive oxygen species, release chemokines and pro-inflammatory cytokines, upregulate phagocytosis, and may migrate toward the pathogens, damage, or aggregates. All of this helps the microglia phagocytose the pathogens, damage, or aggregates, and thereby remove the problem. However, if for whatever reason the pathogens, damage or aggregates are not effectively removed, then the microglia may become chronically activated, and this may result in damage to neurons, due to, for example, excessive cytokine production, excessive reactive oxygen species production, or excessive phagocytosis of synapses and neurons. Thus, chronic neuroinflammation may contribute to neurodegeneration (Ransohoff, 2016; Nichols et al., 2019).
Sialic Acid: Structure, Function, and Signaling
The terms sialylation and desialylation refer, respectively, to the addition and removal of the sugar sialic acid on the non-reducing termini of oligosaccharide chains attached to glycoproteins or glycolipids. In vertebrates, sialic acids are a heterogeneous family of nine-carbon monosaccharides with core structures consisting of either N-acetylneuraminic acid (Neu5Ac), N-glycolylneuraminic acid (Neu5Gc), or deaminoneuraminic acid (Kdn) (Figure 1A). In the human brain, the main sialic acid core structure is Neu5Ac, whereas levels of Neu5Gc and Kdn are very low (Chou et al., 1998; Inoue and Kitajima, 2006; Davies and Varki, 2015). The sialic acid core structures may be further modified by methylation, acetylation, and sulfation at the fourth, seventh, eighth, and ninth positions, generating more than 50 sialic acid species (Angata and Varki, 2002). Sialic acids are synthesized de-novo using the precursor N-acetylmannosamine-6-phosphate (ManNAc-6-P). ManNAc-6-P is the product of glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase (GNE), which is the rate-limiting enzyme for sialic acid synthesis (Keppler et al., 1999). After synthesis, sialic acid is activated by conversion to CMP-sialic acid by the nuclear enzyme CMP-sialic acid synthetase (CMAS) and then transported into the Golgi where it acts as a substrate for sialyl-transferases (STs; Munster-Kuhnel et al., 2004).
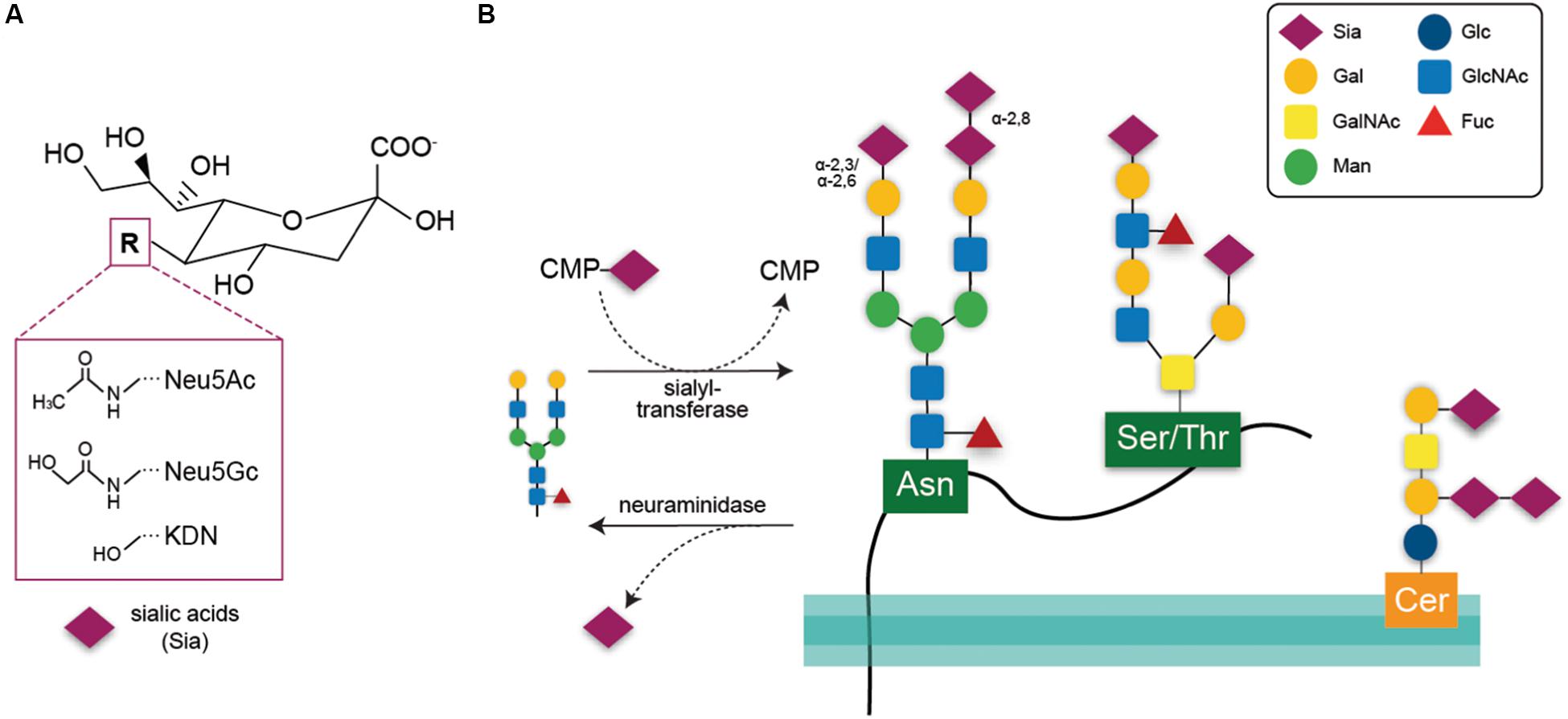
Figure 1. (A) Chemical structure of the three core sialic acids, N-acetylneuraminic acid (Neu5Ac), N-glycolylneuraminic acid (Neu5Gc), and deaminoneuraminic acid (KDN). These core structures may be further modified, e.g., by sulfation or methylation. (B) Sialic acids are transferred onto acceptor glycans via sialyl-transferase enzymes in the Golgi that use the activated cytidine-5’-monophosphate-sialic acid (CMP-Sia) as a donor molecule. Sialyl residues may be added terminally to galactose residues in α-2,3 or α-2,6 linkage or to sialic acid residues in α-2,8 linkage. Such glycan chains may be attached to glycoproteins via asparagine residue (N-glycan) or to serine or threonine residues (O-glycan). Glycosylation of lipids is exemplified here by a ganglioside composed of ceramide (Cer) to an oligosaccharide chain. Sialic acids may be released via hydrolytic enzymes termed neuraminidases. Gal, galactose; GalNAc, N-acetylgalactosamine; Man, mannose; Glc, glucose; GlcNAc, N-acetylglucosamine; Fuc, fucose.
Within the Golgi, sialic acids are attached to the sugars of glycolipids or glycoproteins (Figure 1B) via ST enzymes. In humans, 20 different enzymes transfer sialic acids to galactose (Gal) or N-acetylgalactosamine (GalNAc) acceptor sugars via α-2,3- or α-2,6-bonds (enzymes: ST3Gal, ST6Gal, ST6GalNAc) or to other sialic acid moieties via α-2,8 bonds (ST8Sia) (reviewed by Harduin-Lepers et al., 2001).
A small subset of glycoproteins, including particularly neural cell adhesion molecule (NCAM) are polysialylated, i.e., carry linear chains of 50–150 sialic acid residues linked α2,8, added by polysialyltransferases in the Golgi. Polysialylation is particularly abundant in the brain, and regulates cell adhesion, synaptogenesis, memory, and neurogenesis, as well as binding neurotrophins, growth factors, and neurotransmitters (Sato and Kitajima, 2013; Colley et al., 2014). In mouse brain, polysialylation dramatically decreases 2 weeks after birth, and almost disappears by 8 weeks, except in olfactory bulb, hippocampus, amygdala, suprachiasmatic nucleus, and prefrontal cortex (Abe et al., 2019). Polysialylated NCAM is also present on the surface of microglia, and rapidly decreases in response to LPS activation as a result of the microglial release of sialidase, which then desialylates NCAM (Sumida et al., 2015). In contrast, polysialylated neuropilin-2 is normally present within the microglial Golgi, but is rapidly released to the surface by LPS (Werneburg et al., 2015).
Gangliosides are lipids composed of a glycosphingolipid (ceramide and oligosaccharide) with one or more sialic acids. About 60 different gangliosides are known, which differ mainly in the position and number of sialic acid residues. The most abundant ganglioside in the brain is GM1, which is neuroprotective in multiple brain pathologies (Magistretti et al., 2019).
Sialic acid residues are negatively charged and are major contributors to the charge and hydrophilicity of the cellular surface, which reduces interactions and adhesion between cells (Varki, 1997; Byrne et al., 2007). Moreover, the presence of sialic acid on a glycoprotein or glycolipid may modulate cell signaling: sialic acids themselves serve as ligands for lectins such as selectins and sialic acid-binding immunoglobulin-type lectins (Siglecs). Siglecs form a family of transmembrane proteins that contain an extracellular carbohydrate recognition domain (CRD) that can bind sialic acid residues of the same or different cells, and a cytoplasmic tail that (in most Siglecs) contains an immunoreceptor tyrosine-based inhibition motif (ITIM) domain. Upon binding of the CRD to sialyl residues, the ITIM domain is phosphorylated and recruits and activates protein tyrosine phosphatases, such as Src homology domain-containing phosphatase-1 (SHP-1). These phosphatases reverse the tyrosine phosphorylation of signaling proteins, such as Syk, induced by activating receptors. Thus, Siglecs and the sialylation state of self- or target cells’ glycocalyx act as important negative regulators of cellular activation and phagocytosis (Varki and Angata, 2006). Siglecs are typically expressed by hematopoietic cells (Von and Bochner, 2008) and, in the CNS, are mainly expressed by microglia. Structure, function, and signaling properties of microglial Siglecs (Siglec-1, Siglec-2/CD22, Siglec-3/CD33, Siglec-4/MAG, Siglec-11, and Siglec-E/F/H) have been extensively reviewed elsewhere (Linnartz-Gerlach et al., 2014; Siddiqui et al., 2019).
Some Siglecs have been subject to rapid evolution, and are poorly conserved, potentially resulting in divergent functions. For example, human and mouse CD33 have limited homology in their intracellular domains, such that human CD33 has an ITIM and an ITIM-like domain, whereas mouse CD33 only has an ITIM-like domain (Brinkman-Van der Linden et al., 2003). Thus, human CD33 has been found to inhibit monocyte and microglial phagocytosis, whereas mouse CD33 has been reported to have no effect on phagocytosis (Bhattacherjee et al., 2019).
Desialylation describes the removal of sialyl-residues from glycoconjugates, which is generally mediated by hydrolytic enzymes called sialidases or neuraminidases (Neu) (these terms are synonymous) (Wei and Wang, 2019). Mammalian cells express four different sialidases, Neu1-4. Neu1 is found either in the lysosome or on the surface of plasma membrane and is one of the main enzymes degrading sialo-glycoproteins. Neu1 has the highest expression of the sialidases in human tissue, and may cleave sialic acids linked in α-2,3 and—to a lesser degree—α-2,6 and α-2,8 (Miyagi and Tsuiki, 1984). The cytosolic Neu2 sialidase is expressed at a very low level in humans, and cleaves sialic acids from glycoproteins and glycolipids with a similar linkage-specificity to Neu1 (Miyagi and Tsuiki, 1985). Neu3 is found on the plasma membrane, and has the highest specificity for sialylated gangliosides, cleaving α-2,3, α-2,6, and α-2,8 linked sialic acids equally. Neu4 is found on internal membranes, and has preference for gangliosides (Miyagi and Yamaguchi, 2012; Pshezhetsky and Ashmarina, 2018). Notably, loss of function mutations or knock out of sialidase enzymes gives rise to a variety of brain pathologies in humans and mice. Thus, mutations in the human neuraminidase 1 gene cause sialidosis characterized by the accumulation of sialylated glycans in lysosomes. Brain pathologies associated with this severe lysosomal storage disorder are ataxia, mental retardation, and seizures (Seyrantepe et al., 2003). Double knockout of Neu3 and Neu4 in mice caused accumulation of the GM3 ganglioside inside CNS cells, such as microglia and neurons, causing neuroinflammation, impairment of neurite formation, and memory loss (Pan et al., 2017).
Sialidases are important for degradation of glycoproteins and glycolipids, as well as the recycling of sialic acid. However, the removal of sialic acid residues from the cell surface by sialidases may also trigger cell signaling events as this desialylation: (i) reduces Siglec signaling, (ii) activates other receptors, (iii) decreases some gangliosides and increases others, and (iv) changes binding of lectins, opsonins, and complement. Multiple receptors, such as Toll-like receptor 4 (TLR4), can be activated by desialylation (Pshezhetsky and Ashmarina, 2013). Some lectins, such as Gal-3, are released by inflammatory activated microglia and bind to N-acetyl-lactosamine residues of glycoproteins only when terminal sialic acid residues are removed (Nomura et al., 2017). The removal of sialic acids from the neuronal cell surface encourages binding of the classical complement cascade proteins C1q and C3, which are important opsonins tagging cells for phagocytosis (Linnartz et al., 2012). Inhibition of complement by cell surface sialylation may be mainly mediated by complement factor H, a serum protein recruited to cells by binding sialic acid residues. When recruited by cell surface sialylation, factor H acts as a negative regulator of complement depositions by promoting degradation of C3 convertase and already deposited C3b (Blaum et al., 2015). Mutations in the factor H sialyl-recognition domain can cause atypical hemolytic uremic syndrome (aHUS), a disorder characterized by increased C3b deposition and lysis of blood cells (Hyvarinen et al., 2016) indicating that the factor H-sialic acid interaction is a key regulator of the alternative pathway of complement activation. Therefore, desialylation may promote opsonization of cells by increased binding and decreased degradation of classical complement proteins.
In the following sections, we will use the terms “sialylated” or “desialylated” in a simplified manner to describe the presence or absence, respectively, of sialyl residues on surface glycans, irrespective of linkage or modifications.
Effects of Desialylation/Sialylation in the Brain
Sialic acids are particularly abundant in the brain, including within neuronal gangliosides and as polysialic acid on NCAM (Finne et al., 1983; Pan et al., 2005). Polysialylation of NCAM on neurons regulates neurite outgrowth (Landmesser et al., 1990), axon pathfinding (Tang et al., 1994), synaptogenesis (Dityatev et al., 2004), and long-term potentiation (LTP) in the hippocampus (Becker et al., 1996; Muller et al., 1996; Senkov et al., 2012; Hildebrandt and Dityatev, 2015). Acute stress has been shown to rapidly decrease polysialylation in olfactory bulb and prefrontal cortex in mice, apparently due to sialidases from microglia and astrocytes (Abe et al., 2019). Neural activity has been shown to rapidly increase neuronal (and astrocytic) surface sialidase activity, causing neuronal desialylation, which in turn modifies memory formation (Minami et al., 2016, 2017). Thus, rapid changes in cell-surface sialylation may occur physiologically, usually as a result of transfer of Neu1 or Neu4 to the cell surface, and more dramatic changes may occur as a result of neuroinflammation (see below).
Although sialylation has been found to be dispensable for germ layer formation and early development of the embryo (Abeln et al., 2017), several studies have shown the relevance of sialylation during mammalian development, as demonstrated by the fact that homozygous knockout of either (1) GNE, necessary for sialic acid biosynthesis (Schwarzkopf et al., 2002) or (2) CMAS, necessary for sialic acid activation (Abeln et al., 2019) is embryonically lethal in mice. Interestingly, the phenotype exhibited by CMAS knockout mice was rescued by depleting maternal component C3, indicating that sialylation protects the embryo against attack by maternal complement (Abeln et al., 2019).
In contrast to homozygous GNE knockout mice, heterozygous GNE knockout is not lethal in mice, and reduces sialylation by roughly 50%, and so can be used to investigate the effects of reduced sialylation in mice (Klaus et al., 2020). These mice display microglial activation and neuronal synaptic loss, followed by a slow increase in age-related and complement-dependent neuronal loss, indicating a protective role of sialic acids against microglial phagocytosis and physiological aging in the mouse brain (Klaus et al., 2020). Interestingly, loss of synapses precedes neuronal loss in several neurodegenerative diseases (Fricker et al., 2018; Rajendran and Paolicelli, 2018) and complement-mediated microglial phagocytosis of synapses is aberrantly activated and contributes to synaptic loss in mouse models of Alzheimer’s disease (AD) (Stevens et al., 2007; Schafer and Stevens, 2010; Hong et al., 2016). However, these studies did not determine how complement is binding to synapses during development or neurodegeneration. One intriguing possibility is that complement is binding to synapses because they are desialylated (Figure 2). Indeed, it has been shown that desialylation of neurons facilitates C1q and C3b binding detected by complement receptor 3 (CR3) on microglia surface, which leads to phagocytosis of neuronal dendrites (Linnartz et al., 2012). Thus, there is in vitro and in vivo evidence that desialylation of neurons or neuronal parts can cause complement-mediated microglial phagocytosis of those neurons, synapses, or dendrites.
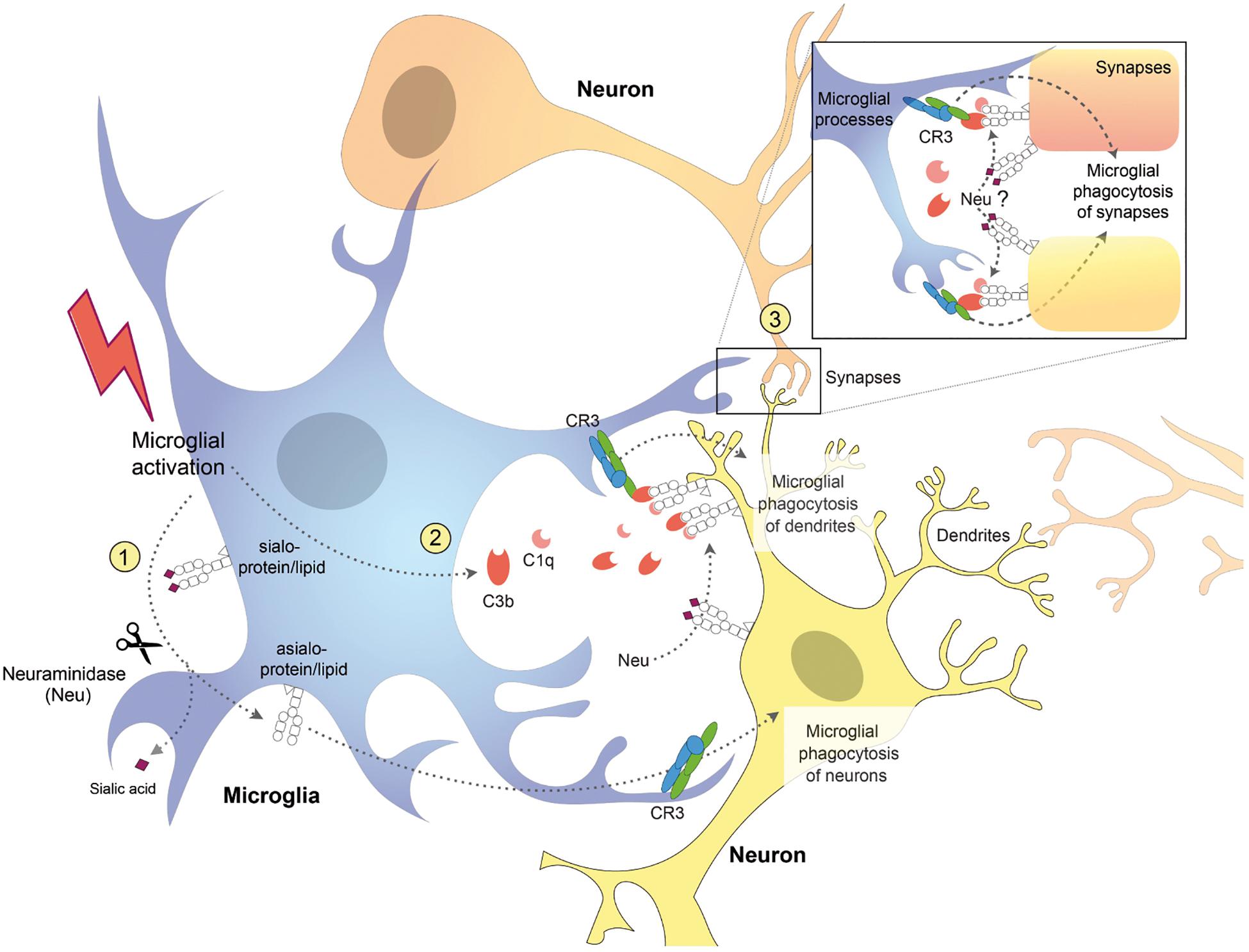
Figure 2. Schematic diagram showing potential mechanisms for complement receptor 3 (CR3)-dependent microglial phagocytosis of neurons, dendrites, and synapses. Activated microglia (1) desialylate their surface via neuraminidase (Neu) which stimulates microglial phagocytosis of neurons via CR3 (Allendorf et al., 2020b) and (2) release complement proteins C1q and C3b, which opsonize desialylated neuronal dendrites and (3) synapses, stimulating their phagocytosis via microglial CR3 (Linnartz et al., 2012). Neuraminidase released from microglia or onto the surface of neurons, desialylates the neuronal surface, and promotes binding of C1q and C3b, stimulating microglial phagocytosis of neurons, dendrites, and synapses.
We have recently found that different stimuli, including LPS, fibrillar amyloid beta (Aβ) and TAU, induced desialylation of the microglial surface (Allendorf et al., 2020b). This desialylation of microglia in turn enhanced microglial phagocytosis via activating CR3, and induced microglia to phagocytose healthy neurons (Figure 2). Addition of LPS or Aβ to glial-neuronal cultures induced neuronal loss that could be blocked by inhibiting sialidases or CR3 (Allendorf et al., 2020b). This suggests that inflammatory stimuli can induce desialylation of microglia, which enhances phagocytosis that may contribute to neurodegeneration. Recent studies suggest that removal of sialyl residues from the microglial cell surface may also activate TLR-mediated signaling. Intracerebral injection of microbial sialidase caused microglial TLR4 and TLR2 activation in vivo (Fernandez-Arjona et al., 2019) and in vitro (Fernandez-Arjona et al., 2019; Allendorf et al., 2020a). Moreover, we found in the BV-2 microglial cell line that LPS causes Neu1 to translocate to the cell surface, where it desialylates TLR4, which enhances and prolongs microglial activation (Allendorf et al., 2020a). We previously reported that LPS-activated BV2 microglia released a sialidase activity that could desialylate neighboring cells (Nomura et al., 2017). Similarly, Sumida et al. (2015) reported that in the Ra2 microglial cell line, LPS caused a rapid and reversible release of a sialidase activity on exovesicles, which removed polysialic acids from the microglial surface. These studies suggest that activated microglia have the potential to desialylate both themselves and surrounding neurons. In vivo, it was shown that LPS injection into the corpus callosum of rat pups induced a dramatic increase in neuraminidase activity (Neu1 and Neu4) leading to a persistent desialylation of glycoproteins and neurons (Demina et al., 2018). Although the authors found that the brain distributions of microglia and neuraminidase activity were different, this does not rule out that neuraminidase activity is being released from microglia (or other cells). Importantly, what this work shows is that neuroinflammation (induced by LPS) can cause desialylation of the brain.
Sialyltransferases in Neuropathology
In humans, mutations in the ganglioside-specific sialyltransferase ST3Gal5 (GM3 synthase) gene cause: infantile-onset epilepsy syndrome (Simpson et al., 2004) and salt and pepper syndrome, with severe intellectual disability (Boccuto et al., 2014). A mutation in another ganglioside-selective biosynthetic gene, B4GALNT1 (GM2 synthase), result in the presence of progressive motor neuropathy accompanied also by cognitive deficits (Harlalka et al., 2013). Consistent with human studies, mice lacking GM2 synthase have been found to develop progressive motor deficits (Chiavegatto et al., 2000). Mutations in the sialyltransferase ST3Gal3 cause: non-syndromic autosomal recessive intellectual disability (Hu et al., 2011) or West syndrome, an age-dependent epileptic encephalopathic syndrome (Edvardson et al., 2013). ST3Gal2/3 double-null mice had decreased myelin, reduced neuronal marker expression, abnormal dendrites, and exhibited cognitive and motor coordination deficits (Yoo et al., 2015). Mice lacking complex gangliosides also exhibited demyelination and axonal degeneration (Sheikh et al., 1999). Interestingly, neuronal, but not glial, expression of complex gangliosides was sufficient to prevent age-dependent degenerative phenotype in mice (Yao et al., 2014). This all suggests that gangliosides maintain myelin and axons but is also compatible with the idea of sialylation preventing microglial activation and microglial phagocytosis of neurons and synapses (Figure 2).
GWAS and other studies have found a positive association between polysialyltransferases and schizophrenia (Maziade et al., 2005; Arai et al., 2006; Tao et al., 2007), psychotic and mood disorders (McAuley et al., 2012), and autism spectrum disorders (Zhiling et al., 2008; Anney et al., 2010). Changes in polysialylation of NCAM1 may also contribute to Parkinson’s disease and AD (Mikkonen et al., 1999; Oizumi et al., 2008; Murray et al., 2016, 2018). A variety of mechanisms by which changes in polysialic acid may contribute to disease have been suggested, including changes in cell–cell interactions, ion channels, neurotrophins (BDNF, NT3, and NGF), neurotransmitters, and growth factors (Sato and Kitajima, 2013). However, the findings are also compatible with the idea that polysialylation inhibits microglial activation and microglial phagocytosis of neurons and synapses, and the reduced polysialylation seen in these neuropathologies induces excessive microglial activation and phagocytosis.
Siglec Receptors in Pathological Processes
Sialic acid-binding Ig-like lectin (Siglec) receptors have been linked to neurodegenerative and aging processes (extensively reviewed in Siddiqui et al., 2019; Duan and Paulson, 2020). Siglec receptors on microglia can be activated by sialic acid residues present on the neuronal surface, which inhibit microglial phagocytosis, while desialylation of neurons leads to microglia phagocytosis of the desialylated neurons or dendrites. One abundant inhibitory Siglec receptor expressed on microglia of the human brain is Siglec-11 (Angata et al., 2002) which has been found to inhibit microglia neurotoxicity upon interaction with sialic acids on the neuronal glycocalyx (Linnartz et al., 2010). Siglec-11 expression in microglia suppressed cytokine release and microglial phagocytosis of polysialylated neurons and neurites (Wang and Neumann, 2010). Interestingly, although both microglia and neurons expressed polysialylated NCAM, only the latter appeared to activate the Siglec-11-mediated neuroprotection.
In mice, Siglec-E recognizes sialic acid residues on the neuronal glycocalyx and has been shown to act as a negative regulator of phagocytosis of neuronal debris and the associated production of superoxide radicals (Claude et al., 2013).
Human microglia abundantly express another inhibitory Siglec receptor, Siglec-3, also called CD33. Several GWAS studies have indicated that CD33 is a risk factor for AD (Bertram et al., 2008; Hollingworth et al., 2011; Naj et al., 2011; Walker et al., 2015). The normal, full-length form of CD33 inhibits microglial phagocytosis of Aβ, while the short form of CD33, lacking exon 2 encoding the sialic acid ligand-binding domain, does not inhibit phagocytosis of Aβ, and may thereby reduce AD risk (Bradshaw et al., 2013; Griciuc et al., 2013; Malik et al., 2013; Raj et al., 2014; Siddiqui et al., 2017; Estus et al., 2019). CD33 is thought to be activated by sialic acid residues on the same cell rather than adjacent cells, so desialylation of microglia may activate microglial phagocytosis in part by removing the CD33 block on phagocytosis.
Interestingly, Siglec-2, also known as CD22, has also been identified as a negative regulator of microglial phagocytosis in the aging brain (Pluvinage et al., 2019). Knockout of CD22 in BV-2 microglia increased phagocytosis, and microglial sialylation inhibited phagocytosis partly by activating CD22 (Pluvinage et al., 2019). Importantly, the authors found that inhibition of CD22 on aged microglia in vivo facilitates the clearance of myelin debris, amyloid-β oligomers, and α-synuclein fibrils. Long-term inhibition of CD22 partially restores the transcriptional state of aged microglia to a younger homeostatic state and improves cognitive function in aged mice. Importantly, CD22 is upregulated not only in aging brains but also in brains of AD (Friedman et al., 2018) amyotrophic lateral sclerosis (Funikov et al., 2018), and Niemann–Pick type C (Cougnoux et al., 2018). Thus, CD22, as well as CD33 and Siglec-11, are potential therapeutic targets to modify neuroinflammation and neurodegeneration.
Importantly, most human Siglecs have undergone rapid, recent evolution, such that there are no clear orthologs between humans and mice, and there are significant differences in ligand specificity (Linnartz-Gerlach et al., 2014). Moreover, while the above Siglec receptors (Siglec-11, CD33) are abundantly expressed on human microglia, mouse microglia abundantly express others, including CD33-related Siglec-E and CD22 (extensively reviewed in Duan and Paulson, 2020). Thus, using mouse models to study the roles of Siglec receptors in human physiology or disease is not always appropriate. Nonetheless, the above studies confirm the functional role of sialic acids in the brain and encourage future studies aiming to investigate the potential of modulating Siglec expression/function on microglia as new therapeutic strategies to delay or prevent neurodegeneration and age-dependent cognitive deficits.
Galectin-3
Galectin-3 is one of the 14 known mammalian galectins, which are lectins (sugar-binding proteins) binding to galactose residues. Gal-3 has a C-terminal carbohydrate-recognition domain that preferentially binds to N-acetyl-lactosamine (a disaccharide of galactose and N-acetyl-glucosamine) residues in glycoproteins or glycolipids (Hirabayashi et al., 2002). Gal-3 is normally a soluble monomer, but when the C-terminal binds N-acetyl-lactosamine on glycoproteins or glycolipids, then the N-terminal oligomerizes with the N-terminal of other Gal-3 units bound to sugars, to form pentamers cross-linking the glycoproteins or glycolipids (Ahmad et al., 2004). Oligomerization of Gal-3 appears to be mediated by liquid-liquid phase separation of aromatic residues in the N-terminal (Chiu et al., 2020). N-acetyl-lactosamine is found in the sugar chains of most glycoproteins or glycolipids; however, binding is normally blocked by a terminal sialic acid residue α2,6-linked to the galactose residue. Thus, Gal-3 binding to cell surface glycoproteins, including receptors, is normally blocked by sialylation of those glycoproteins, and is revealed by desialylation (Zhuo and Bellis, 2011; Nomura et al., 2017) (Figure 3).
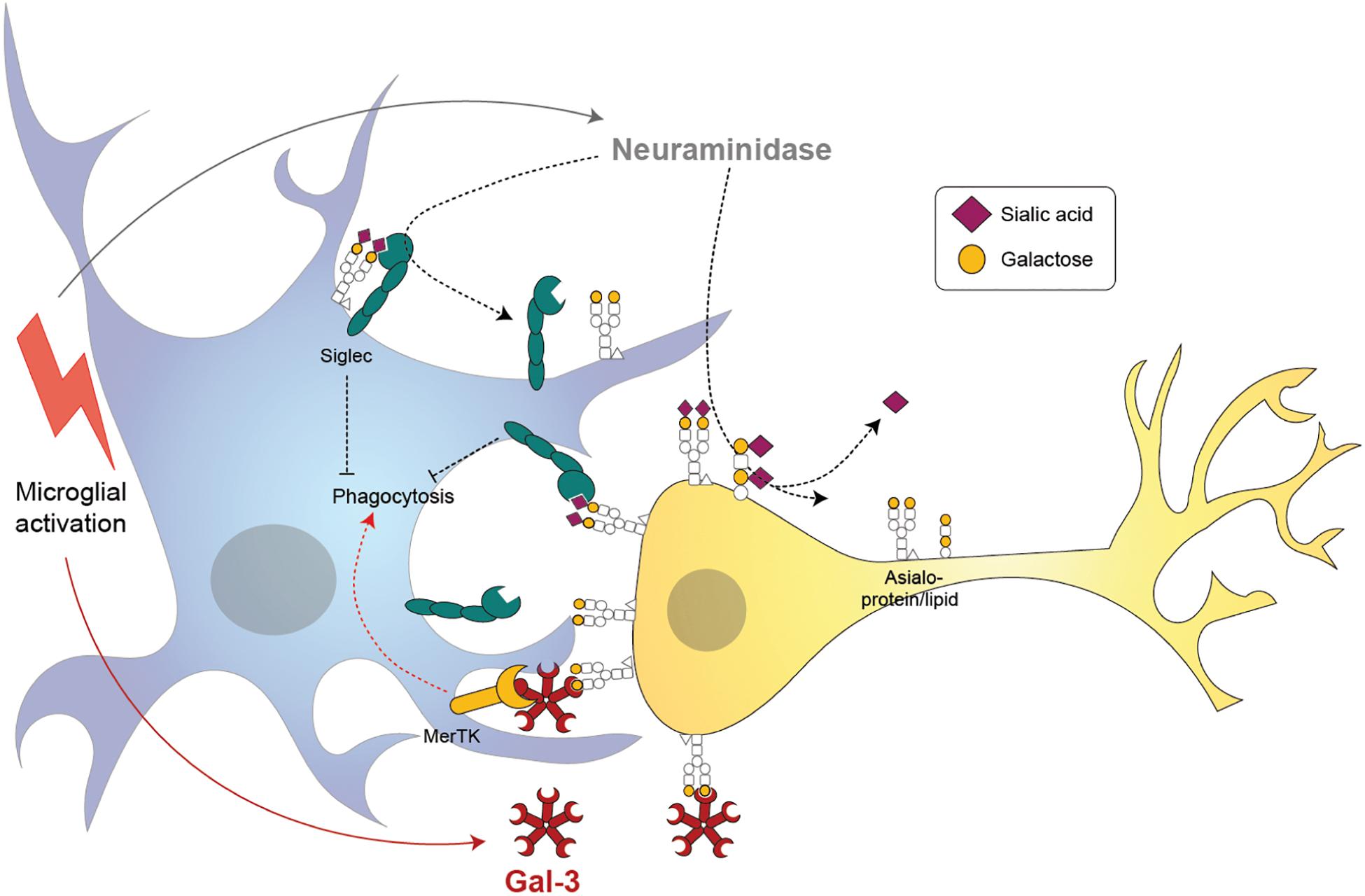
Figure 3. Proposed mechanisms of microglia-induced neurophagy via galectin-3 (Gal-3) and desialylation of microglial and neuronal receptors. Microglia release a neuraminidase after inflammatory activation (i.e., by lipopolysaccharide) that removes sialyl residues on the microglia and surrounding neurons. Desialylation reduces binding of sialic acid binding immunoglobuline-like receptors (Siglecs) in cis and trans, thus potentially increasing microglial phagocytosis. Activated microglia release the soluble lectin Gal-3 which binds to the penultimate sugar on glycan chains, galactose. Gal-3 opsonizes desialylated neurons and induces phagocytosis by microglia via Mer tyrosine kinase (MerTK).
Galectin-3 is highly expressed in myeloid, epithelial and endothelial cells, and fibroblasts. Within the cell, Gal-3 can be found in cytoplasm, nucleus, and membranes, and it can be released from cells following inflammatory stimuli such as LPS. Within the brain, Gal-3 is expressed by microglia, some astrocytes, and weakly by some cortical neurons (Yoo et al., 2017). Gal-3 expression is upregulated in neurodegenerative disease models (Siew et al., 2019; Boza-Serrano et al., 2019). One variant of the Gal-3 gene (LGALS3) is associated with Parkinson’s disease risk at p = 9 × 10–15 (Pickrell et al., 2016) and 4 × 10–16 (Chang et al., 2017). Gal-3 gene variants are also weakly associated with AD (Boza-Serrano et al., 2019).
We and others have shown that LPS-activated microglia release Gal-3 (Burguillos et al., 2015; Nomura et al., 2017). Gal-3 lacks an endoplasmic reticulum-targeting sequence, and therefore does not follow the classical pathway via endoplasmic reticulum and Golgi out of the cell. The mechanism of Gal-3 release from the cytoplasm is unclear, but it appears to be triggered by a rise in cytosolic calcium (Liu et al., 1995). We found that inhibition of calcineurin (a calcium-activated protein phosphatase) blocked LPS-induced Gal-3 release from microglia, suggesting the possibility that dephosphorylation of Gal-3 regulates its release (Nomura et al., 2017). Gal-3 can be phosphorylated on Ser6 and Ser12, which regulates localization and oligomerization. Extracellular Gal-3 can bind to multiple components of the extracellular matrix, and mediate interactions between cells and the extracellular matrix (Dumic et al., 2006).
Extracellular or intracellular Gal-3 levels are elevated in a variety of pathologies, potentially due to neuroinflammation. Extracellular Gal-3 levels were higher in CSF of AD patients (Ashraf and Baeesa, 2018). Amyotrophic lateral sclerosis patients (Ashraf and Baeesa, 2018) after birth asphyxia in humans (Savman et al., 2013) and after brain trauma in mice (Yip et al., 2017). Brain levels of Gal-3 were higher after brain ischemia (Yan et al., 2009) and in Huntington’s disease patients and a mouse model of Huntington’s disease (Siew et al., 2019).
Galectin-3 promotes neuroinflammation by multiple mechanisms (Shin, 2013; Chen et al., 2014). Extracellular Gal-3 can activate microglia apparently by directly activating TLR4 (Burguillos et al., 2015) and can thereby induce neuroinflammation after brain trauma (Yip et al., 2017) and brain ischemia (Rahimian et al., 2018). Consequently, lack of Gal-3 attenuates neuroinflammation, for example, in the retina and optic nerve of diabetic mice (Mendonca et al., 2018). Gal-3 knockout reduced microglial activation in response to brain ischemia in mice (Lalancette-Hebert et al., 2012). And Gal-3 knockout or inhibition reduced microglial activation in response to α-synuclein in culture (Boza-Serrano et al., 2019).
Extracellular Gal-3 can act as an opsonin (Nomura et al., 2017), i.e., it can bind to a cell’s surface and then induce phagocytes to phagocytose that cell by also binding and activating a phagocytic receptor on the phagocyte (Figure 3). Gal-3 binds and activates the phagocytic receptor MerTK (Caberoy et al., 2012) thereby inducing the phagocytosis of Gal-3 opsonized cells, debris, and aggregates by MerTK-expressing phagocytes (Nomura et al., 2017). Moreover, Gal-3 can bind to bacteria and therefore opsonizes bacteria for phagocytosis by microglia (Abreu et al., 2017; Cockram et al., 2019). Extracellular Gal-3 binds to desialylated cells and therefore opsonizes desialylated cells for phagocytosis (Nomura et al., 2017). Gal-3 is released after brain trauma in mice, and increases subsequent neuronal loss, so Gal-3 knockout or anti-Gal-3 antibodies reduce neuronal loss and brain damage (Yip et al., 2017). Similarly, Gal-3 increases after neonatal brain ischemia, and Gal-3–knockout mice are protected from the subsequent neuronal loss (Doverhag et al., 2010). Again, in optic nerve injury, Gal-3 knockout reduced retinal ganglion cell loss (Abreu et al., 2017). In each case, extracellular Gal-3 might promote neuronal loss by opsonizing neurons or by activating microglia—it is not clear which.
Galectin-3 enhances microglial phagocytosis of myelin, which may contribute to myelin-debris clearance (Rotshenker et al., 2008; Rotshenker, 2009). By contrast, extracellular Gal-3, released from microglia, can promote oligodendrocyte differentiation, so Gal-3 knockout mice have reduced axon myelination (Pasquini et al., 2011; Thomas and Pasquini, 2018). However, Gal-3 promotes neuroinflammation in experimental autoimmune encephalomyelitis (EAE) mouse models of multiple sclerosis (MS), so Gal-3 knockout reduces the severity of EAE (Jiang et al., 2009).
We found that Gal-3 was highly upregulated in the brains of AD patients and 5xFAD mice, a mouse model of AD, and this increase was found specifically in the microglia associated with amyloid plaques (Boza-Serrano et al., 2019). Importantly, Gal-3 knockout in 5xFAD mice display reduced microglial expression of pro-inflammatory genes in vivo, decreased amyloid plaques, and improved cognitive performance (Boza-Serrano et al., 2019). In agreement with its detrimental role in AD, co-injection of Gal-3 and Aβ was found to increase amyloid plaque deposition. Gal-3 associated with microglial TREM2 in vivo and bound to pure TREM2 in vitro (Boza-Serrano et al., 2019). Tao et al. (2020) found remarkably similar results: Gal-3 expression was upregulated in the brains of AD patients and APP/PS1 mice, another mouse model of AD; Gal-3 promoted Aβ oligomerization and fibrilization; Gal-3 knockout in APP/PS1 mice reduced amyloid plaque formation and decreased cognitive deficits; and Gal-3 bound microglial TREM2 to induce microglial activation and further Gal-3 expression. Thus, it appears clear that Gal-3 promotes amyloid pathology by amyloid aggregation, whether this has anything to do with the binding to TREM2 is less clear.
Conclusion
Overall, these studies indicate that Gal-3 and sialylation/desialylation are important for neuroinflammation and potentially neurodegeneration. However, several questions remain open, and require further research. 1. Does desialylation of microglia, neurons, or synapses occur in neurodegeneration, and if so, when, and where? 2. Is complement deposition and microglial phagocytosis of synapses mediated by desialylation of synapses? 3. Does inhibition of extracellular sialidases prevent neurodegeneration? 4. Does inhibition of Gal-3 prevent neurodegeneration?
Author Contributions
The authors contributed equally to writing this review.
Funding
This work was supported by the Medical Research Council UK (MR/L010593) and the Innovative Medicines Initiative 2 Joint Undertaking under Grant Agreement No. 115976 (PHAGO consortium).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abe, C., Yi, Y., Hane, M., Kitajima, K., and Sato, C. (2019). Acute stress-induced change in polysialic acid levels mediated by sialidase in mouse brain. Sci. Rep. 9:9950. doi: 10.1038/s41598-019-46240-6
Abeln, M., Albers, I., Peters-Bernard, U., Flachsig-Schulz, K., Kats, E., Kispert, A., et al. (2019). Sialic acid is a critical fetal defense against maternal complement attack. J. Clin. Invest. 129, 422–436. doi: 10.1172/JCI99945
Abeln, M., Borst, K. M., Cajic, S., Thiesler, H., Kats, E., Albers, I., et al. (2017). Sialylation is dispensable for early murine embryonic development in vitro. Chembiochem 18, 1305–1316. doi: 10.1002/cbic.201700083
Abreu, C. A., De Lima, S. V., Mendonca, H. R., Goulart, C. O., and Martinez, A. M. (2017). Absence of galectin-3 promotes neuroprotection in retinal ganglion cells after optic nerve injury. Histol. Histopathol. 32, 253–262. doi: 10.14670/HH-11-788
Ahmad, N., Gabius, H. J., Andre, S., Kaltner, H., Sabesan, S., Roy, R., et al. (2004). Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. J. Biol. Chem. 279, 10841–10847. doi: 10.1074/jbc.m312834200
Allendorf, D. H., Franssen, E. H., and Brown, G. C. (2020a). Lipopolysaccharide activates microglia via neuraminidase 1 desialylation of Toll-like Receptor 4. J Neurochem. doi: 10.1111/jnc.15024 [Epub ahead of print].
Allendorf, D. H., Puigdellivol, M., and Brown, G. C. (2020b). Activated microglia desialylate their surface, stimulating complement receptor 3-mediated phagocytosis of neurons. Glia 68, 989–998. doi: 10.1002/glia.23757
Angata, T., Kerr, S. C., Greaves, D. R., Varki, N. M., Crocker, P. R., and Varki, A. (2002). Cloning and characterization of human Siglec-11. A recently evolved signaling molecule that can interact with SHP-1 and SHP-2 and is expressed by tissue macrophages, including brain microglia. J. Biol. Chem. 277, 24466–24474. doi: 10.1074/jbc.m202833200
Angata, T., and Varki, A. (2002). Chemical diversity in the sialic acids and related alpha-keto acids: an evolutionary perspective. Chem. Rev. 102, 439–469.
Anney, R., Klei, L., Pinto, D., Regan, R., Conroy, J., Magalhaes, T. R., et al. (2010). A genome-wide scan for common alleles affecting risk for autism. Hum. Mol. Genet. 19, 4072–4082. doi: 10.1093/hmg/ddq307
Arai, M., Yamada, K., Toyota, T., Obata, N., Haga, S., Yoshida, Y., et al. (2006). Association between polymorphisms in the promoter region of the sialyltransferase 8B (SIAT8B) gene and schizophrenia. Biol. Psychiatry 59, 652–659. doi: 10.1016/j.biopsych.2005.08.016
Ashraf, G. M., and Baeesa, S. S. (2018). Investigation of Gal-3 expression pattern in serum and cerebrospinal fluid of patients suffering from neurodegenerative disorders. Front. Neurosci. 12:430. doi: 10.3389/fnins.2018.00430
Becker, C. G., Artola, A., Gerardy-Schahn, R., Becker, T., Welzl, H., and Schachner, M. (1996). The polysialic acid modification of the neural cell adhesion molecule is involved in spatial learning and hippocampal long-term potentiation. J. Neurosci. Res. 45, 143–152. doi: 10.1002/(sici)1097-4547(19960715)45:2<143::aid-jnr6>3.0.co;2-a
Bertram, L., Lange, C., Mullin, K., Parkinson, M., Hsiao, M., Hogan, M. F., et al. (2008). Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 83, 623–632. doi: 10.1016/j.ajhg.2008.10.008
Bhattacherjee, A., Rodrigues, E., Jung, J., Luzentales-Simpson, M., Enterina, J. R., Galleguillos, D., et al. (2019). Repression of phagocytosis by human CD33 is not conserved with mouse CD33. Commun. Biol. 2:450. doi: 10.1038/s42003-019-0698-6
Blaum, B. S., Hannan, J. P., Herbert, A. P., Kavanagh, D., Uhrin, D., and Stehle, T. (2015). Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat. Chem. Biol. 11, 77–82. doi: 10.1038/nchembio.1696
Boccuto, L., Aoki, K., Flanagan-Steet, H., Chen, C. F., Fan, X., Bartel, F., et al. (2014). A mutation in a ganglioside biosynthetic enzyme, ST3GAL5, results in salt & pepper syndrome, a neurocutaneous disorder with altered glycolipid and glycoprotein glycosylation. Hum. Mol. Genet. 23, 418–433. doi: 10.1093/hmg/ddt434
Boza-Serrano, A., Ruiz, R., Sanchez-Varo, R., García-Revilla, J., Yang, Y., Jimenez-Ferrer, I., et al. (2019). Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 138, 251–273. doi: 10.1007/s00401-019-02013-z
Bradshaw, E. M., Chibnik, L. B., Keenan, B. T., Ottoboni, L., Raj, T., Tang, A., et al. (2013). CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat. Neurosci. 16, 848–850. doi: 10.1038/nn.3435
Brinkman-Van der Linden, E. C., Angata, T., Reynolds, S. A., Powell, L. D., Hedrick, S. M., and Varki, A. (2003). CD33/Siglec-3 binding specificity, expression pattern, and consequences of gene deletion in mice. Mol. Cell. Biol. 23, 4199–4206. doi: 10.1128/mcb.23.12.4199-4206.2003
Burguillos, M. A., Svensson, M., Schulte, T., Boza-Serrano, A., Garcia-Quintanilla, A., Kavanagh, E., et al. (2015). Microglia-secreted galectin-3 acts as a toll-like receptor 4 Ligand and contributes to microglial activation. Cell Rep. 10, 1626–1638. doi: 10.1016/j.celrep.2015.02.012
Byrne, B., Donohoe, G. G., and O’Kennedy, R. (2007). Sialic acids: carbohydrate moieties that influence the biological and physical properties of biopharmaceutical proteins and living cells. Drug Discov. Today 12, 319–326. doi: 10.1016/j.drudis.2007.02.010
Caberoy, N. B., Alvarado, G., Bigcas, J. L., and Li, W. (2012). Galectin-3 is a new MerTK-specific eat-me signal. J. Cell. Physiol. 227, 401–407. doi: 10.1002/jcp.22955
Chang, D., Nalls, M. A., Hallgrimsdottir, I. B., Hunkapiller, J., van der Brug, M., Cai, F., et al. (2017). A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 49, 1511–1516. doi: 10.1038/ng.3955
Chen, H. L., Liao, F., Lin, T. N., and Liu, F. T. (2014). Galectins and neuroinflammation. Adv. Neurobiol. 9, 517–542. doi: 10.1007/978-1-4939-1154-7_24
Chiavegatto, S., Sun, J., Nelson, R. J., and Schnaar, R. L. (2000). A functional role for complex gangliosides: motor deficits in GM2/GD2 synthase knockout mice. Exp. Neurol. 166, 227–234. doi: 10.1006/exnr.2000.7504
Chiu, Y. P., Sun, Y. C., Qiu, D. C., Lin, Y. H., Chen, Y. Q., Kuo, J. C., et al. (2020). Liquid-liquid phase separation and extracellular multivalent interactions in the tale of galectin-3. Nat. Commun. 11:1229. doi: 10.1038/s41467-020-15007-3
Chou, H. H., Takematsu, H., Diaz, S., Iber, J., Nickerson, E., Wright, K. L., et al. (1998). A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc. Natl. Acad. Sci. U.S.A. 95, 11751–11756. doi: 10.1073/pnas.95.20.11751
Claude, J., Linnartz-Gerlach, B., Kudin, A. P., Kunz, W. S., and Neumann, H. (2013). Microglial CD33-related Siglec-E inhibits neurotoxicity by preventing the phagocytosis-associated oxidative burst. J. Neurosci. 33, 18270–18276. doi: 10.1523/JNEUROSCI.2211-13.2013
Cockram, T. O. J., Puigdellivol, M., and Brown, G. C. (2019). Calreticulin and galectin-3 opsonise bacteria for phagocytosis by microglia. Front. Immunol. 10:2647. doi: 10.3389/fimmu.2019.02647
Colley, K. J., Kitajima, K., and Sato, C. (2014). Polysialic acid: biosynthesis, novel functions and applications. Crit. Rev. Biochem. Mol. Biol. 49, 498–532. doi: 10.3109/10409238.2014.976606
Cougnoux, A., Drummond, R. A., Collar, A. L., Iben, J. R., Salman, A., Westgarth, H., et al. (2018). Microglia activation in Niemann-Pick disease, type C1 is amendable to therapeutic intervention. Hum. Mol. Genet. 27, 2076–2089. doi: 10.1093/hmg/ddy112
Davies, L. R., and Varki, A. (2015). Why is N-Glycolylneuraminic acid rare in the vertebrate brain? Top. Curr. Chem. 366, 31–54. doi: 10.1007/128_2013_419
Demina, E. P., Pierre, W. C., Nguyen, A. L. A., Londono, I., Reiz, B., Zou, C., et al. (2018). Persistent reduction in sialylation of cerebral glycoproteins following postnatal inflammatory exposure. J. Neuroinflammation 15:336. doi: 10.1186/s12974-018-1367-2
Dityatev, A., Dityateva, G., Sytnyk, V., Delling, M., Toni, N., Nikonenko, I., et al. (2004). Polysialylated neural cell adhesion molecule promotes remodeling and formation of hippocampal synapses. J. Neurosci. 24, 9372–9382. doi: 10.1523/jneurosci.1702-04.2004
Doverhag, C., Hedtjarn, M., Poirier, F., Mallard, C., Hagberg, H., Karlsson, A., et al. (2010). Galectin-3 contributes to neonatal hypoxic-ischemic brain injury. Neurobiol. Dis. 38, 36–46. doi: 10.1016/j.nbd.2009.12.024
Duan, S., and Paulson, J. C. (2020). Siglecs as immune cell checkpoints in disease. Annu. Rev. Immunol. 38, 365–395. doi: 10.1146/annurev-immunol-102419-035900
Dumic, J., Dabelic, S., and Flogel, M. (2006). Galectin-3: an open-ended story. Biochim. Biophys. Acta 1760, 616–635. doi: 10.1016/j.bbagen.2005.12.020
Edvardson, S., Baumann, A. M., Muhlenhoff, M., Stephan, O., Kuss, A. W., Shaag, A., et al. (2013). West syndrome caused by ST3Gal-III deficiency. Epilepsia 54, e24–e27. doi: 10.1111/epi.12050
Estus, S., Shaw, B. C., Devanney, N., Katsumata, Y., Press, E. E., and Fardo, D. W. (2019). Evaluation of CD33 as a genetic risk factor for Alzheimer’s disease. Acta Neuropathol. 138, 187–199. doi: 10.1007/s00401-019-02000-4
Fernandez-Arjona, M. D. M., Grondona, J. M., Fernandez-Llebrez, P., and Lopez-Avalos, M. D. (2019). Microglial activation by microbial neuraminidase through TLR2 and TLR4 receptors. J. Neuroinflammation 16:245. doi: 10.1186/s12974-019-1643-9
Finne, J., Finne, U., Deagostini-Bazin, H., and Goridis, C. (1983). Occurrence of alpha 2-8 linked polysialosyl units in a neural cell adhesion molecule. Biochem. Biophys. Res. Commun. 112, 482–487. doi: 10.1016/0006-291x(83)91490-0
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Friedman, B. A., Srinivasan, K., Ayalon, G., Meilandt, W. J., Lin, H., Huntley, M. A., et al. (2018). Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 22, 832–847. doi: 10.1016/j.celrep.2017.12.066
Funikov, S. Y., Rezvykh, A. P., Mazin, P. V., Morozov, A. V., Maltsev, A. V., Chicheva, M. M., et al. (2018). FUS(1-359) transgenic mice as a model of ALS: pathophysiological and molecular aspects of the proteinopathy. Neurogenetics 19, 189–204. doi: 10.1007/s10048-018-0553-9
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. doi: 10.1016/j.neuron.2013.04.014
Harduin-Lepers, A., Vallejo-Ruiz, V., Krzewinski-Recchi, M. A., Samyn-Petit, B., Julien, S., and Delannoy, P. (2001). The human sialyltransferase family. Biochimie 83, 727–737. doi: 10.1016/s0300-9084(01)01301-3
Harlalka, G. V., Lehman, A., Chioza, B., Baple, E. L., Maroofian, R., Cross, H., et al. (2013). Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain 136, 3618–3624. doi: 10.1093/brain/awt270
Hildebrandt, H., and Dityatev, A. (2015). Polysialic acid in brain development and synaptic plasticity. Top. Curr. Chem. 366, 55–96. doi: 10.1007/128_2013_446
Hirabayashi, J., Hashidate, T., Arata, Y., Nishi, N., Nakamura, T., Hirashima, M., et al. (2002). Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochim. Biophys. Acta 1572, 232–254. doi: 10.1016/s0304-4165(02)00311-2
Hollingworth, P., Harold, D., Sims, R., Gerrish, A., Lambert, J. C., Carrasquillo, M. M., et al. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 43, 429–435. doi: 10.1038/ng.803
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hu, H., Eggers, K., Chen, W., Garshasbi, M., Motazacker, M. M., Wrogemann, K., et al. (2011). ST3GAL3 mutations impair the development of higher cognitive functions. Am. J. Hum. Genet. 89, 407–414. doi: 10.1016/j.ajhg.2011.08.008
Hyvarinen, S., Meri, S., and Jokiranta, T. S. (2016). Disturbed sialic acid recognition on endothelial cells and platelets in complement attack causes atypical hemolytic uremic syndrome. Blood 127, 2701–2710. doi: 10.1182/blood-2015-11-680009
Inoue, S., and Kitajima, K. (2006). KDN (deaminated neuraminic acid): dreamful past and exciting future of the newest member of the sialic acid family. Glycoconj. J. 23, 277–290. doi: 10.1007/s10719-006-6484-y
Jiang, H. R., Al, R. Z., Mensah-Brown, E., Shahin, A., Xu, D., Goodyear, C. S., et al. (2009). Galectin-3 deficiency reduces the severity of experimental autoimmune encephalomyelitis. J. Immunol. 182, 1167–1173. doi: 10.4049/jimmunol.182.2.1167
Keppler, O. T., Hinderlich, S., Langner, J., Schwartz-Albiez, R., Reutter, W., and Pawlita, M. (1999). UDP-GlcNAc 2-epimerase: a regulator of cell surface sialylation. Science 284, 1372–1376. doi: 10.1126/science.284.5418.1372
Klaus, C., Hansen, J. N., Ginolhac, A., Gerard, D., Gnanapragassam, V. S., Horstkorte, R., et al. (2020). Reduced sialylation triggers homeostatic synapse and neuronal loss in middle-aged mice. Neurobiol. Aging 88, 91–107. doi: 10.1016/j.neurobiolaging.2020.01.008
Lalancette-Hebert, M., Swarup, V., Beaulieu, J. M., Bohacek, I., Abdelhamid, E., Weng, Y. C., et al. (2012). Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J. Neurosci. 32, 10383–10395. doi: 10.1523/JNEUROSCI.1498-12.2012
Landmesser, L., Dahm, L., Tang, J. C., and Rutishauser, U. (1990). Polysialic acid as a regulator of intramuscular nerve branching during embryonic development. Neuron 4, 655–667. doi: 10.1016/0896-6273(90)90193-j
Linnartz, B., Kopatz, J., Tenner, A. J., and Neumann, H. (2012). Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J. Neurosci. 32, 946–952. doi: 10.1523/JNEUROSCI.3830-11.2012
Linnartz, B., Wang, Y., and Neumann, H. (2010). Microglial immunoreceptor tyrosine-based activation and inhibition motif signaling in neuroinflammation. Int. J. Alzheimers Dis. 2010:587463. doi: 10.4061/2010/587463
Linnartz-Gerlach, B., Kopatz, J., and Neumann, H. (2014). Siglec functions of microglia. Glycobiology 24, 794–799. doi: 10.1093/glycob/cwu044
Liu, F. T., Hsu, D. K., Zuberi, R. I., Kuwabara, I., Chi, E. Y., and Henderson, W. R. (1995). Expression and function of galectin-3, a beta-galactoside-binding lectin, in human monocytes and macrophages. Am. J. Pathol. 147, 1016–1028.
Magistretti, P. J., Geisler, F. H., Schneider, J. S., Li, P. A., Fiumelli, H., and Sipione, S. (2019). Gangliosides: treatment avenues in neurodegenerative disease. Front. Neurol. 10:859. doi: 10.3389/fneur.2019.00859
Malik, M., Simpson, J. F., Parikh, I., Wilfred, B. R., Fardo, D. W., Nelson, P. T., et al. (2013). CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J. Neurosci. 33, 13320–13325. doi: 10.1523/JNEUROSCI.1224-13.2013
Maziade, M., Roy, M. A., Chagnon, Y. C., Cliche, D., Fournier, J. P., Montgrain, N., et al. (2005). Shared and specific susceptibility loci for schizophrenia and bipolar disorder: a dense genome scan in Eastern Quebec families. Mol. Psychiatry 10, 486–499. doi: 10.1038/sj.mp.4001594
McAuley, E. Z., Scimone, A., Tiwari, Y., Agahi, G., Mowry, B. J., Holliday, E. G., et al. (2012). Identification of sialyltransferase 8B as a generalized susceptibility gene for psychotic and mood disorders on chromosome 15q25-26. PLoS One 7:e38172. doi: 10.1371/journal.pone.0038172
Mendonca, H. R., Carvalho, J. N. A., Abreu, C. A., Dos Santos, A., Carvalho, J. R., Marques, S. A., et al. (2018). Lack of Galectin-3 attenuates neuroinflammation and protects the retina and optic nerve of diabetic mice. Brain Res. 1700, 126–137. doi: 10.1016/j.brainres.2018.07.018
Mikkonen, M., Soininen, H., Tapiola, T., Alafuzoff, I., and Miettinen, R. (1999). Hippocampal plasticity in Alzheimer’s disease: changes in highly polysialylated NCAM immunoreactivity in the hippocampal formation. Eur. J. Neurosci. 11, 1754–1764. doi: 10.1046/j.1460-9568.1999.00593.x
Minami, A., Meguro, Y., Ishibashi, S., Ishii, A., Shiratori, M., Sai, S., et al. (2017). Rapid regulation of sialidase activity in response to neural activity and sialic acid removal during memory processing in rat hippocampus. J. Biol. Chem. 292, 5645–5654. doi: 10.1074/jbc.M116.764357
Minami, A., Saito, M., Mamada, S., Ieno, D., Hikita, T., Takahashi, T., et al. (2016). Role of sialidase in long-term potentiation at mossy fiber-CA3 synapses and hippocampus-dependent spatial memory. PLoS One 11:e0165257. doi: 10.1371/journal.pone.0165257
Miyagi, T., and Tsuiki, S. (1984). Rat-liver lysosomal sialidase. Solubilization, substrate specificity and comparison with the cytosolic sialidase. Eur. J. Biochem. 141, 75–81. doi: 10.1111/j.1432-1033.1984.tb08159.x
Miyagi, T., and Tsuiki, S. (1985). Purification and characterization of cytosolic sialidase from rat liver. J. Biol. Chem. 260, 6710–6716.
Miyagi, T., and Yamaguchi, K. (2012). Mammalian sialidases: physiological and pathological roles in cellular functions. Glycobiology 22, 880–896. doi: 10.1093/glycob/cws057
Muller, D., Wang, C., Skibo, G., Toni, N., Cremer, H., Calaora, V., et al. (1996). PSA-NCAM is required for activity-induced synaptic plasticity. Neuron 17, 413–422. doi: 10.1016/s0896-6273(00)80174-9
Munster-Kuhnel, A. K., Tiralongo, J., Krapp, S., Weinhold, B., Ritz-Sedlacek, V., Jacob, U., et al. (2004). Structure and function of vertebrate CMP-sialic acid synthetases. Glycobiology 14, 43R–51R. doi: 10.1093/glycob/cwh113
Murray, H. C., Low, V. F., Swanson, M. E., Dieriks, B. V., Turner, C., Faull, R. L., et al. (2016). Distribution of PSA-NCAM in normal, Alzheimer’s and Parkinson’s disease human brain. Neuroscience 330, 359–375. doi: 10.1016/j.neuroscience.2016.06.003
Murray, H. C., Swanson, M. E. V., Dieriks, B. V., Turner, C., Faull, R. L. M., and Curtis, M. A. (2018). Neurochemical characterization of PSA-NCAM(+) cells in the human brain and phenotypic quantification in Alzheimer’s disease entorhinal cortex. Neuroscience 372, 289–303. doi: 10.1016/j.neuroscience.2017.12.019
Naj, A. C., Jun, G., Beecham, G. W., Wang, L. S., Vardarajan, B. N., Buros, J., et al. (2011). Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 43, 436–441. doi: 10.1038/ng.801
Nichols, M. R., St-Pierre, M. K., Wendeln, A. C., Makoni, N. J., Gouwens, L. K., Garrad, E. C., et al. (2019). Inflammatory mechanisms in neurodegeneration. J. Neurochem. 149, 562–581. doi: 10.1111/jnc.14674
Nomura, K., Vilalta, A., Allendorf, D. H., Hornik, T. C., and Brown, G. C. (2017). Activated microglia desialylate and phagocytose cells via neuraminidase, galectin-3, and Mer tyrosine kinase. J. Immunol. 198, 4792–4801. doi: 10.4049/jimmunol.1502532
Oizumi, H., Hayashita-Kinoh, H., Hayakawa, H., Arai, H., Furuya, T., Ren, Y. R., et al. (2008). Alteration in the differentiation-related molecular expression in the subventricular zone in a mouse model of Parkinson’s disease. Neurosci. Res. 60, 15–21. doi: 10.1016/j.neures.2007.09.004
Pan, B., Fromholt, S. E., Hess, E. J., Crawford, T. O., Griffin, J. W., Sheikh, K. A., et al. (2005). Myelin-associated glycoprotein and complementary axonal ligands, gangliosides, mediate axon stability in the CNS and PNS: neuropathology and behavioral deficits in single- and double-null mice. Exp. Neurol. 195, 208–217. doi: 10.1016/j.expneurol.2005.04.017
Pan, X., De Aragao, C. B. P., Velasco-Martin, J. P., Priestman, D. A., Wu, H. Y., Takahashi, K., et al. (2017). Neuraminidases 3 and 4 regulate neuronal function by catabolizing brain gangliosides. FASEB J. 31, 3467–3483. doi: 10.1096/fj.201601299R
Pasquini, L. A., Millet, V., Hoyos, H. C., Giannoni, J. P., Croci, D. O., Marder, M., et al. (2011). Galectin-3 drives oligodendrocyte differentiation to control myelin integrity and function. Cell Death Differ. 18, 1746–1756. doi: 10.1038/cdd.2011.40
Pickrell, J. K., Berisa, T., Liu, J. Z., Segurel, L., Tung, J. Y., and Hinds, D. A. (2016). Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 48, 709–717. doi: 10.1038/ng.3570
Pluvinage, J. V., Haney, M. S., Smith, B. A. H., Sun, J., Iram, T., Bonanno, L., et al. (2019). CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 568, 187–192. doi: 10.1038/s41586-019-1088-4
Pshezhetsky, A. V., and Ashmarina, L. I. (2013). Desialylation of surface receptors as a new dimension in cell signaling. Biochemistry 78, 736–745. doi: 10.1134/S0006297913070067
Pshezhetsky, A. V., and Ashmarina, M. (2018). Keeping it trim: roles of neuraminidases in CNS function. Glycoconj. J. 35, 375–386. doi: 10.1007/s10719-018-9837-4
Rahimian, R., Beland, L. C., and Kriz, J. (2018). Galectin-3: mediator of microglia responses in injured brain. Drug Discov. Today 23, 375–381. doi: 10.1016/j.drudis.2017.11.004
Raj, T., Ryan, K. J., Replogle, J. M., Chibnik, L. B., Rosenkrantz, L., Tang, A., et al. (2014). CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum. Mol. Genet. 23, 2729–2736. doi: 10.1093/hmg/ddt666
Rajendran, L., and Paolicelli, R. C. (2018). Microglia-mediated synapse loss in Alzheimer’s disease. J. Neurosci. 38, 2911–2919. doi: 10.1523/JNEUROSCI.1136-17.2017
Ransohoff, R. M. (2016). How neuroinflammation contributes to neurodegeneration. Science 353, 777–783. doi: 10.1126/science.aag2590
Rotshenker, S. (2009). The role of Galectin-3/MAC-2 in the activation of the innate-immune function of phagocytosis in microglia in injury and disease. J. Mol. Neurosci. 39, 99–103. doi: 10.1007/s12031-009-9186-7
Rotshenker, S., Reichert, F., Gitik, M., Haklai, R., Elad-Sfadia, G., and Kloog, Y. (2008). Galectin-3/MAC-2, Ras and PI3K activate complement receptor-3 and scavenger receptor-AI/II mediated myelin phagocytosis in microglia. Glia 56, 1607–1613. doi: 10.1002/glia.20713
Sato, C., and Kitajima, K. (2013). Impact of structural aberrancy of polysialic acid and its synthetic enzyme ST8SIA2 in schizophrenia. Front. Cell. Neurosci. 7:61. doi: 10.3389/fncel.2013.00061
Savman, K., Heyes, M. P., Svedin, P., and Karlsson, A. (2013). Microglia/macrophage-derived inflammatory mediators galectin-3 and quinolinic acid are elevated in cerebrospinal fluid from newborn infants after birth asphyxia. Transl. Stroke Res. 4, 228–235. doi: 10.1007/s12975-012-0216-3
Schafer, D. P., and Stevens, B. (2010). Synapse elimination during development and disease: immune molecules take centre stage. Biochem. Soc. Trans. 38, 476–481. doi: 10.1042/BST0380476
Schwarzkopf, M., Knobeloch, K. P., Rohde, E., Hinderlich, S., Wiechens, N., Lucka, L., et al. (2002). Sialylation is essential for early development in mice. Proc. Natl. Acad. Sci. U.S.A. 99, 5267–5270. doi: 10.1073/pnas.072066199
Senkov, O., Tikhobrazova, O., and Dityatev, A. (2012). PSA-NCAM: synaptic functions mediated by its interactions with proteoglycans and glutamate receptors. Int. J. Biochem. Cell Biol. 44, 591–595. doi: 10.1016/j.biocel.2012.01.008
Seyrantepe, V., Poupetova, H., Froissart, R., Zabot, M. T., Maire, I., and Pshezhetsky, A. V. (2003). Molecular pathology of NEU1 gene in sialidosis. Hum. Mutat. 22, 343–352. doi: 10.1002/humu.10268
Sheikh, K. A., Sun, J., Liu, Y., Kawai, H., Crawford, T. O., Proia, R. L., et al. (1999). Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc. Natl. Acad. Sci. U.S.A. 96, 7532–7537. doi: 10.1073/pnas.96.13.7532
Shin, T. (2013). The pleiotropic effects of galectin-3 in neuroinflammation: a review. Acta Histochem. 115, 407–411. doi: 10.1016/j.acthis.2012.11.010
Siddiqui, S. S., Matar, R., Merheb, M., Hodeify, R., Vazhappilly, C. G., Marton, J., et al. (2019). Siglecs in brain function and neurological disorders. Cells 8:1125. doi: 10.3390/cells8101125
Siddiqui, S. S., Springer, S. A., Verhagen, A., Sundaramurthy, V., Alisson-Silva, F., Jiang, W., et al. (2017). The Alzheimer’s disease-protective CD33 splice variant mediates adaptive loss of function via diversion to an intracellular pool. J. Biol. Chem. 292, 15312–15320. doi: 10.1074/jbc.M117.799346
Siew, J. J., Chen, H. M., Chen, H. Y., Chen, H. L., Chen, C. M., Soong, B. W., et al. (2019). Galectin-3 is required for the microglia-mediated brain inflammation in a model of Huntington’s disease. Nat. Commun. 10:3473. doi: 10.1038/s41467-019-11441-0
Simpson, M. A., Cross, H., Proukakis, C., Priestman, D. A., Neville, D. C., Reinkensmeier, G., et al. (2004). Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet. 36, 1225–1229. doi: 10.1038/ng1460
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Sumida, M., Hane, M., Yabe, U., Shimoda, Y., Pearce, O. M., Kiso, M., et al. (2015). Rapid trimming of cell surface polysialic acid (PolySia) by exovesicular sialidase triggers release of preexisting surface neurotrophin. J. Biol. Chem. 290, 13202–13214. doi: 10.1074/jbc.M115.638759
Tang, J., Rutishauser, U., and Landmesser, L. (1994). Polysialic acid regulates growth cone behavior during sorting of motor axons in the plexus region. Neuron 13, 405–414. doi: 10.1016/0896-6273(94)90356-5
Tao, C. C., Cheng, K. M., Ma, Y. L., Hsu, W. L., Chen, Y. C., Fuh, J. L., et al. (2020). Galectin-3 promotes Abeta oligomerization and Abeta toxicity in a mouse model of Alzheimer’s disease. Cell Death Differ. 27, 192–209. doi: 10.1038/s41418-019-0348-z
Tao, R., Li, C., Zheng, Y., Qin, W., Zhang, J., Li, X., et al. (2007). Positive association between SIAT8B and schizophrenia in the Chinese Han population. Schizophr. Res. 90, 108–114. doi: 10.1016/j.schres.2006.09.029
Thomas, L., and Pasquini, L. A. (2018). Galectin-3-mediated Glial crosstalk drives oligodendrocyte differentiation and (Re)myelination. Front. Cell. Neurosci. 12:297. doi: 10.3389/fncel.2018.00297
Varki, A. (1997). Sialic acids as ligands in recognition phenomena. FASEB J. 11, 248–255. doi: 10.1096/fasebj.11.4.9068613
Varki, A., and Angata, T. (2006). Siglecs–the major subfamily of I-type lectins. Glycobiology 16, 1R–27R. doi: 10.1093/glycob/cwj008
Von, G. S., and Bochner, B. S. (2008). Basic and clinical immunology of Siglecs. Ann. N. Y. Acad. Sci. 1143, 61–82. doi: 10.1196/annals.1443.011
Walker, D. G., Whetzel, A. M., Serrano, G., Sue, L. I., Beach, T. G., and Lue, L. F. (2015). Association of CD33 polymorphism rs3865444 with Alzheimer’s disease pathology and CD33 expression in human cerebral cortex. Neurobiol. Aging 36, 571–582. doi: 10.1016/j.neurobiolaging.2014.09.023
Wang, Y., and Neumann, H. (2010). Alleviation of neurotoxicity by microglial human Siglec-11. J. Neurosci. 30, 3482–3488. doi: 10.1523/JNEUROSCI.3940-09.2010
Wei, M., and Wang, P. G. (2019). Desialylation in physiological and pathological processes: new target for diagnostic and therapeutic development. Prog. Mol. Biol. Transl. Sci. 162, 25–57. doi: 10.1016/bs.pmbts.2018.12.001
Werneburg, S., Muhlenhoff, M., Stangel, M., and Hildebrandt, H. (2015). Polysialic acid on SynCAM 1 in NG2 cells and on neuropilin-2 in microglia is confined to intracellular pools that are rapidly depleted upon stimulation. Glia 63, 1240–1255. doi: 10.1002/glia.22815
Yan, Y. P., Lang, B. T., Vemuganti, R., and Dempsey, R. J. (2009). Galectin-3 mediates post-ischemic tissue remodeling. Brain Res. 1288, 116–124. doi: 10.1016/j.brainres.2009.06.073
Yao, D., McGonigal, R., Barrie, J. A., Cappell, J., Cunningham, M. E., Meehan, G. R., et al. (2014). Neuronal expression of GalNAc transferase is sufficient to prevent the age-related neurodegenerative phenotype of complex ganglioside-deficient mice. J. Neurosci. 34, 880–891. doi: 10.1523/JNEUROSCI.3996-13.2014
Yip, P. K., Carrillo-Jimenez, A., King, P., Vilalta, A., Nomura, K., Chau, C. C., et al. (2017). Galectin-3 released in response to traumatic brain injury acts as an alarmin orchestrating brain immune response and promoting neurodegeneration. Sci. Rep. 7:41689. doi: 10.1038/srep41689
Yoo, H. I., Kim, E. G., Lee, E. J., Hong, S. Y., Yoon, C. S., Hong, M. J., et al. (2017). Neuroanatomical distribution of galectin-3 in the adult rat brain. J. Mol. Histol. 48, 133–146. doi: 10.1007/s10735-017-9712-9
Yoo, S. W., Motari, M. G., Susuki, K., Prendergast, J., Mountney, A., Hurtado, A., et al. (2015). Sialylation regulates brain structure and function. FASEB J. 29, 3040–3053. doi: 10.1096/fj.15-270983
Zhiling, Y., Fujita, E., Tanabe, Y., Yamagata, T., Momoi, T., and Momoi, M. Y. (2008). Mutations in the gene encoding CADM1 are associated with autism spectrum disorder. Biochem. Biophys. Res. Commun. 377, 926–929. doi: 10.1016/j.bbrc.2008.10.107
Keywords: sialic acid, desialylation, galectin-3, phagocytosis, microglia, neurodegeneration, aging
Citation: Puigdellívol M, Allendorf DH and Brown GC (2020) Sialylation and Galectin-3 in Microglia-Mediated Neuroinflammation and Neurodegeneration. Front. Cell. Neurosci. 14:162. doi: 10.3389/fncel.2020.00162
Received: 29 March 2020; Accepted: 15 May 2020;
Published: 09 June 2020.
Edited by:
Veronica Ines Brito, Vall d’Hebron University Hospital, SpainReviewed by:
Harald Neumann, University of Bonn, GermanyMyron R. Szewczuk, Queen’s University, Canada
Yijuang Chern, National Research Program for Biopharmaceuticals, Taiwan
Copyright © 2020 Puigdellívol, Allendorf and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mar Puigdellívol, bWRwNDlAY2FtLmFjLnVr; Guy C. Brown, Z2NiM0BjYW0uYWMudWs=