
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Cell. Neurosci. , 08 July 2020
Sec. Cellular Neuropathology
Volume 14 - 2020 | https://doi.org/10.3389/fncel.2020.00150
 Ignazio Cali1,2*
Ignazio Cali1,2* Laura Cracco3†
Laura Cracco3† Dario Saracino4,5†
Dario Saracino4,5† Rossana Occhipinti6
Rossana Occhipinti6 Cinzia Coppola4Brian Stephen Appleby1,2,7,8
Cinzia Coppola4Brian Stephen Appleby1,2,7,8 Gianfranco Puoti4,5*
Gianfranco Puoti4,5*The insertion of additional 168 base pair containing seven octapeptide repeats in the prion protein (PrP) gene region spanning residues 51–91 is associated with inherited prion disease. In 2008, we reported the clinical features of a novel de novo seven-octapeptide repeat insertion (7-OPRI) mutation coupled with codon 129 methionine (M) homozygosity in the PrP gene of a 19-year-old man presenting with psychosis and atypical dementia, and 16-year survival. Here, we describe the histopathological and PrP molecular properties in the autopsied brain of this patient. Histopathological examination revealed widespread brain atrophy, focal spongiform degeneration (SD), cortical PrP plaques, and elongated PrP formations in the cerebellum. Overall, these histopathological features resemble those described in a Belgian pedigree with 7-OPRI mutation except for the presence of PrP plaques in our case, which are morphologically different from the multicore plaques described in some OPRI mutations and in Gerstmann–Sträussler–Scheinker (GSS) syndrome. The comparative characterization of the detergent-soluble and detergent-insoluble PrP in our patient and in sporadic Creutzfeldt–Jakob disease (CJD) revealed distinct molecular signatures. Proteinase K digestion of the pathogenic, disease-associated PrP (PrPD) revealed PrPD type 1 in the cerebral cortex and mixed PrPD types 1 and 2 in the cerebellum. Altogether, the present study outlines the importance of assessing the phenotypical and PrP biochemical properties of these rare conditions, thereby widening the spectrum of the phenotypic heterogeneity of the 7-OPRI insertion mutations. Further studies are needed to determine whether distinct conformers of PrPD are associated with two major clinico-histopathological phenotypes in prion disease with 7-OPRI.
Human prion diseases can be classified into three groups according to etiology: sporadic, genetic, and acquired by infection (Gambetti et al., 2003; Puoti et al., 2012). While the sporadic form represents the most common human prion disease and accounts for about 85–90% of cases, genetic forms have been described in 10–15% of cases and are typically associated with point mutations in the coding region of the prion protein (PrP) gene (Gambetti et al., 2011; Bonda et al., 2016). Although point mutations are the most common cause of pathogenic mutations, deletion and insertion of extra base pairs are continuously reported (Palmer et al., 1993; Beck et al., 2001; Capellari et al., 2002; Xiao et al., 2013; Areškevičìūtė et al., 2019; Piazza et al., 2020). Furthermore, experiments with primate (Goldfarb et al., 1991) and transgenic mice (Mead et al., 2006) have shown that genetic prion diseases with insertion mutations (gPrDIns) are transmissible.
Insertion mutations involve a region consisting of 27 base pairs (bp) nonapeptide (R1) followed by four 24-bp octapeptide repeats (R2, R2, R3, and R4) with slight variations at the nucleotide level (Goldfarb et al., 1991). These repeats lie in the portion of the PrP gene encompassing codons 51 and 91, a region matching the copper-binding domain of the protein.
As new studies on gPrDIns are described, the number of octapeptide repeat insertion (OPRI) variants has increased and encompasses cases with 1- to 12-OPRIs. The broad range of possible OPRIs and the methionine (M)/valine (V) polymorphism at codon 129 of the PrP gene confer phenotypic heterogeneity (Parchi et al., 1999; Kong et al., 2004). In addition, the presence of either one or two types (namely, type 1 and type 2) of the disease-associated PrP (PrPD) and a low molecular fragment of approximately 7–8 kDa (PrP7-8) of Gerstmann–Sträussler–Scheinker (GSS) disease may contribute to this heterogeneity (Puoti et al., 1999; Cali et al., 2009; Cali et al., 2020).
Correlations between the number of OPRI and phenotypic expression have suggested three groups of OPRI-associated genetic prion diseases: (1) cases harboring 2- or 4-OPRI and Creutzfeldt–Jakob disease (CJD) phenotype with fast disease progression; (2) cases with 5- to 7-OPRI with CJD reminiscent phenotype and slower disease progression; and (3) cases with 8- to 12-OPRI with the GSS phenotype (Kim et al., 2018). However, phenotypic expression in subjects with OPRI does not always fit this general classification (Areškevičìūtė et al., 2019). Moreover, great variability in the clinical and histopathological phenotype has been reported within family members.
The phenotypic expression and the allelic origin of PrPD associated with 7-OPRI have been well characterized (Goldfarb et al., 1991; Tateishi, 1991; Brown et al., 1992; Dermaut et al., 2000; Lewis et al., 2003; Wang et al., 2007; Guo et al., 2008; Mauro et al., 2008; Jansen et al., 2011), and two major clinicopathological disease phenotypes have been described. The first one, reported in a Dutch family, resembles GSS with family members presenting with cognitive and motor impairment at around the fifth decade of life and mean disease duration of ~2.5 years. Major histopathological features include uni- and multicentric PrP plaques with amyloid tinctorial properties and the lack of elongated PrP deposits in the cerebellum, a recurrent feature in gPrDIns patients. Genetic analysis available in two family members disclosed cis-V at PrP-codon 129 (Jansen et al., 2011). The second disease phenotype, described in a Belgian kindred, exhibits cognitive decline at mean age of 29 years and mean disease duration of 13 years. Histopathological examination revealed the lack of PrP plaques and the presence of elongated PrP deposits in the cerebellum; these patients were cis-M at PrP-codon 129 (Dermaut et al., 2000). An exception to the aforementioned clinical phenotypes is represented by a Chinese patient with cis-M PrP-codon 129 who exhibited memory impairment at age 44 and relatively short disease duration of 4 years. Histopathological examination did not provide information relative to the presence of plaques or elongated PrP deposits (Wang et al., 2007).
In the present study, we describe the histopathological and molecular findings in a young gPrDIns patient with 7-OPRI mutation and slowly progressive cognitive decline (Mauro et al., 2008). The clinical and most of the histopathological features resemble those described in the Belgium kindred, supporting the diagnosis of genetic CJD. Biochemical characterization of PrP harvested from detergent-soluble and detergent-insoluble fractions unveiled a distinctive signature of the prion protein.
The detailed clinical phenotype and genetic analysis were described in our previous study (Mauro et al., 2008). Briefly, a 19-year-old man presented with psychosis and very slowly progressive atypical dementia, characterized by behavior changes and posterior parietal cognitive signs including visuospatial and constructional deficits, ideomotor apraxia, left–right confusion, and dyscalculia. At the age of 33, the neurological course of the disease changed, becoming rapid and leading the patient to apallic coma in a few months. He died at the age of 34, after 15 years of clinical disease (Supplementary Figure S1).
The diagnosis of genetic prion disease was obtained 8 years from the onset. Sequence analysis on the proband’s DNA demonstrated a 168-bp insertion corresponding to a novel seven extra repeats insertion mutation in the open reading frame of the PrP gene. The 7-OPRI mutation was not present in the patient’s parents.
Although repeats had the same amino acid sequence, they could be discriminated by their DNA sequence. In the mutated allele, repeats were arranged in the following pattern: R1–R2–R2–R3–R2–R2–R3g–R2–R2–R2–R3–R4. Genotype at the polymorphic codon 129 of the PrP gene disclosed methionine homozygosity (Mauro et al., 2008).
Overall, a major histopathological feature of our gPrDIns case was marked atrophy, characterized by neuronal loss and severe astrogliosis throughout the brain (Figure 1A). Spongiform degeneration (SD) was focal with small vacuoles affecting the cerebral neocortex and the parahippocampal gyrus (Figure 1B). Spongiosis was occasionally pronounced in layer II of the cerebral cortex; scattered vacuoles were noted in the cerebellar molecular layer. Unlike other cortical regions, SD in the occipital cortex, and to a lesser extent the parietal cortex, showed few areas with larger and confluent vacuoles associated with more severe gliosis and neuronal loss (Figure 1C). Eosinophilic plaque formations without a dense core were conspicuous in the subiculum and rare in the temporal neocortex (inset of Figure 1F). The cerebellum was characterized by moderate atrophy with loss of Purkinje and granule cells.
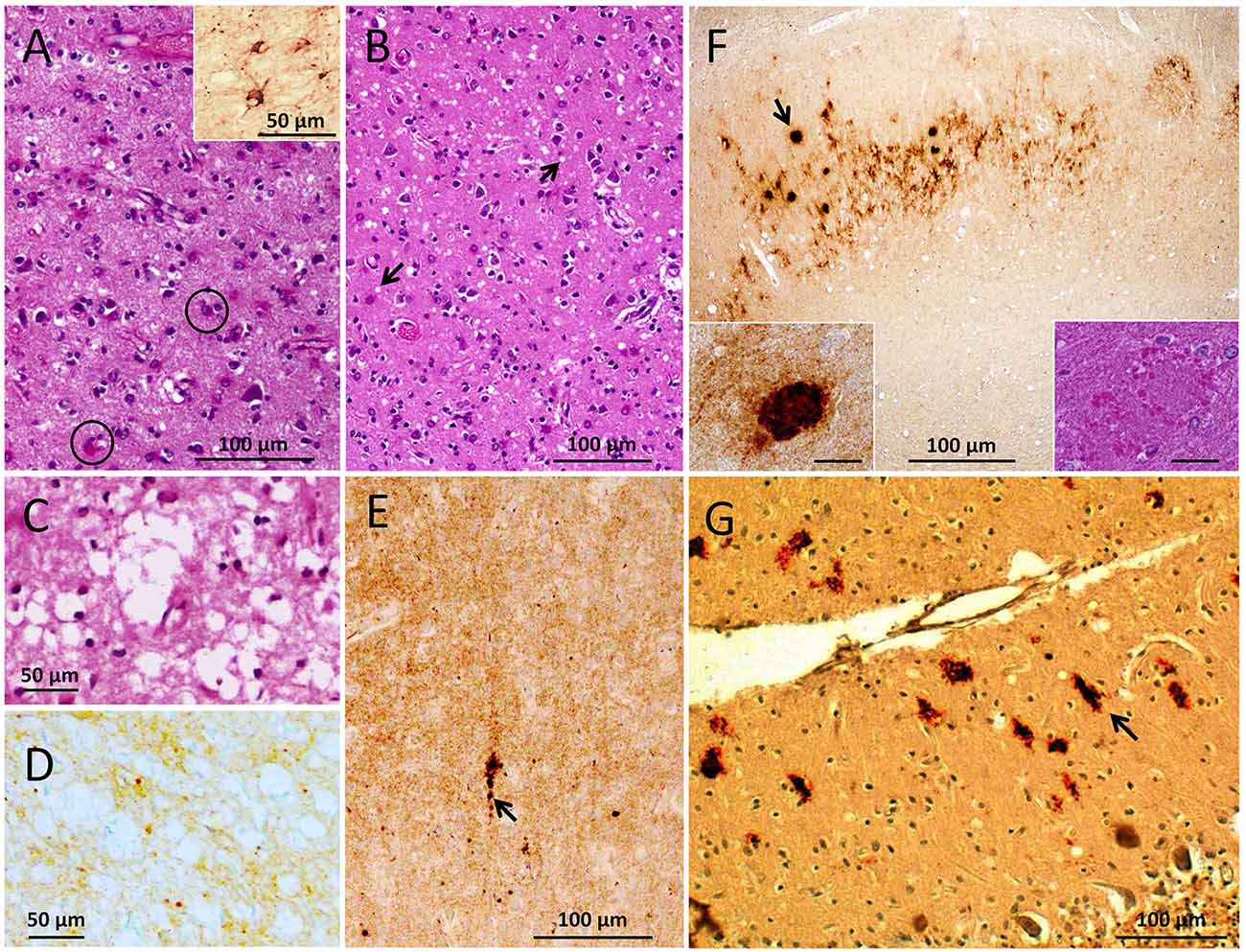
Figure 1. Histopathological phenotype. (A–C) Hematoxylin and eosin (HE) staining. (D–G) Prion protein (PrP) immunohistochemistry (IHC). (A) Severe astrogliosis with reactive astrocytes (circles) affecting the cerebral cortex; inset: glial fibrillary acidic protein (GFAP) immunostaining. (B) Spongiform degeneration (SD, arrows) in the parahippocampal gyrus. (C,D) Large vacuoles SD (C) and diffuse PrP immunostaining (D) of the same region (occipital cortex). (E) Coarser PrP aggregates (arrow) in a background of diffuse PrP (entorhinal cortex). (F) PrP plaque formations (arrow) in a background of diffuse PrP (subiculum); insets: PrP plaque on HE section (right) and after PrP IHC (left). (G) Elongated (arrow) and patchy PrP aggregates in the cerebellar molecular layer. Scale bar insets in (C): 20 μm; antibody: 3F4 (D–G).
Immunohistochemical examination for glial fibrillary acidic protein (GFAP) revealed an intense astrocytic reaction (inset of Figure 1A). Immunostaining for PrP showed scattered small aggregates—sometimes with the appearance of plaque-like formations—in a background of diffuse PrP in the cerebral cortex, including the dentate gyrus of the hippocampus, basal ganglia, and brainstem (Figures 1D,E). The CA4 region of the hippocampus and the entorhinal cortex showed diffuse and perineuronal PrP deposits. In cortical region with large vacuoles PrP immunostaining was of the diffuse type with rare coarser granules (Figure 1D). The eosinophilic plaque formations detected on hematoxylin–eosin sections reacted with an anti-PrP antibody (Figure 1F), while elongated and truncated PrP formations with orientation perpendicular to the leptomeningeal surface populated the molecular layer of the cerebellum (Figure 1G).
Brain homogenates (S1) harvested from gPrDIns showed the typical mono- and unglycosylated PrP isoforms migrating to ~30 and ~27 kDa, respectively, similar to PrP in sCJDMM1—a sCJD subtype with PrP-129MM genotype and PrPD type 1 (Figure 2A). Unlike sCJD, however, PrP from gPrDIns was characterized by the lack of a well-defined diglycosylated isoform. Furthermore, a smear of faint bands in the ~32–40 kDa range migrated above the monoglycosylated PrP isoform in gPrDIns but not in sCJD controls (Figure 2A). PrP fragments ranging between ~17 and ~25 kDa were abundant in gPrDIns (Figure 2A). These fragments were not detected when probing with an antibody (Ab) to an epitope located to the PrP N-terminal region (residues 36–43), indicating that these endogenous fragments are N-terminally truncated (data not shown). In order to have a more detailed picture of the PrP Western blot (WB) profile, S1 preparations were subjected to high-speed centrifugation to separate the detergent-soluble (S2, containing wild-type and mutated PrPC) from detergent-insoluble (P2, containing wild-type and mutated PrPD) fraction (Figures 2B–D). In gPrDIns only, PrPC showed a faint band of ~42–44 kDa in addition to di- (~34 kDa) and mono-glycosylated (~30 kDa) PrP isoforms, while the unglycosylated (~27 kDa) PrP isoform was better visualized at longer signal captures (Figure 2B).
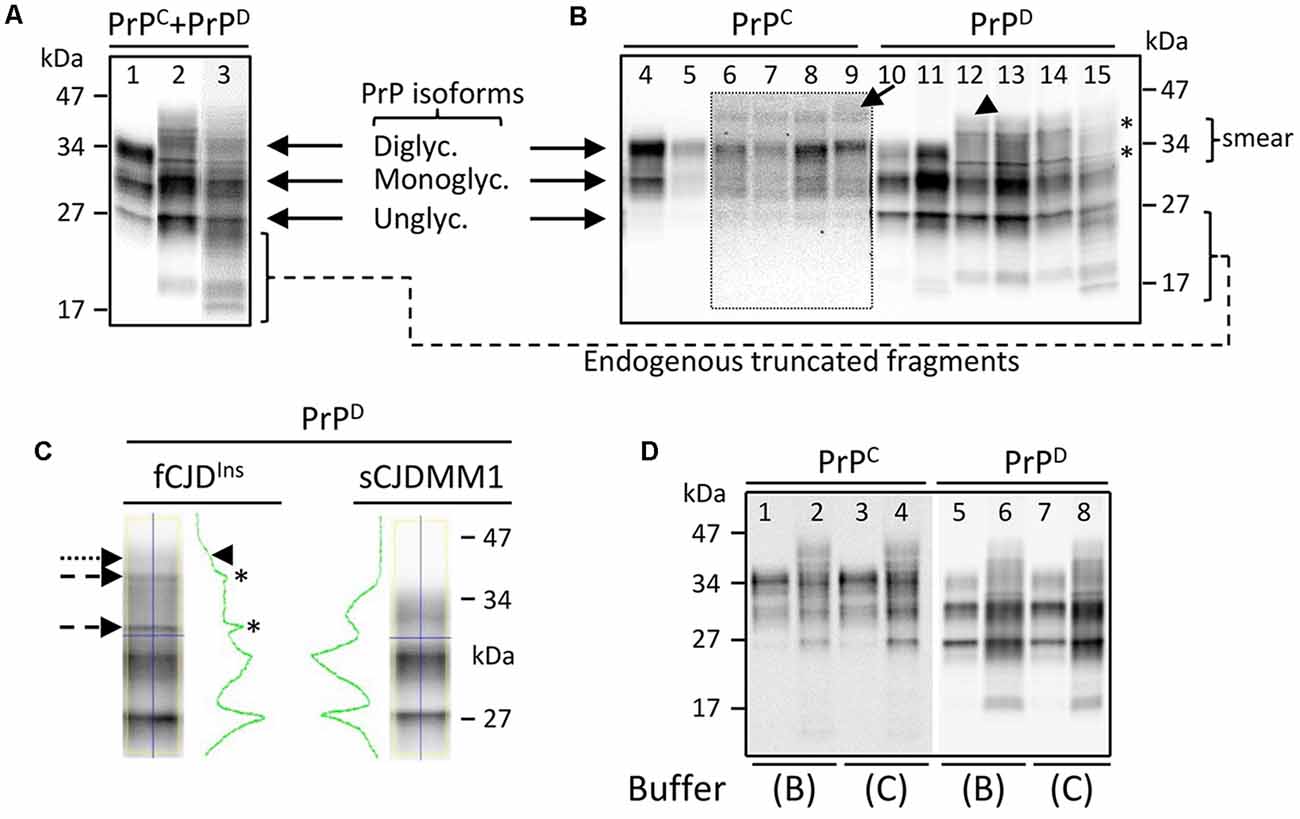
Figure 2. Western blot (WB) profiles of detergent-soluble and detergent-insoluble prion protein (PrP). PrP bands were resolved in a 15% Tris–HCl, 8.7-cm-long gel and visualized with the near-infrared LI-COR system. (A) Brain homogenate (S1) containing both PrPC and PrPD species (PrPC + PrPD) showing three major fragments corresponding to di-, mono- and unglycosylated PrP isoforms in sCJDMM1 (lane 1). A PrP smear in the ~30–45 kDa area of the gel is observed in addition to mono- and unglycosylated PrP isoforms in fCJDIns (lanes 2 and 3). Bands in the ~17–25 kDa regions represent endogenously truncated PrP fragments; frontal cortex (cx; lanes 1 and 2) and cerebellum (lane 3). (B) PrPC shows one band in the ~42–44 kDa region of the gel (arrow) in addition to di-, mono- and unglycosylated PrPC in gPrDIns (lanes 6–9). WB profiles of PrPC from sCJDMM1 (lane 4) and sCJDVV2 (lane 5) show the typical three PrPC isoforms. The inset represents a longer exposure of area highlighted by the dotted rectangle. The detergent-insoluble PrPD shows a smear and two sharp bands (asterisks) between ~32 and ~40 kDa, and a higher faint band of ~42–44 kDa (arrowhead) in gPrDIns (lanes 12–15). PrPD in sCJDMM1 (lane 10) and sCJDVV2 (lane 11) is characterized by the typical three PrPD isoforms; frontal cx (lanes 4–6, 10–12); parietal cx (lanes 7 and 13); occipital cx (lanes 8 and 14); cerebellum (lanes 9 and 15). (C) PrPD harvested from gPrDIns and sCJDMM1 shows distinct WB profiles as highlighted by the green line: the two small peaks (asterisks) and the slope (arrowhead) in PrPD-gPrDIns are generated by the two sharp fragments (dashed arrows) and a smear (dotted arrow), respectively. (D) WB profiles of PrPC and PrPD obtained from gPrDIns (lanes 2, 4, 6, 8) and sCJDMM1 (lanes 1, 3, 5, 7); samples were prepared using two buffers (buffers B and C; see Methods and Supplementary Figure S2) with high percentage of a detergent. The same loading was used in each WB in panels (A–D); antibody: 3F4.
The pathogenic PrPD harvested from the detergent-insoluble fraction showed two sharp fragments in the ~32–40 kDa region lying in a background of PrP smear in gPrDIns but not in sCJD (Figures 2B,C). The ~42–44 kDa high molecular fragment populated also PrPD. Notably, the composition of the buffer and the amount of detergent did not change the molecular profiles of PrPC and PrPD species (Figure 2D).
Proteinase K (PK) digestion of PrPD generated truncated PK-resistant PrPD (resPrPD) corresponding to di- (~30–31 kDa), mono- (~27 kDa), and unglycosylated resPrPD isoforms. The unglycosylated resPrPD band, a surrogate marker for prion strains, migrated as a single band of ~20 kDa in the cerebral cortex of gPrDIns, matching the gel mobility of resPrPD type 1 (T1; Figure 3A; Cali et al., 2006, 2009). On the contrary, two co-existing resPrPD bands of ~20 and ~19 kDa were found in the cerebellum; the lower band comigrated with resPrPD type 2 of sCJDVV2–a sCJD subtype with PrP-129VV genotype and PrPD type 2 (Figure 3A). The T1-selective 12B2 Ab confirmed the presence of resPrPD T1 in gPrDIns and sCJDMM1 but not in sCJDVV2 (Figure 3B), whereas the 1E4 Ab, which preferentially binds to type 2 (T2), detected resPrPD T2 in the cerebellum but not in the cerebral cortex of gPrDIns (Figure 3C). Finally, the 2301 Ab immunoreacting with the C-terminal portion of PrP (residues 220–231) detected similar amounts of ~13 kDa, and possibly ~12 kDa, C-terminal fragments (referred as to PrP-CTF 12/13) in gPrDIns (Figure 3D; Zou et al., 2003).
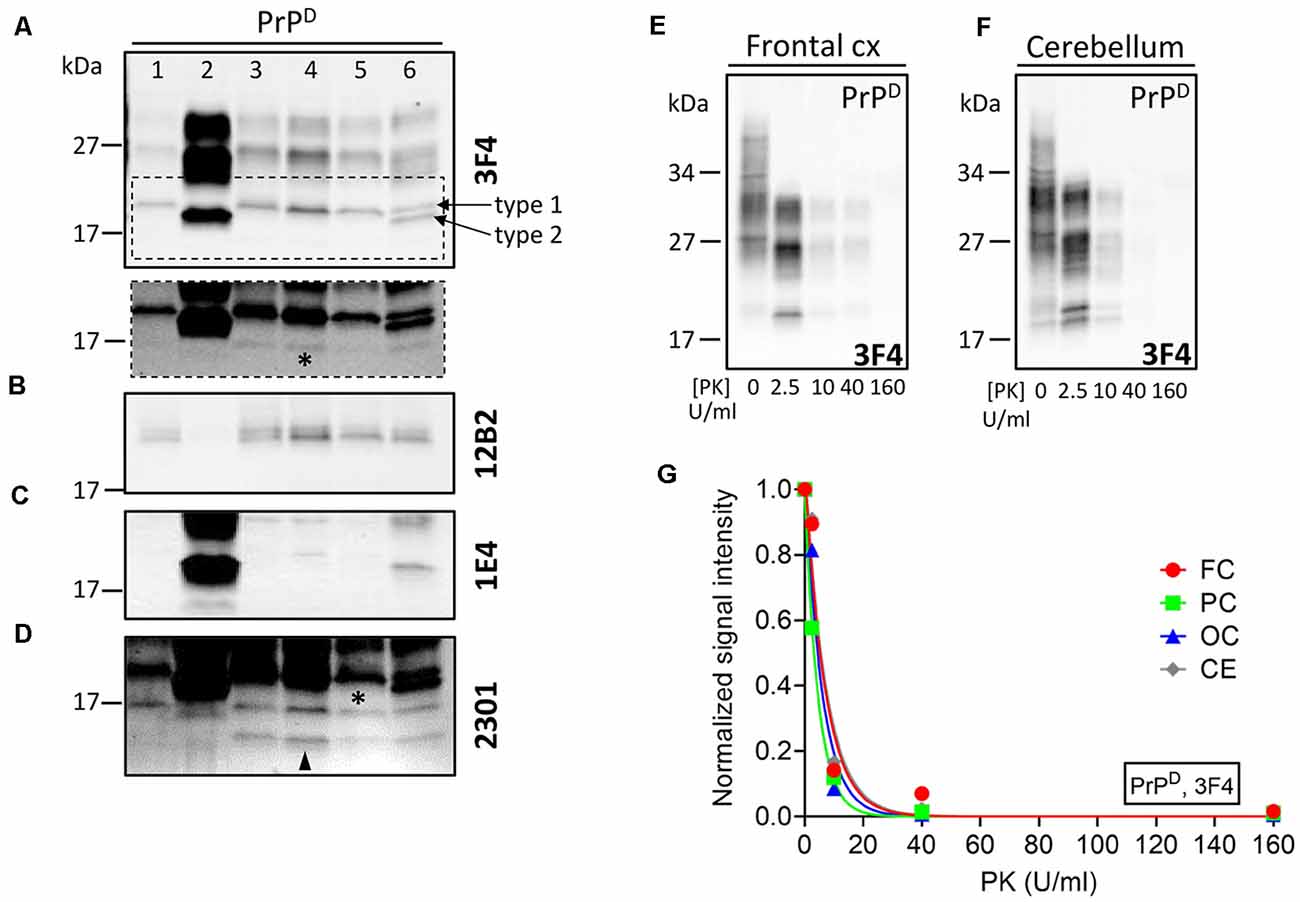
Figure 3. WB profiles of resPrPD and proteinase K (PK)-titration assay of PrPD. PrPD from gPrDIns (lanes 3–6), sCJDMM1 (lane 1), and sCJDVV2 (lane 2) was digested with PK at concentration of 10 U/ml PK and probed with antibodies (A,E–G) 3F4, (B) 12B2, (C) 1E4, and (D) 2301. (A) The unglycosylated resPrPD isoform from the neocortex of gPrDIns (lanes 3–5) and resPrPD T1 from sCJDMM1 (lane 1) comigrates to ~20 kDa. In the cerebellum of gPrDIns (lane 6), the ~20-kDa fragment coexists with a ~19-kDa band matching in mobility resPrPD T2 of sCJDVV2 (lane 2). Dashed rectangle: longer exposure of the area highlighted in the main figure showing low amounts of a ~17-kDa fragment (asterisk) in the cerebral cortex and cerebellum; frontal (lane 3), parietal (lane 4), and occipital (lane 5) cortices. (B) The T1-selective 12B2 Ab immunoreacts with T1 (lanes 1, 3–6) but not with T2 (lanes 2 and 6). (C) The Ab 1E4 efficiently binds to T2 (lanes 2 and 6) and only weakly with T1 (lane 4). (D) The 2301 Ab immunoreacts with resPrPD of ~17 kDa (asterisk) and resPrPD CTF ~12/13 kDa (arrowhead). (E,F) Representative WB showing PrPD-gPrDIns subjected to PK digestion at concentration of 0, 2.5, 10, 40, and 160 U/ml. (G) PK1/2 (corresponding to the PK concentration required to digest half of PrPD) was similar in the brain regions of gPrDIns [4.3 ± 0.4 U/ml expressed as mean ± SEM of frontal cortex (FC), parietal cortex (PC), occipital cortex (OC) and cerebellum (CE)].
PK-titration assay of PrPD harvested from the cerebral cortex and cerebellum showed similar profiles. PK1/2—the amount of PK required to digest 50% of PrPD—ranged between 3 U/ml (parietal cortex) and 5 U/ml (frontal cortex and cerebellum; Figures 3E–G).
Here, we carried out a description of the histopathological and biochemical features of a novel de novo 7-OPRI mutation in a patient presenting with early onset behavioral changes and long survival (Mauro et al., 2008). Overall, the age at onset (19 years) and disease duration (16 years) are in line with the clinical data reported in families with 7-OPRI mutation coupled with methionine at PrP-codon 129 of the mutated allele (cis-M; Dermaut et al., 2000; Lewis et al., 2003). However, we do not know whether the co-occurrence of PrPD types and/or the novel sequence of the extra repeats in our case contributed to further anticipate the age at onset of the youngest patient with 7-OPRI mutation. According to Lewis et al. (2003), the presence of valine in the normal allele (trans-V) coupled with PrPD T2 does not affect age at onset and disease duration clinical features. A marked exception to the abovementioned clinical features is represented by a cis-M member of a Chinese family who presented with memory deficit at older age (46 years) and had relatively shorter disease course (~4 years; Wang et al., 2007). Similar to other patients carrying the 7-OPRI mutation, cognitive decline and apraxia were among the clinical features in our case. However, personality changes with autistic-like behavior at onset, marked parietal atrophy, and absence of cerebellar ataxia represent novelties.
The evaluation of the histopathological phenotype revealed some characteristics matching those described by Dermaut et al. (2000), including: (i) a generalized brain atrophy; (ii) mild spongiosis with preferential distribution in the cortical layer II; (iii) absence of SD in the hippocampus (CA1–CA4); (iv) loss of Purkinje cells; and (v) presence of elongated PrP deposits in the cerebellar molecular layer. However, a distinguishing phenotypical feature is represented by the core-free PrP cortical plaques in our case; these plaques are morphologically different from the unicentric and multicore (Gelpi et al., 2005; Jansen et al., 2011; Xiao et al., 2013), kuru (Tateishi, 1991; Xiao et al., 2013), and florid plaques (Pietrini et al., 2003) reported in cases with various OPRI mutations. Histopathological characteristics as those described by Lewis et al. (2003) in a patient with 7-OPRI mutation only partially resemble those of our and Dermaut’s cases. Genetic differences in the OPRI and the presence of valine in the normal allele may account for the lack of elongated cerebellar PrP deposits (also referred to as “stripes”) in the study of Lewis et al. (2003).
The presence of elongated cerebellar PrP deposits in our and other cases, a pathognomonic feature of the OPRI mutation, has been described in cases with different PrP-codon 129 genotypes and PrPD types (Capellari et al., 1997; Dermaut et al., 2000; Mead et al., 2007; Jansen et al., 2009; Xiao et al., 2013; Areškevičìūtė et al., 2019). Notably, elongated PrP deposits have been reported in cases with 5- and 6-OPRI mutations lacking detectable resPrPD, suggesting that the PK-sensitive portion of PrPD is sufficient to initiate and sustain neurodegeneration in these patients (Mead et al., 2007; Xiao et al., 2013).
Histopathologically, large vacuoles and coarse and/or perivacuolar PrP deposition are cardinal features of sCJDMM2 and sCJDMV2C subtypes (Parchi and Saverioni, 2012; Baiardi et al., 2019) as well as sCJD cases with mixed PrPD types (e.g., sCJDMM1-2 and sCJDMV1-2C; Puoti et al., 1999; Cali et al., 2009; Parchi et al., 2009). Unlike the above sCJD subtypes, PrP immunostaining of cortical regions with large vacuoles was of the diffuse type in our case. The lack of coarse/perivacuolar PrP deposits in regions with large vacuoles may not be uncommon in some prion diseases including gPrDIns (Cali et al., 2018).
The complexity of the electrophoretic profile of PrP from various gPrDIns is undoubtedly generated by the extra octapeptide repeats (Capellari et al., 1997; Lewis et al., 2003; Pietrini et al., 2003; Gelpi et al., 2005; Mead et al., 2007; Xiao et al., 2013). As expected, the WB profiles of PrPC and PrPD differed from those of sCJD. These differences included the presence of a high molecular weight fragment in the mutated PrP, which likely represents the mutated diglycosylated PrP isoform; mutated mono- and unglycosylated PrP isoforms should be interspersed within the wild-type PrP. To determine the glycosylation nature of two sharp PrPD bands in the ~32–40 kDa region and the lack of a well-defined diglycosylated PrPD isoform, additional investigations would be required. Importantly, WB profiles of the mutated PrPC, and of the resulting PrPD, were reproducible under different experimental conditions. Furthermore, the apparently higher levels of endogenously truncated PrPD fragments (spanning ~17–25 kDa) in our case compared to sCJD is in agreement with a previous study (Gelpi et al., 2005) and highlights the propensity of gPrDIns cases to generate truncated PrPD fragments.
Proteinase K digestion of PrPD generated similar ratios of di-, -mono-, and unglycosylated resPrPD in gPrDIns and sCJD. In patients with different OPRI mutations, coexisting resPrPD types T1 and T2 (T1–2) have been reported in the same brain region or separately in different anatomical locations (Pietrini et al., 2003; Jansen et al., 2009). In our case, both resPrPD types were found in the cerebellum, whereas T1, but not T2, was present in the cerebral cortex. The reason for the lack of T2 detection in the cerebral cortex could be due to the limited number of cortical regions assessed and small amount of tissue used (Cali et al., 2009, 2020). Furthermore, the presence of coarse SD in the cerebral cortex, although focal, suggests that a minor component of T2 is present.
As a measure of the conformation of PrPD, we recently determined the resistance to proteolysis by digestion with PK of PrPD T1-2 in patients with sCJDVV1-2 (Cali et al., 2020). From this study, we found that the PK1/2 index of T1–2 was significantly greater than PrPD T1. Here, the PK1/2 of PrPD T1–2 and T1 were virtually identical, suggesting conformational differences of T1–2 between gPrDIns and sCJD. However, the small number of brain regions with T1–2 in our case and some modifications in the protocol may limit this interpretation.
Finally, we did not detect the ~7–8 kDa PrPD fragment described in GSS and in a gPrDIns with 7-OPRI mutation and GSS-like histopathological phenotype (Gelpi et al., 2005; Jansen et al., 2011). The lowest molecular weight PrP fragment in gPrDIns with 6-OPRI mutation described by Gelpi et al. may correspond to the ~7–8 kDa fragment. As demonstrated by these two reports as well as in a GSS study, the ~7–8 kDa PrP band immunoreacts well with an antibody to the central portion of PrP (e.g., 3F4; Parchi et al., 1998; Cracco et al., 2019). Although it is not known with certainty whether the presence of the 7–8 kDa PrP band is invariably associated with the GSS phenotype in gPrDIns (Xiao et al., 2013), the lack of this fragment and the different morphologies of the plaques point towards a novel phenotype of our gPrDIns case.
Overall, the present case study broadens the spectrum of phenotypic expression of the inherited prion disease linked to 7-extra octapeptide repeats insertion mutation. The presence of amorphous PrP plaques in the cerebral cortex adds to the phenotypic variability of cases with the same mutation. Furthermore, this study highlights the importance of determining the biochemical properties of the soluble and insoluble PrP species as the molecular determinant of phenotypic expression in prion diseases.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Written informed consent was obtained from the individuals for the publication of this case report, including any potentially identifiable images or data contained in this article.
IC and GP conceived and designed the experiments. IC, LC, DS, BSA, and GP performed the experiments. IC, LC, DS, RO, CC, and GP analyzed the data. IC and GP wrote the manuscript. All authors reviewed the manuscript.
This study was supported by the Charles S. Britton Fund to P. Gambetti; Centers for Disease Control and Prevention (CDC; 6NU38CK000480-01) to BSA. As a trainee of the research education component (REC) of the Cleveland Alzheimer’s Disease Research Center (CADRC), the work of IC was in part supported by the National Institute of Aging P30 AG062428 01 grant. The work of RO was in part supported by the National Institute of Diabetes and Digestive and Kidney Diseases K01 DK107787 grant.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge the patient’s family and the members of the National Prion Disease Pathology Surveillance Center (NPDPSC) and Prion Disease Diagnosis and Surveillance Center (PDDSC). In particular, we thank Diane Kofskey for her technical support in performing histology and immunohistochemistry. We are grateful to Dr. Man-Sun Sy for kindly providing the antibody 8B4.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2020.00150/full#supplementary-material.
Areškevičìūtė, A., Høgh, P., Bartoletti-Stella, A., Melchior, L. C., Nielsen, P. R., Parchi, P., et al. (2019). A novel eight octapeptide repeat insertion in PRNP causing prion disease in a danish family. J. Neuropathol. Exp. Neurol. 78, 595–604. doi: 10.1093/jnen/nlz037
Baiardi, S., Rossi, M., Capellari, S., and Parchi, P. (2019). Recent advances in the histo-molecular pathology of human prion disease. Brain Pathol. 29, 278–300. doi: 10.1111/bpa.12695
Beck, J. A., Mead, S., Campbell, T. A., Dickinson, A., Wientjens, D. P., Croes, E. A., et al. (2001). Two-octapeptide repeat deletion of prion protein associated with rapidly progressive dementia. Neurology 57, 354–356. doi: 10.1212/wnl.57.2.354
Bonda, D. J., Manjila, S., Mehndiratta, P., Khan, F., Miller, B. R., Onwuzulike, K., et al. (2016). Human prion diseases: surgical lessons learned from iatrogenic prion transmission. Neurosurg. Focus 41:E10. doi: 10.3171/2016.5.focus15126
Brown, P., Goldfarb, L. G., McCombie, W. R., Nieto, A., Squillacote, D., Sheremata, W., et al. (1992). Atypical Creutzfeldt-Jakob disease in an american family with an insert mutation in the PRNP amyloid precursor gene. Neurology 42, 422–427. doi: 10.1212/wnl.42.2.422
Cali, I., Castellani, R., Alshekhlee, A., Cohen, Y., Blevins, J., Yuan, J., et al. (2009). Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt-Jakob disease: its effect on the phenotype and prion-type characteristics. Brain 132, 2643–2658. doi: 10.1093/brain/awp196
Cali, I., Castellani, R., Yuan, J., Al-Shekhlee, A., Cohen, M. L., Xiao, X., et al. (2006). Classification of sporadic Creutzfeldt-Jakob disease revisited. Brain 129, 2266–2277. doi: 10.1093/brain/awl224
Cali, I., Cohen, M. L., Haïk, S., Parchi, P., Giaccone, G., Collins, S. J., et al. (2018). Iatrogenic creutzfeldt-jakob disease with amyloid-β pathology: an international study. Acta Neuropathol. Commun. 6:5. doi: 10.1186/s40478-017-0503-z
Cali, I., Puoti, G., Smucny, J., Curtiss, P. M., Cracco, L., Kitamoto, T., et al. (2020). Co-existence of PrPD types 1 and 2 in sporadic Creutzfeldt-Jakob disease of the VV subgroup: phenotypic and prion protein characteristics. Sci. Rep. 10:1503. doi: 10.1038/s41598-020-58446-0
Capellari, S., Parchi, P., Wolff, B. D., Campbell, J., Atkinson, R., Posey, D. M., et al. (2002). Creutzfeldt-Jakob disease associated with a deletion of two repeats in the prion protein gene. Neurology 59, 1628–1630. doi: 10.1212/01.wnl.0000035533.86833.28
Capellari, S., Vital, C., Parchi, P., Petersen, R. B., Ferrer, X., Jarnier, D., et al. (1997). Familial prion disease with a novel 144-bp insertion in the prion protein gene in a Basque family. Neurology 49, 133–141. doi: 10.1212/wnl.49.1.133
Cracco, L., Xiao, X., Nemani, S. K., Lavrich, J., Cali, I., Ghetti, B., et al. (2019). Gerstmann-Sträussler-Scheinker disease revisited: accumulation of covalently-linked multimers of internal prion protein fragments. Acta Neuropathol. Commun. 7:85. doi: 10.1186/s40478-019-0734-2
Dermaut, B., Cruts, M., Backhovens, H., Lübke, U., Van Everbroeck, B., Sciot, R., et al. (2000). Familial Creutzfeldt-Jakob disease in a patient carrying both a presenilin 1 missense substitution and a prion protein gene insertion. J. Neurol. 247, 364–368. doi: 10.1007/s004150050603
Gambetti, P., Cali, I., Notari, S., Kong, Q., Zou, W.-Q., and Surewicz, W. K. (2011). Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol. 121, 79–90. doi: 10.1007/s00401-010-0761-3
Gambetti, P., Kong, Q., Zou, W., Parchi, P., and Chen, S. G. (2003). Sporadic and familial CJD: classification and characterisation. Br. Med. Bull. 66, 213–239. doi: 10.1093/bmb/66.1.213
Gelpi, E., Kovacs, G. G., Ströbel, T., Koperek, O., Voigtländer, T., Liberski, P. P., et al. (2005). Prion disease with a 144 base pair insertion: unusual cerebellar prion protein immunoreactivity. Acta Neuropathol. 110, 513–519. doi: 10.1007/s00401-005-1073-x
Goldfarb, L. G., Brown, P., McCombie, W. R., Goldgaber, D., Swergold, G. D., Wills, P. R., et al. (1991). Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc. Natl. Acad. Sci. U S A 88, 10926–10930. doi: 10.1073/pnas.88.23.10926
Guo, Y.-J., Wang, X.-F., Han, J., Zhang, B.-Y., Zhao, W.-Q., Shi, Q., et al. (2008). A patient with Creutzfeldt-Jakob disease with an insertion of 7 octa-repeats in the PRNP gene: molecular characteristics and clinical features. Am. J. Med. Sci. 336, 519–523. doi: 10.1097/maj.0b013e3181643e50
Jansen, C., van Swieten, J. C., Capellari, S., Strammiello, R., Parchi, P., and Rozemuller, A. J. M. (2009). Inherited Creutzfeldt-Jakob disease in a Dutch patient with a novel five octapeptide repeat insertion and unusual cerebellar morphology. J. Neurol. Neurosurg. Psychiatry 80, 1386–1389. doi: 10.1136/jnnp.2008.169359
Jansen, C., Voet, W., Head, M. W., Parchi, P., Yull, H., Verrips, A., et al. (2011). A novel seven-octapeptide repeat insertion in the prion protein gene (PRNP) in a Dutch pedigree with Gerstmann-Sträussler-Scheinker disease phenotype: comparison with similar cases from the literature. Acta Neuropathol. 121, 59–68. doi: 10.1007/s00401-010-0656-3
Kim, M.-O., Takada, L. T., Wong, K., Forner, S. A., and Geschwind, M. D. (2018). Genetic PrP prion diseases. Cold Spring Harb. Perspect. Biol. 10:a033134. doi: 10.1101/cshperspect.a033134
Kong, Q., Surewicz, W. Z., and Petersen, R. B. (2004). “Inherited prion diseases,” in Prion Biology and Diseases, Second Edition, (New York, NY: Cold Springs Harbor Laboratory Press), 673.
Lewis, V., Collins, S., Hill, A. F., Boyd, A., McLean, C. A., Smith, M., et al. (2003). Novel prion protein insert mutation associated with prolonged neurodegenerative illness. Neurology 60, 1620–1624. doi: 10.1212/01.wnl.0000065887.14609.0e
Mauro, C., Giaccone, G., Piscosquito, G., Lavorgna, A., Nigro, M., Di Fede, G., et al. (2008). A novel insertional mutation in the prion protein gene: clinical and bio-molecular findings. J. Neurol. Neurosurg. Psychiatry 79, 1395–1398. doi: 10.1136/jnnp.2007.142976
Mead, S., Poulter, M., Beck, J., Webb, T. E. F., Campbell, T. A., Linehan, J. M., et al. (2006). Inherited prion disease with six octapeptide repeat insertional mutation–molecular analysis of phenotypic heterogeneity. Brain 129, 2297–2317. doi: 10.1093/brain/awl226
Mead, S., Webb, T. E. F., Campbell, T. A., Beck, J., Linehan, J. M., Rutherfoord, S., et al. (2007). Inherited prion disease with 5-OPRI: phenotype modification by repeat length and codon 129. Neurology 69, 730–738. doi: 10.1212/01.wnl.0000267642.41594.9d
Palmer, M. S., Mahal, S. P., Campbell, T. A., Hill, A. F., Sidle, K. C., Laplanche, J. L., et al. (1993). Deletions in the prion protein gene are not associated with CJD. Hum. Mol. Genet. 2, 541–544. doi: 10.1093/hmg/2.5.541
Parchi, P., Chen, S. G., Brown, P., Zou, W., Capellari, S., Budka, H., et al. (1998). Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Sträussler-Scheinker disease. Proc. Natl. Acad. Sci. U S A 95, 8322–8327. doi: 10.1073/pnas.95.14.8322
Parchi, P., Giese, A., Capellari, S., Brown, P., Schulz-Schaeffer, W., Windl, O., et al. (1999). Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 46, 224–233. doi: 10.1002/1531-8249(199908)46:2<224::aid-ana12>3.0.co;2-w
Parchi, P., and Saverioni, D. (2012). Molecular pathology, classification and diagnosis of sporadic human prion disease variants. Folia Neuropathol. 50, 20–45.
Parchi, P., Strammiello, R., Notari, S., Giese, A., Langeveld, J. P. M., Ladogana, A., et al. (2009). Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol. 118, 659–671. doi: 10.1007/s00401-009-0585-1
Piazza, M., Prior, T. W., Khalsa, P. S., and Appleby, B. (2020). A case report of genetic prion disease with two different PRNP variants. Mol. Genet. Genomic Med. 8:e1134. doi: 10.1002/mgg3.1134
Pietrini, V., Puoti, G., Limido, L., Rossi, G., Di Fede, G., Giaccone, G., et al. (2003). Creutzfeldt-Jakob disease with a novel extra-repeat insertional mutation in the PRNP gene. Neurology 61, 1288–1291. doi: 10.1212/01.wnl.0000092017.74772.ca
Puoti, G., Bizzi, A., Forloni, G., Safar, J. G., Tagliavini, F., and Gambetti, P. (2012). Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 11, 618–628. doi: 10.1016/s1474-4422(12)70063-7
Puoti, G., Giaccone, G., Rossi, G., Canciani, B., Bugiani, O., and Tagliavini, F. (1999). Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrP(Sc) in the same brain. Neurology 53, 2173–2176. doi: 10.1212/wnl.53.9.2173
Tateishi, J. (1991). Recent advances in the research of Creutzfeldt-Jakob disease (CJD) and Gerstmann-Strüssler syndrome (GSS). Rinsho Shinkeigaku 31, 1306–1308.
Wang, X. F., Guo, Y. J., Zhang, B. Y., Zhao, W. Q., Gao, J. M., Wan, Y. Z., et al. (2007). Creutzfeldt-Jakob disease in a Chinese patient with a novel seven extra-repeat insertion in PRNP. J. Neurol. Neurosurg. Psychiatry 78, 201–203. doi: 10.1136/jnnp.2006.09433
Xiao, X., Cali, I., Dong, Z., Puoti, G., Yuan, J., Qing, L., et al. (2013). Protease-sensitive prions with 144-bp insertion mutations. Aging 5, 155–173. doi: 10.18632/aging.100543
Keywords: genetic, histopathology, Prion protein (PrP), insertion mutation, conformation
Citation: Cali I, Cracco L, Saracino D, Occhipinti R, Coppola C, Appleby BS and Puoti G (2020) Case Report: Histopathology and Prion Protein Molecular Properties in Inherited Prion Disease With a De Novo Seven-Octapeptide Repeat Insertion. Front. Cell. Neurosci. 14:150. doi: 10.3389/fncel.2020.00150
Received: 17 March 2020; Accepted: 05 May 2020;
Published: 08 July 2020.
Edited by:
Ertugrul Kilic, Istanbul Medipol University, TurkeyReviewed by:
Ahmet Burak Caglayan, Istanbul Medipol University, TurkeyCopyright © 2020 Cali, Cracco, Saracino, Occhipinti, Coppola, Appleby and Puoti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ignazio Cali, aXhjMjBAY2FzZS5lZHU=; Gianfranco Puoti, Z2lhbmZyYW5jby5wdW90aUB1bmljYW1wYW5pYS5pdA==
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.