 Chaska C. Walton
Chaska C. Walton David Begelman
David Begelman Julie K. Andersen
Julie K. Andersen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 19 May 2020
Sec. Cellular Neuropathology
Volume 14 - 2020 | https://doi.org/10.3389/fncel.2020.00129
This article is part of the Research TopicThe Role of Cellular Senescence in Health and DiseaseView all 16 articles
Due to their postmitotic status, the potential for neurons to undergo senescence has historically received little attention. This lack of attention has extended to some non-postmitotic cells as well. Recently, the study of senescence within the central nervous system (CNS) has begun to emerge as a new etiological framework for neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). The presence of senescent cells is known to be deleterious to non-senescent neighboring cells via development of a senescence-associated secretory phenotype (SASP) which includes the release of inflammatory, oxidative, mitogenic, and matrix-degrading factors. Senescence and the SASP have recently been hailed as an alternative to the amyloid cascade hypothesis and the selective killing of senescence cells by senolytic drugs as a substitute for amyloid beta (Aß) targeting antibodies. Here we call for caution in rejecting the amyloid cascade hypothesis and to the dismissal of Aß antibody intervention at least in early disease stages, as Aß oligomers (AßO), and cellular senescence may be inextricably linked. We will review literature that portrays AßO as a stressor capable of inducing senescence. We will discuss research on the potential role of secondary senescence, a process by which senescent cells induce senescence in neighboring cells, in disease progression. Once this seed of senescent cells is present, the elimination of senescence-inducing stressors like Aß would likely be ineffective in abrogating the spread of senescence. This has potential implications for when and why AßO clearance may or may not be effective as a therapeutic for AD. The selective killing of senescent cells by the immune system via immune surveillance naturally curtails the SASP and secondary senescence outside the CNS. Immune privilege restricts the access of peripheral immune cells to the brain parenchyma, making the brain a safe harbor for the spread of senescence and the SASP. However, an increasingly leaky blood brain barrier (BBB) compromises immune privilege in aging AD patients, potentially enabling immune infiltration that could have detrimental consequences in later AD stages. Rather than an alternative etiology, senescence itself may constitute an essential component of the cascade in the amyloid cascade hypothesis.
Alzheimer’s disease (AD) is an as of yet incurable neurodegenerative disorder (Selkoe and Hardy, 2016). Its cardinal features are senile plaques formed by nonvascular extracellular deposits of amyloid fibrillary amyloid beta (Aß) and intra-neuronal neurofibrillary tangles (NFT) consisting of aggregates of hyperphosphorylated tau protein (Hyman et al., 2012; Deture and Dickson, 2019). The amyloid cascade hypothesis posits Aβ is the cause of AD, triggering the formation of NFT, neuronal cell loss, vascular damage, and dementia (Selkoe and Hardy, 2016). In the modernized version of the amyloid cascade hypothesis, rather than senile plaques it is now thought that soluble Aβ oligomers (AßO) are the major driver of AD. Aβ peptides result from enzymatic cleavage of the disease-associated amyloidogenic processing of APP (O’Brien and Wong, 2011). Aβ peptides of different lengths can seed formation of AßO, protofibrils, fibrils, and senile plaques (Larson and Lesné, 2012; Karran and De Strooper, 2016; Selkoe and Hardy, 2016; Deture and Dickson, 2019; Panza et al., 2019). The 40 (Aβ40) and 42 (Aβ42) amino acid length peptides are the most intensely studied, with Aβ42 being the most amyloidogenic.
Beyond the fact that extracellular Aβ deposits are a pathological diagnostic hallmark (Hyman et al., 2012), genetic data supporting the amyloid cascade hypothesis is rather powerful (Selkoe and Hardy, 2016). Clinical trial “failures” of Aβ-depleting antibodies are also powerful argument against the hypothesis (Karran and De Strooper, 2016; Panza et al., 2019). There are several antibodies designed to target Aβ, which in turn elicit their putative therapeutic effects by targeting different species of Aβ (Panza et al., 2019). Clearance of amyloid plaques by AN-1792 did not prevent disease progression (Holmes et al., 2008). As noted, neurotoxicity is thought to depend on AβO (Selkoe and Hardy, 2016). Adecanumab targets the neurotoxic effects associated with oligomers as well as prompting the dissolution of Aβ plaques (Sevigny et al., 2016; Panza et al., 2019). Despite this, by the first quarter of 2019 the Aducanumab ENGAGE (NTC02484547) and EMERGE (NTC02477800) trials were halted as a result of futility analysis (Schneider, 2020).
In the wake of this mounting discouraging clinical evidence, encouraging reports simultaneously begun to emerge supporting a new class of drugs known as senolytics as a novel therapeutic avenue for AD (Bussian et al., 2018; Musi et al., 2018; Zhang et al., 2019). Senolytics elicit the selective killing of senescent cells (Kirkland et al., 2017) and there was already proof of concept for their potential use in Parkinson’s disease (PD; Chinta et al., 2018). Most senescent cells develop a senescence associated secretory phenotype (SASP) involving the secretion of cytokines, chemokines, mitogenic factors, and proteases that can damage the surrounding microenvironment (Acosta et al., 2008; Coppé et al., 2008, 2010a,b; Kuilman et al., 2008; Neves et al., 2015). Senolytics eliminate senescent cells and therefore the SASP (Kirkland et al., 2017). Within the context of AD, proof of concept for senolytic therapies was first provided in mice expressing wild type or mutated human tau isoforms but lacking Aβ pathology (Bussian et al., 2018; Musi et al., 2018). A role for Aβ in the development of cellular senescence was rejected (Musi et al., 2018). Paradoxically a senolytic intervention was later proven successful in transgenic mice presenting Aβ without tau pathology (Zhang et al., 2019), underscoring that each of the proposed mechanisms of action of senolytic intervention are radically different.
Unexpectedly, by the last quarter of 2019 a subgroup of patients receiving high-dose Aducanumab treatments were claimed to have met its target outcomes in prodromal and early AD patients. Although not free of skepticism (Schneider, 2020), this has prompted the re-launch of a clinical trial to assess high dose Adecanumab to begin in March 2020 (NCT04241068). It is possible that Aβ may trigger a pathological cascade of events that may evolve on its own independent of the continued presence of Aβ and therefore its clearance after the cascade has already been set in motion may be too late (Selkoe and Hardy, 2016). Implementing early intervention in AD is particularly complex considering that the first signs of Aβ pathology can precede clinical AD by 15 to 25 years (Bateman et al., 2012; Villemagne et al., 2013; Vermunt et al., 2019). Both proponents and detractors of the amyloid cascade hypothesis seem to be in agreement on one thing; Aβ clearance is not likely effective in mild-to-moderate clinical AD. Antibodies such as Aducanumab may therefore only be effective at early stages, prior to the activation of the cascade. We arguably need to shift to therapies suppressing the cascade and yet we do not know what the cascade is. As we will review, the cascade may be senescence itself.
Senescence is traditionally regarded as an oncosuppressive mechanism that imposes an irreversible cell cycle withdrawal (Gorgoulis et al., 2019). The classical senescence-inducing stressor is DNA damage signaling associated to telomere attrition, better known as replicative senescence (RS; Hayflick and Moorhead, 1961; Gorgoulis et al., 2019). RS marks the end of the replicative lifespan of the cell, but cells can undergo senescence before reaching it. This is often termed stress-induced premature senescence (SIPS). Multiple stressors can result in SIPS including reactive oxygen species (ROS), oncogenes, and ionizing radiation (IR; Gorgoulis et al., 2019). Although there are exceptions, most stressors result in SIPS by causing persistent DNA damage either directly or indirectly (Alimonti et al., 2010; Freund et al., 2011; Ziegler et al., 2015; Wiley et al., 2016; Gorgoulis et al., 2019). ROS and IR can directly elicit DNA damage, while oncogenes often–albeit not invariantly-do so indirectly by aberrantly activating the DNA replication machinery (Halazonetis et al., 2008; Lecot et al., 2016; Gorgoulis et al., 2019). SIPS is often associated with cell cycle dysregulation but nevertheless cells that are not mitotically active can undergo senescence (Toledo et al., 2008; Alimonti et al., 2010).
Evidence for SIPS in neurons and non-neuronal cell types has been provided in in vitro and in vivo models of AD (Bhat et al., 2012; He et al., 2013; Zhang et al., 2019), and senescence markers have been described in neurons of AD patients (Arendt et al., 1996, 1998; McShea et al., 1997; Lüth et al., 2000). As we will argue, there is reasonable evidence that AβO is a SIPS-inducing stressor. Senescence is a largely irreversible phenotype (Gorgoulis et al., 2019). It follows that the clearance of AβO should prevent the onset of cellular senescence but not revert it once it is established. If senescence is the actual cascade of the amyloid cascade hypothesis it may be largely irrevocable, potentially explaining the failure of some Aβ-targeting antibodies in clinical trials.
Although it is not always the case, when it comes to neurons it is common to see use of the term “senescent-like phenotype” (Walton and Andersen, 2019). Senescent-like is a conservative denomination that reflects potentially insurmountable challenges in the study of senescence in neurons.
There are no universal markers of senescence and therefore use of a single senescent marker is not a reliable mean of proving senescence in any cell type (Hernandez-Segura et al., 2017, 2018; Gorgoulis et al., 2019). For example, a widely used senescence marker in non-neuronal cells is senescence-associated-beta-galactosidase (SA-ß-Gal; Debacq-Chainiaux et al., 2009). However, SA-ß-Gal has been shown to be up-regulated in neurons that lack other senescence markers (Piechota et al., 2016; Musi et al., 2018; Walton and Andersen, 2019). SA-ß-Gal is lysosomal and reflects the increased lysosomal mass in senescent cells but is not necessary nor causes senescence (Kurz et al., 2000; Lee et al., 2006; Hernandez-Segura et al., 2018; Gorgoulis et al., 2019). SA-ß-Gal in neurons has indeed been argued to simply reflect senescence-unrelated lysosome biogenesis (Piechota et al., 2016; Musi et al., 2018; Walton and Andersen, 2019). In order to prove neuronal senescence, multiple markers of senescence should be used which may include p16INK4A, p21CIP1, Lamin B1, HMGB1, and amongst others (Hernandez-Segura et al., 2018; Gorgoulis et al., 2019). The phenotype should also be relatively stable, as cellular senescence is considered an irreversible phenotype. With the aforementioned in mind, we propose that: 1. Multiple senescence markers need to be used to assess senescence in neurons; 2. The mechanism of action of any identified senescence-inducing stressor should be consistent with that in mitotically-competent cells; and 3. The phenotype should still persist after the senescence-inducing stressor has been removed. If successfully demonstrated, this would provide a convincing characterization of neuronal senescence.
Arguably the gold standard for identifying cellular senescence is demonstrating an irreversible block on cellular proliferation. Normally differentiated neurons never proliferate under physiological conditions (Frade and Ovejero-Benito, 2015). When non-physiological means are used to force neuronal cell division, the rate of success is under 5% and thus far entails detection of only a single cell division (Walton et al., 2019). Because neurons in general do not proliferate, it is not possible to prove that an irreversible block on proliferation is caused by senescence rather than their native postmitotic state. The best we can hope for is to characterize a senescent-like phenotype that is consistent with senescence. Nevertheless, given that this review concerns several cell types, to avoid cumbersome phrasing we will hereafter use the term “senescence” for neurons, and glial cells alike.
Aberrant cell cycle entry in neurons of AD patients is well established (Frade and Ovejero-Benito, 2015). It is important to distinguish this from adult neurogenesis. Adult neurogenesis entails the physiological proliferation of neuronal precursor cells (NPCs), which latter differentiate into two specific neuronal types in very restricted niches (Gross, 2000; Kempermann et al., 2018). In contrast, aberrant activation of the cell cycle in AD patients takes place within neurons and is strictly pathological (Frade and Ovejero-Benito, 2015). Cell cycle reactivation in neurons of AD patients does not result in cell division (Frade and Ovejero-Benito, 2015). Neurons in the brains of AD patients have been shown to survive for extended periods of time after cell cycle entry (Arendt et al., 2010). This is consistent with cells having undergone cellular senescence after an abortive cell cycle (Kastan and Bartek, 2004; Halazonetis et al., 2008; Vitale et al., 2011; Johmura et al., 2014). In support of this, up-regulation of the senescence marker p16INK4A was reported within pyramidal neurons of AD patients in several older historic studies, suggesting the potential involvement of senescence in AD as early as two decades ago (Arendt et al., 1996, 1998; McShea et al., 1997; Lüth et al., 2000).
Given that potential evidence for senescence in AD patients has existed for years, it is somewhat curious that research of senescence in neurobiology is only now blooming (Walton and Andersen, 2019). The historic paucity of studies of senescence in AD may be explained by a misunderstanding of mitotic cell biology. For many years, forcing primary neurons in culture to enter the cell cycle resulted in cell death (Frade and Ovejero-Benito, 2015). In one way or another, many thought that cell death was a consequence of the postmitotic status of neurons, likely influenced by the erroneous presumption that mitotically-competent cells never undergo cell death upon cell cycle entry. Oncogenes can and do force cell cycle entry in mitotically-competent cells, where cell death is an indispensable response to prevent carcinogenic cell division (Kastan and Bartek, 2004; Brito and Rieder, 2006; Halazonetis et al., 2008; Vitale et al., 2011; Johmura et al., 2014). The first in vitro models of an abortive cell cycle with viable exit were achieved by inactivating the same machinery that causes oncosuppressive cell death and senescence in mitotically-competent cells (Barrio-Alonso et al., 2018; Walton et al., 2019). Hence, cell death after forced cell cycle suggests that neurons possess at least one of two major oncosuppressive mechanisms present in virtually all mitotically-competent cells. The other described mechanism for oncogenic suppression is cellular senescence (Kastan and Bartek, 2004; Halazonetis et al., 2008; Vitale et al., 2011; Childs et al., 2014; Johmura et al., 2014; Lecot et al., 2016; Gorgoulis et al., 2019).
As noted, senolytics have been demonstrated to have a therapeutic effect in a tau transgenic mice models (Bussian et al., 2018; Musi et al., 2018). Whether it is the killing of senescent neurons or senescent glia remains unclear (Walton and Andersen, 2019). In the case in which it was argued that senescent neurons were killed, it was speculated that senescence was caused by NFT mediated cell cycle entry (Musi et al., 2018). Indeed, there are some reports of tau-induced cell cycle entry in neurons (Andorfer et al., 2005; Bonda et al., 2009; Jaworski et al., 2009; Seward et al., 2013a; Hradek et al., 2014), albeit entry mediated by NFT has been contested (Jaworski et al., 2009), and at least in some cases it involves Aβ (Seward et al., 2013a; Hradek et al., 2014). When compared to tau-related models, cell cycle deregulation has been vigorously researched in models of Aβ pathology (Copani et al., 1999, 2006; Giovanni et al., 1999, 2000; Park et al., 2000; Wu et al., 2000; Kruman et al., 2004; Sortino et al., 2004; Yang et al., 2006; Caraci et al., 2008; Majd et al., 2008; Varvel et al., 2008, 2009; Bhaskar et al., 2009; Lopes et al., 2009, 2010; Li et al., 2011; Modi et al., 2012, 2015; Seward et al., 2013b; Hradek et al., 2014; Merlo et al., 2015; Caraci et al., 2016; Leggio et al., 2016; Table 1). Virtually all reports report cell cycle related cell death, which is of relevance for senescence. As noted above, aberrant cell cycle activation should result in cell death or senescence. For example, the E2F1 transcription factor, a major driver of G1/S transition, can also result in cell death or senescence if aberrantly activated (Johnson and DeGregori, 2012). Fittingly, Aβ-mediated cell death is at least in part mediated by E2F1 (Giovanni et al., 1999, 2000). It may be argued that if aberrant cell cycle entry is the means by which neurons senesce in AD, Aβ is the most likely cause.
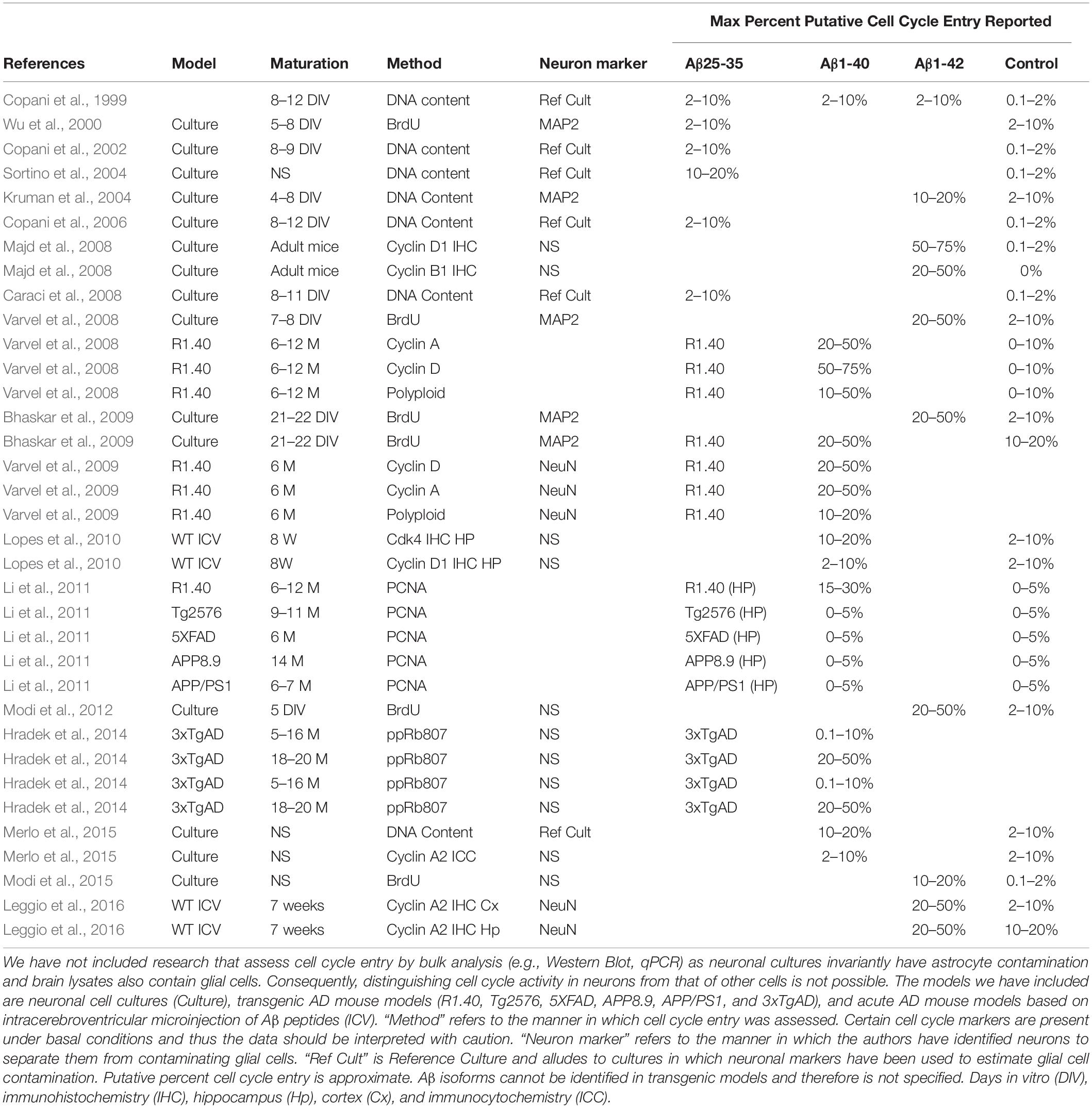
Table 1. Literature describing cell cycle entry downstream of Aβ treatment or in mouse models of AD.
Amyloid beta pathology models have been used to study senescence despite the lack of studies in neuronal cell cycle dysregulation. Aβ peptide administration is shown to induce senescence in astrocytes in vitro and the number of astrocytes with a senescent phenotype are increased in AD patients (Bhat et al., 2012). Increased expression of p16 is reported in neurons of an AD mouse model and AβO reportedly increase p16 levels in vitro (Wei et al., 2016). Mechanisms involving ROS are known to induce senescence in mitotically competent cells (Gorgoulis et al., 2019). AβO exposure induces senescence via ROS in NPCs from wild type (WT) and AD mouse models (He et al., 2013). DNA damage can result in persistent DDR and p21-mediated mitochondrial dysfunction leading to increased ROS production (Passos et al., 2010). ROS regenerates DNA damage, locking cells into senescence. A similar senescent-like phenotype has been described in the neurons of aging mice (Jurk et al., 2012). Thus, increases in ROS downstream of AβO is a plausible mechanism for senescence induction.
There is extensive research linking Aβ to ROS in neurons. Extracellular actions of AβO elicits neurotoxic effects via ROS production and Ca2+ dysregulation by binding to N-Methyl-D-Aspartate receptor (NMDAr) on excitatory synapses (De Felice et al., 2007; Lacor et al., 2007; Shelat et al., 2008; Gunn et al., 2016; Smith and Strittmatter, 2017). In fit, toxic, or physiological ROS levels are produced downstream of NMDAr excitotoxic or normal activation, respectively (Dugan et al., 1995; Reynolds and Hastings, 1995; Ward et al., 2000; Brennan et al., 2009). AβO exposure in vitro induces ROS and JNK pathway activation (Kadowaki et al., 2005), up-regulation of p38, and Ca2+ influx (Drews et al., 2016). Intracellularly-acting AβO has been associated with oxidative stress and ROS production via mitochondrial dysfunction, Ca2+ perturbation, and trace element interactions. AβO impacts mitochondrial dysfunction via inhibition of nuclear protein import (Sirk et al., 2007; Cenini et al., 2016), abnormal fission and fusion dynamics (Barsoum et al., 2006; Zhang et al., 2008; Hung et al., 2018), ATP synthase activity impairment (Schmidt et al., 2008; Cha et al., 2015; Beck et al., 2016), and an up-regulation of mitochondrial production of ROS (Rhein et al., 2009; Mao and Reddy, 2011; Mossmann et al., 2014; Kaminsky et al., 2015; Hung et al., 2018; Fang et al., 2019). AβO accumulation within the mitochondria directly interacts with ABAD and cyclophilin D, promoting ROS leakage, membrane potential change, and Ca2+ dysregulation (Lustbader et al., 2004; Hemmerová et al., 2019; Morsy and Trippier, 2019). Perturbation of mitochondrial or endoplasmic reticulum (ER)-mediated Ca2+ homeostasis may underlie intracellular-mediated Aβ excitotoxicity. Intracellular AβO modulates resting cytosolic free Ca2+ levels (Sanz-Blasco et al., 2008; Demuro and Parker, 2013; Müller et al., 2018; Jadiya et al., 2019), remodels intra-organellar Ca2+ by disruption of mitochondria-associated ER membranes (MAMs; Müller et al., 2018), and alters Ca2+ release from internal stores (Sanz-Blasco et al., 2008; Müller et al., 2018; Calvo-Rodriguez et al., 2019; Jadiya et al., 2019), which can lead to ROS formation and further pathological oligomerization of Aβ (Kadowaki et al., 2005; Meli et al., 2014; Kaminsky et al., 2015; Boyman et al., 2020). Further, production of ROS can be mediated by Aβ interaction with transition metals, specifically copper or iron, to produce hydrogen peroxide, and superoxide via Fenton reaction (Jomova et al., 2010; Cheignon et al., 2018; Masaldan et al., 2018; Butterfield and Halliwell, 2019; Gomes et al., 2019). AβO-dependent increase in ROS and activation of the cell cycle machinery are alternative ways in which AβO can induce potentially senescence in several cell types (Figure 1).
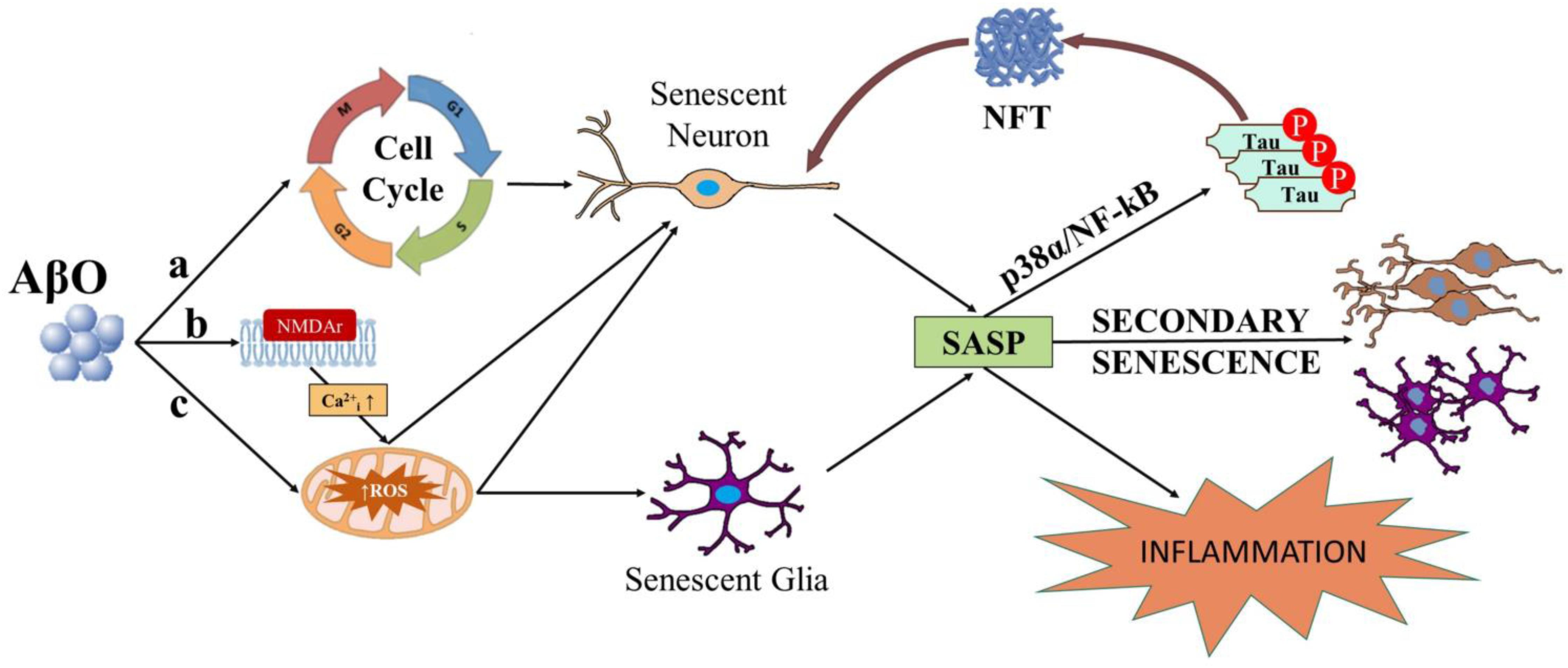
Figure 1. Diagram depicting possible ways in which AβO can induce senescence in neurons and or glial cells. AβO can induce unscheduled cell cycle entry in neurons leading to senescence (a). AβO can also induce ROS, either downstream of aberrant activation of post-synaptic receptors (e.g., NMDAr) in neurons (b) or independent of receptor activation in neurons and or glial cells (c). Senescent neurons and or glial cells may develop the SASP. SASP factors released from senescent neurons and or glial cells can result in inflammation and secondary senescence, spreading the disease. Major SASP regulators p38 and NF-kB may also elicit tau hyperphosphorylation, potentially linking the SASP to NFTs in neurons.
Within a senescence-based hypothesis of AD, the consensus is that the SASP is the culprit for subsequent observed disease phenotypes (Bussian et al., 2018; Musi et al., 2018; Walton and Andersen, 2019; Zhang et al., 2019). Although the SASP is a very heterogeneous phenotype (Basisty et al., 2020), secreted components often include interleukin IL-6, chemokine IL-8, or TGFβ (Coppé et al., 2010a; Neves et al., 2015; Gorgoulis et al., 2019). Prominent SASP regulators include p38MAPK, NF-kB C/EBPβ, GATA4, and mammalian target of rapamycin (mTOR). Senescent markers are up-regulated in the astrocytes of AD patients and Aβ has been reported to elicit senescence in astrocytes in vitro via ROS accompanied by p38, IL-6, and IL-8 up-regulation (Bhat et al., 2012). Further, oligodendrocyte precursor cells (OPC) in an AD mouse model show a pro-inflammatory phenotype along with increases p16 and p21 expression near Aβ plaques and Aβ can induce senescence in cultured OPCs (Zhang et al., 2019). IL-6 and TGFβ mRNA are upregulated in AD patients (Luterman et al., 2000; Gruol, 2015), but inflammatory response from resident immune cells is also prominent in AD and cannot be ruled out (Heneka et al., 2015). However, active p38 overlaps and immunoprecipitates with NFT from neurons of AD patients, but not from healthy controls (Zhu et al., 2000). Aβ activation of p38 has been placed upstream of pathological tau phosphorylation in neuron cultures (Origlia et al., 2008; Munoz and Ammit, 2010). Aβ activates NF-kB in primary neurons and is found in astrocytes and neurons in proximity to senile or diffuse plaques as well as NFT-positive neurons in AD patients, but not in healthy controls (Terai et al., 1996; Kaltschmidt et al., 1997; Ferrer et al., 1998; Snow and Albensi, 2016). A “senescent-like” phenotype described in neurons of aging mice involves p38, ROS and intraneuronal IL-6 suggesting neurons may develop a “SASP-like” phenotype during normal aging (Jurk et al., 2012), which, in light of aforementioned evidence, could be exacerbated in AD.
It is plausible that AβO can result in senescence, with subsequent increases in p38 and NF-kB activity reflecting the development of the SASP. Given the association of active p38 and NF-kB with NFTs (Munoz and Ammit, 2010; Gruol, 2015), tau pathology may be part of the SASP and therefore a feature of senescence whether the latter is caused by Aβ or not (Figure 1). Hence, the cascade of the amyloid cascade hypothesis may be irreversible if it is indeed cellular senescence and deleterious if it also involves the SASP.
Senescent cells can induce senescence in neighboring non-senescent cells via a process known as paracrine senescence or secondary senescence (hereafter secondary senescence; Gorgoulis et al., 2019). Secondary senescence is dependent on SASP factors and has been shown to be transmitted by either diffusible factors, gap-junctions, or both (Hubackova et al., 2012; Nelson et al., 2012, 2018; Acosta et al., 2013; Jurk et al., 2014; da Silva et al., 2019). It may be expected that a population of senescent astrocytes, microglia, OPCs, and or NPCs radiate away from Aβ foci by secondary senescence. Consequently, these senescent cells would be expected to be found near regions of Aβ burden but not necessarily elsewhere, which does not appear to be consistent with what is known about AD progression. Senile plaques and NFT in AD largely follow a stereotypical pattern of neuroanatomical distribution (Hyman et al., 2012; Deture and Dickson, 2019). This has led to the Thal staging of amyloid phases (Thal et al., 2002) and the Braak staging based on hyperphosphorylated tau and NFTs (Braak and Braak, 1991; Braak et al., 2006). In Braak stages I and II, NFTs first develop in the trans-entorhinal cortex and layers two and four of the entorhinal cortex. In stages III and IV, NFT burden is also present in the hippocampus. In the last phase, and stages V and VI, tau pathology is spread to the neocortex. Paradoxically, the neocortex is the first site of Aβ deposits in Thal phase 1 (Thal et al., 2002). In phase 2/3 there is a spread to allocortical brain regions, including the hippocampus and entorhinal cortex. Stage 3, 4, and 5 entail a spread into subcortical and cerebellar regions. Albeit not impossible, this progression is hard to reconcile with the spread of AD neuropathology being driven by senescent astrocyte, microglia, OPCs, and or NPCs. In contrast, neurons emit long-range axonal projections that can span the entire brain. If cortical neurons senescence and develop the SASP in response to AβO, then SASP and secondary senescence can reach distant regions that are free of Aβ. Importantly, the trans-entorhinal region converges widespread afferent projections from the neocortex (Vismer et al., 2015). At least in theory, a sparse population of neocortical senescent neurons spawned by diffuse AβO can converge their axonal projections into trans-entorhinal and entorhinal cortices, potentially allowing spread of the SASP and secondary senescence from the entire cortex. While cellular senescence and the SASP may, respectively, render the amyloid cascade irreversible and deleterious, secondary senescence can explain why the cascade results in the striking topographical spread of AD neuropathology.
Outside the central nervous system (CNS) senescent cells are normally cleared by the innate immune system (Lujambio, 2016). Senescent cells have been shown to be cleared by natural killer cells (NKs; Iannello et al., 2013; Eggert et al., 2016; Sagiv et al., 2016; Antonangeli et al., 2019; Muñoz et al., 2019; Pereira et al., 2019) and macrophages (Xue et al., 2007; Krizhanovsky et al., 2008; Kang et al., 2011; Muñoz-Espín et al., 2013). The way in which NKs kill senescent cells in the periphery is well-understood (Antonangeli et al., 2019). The activation of NKs depends on a complex interplay between their activator and inhibitory receptors. Specifically, human senescent cells up-regulate MICA, and ULBP2, ligands for the stimulatory receptor NKG2D (Antonangeli et al., 2019). Given the CNS is under immune privilege, T-cells, NKs, and peripheral macrophages normally have limited access to the meninges and choroid plexus and far-limited access to the CNS parenchyma (Galea et al., 2007; Korin et al., 2017; Benakis et al., 2018). Thus, regardless of which CNS cell types undergo senescence, the clearance of senescent cells is likely limited in healthy non-aged individuals. Senescence cells, the SASP and secondary senescence, may therefore continue in the brain for years to decades.
A feature of aging that is exacerbated in AD is the progressive dysfunction of the blood brain barrier (BBB) resulting in immune cell infiltration (Gorlé et al., 2016; Sweeney et al., 2018; Nation et al., 2019). In AD, the question arises as to whether loss of immune privilege with advancing age eventually enables immune cells to kill senescent that have accumulated for years in the CNS. Infiltrating monocytes and their derived macrophages have been studied in AD (Herz et al., 2017), albeit their role is unlikely linked to senescent cell killing. In as far as microglia are considered CNS resident macrophages, a case for microglia-mediated killing of senescent cells could potentially be made. This would require infiltrating CD4+ T-cells, as peripheral macrophages appear to depend on these cells to kill senescent cells outside the CNS (Kang et al., 2011). To the best of our knowledge, there is no evidence that microglia selectively kill senescent cells. Studies have focused on infiltrating CD8+ T-cells in AD (Lindestam Arlehamn et al., 2019), some with surprising results (Gate et al., 2020), yet these are cells from the adaptive immune system that do not seem to play a relevant role in senescence immune surveillance (Antonangeli et al., 2019). With regards to NK cells, studies in AD patients and AD mouse models have assessed peripheral but not infiltrating NKs (Solana et al., 2018). Unfortunately, from these studies it is hard to infer what may be happening within the brain parenchyma.
There is abundant evidence that NK cells can infiltrate and kill brain cells under other pathological conditions such as ischemia and NK are known to kill stressed neurons in co-culture (Backström et al., 2003, 2007; Poli et al., 2013; Gan et al., 2014; Zhang et al., 2014; Li et al., 2017; Wang et al., 2018). Interestingly, under non-stressed conditions primary hippocampal neurons have been reported to be protected against NK cell killing by the lack of expression of NKG2D ligands (Backström et al., 2003, 2007). These same stress ligands are up-regulated in senescent cells and target them for killing by NKs (Antonangeli et al., 2019). Whether NKs can infiltrate the brain at latter stages of the disease and selectively eliminate senescent neurons, astrocytes or other CNS cell types will require further studies, although existing data fits with this possibility.
Evidence that senolytic intervention may be an effective treatment for AD was first reported in tau transgenic mice models of frontotemporal dementia (FTD; Bussian et al., 2018; Musi et al., 2018), which do not present amyloid plaques but are arguably an AD-like tau pathology model. Shortly thereafter, senolytics were proven therapeutic in APP/PSEN1 AD transgenic models which present plaques but not tau pathology (Zhang et al., 2019). The beneficial effects of senolytic intervention are attributed to either the selective killing of senescent astrocytes and microglia–not neurons-(Bussian et al., 2018) or the selective killing of neurons–not astrocytes-(Musi et al., 2018) in these tau models and to the selective killing of senescent OPCs –not astrocytes nor microglia-in the APP/PSEN1 AD model (Zhang et al., 2019). The use of AD and FTD models may explain why senescent OPCs appear to be the culprit in the AD mouse model but not in the FTD models. The selective killing of neurons versus glial cells within the FTD models is harder to reconcile, albeit different transgenic tau models were used (Bussian et al., 2018; Musi et al., 2018). The future of senolytics requires resolving the cell type that is killed, but more importantly whether they kill neurons or not (Walton and Andersen, 2019). A prime objective in future studies should include a thorough brain-wide assessment of neuronal cell death after senolytic intervention.
Cdkn2a up-regulation as measured by qPCR was reported to be absent in 15 month old 3xTg-AD mice by Musi et al. (2018). Cdkn2a encodes not only the widely used senescence marker p16INK4A but also p19ARF (p14ARF in humans), the latter of which can have opposing functions (Baker et al., 2008). Zhang et al. (2019) report an increase in Cdkn2a in Aβ-producing APP/PS1 transgenic mice crossed with the INK-ATTAC mice. The INK-ATTAC transgene expresses a fluorescent reporter from the p16 promoter which, contrary to qPCR, and allows identification of specific cell types transcribing p16. The conflicting results between the 3xTg-AD (Musi et al., 2018) and the APP/PS1 (Zhang et al., 2019) mice can be resolved by considering reported temporal expression of intraneuronal versus extraneuronal Aβ species in the 3xTg-AD mice. Intraneuronal AβO is present at 6 months, followed by a dip in expression that is only fully restored at 20 months (Oddo et al., 2006). While intraneuronal Aβ peptides are present at 6 months of age, extracellular Aβ is not readily evident until 18 months of age (Oddo et al., 2008). Hence, at 15 month of age, neither extracellular nor intracellular AβO pathology is fully developed in the 3xTg-AD. Attention should be paid to the presence of monomeric, oligomeric, and fibrillary Aβ as well as whether it is intracellular and or extracellular. Particularly considering that extracellular amyloid plaques are a diagnostic hallmark of AD (Hyman et al., 2012; Deture and Dickson, 2019), some AβO act exclusively extracellularly (Larson and Lesné, 2012), extracellular AβO preparations can interact with receptors and the plasma membrane itself (Chen et al., 2017), and kill primary neurons (Yankner et al., 1990), alter synaptic functions (Verdier et al., 2004), induce tau-hyperphosphorylation (De Felice et al., 2008), and the vast majority of the in vitro experiments referenced above regarding ROS and the cell cycle are based on extracellular Aβ administration. Future work is needed to assess senolytic interventions in transgenic mice with combined Aβ and tau pathology at stages in which both extracellular and intracellular Aβ pathologies are fully developed.
Failure of Aβ antibody-mediated clearance in clinical trials challenging the amyloid cascade hypothesis occurred around the same time that senolytic interventions began emerging as a therapeutic alternative for the treatment of AD. Senescence and the amyloid cascade hypothesis have generally been presented as separate etiological phenomena during the progression of AD. This unfortunately has led to a disregard of the abundant literature that directly and indirectly supports the ability of AβO to induce cellular senescence. Aβ pathology models have been shown to induce senescence in astrocytes (Bhat et al., 2012), OPCs (Zhang et al., 2019), and NPCs (He et al., 2013). As discussed above, AβO has also been shown to induce aberrant cell cycle entry and ROS in neurons, placing it upstream of stressors that are known to induce senescence in mitotically-competent CNS cell types (Gorgoulis et al., 2019). Rather than senescence being an alternative etiology to the amyloid cascade hypothesis, we describe aspects of senescence that potentially allow substitution of the term “senescence” for “cascade” which we propose as a novel amyloid-senescence hypothesis (Figure 2). Future studies will be required to determine whether senescence provides the “cascade” in the amyloid cascade hypothesis. However, based on the current literature, it is likely too early to reject an amyloid-senescence hypothesis out of hand.
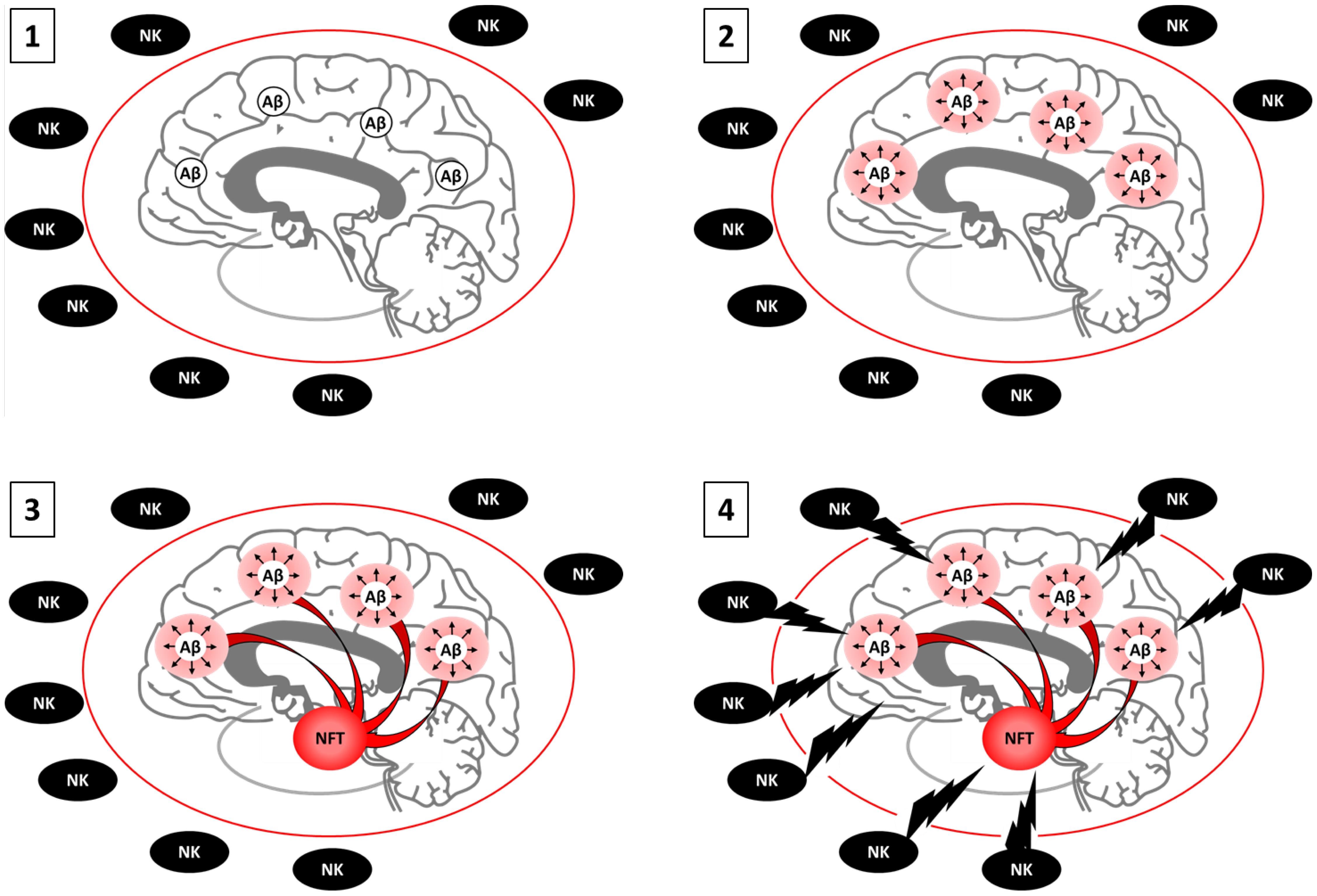
Figure 2. Proposed model of the amyloid-senescence hypothesis. 1. AβO burden appears 15 to 25 years prior to clinical onset of AD. At this point, antibody-mediated clearance is effective. 2. AβO causes foci of senescent neurons, astrocytes, microglia, OPCs, and or NPCs by inducing aberrant cell cycle entry and or persistent ROS and DNA damage. Senescence cells and the SASP cause oxidative stress, inflammation, and initial stages of cognitive impairment. At early stages, the BBB is healthy. NKs cannot access the brain parenchyma to clear senescent cells, which remain viable. Antibody mediated clearance of AβO is no longer effective because senescence is irreversible. Senolytics can stop disease progression 3. Foci of senescent neurons from across the cortex project into the transentorhinal cortex, where deleterious SASP and secondary senescence builds for years to decades. The SASP is accompanied by tau hyperphosphorylation and NFT. Cognitive impairment is accentuated. If too many senescent neurons are present, senolytics may prove fatal as neurons cannot regenerate. Senostatics, which target the SASP without killing cells, are an alternative. 4. Age-associated disruption of the BBB enables NK cell infiltration and the killing of a large pool of senescent neurons, astrocyte, microglia, OPCs, and or NPCs, increasing inflammation and marking the onset of clinical AD. Immune suppression may be the only viable alternative at this stage. Boosting acetylcholine levels can help suppress the NK cell response. Individuals with senile plaques and NFT with a healthy BBB may remain relatively spared.
CW, DB, and WN wrote the manuscript. JA edited the manuscript.
This study was supported by awards from the Pittsburgh Foundation and the Michael J. Fox Foundation (JA), a T32 fellowship (CW), and SENS Research Foundation Post-Bac Scholarships (DB and WN).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acosta, J. C., Banito, A., Wuestefeld, T., Georgilis, A., Janich, P., Morton, J. P., et al. (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990. doi: 10.1038/ncb2784
Acosta, J. C., O’Loghlen, A., Banito, A., Guijarro, M. V., Augert, A., Raguz, S., et al. (2008). Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018. doi: 10.1016/j.cell.2008.03.038
Alimonti, A., Nardella, C., Chen, Z., Clohessy, J. G., Carracedo, A., Trotman, L. C., et al. (2010). A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J. Clin. Invest. 120, 681–693. doi: 10.1172/JCI40535
Andorfer, C., Acker, C. M., Kress, Y., Hof, P. R., Duff, K., and Davies, P. (2005). Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 25, 5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005
Antonangeli, F., Zingoni, A., Soriani, A., and Santoni, A. (2019). Senescent cells: living or dying is a matter of NK cells. J. Leukoc. Biol. 105, 1275–1283. doi: 10.1002/JLB.MR0718-299R
Arendt, T., Brückner, M. K., Mosch, B., and Lösche, A. (2010). Selective cell death of hyperploid neurons in Alzheimer’s disease. Am. J. Pathol. 177, 15–20. doi: 10.2353/ajpath.2010.090955
Arendt, T., Holzer, M., and Gärtner, U. (1998). Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer’s disease. J. Neural Transm. 105, 949–960. doi: 10.1007/s007020050104
Arendt, T., Rödel, L., Gärtner, U., and Holzer, M. (1996). Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer’s disease. Neuroreport 7, 3047–3049. doi: 10.1097/00001756-199611250-00050
Backström, E., Chambers, B. J., Ho, E. L., Naidenko, O. V., Mariotti, R., Fremont, D. H., et al. (2003). Natural killer cell-mediated lysis of dorsal root ganglia neurons via RAE1/NKG2D interactions. Eur. J. Immunol. 33, 92–100. doi: 10.1002/immu.200390012
Backström, E., Ljunggren, H. G., and Kristensson, K. (2007). NK cell-mediated destruction of influenza A virus-infected peripheral but not central neurones. Scand. J. Immunol. 65, 353–361. doi: 10.1111/j.1365-3083.2007.01912.x
Baker, D. J., Perez-Terzic, C., Jin, F., Pitel, K., Niederländer, N. J., Jeganathan, K., et al. (2008). Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat. Cell Biol. 10, 825–836. doi: 10.1038/ncb1744
Barrio-Alonso, E., Hernández-Vivanco, A., Walton, C. C., Perea, G., and Frade, J. M. (2018). Cell cycle reentry triggers hyperploidization and synaptic dysfunction followed by delayed cell death in differentiated cortical neurons. Sci. Rep. 8:14316. doi: 10.1038/s41598-018-32708-4
Barsoum, M. J., Yuan, H., Gerencser, A. A., Liot, G., Kushnareva, Y., Gräber, S., et al. (2006). Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 25, 3900–3911. doi: 10.1038/sj.emboj.7601253
Basisty, N., Kale, A., Jeon, O. H., Kuehnemann, C., Payne, T., Rao, C., et al. (2020). A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 18:e3000599. doi: 10.1371/journal.pbio.3000599
Bateman, R. J., Xiong, C., Benzinger, T. L. S., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Beck, S. J., Guo, L., Phensy, A., Tian, J., Wang, L., Tandon, N., et al. (2016). Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 7:11483. doi: 10.1038/ncomms11483
Benakis, C., Llovera, G., and Liesz, A. (2018). The meningeal and choroidal infiltration routes for leukocytes in stroke. Ther. Adv. Neurol. Disord. 11:1756286418783708. doi: 10.1177/1756286418783708
Bhaskar, K., Miller, M., Chludzinski, A., Herrup, K., Zagorski, M., and Lamb, B. T. (2009). The PI3K-Akt-mTOR pathway regulates a oligomer induced neuronal cell cycle events. Mol. Neurodegener. 4:14. doi: 10.1186/1750-1326-4-14
Bhat, R., Crowe, E. P., Bitto, A., Moh, M., Katsetos, C. D., Garcia, F. U., et al. (2012). Astrocyte senescence as a component of Alzheimer’s disease. PLoS One 7:45069. doi: 10.1371/journal.pone.0045069
Bonda, D. J., Evans, T. A., Santocanale, C., Llosá, J. C., Viña, J., Bajic, V. P., et al. (2009). Evidence for the progression through S-phase in the ectopic cell cycle re-entry of neurons in Alzheimer disease. Aging 1, 382–388. doi: 10.18632/aging.100044
Boyman, L., Karbowski, M., and Lederer, W. J. (2020). Regulation of mitochondrial ATP production: Ca2+ signaling and quality control. Trends Mol. Med. 26, 21–39. doi: 10.1016/j.molmed.2019.10.007
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H., and Tredici, K. (2006). Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404. doi: 10.1007/s00401-006-0127-z
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Brennan, A. M., Won Suh, S., Joon Won, S., Narasimhan, P., Kauppinen, T. M., Lee, H., et al. (2009). NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 12, 857–863. doi: 10.1038/nn.2334
Brito, D. A., and Rieder, C. L. (2006). Mitotic checkpoint slippage in humans occurs via cyclin b destruction in the presence of an active checkpoint. Curr. Biol. 16, 1194–1200. doi: 10.1016/j.cub.2006.04.043
Bussian, T. J., Aziz, A., Meyer, C. F., Swenson, B. L., van Deursen, J. M., and Baker, D. J. (2018). Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582. doi: 10.1038/s41586-018-0543-y
Butterfield, D. A., and Halliwell, B. (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 20, 148–160. doi: 10.1038/s41583-019-0132-6
Calvo-Rodriguez, M., Hernando-Perez, E., Nuñez, L., and Villalobos, C. (2019). Amyloid β oligomers increase ER-mitochondria Ca2+ cross talk in young hippocampal neurons and exacerbate aging-induced intracellular Ca2+ remodeling. Front. Cell. Neurosci. 13:22. doi: 10.3389/fncel.2019.00022
Caraci, F., Battaglia, G., Busceti, C., Biagioni, F., Mastroiacovo, F., Bosco, P., et al. (2008). TGF-β1 protects against Aβ-neurotoxicity via the phosphatidylinositol-3-kinase pathway. Neurobiol. Dis. 30, 234–242. doi: 10.1016/j.nbd.2008.01.007
Caraci, F., Tascedda, F., Merlo, S., Benatti, C., Spampinato, S. F., Munafò, A., et al. (2016). Fluoxetine prevents Aβ1-42-induced toxicity via a paracrine signaling mediated by transforming-growth-factor-β1. Front. Pharmacol. 7:389. doi: 10.3389/fphar.2016.00389
Cenini, G., Rub, C., Bruderek, M., and Voos, W. (2016). Amyloid β-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol. Biol. Cell 27, 3257–3272. doi: 10.1091/mbc.E16-05-0313
Cha, M. Y., Cho, H. J., Kim, C., Jung, Y. O., Kang, M. J., Murray, M. E., et al. (2015). Mitochondrial ATP synthase activity is impaired by suppressed O-GlcNAcylation in Alzheimer’s disease. Hum. Mol. Genet. 24, 6492–6504. doi: 10.1093/hmg/ddv358
Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018). Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14, 450–464. doi: 10.1016/j.redox.2017.10.014
Chen, G. F., Xu, T. H., Yan, Y., Zhou, Y. R., Jiang, Y., Melcher, K., et al. (2017). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235. doi: 10.1038/aps.2017.28
Childs, B. G., Baker, D. J., Kirkland, J. L., Campisi, J., and van Deursen, J. M. (2014). Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 15, 1139–1153. doi: 10.15252/embr.201439245
Chinta, S. J., Woods, G., Demaria, M., Rane, A., Zou, Y., McQuade, A., et al. (2018). Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 22, 930–940. doi: 10.1016/j.celrep.2017.12.092
Copani, A., Condorelli, F., Caruso, A., Vancheri, C., Sala, A., Giuffrida Stella, A. M., et al. (1999). Mitotic signaling by beta-amyloid causes neuronal death. FASEB J. 13, 2225–2234.
Copani, A., Hoozemans, J. J. M., Caraci, F., Calafiore, M., Van Haastert, E. S., Veerhuis, R., et al. (2006). DNA polymerase-β is expressed early in neurons of Alzheimer’s disease brain and is loaded into DNA replication forks in neurons challenged with β-amyloid. J. Neurosci. 26, 10949–10957. doi: 10.1523/JNEUROSCI.2793-06.2006
Copani, A., Melchiorri, D., Caricasole, A., Martini, F., Sale, P., Carnevale, R., et al. (2002). Beta-amyloid-induced synthesis of the ganglioside GD3 is a requisite for cell cycle reactivation and apoptosis in neurons. J. Neurosci. 22, 3963–3968. doi: 10.1523/JNEUROSCI.22-10-03963.2002
Coppé, J.-P., Desprez, P.-Y., Krtolica, A., and Campisi, J. (2010a). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118. doi: 10.1146/annurev-pathol-121808-102144
Coppé, J. P., Patil, C. K., Rodier, F., Krtolica, A., Beauséjour, C. M., Parrinello, S., et al. (2010b). A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One 5:e9188. doi: 10.1371/journal.pone.0009188
Coppé, J. P., Patil, C. K., Rodier, F., Sun, Y., Muñoz, D. P., Goldstein, J., et al. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6:e301. doi: 10.1371/journal.pbio.0060301
da Silva, P. F. L., Ogrodnik, M., Kucheryavenko, O., Glibert, J., Miwa, S., Cameron, K., et al. (2019). The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 18:e12848. doi: 10.1111/acel.12848
De Felice, F. G., Velasco, P. T., Lambert, M. P., Viola, K., Fernandez, S. J., Ferreira, S. T., et al. (2007). Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282, 11590–11601. doi: 10.1074/jbc.M607483200
De Felice, F. G., Wu, D., Lambert, M. P., Fernandez, S. J., Velasco, P. T., Lacor, P. N., et al. (2008). Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers. Neurobiol. Aging 29, 1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029
Debacq-Chainiaux, F., Erusalimsky, J. D., Campisi, J., and Toussaint, O. (2009). Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 4, 1798–1806. doi: 10.1038/nprot.2009.191
Demuro, A., and Parker, I. (2013). Cytotoxicity of intracellular Aβ42 amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J. Neurosci. 32, 11820–11834. doi: 10.1523/JNEUROSCI.4367-12.2013
Deture, M. A., and Dickson, D. W. (2019). The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 14:32. doi: 10.1186/s13024-019-0333-5
Drews, A., Flint, J., Shivji, N., Jönsson, P., Wirthensohn, D., De Genst, E., et al. (2016). Individual aggregates of amyloid beta induce temporary calcium influx through the cell membrane of neuronal cells. Sci. Rep. 6:31910. doi: 10.1038/srep31910
Dugan, L. L., Sensi, S. L., Canzoniero, L. M., Handran, S. D., Rothman, S. M., Lin, T. S., et al. (1995). Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-Aspartate. J. Neurosci. 15, 6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995
Eggert, T., Wolter, K., Ji, J., Ma, C., Yevsa, T., Klotz, S., et al. (2016). Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell 30, 533–547. doi: 10.1016/j.ccell.2016.09.003
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412. doi: 10.1038/s41593-018-0332-9
Ferrer, I., Martí, E., López, E., and Tortosa, A. (1998). NF-κB immunoreactivity is observed in association with βA4 diffuse plaques in patients with Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 24, 271–277. doi: 10.1046/j.1365-2990.1998.00116.x
Frade, J. M., and Ovejero-Benito, M. C. (2015). Neuronal cell cycle: the neuron itself and its circumstances. Cell Cycle 14, 712–720. doi: 10.1080/15384101.2015.1004937
Freund, A., Patil, C. K., and Campisi, J. (2011). p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 30, 1536–1548. doi: 10.1038/emboj.2011.69
Galea, I., Bechmann, I., and Perry, V. H. (2007). What is immune privilege (not)? Trends Immunol. 28, 12–18. doi: 10.1016/j.it.2006.11.004
Gan, Y., Liu, Q., Wu, W., Yin, J. X., Bai, X. F., Shen, R., et al. (2014). Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc. Natl. Acad. Sci. U.S.A. 111, 2704–2709. doi: 10.1073/pnas.1315943111
Gate, D., Saligrama, N., Leventhal, O., Yang, A. C., Unger, M. S., Middeldorp, J., et al. (2020). Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577, 399–404. doi: 10.1038/s41586-019-1895-7
Giovanni, A., Keramaris, E., Morris, E. J., Hou, S. T., O’Hare, M., Dyson, N., et al. (2000). E2F1 mediates death of B-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J. Biol. Chem. 275, 11553–11560. doi: 10.1074/jbc.275.16.11553
Giovanni, A., Wirtz-Brugger, F., Keramaris, E., Slack, R., and Park, D. S. (1999). Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F x DP, in B-amyloid-induced neuronal death. J. Biol. Chem. 274, 19011–19016. doi: 10.1074/jbc.274.27.19011
Gomes, L. M. F., Mahammed, A., Prosser, K. E., Smith, J. R., Silverman, M. A., Walsby, C. J., et al. (2019). A catalytic antioxidant for limiting amyloid-beta peptide aggregation and reactive oxygen species generation. Chem. Sci. 10, 1634–1643. doi: 10.1039/c8sc04660c
Gorgoulis, V., Adams, P. D., Alimonti, A., Bennett, D. C., Bischof, O., Bishop, C., et al. (2019). Cellular senescence: defining a path forward. Cell 179, 813–827. doi: 10.1016/j.cell.2019.10.005
Gorlé, N., Van Cauwenberghe, C., Libert, C., and Vandenbroucke, R. E. (2016). The effect of aging on brain barriers and the consequences for Alzheimer’s disease development. Mamm. Genome 27:407. doi: 10.1007/s00335-016-9637-8
Gross, C. (2000). OPINION: neurogenesis in the adult brain: death of a dogma. Nat. Rev. Neurosci. 1, 67–73.
Gruol, D. L. (2015). IL-6 regulation of synaptic function in the CNS. Neuropharmacology 96, 42–54. doi: 10.1016/j.neuropharm.2014.10.023
Gunn, A. P., Wong, X. B. X., Johanssen, X. T., Griffith, X. J. C., Masters, C. L., Bush, A. I., et al. (2016). Amyloid- peptide A-3pE-42 induces lipid peroxidation, membrane permeabilization, and calcium influx in neurons. J. Biol. Chem. 291, 6134–6145. doi: 10.1074/jbc.M115.655183
Halazonetis, T. D., Gorgoulis, V. G., and Bartek, J. (2008). An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355. doi: 10.1126/science.1140735
Hayflick, L., and Moorhead, P. S. (1961). The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621. doi: 10.1016/0014-4827(61)90192-6
He, N., Jin, W.-L., Lok, K.-H., Wang, Y., Yin, M., and Wang, Z.-J. (2013). Amyloid-β1–42 oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis. 4:e924. doi: 10.1038/cddis.2013.437
Hemmerová, E., Špringer, T., Krištofiková, Z., and Homola, J. (2019). In vitro study of interaction of 17β-hydroxysteroid dehydrogenase type 10 and cyclophilin D and its potential implications for Alzheimer’s disease. Sci. Rep. 9:16700. doi: 10.1038/s41598-019-53157-7
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Hernandez-Segura, A., de Jong, T. V., Melov, S., Guryev, V., Campisi, J., and Demaria, M. (2017). Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 27, 2652–2660.e4. doi: 10.1016/j.cub.2017.07.033
Hernandez-Segura, A., Nehme, J., and Demaria, M. (2018). Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453. doi: 10.1016/j.tcb.2018.02.001
Herz, J., Filiano, A. J., Smith, A., Yogev, N., and Kipnis, J. (2017). Myeloid cells in the central nervous system. Immunity 161, 197–213. doi: 10.1016/j.immuni.2017.06.007
Holmes, C., Boche, D., Wilkinson, D., Yadegarfar, G., Hopkins, V., Bayer, A., et al. (2008). Long-term effects of Aβ42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372, 216–223. doi: 10.1016/S0140-6736(08)61075-2
Hradek, A. C., Lee, H. P., Siedlak, S. L., Torres, S. L., Jung, W., Han, A. H., et al. (2014). Distinct chronology of neuronal cell cycle Re-entry and Tau pathology in the 3xTg-AD mouse model and Alzheimer’s disease patients. J. Alzheimers Dis. 43, 57–65. doi: 10.3233/JAD-141083
Hubackova, S., Krejcikova, K., Bartek, J., and Hodny, Z. (2012). IL1-and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine “Bystander senescence.”. Aging 4, 932–951. doi: 10.18632/aging.100520
Hung, C. H. L., Cheng, S. S. Y., Cheung, Y. T., Wuwongse, S., Zhang, N. Q., Ho, Y. S., et al. (2018). A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox Biol. 14, 7–19. doi: 10.1016/j.redox.2017.08.010
Hyman, B. T., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Carrillo, M. C., et al. (2012). National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 8, 1–13. doi: 10.1016/j.jalz.2011.10.007
Iannello, A., Thompson, T. W., Ardolino, M., Lowe, S. W., and Raulet, D. H. (2013). p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 210, 2057–2069. doi: 10.1084/jem.20130783
Jadiya, P., Kolmetzky, D. W., Tomar, D., Di Meco, A., Lombardi, A. A., Lambert, J. P., et al. (2019). Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 10:3885. doi: 10.1038/s41467-019-11813-6
Jaworski, T., Dewachter, I., Lechat, B., Croes, S., Termont, A., Demedts, D., et al. (2009). AAV-tau mediates pyramidal neurodegeneration by cell-cycle re-entry without neurofibrillary tangle formation in wild-type mice. PLoS One 4:e7280. doi: 10.1371/journal.pone.0007280
Johmura, Y., Shimada, M., Misaki, T., Naiki-Ito, A., Miyoshi, H., Motoyama, N., et al. (2014). Necessary and sufficient role for a mitosis skip in senescence induction. Mol. Cell 55, 73–84. doi: 10.1016/j.molcel.2014.05.003
Johnson, D. G., and DeGregori, J. (2012). Putting the oncogenic and tumor suppressive activities of E2F into context. Curr. Mol. Med. 6, 731–738. doi: 10.2174/156652406778773493
Jomova, K., Vondrakova, D., Lawson, M., and Valko, M. (2010). Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 345, 91–104. doi: 10.1007/s11010-010-0563-x
Jurk, D., Wang, C., Miwa, S., Maddick, M., Korolchuk, V., Tsolou, A., et al. (2012). Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004. doi: 10.1111/j.1474-9726.2012.00870.x
Jurk, D., Wilson, C., Passos, J. F., Oakley, F., Correia-Melo, C., Greaves, L., et al. (2014). Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 5:4172. doi: 10.1038/ncomms5172
Kadowaki, H., Nishitoh, H., Urano, F., Sadamitsu, C., Matsuzawa, A., Takeda, K., et al. (2005). Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 12, 19–24. doi: 10.1038/sj.cdd.4401528
Kaltschmidt, B., Uherek, M., Volk, B., Baeuerle, P. A., and Kaltschmidt, C. (1997). Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 94, 2642–2647. doi: 10.1073/pnas.94.6.2642
Kaminsky, Y. G., Tikhonova, L. A., and Kosenko, E. A. (2015). Critical analysis of Alzheimer’s amyloid-beta toxicity to mitochondria. Front. Biosci. Landmark 20, 173–197. doi: 10.2741/4304
Kang, T. W., Yevsa, T., Woller, N., Hoenicke, L., Wuestefeld, T., Dauch, D., et al. (2011). Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551. doi: 10.1038/nature10599
Karran, E., and De Strooper, B. (2016). The amyloid cascade hypothesis: are we poised for success or failure? J. Neurochem. 139, 237–252. doi: 10.1111/jnc.13632
Kastan, M. B., and Bartek, J. (2004). Cell-cycle checkpoints and cancer. Nature 432, 316–323. doi: 10.1038/nature03097
Kempermann, G., Gage, F. H., Aigner, L., Song, H., Curtis, M. A., Thuret, S., et al. (2018). Human adult neurogenesis: evidence and remaining questions. Cell Stem Cell 23, 25–30. doi: 10.1016/j.stem.2018.04.004
Kirkland, J. L., Tchkonia, T., Zhu, Y., Niedernhofer, L. J., and Robbins, P. D. (2017). The clinical potential of senolytic drugs. J. Am. Geriatr. Soc. 65, 2297–2301. doi: 10.1111/jgs.14969
Korin, B., Ben-Shaanan, T. L., Schiller, M., Dubovik, T., Azulay-Debby, H., Boshnak, N. T., et al. (2017). High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 20, 1300–1309. doi: 10.1038/nn.4610
Krizhanovsky, V., Yon, M., Dickins, R. A., Hearn, S., Simon, J., Miething, C., et al. (2008). Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667. doi: 10.1016/j.cell.2008.06.049
Kruman, I. I., Wersto, R. P., Cardozo-Pelaez, F., Smilenov, L., Chan, S. L., Chrest, F. J., et al. (2004). Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 41, 549–561. doi: 10.1016/S0896-6273(04)00017-0
Kuilman, T., Michaloglou, C., Vredeveld, L. C. W., Douma, S., van Doorn, R., Desmet, C. J., et al. (2008). Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031. doi: 10.1016/j.cell.2008.03.039
Kurz, D., Decary, S., Hong, Y., and Erusalimsky, J. (2000). Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 113(Pt 20), 3613–3622.
Lacor, P. N., Buniel, M. C., Furlow, P. W., Clemente, A. S., Velasco, P. T., Wood, M., et al. (2007). Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 27, 796–807. doi: 10.1523/JNEUROSCI.3501-06.2007
Larson, M. E., and Lesné, S. E. (2012). Soluble Aβ oligomer production and toxicity. J. Neurochem. 120(Suppl. 1), 125–139. doi: 10.1111/j.1471-4159.2011.07478.x
Lecot, P., Alimirah, F., Desprez, P. Y., Campisi, J., and Wiley, C. (2016). Context-dependent effects of cellular senescence in cancer development. Br. J. Cancer 114, 1180–1184. doi: 10.1038/bjc.2016.115
Lee, B. Y., Han, J. A., Im, J. S., Morrone, A., Johung, K., Goodwin, E. C., et al. (2006). Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 5, 187–195. doi: 10.1111/j.1474-9726.2006.00199.x
Leggio, G. M., Catania, M. V., Puzzo, D., Spatuzza, M., Pellitteri, R., Gulisano, W., et al. (2016). The antineoplastic drug flavopiridol reverses memory impairment induced by Amyloid-ß1-42 oligomers in mice. Pharmacol. Res. 106, 10–20. doi: 10.1016/j.phrs.2016.02.007
Li, L., Cheung, T., Chen, J., and Herrup, K. (2011). A comparative study of five mouse models of Alzheimer’s disease: cell cycle events reveal new insights into neurons at risk for death. Int. J. Alzheimers Dis. 2001:171464. doi: 10.4061/2011/171464
Li, M., Li, Z., Yao, Y., Jin, W. N., Wood, K., Liu, Q., et al. (2017). Astrocyte-derived interleukin-15 exacerbates ischemic brain injury via propagation of cellular immunity. Proc. Natl. Acad. Sci. U.S.A. 114, E396–E405. doi: 10.1073/pnas.1612930114
Lindestam Arlehamn, C. S., Garretti, F., Sulzer, D., and Sette, A. (2019). Roles for the adaptive immune system in Parkinson’s and Alzheimer’s diseases. Curr. Opin. Immunol. 59, 115–120. doi: 10.1016/j.coi.2019.07.004
Lopes, J. P., Oliveira, C. R., and Agostinho, P. (2009). Cdk5 acts as a mediator of neuronal cell cycle re-entry triggered by amyloid-β and prion peptides. Cell Cycle 8, 97–104. doi: 10.4161/cc.8.1.7506
Lopes, J. P., Oliveira, C. R., and Agostinho, P. (2010). Neurodegeneration in an Abeta-induced model of Alzheimer’s disease: the role of Cdk5. Aging Cell 9, 64–77. doi: 10.1111/j.1474-9726.2009.00536.x
Lujambio, A. (2016). To clear, or not to clear (senescent cells)? That is the question. Bioessays 38, S56–S64. doi: 10.1002/bies.201670910
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452. doi: 10.1126/science.1091230
Luterman, J. D., Haroutunian, V., Yemul, S., Ho, L., Purohit, D., Aisen, P. S., et al. (2000). Cytokine gene expression as a function of the clinical progression of Alzheimer disease dementia. Arch. Neurol. 57, 1153–1160. doi: 10.1001/archneur.57.8.1153
Lüth, H. J., Holzer, M., Gertz, H. J., and Arendt, T. (2000). Aberrant expression of nNOS in pyramidal neurons in Alzheimer’s disease is highly co-localized with p21(ras) and p16(INK4a). Brain Res. 852, 45–55. doi: 10.1016/S0006-8993(99)02178-2
Majd, S., Zarifkar, A., Rastegar, K., and Takhshid, M. A. (2008). Different fibrillar Aβ 1-42 concentrations induce adult hippocampal neurons to reenter various phases of the cell cycle. Brain Res. 1218, 224–229. doi: 10.1016/j.brainres.2008.04.050
Mao, P., and Reddy, P. H. (2011). Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: implications for early intervention and therapeutics. Biochim. Biophys. Acta 1812, 1359–1370. doi: 10.1016/j.bbadis.2011.08.005
Masaldan, S., Clatworthy, S. A. S., Gamell, C., Meggyesy, P. M., Rigopoulos, A. T., Haupt, S., et al. (2018). Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 14, 100–115. doi: 10.1016/j.redox.2017.08.015
McShea, A., Harris, P. L., Webster, K. R., Wahl, A. F., and Smith, M. A. (1997). Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am. J. Pathol. 150, 1933–1939.
Meli, G., Lecci, A., Manca, A., Krako, N., Albertini, V., Benussi, L., et al. (2014). Conformational targeting of intracellular A’ 2 oligomers demonstrates their pathological oligomerization inside the endoplasmic reticulum. Nat. Commun. 5:3867. doi: 10.1038/ncomms4867
Merlo, S., Basile, L., Giuffrida, M. L., Sortino, M. A., Guccione, S., and Copani, A. (2015). Identification of 5-methoxyflavone as a novel DNA polymerase-beta inhibitor and neuroprotective agent against beta-amyloid toxicity. J. Nat. Prod. 78, 2704–2711. doi: 10.1021/acs.jnatprod.5b00621
Modi, P. K., Jaiswal, S., and Sharma, P. (2015). Regulation of neuronal cell cycle and apoptosis by miR-34a. Mol. Cell. Biol. 36, 84–94. doi: 10.1128/MCB.00589-15
Modi, P. K., Komaravelli, N., Singh, N., and Sharma, P. (2012). Interplay between MEK-ERK signaling, cyclin D1, and cyclin-dependent kinase 5 regulates cell cycle reentry and apoptosis of neurons. Mol. Biol. Cell 23, 3722–3730. doi: 10.1091/mbc.E12-02-0125
Morsy, A., and Trippier, P. C. (2019). Amyloid-binding alcohol dehydrogenase (ABAD) inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 62, 4252–4264. doi: 10.1021/acs.jmedchem.8b01530
Mossmann, D., Vögtle, F. N., Taskin, A. A., Teixeira, P. F., Ring, J., Burkhart, J. M., et al. (2014). Amyloid-β peptide induces mitochondrial dysfunction by inhibition of preprotein maturation. Cell Metab. 20, 662–669. doi: 10.1016/j.cmet.2014.07.024
Müller, M., Ahumada-Castro, U., Sanhueza, M., Gonzalez-Billault, C., Court, F. A., and Cárdenas, C. (2018). Mitochondria and calcium regulation as basis of neurodegeneration associated with aging. Front. Neurosci. 12:470. doi: 10.3389/fnins.2018.00470
Muñoz, D. P., Yannone, S. M., Daemen, A., Sun, Y., Vakar-Lopez, F., Kawahara, M., et al. (2019). Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight 4:e124716. doi: 10.1172/jci.insight.124716
Munoz, L., and Ammit, A. J. (2010). Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 58, 561–568. doi: 10.1016/j.neuropharm.2009.11.010
Muñoz-Espín, D., Cañamero, M., Maraver, A., Gómez-López, G., Contreras, J., Murillo-Cuesta, S., et al. (2013). XProgrammed cell senescence during mammalian embryonic development. Cell 155, 1104–1118. doi: 10.1016/j.cell.2013.10.019
Musi, N., Valentine, J. M., Sickora, K. R., Baeuerle, E., Thompson, C. S., Shen, Q., et al. (2018). Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 17:e12840. doi: 10.1111/acel.12840
Nation, D. A., Sweeney, M. D., Montagne, A., Sagare, A. P., D’Orazio, L. M., Pachicano, M., et al. (2019). Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 25, 270–276. doi: 10.1038/s41591-018-0297-y
Nelson, G., Kucheryavenko, O., Wordsworth, J., and von Zglinicki, T. (2018). The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech. Ageing Dev. 170, 30–36. doi: 10.1016/j.mad.2017.08.005
Nelson, G., Wordsworth, J., Wang, C., Jurk, D., Lawless, C., Martin-Ruiz, C., et al. (2012). A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11, 345–349. doi: 10.1111/j.1474-9726.2012.00795.x
Neves, J., Demaria, M., Campisi, J., and Jasper, H. (2015). Of flies, mice, and men: evolutionarily conserved tissue damage responses and aging. Dev. Cell 32, 9–18. doi: 10.1016/j.devcel.2014.11.028
O’Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204.
Oddo, S., Caccamo, A., Tran, L., Lambert, M. P., Glabe, C. G., Klein, W. L., et al. (2006). Temporal profile of amyloid-β (Aβ) oligomerization in an in vivo model of Alzheimer disease: a link between Aβ and tau pathology. J. Biol. Chem. 81, 1599–1604. doi: 10.1074/jbc.M507892200
Oddo, S., Caccamo, A., Tseng, B., Cheng, D., Vasilevko, V., Cribbs, D. H., et al. (2008). Blocking Aβ42 accumulation delays the onset and progression of tau pathology via the C terminus of heat shock protein70-interacting protein: a mechanistic link between Aβ and tau pathology. J. Neurosci. 28, 12163–12175. doi: 10.1523/JNEUROSCI.2464-08.2008
Origlia, N., Righi, M., Capsoni, S., Cattaneo, A., Fang, F., Stern, D. M., et al. (2008). Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-β-mediated cortical synaptic dysfunction. J. Neurosci. 28, 3521–3530. doi: 10.1523/JNEUROSCI.0204-08.2008
Panza, F., Lozupone, M., Logroscino, G., and Imbimbo, B. P. (2019). A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 15, 73–88. doi: 10.1038/s41582-018-0116-6
Park, D. S., Obeidat, A., Giovanni, A., and Greene, L. A. (2000). Cell cycle regulators in neuronal death evoked by excitotoxic stress: implications for neurodegeneration and its treatment. Neurobiol. Aging 21, 771–781. doi: 10.1016/S0197-4580(00)00220-7
Passos, J. F., Nelson, G., Wang, C., Richter, T., Simillion, C., Proctor, C. J., et al. (2010). Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6:347. doi: 10.1038/msb.2010.5
Pereira, B. I., Devine, O. P., Vukmanovic-Stejic, M., Chambers, E. S., Subramanian, P., Patel, N., et al. (2019). Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell inhibition. Nat. Commun. 10:2387. doi: 10.1038/s41467-019-10335-5
Piechota, M., Sunderland, P., Wysocka, A., Nalberczak, M., Sliwinska, M. A., Radwanska, K., et al. (2016). Is senescence-associated β-galactosidase a marker of neuronal senescence? Oncotarget 81099–81109. doi: 10.18632/oncotarget.12752
Poli, A., Kmiecik, J., Domingues, O., Hentges, F., Bléry, M., Chekenya, M., et al. (2013). NK cells in central nervous system disorders. J. Immunol. 190, 5355–5362. doi: 10.4049/jimmunol.1203401
Reynolds, I. J., and Hastings, T. G. (1995). Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J. Neurosci. 15, 3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995
Rhein, V., Song, X., Wiesner, A., Ittner, L. M., Baysang, G., Meier, F., et al. (2009). Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. U.S.A. 106, 20057–20062. doi: 10.1073/pnas.0905529106
Sagiv, A., Burton, D. G. A., Moshayev, Z., Vadai, E., Wensveen, F., Ben-Dor, S., et al. (2016). NKG2D ligands mediate immunosurveillance of senescent cells. Aging 8, 328–344. doi: 10.18632/aging.100897
Sanz-Blasco, S., Valero, R. A., Rodríguez-Crespo, I., Villalobos, C., and Núñez, L. (2008). Mitochondrial Ca2+ overload underlies Aβ oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS One 3:e2718. doi: 10.1371/journal.pone.0002718
Schmidt, C., Lepsverdize, E., Chi, S. L., Das, A. M., Pizzo, S. V., Dityatev, A., et al. (2008). Amyloid precursor protein and amyloid β-peptide bind to ATP synthase and regulate its activity at the surface of neural cells. Mol. Psychiatry 13, 953–969. doi: 10.1038/sj.mp.4002077
Schneider, L. (2020). A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 19, 111–112. doi: 10.1016/S1474-4422(19)30480-6
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Sevigny, J., Chiao, P., Bussière, T., Weinreb, P. H., Williams, L., Maier, M., et al. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56. doi: 10.1038/nature19323
Seward, M. E., Swanson, E., Norambuena, A., Reimann, A., Cochran, J. N., Li, R., et al. (2013a). Amyloid-signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J. Cell Sci. 126, 1278–1286. doi: 10.1242/jcs.1125880
Seward, M. E., Swanson, E., Norambuena, A., Reimann, A., Nicholas Cochran, J., Li, R., et al. (2013b). Amyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in alzheimer’s disease. J. Cell Sci. 126, 1278–1286. doi: 10.1242/jcs.1125880
Shelat, P. B., Chalimoniuk, M., Wang, J. H., Strosznajder, J. B., Lee, J. C., Sun, A. Y., et al. (2008). Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J. Neurochem. 106, 45–55. doi: 10.1111/j.1471-4159.2008.05347.x
Sirk, D., Zhu, Z., Wadia, J. S., Shulyakova, N., Phan, N., Fong, J., et al. (2007). Chronic exposure to sub-lethal beta-amyloid (Aβ) inhibits the import of nuclear-encoded proteins to mitochondria in differentiated PC12 cells. J. Neurochem. 103, 1989–2003. doi: 10.1111/j.1471-4159.2007.04907.x
Smith, L. M., and Strittmatter, S. M. (2017). Binding sites for amyloid-β oligomers and synaptic toxicity. Cold Spring Harb. Perspect. Med. 7:a024075. doi: 10.1101/cshperspect.a024075
Snow, W. M., and Albensi, B. C. (2016). Neuronal gene targets of NF-κB and their dysregulation in Alzheimer’s disease. Front. Mol. Neurosci. 9:118. doi: 10.3389/fnmol.2016.00118
Solana, C., Tarazona, R., and Solana, R. (2018). Immunosenescence of natural killer cells, inflammation, and Alzheimer’s disease. Int. J. Alzheimers Dis. 2018:3128758. doi: 10.1155/2018/3128758
Sortino, M. A., Chisari, M., Merlo, S., Vancheri, C., Caruso, M., Nicoletti, F., et al. (2004). Glia mediates the neuroprotective action of estradiol on β-amyloid-induced neuronal death. Endocrinology 145, 5080–5086. doi: 10.1210/en.2004-0973
Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2018). Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150. doi: 10.1038/nrneurol.2017.188
Terai, K., Matsuo, A., and McGeer, P. L. (1996). Enhancement of immunoreactivity for NF-κB in the hippocampal formation and cerebral cortex of Alzheimer’s disease. Brain Res. 735, 159–168. doi: 10.1016/0006-8993(96)00310-1
Thal, D. R., Rüb, U., Orantes, M., and Braak, H. (2002). Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. doi: 10.1212/WNL.58.12.1791
Toledo, L. I., Murga, M., Gutierrez-Martinez, P., Soria, R., and Fernandez-Capetillo, O. (2008). ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 22, 297–302. doi: 10.1101/gad.452308
Varvel, N. H., Bhaskar, K., Kounnas, M. Z., Wagner, S. L., Yang, Y., Lamb, B. T., et al. (2009). NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease. J. Clin. Invest. 119, 3692–3702. doi: 10.1172/JCI39716
Varvel, N. H., Bhaskar, K., Patil, A. R., Pimplikar, S. W., Herrup, K., and Lamb, B. T. (2008). Aβ oligomers induce neuronal cell cycle events in Alzheimer’s disease. J. Neurosci. 28, 10786–10793. doi: 10.1523/JNEUROSCI.2441-08.2008
Verdier, Y., Zarándi, M., and Penke, B. (2004). Amyloid β-peptide interactions with neuronal and glial cell plasma membrane: binding sites and implications for Alzheimer’s disease. J. Pept. Sci. 10, 229–248. doi: 10.1002/psc.573
Vermunt, L., Sikkes, S. A. M., van den Hout, A., Handels, R., Bos, I., van der Flier, W. M., et al. (2019). Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement. 15, 888–898. doi: 10.1016/j.jalz.2019.04.001
Villemagne, V. L., Burnham, S., Bourgeat, P., Brown, B., Ellis, K. A., Salvado, O., et al. (2013). Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 12, 357–367. doi: 10.1016/S1474-4422(13)70044-9
Vismer, M. S., Forcelli, P. A., Skopin, M. D., Gale, K., and Koubeissi, M. Z. (2015). The piriform, perirhinal, and entorhinal cortex in seizure generation. Front. Neural Circuits 9:27. doi: 10.3389/fncir.2015.00027
Vitale, I., Galluzzi, L., Castedo, M., and Kroemer, G. (2011). Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 12, 385–392. doi: 10.1038/nrm3115
Walton, C. C., and Andersen, J. K. (2019). Unknown fates of (brain) oxidation or UFO: close encounters with neuronal senescence. Free Radic. Biol. Med. 134, 695–701. doi: 10.1016/J.FREERADBIOMED.2019.01.012
Walton, C. C., Zhang, W., Patiño-Parrado, I., Barrio-Alonso, E., Garrido, J. J., and Frade, J. M. (2019). Primary neurons can enter M-phase. Sci. Rep. 9:4594. doi: 10.1038/s41598-019-40462-4
Wang, J., Gan, Y., Han, P., Yin, J., Liu, Q., Ghanian, S., et al. (2018). Ischemia-induced neuronal cell death is mediated by chemokine receptor CX3CR1. Sci. Rep. 8:556. doi: 10.1038/s41598-017-18774-0
Ward, M. W., Rego, A. C., Frenguelli, B. G., and Nicholls, D. G. (2000). Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurosci. 20, 7208–7219. doi: 10.1523/JNEUROSCI.20-19-07208.2000
Wei, Z., Chen, X. C., Song, Y., Pan, X. D., Dai, X. M., Zhang, J., et al. (2016). Amyloid β protein aggravates neuronal senescence and cognitive deficits in 5XFAD mouse model of Alzheimer’s disease. Chin. Med. J. 129, 1835–1844. doi: 10.4103/0366-6999.186646
Wiley, C. D., Velarde, M. C., Lecot, P., Liu, S., Sarnoski, E. A., Freund, A., et al. (2016). Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 23, 303–314. doi: 10.1016/j.cmet.2015.11.011
Wu, Q., Combs, C., Cannady, S. B., Geldmacher, D. S., and Herrup, K. (2000). Beta-amyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol. Aging 21, 797–806. doi: 10.1016/S0197-4580(00)00219-0
Xue, W., Zender, L., Miething, C., Dickins, R. A., Hernando, E., Krizhanovsky, V., et al. (2007). Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660. doi: 10.1038/nature05529
Yang, Y., Varvel, N. H., Lamb, B. T., and Herrup, K. (2006). Ectopic cell cycle events link human Alzheimer’s disease and amyloid precursor protein transgenic mouse models. J. Neurosci. 26, 775–784. doi: 10.1523/JNEUROSCI.3707-05.2006
Yankner, B., Duffy, L., and Kirschner, D. (1990). Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 250, 279–282. doi: 10.1126/science.2218531
Zhang, J., Cicero, S. A., Wang, L., Romito-Digiacomo, R. R., Yang, Y., and Herrup, K. (2008). Nuclear localization of Cdk5 is a key determinant in the postmitotic state of neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 8772–8777. doi: 10.1073/pnas.0711355105
Zhang, P., Kishimoto, Y., Grammatikakis, I., Gottimukkala, K., Cutler, R. G., Zhang, S., et al. (2019). Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 22, 719–728. doi: 10.1038/s41593-019-0372-9
Zhang, Y., Gao, Z., Wang, D., Zhang, T., Sun, B., Mu, L., et al. (2014). Accumulation of natural killer cells in ischemic brain tissues and the chemotactic effect of IP-10. J. Neuroinflammation 11:79. doi: 10.1186/1742-2094-11-79
Zhu, X., Rottkamp, C. A., Boux, H., Takeda, A., Perry, G., and Smith, M. A. (2000). Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J. Neuropathol. Exp. Neurol. 59, 880–888. doi: 10.1046/J.0022-3042.2001.00729.X
Keywords: Alzheimer’s disease, Neurodegeneration, senescence, senescence associated secretory phenotype, cell cycle, reactive oxygen species
Citation: Walton CC, Begelman D, Nguyen W and Andersen JK (2020) Senescence as an Amyloid Cascade: The Amyloid Senescence Hypothesis. Front. Cell. Neurosci. 14:129. doi: 10.3389/fncel.2020.00129
Received: 25 February 2020; Accepted: 20 April 2020;
Published: 19 May 2020.
Edited by:
Vassilis G. Gorgoulis, National and Kapodistrian University of Athens, GreeceReviewed by:
Sharon DeMorrow, University of Texas at Austin, United StatesCopyright © 2020 Walton, Begelman, Nguyen and Andersen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chaska C. Walton, Y3dhbHRvbkBidWNraW5zdGl0dXRlLm9yZw==; Julie K. Andersen, amthbmRlcnNlbkBidWNraW5zdGl0dXRlLm9yZw==; amFuZGVyc2VuQGJ1Y2tpbnN0aXR1dGUub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.