 Osvaldo Flores-Bastías
Osvaldo Flores-Bastías Alfredo Adriasola-Carrasco
Alfredo Adriasola-Carrasco Eduardo Karahanian
Eduardo Karahanian
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci., 28 January 2020
Sec. Cellular Neuropathology
Volume 14 - 2020 | https://doi.org/10.3389/fncel.2020.00005
This article is part of the Research TopicIs Early Onset of Alcohol Use Associated with Later Alcohol Use?View all 14 articles
The concept that neuroinflammation induced by excessive alcohol intake in adolescence triggers brain mechanisms that perpetuate consumption has strengthened in recent years. The melanocortin system, composed of the melanocortin 4 receptor (MC4R) and its ligand α-melanocyte-stimulating hormone (α-MSH), has been implicated both in modulation of alcohol consumption and in ethanol-induced neuroinflammation decrease. Chronic alcohol consumption in adolescent rats causes a decrease in an α-MSH release by the hypothalamus, while the administration of synthetic agonists of MC4R causes a decrease in neuroinflammation and a decrease in voluntary alcohol consumption. However, the mechanism that connects the activation of MC4R with the decrease of both neuroinflammation and voluntary alcohol consumption has not been elucidated. Brain-derived neurotrophic factor (BDNF) has been implicated in alcohol drinking motivation, dependence and withdrawal, and its levels are reduced in alcoholics. Deficiencies in BDNF levels increased ethanol self-administration in rats. Further, BDNF triggers important anti-inflammatory effects in the brain, and this could be one of the mechanisms by which BDNF reduces chronic alcohol intake. Interestingly, MC4R signaling induces BDNF expression through the activation of the cAMP-responsive element-binding protein (CREB). We hypothesize that ethanol exposure during adolescence decreases the expression of α-MSH and hence MC4R signaling in the hippocampus, leading to a lower BDNF activity that causes dramatic changes in the brain (e.g., neuroinflammation and decreased neurogenesis) that predispose to maintain alcohol abuse until adulthood. The activation of MC4R either by α-MSH or by synthetic agonist peptides can induce the expression of BDNF, which would trigger several processes that lead to lower alcohol consumption.
Alcohol is the most commonly abused drug worldwide. The World Health Organization (WHO) placed alcohol abuse disorder (AUD) among the top risk factors for diseases leading to death worldwide (Lim et al., 2012; World Health Organization, 2014). As reported by WHO, the global effect of alcohol abuse is near 3.3 million deaths per year (approximately 5.9% of all deaths worldwide; World Health Organization, 2014). A study in the United Kingdom concluded that alcohol is the most noxious drug to society, above heroin and cocaine (Nutt et al., 2010). Alcohol abuse is associated with an increased risk of developing a variety of health issues including cancer and liver cirrhosis (Baan et al., 2007; Shield et al., 2013).
In addition to the various health problems associated with AUD, the excessive consumption of alcohol generates significant consequences in the brain such as neurodegeneration (Crews and Nixon, 2009), depression (Whiteford et al., 2013), anxiety (Kushner et al., 2000), memory impairment (Rose and Grant, 2010) and malfunction of the prefrontal cortex that leads to impaired executive function (e.g., planning, thinking, and judgment; Peterson et al., 1990). These alterations are especially important if excessive alcohol consumption occurs during brain development in adolescence, which would lead to long-term damage to brain structure and function (Crews et al., 2007). There is extensive evidence supporting the idea that the beginning of alcohol consumption in adolescence increases the risk of developing AUD in adulthood (DeWit et al., 2000; Dawson et al., 2008).
Although there are many neurobiological mechanisms involved in the development of AUD (i.e., motivation for alcohol consumption, establishment of chronic alcohol intake and relapse), the idea that neuroinflammation and oxidative stress play a role in the maintenance of chronic alcohol intake has recently gained relevance (Israel et al., 2017). It has been proposed that the activation of genes related to the innate immune system in the brain and neuroinflammation play a significant role in the establishment of addiction to alcohol (Coller and Hutchinson, 2012; Flores-Bastías and Karahanian, 2018). The activation of neuroinflammation would produce long-lasting neurobiological changes, as increased glutamate-induced hyperexcitability and excitotoxicity and decreased neurogenesis, which are related to addictive substances abuse; all these neurobiological changes are more evident if alcohol abuse begins at an early age (Crews et al., 2011). Although it was already known that alcohol consumption produced neuroinflammation (Blanco and Guerri, 2007), the first evidence that neuroinflammation would be directly related to alcohol addiction came in 2011 from experiments by Blednov et al. (2011), who demonstrated that the systemic administration of bacterial lipopolysaccharide (LPS) to mice increases voluntary ethanol intake for a prolonged period. Ethanol consumption disturb the intestinal barrier, allowing the passage of bacterial LPS into circulation (Ferrier et al., 2006). LPS inside the body induces the synthesis of tumor necrosis factor-α (TNF-α) by Kupffer cells, which is released from liver to the blood; TNF-α then crosses the blood-brain barrier and activates microglia and astrocytes TNF receptors to induce neuroinflammation (Qin et al., 2007; Crews and Vetreno, 2016). This state of neuroinflammation is maintained for several months (Qin et al., 2007), insinuating that there exist long-lasting mechanisms that perpetuate it. Accordingly, ethanol-induced neuroinflammation persists upregulated for a long period after alcohol withdrawal, whereas levels of peripheral inflammatory cytokines rapidly return to basal, indicating that neuroinflammation is maintained by a strong mechanism of self-perpetuation (Qin et al., 2008).
Besides the neuroinflammatory processes triggered by systemic LPS, the generation of reactive oxygen species (ROS) from ethanol metabolism in the brain may also contribute to neuroinflammation (Flores-Bastías and Karahanian, 2018). Microglia and astrocytes are key cells in the neuroinflammatory process, and they are highly reactive to ethanol exposure (Orellana et al., 2017), releasing proinflammatory cytokines and nitric oxide (Blanco and Guerri, 2007). Ethanol intake produces a potent activation of glial cells in both adult and adolescent rodents. Interestingly, withdrawal of alcohol intake by adolescent rodents does not completely reverse such activation when they reach adulthood (Evrard et al., 2006). The hippocampus is the brain region that suffers major pathological alterations due to chronic alcohol consumption (Franke et al., 1997). Astrocytes and microglia are highly activated in the hippocampus from alcoholics (He and Crews, 2008), increasing the release of pro-inflammatory cytokines which can lead to neuron death (Ward et al., 2009). In the case of adolescents, their hippocampus suffers greater damage than that of adults, since they are more sensitive to the EtOH-induced alterations of memory processes requiring hippocampal integrity (Markwiese et al., 1998; White and Swartzwelder, 2004).
Microglia, astrocytes, and neurons abundantly express NF-κB, an essential transcription factor that activates the genes of the innate immune response. NF-κB is also involved in ethanol-induced neuroinflammation (Zou and Crews, 2010). Ethanol increases cytochrome P4502E1 (Cyp2E1) activity (Lieber, 1999), and in turn, Cyp2E1 oxidation of ethanol increases ROS levels that activate NF-κB directly (Chandel et al., 2000), leading to the synthesis of pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-α (Cao et al., 2005). In addition, NF-κB activation induces NADPH oxidase expression (Cao et al., 2005), leading to the production of more ROS, which creates an “activation loop” that potentiates the innate immune response (Crews et al., 2011; Flores-Bastías and Karahanian, 2018). Accordingly, the daily administration of N-acetyl cysteine (a precursor in the formation of the antioxidant glutathione) to rats that have chronically consumed ethanol, has been shown to significantly reduce voluntary ethanol intake (Israel et al., 2017). Along this line, ibudilast, a drug that reduces TNF-α activity, inhibits by 50% chronic ethanol intake in high alcohol drinking rats (Bell et al., 2015). In addition to the intrinsic damage induced by ROS, leading to neuron death, NF-κB activation increase TNF-α, IL-1β, IL-6 and transforming growth factor β1 (TGF-β1) release, resulting in neuronal apoptosis (Crews et al., 2015). “The constant activation of neuroinflammation, together with the exacerbated production of ROS, would lead to neurodegeneration of key areas involved in excessive alcohol consumption. It has been proposed that, instead of simply being a side effect of excessive alcohol consumption, neuronal damage associated with drinking may actually underlie some of the mechanisms that regulate the development of alcohol abuse disorder (Crews et al., 2015). Alcohol-induced cell death in regions such as the prefrontal cortex may lead to a lack of inhibition in subcortical reward areas such as the striatum, which in turn may reduce behavioral inhibition and increase motivation to drink. Repeated stimulation of the innate immune system during chronic or binge alcohol consumption may facilitate this process by decreasing inhibition of the mesolimbic reward system, thus increasing drinking (Crews et al., 2011, 2015; Flores-Bastías and Karahanian, 2018).
The hypothalamus is one of the brain regions that undergoes more pathophysiological changes during excessive alcohol intake (Barson and Leibowitz, 2016). In the hypothalamus, several orexigenic peptides (such as orexin, encephalin, and galanin) stimulate alcohol consumption (Rada et al., 2004; Schneider et al., 2007; Barson et al., 2010); on the other hand, anorexigenic hormones such as dynorphin, corticotropin-releasing factor and melanocortins inhibit alcohol drinking (Thorsell et al., 2005; Navarro et al., 2008; Barson et al., 2010). Melanocortins (MC) is a group of neuropeptides that are produced in the arcuate nucleus of the hypothalamus (Arc) and that include α-, β-, and γ- melanocyte-stimulating hormones (α-MSH, β-MSH and γ-MSH; Hadley and Haskell-Luevano, 1999). α-MSH is involved in the regulation of sexual behavior, food appetite and memory through its agonist activity on MC3R and melanocortin 4 receptor (MC4R) melanocortin receptors present in the hippocampus, paraventricular nucleus of the hypothalamus (PVN), ventral tegmental area (VTA) and nucleus accumbens (NAc; Mountjoy, 2010; Caruso et al., 2014). Interestingly, α-MSH has also been implicated in the modulation of ethanol intake (Olney et al., 2014). The administration of a nonspecific agonist of both MC3R and MC4R (Melanotan-II) reduces voluntary alcohol intake in mice (Navarro et al., 2003) and in alcohol-preferring rats (Ploj et al., 2002). However, when this agonist was administered to knock-out mice that lack MC4R no effect was observed, revealing that MC4R (and not MC3R) is responsible for reducing alcohol intake (Navarro et al., 2011). Accordingly, the administration of a selective MC4R-agonist synthetic peptide at the NAc and VTA lowered voluntary alcohol intake in rats (Lerma-Cabrera et al., 2012). Ethanol has direct effects on the levels of α-MSH in the hypothalamus, and these effects appear to be opposite depending on whether the exposure to ethanol is chronic or acute. Evidence indicates that chronic ethanol consumption reduces α-MSH levels in the hypothalamus (Rainero et al., 1990; Navarro et al., 2008; Lerma-Cabrera et al., 2013; Sprow et al., 2016), while an increase in α-MSH was observed in acute ethanol-treated rats (Shelkar et al., 2015).
The MC system has been linked for many years with anti-inflammatory effects in the brain (Macaluso et al., 1994; Orellana et al., 2017). MC4R is expressed in microglia and astrocytes (Caruso et al., 2007; Selkirk et al., 2007; Benjamins et al., 2013), suggesting that α-MSH may produce its anti-inflammatory effects via MC4R in these neuroinflammation-related cells (Caruso et al., 2004). The effect of MC4R activation on the reduction of neuroinflammation has been studied in models of brain injury other than ethanol intake, e.g., LPS-induced brain inflammation (Ichiyama et al., 1999a,b; Muceniece et al., 2005), cerebral ischemia damage (Giuliani et al., 2006; Spaccapelo et al., 2011) and spinal cord injury (van de Meent et al., 1997; Lankhorst et al., 1999). There is evidence that this ability resides in its capacity to decrease glial activation of NF-κB. Specifically, α-MSH and other synthetic melanocortin receptor ligands prevent IκBα phosphorylation and, consequently, inhibit NF-κB activation (Catania, 2008). In agreement, α-MSH decreases the release of pro-inflammatory cytokines such as TNF-α (Rajora et al., 1997; Wong et al., 1997; Delgado et al., 1998; Giuliani et al., 2006; Forslin Aronsson et al., 2007; Spaccapelo et al., 2011), enzymes as iNOS and COX-2 (Caruso et al., 2004) and inflammation mediators, like NO (Galimberti et al., 1999; Muceniece et al., 2006) in the brain. In addition to the inhibition of NF-κB activity, α-MSH also stimulates the production of the anti-inflammatory cytokines IL-10 from microglia and TGF-β from astrocytes, mediated by an increased expression of peroxisome proliferator-activated receptor-gamma (PPARγ; Carniglia et al., 2013). In recent work, we demonstrated that the activation of MC4R by a synthetic agonist peptide inhibits ethanol-induced neuroinflammation in rats (Flores-Bastías et al., 2019). However, the mechanism underlying this effect has not yet been elucidated.
Although we hypothesized that the activation of MC4R in the brain inhibits NF-κB activity and neuroinflammation induced by ethanol (Flores-Bastías and Karahanian, 2018; Flores-Bastías et al., 2019), MC4R elicits additional mechanisms that would help explain the effects of α-MSH or its synthetic analogs in reducing alcohol intake. After the pioneering work of Xu et al. (2003) who reported that the activation of MC4R induces brain-derived neurotrophic factor (BDNF) expression in the hypothalamus, it was determined that BDNF is a downstream effector of MC4R signaling in food intake control (Nicholson et al., 2007; Bariohay et al., 2009). Such signaling occurs through the cAMP-PKA-CREB pathway (Caruso et al., 2012); MC4R is a G protein-coupled receptor that activates adenylate cyclase leading to an increase in cyclic AMP (cAMP) levels, which activates protein kinase A (PKA). PKA then phosphorylates and activates the cAMP-responsive element-binding protein (CREB), which is a transcription factor. CREB mediates in several physiological processes in the central nervous system by the induction of different genes, including BDNF (Tao et al., 1998). Accordingly, CREB is activated by α-MSH-treatment in the hypothalamus (Sarkar et al., 2002; Sutton et al., 2005; Caruso et al., 2010).
BDNF is a neurotrophic factor that plays several roles in neurons growth and function. BDNF, through its receptor tropomyosin-related kinase B (TrkB), regulates CREB phosphorylation via the extracellular-signal-regulated kinases (Erk1/2) pathway (Bibel and Barde, 2000; Schinder and Poo, 2000), playing a key role in synaptic plasticity. BDNF signaling has also been implicated in alcohol drinking behavior, dependence and withdrawal (Davis, 2008), and its levels are altered in alcoholics (Heberlein et al., 2010). A BDNF gene polymorphism has been linked to a greater predisposition to develop AUD in humans (Uhl et al., 2001), and a deficiency of the Bdnf gene causes increased alcohol intake in mice (Hensler et al., 2003). Accordingly, siRNA-mediated downregulation of endogenous BDNF in the striatum increased alcohol self-administration in rats, and subsequent infusion of exogenous BDNF reverted this response (Jeanblanc et al., 2009). It was suggested that BDNF would serve as a homeostatic factor in the hippocampus and striatum that somehow regulates ethanol intake (McGough et al., 2004). Changes in BDNF expression in the brain following chronic ethanol exposure play a role in the regulation of protracted alcohol consumption and withdrawal-induced anxiety (Pandey et al., 2006). Similar to that described above for α-MSH, exposure to ethanol has opposite effects on BDNF levels depending on whether the exposure is chronic or acute: after chronic alcohol consumption, BDNF gene expression in corticostriatal areas is downregulated (Logrip et al., 2009; Melendez et al., 2012). However, in the case of acute alcohol exposure, BDNF levels are increased in striatal and hippocampal brain regions (McGough et al., 2004; Jeanblanc et al., 2009; Logrip et al., 2009). Thus, it is conceivable that prolonged exposure to ethanol reduces α-MSH/MC4R signaling which would decrease BDNF expression, eventually leading to the establishment of a protracted alcohol consumption behavior.
What would be the mechanisms by which BDNF could regulate alcohol consumption? As mentioned above, neuroinflammation induced by ethanol is a process associated with the perpetuation of chronic alcohol intake, and the inhibition of such neuroinflammation lowers voluntary alcohol intake in animal models (Bell et al., 2015; Israel et al., 2017; Ezquer et al., 2018). Interestingly, BDNF triggers important anti-inflammatory and anti-apoptotic mechanisms in glial cells and neurons, respectively, and this could be one of the actions by which BDNF reduces chronic alcohol intake. It was reported that BDNF treatment decreases the levels of the proinflammatory cytokines TNF-α, IL-1β and IL-6 induced by bacterial components in the brain and increases the expression of the anti-inflammatory cytokine IL-10 (Xu et al., 2017). Similarly, BDNF suppressed TNF-α expression while increased IL-10 expression in the brain after ischemic injury (Jiang et al., 2011). Chronic alcohol consumption, in addition to producing neuroinflammation as we have already described, also leads to other important changes in the brain such as a reduction of neurogenesis in the hippocampus (Herrera et al., 2003; Crews et al., 2006; Zou and Crews, 2010). The hippocampus has been extensively examined because of its role in memory consolidation, along with the deterioration of cognitive function seen in alcoholics; interestingly, recovery from alcoholism is associated with increased neurogenesis in this brain region (Crews and Nixon, 2009). Because BDNF plays a fundamental role in neuroinflammation and neurogenesis, it is possible to speculate that activation of MC4R leads to the upregulation of BDNF in glia, which would decrease the neuroinflammation response (Figure 1). Also, BDNF secreted by glial cells would activate TrkB signaling in neurons, through the MAPK/ERK and PLCγ/IP3 pathways that upregulate CREB, leading to increased neurogenesis and enhanced long-term potentiation (LTP), which were reduced by chronic alcohol consumption (Figure 1). The latter would contribute significantly to improve cognitive disorders associated with alcohol consumption. Finally, BDNF signaling in neurons also activates the PI3/Akt pathway, which inhibits the pro-apoptotic protein Bad, thus decreasing ethanol-induced apoptosis and increasing neuronal survival (Figure 1). Additionally, the activation of MC4R in neurons would lead to an increased expression of BDNF through the PKA/CREB pathway in these cells. In this way, α-MSH/MC4R signaling increases BDNF secretion by neurons themselves, which would bind to their own TrkB receptors creating a self-activation loop that will potentiate the described neuroprotection mechanisms.
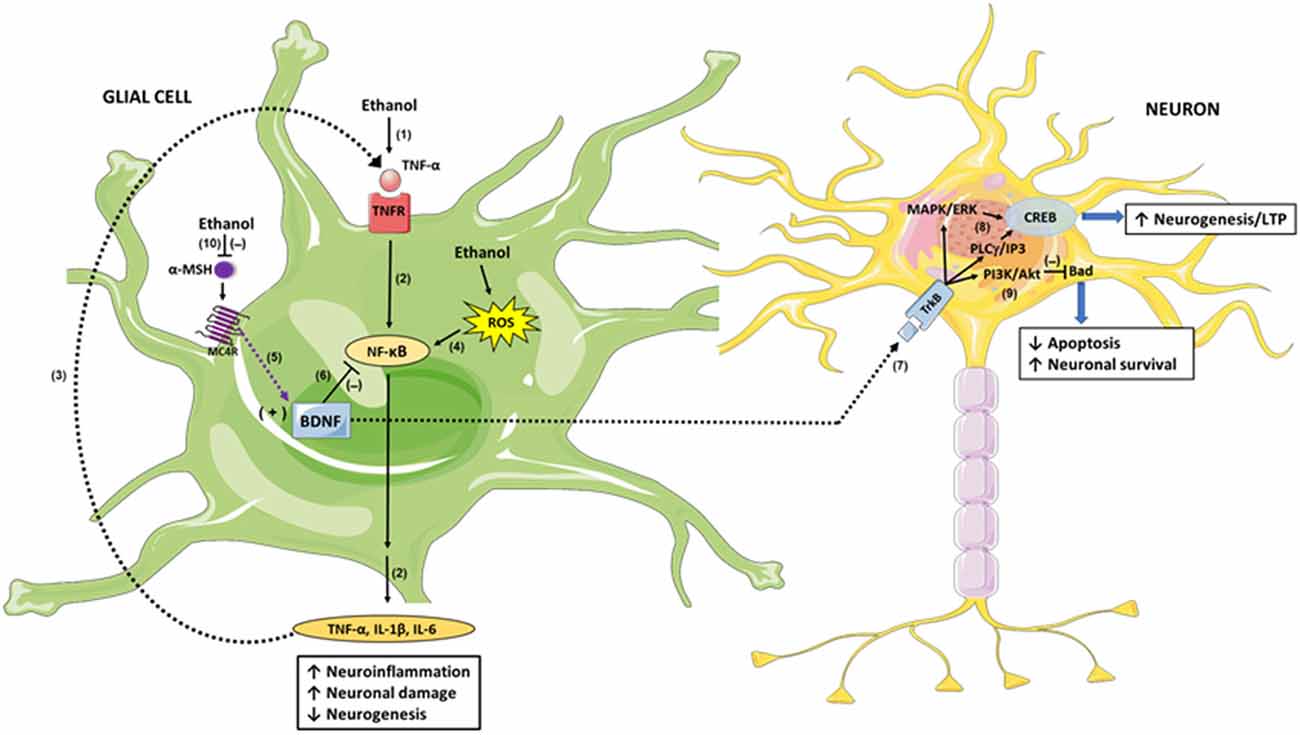
Figure 1. Brain-derived neurotrophic factor (BDNF) mediation in the anti-inflammatory and neuroprotective response of α-melanocyte-stimulating hormone/melanocortin 4 receptor α-MSH/MC4R against damage produced by excessive alcohol consumption. Ethanol intake increases hepatic tumor necrosis factor-α (TNF-α) release into the blood (1); TNF-α is then transported across the blood-brain-barrier to glial cells and signals through Tumor necrosis factor receptor/Nuclear factor-κB (TNFR/NF-κB), inducing the expression of inflammation mediators such as IL-1β, IL-6 and TNF-α itself (2); these proinflammatory cytokines produce neuronal damage and reduce neurogenesis. The glial-secreted TNF-α binds to TNFR, creating an activation loop that potentiates the initial neuroinflammation response (3). Additionally, ethanol-generated reactive oxygen species (ROS) activates NF-κB directly, exacerbating the inflammatory damage (4). α-MSH/MC4R signaling induces BDNF expression (5), and BDNF inhibits NF-κB activity (6) thus inhibiting the neuroinflammatory response. In addition, BDNF secreted by glial cells acts in neurons through its TrkB receptor (7), signaling through MAPK/ERK and PLCγ/IP3 pathways (8) which in turn activate CREB, leading to increased neurogenesis and enhanced long-term potentiation (LTP). Additionally, BDNF/TrkB signaling activates PI3K/Akt pathway which inhibits the pro-apoptotic protein Bad, increasing neuronal survival (9). Ethanol decreases the activity of the central melanocortins system, reducing α-MSH levels and therefore MC4R activity (10); thereby, BDNF expression is downregulated averting its anti-inflammatory and neuroprotective activities.
In summary, based on the background presented, we hypothesize that ethanol exposure during adolescence decreases the expression of α-MSH and hence MC4R signaling in the hippocampus, leading to a lower BDNF activity that causes dramatical changes in the brain (e.g., neuroinflammation, neuronal death and decreased neurogenesis) that predispose to maintain alcohol abuse until adulthood. The activation of MC4R either by α-MSH or by synthetic agonist peptides is able to induce the expression of BDNF, which would trigger several processes that lead to lower both neuroinflammation and alcohol consumption in adulthood.
OF-B and EK wrote the article. AA-C designed the figure.
This work was supported by DIUA149-2019 (Universidad Autónoma de Chile) and ACT1411 (PIA CONICYT).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Baan, R., Straif, K., Grosse, Y., Secretan, B., El Ghissassi, F., Bouvard, V., et al. (2007). Carcinogenicity of alcoholic beverages. Lancet Oncol. 8, 292–293. doi: 10.1016/s1470-2045(07)70099-2
Bariohay, B., Roux, J., Tardivel, C., Trouslard, J., Jean, A., and Lebrun, B. (2009). Brain-derived neurotrophic factor/tropomyosin-related kinase receptor type B signaling is a downstream effector of the brainstem melanocortin system in food intake control. Endocrinology 150, 2646–2653. doi: 10.1210/en.2008-1184
Barson, J. R., Carr, A. J., Soun, J. E., Sobhani, N. C., Rada, P., Leibowitz, S. F., et al. (2010). Opioids in the hypothalamic paraventricular nucleus stimulate ethanol intake. Alcohol. Clin. Exp. Res. 34, 214–222. doi: 10.1111/j.1530-0277.2009.01084.x
Barson, J. R., and Leibowitz, S. F. (2016). Hypothalamic neuropeptide signaling in alcohol addiction. Prog. Neuropsychopharmacol. Biol. Psychiatry 65, 321–329. doi: 10.1016/j.pnpbp.2015.02.006
Bell, R. L., Lopez, M. F., Cui, C., Egli, M., Johnson, K. W., Franklin, K. M., et al. (2015). Ibudilast reduces alcohol drinking in multiple animal models of alcohol dependence. Addict. Biol. 20, 38–42. doi: 10.1111/adb.12106
Benjamins, J. A., Nedelkoska, L., Bealmear, B., and Lisak, R. P. (2013). ACTH protects mature oligodendroglia from excitotoxic and inflammation-related damage in vitro. Glia 61, 1206–1217. doi: 10.1002/glia.22504
Bibel, M., and Barde, Y. A. (2000). Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 14, 2919–2937. doi: 10.1101/gad.841400
Blanco, A. M., and Guerri, C. (2007). Ethanol intake enhances inflammatory mediators in brain: role of glial cells and TLR4/IL-1RI receptors. Front. Biosci. 12, 2616–2630. doi: 10.2741/2259
Blednov, Y. A., Benavidez, J. M., Geil, C., Perra, S., Morikawa, H., and Harris, R. A. (2011). Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain. Behav. Immun. 25, S92–S105. doi: 10.1016/j.bbi.2011.01.008
Cao, Q., Mak, K. M., and Lieber, C. S. (2005). Cytochrome P4502E1 primes macrophages to increase TNF-α production in response to lipopolysaccharide. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G95–G107. doi: 10.1152/ajpgi.00383.2004
Carniglia, L., Durand, D., Caruso, C., and Lasaga, M. (2013). Effect of NDP-α-MSH on PPAR-γ and -β expression and anti-inflammatory cytokine release in rat astrocytes and microglia. PLoS One 8:e57313. doi: 10.1371/journal.pone.0057313
Caruso, C., Carniglia, L., Durand, D., Gonzalez, P. V., Scimonelli, T. N., and Lasaga, M. (2012). Melanocortin 4 receptor activation induces brain-derived neurotrophic factor expression in rat astrocytes through cyclic AMP-protein kinase A pathway. Mol. Cel. Endocrinol. 348, 47–54. doi: 10.1016/j.mce.2011.07.036
Caruso, C., Durand, D., Schiöth, H. B., Rey, R., Seilicovich, A., and Lasaga, M. (2007). Activation of melanocortin 4 receptors reduces the inflammatory response and prevents apoptosis induced by lipopolysaccharide and interferon γ in astrocytes. Endocrinology 148, 4918–4926. doi: 10.1210/en.2007-0366
Caruso, C., Mohn, C., Karara, A. L., Rettori, V., Watanobe, H., Schiöth, H. B., et al. (2004). α-Melanocyte-stimulating hormone through melanocortin-4 receptor inhibits nitric oxide synthase and cyclooxygenase expression in the hypothalamus of male rats. Neuroendocrinology 79, 278–286. doi: 10.1159/000079321
Caruso, C., Sanchez, M., Durand, D., Perez M de, L., Gonzalez, P. V., Lasaga, M., et al. (2010). α-melanocyte-stimulating hormone modulates lipopolysaccharide plus interferon-γ-induced tumor necrosis factor-α expression but not tumor necrosis factor-α receptor expression in cultured hypothalamic neurons. J. Neuroimmunol. 227, 52–59. doi: 10.1016/j.jneuroim.2010.06.013
Caruso, V., Lagerström, M. C., Olszewski, P. K., Fredriksson, R., and Schiöth, H. B. (2014). Synaptic changes induced by melanocortin signalling. Nat. Rev. Neurosci. 15, 98–110. doi: 10.1038/nrn3657
Catania, A. (2008). Neuroprotective actions of melanocortins: a therapeutic opportunity. Trends Neurosci. 31, 353–360. doi: 10.1016/j.tins.2008.04.002
Chandel, N. S., Trzyna, W. C., McClintock, D. S., and Schumacker, P. T. (2000). Role of oxidants in NF-κB activation and TNF-α gene transcription induced by hypoxia and endotoxin. J. Immunol. 165, 1013–1021. doi: 10.4049/jimmunol.165.2.1013
Coller, J. K., and Hutchinson, M. R. (2012). Implications of central immune signaling caused by drugs of abuse: mechanisms, mediators and new therapeutic approaches for prediction and treatment of drug dependence. Pharmacol. Ther. 134, 219–245. doi: 10.1016/j.pharmthera.2012.01.008
Crews, F., He, J., and Hodge, C. (2007). Adolescent cortical development: a critical period of vulnerability for addiction. Pharmacol. Biochem. Behav. 86, 189–199. doi: 10.1016/j.pbb.2006.12.001
Crews, F. T., Mdzinarishvili, A., Kim, D., He, J., and Nixon, K. (2006). Neurogenesis in adolescent brain is potently inhibited by ethanol. Neuroscience 137, 437–445. doi: 10.1016/j.neuroscience.2005.08.090
Crews, F. T., and Nixon, K. (2009). Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol. 44, 115–127. doi: 10.1093/alcalc/agn079
Crews, F. T., Sarkar, D. K., Qin, L., Zou, J., Boyadjieva, N., and Vetreno, R. P. (2015). Neuroimmune function and the consequences of alcohol exposure. Alcohol Res. 37, 331–341.
Crews, F. T., and Vetreno, R. P. (2016). Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology 233, 1543–1557. doi: 10.1007/s00213-015-3906-1
Crews, F. T., Zou, J., and Qin, L. (2011). Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav. Immun. 25, S4–S12. doi: 10.1016/j.bbi.2011.03.003
Davis, M. I. (2008). Ethanol-BDNF interactions: still more questions than answers. Pharmacol. Ther. 118, 36–57. doi: 10.1016/j.pharmthera.2008.01.003
Dawson, D. A., Goldstein, R. B., Chou, S. P., Ruan, W. J., and Grant, B. F. (2008). Age at first drink and the first incidence of adult-onset DSM-IV alcohol use disorders. Alcohol. Clin. Exp. Res. 32, 2149–2160. doi: 10.1111/j.1530-0277.2008.00806.x
Delgado, R., Carlin, A., Airaghi, L., Demitri, M. T., Meda, L., Galimberti, D., et al. (1998). Melanocortin peptides inhibit production of proinflammatory cytokines and nitric oxide by activated microglia. J. Leukoc. Biol. 63, 740–745. doi: 10.1002/jlb.63.6.740
DeWit, D. J., Adlaf, E. M., Offord, D. R., and Ogborne, A. C. (2000). Age at first alcohol use: a risk factor for the development of alcohol disorders. Am. J. Psychiatry 157, 745–750. doi: 10.1176/appi.ajp.157.5.745
Evrard, S. G., Duhalde-Vega, M., Tagliaferro, P., Mirochnic, S., Caltana, L. R., and Brusco, A. (2006). A low chronic ethanol exposure induces morphological changes in the adolescent rat brain that are not fully recovered even after a long abstinence: an immunohistochemical study. Exp. Neurol. 200, 438–459. doi: 10.1016/j.expneurol.2006.03.001
Ezquer, F., Morales, P., Quintanilla, M. E., Santapau, D., Lespay-Rebolledo, C., Ezquer, M., et al. (2018). Intravenous administration of anti-inflammatory mesenchymal stem cell spheroids reduces chronic alcohol intake and abolishes binge-drinking. Sci. Rep. 8:4325. doi: 10.1038/s41598-018-22750-7
Ferrier, L., Bérard, F., Debrauwer, L., Chabo, C., Langella, P., Buéno, L., et al. (2006). Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am. J. Pathol. 168, 1148–1154. doi: 10.2353/ajpath.2006.050617
Flores-Bastías, O., Gómez, G. I., Orellana, J. A., and Karahanian, E. (2019). Activation of melanocortin-4 receptor by a synthetic agonist inhibits ethanol-induced neuroinflammation in rats. Curr. Pharm. Des. 25, 4799–4805. doi: 10.2174/138161282566619121614515
Flores-Bastías, O., and Karahanian, E. (2018). Neuroinflammation produced by heavy alcohol intake is due to loops of interactions between Toll-like 4 and TNF receptors, peroxisome proliferator-activated receptors and the central melanocortin system: a novel hypothesis and new therapeutic avenues. Neuropharmacology 128, 401–407. doi: 10.1016/j.neuropharm.2017.11.003
Forslin Aronsson, A., Spulber, S., Oprica, M., Winblad, B., Post, C., and Schultzberg, M. (2007). α-MSH rescues neurons from excitotoxic cell death. J. Mol. Neurosci. 33, 239–251. doi: 10.1007/s12031-007-0019-2
Franke, H., Kittner, H., Berger, P., Wirkner, K., and Schramek, J. (1997). The reaction of astrocytes and neurons in the hippocampus of adult rats during chronic ethanol treatment and correlations to behavioral impairments. Alcohol 14, 445–454. doi: 10.1016/s0741-8329(96)00209-1
Galimberti, D., Baron, P., Meda, L., Prat, E., Scarpini, E., Delgado, R., et al. (1999). α-MSH peptides inhibit production of nitric oxide and tumor necrosis factor-α by microglial cells activated with β amyloid and interferon γ. Biochem. Biophys. Res. Commun. 263, 251–256. doi: 10.1006/bbrc.1999.1276
Giuliani, D., Mioni, C., Altavilla, D., Leone, S., Bazzani, C., Minutoli, L., et al. (2006). Both early and delayed treatment with melanocortin 4 receptor stimulating melanocortins produces neuroprotection in cerebral ischemia. Endocrinology 147, 1126–1135. doi: 10.1210/en.2005-0692
Hadley, M. E., and Haskell-Luevano, C. (1999). The proopiomelanocortin system. Ann. N Y Acad. Sci. 885, 1–21. doi: 10.1111/j.1749-6632.1999.tb08662.x
He, J., and Crews, F. T. (2008). Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp. Neurol. 210, 349–358. doi: 10.1016/j.expneurol.2007.11.017
Heberlein, A., Muschler, M., Wilhelm, J., Frieling, H., Lenz, B., Gröschl, M., et al. (2010). BDNF and GDNF serum levels in alcohol-dependent patients during withdrawal. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 1060–1064. doi: 10.1016/j.pnpbp.2010.05.025
Hensler, J. G., Ladenheim, E. E., and Lyons, W. E. (2003). Ethanol consumption and serotonin-1A (5-HT1A) receptor function in heterozygous BDNF (+/−) mice. J. Neurochem. 85, 1139–1147. doi: 10.1046/j.1471-4159.2003.01748.x
Herrera, D. G., Yague, A. G., Johnsen-Soriano, S., Bosch-Morell, F., Collado-Morente, L., Muriach, M., et al. (2003). Selective impairment of hippocampal neurogenesis by chronic alcoholism: protective effects of an antioxidant. Proc. Natl. Acad. Sci. U S A 100, 7919–7924. doi: 10.1073/pnas.1230907100
Ichiyama, T., Sakai, T., Catania, A., Barsh, G. S., Furukawa, S., and Lipton, J. M. (1999a). Systemically administered α-melanocyte-stimulating peptides inhibit NF-κB activation in experimental brain inflammation. Brain Res. 836, 31–37. doi: 10.1016/s0006-8993(99)01584-x
Ichiyama, T., Zhao, H., Catania, A., Furukawa, S., and Lipton, J. M. (1999b). α-melanocyte-stimulating hormone inhibits NF-κB activation and IκBα degradation in human glioma cells and in experimental brain inflammation. Exp. Neurol. 157, 359–365. doi: 10.1006/exnr.1999.7064
Israel, Y., Karahanian, E., Ezquer, F., Morales, P., Ezquer, M., Rivera-Meza, M., et al. (2017). Acquisition, maintenance and relapse-like alcohol drinking: lessons from the UChB rat line. Front. Behav. Neurosci. 11:57. doi: 10.3389/fnbeh.2017.00057
Jeanblanc, J., He, D. Y., Carnicella, S., Kharazia, V., Janak, P. H., and Ron, D. (2009). Endogenous BDNF in the dorsolateral striatum gates alcohol drinking. J. Neurosci. 29, 13494–13502. doi: 10.1523/JNEUROSCI.2243-09.2009
Jiang, Y., Wei, N., Lu, T., Zhu, J., Xu, G., and Liu, X. (2011). Intranasal brain-derived neurotrophic factor protects brain from ischemic insult via modulating local inflammation in rats. Neuroscience 172, 398–405. doi: 10.1016/j.neuroscience.2010.10.054
Kushner, M. G., Abrams, K., and Borchardt, C. (2000). The relationship between anxiety disorders and alcohol use disorders: a review of major perspectives and findings. Clin. Psychol. Rev. 20, 149–171. doi: 10.1016/s0272-7358(99)00027-6
Lankhorst, A. J., Duis, S. E., ter Laak, M. P., Joosten, E. A., Hamers, F. P., and Gispen, W. H. (1999). Functional recovery after central infusion of α-melanocyte-stimulating hormone in rats with spinal cord contusion injury. J. Neurotrauma 16, 323–331. doi: 10.1089/neu.1999.16.323
Lerma-Cabrera, J. M., Carvajal, F., Alcaraz-Iborra, M., de la Fuente, L., Navarro, M., Thiele, T. E., et al. (2013). Adolescent binge-like ethanol exposure reduces basal α-MSH expression in the hypothalamus and the amygdala of adult rats. Pharmacol. Biochem. Behav. 110, 66–74. doi: 10.1016/j.pbb.2013.06.006
Lerma-Cabrera, J. M., Carvajal, F., de la Torre, L., de la Fuente, L., Navarro, M., Thiele, T. E., et al. (2012). Control of food intake by MC4-R signaling in the lateral hypothalamus, nucleus accumbens shell and ventral tegmental area: interactions with ethanol. Behav. Brain Res. 234, 51–60. doi: 10.1016/j.bbr.2012.06.006
Lieber, C. S. (1999). Microsomal ethanol-oxidizing system (MEOS): the first 30 years (1968–1998)—a review. Alcohol. Clin. Exp. Res. 23, 991–1007. doi: 10.1097/00000374-199906000-00006
Lim, S. S., Vos, T., Flaxman, A. D., Danaei, G., Shibuya, K., Adair-Rohani, H., et al. (2012). A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2224–2260. doi: 10.1016/S0140-6736(12)61766-8
Logrip, M. L., Janak, P. H., and Ron, D. (2009). Escalating ethanol intake is associated with altered corticostriatal BDNF expression. J. Neurochem. 109, 1459–1468. doi: 10.1111/j.1471-4159.2009.06073.x
Macaluso, A., McCoy, D., Ceriani, G., Watanabe, T., Biltz, J., Catania, A., et al. (1994). Anti-inflammatory influences of α-MSH molecules: central neurogenic and peripheral actions. J. Neurosci. 14, 2377–2382. doi: 10.1523/JNEUROSCI.14-04-02377.1994
Markwiese, B. J., Acheson, S. K., Levin, E. D., Wilson, W. A., and Swartzwelder, H. S. (1998). Differential effects of ethanol on memory in adolescent and adult rats. Alcohol. Clin. Exp. Res. 22, 416–421. doi: 10.1111/j.1530-0277.1998.tb03668.x
McGough, N. N., He, D. Y., Logrip, M. L., Jeanblanc, J., Phamluong, K., Luong, K., et al. (2004). RACK1 and brain-derived neurotrophic factor: a homeostatic pathway that regulates alcohol addiction. J. Neurosci. 24, 10542–10552. doi: 10.1523/JNEUROSCI.3714-04.2004
Melendez, R. I., McGinty, J. F., Kalivas, P. W., and Becker, H. C. (2012). Brain region-specific gene expression changes after chronic intermittent ethanol exposure and early withdrawal in C57BL/6J mice. Addict. Biol. 17, 351–364. doi: 10.1111/j.1369-1600.2011.00357.x
Mountjoy, K. G. (2010). Distribution and function of melanocortin receptors within the brain. Adv. Exp. Med. Biol. 681, 29–48. doi: 10.1007/978-1-4419-6354-3_3
Muceniece, R., Zvejniece, L., Kirjanova, O., Liepinsh, E., Krigere, L., Vilskersts, R., et al. (2005). β-MSH inhibits brain inflammation via MC3/4 receptors and impaired NF-κB signaling. J. Neuroimmunol. 169, 13–19. doi: 10.1016/j.jneuroim.2005.07.024
Muceniece, R., Zvejniece, L., Liepinsh, E., Kirjanova, O., Baumane, L., Petrovska, R., et al. (2006). The MC3 receptor binding affinity of melanocortins correlates with the nitric oxide production inhibition in mice brain inflammation model. Peptides 27, 1443–1450. doi: 10.1016/j.peptides.2005.12.002
Navarro, M., Cubero, I., Knapp, D. J., Breese, G. R., and Thiele, T. E. (2008). Decreased immunoreactivity of the melanocortin neuropeptide α-melanocyte- stimulating hormone (α-MSH) after chronic ethanol exposure in Sprague-Dawley rats. Alcohol. Clin. Exp. Res. 32, 266–276. doi: 10.1111/j.1530-0277.2007.00578.x
Navarro, M., Cubero, I., Knapp, D. J., and Thiele, T. E. (2003). MTII-induced reduction of voluntary ethanol drinking is blocked by pretreatment with AgRP-(83–132). Neuropeptides 37, 338–344. doi: 10.1016/j.npep.2003.10.003
Navarro, M., Lerma-Cabrera, J. M., Carvajal, F., Lowery, E. G., Cubero, I., and Thiele, T. E. (2011). Assessment of voluntary ethanol consumption and the effects of a melanocortin (MC) receptor agonist on ethanol intake in mutant C57BL/6J mice lacking the MC-4 receptor. Alcohol. Clin. Exp. Res. 35, 1058–1066. doi: 10.1111/j.1530-0277.2011.01438.x
Nicholson, J. R., Peter, J. C., Lecourt, A. C., Barde, Y. A., and Hofbauer, K. G. (2007). Melanocortin-4 receptor activation stimulates hypothalamic brain-derived neurotrophic factor release to regulate food intake, body temperature and cardiovascular function. J. Neuroendocrinol. 19, 974–982. doi: 10.1111/j.1365-2826.2007.01610.x
Nutt, D. J., King, L. A., and Phillips, L. D. (2010). Drug harms in the UK: a multicriteria decision analysis. Lancet 376, 1558–1565. doi: 10.1016/s0140-6736(10)61462-6
Olney, J. J., Navarro, M., and Thiele, T. E. (2014). Targeting central melanocortin receptors: a promising novel approach for treating alcohol abuse disorders. Front. Neurosci. 8:128. doi: 10.3389/fnins.2014.00128
Orellana, J. A., Cerpa, W., Carvajal, M. F., Lerma-Cabrera, J. M., Karahanian, E., Osorio-Fuentealba, C., et al. (2017). New implications for the melanocortin system in alcohol drinking behavior in adolescents: the glial dysfunction hypothesis. Front. Cell. Neurosci. 11:90. doi: 10.3389/fncel.2017.00090
Pandey, S. C., Zhang, H., Roy, A., and Misra, K. (2006). Central and medial amygdaloid brain-derived neurotrophic factor signaling plays a critical role in alcohol-drinking and anxiety-like behaviors. J. Neurosci. 26, 8320–8331. doi: 10.1523/JNEUROSCI.4988-05.2006
Peterson, J. B., Rothfleisch, J., Zelazo, P. D., and Pihl, R. O. (1990). Acute alcohol intoxication and cognitive functioning. J. Stud. Alcohol 51, 114–122. doi: 10.15288/jsa.1990.51.114
Ploj, K., Roman, E., Kask, A., Hyytiä, P., Schiöth, H. B., Wikberg, J. E., et al. (2002). Effects of melanocortin receptor ligands on ethanol intake and opioid peptide levels in alcohol-preferring AA rats. Brain Res. Bull. 59, 97–104. doi: 10.1016/s0361-9230(02)00844-4
Qin, L., He, J., Hanes, R. N., Pluzarev, O., Hong, J. S., and Crews, F. T. (2008). Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J. Neuroinflammation 5:10. doi: 10.1186/1742-2094-5-10
Qin, L., Wu, X., Block, M. L., Liu, Y., Breese, G. R., Hong, J. S., et al. (2007). Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55, 453–462. doi: 10.1002/glia.20467
Rada, P., Avena, N. M., Leibowitz, S. F., and Hoebel, B. G. (2004). Ethanol intake is increased by injection of galanin in the paraventricular nucleus and reduced by a galanin antagonist. Alcohol 33, 91–97. doi: 10.1016/s0741-8329(04)00097-7
Rainero, I., Degennaro, T., Visentin, G., Brunetti, E., Cerrato, P., Torre, E., et al. (1990). Effects of chronic ethanol treatment on α-msh concentrations in rat-brain and pituitary. Neuropeptides 15, 139–141. doi: 10.1016/0143-4179(90)90145-o
Rajora, N., Boccoli, G., Burns, D., Sharma, S., Catania, A. P., and Lipton, J. M. (1997). α-MSH modulates local and circulating tumor necrosis factor in experimental brain inflammation. J. Neurosci. 17, 2181–2186. doi: 10.1523/JNEUROSCI.17-06-02181.1997
Rose, M. E., and Grant, J. E. (2010). Alcohol-induced blackout. Phenomenology, biological basis, and gender differences. J. Addict. Med. 4, 61–73. doi: 10.1097/ADM.0b013e3181e1299d
Sarkar, S., Légrádi, G., and Lechan, R. M. (2002). Intracerebroventricular administration of α-melanocyte stimulating hormone increases phosphorylation of CREB in TRH- and CRH-producing neurons of the hypothalamic paraventricular nucleus. Brain Res. 945, 50–59. doi: 10.1016/s0006-8993(02)02619-7
Schinder, A. F., and Poo, M. (2000). The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 23, 639–645. doi: 10.1016/s0166-2236(00)01672-6
Schneider, E. R., Rada, P., Darby, R. D., Leibowitz, S. F., and Hoebel, B. G. (2007). Orexigenic peptides and alcohol intake: differential effects of orexin, galanin, and ghrelin. Alcohol. Clin. Exp. Res. 31, 1858–1865. doi: 10.1111/j.1530-0277.2007.00510.x
Selkirk, J. V., Nottebaum, L. M., Lee, J., Yang, W., Foster, A. C., and Lechner, S. M. (2007). Identification of differential melanocortin 4 receptor agonist profiles at natively expressed receptors in rat cortical astrocytes and recombinantly expressed receptors in human embryonic kidney cells. Neuropharmacology 52, 459–466. doi: 10.1016/j.neuropharm.2006.08.015
Shelkar, G. P., Kale, A. D., Singh, U., Singru, P. S., Subhedar, N. K., and Kokare, D. M. (2015). α-melanocyte stimulating hormone modulates ethanol self-administration in posterior ventral tegmental area through melanocortin-4 receptors. Addict. Biol. 20, 302–315. doi: 10.1111/adb.12126
Shield, K. D., Parry, C., and Rehm, J. (2013). Chronic diseases and conditions related to alcohol use. Alcohol Res. 35, 155–173.
Spaccapelo, L., Bitto, A., Galantucci, M., Ottani, A., Irrera, N., Minutoli, L., et al. (2011). Melanocortin MC4 receptor agonists counteract late inflammatory and apoptotic responses and improve neuronal functionality after cerebral ischemia. Eur. J. Pharmacol. 670, 479–486. doi: 10.1016/j.ejphar.2011.09.015
Sprow, G. M., Rinker, J. A., Lowery-Gointa, E. G., Sparrow, A. M., Navarro, M., and Thiele, T. E. (2016). Lateral hypothalamic melanocortin receptor signaling modulates binge-like ethanol drinking in C57BL/6J mice. Addict. Biol. 21, 835–846. doi: 10.1111/adb.12264
Sutton, G. M., Duos, B., Patterson, L. M., and Berthoud, H. R. (2005). Melanocortinergic modulation of cholecystokinin-induced suppression of feeding through extracellular signal-regulated kinase signaling in rat solitary nucleus. Endocrinology 146, 3739–3747. doi: 10.1210/en.2005-0562
Tao, X., Finkbeiner, S., Arnold, D. B., Shaywitz, A. J., and Greenberg, M. E. (1998). Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20, 709–726. doi: 10.1016/s0896-6273(00)81010-7
Thorsell, A., Slawecki, C. J., and Ehlers, C. L. (2005). Effects of neuropeptide Y and corticotropin-releasing factor on ethanol intake inWistar rats: interaction with chronic ethanol exposure. Behav. Brain Res. 161, 133–140. doi: 10.1016/j.bbr.2005.01.016
Uhl, G. R., Liu, Q. R., Walther, D., Hess, J., and Naiman, D. (2001). Polysubstance abuse-vulnerability genes: genome scans for association, using 1,004 subjects and 1,494 single-nucleotide polymorphisms. Am. J. Hum. Genet. 69, 1290–1300. doi: 10.1086/324467
van de Meent, H., Hamers, F. P., Lankhorst, A. J., Joosten, E. A., and Gispen, W. H. (1997). Beneficial effects of the melanocortin α-melanocyte-stimulating hormone on clinical and neurophysiological recovery after experimental spinal cord injury. Neurosurgery 40, 122–130. doi: 10.1097/00006123-199701000-00028
Ward, R. J., Colivicchi, M. A., Allen, R., Schol, F., Lallemand, F., de Witte, P., et al. (2009). Neuro-inflammation induced in the hippocampus of ‘binge drinking’ rats may be mediated by elevated extracellular glutamate content. J. Neurochem. 111, 1119–1128. doi: 10.1111/j.1471-4159.2009.06389.x
White, A. M., and Swartzwelder, H. S. (2004). Hippocampal function during adolescence: a unique target of ethanol effects. Ann. N Y Acad. Sci. 1021, 206–220. doi: 10.1196/annals.1308.026
Whiteford, H. A., Degenhardt, L., Rehm, J., Baxter, A. J., Ferrari, A. J., Erskine, H. E., et al. (2013). Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet 382, 1575–1586. doi: 10.1016/S0140-6736(13)61611-6
Wong, K. Y., Rajora, N., Boccoli, G., Catania, A., and Lipton, J. M. (1997). A potential mechanism of local anti-inflammatory action of α-melanocyte-stimulating hormone within the brain: modulation of tumor necrosis factor-α production by human astrocytic cells. Neuroimmunomodulation 4, 37–41. doi: 10.1159/000097313
World Health Organization. (2014). Global Status Report on Alcohol and Health 2014. Available online at: www.who.int/substance_abuse/publications/global_alcohol_report/en/. Accessed October 17, 2019.
Xu, B., Goulding, E. H., Zang, K., Cepoi, D., Cone, R. D., Jones, K. R., et al. (2003). Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci. 6, 736–742. doi: 10.1038/nn1073
Xu, D., Lian, D., Wu, J., Liu, Y., Zhu, M., Sun, J., et al. (2017). Brain-derived neurotrophic factor reduces inflammation and hippocampal apoptosis in experimental Streptococcus pneumoniae meningitis. J. Neuroinflammation 14:156. doi: 10.1186/s12974-017-0930-6
Keywords: melanocyte-stimulating hormone, MC4R, brain-derived neurotrophic factor, alcohol use disorder, alcoholism, α-MSH
Citation: Flores-Bastías O, Adriasola-Carrasco A and Karahanian E (2020) Activation of Melanocortin-4 Receptor Inhibits Both Neuroinflammation Induced by Early Exposure to Ethanol and Subsequent Voluntary Alcohol Intake in Adulthood in Animal Models: Is BDNF the Key Mediator? Front. Cell. Neurosci. 14:5. doi: 10.3389/fncel.2020.00005
Received: 28 November 2019; Accepted: 10 January 2020;
Published: 28 January 2020.
Edited by:
Ricardo Marcos Pautassi, National Council for Scientific and Technical Research (CONICET), ArgentinaReviewed by:
Mercedes Lasaga, University of Buenos Aires, ArgentinaCopyright © 2020 Flores-Bastías, Adriasola-Carrasco and Karahanian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eduardo Karahanian, ZWR1YXJkby5rYXJhaGFuaWFuQHVhdXRvbm9tYS5jbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.